
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe selectivity behaviour of 9,9′-bifluorenyl-9,9′-diol as a host compound for highly efficient separations of mixed pyridines†
Jaime-lee Groenewaldt *,
Benita Barton and
Eric C. Hosten
*,
Benita Barton and
Eric C. Hosten
Department of Chemistry, Nelson Mandela University, PO Box 77000, Gqeberha (Port Elizabeth), 6031, South Africa. E-mail: s224525069@mandela.ac.za
First published on 28th May 2025
Abstract
This investigation explored the selectivity behaviour of 9,9′-bifluorenyl-9,9′-diol (H) as a host compound for the separation, through supramolecular chemistry strategies, of mixed pyridines (pyridine (PYR) and its methylated derivatives, 2-, 3- and 4-methylpyridine (2MP, 3MP and 4MP)). Initial single-solvent crystallization experiments demonstrated that H formed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest inclusion complexes with each of PYR, 3MP and 4MP while, in 2MP, no crystallization occurred, and a gel remained in the glass vessel. Equimolar guest competition experiments revealed a clear host preference for 3MP and 4MP, while selectivity profiles employing binary mixed guest solutions indicated that H possessed remarkable separation potential for PYR/4MP, 2MP/3MP and 3MP/4MP mixtures, amongst numerous others. Single crystal X-ray diffraction (SCXRD) analyses corroborated these findings, revealing that 3MP and 4MP engaged in more linear hydrogen bonding interactions with H, which likely contributed to their preferential inclusion. Furthermore, these two guests also formed complexes with greater crystal density compared to H·PYR. Hirshfeld surface analyses substantiated these observations, denoting a greater percentage of hydrogen atom interactions in the H complexes with 3MP and 4MP. Further support was provided by thermal analyses, where the H·4MP complex possessed the highest thermal stability, followed by H·3MP, while H·PYR was the least stable one. These results underscore 9,9′-bifluorenyl-9,9′-diol to be a highly effective host compound for the selective separation of various PYR/MP mixtures, offering an alternative separatory strategy that is efficient and, moreover, environmentally friendly, compared with more conventional approaches.
:1 host–guest inclusion complexes with each of PYR, 3MP and 4MP while, in 2MP, no crystallization occurred, and a gel remained in the glass vessel. Equimolar guest competition experiments revealed a clear host preference for 3MP and 4MP, while selectivity profiles employing binary mixed guest solutions indicated that H possessed remarkable separation potential for PYR/4MP, 2MP/3MP and 3MP/4MP mixtures, amongst numerous others. Single crystal X-ray diffraction (SCXRD) analyses corroborated these findings, revealing that 3MP and 4MP engaged in more linear hydrogen bonding interactions with H, which likely contributed to their preferential inclusion. Furthermore, these two guests also formed complexes with greater crystal density compared to H·PYR. Hirshfeld surface analyses substantiated these observations, denoting a greater percentage of hydrogen atom interactions in the H complexes with 3MP and 4MP. Further support was provided by thermal analyses, where the H·4MP complex possessed the highest thermal stability, followed by H·3MP, while H·PYR was the least stable one. These results underscore 9,9′-bifluorenyl-9,9′-diol to be a highly effective host compound for the selective separation of various PYR/MP mixtures, offering an alternative separatory strategy that is efficient and, moreover, environmentally friendly, compared with more conventional approaches.
