
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis and 35Cl NMR crystallography of ionic cocrystals of phenothiazine drugs†
Zachary T.
Dowdell
 ab,
Austin A.
Peach
ab,
John P.
Purdie
c,
Sean T.
Holmes
ab,
Lara K.
Watanabe
c,
Jeremy M.
Rawson
c and
Robert W.
Schurko
*ab
ab,
Austin A.
Peach
ab,
John P.
Purdie
c,
Sean T.
Holmes
ab,
Lara K.
Watanabe
c,
Jeremy M.
Rawson
c and
Robert W.
Schurko
*ab
aDepartment of Chemistry & Biochemistry, Florida State University, Tallahassee, FL 32306, USA. E-mail: rschurko@fsu.edu
bNational High Magnetic Field Laboratory, Tallahassee, FL 32310, USA
cDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, ON N9B 3P4, Canada
First published on 29th April 2025
Abstract
The solid forms of active pharmaceutical ingredients (APIs), such as polymorphs, cocrystals, and hydrates, possess unique physicochemical properties, including stability, bioavailability, and solubility. These properties are closely tied to their structures, which can be specifically tailored through rational design of novel forms. HCl salts are among the most common solid forms of APIs; however, the formation of pharmaceutical cocrystals of HCl APIs with pharmaceutically acceptable coformers, while relatively rare, can vastly expand the landscape of solid forms. This study explores the mechanochemical synthesis of known cocrystals of promethazine HCl and novel cocrystals of promethazine HCl and chlorpromazine HCl, along with the use of 35Cl solid-state NMR (SSNMR) spectroscopy and density functional theory (DFT) calculations for their structural characterization. Mechanochemical synthesis methods were employed, offering higher yields and better scalability than conventional methods. However, the nature of the products of mechanochemical reactions, which are often powders, presents significant challenges for crystal structure determination. 35Cl SSNMR experiments were used to reveal the chloride environments with distinct hydrogen bonding networks in each solid form, identified through unique quadrupolar interaction parameters. Experimental 35Cl electric field gradient (EFG) tensor data were compared with those from DFT calculations, and used to identify a series of unique chloride ion hydrogen bonding configurations. This work highlights the potential of NMR crystallography for advancing the rational design of cocrystals, improving synthetic methodologies, and providing deeper insights into their solid-state properties.
1. Introduction
Approximately 80% of active pharmaceutical ingredients (APIs) are manufactured and administered as solids,1 which can take on a multitude of solid forms such as salts, solvates, polymorphs, cocrystals, amorphous solid dispersions, and combinations thereof (e.g., ionic cocrystals).2 Currently, more than 50% of APIs are manufactured as solid, organic HCl salts, in which the chloride ions are involved in intricate hydrogen bonding networks that have been demonstrated to impart improved solubility and stability.3,4 Each solid form of an API can have disparate physicochemical properties (e.g., solubility, stability, bioavailability, etc.); as such, the ability to synthesize a variety of solid forms in high purity can permit the manufacture of more efficacious drugs with desirable properties, which is of paramount interest to the pharmaceutical industry as well as consumers of pharmaceutical products.Pharmaceutical cocrystals, which are crystalline, single-phase materials composed of two or more distinct pharmaceutically acceptable molecules within a unit cell (not including hydrates, solvates, or simple salts),5,6 are of particular interest due to the sheer number of coformers that can be used to elicit a wide range of structures and concomitant physicochemical properties.7–9 Cocrystals are often formed via slow evaporation from solution;6,10,11 however, this has several drawbacks, such as the use of relatively large volumes of solvents, high energy inputs, long waiting times (e.g., weeks to months), and difficulties with scaling up output for industrial production.12–18 Despite the widespread use of HCl salts of APIs and the significant potential for ionic cocrystal synthesis, studies on cocrystals of HCl APIs are surprisingly scarce, but are gaining much interest in pharmaceutical research, since they offer the enticing prospects of tailoring different solid forms with modified stability, solubility, or controlled release properties. However, their synthesis using conventional solution-based recrystallization is challenging, since they are often less likely to form, either because of preferred protonation of basic functional groups or less favorable thermodynamic properties, both of which often lead to the formation of a simple HCl salt.
Mechanochemistry offers alternative routes for cocrystal synthesis with quantitative yields, minimal impurities, short timescales (i.e., minutes to hours), and straightforward scalability.19–25 One of the most common mechanochemical synthetic techniques for small scale reactions is ball milling,26–28 in which the reagents (or educts) are placed into a milling jar with ball bearings and ground at a specific frequency for a fixed duration.27,29 A small amount of liquid can be used as well, typically no greater than 2 μL mg−1 of solid, to speed up or facilitate the reaction – this is known as liquid assisted grinding (LAG), while ball milling in the absence of solvent is referred to as neat grinding (NG).30,31 The products and/or mixtures of species that emerge from ball milling reactions often come in the form of microcrystalline powders, which often restricts the application of single-crystal X-ray diffraction (SCXRD) methods (i.e., crystals of suitable size and/or quality cannot be extracted). Fortunately, there are numerous other analytical techniques for characterizing the molecular structures of these products, with powder X-ray diffraction (PXRD), IR/Raman spectroscopy, and solid-state NMR (SSNMR) spectroscopy at the forefront. SSNMR spectroscopy is of particular value for its ability to resolve molecular structures in an atom-by-atom fashion, while also providing “fingerprints” for each solid form, akin to those of PXRD, IR, and Raman methods.
35Cl (spin I = 3/2) SSNMR is well established for characterizing HCl APIs and their cocrystals, since spectra have broad, central transition (CT, +1/2 ↔ −1/2) powder patterns that are tens to hundreds of kHz in breadth, due to influences from the second-order quadrupolar interaction (SOQI) and chemical shift anisotropy (CSA).32–41 The quadrupolar interaction is the interaction between the nuclear quadrupole moment and the electric field gradient (EFG) at the nucleus, the latter of which is described by a second-rank tensor. Measurement of 35Cl EFG tensors is especially useful for characterizing chloride ion environments, since they are very sensitive to their local ground-state electronic environments and can distinguish even minute differences or changes between hydrogen bonding networks.3,42–44 With the aid of dispersion-corrected density functional theory (DFT) geometry optimizations and computation of 35Cl EFG tensors, further insight into the molecular-level structures of materials without known crystal structures can be gained.32,45,46
Given the relative scarcity of ionic HCl cocrystals of APIs and their potential importance in the design of new solid forms with useful physicochemical properties, herein, we discuss mechanochemical syntheses of eleven such cocrystals featuring the phenothiazine drugs promethazine HCl (Ptz), chlorpromazine HCl (Cpz), or promazine HCl (Pmz) (Scheme 1) and a series of neutral coformers (see ESI,† Scheme S1) as starting reagents, which yield 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 promethazine HCl:fumaric acid (Ptz2F), 2:1 promethazine HCl:succinic acid (Ptz2S), 2:1 promethazine HCl:adipic acid (Ptz2A), 2:1 promethazine HCl:oxalic acid (Ptz2O), 2:1 promethazine HCl:glutaric acid (Ptz2G), 1:1 promethazine HCl:malonic acid (PtzMo), 1:1 promethazine HCl:malic acid (PtzMi), 2:1 chlorpromazine HCl:succinic acid (Cpz2S), 2:1 chlorpromazine HCl:fumaric acid (Cpz2F), and 1:1 chlorpromazine HCl:terephthalic acid (CpzT) (see Scheme 2 for examples). We demonstrate (i) the advantages of mechanochemical methods over previously reported synthetic procedures, (ii) the optimization of mechanochemical syntheses to maximize efficiency and yield, (iii) the application of these optimized protocols for the discovery of novel cocrystals, and (iv) the use of 35Cl SSNMR to investigate the structures of known Ptz cocrystals,32 and to aid in the structural elucidation of the novel Ptz and Cpz cocrystals, via NMR crystallography (NMRX).47–49 Lastly, experimental 35Cl EFG tensors are compared to those calculated from structural models that have been geometry optimized using dispersion-corrected plane-wave DFT methods.46 This helps assess how 35Cl EFG tensors of chloride ions are influenced by hydrogen bonding networks, which is key for future applications of quadrupolar NMR-guided crystallographic crystal structure prediction methods to HCl APIs.50,51
