
Navigating phase behaviour in pharmaceuticals to enable phases with desired properties
Ivo B.
Rietveld
 *,
Cheng-long
Stephan
and
Gabin
Gbabode
*,
Cheng-long
Stephan
and
Gabin
Gbabode
SMS Laboratory (UR 3233), University of Rouen Normandy, Normandy University, F-76000 Rouen, France. E-mail: ivo.rietveld@univ-rouen.fr
First published on 11th June 2025
Abstract
Many active pharmaceutical ingredients (APIs) exhibit crystalline polymorphism and only one of those polymorphs is the most stable one. Moreover, the solubility of recently developed APIs is often limited, leading to formulations containing metastable polymorphs, amorphous material or stabilised supersaturated solutions. Before marketing such formulations, it must be ensured that they persist up to their expiration date, on average about three years. Despite considerable progress in crystal structure prediction (CSP), it remains difficult to foresee which of the predicted crystalline forms will be found experimentally. In part, this is due to difficulties in predicting the crystallisation kinetics of the different polymorphs and therefore the ability to assess crystallisation kinetics needs to be improved. Each molecule remains to be tested experimentally and if necessary unary and binary phase diagrams need to be constructed for a complete picture of their phase behaviour, which will provide a basis for formulation design and risk assessment in case a metastable state is chosen for the formulation. The COST action BEST-CSP is contributing to calibrate stability calculations in CSP by preparing a benchmark of experimental physical data on the organic solid state. Hopefully, this will improve the calculation of the Gibbs free energy of the different polymorphs and therefore predictions on the phase behaviour of an API. Still, for now, each molecule remains a separate case with its particularities, which requires experimental study of its thermodynamic and kinetic behaviour before the stability assessment of its solid state can be completed.
 Ivo B. Rietveld | Ivo Rietveld obtained his PhD in 2000 from the University of Leiden. He worked on electrospray, photoluminescent dendrimers and polymer thin films in the USA and Japan. In 2007, he started working on the thermodynamic stability of APIs in France. He became full professor in September 2024 in the SMS laboratory of the University of Rouen Normandy. His research subjects cover thermodynamic stability, resolution of enantiomer mixtures, crystallisation, and the stabilisation of metastable forms. Since September 2023, he has been chair of a European network to establish a benchmark on physical properties of organic molecular materials. |
 Cheng-long Stephan | With a background of two years in the pharmaceutical industry in solid oral dosage form development, including solid-state characterization and formulation of poorly soluble APIs, Cheng-long Stephan, PharmD, is currently pursuing a PhD at the SMS laboratory. His research focuses on the polymorphism of active pharmaceutical ingredients. He aims to achieve tailor-made control of polymorphic forms using additives while combining experimental and analytical approaches to better understand and guide the design of drug substances with optimized performance and stability. |
 Gabin Gbabode | Dr Gabin Gbabode obtained his PhD degree in Physics from the University of Bordeaux, France under the supervision of Dr Denise Mondieig and Dr Philippe Négrier in 2005. After 2 post-doctoral positions, including one in the group of Yves Geerts at ULB (Brussels, Belgium) from 2007 to 2012, he was hired as an associate professor at Rouen Normandie University in September 2012. His current research focuses on the study of polymorphism of organic compounds (in particular active pharmaceutical ingredients) under various conditions (temperature, pressure,…) and particularly on polymorph selection in confined geometry (thin films, nano-porous particles). |
1 Navigating the complexities of phase behaviour in drug development
1.1 Pharmaceutical solids
Many molecules interfere with the human body inducing pharmacological, immunological, or metabolic effects.1 These molecules are known as active pharmaceutical ingredients (APIs). APIs may crystallise in different crystal structures with different solid state properties, affecting the behaviour of the drug.2 Crystals are the predominant form in pharmaceutical solids for purity reasons. Their structure can be represented by a single unit cell in which molecules are bound by physical interactions and its replication in three dimensions gives rise to the crystal.3 An API may exhibit different packing arrangements in the crystalline state: this phenomenon is known as polymorphism and it may have an impact on the solid state properties of the API, which is reflected by the different crystal habits of polymorphs (see Fig. 1).2 When ritonavir, an antiretroviral prescribed against HIV infection, was launched on the market, polymorphic form I was the only one known for this API. However, two years later, a more stable polymorph (form II) was found in the drug formulation. Form II was described as having a lower aqueous solubility, considerably affecting bioavailability and thus the drug's efficiency.4 | ||
| Fig. 1 Typical crystal habits of 7αMNa. (a) The stable polymorph I in acetone and (b) the metastable polymorph II in hexane. Reproduced from ref. 5 with permission from Elsevier, copyright 2006. | ||
Some crystalline APIs include solvent molecules in their crystal lattice, so-called hydrates or solvates depending on the nature of the solvent molecule. Because of regulatory requirements, the use of solvent is restricted as well as residual solvent in the final product.6 Thus, if solvates are used, it will mainly be a hydrate. Prednisolone is an example of an API exhibiting two polymorphs (forms I and II) and a sesquihydrate. In a study of hydration–dehydration of prednisolone,7 a new crystalline phase was observed, which was the isomorphic anhydrous form of the hydrate. If instead of a solvent another molecule, called coformer, is part of the crystal structure with the API, the system is called a cocrystal. Although the naming is quite arbitrary, as a cocrystal implies that the pure coformer is a crystalline solid (instead of a liquid for a solvate), cocrystals tend to be more stable than solvates and hydrates. Cocrystals too, like any other crystalline combinations, exhibit the potential to form polymorphs.
The pharmaceutical industry has recently taken an interest in amorphous solids since they improve the solubilization of poorly soluble drugs.8,9 However, because of high Gibbs free energies, amorphous drugs are thermodynamically unstable, potentially leading to reorganisation into a more stable crystalline form. Spray-drying or hot-melt extrusion in the combination of polymer excipients facilitates the manufacturing of so-called amorphous solid dispersions (ASD).8 These are metastable but highly persistent amorphous materials and thus become suitable for pharmaceutical use.
1.2 Crystal structure and drug properties
New drug substances subject to polymorphism must follow the guideline Q6A from the International Conference on Harmonization.10 It states “differences in these forms could, in some cases, affect the quality or performance of the new drug products. In cases where differences exist which have been shown to affect drug product performance, bioavailability or stability, then the appropriate solid state should be specified”. Polymorphs possess the same chemical composition but have different physicochemical properties, with polymorphism affecting, among other properties, the melting point, solubility, stability, hygroscopicity and chemical reactivity.3Polymorphism impacts the melting temperature of the API, as the molecular arrangement in the unit cell differs between polymorphs, affecting intermolecular interactions and the parts of the molecule exposed at the crystal faces. The melting point can be determined using differential scanning calorimetry (DSC) as melting of a crystalline state unmistakably results in a large endothermic peak. Pyrazinamide, an antibiotic primarily used in the treatment of tuberculosis, is known for its polymorphism.11 Melting points of two out of four known polymorphs have been obtained at 457 K for form I (α form) and 462 K for form IV (γ form).12
As polymorphism may impact drug stability, investigations are required by health authorities in case of market authorization demand. Stability directly impacts safety, efficacy, and shelf life. Drug instability produces degradation impurities and a decrease of drug content, involving patients' lives. Unstable drugs may require strict storage conditions or shorter shelf lives, complicating supply and use. Focusing on drug product stability, Corrêa et al. investigated the polymorphic stability of darunavir and its formulation.13 As this antiretroviral is administered all over the world, it encounters many different environmental conditions. The authors found that the crystal lattice alters under stress (55 °C, up to 90 days) without specifying the polymorphic changes of the API. Hygroscopicity is also a common stability issue for polymorphic pro-drugs such as theophylline,14 causing dissolution or hydrates when it is not intended.
APIs must dissolve in human fluids (mainly water-based), cross the intestinal barrier, and follow the blood circulation to reach their target (enzyme, protein, etc.). So, to be effective, interactions between the API and water need to be identified and understood. Solubility is a key parameter for API candidates as it determines their bioavailability (fraction of the dose that reaches the systemic blood circulation). The biopharmaceutical classification system (BCS)15 offers a classification into four classes for APIs based on solubility and permeability. It is a predictive tool to determine the resorption behaviour of a drug and adapt the formulation strategy for enhancing solubility if needed. Drugs in class I offer high solubility and high permeability, while class II exhibits low solubility and high permeability, class III high solubility and low permeability, and class IV possesses low solubility and low permeability (most complex class). Considering that crystalline structure competes with dissolved state, and thus controls solvation and dissolution of a drug,2 the thermodynamically stable polymorph presents the lowest solubility.2 Chloramphenicol palmitate (bacteriostatic pro-drug) exhibits three polymorphic forms: stable form A, metastable form B and unstable form C. Form A has been described as having lower solubility than form B, thus the latter dissolves faster and possesses better intestinal absorption and bioavailability than form A.16
1.3 Solid form in pharmaceutical processes
In the pharmaceutical industry, all processes should be precisely controlled, as any variation in the physical state may impact drug manufacturing, increasing the risk of deviation from the production criteria and subsequent batch rejection. Solid dosage forms (i.e. tablet, capsule, implants) rely on a solid active ingredient, excipients, and established manufacturing processes. During the many manufacturing steps, the API would need to align with the necessary physicochemical properties to ensure manufacturability and proper drug product quality for pharmaceutical use. Excipients are generally designed for specific purposes, and their physicochemical properties are well characterized. If these properties do not meet the formulator's requirements, alternative manufacturers can be considered, an option that is often not available for the active ingredient. Polymorphism affects the crystal habit and therefore the manufacturing process. Hence, control over the crystal morphology is essential because it impacts density, flowability, blending and therefore the final drug product quality.A modification of the physical state of the API due to processing is called process-induced phase transformation.17 Each processing stage (i.e. mixing, granulation, drying, tabletting) could be a source of mechanical and/or thermal stress for the powder and thus the crystalline drug. The following paragraphs are ordered according to a typical tablet manufacturing process summarised in Fig. 2.
 | ||
| Fig. 2 Flow chart for the wet granulation tabletting process. Green hexagons: API, grey squares: excipients, black rectangles: lubricant, thermometers: thermal stress, gears: mechanical stress, drop: solvent. | ||
It starts with particle morphology (size, shape, density). Homogenization of particles limits segregation, leading to optimal filling of all equipment (i.e. hopper and compression chamber). Rossman et al. obtained flat crystals, while manipulating the size, the morphology and the polymorphism of acetaminophen (paracetamol) using supercritical antisolvent precipitation.18 However, angular or needle-like shapes were obtained when crystalized from ethanol, planar cuboidal shapes from acetone and cuboidal/orthorhombic shapes from a mixture of ethanol and acetone. It demonstrates how crystallisation processes can change the particle shape. At the industrial scale, needle-like crystals are difficult to homogenize with excipients. Nevertheless, due to a larger surface to bulk ratio, blended powder could be more stable as elongated particles act as a particle-immobilising matrix. Particle size reduction processes involve the use of a grinder or a ball mill and may induce mechanical and thermal stresses. While increasing temperature may induce phase conversion,2 heat production coupled with vibrational and mechanical energy offers perfect conditions to amorphization.17 This highlights the need to work under conditions that do not affect the phase of the API.