1. Introduction
Pyridine (PYR) and its methylated derivatives 2-methylpyridine (2MP), 3-methylpyridine (3MP) and 4-methylpyridine (4MP), collectively known as the picolines and pyridine bases, are indispensable heterocyclic compounds with broad industrial applications.1,2 PYR is commonly used as an effective solvent and a precursor towards pharmaceuticals and agrochemicals, including herbicides, insecticides and fungicides. 2MP is primarily used to produce 2-vinylpyridine, which forms an adhesive for textile tire cords. Additionally, it serves as a key intermediate in the synthesis of chemicals such as nitrapyrin, which mitigates ammonia loss from fertilizers. A substantial amount of 3MP is employed as a starting material for the herbicide fluazifop-butyl, insecticide chlorpyrifos, and feed additives, including nicotinic acid and nicotine carboxamide. 4MP is essential for synthesising isonicotinic hydrazide, an anti-tuberculosis drug, as well as polymers for anion exchange.3–6While PYR and the MPs were historically extracted in trace amounts from coal tar (the condensate from coking ovens),7 synthetic methodologies have largely supplanted natural sources due to scalability and efficiency. Among these, the Chichibabin pyridine synthesis remains the predominant industrial route, wherein aldehydes or ketones undergo condensation with ammonia in the presence of oxide catalysts such as alumina or silica.8–10 Variations of this process utilise zeolitic catalysts (ZSM-5, H-ZSM-5, H-beta) or metal-doped aluminosilicates incorporating Ni, Cr, Zn or Th to enhance conversion rates.11–15 However, the synthesis of PYR and the MPs often results in complex mixtures, which demand challenging post-synthetic separation techniques.13
Conventional methods such as fractional and azeotropic distillations, and adsorptions employing zeolites or crystalline macrocycles, like the cucurbiturils, have been explored for their ability to separate these pyridine mixtures; however, many of these approaches are inherently limited.12,16–18 For example, cucurbit[6]uril has been employed to selectively encapsulate PYR from a PYR/3MP mixture. Since PYR has a very different boiling point (115 °C) compared with 3MP (144 °C), the same result may be achieved through a simple and less costly fractional distillation. Furthermore, during the synthesis of cucurbit[6]uril, other byproducts are also obtained, necessitating a number of additional purification steps before the host compound is isolated in pure form. The ripple effect of this challenge is exorbitant cucurbit[6]uril costs when purchasing these types of compounds. The separation of the MPs, in particular, remains the more formidable challenge in the industry due to their comparable boiling points (2MP boils at 129 °C, 3MP at 144 °C and 4MP at 145 °C). Fractional distillation, despite being a widely employed technique, is highly energy-intensive and requires an impractically large number of theoretical plates to achieve meaningful separations. This is especially applicable to 3MP and 4MP, which differ in boiling point by only ∼1 °C. Azeotropic distillation has been shown to enhance selectivity, but its overall efficiency is often characterised by low yields.16 The primary drawback of these methods is their tendency to produce mixtures rather than pure compounds, entailing additional separation steps that further increase processing costs and energy consumption.
Host–guest chemistry, a branch of supramolecular chemistry,19,20 presents an innovative alternative for the separation of mixed pyridines, overcoming many of the limitations associated with conventional methodologies. This approach leverages the structural attributes of both host and guest, whereby the host participates in noncovalent interactions (such as hydrogen bonding, π⋯π stacking and C–H⋯π close contacts) with specific guest molecules to obtain stable inclusion complexes. The solid nature of these complexes enables facile isolation of the host–guest inclusion compound from the other isomers in solution via vacuum filtration. Thereafter, the selectively encapsulated guest can be collected through mild thermal treatment and the guest-free host compound may then be recycled and reused. This non-destructive and recyclable separation strategy not only enhances efficiency but also aligns with green chemistry principles by minimizing solvent waste and energy consumption. Numerous studies in literature have explored the use of host–guest chemistry for separating pyridine mixtures, where the host demonstrated a high degree of selectivity, highlighting the viability of efficient separations through this approach.21,22 Our laboratory has been actively engaged in the quest to identify optimal host compounds for the efficient separation and purification of MP isomers through supramolecular strategies. While, for example, the cucurbiturils (as mentioned earlier) have an intrinsic cavity in both the solid and liquid phases into which guest species fit, our laboratory employs host compounds that form only extrinsic cavities and only in the solid state to furnish co-crystals with the various guest molecules. We have thus explored host systems derived from tartaric acid,23–25 xanthone and thioxanthone,26–29 as well as host molecules having a roof-shaped geometry30,31 and, also, those with a wheel-and-axle design.32 The selectivity and efficacy of these host species varied significantly, governed by their structural attributes, with distinct and often complementary selectivity trends emerging. For example, the two compounds, (4R,5R)-bis(diphenylhydroxymethyl)-2-spiro-1′-cyclohexane-1,3-dioxolane (TADDOL6) and (4R,5R)-bis(diphenylhydroxymethyl)-2-spiro-1′-cyclopentane-1,3-dioxolane (TADDOL5),25,33 though closely related in terms of molecular structure, possessed very different host selectivities when presented with the pyridine mixtures: TADDOL6 preferred PYR and then 3MP while this order was reversed for TADDOL5. It is therefore imperative that the behaviour of other host compounds not yet explored in such conditions be scrutinized carefully, since predicting selectivity is not trivial and differs widely amongst the various host species.