:1 promethazine HCl:fumaric acid (Ptz2F), 2:1 promethazine HCl:succinic acid (Ptz2S), 2:1 promethazine HCl:adipic acid (Ptz2A), 2:1 promethazine HCl:oxalic acid (Ptz2O), 2:1 promethazine HCl:glutaric acid (Ptz2G), 1:1 promethazine HCl:malonic acid (PtzMo), 1:1 promethazine HCl:malic acid (PtzMi), 2:1 chlorpromazine HCl:succinic acid (Cpz2S), 2:1 chlorpromazine HCl:fumaric acid (Cpz2F), and 1:1 chlorpromazine HCl:terephthalic acid (CpzT) (see Scheme 2 for examples). We demonstrate (i) the advantages of mechanochemical methods over previously reported synthetic procedures, (ii) the optimization of mechanochemical syntheses to maximize efficiency and yield, (iii) the application of these optimized protocols for the discovery of novel cocrystals, and (iv) the use of 35Cl SSNMR to investigate the structures of known Ptz cocrystals,32 and to aid in the structural elucidation of the novel Ptz and Cpz cocrystals, via NMR crystallography (NMRX).47–49 Lastly, experimental 35Cl EFG tensors are compared to those calculated from structural models that have been geometry optimized using dispersion-corrected plane-wave DFT methods.46 This helps assess how 35Cl EFG tensors of chloride ions are influenced by hydrogen bonding networks, which is key for future applications of quadrupolar NMR-guided crystallographic crystal structure prediction methods to HCl APIs.50,51
 | ||
| Scheme 1 Molecular structures of (A) promethazine HCl (Ptz), (B) chlorpromazine HCl (Cpz), and (C) promazine HCl (Pmz). | ||
 | ||
| Scheme 2 Schematic molecular structures of (A) promethazine HCl:fumaric acid 2:1 cocrystal (Ptz2F), (B) promethazine HCl:glutaric acid 2:1 cocrystal (Ptz2G), (C) chlorpromazine HCl:succinic acid 2:1 cocrystal (Cpz2S), and (D) promethazine HCl:malic acid 1:1 cocrystal (PtzMi). The coformers fumaric acid, glutaric acid, succinic acid, and malic acid are highlighted in red, yellow, blue, and green respectively. | ||
2. Methods
2.1 Materials
Promethazine HCl (Ptz), chlorpromazine HCl (Cpz), promazine HCl (Pmz), and coformers succinic acid (S), oxalic acid (O), malonic acid (Mo), malic acid (Mi), glutaric acid (G), adipic acid (A), terephthalic acid, (T) and fumaric acid (F), were purchased from Millipore Sigma and used as received without further purification. PXRD patterns were acquired for all starting reagents and compared to simulated PXRD patterns of known structures to assess identity and purity.2.2 Cocrystal synthesis
Cocrystals were synthesized using slow evaporation and/or mechanochemically via ball milling. Initial mechanochemical syntheses were carried out following the procedures reported by Borodi et al.10 Mechanochemical syntheses involved either LAG or NG, and were conducted using a Retsch Mixer Mill 400 using two 15 mL stainless steel jars with two 7 mm stainless steel grinding balls, a milling frequency of 30 or 35 Hz, and milling times ranging from 5 to 360 minutes (Tables 1 and 2). No further grinding of samples was needed prior to analyses by SSNMR or PXRD, since all were produced as fine, microcrystalline powders. For syntheses of cocrystals via slow evaporation, supersaturated solutions were prepared in a 20 mL glass vial by dissolving one of three APIs and one of the eight coformers in 2 mL of acetonitrile (MeCN), with gentle heating (ca. 55 °C) to completely dissolve the solids. The solutions were placed in a fume hood to evaporate over a period of 5 days (see Table 3 for details). The resulting crystals were analyzed by SCXRD.| Cocrystal | Original milling timea (minutes) at 27 Hz | Optimized milling timeb (minutes) at 30 Hz | Acetonitrile added for optimized millingb (μL) |
|---|---|---|---|
| a Reported by Borodi et al., unspecified amount of solvent, see ref. 10. b Reported in this work. | |||
| Ptz 2 F | 90 | 30 | 20 |
| Ptz 2 S | n/a | 5 | 15 |
| Ptz 2 O | 180 | 5 | 15 |
| Ptz 2 A | 90 | 10 | 10 |
| Cocrystal | Milling time (minutes) at 30 Hz | Milling timea (minutes) at 35 Hz | Acetonitrileb (μL) |
|---|---|---|---|
| a Ball milling at a rate of 35 Hz was carried out using an MM500 Vario, further optimizations were not attempted. b Small amounts of acetonitrile were added to aid in liquid assisted grinding (LAG), the same amount of acetonitrile was used for syntheses at both milling rates of 30 and 35 Hz. | |||
| PtzMo | 30 | 15 | 20 |
| PtzMi | 10 | 5 | 10 |
| Ptz 2 G | 360 | 180 | 5 |
| Cpz 2 S | 30 | 15 | 15 |
| CpzT | 30 | 15 | 10 |
| Cpz 2 F | 30 | 15 | 10 |
| Cocrystal | Mass of API (mg) | Mol of API (mmol) | Mass of coformer (mg) | Mol of coformer (mmol) | Acetonitrile added (mL) | Time for suitable crystals (days) |
|---|---|---|---|---|---|---|
| Cpz 2 F | 61 | 0.17 | 12 | 0.10 | 2 | 5 |
| Cpz 2 S | 62 | 0.17 | 11 | 0.09 | 2 | 5 |
2.3 Powder X-ray diffraction
PXRD patterns were acquired on a Rigaku Miniflex with a Cu Kα (λ = 1.540593 Å) radiation source and a HyPix-400 MF 2D hybrid pixel array detector. The X-ray tube voltage and amperage were set to 40 kV and 15 mA. Experiments were run with the detector scanning 2θ angles from 5° to 50° with a step size of 0.03° at a speed of 5° min−1. The PXRD patterns for all reagents and cocrystals were compared to those simulated from reported structures10,52–56 using CrystalDiffract software,57 to confirm crystallinity, purity, and the presence of novel phases.2.4 Single-crystal XRD
Single crystals of CpzF and CpzS were mounted on a cryoloop with paratone oil and examined on a Bruker Dual Source D8 Venture diffractometer equipped with Photon 100 CCD area detector using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). An Oxford Cryostream cooler was used to maintain cryogenic temperatures of 170 K for these studies. Data were collected using the APEX-III software, integrated using SAINT58 and corrected for absorption using a multi-scan approach (SADABS).59 Final cell constants were determined from full least squares refinement of all observed reflections. The structures were solved using intrinsic phasing (SHELXT)60 and refined with full least squares refinement on F2 using SHELXL61 in Olex2–1.5 software.62 Simulated PXRD patterns derived from the single-crystal structures were found to be identical to those of the bulk, microcrystalline samples.2.5 Solid-state NMR spectroscopy
2.6 DFT calculations
Quantum chemical calculations were performed on structural models obtained from previous SCXRD studies,10,52,53,56 or the two novel cocrystals obtained through SCXRD analyses presented herein. Geometry optimizations and subsequent calculations of 35Cl EFG and magnetic shielding tensors were performed with plane-wave DFT methods using the CASTEP module within Materials Studio 2020.78,79 These calculations used the generalized gradient approximation (GGA) with the RPBE functional,80 ultrasoft pseudopotentials generated on-the-fly,81 a k-point spacing of 0.05 Å−1, a plane-wave cut-off energy of 800 eV, and an SCF convergence threshold of 5 × 10−7 eV. Geometry optimizations employed the LBFGS method,82 in which the positions of all atoms were relaxed with unit cell parameters remaining fixed at their experimental values. The thresholds for structural convergence included a maximum change in energy of 5 × 10−6 eV per atom, a maximum displacement of 5 × 10−4 Å per atom, and a maximum Cartesian force of 10−2 eV Å−1. A correction to the dispersion energy was introduced using Grimme's semi-empirical two-body dispersion force field,83,84 with an empirical reparameterization introduced by our group (DFT-D2*).46,85–8835Cl magnetic shielding tensors were calculated using the GIPAW method.8935Cl chemical shift values were referenced with respect to NaCl(s) (δiso = −41.1 ppm), which has an absolute isotropic chemical shielding value of 952.38 ppm. EFGShield90 was used to extract the Euler angles describing the relative orientations of magnetic shielding and EFG tensors, which are reported using the ZY′Z′′ convention.3. Results and discussion
3.1 Overview
In this section, we discuss (i) the optimized mechanochemical syntheses of previously reported Ptz cocrystals; (ii) the optimized mechanochemical syntheses of novel Ptz and Cpz cocrystals with various coformers; (iii) the characterization of these cocrystals using a combination of SSNMR, PXRD, and SCXRD; and (iv) the use of DFT calculations to gain insight into relationships between 35Cl EFG tensors and molecular-level structure, as well as the application of NMRX methods for the preliminary characterization of materials without known crystal structures. All cocrystals were characterized by SSNMR; in particular, 35Cl SSNMR is highlighted as the central characterization method due to the unique 35Cl central transition (CT, +1/2 ↔ −1/2) powder patterns resulting from the multifarious hydrogen bonding arrangements of the Cl− anions in both the HCl APIs and their cocrystals. For cocrystals with known crystal structures, DFT-D2* geometry optimizations and calculations of 35Cl EFG tensors are used to correlate the 35Cl EFG tensor parameters and orientations to the local hydrogen bonding environments of the chloride ions – this is crucial for understanding the roles of the chloride ions in stabilizing these novel ionic cocrystals.3.2 General observations
The optimized ball milling protocols served as a starting point for the syntheses or attempted syntheses of novel cocrystals with Ptz, Cpz, and Pmz as starting reagents. Six new cocrystals were synthesized, including three with Ptz and three with Cpz. Most of these cocrystals can be synthesized in less than 30 minutes under LAG conditions, with the exception of Ptz2G, which takes six hours of milling before formation of the cocrystal is completed (Table 2). Attempts to synthesize other novel cocrystals of Ptz, Cpz, and Pmz with different coformers (i.e., tartaric acid, salicylic acid, benzoic acid, benzamide, gentisic acid, xinafoic acid, maleic acid, and aspartic acid) were unsuccessful, despite efforts with various stoichiometric ratios to API to coformer, solvent identity and volume, milling media (i.e., stainless steel and Teflon), and milling times (e.g., 30 minutes and up to five hours). Details on attempts to produce Pmz cocrystals are reported in Table S8.†
425375 (Cpz2F) and 2425376 (Cpz2S).