Granulation ensures the correct flowability of the powder and the homogeneous distribution of the API thanks to particle rounding and densification. High shear granulators or spray-dryers are commonly used with solvents such as water and sometimes ethanol for the manufacturing of granulated powder. Here again, the API is subjected to high energy (heat, shear) and solvent. As polymorphs have different aqueous solubilities depending on their crystalline organisation, wetting and therefore granulation efficiency are directly impacted by the solubility of the polymorph. APIs, such as indomethacin, nimodipine, and carbamazepine, convert into a more stable polymorph with lower solubility during wetting in granulation.19 Anhydrous forms can also convert into hydrates, and hydrates into di-, tri- or higher hydrate forms.2
Tabletting is another processing step affected by and affecting the phase behaviour of the API. Granulated powder is compressed into tablets using high mechanical stress and recrystallization on decompression is one of the mechanisms of tablet consolidation.17 Compaction can disrupt the crystal structure, creating dislocations and nucleation sites for a more stable phase within the initial solid phase.2 Depending on the crystal lattice, response to compaction differs. This is well-illustrated with paracetamol (acetaminophen) in which the monoclinic form I exhibits W-pleated sheets whereas form II possesses a planar sheet-like organization. The latter is suitable for tableting as it easily undergoes plastic deformation, essential in direct compression20 while the W-sheet arrangement leads to elastic deformation: unsuitable for direct compression. However, form I is the thermodynamically stable polymorph, and the reason why despite its rather unfavourable compressibility properties, it is the commercially used form and its tabletting properties were investigated.21 Likewise, for carbamazepine, Mohapatra et al. explored the mechanical properties of the monoclinic form III using Brillouin scattering.22 They determined that these properties are controlled by “nondirected dispersive type interactions similar to aromatic systems with delocalized π bonds”. Gabriele et al. determined the anisotropic properties using nanoindentation measurements.23 The butterflylike shape of the carbamazepine molecule and its crystal packing offer a higher degree of molecular flexibility. Simulating the deformation of the crystal structure under compression, they found that elastic deformation was dominant over the plastic one. In the case of indomethacin, Khomane et al. investigated the compaction behaviour of two polymorphs using a tableting press.24 The α-form was described as having compaction capability whereas the γ-form shows better compressible performance and lower porosity. Higher tensile strength was measured for the α-form. Young et al. reported similar results using Brillouin light scattering.25 They described a higher elastic anisotropy (meaning stronger intermolecular interactions) for the α form. Using a rotary tablet press, they highlighted the plastic behaviour of both forms. However, they found that the γ form is more compressible and the α form has better compactibility properties, confirming the results by Khomane et al.24
As we demonstrate above, drug manufacturing involves different solid-state properties and risks related to API polymorphism and other solid forms. Pharmaceutical processes often involve mechanical and thermal stress, potentially leading to phase conversion. As each polymorph possesses its own physical properties, it is necessary to understand the phase behaviour of a drug molecule, so that intended properties can be guaranteed and unintended phase transitions can be avoided.
2 Phase behaviour of drug molecules
2.1 Stability
Solid form screening is routinely carried out at the early stage of drug development.2 On the one hand, it involves in silico crystal structure prediction and on the other hand, physical experiments to obtain all the relevant forms found in the prediction. There is still a disconnect between the two methods and predicted forms are not all found in experiments, however, the experimental forms are mostly present in the predicted forms, although not always among the more obvious polymorphs with lower free energies and their relevance is therefore not always evident. After the computational and experimental solid form screening, the most pertinent polymorphs and solvates of an API, and possibly some cocrystals, will have been discovered. The ideal solid form possesses a high solubility and a high stability, although these two properties are thermodynamically mutually exclusive for compounds with an inherently low solubility. There are various types of pharmaceutical formulations, involving either the crystalline state, the amorphous state, or the liquid state; however, even in the liquid state, the solution is formulated against the most stable crystalline state, also if this form is unknown as was the case for ritonavir4 and rotigotine.26It may be important to define “stability”, as both thermodynamic and kinetic stability play a role and are often used interchangeably in the literature. If a solid form is said to be the most stable, then often thermodynamic stability is meant or in other words, its Gibbs free energy is the lowest among the known solid forms under the given conditions. Because new forms may be discovered with even lower energy, the stable form may end up being a metastable form, as what happened with ritonavir and rotigotine.4,26–28 In a similar way, a hydrate may be the most stable form in an aqueous solution, because the Gibbs free energy happens to be minimal for the hydrate in the presence of water. The meaning of thermodynamic stability is clearcut in terms of Gibbs free energy, although it may be difficult to determine the Gibbs energies for the solid forms involved. For patenting and formulation, it is important to ensure that the solid-state landscape is sufficiently mapped, so that valid choices can be made, although from a scientific point of view any new form being discovered even a hundred years from now is welcome new information.
Harder to put a finger on is “kinetic stability”, which depends on a high activation energy. It implies that if the system finds a way around the activation energy, it may relax unimpededly into a lower energetic state. An example is when the presence of humidity, which increases the overall mobility of the molecules, may cause an amorphous sample to crystallise. Thus, although kinetic stability implies high energy barriers, it does not imply inherent thermodynamic stability and therefore the authors prefer the word persistence instead of stability when speaking of kinetic stability, while the word stability will be reserved for thermodynamic stability (i.e. with the lowest Gibbs free energy among the different solid forms in the system to the best of our knowledge…).
2.2 Charting and controlling phase behaviour
Once an understanding of the available polymorphs, solvates and cocrystals is obtained for a given API, their structural and Gibbs-energy landscape can be investigated leading to a phase diagram. Ideally, one phase diagram will describe the entire phase behaviour of an API, but for readability, most phase diagrams consist of two parameters such as pressure and temperature, temperature and composition, or composition and composition (for ternary phase diagrams at a single temperature, for example). Although these two- or three-dimensional phase diagrams are helpful to quickly gauge stability behaviour for a given set of conditions (temperature, composition), it may limit our view over the entire phase stability landscape. This may be the case for hydrates, in which humidity plays an important role; however, the vapour phase is not part of a binary temperature–composition phase diagram (cf.Fig. 3). In this respect, machine learning or artificial intelligence (here used as synonyms) may provide a way to rapidly interpret complex phase behaviour, although these approaches will need to be developed and be based on reliable data, which for now does not exist or may be too scattered over many scientific contributions in formats that are not readily accessible. It is therefore estimated that a lot of effort will be necessary on producing reliable data before machine learning will be able to make any significant difference in the way that we deal with phase behaviour. | ||
| Fig. 3 Different types of phase behaviour in binary mixtures depicted in temperature–composition phase diagrams. Eutectic: a mixed liquid forms at a temperature below the melting point of the pure pharmaceutical. The eutectic temperature and the eutectic concentration depend on the melting points of the two constituents and on their interactions in the liquid phase. Cocrystal: formation of a binary compound with a lower (or higher, but in drug formulations that would beat the purpose) melting point than the pharmaceutical, while stability is in part guaranteed by crystallinity. Formation of a cocrystal depends on the interactions between the two constituents; however, its melting point does not have a direct relationship with the melting points of the pure components. Hydrate: special type of cocrystal which often (but not necessarily) exhibits incongruent melting (i.e., the melting hydrate forms a liquid different in concentration from the constituent ratio in the hydrate). Regular cocrystals may demonstrate incongruent melting as some hydrates or solvates may exhibit congruent melting depicted in the “cocrystal” panel (congruent melting indicates that the concentration of the formed liquid is the same as the constituent ratio in the cocrystal or hydrate). Liquid–liquid demixing: rare, however, it results into two liquids of which one with a high concentration of pharmaceutical, whereas the other liquid is a dilute solution in the solvent (water). Blue line: liquidus, horizontal grey line: eutectic equilibrium, purple vertical lines: cocrystal or hydrate, dashed lines: equilibria with metastable forms. In the liquid–liquid demixing, the grey line represents a monotectic invariant. | ||
In the rest of this section, we will discuss recent papers in which the conditions have been studied leading to different solid forms of a given chemical compound or API, which potentially can be used to obtain specific material properties. The sections below have been divided into “controlling polymorphism of unary systems” and “solid phases of binary and higher systems”. Thus, the section on unary systems focuses on API polymorphism even if it is obtained from solution (a binary system). As far as solvates are concerned, we will mainly consider the hydrate subgroup as they are most important in pharmaceutical applications as mentioned in section 1.1. Solvates will behave like hydrates and follow the phase diagrams of hydrates or cocrystals (Fig. 3) depending on the strength of interaction between the solvate molecules and the other constituent in the crystal.
The crystallisation kinetics of L-glutamic acid clearly demonstrates that different habits of the stable β form can be obtained depending on the crystallisation conditions. While stable β glutamic acid tends to crystallise as needles, if the supersaturation is modified, platelike crystals of the same polymorph can be obtained too, which may therefore improve processability.38,39 While in the case of glutamic acid the stable form crystallises with different habits, it may also be possible that polymorphs crystallise concomitantly,40 or that first a metastable form appears, which then will convert into a more stable form through a liquid mediated transformation. Depending on the form of interest, population balance modelling using empirical data can help in finding the optimal conditions to obtain the most desirable polymorph or habit.41 It has been shown for continuous crystallisation that the crystallising polymorph may be controlled in the steady state.42 In the case that nucleation rates and crystal growth rates are known as in the case of the polymorphs of L-glutamic acid and of p-aminobenzoic acid the steady state crystallisation conditions for a given polymorph can be reliably predicted. Despite a relatively straightforward mathematical description of steady state crystallisation of almost any polymorph, reality can be harder due to physical properties of crystals, even as simple as its crystalline form. In experimental continuous crystallisation with L-glutamic acid, while modelling provided favourable steady state conditions, the platelike crystals of the α form caused aggregation leading to a loss of the steady state.43
Antisolvents can be used to obtain metastable forms such as in the case of L-histidine for which the metastable from B is obtained by using the antisolvent ethanol or acetonitrile in combination with an L-histidine solution in water, in particular at high supersaturation concentrations.44,45
Additives in the form of small molecules may help to affect crystallisation rates46 or to obtain metastable forms.47,48 Crystallisation of the metastable form α of DL-methionine is controlled by adding DL-leucine. From aqueous solution, generally the β form crystallises directly or in a mixture with the α form. DL-Leucine appears to bind more strongly to the β form faces, preventing this form to develop any further. At high enough concentrations of DL-leucine, only α form DL-methionine crystallises out, while its habit changes too due to the interaction with leucine.49 Polymers can also be used as substrates, control nucleation and lead to different polymorphs such as in the case of flufenamic acid,50ortho-aminobenzoic acid,51 or 2,4-dichlorophenoxyacetic acid.52
D-Mannitol was shown to crystallise either in the α form or in the δ form in the presence of NaCl depending on the total sample size. Small concentrations of NaCl promoted the crystallisation of the metastable δ form, whereas large concentrations of NaCl resulted in form α. Phase diagrams involving the eutectic temperatures of D-mannitol with NaCl and also with other salts such as KCl were determined.53 The eutectic temperature between the δ form and NaCl is about 10 degrees lower than the eutectic between the α form and NaCl demonstrating a relatively increased stability for the δ form in the presence of NaCl.