In the current investigation, the potential of 9,9′-bifluorenyl-9,9′-diol (H) to behave as a selective host compound for the separation of PYR and its methylated derivatives (2MP, 3MP, and 4MP) through supramolecular complexation was assessed (Scheme 1). All of the inclusion compounds were examined using SCXRD experiments, while Hirshfeld surfaces and thermoanalytical analyses were also considered. This host compound has not been previously applied in this context, and all findings that were obtained are reported herein.
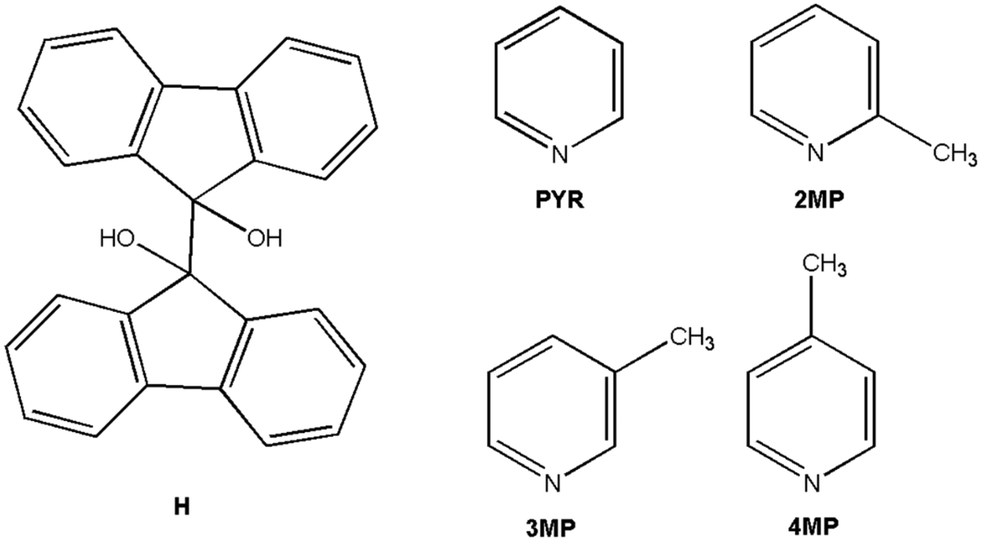 | ||
| Scheme 1 The molecular structures of the host compound 9,9′-bifluorenyl-9,9′-diol (H) as well as the proposed guest solvents pyridine (PYR) and 2, 3 and 4-methylpyridine (2MP, 3MP and 4MP). | ||
2. Experimental
2.1 General
All reagents required for the synthesis of H, along with the pyridyl solvents, were procured from Merck (South Africa) and employed as such without further purification.1H-NMR experiments were carried out by means of a Bruker Ultrashield Plus 400 MHz spectrometer with CDCl3 as the deuterated solvent. The obtained spectral data were analysed using Topspin 4.4.1 software.