| Material | Contact typea | H⋯Cl− distanceb (Å) | X⋯Cl− distancec (Å) | X–H⋯Cl− angled (°) | X1–Cl−–X2 anglee (°) | C Q (MHz) | η Q f |
|---|---|---|---|---|---|---|---|
| APIs with a single H⋯Cl− short contact | |||||||
| Ptz | RR′R′′NH+⋯Cl− | 1.964 | 2.969 | 173.8 | n/a | −5.88 | 0.21 |
| Cpz | RR′R′′NH+⋯Cl− | 2.035 | 3.024 | 169.9 | n/a | −4.91 | 0.20 |
| Pmz | RR′R′′NH+⋯Cl− | 2.001 | 2.994 | 171.0 | n/a | −4.79 | 0.38 |
| Cocrystals with two H⋯Cl− short contacts (type 1) | |||||||
|---|---|---|---|---|---|---|---|
| Ptz 2 F | ROOH⋯Cl− | 2.033 | 2.975 | 169.2 | 154.1 | −9.90 | 0.08 |
| RR′R′′NH+⋯Cl− | 2.055 | 3.045 | 174.1 | ||||
| Ptz 2 S | RR′R′′NH+⋯Cl− | 2.053 | 3.046 | 175.1 | 152.0 | −9.47 | 0.11 |
| ROOH⋯Cl− | 2.068 | 3.006 | 168.9 | ||||
| Ptz 2 A | RR′R′′NH+⋯Cl− | 2.039 | 3.034 | 177.5 | 156.8 | −9.00 | 0.19 |
| ROOH⋯Cl− | 2.087 | 3.027 | 169.9 | ||||
| Cocrystals with two H⋯Cl− short contacts (type 2) | |||||||
|---|---|---|---|---|---|---|---|
| a Indicates the functional group involved in the H⋯Cl− bond (i.e., ROOH⋯Cl− signifies a carboxylic acid and RR′R′′NH⋯Cl− signifies a charged tertiary amine contact). b The shortest H⋯Cl− hydrogen bonds (<2.6 Å), as determined via energy minimization and geometry optimization with DFT plane wave calculations. c The distance between the chloride ion and the hydrogen-bond donor atom (X = N, O). d Angle between the hydrogen-bond donor atom (X = N, O), the hydrogen atom, and the chloride ion. e Angle between the first short contact atom (X1), the chloride ion, and the second short contact atom (X2). f DFT calculations yield the sign of the quadrupolar coupling constant, CQ, which is unobtainable from 35Cl SSNMR experiments. It is noted that since Q(35Cl) = −8.165 fm2, positive and negative values of CQ correspond to negative and positive EFGs, respectively. Definitions of CQ and ηQ are given in Table 5. | |||||||
| Ptz 2 O | ROOH⋯Cl− | 2.029 | 2.975 | 168.6 | 99.8 | 5.19 | 0.13 |
| RR′R′′NH+⋯Cl− | 2.065 | 3.050 | 169.6 | ||||
| Cpz 2 F | ROOH⋯Cl− | 2.068 | 3.002 | 166.8 | 101.4 | 5.03 | 0.52 |
| RR′R′′NH+⋯Cl− | 2.093 | 3.069 | 167.3 | ||||
| Cpz 2 S | ROOH⋯Cl− | 2.051 | 2.990 | 167.6 | 102.1 | 5.24 | 0.53 |
| RR′R′′NH+⋯Cl− | 2.094 | 3.075 | 169.5 | ||||
| Material | C Q (MHz) | η Q | δ iso (ppm) | Ω (ppm) | κ | α (°) | β (°) | γ (°) | |
|---|---|---|---|---|---|---|---|---|---|
| a 35Cl SSNMR spectra were also acquired under MAS conditions for these samples. b The principal components of the EFG tensors are defined such that |V33| ≥ |V22| ≥ |V11|. The quadrupolar coupling constant and asymmetry parameter are given by CQ = eQV33/h, and ηQ = (V11 – V22)/V33, respectively. The sign of CQ cannot be determined from the experimental 35Cl spectra. c The principal components of the chemical shift tensors are defined using the frequency-ordered convention, with δ11 ≥ δ22 ≥ δ33. The isotropic chemical shift, span, and skew are given by δiso = (δ11 + δ22 + δ33)/3, Ω = δ11 − δ33, and κ = 3(δ22 − δiso)/Ω, respectively. d The Euler angles α, β, and γ define the relative orientation of the EFG and chemical shift tensors using the ZY′Z′′ convention for rotation. The experimental angles derived from ssNake (which uses the ZX′Z′′ convention) are adjusted to match the calculated values extracted by EFGShield. See ref. 90. e This parameter is not reported since it has little to no effect on the appearance of simulated powder patterns. | |||||||||
| Ptz | Exp | 6.49(4) | 0.25(2) | 66(9) | n/ae | n/ae | n/ae | n/ae | n/ae |
| DFT | −5.88 | 0.21 | 52 | 17 | 0.07 | 28 | 12 | 62 | |
| Cpz | Exp | 5.66(5) | 0.23(3) | 41(2) | n/ae | n/ae | n/ae | n/ae | n/ae |
| DFT | −4.91 | 0.20 | 34 | 73 | 0.01 | 13 | 19 | 189 | |
| Pmz | Exp | 5.11(5) | 0.23(2) | 60(6) | 72(30) | n/ae | 152(20) | 0(45) | 0(45) |
| DFT | −4.79 | 0.38 | 55 | 101 | 0.16 | 214 | 13 | 189 | |
| Ptz 2 F | Exp | 9.42(7) | 0.09(2) | 14(2) | 184(40) | 0.3(2) | 90(10) | 4(8) | 27(20) |
| DFT | −9.90 | 0.08 | 44 | 118 | 0.48 | 45 | 14 | 130 | |
| Ptz 2 S | Exp | 9.41(5) | 0.09(2) | 44(1) | 90(20) | 0.55(20) | 45(15) | 10(10) | 30(30) |
| DFT | −9.47 | 0.11 | 34 | 116 | 0.55 | 38 | 14 | 132 | |
| Ptz 2 O | Exp | 5.07(5) | 0.20(2) | 39(2) | 85(10) | n/ae | 65(12) | 80(5) | 15(7) |
| DFT | 5.19 | 0.13 | 36 | 78 | −0.25 | 69 | 87 | 172 | |
| Ptz 2 A | Exp | 8.9(1) | 0.15(2) | 42(2) | 100(20) | −0.7(3) | 60(5) | 10(10) | 60(10) |
| DFT | −9.00 | 0.19 | 36 | 118 | 0.58 | 55 | 10 | 102 | |
| PtzMo | Exp | 8.64(5) | 0.20(2) | 10(10) | n/ae | n/ae | n/ae | n/ae | n/ae |
| PtzMi | Exp | 4.4(1) | 0.62(4) | 70(2) | 150(20) | −0.5(4) | 85(15) | 75(20) | 20(10) |
| Ptz 2 G | Exp | 8.8(1) | 0.12(2) | 31(8) | 100(20) | 0.7(3) | n/ae | 25(10) | n/ae |
| Cpz 2 S | Exp | 4.95(5) | 0.57(4) | 38(2) | 150(30) | −0.6(3) | 90(15) | 90(10) | 0(10) |
| DFT | 5.24 | 0.53 | 30 | 89 | −0.11 | 129 | 88 | 182 | |
| Cpz 2 F | Exp | 5.13(5) | 0.51(2) | 42(2) | 100(20) | −0.7(4) | 90(20) | 90(20) | 0(10) |
| DFT | 5.03 | 0.52 | 36 | 89 | −0.10 | 137 | 87 | 180 | |
| CpzT | Exp | 4.50(8) | 0.10(2) | 27(1) | 65(15) | −0.8(2) | 85(25) | 90(15) | 0(10) |
3.3 35Cl SSNMR
35Cl SSNMR spectra were acquired for all APIs and cocrystals under static conditions at two magnetic fields, 9.4 T and 18.8 T, as well as under MAS conditions at 18.8 T for some materials (even at 18.8 T, certain samples with large magnitudes of CQ are not amenable to MAS NMR experiments). These data aid in the accurate determination of both the CS and EFG tensors (Table 5), since the effects of the CSA and SOQI on the CT patterns scale proportional and inversely proportional to the magnitude of B0, respectively. MAS can completely or partially average the contributions from CSA, but only partially average the SOQI contributions; this enables facile determination of the quadrupolar coupling constant, asymmetry parameter, and isotropic chemical shift (CQ, ηQ, and δiso, respectively) and simplifies simulations of corresponding static NMR spectra (the latter require eight parameters to fit, including the CS tensor parameters and Euler angles that describe the relative orientation of the EFG and CS tensors; see Table 5 for definitions of these parameters).Unlike the 13C SSNMR spectra, which appear quite similar to one another, the 35Cl SSNMR patterns for each solid form are markedly different (cf.Fig. 1–4). This is due to the great sensitivity of the 35Cl EFG tensors to the local hydrogen-bonding environments of the chloride ions. The 35Cl spectra of each API and cocrystal feature a unique CT patterns that can be simulated to yield distinct sets of quadrupolar and chemical shift parameters (Table 5). The former are easily interpretable in terms of electronic structure: the absolute magnitudes of CQ reflect the degree of spherical symmetry of the ground state electron density about the Cl− ion (increased magnitudes indicate less spherical symmetry), while the ηQ values describe the axial asymmetry of the EFG tensor (i.e., 0 ≤ ηQ ≤ 1, where ηQ = 0 is perfectly axial). All of this, combined with the relatively rapid acquisition times (Tables S1–S5†), makes 35Cl SSNMR an ideal technique for obtaining spectral fingerprints and studying the relationships between the 35Cl EFG tensors and hydrogen bonding environments (see §3.4 for the latter).