Artemisinin was investigated by Horosanskaia et al. demonstrating two enantiotropically related polymorphs with an equilibrium temperature at 130 °C. In the case of artemisinin, form II cannot be kept at room temperature as it slowly transforms into form I below 130 °C.54
The transition temperature between two polymorphs can change because of a solid solution. This has been shown for benzocaine, in which form I becomes less stable due to the incorporation of water in the crystal structure. It lowers the transition temperature between form I and form II with almost 10 degrees.55 A very similar effect has been observed for dimethylurea in which only a little amount of water in the system, in terms of ppm, changes the phase equilibrium temperature between the two polymorphs with more than 25 °C.56
Epitaxial nucleation of the stable form of the steroid 7αMNa on its metastable form (see Fig. 4) clearly precludes the metastable form from being stored for long periods of time. Although nucleation of the stable form is accelerated in the presence of a solution, the presence of humidity or even the vapour phase itself could already initiate this type of crystallisation.5 Also for the beforementioned D-mannitol, epitaxial growth of the stable form α on the more rapidly crystallizing, metastable form δ has been observed.53
 | ||
| Fig. 4 Concomitant growth for intermediate Ostwald ratios of the stable form of 7αMna and its metastable form (two patches on the top crystal); whether the metastable form grows depends on the supersaturation. Reproduced from ref. 5 with permission from Elsevier, copyright 2006. | ||
Aripiprazole is a second-generation antipsychotic drug. Five polymorphs have been shown to exist that can be prepared under different conditions.57 Form I, the high temperature one, can be obtained by heating the other forms, although using a suspension in butanol above 80 °C leads to the purest crystals. Form II can be obtained from a suspension with 1-butanol or acetonitrile between 65 and 75 °C. Form III can be obtained through the supercooled melt, desolvation of several solvates, and direct crystallisation from several solvents such as ethyl acetate or n-hexane. Form IV can be obtained from solutions in toluene or dioxane. Form X° can be obtained from stirred suspensions of any other polymorph in solvents like acetone, 1-propanol, 2-propanol, acetonitrile or 1-butanol, all kept below 65 °C. It is the stable form at room temperature. Except for form I, the polymorphs possess high persistency (kinetic stability) and no conversion in more stable polymorphs occurs for over a year.57
Similar studies have been carried out with pyrazinamide, which possesses four polymorphs.11,58,59 This has eventually led to a pressure–temperature phase diagram demonstrating that each of the polymorphs possesses a stable temperature domain (cf.Fig. 5). It does not necessarily mean that at the appropriate temperature, the stable polymorph will immediately appear, but it does imply that if the stable polymorph is obtained under its stable conditions, it will not change if the pressure and temperature conditions are not changed. It has been shown for example that form β can be obtained at crystallisation temperatures below −20 °C.59 Form γ on the other hand crystallizes out in most cases and in particular if the crystallisation process is rapid.60 However, this form is stable at high temperature above 119 °C. Below 119 °C, the γ form can be maintained if crystallised with dimethylurea.12 The reason for this is not entirely understood yet, but it must have to do with the quality of the crystals of form γ obtained in the presence of dimethylurea.60
 | ||
| Fig. 5 The pressure–temperature phase diagram of pyrazinamide. Reproduced from ref. 59 with permission from the Royal Society of Chemistry, copyright 2022. | ||
It has also been shown that the metastable polymorph of ritonavir is stable at high pressure, and this pressure is actually quite accessible at 17.5 MPa (Fig. 6).61 Once again, it does not mean that form I, which is metastable with respect to form II at atmospheric pressure, will form if the system's pressure is increased up to 17.5 MPa, but if the polymorph is obtained under these conditions, it can be maintained, although for a drug formulation this may be of less interest if samples have to remain pressurised. In fact, Sacchi et al.62 recently showed that the necessary pressure to obtain form I is easily achieved by grinding the sample, in line with the low pressure of 17.5 MPa.61
 | ||
| Fig. 6 Schematic pressure–temperature phase diagram of the dimorphism of ritonavir. The triple point II-I-L is located at 17.5 MPa, which can be easily reached by grinding and tabletting as demonstrated in ref. 62. Reproduced from ref. 61 with permission from EM Consulte Elsevier, copyright 2015. | ||
The chiral muscle relaxant metaxalone possesses at least five crystalline forms of which some are racemic compounds and others pure single enantiomer crystals obtained through enantiospecific synthesis.63 Crystallisation of the related conglomerate is possible through eutectic systems with highly volatile solvents. Due to rapid evaporation of the solvent, small needle-like conglomerate solvate crystals are formed that, after desolvation, become unary conglomerate crystals.64 The racemic B form is thermodynamically the more stable one, while the racemic A form tends to crystallize out first in line with the Ostwald rule of stages.63
It should be kept in mind that for a given set of polymorphs, I and II, of an API and for a given temperature, at which form I is more stable than form II, the solubility of II (SII) is higher than that of I (SI). For a given concentration C of the API, the supersaturation ratio C/SI for the stable form I will be higher than the supersaturation ratio for the metastable II at the same concentration C/SII, because SI < SII and therefore C/SI > C/SII.
Cardew and Davey proposed an Ostwald ratio, which compares nucleation rates and growth rates between stable and metastable forms and allows an analysis based on the supersaturation ratios between different polymorphs. Low ratios favour the crystallisation of the stable form, whereas high ratios favour the crystallisation of a metastable form. Concomitant crystallisation is found for intermediate Ostwald ratios (see also Fig. 4).65 It follows that the Ostwald rule of stages is not particularly valid and simply depends on crystallisation conditions. It allows a certain amount of control over the phase that crystallises by selecting a temperature range in which the desired polymorph is stable as for example in the phase diagram of pyrazinamide mentioned above. To ensure the stable form to crystallise the Ostwald ratio needs to be kept low. It is therefore clearly important to understand the thermodynamic stability behaviour of a system as well as its nucleation and growth kinetics if one needs a good level of control over the crystallization process and the resulting polymorph.
Racemic fluoxetine nitrate is a monotropic system66,67 in which the metastable form can be obtained by slow evaporation from a methanol solution at room temperature, whereas the stable form is obtained at −5 °C from a solution in 95% ethanol.67 It implies that in particular the nucleation and growth rates play an important role in these crystallizations.
Kinetic trapping of metastable forms can be a useful approach to find conglomerates.68 The pre-exponential term of the nucleation rate equation (eqn (1)) is important in kinetically trapping metastable polymorphs. In particular if the interfacial energies of the two polymorphs are relatively low, high values of the pre-exponential term A give ready access to metastable forms,69 although concomitant crystallisation cannot be excluded as this depends on the Gibbs free energy term too:
| J = Ae(−ΔG*c/RT) | (1) |
A different example of kinetic trapping of metastable polymorphs can be observed for the crystallisation of the δ form of D-mannitol at the solution–substrate contact line of an evaporating droplet, while in the core of the droplet the stable form crystallises. The crystallisation of the metastable form may be due to higher supersaturations owing to higher evaporation rates at the rim of the droplet; however, an alternative explanation is that the Marangoni effect causes an increase in the concentration at the droplet rim with an increase in the supersaturation as a result.70 Similar behaviour has been observed for the β form of glycine, however, this form could not be prevented from transforming into the stable polymorph.70 Another interesting way to kinetically trap crystallisation kinetics is by using polymer melts as was shown for paracetamol in PEG melts in which the polymorphic transformation of form II into form I could be drastically slowed down.71
Sacchi et al. studied the nucleation and growth kinetics of three polymorphs of tolfenamic acid. They came to the conclusion that three nucleation and growth scenarios exist governing the possible observation of metastable polymorphs.72 First, if the metastable form nucleates more rapidly than the stable form, it should be observed in solution, as it appears first. Second, if the metastable and stable forms nucleate concomitantly, but the growth rate of the metastable form is higher, it may still be possible to observe the metastable form under conditions of supersaturation, while growth is taking place. Last, for metastable forms that are not nucleating faster than the stable form, observation will be very difficult, and these forms will be elusive polymorphs, which may have been computationally predicted as viable crystal structures but are not observed experimentally. Currently, it is still difficult to predict nucleation and growth rates of different forms, which makes it difficult to foresee the appearance of all predicted polymorphs. However, if pressure and temperature conditions can be found in which the slowly nucleating metastable polymorph is stable, which necessitates knowledge of its unary and possibly binary phase diagrams, access to this polymorph may nonetheless be possible. In the case of piracetam, it was shown that the metastable polymorph form II exhibits faster crystal growth than the stable form III.73 This is valid both in ethanol and in isopropanol, even if the overall kinetics in the two solvents differed, that in ethanol being faster. Moreover, it has been shown that for two metastable forms of piracetam, forms VI and II, a lower temperature and the use of isopropanol favour the formation of form VI, but in each nucleation event any of the forms may nucleate, independent of solvent or temperature, only the relative occurrences change.74
Seeding is the method to closely control the crystallisation outcome as the API GENE-A demonstrates. It exhibits monotropic dimorphism; however, the Gibbs free energy difference between the two forms is very small, leading to either form appearing depending on the crystallisation conditions. Seeding remains the easiest way to obtain the desired form as concomitant crystallisation occurs frequently.75
A different way of seeding is templating, by using surfaces that are different from the material to crystallise. Templating can be very powerful as a family of acids demonstrates. Mefenamic acid, tolfenamic acid, and flufenamic acid have been studied and used as template crystals to induce the formation of a different polymorph among the other two molecules. In particular tolfenamic acid exhibits sensitivity to adapt itself to the different phases and solid solutions that these acids can form and three new phases for tolfenamic acid were found through templating of tolfenamic acid on mefenamic acid and flufenamic acid.76 Nonetheless, it does not always work as mefenamic acid and flufenamic acid demonstrate, as they did not crystallise in new, previously unobserved phases in the presence of the other two molecules.
In the case of continuous crystallisation, it can be difficult to maintain the crystallisation of a metastable form, as was shown for paracetamol as a model system.77 In this case, adding 1% of metacetamol as an impurity prevented the crystallisation of the stable form and a steady state of metastable form II could be maintained. This was interpreted as a modification of the crystallisation kinetics by preventing form I from crystallising, because metacetamol preferentially limits crystal growth on form I crystal faces. A disadvantage of adding an impurity is the incorporation of metacetamol in the form II crystals too.77 In this particular case, metacetamol has a similar pharmaceutical activity to paracetamol and quantities remain small; however, for pharmaceutical applications nontoxicity would need to be demonstrated.
2.2.2.1 Hydrates. Hydrates are common among API crystals, which should not fully come as a surprise because solubility in water is one desired aspect of drug molecules. It implies that functional groups with a propensity to interact with water may be present in the molecule and their presence increases the tendency to form crystalline combinations involving water. Hydrates, or more general, solvates, can be considered cocrystals, as the API cocrystallises with water or another solvent and the fact that one of the cocrystal components is liquid at room temperature is a rather arbitrary condition. Hydrates lower the solubility of an API, as the hydrate inherently contains interactions between the API and water, lowering the Gibbs free energy driving force for the API to fully go into solution. This can be seen in the phase diagram of triethylenetetramine-dihydrochloride in Fig. 7,78 which has been simplified in the hydrate panel in Fig. 3 where the blue dashed line is the solubility of the anhydrous form, whereas the hydrate solubility is given by the curved liquidus line on its left (at lower API concentrations). A hydrate possesses a complex stability behaviour as it depends on the relative humidity in the air and on the temperature and both are subject to continuous change. One should not forget that a relative humidity of 80% at 0 °C has much lower water content than a relative humidity of 80% at 40 °C. The absolute partial pressure of water will be much lower at 0 °C, which may trigger the water molecules to leave the hydrate. This has been extensively reported in the paper on triethylenetetramine-dihydrochloride (Fig. 7).78 Dehydrated crystals may collapse and become amorphous, could recrystallise into an anhydrous crystalline form, which might not necessarily be the most stable form, or they may stay in a metastable structure that reflects that of the hydrate. Most hydrates are difficult to process and to use in manufacturing due to their sensitivity to temperature and humidity leading to full or partial dehydration and therefore they are mostly avoided if possible. On the other hand, desolvation may be a way to obtain metastable forms that are otherwise difficult to obtain from solution, such as in the case of prednisolone.79,80
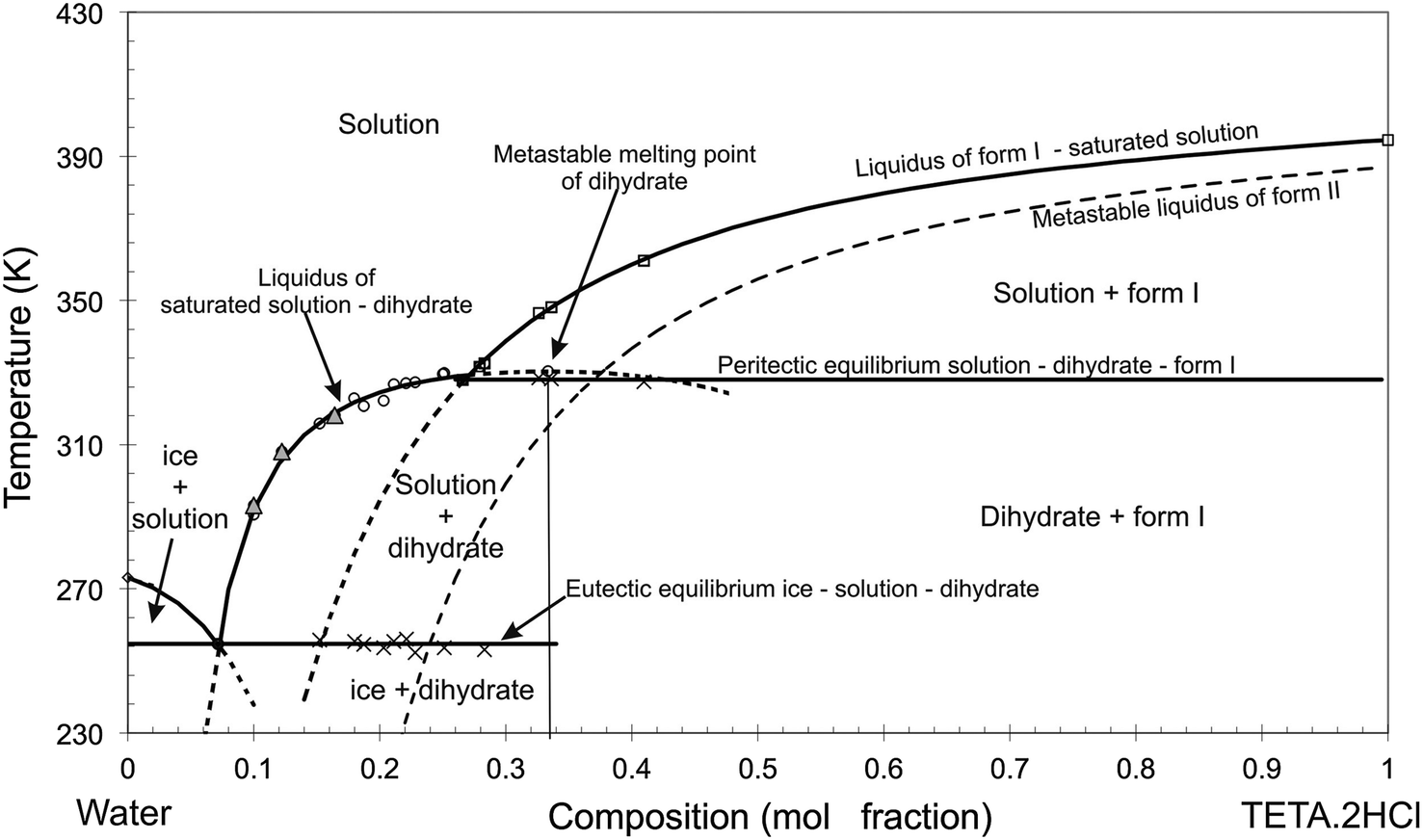 | ||
| Fig. 7 The binary phase diagram of triethylenetetramine-dihydrochloride and water demonstrating the differences in solubility between the hydrate (solid curve at about 0.1 mol fraction), the stable anhydrous form (dashed curve at 0.2 mol fraction), and the metastable anhydrous form (dashed curve at 0.25 mol fraction). Reproduced from ref. 78 with permission from Elsevier, copyright 2016. | ||
2.2.2.2 Liquid–liquid demixing. A particularly interesting but also rare phenomenon (for drug molecules) is liquid–liquid demixing. It exists in mixtures of prilocaine and water,81 and lidocaine and water.82 They are two very similar local anaesthetics drugs used in the composition of the EMLA® princeps.83–85 Both compounds form dilute solutions with water, which are not convenient to deliver the anaesthetics as their concentrations are too low. However, by increasing the concentration of the API, the systems phase-separate into two liquids (Fig. 3 liquid–liquid demixing): one high in anaesthetics and one a dilute solution. This occurs above room temperature. However, by mixing lidocaine and prilocaine, the temperature at which the API rich solution occurs descends below the body temperature, producing a useful vehicle for local anaesthetics on the skin. Even the single anaesthetic-water systems remain liquid at room temperature once liquified due to a strong persistent metastability and lack of crystallisation of the APIs from solution.81,82 The mixing of the two compounds has led to an excipient free liquid mixture called EMLA (eutectic mixture of local anaesthetics), which in fact is based on a monotectic invariant (Fig. 3) with liquid–liquid separation.
2.2.2.3 Cocrystals and eutectic systems. If the API possesses low solubility, such as the BCS class II nebivolol hydrochloride, adding GRAS (generally regarded as safe) compounds such as 4-hydroxy benzoic acid or nicotinamide may result in cocrystals with increased solubility and solubilization behaviour (Fig. 3). For the current example a threefold increase in the solubility could be obtained.86 Obviously, in the case of cocrystal engineering, logical choices for coformers are those with a lower melting point to try and increase solubility, but whether or not a specific cocrystal possesses the right properties for applications, such as sufficient stability, solubility, and processability, remains a question of trial and error.