 | ||
| Fig. 1 35Cl SSNMR spectra of Ptz, Cpz, and Pmz acquired under static conditions at 18.8 T and 9.4 T (blue) and corresponding simulated spectra (black). | ||
 | ||
| Fig. 2 35Cl SSNMR spectra and simulations of several Ptz cocrystals acquired under static conditions at 9.4 T and static and MAS conditions at 18.8 T. | ||
 | ||
| Fig. 3 35Cl SSNMR spectra and simulations of novel Ptz cocrystals acquired under static conditions at 9.4 T and static and MAS conditions at 18.8 T. | ||
 | ||
| Fig. 4 35Cl SSNMR spectra and simulations of novel Cpz cocrystals acquired under static conditions at 9.4 T and static and MAS conditions at 18.8 T. | ||
In most cases, accurately measuring the chlorine CSA is difficult due to the dominant effects of the SOQI on the 35Cl CT patterns. As a result, the uncertainties in the CS tensor parameters (i.e., δiso, Ω, and κ) and Euler angles, which describe the relative orientations of the different tensors, are often high. This means that unlike the 35Cl EFG tensors, we cannot reliably identify simple correlations between the chlorine CS tensor parameters and the local chloride ion environments.92 Nonetheless, inclusion of chlorine CS tensor parameters in the simulations aids in differentiating the powder patterns of the different solid forms.
The CQ values of Ptz2A, Ptz2F, and Ptz2S are large in comparison to that of Ptz and are at the upper end of those typically observed for chloride ions (i.e., approaching 9–10 MHz), suggesting low spherical symmetries in the surrounding electron density. The range of ηQ values is from 0.05 to 0.25, indicating EFG tensors with high degrees of axial symmetry. Such quadrupolar parameters for chloride ions are usually indicative of hydrogen bonding arrangements featuring two short H⋯Cl− contacts (i.e., r(H⋯Cl−) ≲ 2.2 Å) with an almost linear H⋯Cl−⋯H arrangement (i.e., ∠(H⋯Cl−⋯H) ≈ 180°, see §3.4).42,93,94 Due to the large CQ values, accurate CS tensor parameters are difficult to obtain, even with data at 18.8 T. In this series, Ptz2O is the exception, having a CQ smaller than that of Ptz (i.e., 5.07 and 6.49 MHz, respectively), and a span Ω = 85 ppm. Ptz2F and Ptz2S have very similar CQ and ηQ parameters, which is expected because these cocrystals are isostructural; hence, these cocrystals are most easily differentiated in these spectra by their distinct δiso values (14 and 44 ppm, respectively). Although these cocrystals are isostructural, subtle differences in hydrogen bond lengths influence the local electronic structure, resulting in distinct 35Cl chemical shifts.
3.4 NMR crystallography using 35Cl EFG tensors
Plane-wave DFT calculations were conducted on structural models based on the crystal structures of the three APIs (Ptz, Pmz, and Cpz), four previously reported cocrystals (Ptz2F, Ptz2S, Ptz2A, and Ptz2O), and two novel cocrystals (Cpz2F and Cpz2S). The structural models were geometry optimized using dispersion-corrected plane-wave DFT calculations (DFT-D2*), which are necessary to obtain low-energy configurations of atoms involved in intermolecular hydrogen-bonding interactions.45,8535Cl EFG and CS tensors were then calculated for the refined structural models (Table 5).Since there is generally good agreement between experimental and calculated 35Cl EFG tensors (Fig. 5), it is possible to explore their relationships to the structural environments of the chloride ions, which are sensitive to variations in the number and arrangement of short H⋯Cl− contacts and the nature of the hydrogen-bonding moieties (Table 6).32,34,39,41,43,92,94,99 Key interatomic distances and interbond angles obtained from DFT-D2* geometry optimized structures are summarized in Table 4.
 | ||
| Fig. 5 Relationships between principal components of the 35Cl EFG tensors (i.e., Vkk, where k = 1, 2, or 3 – see Table 5 for definitions) that have been measured experimentally and calculated from DFT-D2* geometry-optimized structures. The grey dotted line represents perfect correlation. | ||
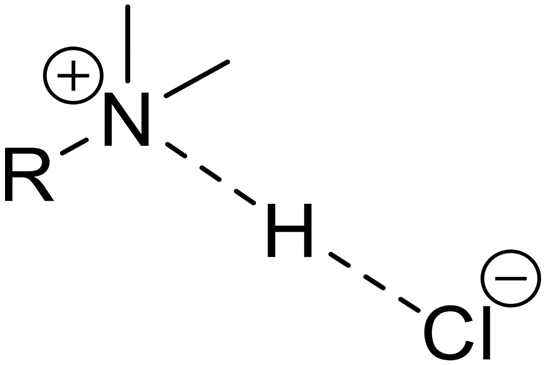
| Hydrogen bonding arrangement | Systems | Number of short H⋯Cl contacts | Range of CQ values | Range of ηQ values | Respective signs of V33 and CQ | Direction of V33 |
|---|---|---|---|---|---|---|
| a These materials do not have a known crystal structure. | ||||||
|
Ptz, Cpz, Pmz | 1 | ∼4.5–6.0 | ∼0.0–0.4 | +/— | Along/near the hydrogen bond |
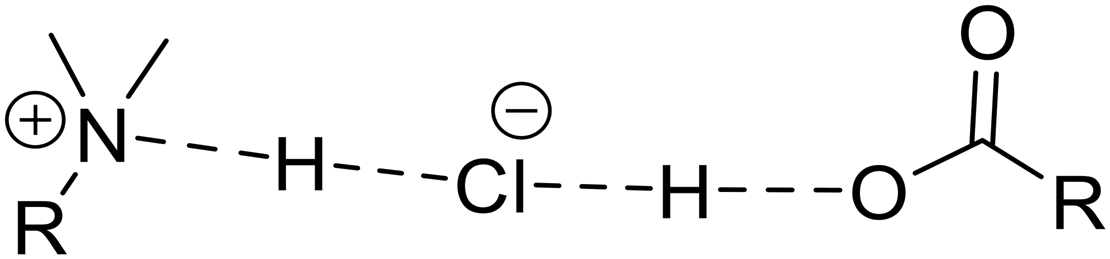
|
Ptz2F, Ptz2S, Ptz2A, Ptz2G,a PtzMoa | 2 | ∼8.5–10.0 | ∼0.0–0.2 | +/— | Along/near the hydrogen bond (type 1) |

|
Ptz2O, Cpz2S, Cpz2F, CpzT,a PtzMia | 2 | ∼4.0–6.0 | ∼0.1–0.7 | —/+ | Perpendicular to the H⋯Cl⋯H plane (type 2) |
The chloride ions of Ptz, Pmz, and Cpz have moderate magnitudes of |CQ| and low values of ηQ (i.e., the largest component of the EFG tensor, V33, is the distinct component, and V11 and V22 are similar in magnitude to one another and opposite in sign to V33). Based on the growing database of 35Cl EFG tensor data for cationic organic chloride salts and HCl APIs,32,34,41,43 these values indicate the presence of a single, short H⋯Cl− contact of ≲ 2.2 Å, with V33 oriented along or near its direction. The DFT-D2* refined model structures confirm this, revealing that (i) each has a single RR′R′′NH+⋯Cl− short contact; (ii) each V33 is oriented close to the short contact, with ∠(V33⋯Cl−⋯H) ranging from ca. 5° to 11°; (iii) the sign of CQ is calculated to be negative in all cases (the sign cannot be determined from the experimental 35Cl NMR spectra, but is useful in structural interpretation), which is a result of the negative quadrupole moment, meaning that if V33 > 0 then CQ < 0; and (iv) the magnitude of |CQ| would normally be expected to increase with decreasing contact length in the order Ptz > Cpz > Pmz – although Ptz has the shortest contact and largest |CQ|, a clear relationship is not discerned for Cpz and Pmz due to variations in ∠(V33⋯Cl−⋯H) (Fig. S11†).
The Ptz cocrystals with known structures are now considered. Ptz2F, Ptz2S, and Ptz2A have large magnitudes of |CQ| and low values of ηQ. This indicates two short H⋯Cl− contacts (i.e., r(H⋯Cl−) ≲ 2.2 Å) with ∠(H⋯Cl−⋯H) approaching 180°, with V33 oriented along or near these vectors (Fig. S12†).100–103 This is borne out by the DFT-D2* calculations, which predict two short H⋯Cl− contacts in each system (RR′R′′NH+⋯Cl− and COOH⋯Cl−), with the functional groups sitting approximately on opposite sides of the chloride ion (as indicated by the H⋯Cl−⋯H angles, which range from ca. 153° to 156°), and 35Cl EFG tensors of high axial symmetry with V33 (that are positive in sign) oriented near the H⋯Cl− contacts.104
Ptz 2 O stands out in this series, with values of CQ = 5.07 MHz and ηQ = 0.20, akin to those of the APIs with single short H⋯Cl− contacts. Examination of the DFT-D2* structure confirms the presence of R′R2NH+⋯Cl− and COOH⋯Cl− short contacts, but in an arrangement such that the H⋯Cl−⋯H angle is 99.8° (i.e., the moieties are not on opposite sides of the chloride ion). In this instance, V33 is not oriented near the short contacts, but rather, approximately perpendicular to the H⋯Cl−⋯H plane (Fig. S12†). Furthermore, V11 and V22, which reside in the H⋯Cl−⋯H plane, are similar in magnitude and positive (V33 is negative, unlike the previous systems), consistent with observations for many other systems.39,42,88,92,94,105,106
Finally, Cpz2F and Cpz2S feature moderate |CQ| values and larger ηQ values (the latter indicate EFG tensors of non-axial symmetry, with V11 as the distinct component) (Fig. S13†). As in the case of Ptz2O, the 35Cl EFG tensors are oriented such that V33 (negative values) are approximately perpendicular to the H⋯Cl−⋯H planes (V11 and V22 are positive and approximately in the planes). Unlike Ptz2O, the RR′R′′NH+⋯Cl− and COOH⋯Cl− contacts have different lengths, with V22 aligned nearer to the longer RR′R′′NH+⋯Cl− contact.