A cocrystal is part of a binary phase diagram (Fig. 3), implying that its phase behaviour can be shifted to a eutectic equilibrium, which will liquefy at a lower temperature (compare the congruent melting temperature of the cocrystal and the eutectic liquid temperature in the cocrystal pane of Fig. 3). However, this liquid will contain more coformer than API as the lower eutectic will be located in the coformer-rich part of the phase diagram. This type of behaviour is demonstrated in a paper by Évora et al.87 Diflunisal is cocrystallised with nicotinamide and the binary phase diagram demonstrates a diminished stabilisation of the cocrystal (Tfus = 193 °C) in comparison with pure diflunisal (Tfus = 212 °C). If even more nicotinamide is added a eutectic occurs with a temperature of 117 °C and a eutectic concentration with about 10% of diflunisal. Using this eutectic temperature decreases the effective melting temperature of the API with almost 100 degrees and it will promote solubilisation of the drug as the solubility of the mixture will depend on the eutectic point as illustrated in Fig. 8 for a simple eutectic system. However, if the eutectic within a cocrystal system is used to increase solubility, one may want to consider using a simple eutectic binary system as depicted in the first panel on the left in Fig. 3. In principle, this would lead to an even lower eutectic temperature promoting solubilisation of the API in the aqueous phase, as observed for binary systems with levetiracetam.88 The use of a coformer to generate a eutectic equilibrium would not be recommended as in that case the overall eutectic equilibrium between the API and the coformer would be metastable; thus, such mixtures could at any time form for example the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diflunisal – nicotinamide cocrystal mentioned above. Therefore, GRAS compounds should be selected with a low melting point, that easily dissolve in water, and also have a good interaction with the API in the liquid state.89 The ternary combination of the API, excipient and water may lead to a higher concentration of the API in solution, than its binary solubility in water as shown in Fig. 8. Thorough mixing between the API and the excipient is important to ensure that both components dissolve simultaneously. This may be the most difficult part in the design of eutectic systems, as it remains complicated to prepare reproducible eutectic microstructures that melt and dissolve evenly.89
:1 diflunisal – nicotinamide cocrystal mentioned above. Therefore, GRAS compounds should be selected with a low melting point, that easily dissolve in water, and also have a good interaction with the API in the liquid state.89 The ternary combination of the API, excipient and water may lead to a higher concentration of the API in solution, than its binary solubility in water as shown in Fig. 8. Thorough mixing between the API and the excipient is important to ensure that both components dissolve simultaneously. This may be the most difficult part in the design of eutectic systems, as it remains complicated to prepare reproducible eutectic microstructures that melt and dissolve evenly.89
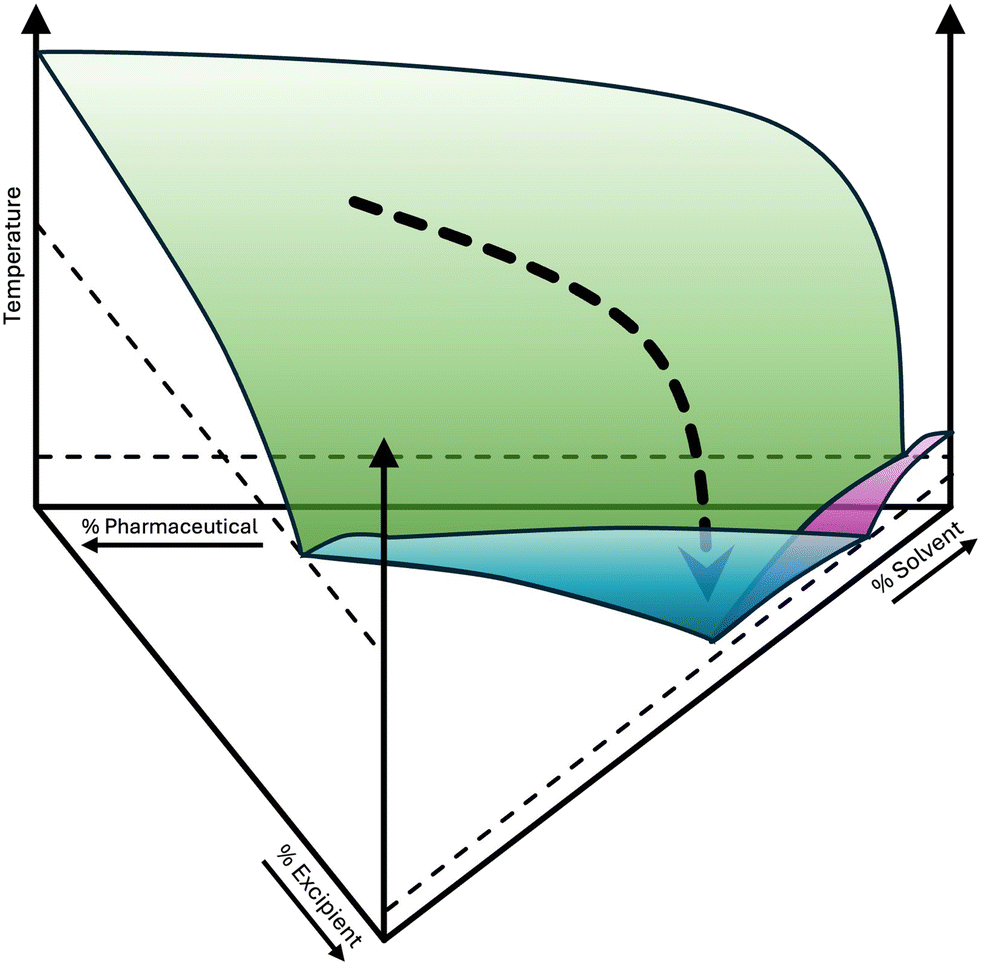 | ||
| Fig. 8 How a eutectic mixture may increase overall solubility. The API generally possesses poor solubility in water: the eutectic equilibrium is found at the water-rich side. Mixing between the API and excipient may be more favourable: the eutectic equilibrium is located at higher API concentrations than in water. The combination of the API, excipient and water may lead to an overall favourable concentration for the API in solution: curved black arrow leading to a global minimum in the ternary system with a relatively high concentration of the API due to interactions between the API, excipient, and water. | ||
The relative stability of cocrystals in relation to a particular solvent is nicely demonstrated in the paper by Ainouz et al. through the use of ternary phase diagrams.90 While grinding may lead to cocrystals that are part of the binary phase diagram, in the presence of a third phase such as a solvent, these cocrystals may become metastable. This will depend on the individual solubilities of the API and the coformer. It may be advantageous to select highly water-soluble coformers leading to cocrystals that have low stability in water and dissolve therefore rapidly, while they may be harvested by grinding or from another solvent in which the cocrystals are stable.90 This approach has been worked out further by Codan et al. among others (Fig. 9).91 It should be said, however, that much depends on the strength of interaction between the coformer and API and good solubility of the cocrystal is not guaranteed if only the coformer is very soluble; the balance in interactions between all three constituents is key.
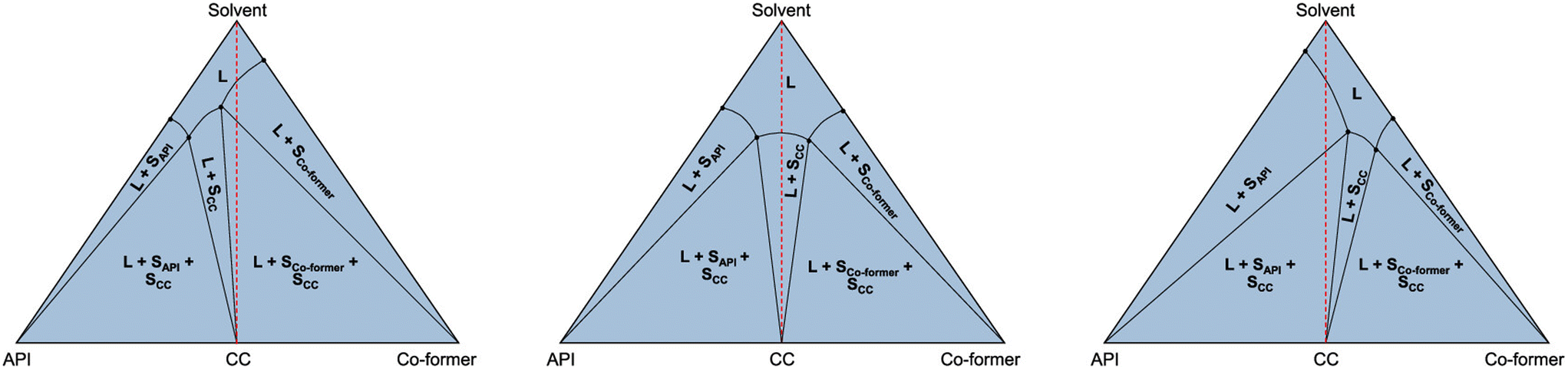 | ||
| Fig. 9 Different combinations of solubility of a cocrystal (SCC). In the left-hand case, the solution needs to be enriched in API, while in the right-hand figure, the solution needs to be enriched in coformer. The case in the centre is the ternary system equivalent to the cocrystal in Fig. 3 with congruent melting/dissolution. Reproduced from ref. 91 with permission from the American Chemical Society, copyright 2023. | ||
2.2.2.4 Cocrystals, conglomerates, and chirality. To separate racemic compounds, conglomerates are important. Conglomerates are crystalline mixtures forming a eutectic in which an individual crystal only contains one type of enantiomer R or S. In the case of conglomerates, the two enantiomers can be separated using preferential crystallisation.92,93 However, depending on the level of required separation, enrichment of the API in one of the enantiomers may be sufficient. This can either be enrichment through the solid phase or through the liquid phase according to which of the two phases contains most of the required enantiomer with the desired therapeutic effect. The use of ternary phase diagrams involving both enantiomers and a solvent are essential in choosing the optimal conditions.94 Another approach may be kinetic trapping of conglomerate systems for which inkjet printers can be used as they produce very small droplets that evaporate rapidly, providing an excellent environment for rapid crystallisation and trapping of metastable conglomerate forms.68
Praziquantel is a drug used against schistosomiasis and consists of an enantiomer system in which the R form is the active agent, whereas the S form gives the drug a bitter taste.95 The stable solid of praziquantel is a racemic compound; however, the API forms two different cocrystals with vanillin. One of these cocrystals, 1:2 praziquantel/vanillin, is a conglomerate, which allows the separation of praziquantel enantiomers by preferential crystallisation. Thus, control over the required crystal form involves extending the variables with a suitable co-former, here vanillin, to obtain a conglomerate system. Even within this system, the concentration of vanillin needs to be chosen high enough to stabilize the 1:2 cocrystals instead of the 1:1 cocrystal, which is racemic.95
2.2.2.5 Mesophases, amorphous systems, and amorphous solid dispersions. Mesophases, which include plastic crystals, liquid crystals and also conformationally disordered crystals,96 make up part of the stable phase diagram of organic molecular species. Plastic crystals and liquid crystals can be found around the melting point of the API and form therefore stable crystalline states or stable liquid states that contain a certain level of order. Moreover, conformational disorder may be inherent to the molecular system.97 These types of phases possess overall weaker interactions, which facilitates dissolution of the API in comparison to a fully crystalline compound. If the phase diagram of an API is known, it is in principle straightforward to maintain the desired phase under the conditions, in which it is stable. Maintaining that the phase under processing conditions and storage conditions is obviously the more important challenge. Atorvastatin calcium is a dream come true in the sense that it only forms a suitable crystalline phase in the form of a trihydrate. It implies that once atorvastatin calcium is brought into its amorphous phase, which is in fact a mesophase (liquid crystalline phase), no recrystallisation occurs as long as water is kept out of the system.98 It is therefore no problem to cool the mesophase down and keep it at room temperature for years without the risk of recrystallisation. The same is valid for systems in which the disorder in the crystalline phase is part of its thermodynamic stable structure.97
The amorphous state can also be used as the medium to prepare metastable polymorphs. In the case of ranolazine, the amorphous state exhibits a glass transition temperature below room temperature, which implies that it is not immediately possible to prevent the amorphous state from crystallising. However, crystallisation leads to metastable forms that unfortunately rapidly convert towards the low energy stable form.99 Nonetheless, because crystallisation kinetics in solid amorphous systems is relatively slow in comparison to crystallisation from solution, some control exists over the kinetics of crystallisation.
Going one step further and stabilizing the amorphous phases using polymer resulting in amorphous molecular solid dispersions, in which the API is molecularly dispersed in a polymer matrix, processing parameters can be determined based on thermodynamic and kinetic analysis.100 Thermodynamics involve the eutectic temperature between the API and the polymer, which is considered the critical minimum temperature for the processing to take place. Kinetics are necessary to determine residence time in extruders to ensure that all drug material has melted and is dispersed in the polymer matrix to avoid crystalline residues. Once the amorphous molecular solid dispersion has been obtained and is brought below the glass transition temperature, these metastable, or even unstable states can persist for sufficiently long times to be used in drug formulations. In the case that solvents are used for mixing, miscibility between the polymer and the solvent is an important factor to ensure full dissolution of the polymers to allow, in a next step, mixing with the API. Multiple systems involving among others PVP K90 and solvents such as acetone and ethanol demonstrated that predictions with PC-SAFT (perturbed-chain statistical associating fluid theory) are reliable to predict stability behaviour.101 Once mixed with the API and the solvent evaporated, amorphous molecular solid dispersions remain that are persistent enough to be used for formulations. Taking into consideration the hygroscopicity of PVPK90, HPMCAS (hydroxypropylmethylcellulose acetate succinate), a polymer that is less sensitive to humidity, is most likely a better candidate to stabilise amorphous dispersions.102
3 Conclusions
Two main pathways exist to stabilize solid forms with the required properties, thermodynamic and kinetic. The thermodynamic pathway implies that the desired solid form is stable under the given conditions and can be crystallised under the most convenient conditions from solution, by sublimation, or from the melt. Although temperature is the most obvious parameter, concentration should not be forgotten both in terms of concentration in solution (saturation or supersaturation) and in terms of cofactors leading to hydrates, cocrystals or even liquid–liquid demixing. The kinetic pathway implies control over the Ostwald rule of stages, making sure that the obtained form does not immediately transform into a more stable form under the conditions that the system is subjected to. The kinetic example involving pyrazinamide is the crystallisation of the γ form, which can be obtained between room temperature and about 100 °C in the presence of 1,3-dimethylurea providing conditions that considerably slow down the transformation from γ into the stable α form for up to 12 months.60 A thermodynamic pathway example is the crystallisation of the β form of pyrazinamide at temperatures below −20 °C, where β is stable and can be obtained pure, while at higher temperatures it only crystallises concomitantly.59Crystal structure prediction has clearly provided a means to determine which crystal structures may be important103–106 and they are also to a certain extent capable of providing pathways towards the crystallisation of forms that have not been obtained experimentally76,107 in which the use of impurities or templates absolutely has its place,49,60,76 epitaxial growth on surfaces can be very useful,108,109 and seeding with closely related molecules too.75,110 Although with epitaxial growth, the extent of the new phase may only be a few molecular layers thick, separation of the crystal from the surface may be relatively easy, whereas in the case of seeding, the seeds are bound to remain as impurities in the newly obtained crystals, if those seeds are not the same molecules.75
It remains difficult to crystallize all predicted low energy forms. Most of the experiments to obtain different polymorphs or other solid forms are based on trial and error and have been robotised using many different solvents and crystallisation conditions, but there is no clear way yet to reliably predict nucleation and growth kinetics beyond the energy attachment (Perdok–Hartman) method111 and the Bravais–Friedel–Donnay–Harker (BFDH) rule.112 Obviously, kinetics play an important role. Faster crystallising polymorphs will be observed, whether they are metastable or stable, while the more slowly crystalising polymorphs only have a chance to be observed if they are more stable than the already crystallised form. This explains why experiments do not result in all predicted low energy polymorphs, but it would be useful if the kinetics in combination with the thermodynamics can be predicted so that a complete risk assessment exists for a given API and its most desirable polymorphs.
For metastable forms with useful properties, it also remains difficult to predict whether they can be stabilized kinetically. Some molecules and their structures do not convert easily from one polymorph to another, whereas other molecules do not sustain metastable polymorphs for any lengths of time.99 Even for a single molecule and a single metastable polymorph considerable differences in lifetimes of metastable forms are observed simply depending on the crystallisation conditions.113 The causes are often complex as crystallisation conditions are a multidimensional space in which various factors play a role such as temperature, pressure, solvent, co-formers, and the crystallising molecule, and these all have their impact on the thermodynamic stability and the crystallisation kinetics of a solid form. Crystallisation kinetics are for example influenced by the viscosity, diffusion, or a tendency to exhibit disorder in the crystal. Thus, for each molecule a case-by-case analysis is necessary in which simulation provides a theoretical outline for crystallisation strategies; however, no complete answers are provided as to which polymorph can be crystallized, with which kinetics, and how stable or persistent the obtained polymorph finally is. Even thermodynamic stability is still not equivocally solved in the computational domain, and it remains a case-by-case experimental study to obtain data to confirm or determine the final stability behaviour of the observed polymorphs of a molecule.
As molecules will always have their individual physical behaviour and crystal structure prediction provides sufficient crystal structures, efforts should be aimed at nucleation and growth kinetics of polymorphs. First, in terms of preparation of the polymorphs, but also in terms of polymorph conversion kinetics. With this information, prediction of the polymorphs that really matter may be easier and it will become possible to predict which of the metastable polymorphs can be reliably developed in industry. This should go hand in hand with the prediction of the physical properties of molecular materials for which the COST Action BEST-CSP114–116 is establishing a new benchmark with experimental physical properties of organic solid materials, so that polymorphs with useful physical properties can eventually be predicted, obtained, and maintained. Other efforts on the computational site using crystal structure prediction-informed evolutionary optimisation are also currently underway.117
Data availability
We have not generated any new data for this paper.Author contributions
Conceptualization: IBR, investigation: IBR, C-LS, GG, writing original Draft: IBR, C-LS, GG, visualisation: IBR, C-LS, writing review & editing: IBR, C-LS, GG.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C-LS thanks the Normandy region (France) for a thesis scholarship.References
- European Pharmacopoeia Commission, European Pharmacopoeia, ed. European Directorate for the Quality of Medicines & HealthCare (EDQM), 2024, vol. 11, p. 4 Search PubMed.
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, CRC Press, Boca Raton, 2nd edn, 2009, vol. 192 Search PubMed.
- A. Bhatia, S. Chopra, K. Nagpal, P. K. Deb, M. Tekade and R. K. Tekade, in Dosage Form Design Parameters, ed. R. K. Tekade, Academic Press, 2018, pp. 31–65 Search PubMed.
- J. F. Bauer, J. A. Bauer, S. G. Spanton, S. G. Spanton, R. F. Henry, R. Henry, R. F. Henry, J. John Quick, W. D. Quick, W. Dziki, W. R. Porter, P. William, W. Porter, J. B. Morris and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS PubMed.
- C. Stoica, P. Verwer, H. Meekes, E. Vlieg, P. J. C. M. Van Hoof and F. M. Kaspersen, Int. J. Pharm., 2006, 309, 16–24 CrossRef CAS PubMed.
- European Medicines Agency (EMA), 2024.
- A. Lemercier, N. Couvrat, Y. Cartigny, M. Sanselme, Y. Corvis, P. Espeau and G. Coquerel, Pharmaceutics, 2023, 15, 1694 CrossRef CAS PubMed.
- N. Shah, H. Sandhu, D. S. Choi, H. Chokshi and A. W. Malick, Amorphous Solid Dispersions: Theory and Practice, Springer, New York, NY, 2014 Search PubMed.
- S. Tambe, D. Jain, S. K. Meruva, G. Rongala, A. Juluri, G. Nihalani, H. K. Mamidi, P. K. Nukala and P. K. Bolla, Pharmaceutics, 2022, 14, 2203 CrossRef CAS PubMed.
- J. Abraham, in Handbook of Transnational Economic Governance Regimes, ed. C. Tietje and A. Brouder, Brill, Nijhoff, 2010, pp. 1041–1053 Search PubMed.
- R. A. E. Castro, T. M. R. Maria, A. O. L. Évora, J. C. Feiteira, M. R. Silva, A. M. Beja, J. Canotilho and M. E. S. Eusébio, Cryst. Growth Des., 2010, 10, 274–282 CrossRef CAS.
- K. Li, G. Gbabode, M. Barrio, J.-L. Tamarit, M. Vergé-Depré, B. Robert and I. B. Rietveld, Int. J. Pharm., 2020, 580, 119230 CrossRef CAS PubMed.
- J. C. R. Corrêa, A. G. Perissinato, C. H. dos Reis Serra, M. G. Trevisan and H. R. N. Salgado, J. Therm. Anal. Calorim., 2016, 123, 2185–2190 CrossRef.
- S. A. Stanton, J. J. Du, F. Lai, G. Stanton, B. A. Hawkins, J. A. Ong, P. W. Groundwater, J. A. Platts and D. E. Hibbs, J. Phys. Chem. A, 2021, 125, 9736–9756 CrossRef CAS PubMed.
- M. Mehta, Biopharmaceutics Classification System BCS: Development, Implementation, and Growth, Wiley, 2017 Search PubMed.
- R. Censi and P. Di Martino, Molecules, 2015, 20, 18759–18776 CrossRef CAS PubMed.
- R. Hilfiker, Polymorphism in the pharmaceutical industry, WILEY-VCH, Weinheim, Germany, 2006 Search PubMed.
- M. Rossmann, A. Braeuer, A. Leipertz and E. Schluecker, J. Supercrit. Fluids, 2013, 82, 230–237 CrossRef CAS.
- J. Sood, B. Sapra, S. Bhandari, M. Jindal and A. K. Tiwary, Ther. Delivery, 2014, 5, 1123–1142 CrossRef CAS PubMed.
- S. J. Smith, M. M. Bishop, J. M. Montgomery, T. P. Hamilton and Y. K. Vohra, J. Phys. Chem. A, 2014, 118, 6068–6077 CrossRef CAS PubMed.
- D.-K. Bučar, J. A. Elliott, M. D. Eddleston, J. K. Cockcroft and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 249–253 CrossRef PubMed.
- H. Mohapatra and C. J. Eckhardt, J. Phys. Chem. B, 2008, 112, 2293–2298 CrossRef CAS PubMed.
- B. P. A. Gabriele, C. J. Williams, M. E. Lauer, B. Derby and A. J. Cruz Cabeza, CrystEngComm, 2021, 5826–5838 RSC.
- K. S. Khomane, P. K. More, G. Raghavendra and A. K. Bansal, Mol. Pharmaceutics, 2013, 10, 631–639 CrossRef CAS PubMed.
- B. A. Young, D. Bahl and L. L. Stevens, Pharm. Res., 2019, 36, 150 CrossRef PubMed.
- I. B. Rietveld and R. Céolin, J. Pharm. Sci., 2015, 104, 4117–4122 CrossRef CAS PubMed.
- D. Bučar, R. W. Lancaster and J. Bernstein, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef PubMed.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- Y. Zhou, C. Lv, X. Liu, J. Gao, Y. Gao, T. Wang and X. Huang, Cryst. Growth Des., 2024, 24, 584–600 CrossRef CAS.
- Q. Zhu, A. G. Shtukenberg, D. J. Carter, T.-Q. Yu, J. Yang, M. Chen, P. Raiteri, A. R. Oganov, B. Pokroy, I. Polishchuk, P. J. Bygrave, G. M. Day, A. L. Rohl, M. E. Tuckerman and B. Kahr, J. Am. Chem. Soc., 2016, 138, 4881–4889 CrossRef CAS PubMed.
- S. Li, B. Liu, Z. Chen, X. Ou, H. Rong and M. Lu, Mol. Pharmaceutics, 2023, 20, 3854–3863 CrossRef CAS PubMed.