These insights lead to four simple relationships that can be used to make preliminary predictions about the chloride ion environments in cocrystals without known crystal structures and allow us to classify the systems described in this work into one of three possible geometric arrangements (Table 6). Ptz2G and PtzMo have large values of CQ (>8.8 MHz) and small ηQ values (<0.16), suggesting Cl− ion environments similar to those of Ptz2F, Ptz2S, and Ptz2A, with two short contacts (RR′R′′NH+⋯Cl− and COOH⋯Cl−) sitting on opposite sides of the chloride ion with an ∠(H⋯Cl⋯H) approaching 180°. CpzT has an intermediate value of CQ (4.41 MHz) and small ηQ value (0.04), similar to Ptz2O, which features two short contacts (RR′R′′NH+⋯Cl− and COOH⋯Cl−) with moieties not on opposite sides of the chloride ion and V33 orientated ca. 90° to the plane of these contacts. PtzMi and PtzMo are interesting cases, since both have a 1:1 ratio of API:coformer, possibly suggesting dimeric structures.107–109 However, the quadrupolar parameters of PtzMo are similar to those of Ptz2F, Ptz2S, Ptz2G and Ptz2A, suggesting an analogous “near-linear” H⋯Cl⋯H configuration, whereas for PtzMi, its distinct values of CQ = 4.42 MHz and ηQ = 0.62 suggests an arrangement of contacts similar to Cpz2F and Cpz2S (i.e., ∠(H⋯Cl−⋯H) of ca. 90–100°, with longer RR′R′′NH+⋯Cl− and shorter COOH⋯Cl− contacts). In the case of PtzMo, we propose four possible arrangements based on crystal structures of pure malonic acid and several concomitant solid forms (in a 1:1 stoichiometric ratio) found in the literature (Scheme S2†).110–113 Of these, we consider arrangement B the most plausible, as it would likely result in the densest crystal packing.
Without geometry-optimized structures and corresponding 35Cl EFG tensors that can be obtained from DFT calculations, we are currently limited to these simple structural interpretations. However, these experimental data, along with the combined experimental and calculated NMR data from well-characterized systems and high-quality PXRD data for Rietveld refinement, may inform future NMRX solutions of these structures and others. This will be pursued using the data herein along with our separate and continuing efforts on 35Cl SSNMR-based NMRX methods, including quadrupolar-guided NMR crystallography crystal structure prediction (QNMRX-CSP) methods.3,41,44,51,92
4. Conclusions
This study has demonstrated: (i) the rapid and quantitative mechanochemical synthesis of eleven ionic HCl cocrystals featuring two cationic APIs, a number of neutral, pharmaceutically acceptable coformers, and chloride ions; (ii) the advantages of mechanochemical methods, including their optimization for efficiency and application for discovering novel cocrystals; (iii) the value of 35Cl SSNMR spectroscopy for the analysis of known Ptz cocrystals and elucidation of novel Ptz and Cpz cocrystal structures; and (iv) the use of plane-wave DFT calculations of 35Cl EFG tensors to investigate their relationships to the complex hydrogen bonding networks of the chloride ions.This combination of mechanochemical synthesis and subsequent characterization has extended our knowledge in terms of what is possible for synthesizing cocrystals of this largely unexplored class of ionic HCl API cocrystals, which may allow us to rationally choose coformers that will lead to an increased number of novel pharmaceutical cocrystals, and perhaps even achieve novel forms of promazine HCl cocrystals, which have been unsuccessful to date – at this point, we are unclear why this is the case for Pmz, since the only difference between the molecular structures of Cpz and Pmz is a covalently-bound chlorine atom in the former. To determine and refine crystal structures for novel cocrystals using SSNMR of quadrupolar nuclides, we have two promising options going forward: (i) our new QNMRX-CSP protocol and (ii) NMR-guided Rietveld refinement.50,51,114–116
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the CCDC under 2425375 and 2425376.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Florida State University and the National High Magnetic Field Laboratory for funding this research. The National High Magnetic Field Laboratory was supported by the National Science Foundation through NSF/DMR-1644779, DMR-2128556 and the State of Florida. The authors also thank the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery grants 2016-06642 and 2020-04627 for RWS and JMR, respectively), the Canadian Foundation for Innovation, the Ontario Innovation Trust, the Ontario Research Fund, and the University of Windsor for supporting the initial stages of this project. A portion of this research used resources provided by the X-ray Crystallography Center at the FSU Department of Chemistry and Biochemistry. The authors also acknowledge use of the SSNMR and SCXRD facilities at the University of Windsor, supported by the Canadian Foundation for Innovation, Ontario Innovation Trust, and the University of Windsor.References
- M. Geppi, G. Mollica, S. Borsacchi and C. A. Veracini, Solid-State NMR Studies of Pharmaceutical Systems, Appl. Spectrosc. Rev., 2008, 43(3), 202–302, DOI:10.1080/05704920801944338.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Pharmaceutical Cocrystals: Along the Path to Improved Medicines, Chem. Commun., 2016, 52(4), 640–655, 10.1039/C5CC08216A.
- H. Hamaed, J. M. Pawlowski, B. F. T. T. Cooper, R. Fu, S. H. Eichhorn and R. W. Schurko, Application of Solid-State 35Cl NMR to the Structural Characterization of Hydrochloride Pharmaceuticals and Their Polymorphs, J. Am. Chem. Soc., 2008, 130(33), 11056–11065, DOI:10.1021/ja802486q.
- G. S. Paulekuhn, J. B. Dressman and C. Saal, Trends in Active Pharmaceutical Ingredient Salt Selection Based on Analysis of the Orange Book Database, J. Med. Chem., 2007, 50(26), 6665–6672, DOI:10.1021/JM701032Y.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar and M. J. Zaworotko, Salts, and Cocrystals: What's in a Name?, Cryst. Growth Des., 2012, 12(5), 2147–2152, DOI:10.1021/CG3002948.
- S. L. Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly and G. P. Stahly, Crystal Engineering Approach To Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids, J. Am. Chem. Soc., 2004, 126(41), 13335–13342, DOI:10.1021/ja048114o.
- S. Datta and D. J. W. Grant, Crystal Structures of Drugs: Advances in Determination, Prediction and Engineering, Nat. Rev. Drug Discovery, 2004, 3(1), 42–57, DOI:10.1038/nrd1280.
- A. M. Healy, Z. A. Worku, D. Kumar and A. M. Madi, Pharmaceutical Solvates, Hydrates and Amorphous Forms: A Special Emphasis on Cocrystals, Adv. Drug Delivery Rev., 2017, 117, 25–46, DOI:10.1016/J.ADDR.2017.03.002.
- E. Pindelska, A. Sokal and W. Kolodziejski, Pharmaceutical Cocrystals, Salts and Polymorphs: Advanced Characterization Techniques, Adv. Drug Delivery Rev., 2017, 117, 111–146, DOI:10.1016/J.ADDR.2017.09.014.
- G. Borodi, A. Turza, O. Onija and A. Bende, Succinic, Fumaric, Adipic and Oxalic Acid Cocrystals of Promethazine Hydrochloride, Acta Crystallogr., Sect. C:Struct. Chem., 2019, 75(2), 107–119, DOI:10.1107/S2053229618017904.
- A. Olejniczak, K. Krukle-Berziņa and A. Katrusiak, Pressure-Stabilized Solvates of Xylazine Hydrochloride, Cryst. Growth Des., 2016, 16(7), 3756–3762, DOI:10.1021/ACS.CGD.6B00264.
- A. Saha, A. A. Ahangar, A. A. Dar, S. Thirunahari and J. V. Parambil, Pharmaceutical Cocrystals: A Perspective on Development and Scale-up of Solution Cocrystallization, Cryst. Growth Des., 2023, 23(11), 7558–7581, DOI:10.1021/acs.cgd.2c01553.
- N. Pawar, A. Saha, N. Nandan and J. V. Parambil, Solution Cocrystallization: A Scalable Approach for Cocrystal Production, Crystals, 2021, 11(3), 303, DOI:10.3390/CRYST11030303.
- S. N. Madanayake, A. Manipura, R. Thakuria and N. M. Adassooriya, Opportunities and Challenges in Mechanochemical Cocrystallization toward Scaled-Up Pharmaceutical Manufacturing, Org. Process Res. Dev., 2023, 27(3), 409–422, DOI:10.1021/acs.oprd.2c00314.
- M. S. Hossain Mithu, S. Economidou, V. Trivedi, S. Bhatt and D. Douroumis, Advanced Methodologies for Pharmaceutical Salt Synthesis, Cryst. Growth Des., 2021, 21(2), 1358–1374, DOI:10.1021/acs.cgd.0c01427.
- A. R. Buist, A. R. Kennedy and C. Manzie, Four Salt Phases of Theophylline, Acta Crystallogr., Sect. C:Struct. Chem., 2014, 70(2), 220–224, DOI:10.1107/S2053229614000825.
- M. R. J. Elsegood, D. A. Hussain and N. M. Sanchez-Ballester, Redetermination of Picolinic Acid Hydro-Chloride at 150 K, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63(10), o4044–o4044, DOI:10.1107/S1600536807044030.
- H. Ferjani, H. Chebbi, A. Guesmi, O. S. Alruqi and S. A. Al-Hussain, Two-Dimensional Hydrogen-Bonded Crystal Structure, Hirshfeld Surface Analysis and Morphology Prediction of a New Polymorph of 1H-Nicotineamidium Chloride Salt, Crystals, 2019, 9(11), 571, DOI:10.3390/CRYST9110571.
- D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Organic Synthesis by Twin Screw Extrusion (TSE): Continuous, Scalable and Solvent-Free, Green Chem., 2017, 19(6), 1507–1518, 10.1039/C6GC03413F.
- D. J. Berry, C. C. Seaton, W. Clegg, R. W. Harrington, S. J. Coles, P. N. Horton, M. B. Hursthouse, R. Storey and N. Blagden, Applying Hot-Stage Microscopy to Co-Crystal Screening: A Study of Nicotinamide with Seven Active Pharmaceutical Ingredients, Cryst. Growth Des., 2008, 8(5), 1697–1712, DOI:10.1021/cg800035w.
- T. Stolar and K. Užarević, Mechanochemistry: An Efficient and Versatile Toolbox for Synthesis, Transformation, and Functionalization of Porous Metal–Organic Frameworks, CrystEngComm, 2020, 22(27), 4511–4525, 10.1039/D0CE00091D.
- A. Bodach, A. Portet, F. Winkelmann, B. Herrmann, F. Gallou, E. Ponnusamy, D. Virieux, E. Colacino and M. Felderhoff, Scalability of Pharmaceutical Co-Crystal Formation by Mechanochemistry in Batch, ChemSusChem, 2024, 17(6), e202301220, DOI:10.1002/CSSC.202301220.
- K. Trzeciak, M. K. Dudek and M. J. Potrzebowski, Mechanochemical Transformations of Pharmaceutical Cocrystals: Polymorphs and Coformer Exchange, Chem. – Eur. J., 2024, 30(71), e202402683, DOI:10.1002/CHEM.202402683.
- J.-H. Schöbel, F. Winkelmann, J. Bicker and M. Felderhoff, Mechanochemical Kilogram-Scale Synthesis of Rac -Ibuprofen:Nicotinamide Co-Crystals Using a Drum Mill, RSC Mechanochem., 2025, 2, 224–229, 10.1039/D4MR00096J.
- D. Šilić, B. Cetina-Čižmek, M. Antonijevic and D. Douroumis, One-Step Synthesis of a Drug-Drug Cocrystal Hydrate Using Hot Melt Extrusion, Cryst. Growth Des., 2024, 24(19), 8044–8055, DOI:10.1021/acs.cgd.4c00974.
- T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for Synthesis, Angew. Chem., Int. Ed., 2019, 1018–1029, DOI:10.1002/anie.201906755.
- L. S. Germann, M. Arhangelskis, M. Etter, R. E. Dinnebier and T. Friščić, Challenging the Ostwald Rule of Stages in Mechanochemical Cocrystallisation, Chem. Sci., 2020, 11, 10092–10100, 10.1039/d0sc03629c.
- F. Fischer, D. Lubjuhn, S. Greiser, K. Rademann and F. Emmerling, Supply and Demand in the Ball Mill: Competitive Cocrystal Reactions, Cryst. Growth Des., 2016, 16(10), 5843–5851, DOI:10.1021/acs.cgd.6b00928.
- D. Margetić and V. Štrukil, Practical Considerations in Mechanochemical Organic Synthesis, 2016, DOI:10.1016/b978-0-12-802184-2.00001-7.
- T. Friić, S. L. Childs, S. A. A. Rizvi and W. Jones, The Role of Solvent in Mechanochemical and Sonochemical Cocrystal Formation: A Solubility-Based Approach for Predicting Cocrystallisation Outcome, CrystEngComm, 2009, 11(3), 418–426, 10.1039/B815174A.
- D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Mechanochemical Preparation of Molecular and Supramolecular Organometallic Materials and Coordination Networks, Dalton Trans., 2006,(10), 1249–1263, 10.1039/B516165G.
- H. Hamaed, J. M. Pawlowski, B. F. T. Cooper, R. Fu, S. H. Eichhorn and R. W. Schurko, Application of Solid-State 35 Cl NMR to the Structural Characterization of Hydrochloride Pharmaceuticals and Their Polymorphs, J. Am. Chem. Soc., 2008, 130(33), 11056–11065, DOI:10.1021/ja802486q.
- D. A. Hirsh, A. J. Rossini, L. Emsley and R. W. Schurko, 35Cl Dynamic Nuclear Polarization Solid-State NMR of Active Pharmaceutical Ingredients, Phys. Chem. Chem. Phys., 2016, 18(37), 25893–25904, 10.1039/c6cp04353d.
- D. L. Bryce and G. D. Sward, Chlorine-35/37 NMR Spectroscopy of Solid Amino Acid Hydrochlorides: Refinement of Hydrogen-Bonded Proton Positions Using Experiment and Theory, J. Phys. Chem. B, 2006, 110(51), 26461–26470, DOI:10.1021/jp065878c.
- P. M. J. Szell and D. L. Bryce, Cl Solid-State NMR and Computational Study of Chlorine Halogen Bond Donors in Single-Component Crystalline Chloronitriles, J. Phys. Chem. C, 2016, 120(20), 11121–11130, DOI:10.1021/acs.jpcc.6b02806.
- J. Struppe, C. M. Quinn, S. Sarkar, A. M. Gronenborn and T. Polenova, Ultrafast 1 H MAS NMR Crystallography for Natural Abundance Pharmaceutical Compounds, Mol. Pharmaceutics, 2020, 17(2), acs.molpharmaceut.9b01157, DOI:10.1021/acs.molpharmaceut.9b01157.
- F. G. Vogt, G. R. Williams, M. Strohmeier, M. N. Johnson and R. C. B. Copley, Solid-State NMR Analysis of a Complex Crystalline Phase of Ronacaleret Hydrochloride, J. Phys. Chem. B, 2014, 118(34), 10266–10284, DOI:10.1021/jp505061j.
- M. K. Pandey, H. Kato, Y. Ishii and Y. Nishiyama, Two-Dimensional Proton-Detected 35Cl/1H Correlation Solid-State NMR Experiment under Fast Magic Angle Sample Spinning: Application to Pharmaceutical Compounds, Phys. Chem. Chem. Phys., 2016, 18(8), 6209–6216, 10.1039/C5CP06042G.
- L. M. Abdulla, A. A. Peach, S. T. Holmes, Z. T. Dowdell, L. K. Watanabe, E. M. Iacobelli, D. A. Hirsh, J. M. Rawson and R. W. Schurko, Synthesis and Characterization of Xylazine Hydrochloride Polymorphs, Hydrates, and Cocrystals: A 35Cl Solid-State NMR and DFT Study, Cryst. Growth Des., 2023, 23(5), 3412–3426, DOI:10.1021/acs.cgd.2c01539.
- A. J. Stirk, S. T. Holmes, F. E. S. Souza, I. Hung, Z. Gan, J. F. Britten, A. W. Rey and R. W. Schurko, An Unusual Ionic Cocrystal of Ponatinib Hydrochloride: Characterization by Single-Crystal X-Ray Diffraction and Ultra-High Field NMR Spectroscopy, CrystEngComm, 2024, 26(9), 1219–1233, 10.1039/D3CE01062G.
- M. Hildebrand, H. Hamaed, A. M. Namespetra, J. M. Donohue, R. Fu, I. Hung, Z. Gan and R. W. Schurko, 35 Cl Solid-State NMR of HCl Salts of Active Pharmaceutical Ingredients: Structural Prediction, Spectral Fingerprinting and Polymorph Recognition, CrystEngComm, 2014, 16(31), 7334–7356, 10.1039/C4CE00544A.
- A. A. Peach, D. A. Hirsh, S. T. Holmes and R. W. Schurko, Mechanochemical Syntheses and 35 Cl Solid-State NMR Characterization of Fluoxetine HCl Cocrystals, CrystEngComm, 2018, 20(20), 2780–2792, 10.1039/C8CE00378E.
- A. M. Namespetra, D. A. Hirsh, M. P. Hildebrand, A. R. Sandre, H. Hamaed, J. M. Rawson and R. W. Schurko, 35Cl Solid-State NMR Spectroscopy of HCl Pharmaceuticals and Their Polymorphs in Bulk and Dosage Forms, CrystEngComm, 2016, 18(33), 6213–6232, 10.1039/c6ce01069e.