- A. G. Shtukenberg, M. Tan, L. Vogt-Maranto, E. J. Chan, W. Xu, J. Yang, M. E. Tuckerman, C. T. Hu and B. Kahr, Cryst. Growth Des., 2019, 19, 4070–4080 CrossRef CAS.
- A. G. Shtukenberg, C. T. Hu, Q. Zhu, M. U. Schmidt, W. Xu, M. Tan and B. Kahr, Cryst. Growth Des., 2017, 17, 3562–3566 CrossRef CAS.
- S. Toscani, H. Allouchi, R. Ceolin, L. Villalobos, M. Barrio, J.-L. Tamarit and I. B. Rietveld, Int. J. Pharm., 2025, 669, 125060 CrossRef CAS PubMed.
- M. Romanini, B. Ivo, M. Rietveld, P. Barrio, D. Negrier, R. Mondieig, R. C. Macovez and J.-L. Tamarit, Cryst. Growth Des., 2021, 21, 2167–2175 CrossRef CAS.
- J. Lombard, D. A. Haynes and T. le Roex, Cryst. Growth Des., 2020, 20, 7840–7849 CrossRef CAS.
- P. McArdle and A. Erxleben, CrystEngComm, 2021, 23, 5965–5975 RSC.
- M. Kitamura, J. Cryst. Growth, 1989, 96, 541–546 CrossRef CAS.
- Y. Tahri, E. Gagnière, E. Chabanon, T. Bounahmidi and D. Mangin, J. Cryst. Growth, 2016, 435, 98–104 CrossRef CAS.
- J. Bernstein, R. J. Davey and J.-O. Henck, Angew. Chem., Int. Ed., 1999, 38, 3440–3461 CrossRef PubMed.
- A. E. Flood and L. Wantha, J. Cryst. Growth, 2013, 373, 7–12 CrossRef CAS.
- T. C. Farmer, C. L. Carpenter and M. F. Doherty, AIChE J., 2016, 62, 3505–3514 CrossRef CAS.
- R. Achermann, A. Košir, B. Bodák, L. Bosetti and M. Mazzotti, Cryst. Growth Des., 2023, 23, 2485–2503 CrossRef CAS PubMed.
- L. Wantha, N. Punmalee, V. Sawaddiphol and A. E. Flood, J. Cryst. Growth, 2018, 490, 65–70 CrossRef CAS.
- P. Kongsamai, L. Wantha, A. E. Flood and C. Tangsathitkulchai, J. Cryst. Growth, 2019, 525, 125209 CrossRef CAS.
- H. Wu, J. Wang, Q. Liu, S. Zong, B. Tian, X. Huang, T. Wang, Q. Yin and H. Hao, Cryst. Growth Des., 2020, 20, 973–983 CrossRef CAS.
- J. Aarthi, P. Dhanasekaran, T. S. Senthil and N. M. Ganesan, Cryst. Res. Technol., 2018, 53, 1700190 CrossRef.
- S. Manivel and S. Karuppannan, Mol. Cryst. Liq. Cryst., 2022, 745, 68–83 CrossRef CAS.
- Z. Li, P. Shi, Y. Yang, P. Sun, Y. Wang, S. Xu and J. Gong, CrystEngComm, 2019, 21, 3731–3739 RSC.
- V. López-Mejías, J. W. Kampf and A. J. Matzger, J. Am. Chem. Soc., 2012, 134, 9872–9875 CrossRef PubMed.
- E. Simone, M. V. Cenzato and Z. K. Nagy, J. Cryst. Growth, 2016, 446, 50–59 CrossRef CAS.
- L. Fang, J. Liu, D. Han, Z. Gao and J. Gong, Powder Technol., 2023, 413, 118077 CrossRef CAS.
- C. Telang, R. Suryanarayanan and L. Yu, Pharm. Res., 2003, 20, 1939–1945 CrossRef CAS PubMed.
- E. Horosanskaia, A. Seidel-Morgenstern and H. Lorenz, Thermochim. Acta, 2014, 578, 74–81 CrossRef CAS.
- M. A. Patel, M. H. H. AbouGhaly and K. Chadwick, Int. J. Pharm., 2017, 532, 166–176 CrossRef CAS PubMed.
- G. Baaklini, M. Schindler, L. Yuan, C. D. S. Jores, M. Sanselme, N. Couvrat, S. Clevers, P. Négrier, D. Mondieig, V. Dupray, Y. Cartigny, G. Gbabode and G. Coquerel, Molecules, 2023, 28, 7061 CrossRef CAS PubMed.
- D. E. Braun, T. Gelbrich, V. Kahlenberg, R. Tessadri, J. Wieser and U. J. Griesser, J. Pharm. Sci., 2009, 98, 2010–2026 CrossRef CAS PubMed.
- S. Cherukuvada, R. Thakuria and A. Nangia, Cryst. Growth Des., 2010, 10, 3931–3941 CrossRef CAS.
- K. Li, G. Gbabode, M. Vergé-Depré, B. Robert, M. Barrio, J.-P. Itié, J.-L. Tamarit and I. B. Rietveld, CrystEngComm, 2022, 24, 5041–5051 RSC.
- G. Baaklini, V. Dupray and G. Coquerel, Int. J. Pharm., 2015, 479, 163–170 CrossRef CAS PubMed.
- R. Céolin and I. B. Rietveld, Ann. Pharm. Fr., 2015, 73, 22–30 CrossRef.
- P. Sacchi, S. E. Wright, P. Neoptolemou, G. I. Lampronti, A. K. Rajagopalan, W. Kras, C. L. Evans, P. Hodgkinson and A. J. Cruz-Cabeza, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2319127121 CrossRef CAS PubMed.
- A. A. Bredikhin, D. V. Zakharychev, A. T. Gubaidullin and Z. A. Bredikhina, Cryst. Growth Des., 2018, 18, 6627–6639 CrossRef CAS.
- V. Hamilton, I. Andrusenko, J. Potticary, C. Hall, R. Stenner, E. Mugnaioli, A. E. Lanza, M. Gemmi and S. R. Hall, Cryst. Growth Des., 2020, 20, 4731–4739 CrossRef CAS.
- P. T. Cardew and R. J. Davey, Cryst. Growth Des., 2019, 19, 5798–5810 CrossRef CAS.
- R. Céolin and I. B. Rietveld, Eur. Phys. J.: Spec. Top., 2017, 226, 881–888 Search PubMed.
- P. S. Carvalho, J. Ellena, D. S. Yufit and J. A. K. Howard, Cryst. Growth Des., 2016, 16, 3875–3883 CrossRef CAS.
- A. U. Chowdhury, C. M. Dettmar, S. Z. Sullivan, S. Zhang, K. T. Jacobs, D. J. Kissick, T. Maltais, H. G. Hedderich, P. A. Bishop and G. J. Simpson, J. Am. Chem. Soc., 2014, 136, 2404–2412 CrossRef CAS PubMed.
- V. Verma and B. K. Hodnett, CrystEngComm, 2022, 24, 3088–3095 RSC.
- S. K. Poornachary, J. V. Parambil, P. S. Chow, R. B. H. Tan and J. Y. Y. Heng, Cryst. Growth Des., 2013, 13, 1180–1186 CrossRef CAS.
- J. R. Hernández Espinell, V. Toro, X. Yao, L. Yu, V. Lopéz-Mejías and T. Stelzer, Mol. Pharmaceutics, 2022, 19, 2183–2190 CrossRef PubMed.
- P. Sacchi, P. Neoptolemou, R. J. Davey, S. M. Reutzel-Edens and A. J. Cruz-Cabeza, Chem. Sci., 2023, 14, 11775–11789 RSC.
- A. Lynch, R. Soto and Å. Rasmuson, Cryst. Growth Des., 2021, 21, 5631–5640 CrossRef CAS.
- S. Kakkar, L. Zeng, M. Svärd and Å. C. Rasmuson, Cryst. Growth Des., 2022, 22, 2964–2973 CrossRef CAS PubMed.
- P. Chakravarty, A. G. DiPasquale, A. Stumpf, J. W. Lubach and K. Nagapudi, Mol. Pharmaceutics, 2018, 15, 5072–5080 CrossRef CAS PubMed.
- D. H. Case, V. K. Srirambhatla, R. Guo, R. E. Watson, L. S. Price, H. Polyzois, J. K. Cockcroft, A. J. Florence, D. A. Tocher and S. L. Price, Cryst. Growth Des., 2018, 18, 5322–5331 CrossRef CAS.
- L. Nicoud, F. Licordari and A. S. Myerson, Org. Process Res. Dev., 2019, 23, 794–806 CrossRef CAS.
- T. Henriet, I. Gana, C. Ghaddar, M. Barrio, Y. Cartigny, N. Yagoubi, B. Do, J.-L. Tamarit and I. B. Rietveld, Int. J. Pharm., 2016, 511, 312–321 CrossRef CAS PubMed.
- D. Giron, C. Goldbronn, M. Mutz, S. Pfeffer, P. Piechon and P. Schwab, J. Therm. Anal. Calorim., 2002, 68, 453–465 CrossRef CAS.
- J. Mahieux, M. Sanselme and G. Coquerel, Cryst. Growth Des., 2016, 16, 396–405 CrossRef CAS.
- I. B. Rietveld, M.-A. Perrin, S. Toscani, M. Barrio, B. Nicolai, J.-L. Tamarit and R. Ceolin, Mol. Pharmaceutics, 2013, 10, 1332–1339 CrossRef CAS PubMed.
- R. Ceolin, M. Barrio, J.-L. Tamarit, N. Veglio, M.-A. Perrin and P. Espeau, J. Pharm. Sci., 2010, 99, 2756–2765 CrossRef CAS PubMed.
- A. Brodin, A. Nyqvist-Mayer, T. Wadsten, B. Forslund and F. Broberg, J. Pharm. Sci., 1984, 73, 481–484 CrossRef CAS PubMed.
- A. A. Nyqvist-Mayer, A. F. Brodin and S. G. Frank, J. Pharm. Sci., 1985, 74, 1192–1195 CrossRef CAS PubMed.
- A. A. Nyqvist-Mayer, A. F. Brodin and S. G. Frank, J. Pharm. Sci., 1986, 75, 365–373 CrossRef CAS PubMed.
- V. J. Nikam and S. B. Patil, J. Cryst. Growth, 2020, 534, 125488 CrossRef CAS.
- A. O. L. Évora, R. A. E. Castro, T. M. R. Maria, M. R. Silva, J. H. Ter Horst, J. Canotilho and M. E. S. Eusébio, Int. J. Pharm., 2014, 466, 68–75 CrossRef PubMed.
- S. M. T. Machado, R. A. E. Castro, T. M. R. Maria, J. Canotilho and M. E. S. Eusébio, Int. J. Pharm., 2017, 533, 1–13 CrossRef CAS PubMed.