- C. S. Vojvodin, S. T. Holmes, L. K. Watanabe, J. M. Rawson and R. W. Schurko, Multi-Component Crystals Containing Urea: Mechanochemical Synthesis and Characterization by 35 Cl Solid-State NMR Spectroscopy and DFT Calculations, CrystEngComm, 2022, 24(14), 2626–2641, 10.1039/D1CE01610E.
- S. T. Holmes, C. S. Vojvodin and R. W. Schurko, Dispersion-Corrected DFT Methods for Applications in Nuclear Magnetic Resonance Crystallography, J. Phys. Chem. A, 2020, 124(49), 10312–10323, DOI:10.1021/acs.jpca.0c06372.
- S. T. Holmes and R. W. Schurko, Refining Crystal Structures with Quadrupolar NMR and Dispersion-Corrected Density Functional Theory, J. Phys. Chem. C, 2018, 122(3), 1809–1820, DOI:10.1021/acs.jpcc.7b12314.
- D. L. Bryce, NMR Crystallography: Structure and Properties of Materials from Solid-State Nuclear Magnetic Resonance Observables, IUCrJ, 2017, 4(4), 350–359, DOI:10.1107/S2052252517006042.
- P. Hodgkinson, NMR Crystallography of Molecular Organics, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53, DOI:10.1016/J.PNMRS.2020.03.001.
- F. Taulelle, Fundamental Principles of NMR Crystallography, John Wiley & Sons, Ltd, Chichester, UK, 2009, DOI:10.1002/9780470034590.emrstm1003.
- A. A. Peach, C. H. Fleischer, K. Levin, S. T. Holmes, J. E. Sanchez and R. W. Schurko, Quadrupolar NMR Crystallography Guided Crystal Structure Prediction (QNMRX-CSP), CrystEngComm, 2024, 26(35), 4782–4803, 10.1039/D3CE01306E.
- C. H. Fleischer, S. T. Holmes, K. Levin, S. L. Veinberg and R. W. Schurko, Characterization of Ephedrine HCl and Pseudoephedrine HCl Using Quadrupolar NMR Crystallography Guided Crystal Structure Prediction, Faraday Discuss., 2025, 255(0), 88–118, 10.1039/D4FD00089G.
- D. Feil, M. H. Linck and J. J. H. McDowell, Preliminary X-Ray Data on Phenothiazine and Certain of Its Derivatives, Nature, 1965, 207(4994), 285–286, DOI:10.1038/207285a0 DOI:10.1038/207285a0.
- W. I. F. David, K. Shankland and N. Shankland, Routine Determination of Molecular Crystal Structures from Powder Diffraction Data, Chem. Commun., 1998, 931–932, 10.1039/A800855H.
- C. J. Brown and IUCr., The Crystal Structure of Fumaric Acid, Acta Crystallogr., 1966, 21(1), 1–5, DOI:10.1107/S0365110X66002226.
- V. R. Thalladi, M. Nüsse and R. Boese, The Melting Point Alternation in α,ω-Alkanedicarboxylic Acids, J. Am. Chem. Soc., 2000, 122(38), 9227–9236, DOI:10.1021/JA0011459.
- G. Borodi, M. M. Pop, O. Onija and X. Filip, Distinct Disordered Forms of Promethazine Hydrochloride: A Case of Intergrowth of Polymorphic Domains?, Cryst. Growth Des., 2012, 12(12), 5846–5851, DOI:10.1021/cg300943b DOI:10.1021/cg300943b.
- CrystalDiffract Overview, http://crystalmaker.com/crystaldiffract/ (accessed 2022-11-11) Search PubMed.
- Bruker, SAINT, Bruker AXS Inc, Bruker AXS Inc., Madison, WI, USA, 2002 Search PubMed.
- Bruker, SADABS, Bruker AXS Inc., Madison, WI, USA, 2002 Search PubMed.
- G. M. Sheldrick, IUCr, SHELXT – Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., 2015, 71(1), 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., 2015, 71(1), 3–8, DOI:10.1107/S2053229614024218.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A Complete Structure Solution, Refinement and Analysis Program, J. Appl. Crystallogr., 2009, 42(2), 339–341, DOI:10.1107/S0021889808042726.
- L. A. O'Dell, A. J. Rossini and R. W. Schurko, Acquisition of Ultra-Wideline NMR Spectra from Quadrupolar Nuclei by Frequency Stepped WURST–QCPMG, Chem. Phys. Lett., 2009, 468(4–6), 330–335, DOI:10.1016/j.cplett.2008.12.044.
- L. A. O'Dell and R. W. Schurko, QCPMG Using Adiabatic Pulses for Faster Acquisition of Ultra-Wideline NMR Spectra, Chem. Phys. Lett., 2008, 464(1–3), 97–102, DOI:10.1016/J.CPLETT.2008.08.095.
- R. Bhattacharyya and L. Frydman, Quadrupolar Nuclear Magnetic Resonance Spectroscopy in Solids Using Frequency-Swept Echoing Pulses, J. Chem. Phys., 2007, 127(19), 194503, DOI:10.1063/1.2793783.
- Ē. Kupče and R. Freeman, Adiabatic Pulses for Wideband Inversion and Broadband Decoupling, J. Magn. Reson., Ser. A, 1995, 115(2), 273–276, DOI:10.1006/JMRA.1995.1179.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, QCPMG-MAS NMR of Half-Integer Quadrupolar Nuclei, J. Magn. Reson., 1998, 131(1), 144–147, DOI:10.1006/JMRE.1997.1341.
- O. B. Peersen, X. Wu, I. Kustanovich and S. O. Smith, Variable-Amplitude Cross-Polarization MAS NMR, J. Magn. Reson., Ser. A, 1993, 104(3), 334–339, DOI:10.1006/JMRA.1993.1231.
- G. Metz, X. Wu and S. O. Smith, Ramped-Amplitude Cross Polarization in Magic-Angle-Spinning NMR, J. Magn. Reson., Ser. A, 1994, 110(2), 219–227, DOI:10.1006/JMRA.1994.1208.
- J. Schaefer and E. O. Stejskal, Carbon-13 Nuclear Magnetic Resonance of Polymers Spinning at the Magic Angle, J. Am. Chem. Soc., 1976, 98(4), 1031–1032, DOI:10.1021/JA00420A036.
- A. Pines, M. G. Gibby and J. S. Waugh, Proton-Enhanced Nuclear Induction Spectroscopy 13C Chemical Shielding Anisotropy in Some Organic Solids, Chem. Phys. Lett., 1972, 15(3), 373–376, DOI:10.1016/0009-2614(72)80191-X.
- A. Pines, M. G. Gibby and J. S. Waugh, Proton-enhanced NMR of Dilute Spins in Solids, J. Chem. Phys., 1973, 59(2), 569–590, DOI:10.1063/1.1680061.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids, J. Magn. Reson., 2000, 142(1), 97–101, DOI:10.1006/JMRE.1999.1896.
- R. E. Taylor, 13C CP/MAS: Application to Glycine, Concepts Magn. Reson., Part A, 2004, 22A(2), 79–89, DOI:10.1002/CMR.A.20015.
- M. J. Potrzebowski, P. Tekely and Y. Dusausoy, Comment to 13C-NMR Studies of α and γ Polymorphs of Glycine, Solid State Nucl. Magn. Reson., 1998, 11(3–4), 253–257, DOI:10.1016/S0926-2040(98)00028-9.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, SsNake: A Cross-Platform Open-Source NMR Data Processing and Fitting Application, J. Magn. Reson., 2019, 301, 56–66, DOI:10.1016/J.JMR.2019.02.006.
- K. Eichele WSolids1 ver. 1.21.7, http://anorganik.uni-tuebingen.de/klaus/soft/index.php?p=wsolids1/wsolids1 (accessed 2025-01-12) Search PubMed.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Iterative Minimization Techniques for Ab Initio Total-Energy Calculations: Molecular Dynamics and Conjugate Gradients, Rev. Mod. Phys., 1992, 64(4), 1045, DOI:10.1103/RevModPhys.64.1045.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First Principles Methods Using CASTEP, Z. Kristallogr. - Cryst. Mater., 2005, 220(5–6), 567–570, DOI:10.1524/zkri.220.5.567.65075.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(11), 7413, DOI:10.1103/PhysRevB.59.7413.
- J. R. Yates, C. J. Pickard and F. Mauri, Calculation of NMR Chemical Shifts for Extended Systems Using Ultrasoft Pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76(2), 024401, DOI:10.1103/PhysRevB.76.024401.
- B. G. Pfrommer, M. Côté, S. G. Louie and M. L. Cohen, Relaxation of Crystals with the Quasi-Newton Method, J. Comput. Phys., 1997, 131(1), 233–240, DOI:10.1006/JCPH.1996.5612.
- S. Grimme, Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 27(15), 1787–1799, DOI:10.1002/jcc.20495.
- E. R. McNellis, J. Meyer and K. Reuter, Azobenzene at Coinage Metal Surfaces: Role of Dispersive van Der Waals Interactions, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80(20), 205414, DOI:10.1103/PHYSREVB.80.205414.
- S. T. Holmes and R. W. Schurko, Refining Crystal Structures with Quadrupolar NMR and Dispersion-Corrected Density Functional Theory, J. Phys. Chem. C, 2018, 122(3), 1809–1820, DOI:10.1021/acs.jpcc.7b12314.
- S. T. Holmes, R. J. Iuliucci, K. T. Mueller and C. Dybowski, Semi-Empirical Refinements of Crystal Structures Using 17O Quadrupolar-Coupling Tensors, J. Chem. Phys., 2017, 146(6), 064201, DOI:10.1063/1.4975170.