- G. C. Bazzo, B. R. Pezzini and H. K. Stulzer, Int. J. Pharm., 2020, 588, 119741 CrossRef CAS PubMed.
- A. Ainouz, J.-R. Authelin, P. Billot and H. Lieberman, Int. J. Pharm., 2009, 374, 82–89 CrossRef CAS PubMed.
- L. Codan, L. Daza and E. Sirota, Org. Process Res. Dev., 2023, 27, 513–522 CrossRef CAS.
- G. Coquerel, in Novel Optical Resolution Technologies, ed. K. Sakai, N. Hirayama and R. Tamura, Springer, Berlin, Heidelberg, 2007, pp. 1–51 Search PubMed.
- G. Levilain and G. Coquerel, CrystEngComm, 2010, 12, 1983–1992 RSC.
- A. M. Chen, Y. Wang and R. M. Wenslow, Org. Process Res. Dev., 2008, 12, 271–281 CrossRef CAS.
- C. J. J. Gerard, C. Pinetre, H. Cercel, M. D. Charpentier, M. Sanselme, N. Couvrat, C. Brandel, Y. Cartigny, V. Dupray and J. H. Ter Horst, Cryst. Growth Des., 2024, 24, 3378–3387 CrossRef CAS.
- E. Shalaev, K. Wu, S. Shamblin, J. F. Krzyzaniak and M. Descamps, Adv. Drug Delivery Rev., 2016, 100, 194–211 CrossRef CAS.
- G. R. Woollam, M. A. Neumann, T. Wagner and R. J. Davey, Faraday Discuss., 2018, 211, 209–234 RSC.
- C. Tizaoui, H. Galai, M. Barrio, S. Clevers, N. Couvrat, V. Dupray, G. Coquerel, J.-L. Tamarit and I. B. Rietveld, Eur. J. Pharm. Sci., 2020, 148, 105334 CrossRef CAS PubMed.
- J. F. C. Silva, P. S. Pereira Silva, M. Ramos Silva, E. Fantechi, L. Chelazzi, S. Ciattini, M. E. S. Eusébio and M. T. S. Rosado, Cryst. Growth Des., 2023, 23, 6679–6691 CrossRef CAS.
- D. E. Moseson and L. S. Taylor, Int. J. Pharm., 2018, 553, 454–466 CrossRef CAS PubMed.
- S. Dohrn, C. Luebbert, K. Lehmkemper, S. O. Kyeremateng, M. Degenhardt and G. Sadowski, Int. J. Pharm., 2020, 577, 119065 CrossRef CAS PubMed.
- A. Butreddy, Eur. J. Pharm. Biopharm., 2022, 177, 289–307 CrossRef CAS PubMed.
- Z. Falls, P. Avery, X. Wang, K. P. Hilleke and E. Zurek, J. Phys. Chem. C, 2021, 125, 1601–1620 CrossRef CAS.
- D. H. Bowskill, I. J. Sugden, S. Konstantinopoulos, C. S. Adjiman and C. C. Pantelides, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 593–623 CrossRef CAS PubMed.
- L. M. Hunnisett, J. Nyman, N. Francia, N. S. Abraham, C. S. Adjiman, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli, D. M. Anstine, J. E. Anthony, J. E. Arnold, F. Bahrami, M. A. Bellucci, R. M. Bhardwaj, I. Bier, J. A. Bis, A. D. Boese, D. H. Bowskill, J. Bramley, J. G. Brandenburg, D. E. Braun, P. W. V. Butler, J. Cadden, S. Carino, E. J. Chan, C. Chang, B. Cheng, S. M. Clarke, S. J. Coles, R. I. Cooper, R. Couch, R. Cuadrado, T. Darden, G. M. Day, H. Dietrich, Y. Ding, A. DiPasquale, B. Dhokale, B. P. Van Eijck, M. R. J. Elsegood, D. Firaha, W. Fu, K. Fukuzawa, J. Glover, H. Goto, C. Greenwell, R. Guo, J. Harter, J. Helfferich, D. W. M. Hofmann, J. Hoja, J. Hone, R. Hong, G. Hutchison, Y. Ikabata, O. Isayev, O. Ishaque, V. Jain, Y. Jin, A. Jing, E. R. Johnson, I. Jones, K. V. J. Jose, E. A. Kabova, A. Keates, P. F. Kelly, D. Khakimov, S. Konstantinopoulos, L. N. Kuleshova, H. Li, X. Lin, A. List, C. Liu, Y. M. Liu, Z. Liu, Z.-P. Liu, J. W. Lubach, N. Marom, A. A. Maryewski, H. Matsui, A. Mattei, R. A. Mayo, J. W. Melkumov, S. Mohamed, Z. Momenzadeh Abardeh, H. S. Muddana, N. Nakayama, K. S. Nayal, M. A. Neumann, R. Nikhar, S. Obata, D. O'Connor, A. R. Oganov, K. Okuwaki, A. Otero-de-la-Roza, C. C. Pantelides, S. Parkin, C. J. Pickard, L. Pilia, T. Pivina, R. Podeszwa, A. J. A. Price, L. S. Price, S. L. Price, M. R. Probert, A. Pulido, G. R. Ramteke, A. U. Rehman, S. M. Reutzel-Edens, J. Rogal, M. J. Ross, A. F. Rumson, G. Sadiq, Z. M. Saeed, A. Salimi, M. Salvalaglio, L. Sanders De Almada, K. Sasikumar, S. Sekharan, C. Shang, K. Shankland, K. Shinohara, B. Shi, X. Shi, A. G. Skillman, H. Song, N. Strasser, J. Van De Streek, I. J. Sugden, G. Sun, K. Szalewicz, B. I. Tan, L. Tan, F. Tarczynski, C. R. Taylor, A. Tkatchenko, R. Tom, M. E. Tuckerman, Y. Utsumi, L. Vogt-Maranto, J. Weatherston, L. J. Wilkinson, R. D. Willacy, L. Wojtas, G. R. Woollam, Z. Yang, E. Yonemochi, X. Yue, Q. Zeng, Y. Zhang, T. Zhou, Y. Zhou, R. Zubatyuk and J. C. Cole, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2024, 80, 517–547 CrossRef CAS PubMed.
- L. M. Hunnisett, N. Francia, J. Nyman, N. S. Abraham, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli, D. M. Anstine, J. E. Anthony, J. E. Arnold, F. Bahrami, M. A. Bellucci, G. J. O. Beran, R. M. Bhardwaj, R. Bianco, J. A. Bis, A. D. Boese, J. Bramley, D. E. Braun, P. W. V. Butler, J. Cadden, S. Carino, C. Červinka, E. J. Chan, C. Chang, S. M. Clarke, S. J. Coles, C. J. Cook, R. I. Cooper, T. Darden, G. M. Day, W. Deng, H. Dietrich, A. DiPasquale, B. Dhokale, B. P. Van Eijck, M. R. J. Elsegood, D. Firaha, W. Fu, K. Fukuzawa, N. Galanakis, H. Goto, C. Greenwell, R. Guo, J. Harter, J. Helfferich, J. Hoja, J. Hone, R. Hong, M. Hušák, Y. Ikabata, O. Isayev, O. Ishaque, V. Jain, Y. Jin, A. Jing, E. R. Johnson, I. Jones, K. V. J. Jose, E. A. Kabova, A. Keates, P. F. Kelly, J. Klimeš, V. Kostková, H. Li, X. Lin, A. List, C. Liu, Y. M. Liu, Z. Liu, I. Lončarić, J. W. Lubach, J. Ludík, N. Marom, H. Matsui, A. Mattei, R. A. Mayo, J. W. Melkumov, B. Mladineo, S. Mohamed, Z. M. Abardeh, H. S. Muddana, N. Nakayama, K. S. Nayal, M. A. Neumann, R. Nikhar, S. Obata, D. O'Connor, A. R. Oganov, K. Okuwaki, A. Otero-de-la-Roza, S. Parkin, A. Parunov, R. Podeszwa, A. J. A. Price, L. S. Price, S. L. Price, M. R. Probert, A. Pulido, G. R. Ramteke, A. U. Rehman, S. M. Reutzel-Edens, J. Rogal, M. J. Ross, A. F. Rumson, G. Sadiq, Z. M. Saeed, A. Salimi, K. Sasikumar, S. Sekharan, K. Shankland, B. Shi, X. Shi, K. Shinohara, A. G. Skillman, H. Song, N. Strasser, J. Van De Streek, I. J. Sugden, G. Sun, K. Szalewicz, L. Tan, K. Tang, F. Tarczynski, C. R. Taylor, A. Tkatchenko, R. Tom, P. Touš, M. E. Tuckerman, P. A. Unzueta, Y. Utsumi, L. Vogt-Maranto, J. Weatherston, L. J. Wilkinson, R. D. Willacy, L. Wojtas, G. R. Woollam, Y. Yang, Z. Yang, E. Yonemochi, X. Yue, Q. Zeng, T. Zhou, Y. Zhou, R. Zubatyuk and J. C. Cole, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2024, 80, 548–574 CrossRef CAS PubMed.
- M. A. Neumann, J. Van De Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- S. Trabattoni, M. Moret, M. Campione, L. Raimondo and A. Sassella, Cryst. Growth Des., 2013, 13, 4268–4278 CrossRef CAS.
- A. Hoshino, S. Isoda and T. Kobayashi, J. Cryst. Growth, 1991, 115, 826–830 CrossRef CAS.
- C. A. Mitchell, L. Yu and M. D. Ward, J. Am. Chem. Soc., 2001, 123, 10830–10839 CrossRef CAS PubMed.
- P. Hartman and W. G. Perdok, Acta Crystallogr., 1955, 8, 49–52 CrossRef.
- J. D. H. Donnay and D. Harker, Am. Mineral., 1937, 22, 446–467 CAS.
- M. M. H. Smets, G. Baaklini, A. Tijink, L. Sweers, C. H. F. Vossen, C. Brandel, H. Meekes, H. M. Cuppen and G. Coquerel, Cryst. Growth Des., 2018, 18, 1109–1116 CrossRef CAS PubMed.
- The COST Action BEST-CSP ‘Bringing Experiment and Simulation Together in Crystal Structure Prediction’ is a European network of academic and industrial laboratories and simulation groups that are collaborating to establish a benchmark for physical properties of the organic solid state to help guide simulation efforts find the most reliable predictions, The network receives support from the European Union through the COST Association.
- Cost Action CA22107: Bringing Experiment and Simulation Together in Crystal Structure Prediction (BEST-CSP), https://www.cost.eu/actions/CA22107, (accessed May 28, 2025).
- Bringing Experiment and Simulation Together in Crystal Structure Prediction, https://best-csp.eu/, (accessed May 28, 2025).
- J. Johal and G. Day, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-v8692.
| This journal is © The Royal Society of Chemistry 2025 |