- S. T. Holmes, C. S. Vojvodin and R. W. Schurko, Dispersion-Corrected DFT Methods for Applications in Nuclear Magnetic Resonance Crystallography, J. Phys. Chem. A, 2020, 124(49), 10312–10323, DOI:10.1021/acs.jpca.0c06372.
- S. T. Holmes, O. G. Engl, M. N. Srnec, J. D. Madura, R. Quiñones, J. K. Harper, R. W. Schurko and R. J. Iuliucci, Chemical Shift Tensors of Cimetidine Form A Modeled with Density Functional Theory Calculations: Implications for NMR Crystallography, J. Phys. Chem. A, 2020, 124(16), 3109–3119, DOI:10.1021/acs.jpca.0c00421.
- C. J. Pickard and F. Mauri, All-Electron Magnetic Response with Pseudopotentials: NMR Chemical Shifts, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63(24), 245101, DOI:10.1103/PhysRevB.63.245101.
- S. Adiga, D. Aebi and D. L. Bryce, EFGShield — A Program for Parsing and Summarizing the Results of Electric Field Gradient and Nuclear Magnetic Shielding Tensor Calculations, Can. J. Chem., 2011, 85(7–8), 496–505, DOI:10.1139/V07-069.
- T. Friščić, New Opportunities for Materials Synthesis Using Mechanochemistry, J. Mater. Chem., 2010, 20(36), 7599–7605, 10.1039/C0JM00872A.
- S. T. Holmes, J. M. Hook and R. W. Schurko, Nutraceuticals in Bulk and Dosage Forms: Analysis by 35Cl and 14N Solid-State NMR and DFT Calculations, Mol. Pharmaceutics, 2022, 19(2), 440–455, DOI:10.1021/ACS.MOLPHARMACEUT.1C00708.
- A. A. Peach, S. T. Holmes, L. R. MacGillivray and R. W. Schurko, The Formation and Stability of Fluoxetine HCl Cocrystals Investigated by Multicomponent Milling, CrystEngComm, 2023, 25(2), 213–224, 10.1039/D2CE01341J.
- A. A. Peach, D. A. Hirsh, S. T. Holmes and R. W. Schurko, Mechanochemical Syntheses and 35 Cl Solid-State NMR Characterization of Fluoxetine HCl Cocrystals, CrystEngComm, 2018, 20(20), 2780–2792, 10.1039/C8CE00378E.
- O. Al Rahal, P. Andrew Williams, C. E. Hughes, B. M. Kariuki and K. D. M. Harris, Structure Determination of Multicomponent Crystalline Phases of (S)-Ibuprofen and L-Proline from Powder X-Ray Diffraction Data, Augmented by Complementary Experimental and Computational Techniques Published as Part of a Crystal Growth and Design Virtual Special Issue on The Rietveld Refinement Method: Half of a Century Anniversary, Cite This, Cryst. Growth Des., 2021, 21(4), 2498–2507, DOI:10.1021/acs.cgd.1c00160.
- A. A. Peach, C. H. Fleischer, K. Levin, S. T. Holmes, J. E. Sanchez and R. W. Schurko, Quadrupolar NMR Crystallography Guided Crystal Structure Prediction (QNMRX-CSP), CrystEngComm, 2024, 26(35), 4782–4803, 10.1039/D3CE01306E.
- M. Baias, C. M. Widdifield, J.-N. Dumez, H. P. G. Thompson, T. G. Cooper, E. Salager, S. Bassil, R. S. Stein and L. Emsley, Powder Crystallography of Pharmaceutical Materials by Combined Crystal Structure Prediction and Solid-State 1H NMR Spectroscopy, Phys. Chem. Chem. Phys., 2013, 15(21), 8069, 10.1039/c3cp41095a.
- J. K. Harper and D. M. Grant, Enhancing Crystal-Structure Prediction with NMR Tensor Data, Cryst. Growth Des., 2006, 6(10), 2315–2321, DOI:10.1021/cg060244g.
- C. S. Vojvodin, S. T. Holmes, L. K. Watanabe, J. M. Rawson and R. W. Schurko, Multi-Component Crystals Containing Urea: Mechanochemical Synthesis and Characterization by 35 Cl Solid-State NMR Spectroscopy and DFT Calculations, CrystEngComm, 2022, 24(14), 2626–2641, 10.1039/D1CE01610E.
- J. Autschbach, S. Zheng and R. W. Schurko, Analysis of Electric Field Gradient Tensors at Quadrupolar Nuclei in Common Structural Motifs, Concepts Magn. Reson., Part A, 2010, 36A(2), 84–126, DOI:10.1002/cmr.a.20155.
- J. W. Akitt and W. S. McDonald, Arrangements of Ligands Giving Low Electric Field Gradients, J. Magn. Reson., 1984, 58(3), 401–412, DOI:10.1016/0022-2364(84)90144-6.
- O. Knop, E. M. Palmer and R. W. Robinson, Arrangements of Point Charges Having Zero Electric-Field Gradient, Acta Crystallogr., 1975, 31(1), 19–31, DOI:10.1107/S0567739475000046.
- O. Knop, Arrangements of Point Charges Having Zero Electric-Field Gradient, Acta Crystallogr., 1976, 32(1), 147–149, DOI:10.1107/S0567739476000272.
- P. Pyykkö, Year-2017 Nuclear Quadrupole Moments, Mol. Phys., 2018, 116(10), 1328–1338, DOI:10.1080/00268976.2018.1426131.
- D. A. Hirsh, A. J. Rossini, L. Emsley and R. W. Schurko, 35Cl Dynamic Nuclear Polarization Solid-State NMR of Active Pharmaceutical Ingredients, Phys. Chem. Chem. Phys., 2016, 18(37), 25893–25904, 10.1039/c6cp04353d.
- H. Hamaed, J. M. Pawlowski, B. F. T. T. Cooper, R. Fu, S. H. Eichhorn and R. W. Schurko, Application of Solid-State 35Cl NMR to the Structural Characterization of Hydrochloride Pharmaceuticals and Their Polymorphs, J. Am. Chem. Soc., 2008, 130(33), 11056–11065, DOI:10.1021/ja802486q.
- J. A. Goedkoop and C. H. MacGillavry, IUCr, The Crystal Structure of Malonic Acid, Acta Crystallogr., 1957, 10(2), 125–127, DOI:10.1107/S0365110X57000353.
- I. Tsivintzelis, G. M. Kontogeorgis and C. Panayiotou, Dimerization of Carboxylic Acids: An Equation of State Approach, J. Phys. Chem. B, 2017, 121(9), 2153–2163, DOI:10.1021/acs.jpcb.6b10652.
- R. Prohens, D. De Sande, M. Font-Bardia, A. Franconetti, J. F. González and A. Frontera, Gallic Acid Dimer As a Double Ï€-Hole Donor: Evidence from X-Ray, Theoretical Calculations, and Generalization from the Cambridge Structural Database, Cryst. Growth Des., 2019, 19(7), 3989–3997, DOI:10.1021/acs.cgd.9b00387.
- H. C. S. Chan, G. R. Woollam, T. Wagner, M. U. Schmidt and R. A. Lewis, Can Picolinamide Be a Promising Cocrystal Former?, CrystEngComm, 2014, 16(21), 4365–4368, 10.1039/C4CE00265B.
- I. S. Divya, S. Amrutha, S. Seethalekshmi and S. Varughese, Molecular Salts of Quinine: A Crystal Engineering Route to Enhance the Aqueous Solubility, CrystEngComm, 2021, 23(39), 6942–6951, 10.1039/D1CE00791B.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Physical Stability Enhancement of Theophylline via Cocrystallization, Int. J. Pharm., 2006, 320(1–2), 114–123, DOI:10.1016/J.IJPHARM.2006.04.018.
- N. R. Jagannathan, S. S. Rajan and E. Subramanian, Refinement of the Crystal Structure of Malonic Acid, C3H4O4, J. Chem. Crystallogr., 1994, 24(1), 75–78, DOI:10.1007/BF01665349.
- C. E. Hughes, G. N. M. Reddy, S. Masiero, S. P. Brown, P. A. Williams and K. D. M. Harris, Determination of a Complex Crystal Structure in the Absence of Single Crystals: Analysis of Powder X-Ray Diffraction Data, Guided by Solid-State NMR and Periodic DFT Calculations, Reveals a New 2′-Deoxyguanosine Structural Motif, Chem. Sci., 2017, 8(5), 3971–3979, 10.1039/C7SC00587C.
- C. A. O'Keefe, C. Mottillo, J. Vainauskas, L. Fábián, T. Friščić and R. W. Schurko, NMR-Enhanced Crystallography Aids Open Metal-Organic Framework Discovery Using Solvent-Free Accelerated Aging, Chem. Mater., 2020, 32(10), 4273–4281, DOI:10.1021/acs.chemmater.0c00894.
- C. S. Vojvodin, S. T. Holmes, C. E. A. Kirschhock, D. A. Hirsh, I. H. Huskić, S. Senanayake, L. Betancourt, W. Xu and R. W. Schurko, Rietveld Refinement and NMR Crystallographic Investigations of Multicomponent Crystals Containing Alkali Metal Chlorides and Urea, J. Appl. Crystallogr., 2025, 58(2), 58, DOI:10.1107/S1600576725001360.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2425375 and 2425376. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00426h |
| This journal is © The Royal Society of Chemistry 2025 |