The crystal quality of the complexes H·PYR, H·3PYR and H·4PYR was adequate for their structural elucidation through SCXRD experiments. Two instruments were employed. The first was a Bruker Kappa Apex II diffractometer utilising graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). X-Ray data acquisition and analysis were performed using APEXII software, and subsequent cell refinement and data reduction were executed using the SAINT program. Numerical absorption corrections were applied with the SADABS software.34 The crystal structures were solved using SHELXT-2018/2,35 followed by least-squares refinement using SHELXL-2018/3 (ref. 36) in conjunction with SHELXLE37 as the graphical user interface. The second instrument was a Bruker D8 Quest diffractometer, featuring a Photon II CPAD detector and IμS 3.0 Mo source (Kα, λ = 0.71073 Å). Diffraction data acquisition was conducted with APEX4,38 while SAINT facilitated cell refinement and data reduction. Absorption effects were accounted for by using the numerical correction method in SADABS.38 Crystal structures were solved by means of the dual-space approach in SHELXT-2018/2 (ref. 35) and subsequently refined via the least-squares techniques in SHELXL-2019/3.36 Graphical visualization was achieved with SHELXLE,37 and structural diagrams were created using ORTEP-3 for Windows (version 2023.1).39 Anisotropic refinement was applied to all non-hydrogen atoms, while carbon-bound hydrogen atoms were positioned according to standard geometric constraints (C–H bond lengths: 0.95 Å for aromatic carbons, 1.00 Å for methine, and 0.99 Å for methylene) and refined using the riding model approximation, with Uiso(H) fixed at 1.2Ueq(C). Hydrogen atoms of the methyl groups were modelled with rotational freedom about the C–C bond to best match the experimental electron density (HFIX 137 in SHELXL36 program). The Uiso(H) parameters were fixed at 1.5Ueq(C), with C–H bond lengths restrained to 0.98 Å. Hydroxyl hydrogen atoms were refined with rotational freedom about the C–O bond to achieve optimal fit with the experimental electron density (HFIX 147 in SHELXL36 program). Uiso(H) was fixed at 1.5Ueq(O), with O–H bond lengths restrained to 0.84 Å. Whenever feasible, nitrogen-bound hydrogen atoms were located on the difference Fourier maps and refined freely. If not located, they were assigned calculated positions and refined with a riding model (Uiso(H) was fixed at 1.2Ueq(N), with N–H bond lengths restrained to 0.88 Å). The crystal structures of these complexes (H·PYR, H·3PYR, and H·4PYR) were deposited at the Cambridge Crystallographic Data Centre, and their respective CCDC numbers are 2409400, 2409401 and 2409402.
Gas chromatography (GC) was employed for the quantification of the pyridyl guest compounds in any mixed complexes prepared in this study. Analyses were carried out using an Agilent Technologies 7890A gas chromatograph paired with an Agilent 5975C VL mass spectrometer (GC-MS), which was equipped with a calibrated Agilent J&W GC Cyclosil-B column (30 m × 250 μm × 0.25 μm). The method involved an initial 5 min hold time at 50 °C, followed by an immediate heating ramp of 10 °C min−1 until 100 °C was attained. The total run time was 10 min, with a split ratio of 1:80 and a flow rate of 1.5 mL min−1. For all of these GC analyses, dichloromethane served as the dissolution solvent.
The relative thermal stabilities of the single-solvent complexes were assessed through thermal analyses. A Perkin Elmer STA6000 Simultaneous Thermal Analyser was employed for these experiments, and the so-obtained data were analysed using Perkin Elmer Pyris 13 Thermal Analysis software. The solids (complexes) were recovered from solutions by vacuum filtration and washed with petroleum ether (bp 40–60 °C) under suction. The crystals were then patted dry in folded filter paper and transferred to a ceramic pan for analysis. The reference measurement was made using an empty ceramic pan, with the same open pan subsequently employed for the sample run. The system used high purity nitrogen as the purge gas and all samples were heated from approximately 50 to 280 °C with a heating rate of 10 °C min−1.
2.2 Synthesis of 9,9′-bifluorenyl-9,9′-diol (H)
The host compound was synthesised according to a previously reported procedure.402.3 Single-solvent host crystallization experiments
These experiments were undertaken to evaluate the complexation ability of H for the pyridyl guest solvents under investigation. The solid host compound (0.05 g, 0.14 mmol) was dissolved in each of the liquid guest solvents (5–10 mmol) with gentle heating being applied to promote complete dissolution. The glass vials were then left open at ambient temperature, allowing slow solvent evaporation to progressively increase solution saturation, thereby inducing nucleation and subsequent crystallization. The resulting crystals were collected by vacuum filtration, washed with petroleum ether (bp 40–60 °C) to remove any residual guest solvent on the crystal surfaces, and dissolved in CDCl3 for 1H-NMR spectroscopy analysis. The host-to-guest (H:G) ratios of the successfully formed complexes were determined by comparing the integrals of relevant host and guest resonance signals.
2.4 Equimolar guest competition experiments
Equimolar guest competition experiments were performed to determine whether the host compound possessed an affinity towards a particular guest compound within various guest solvent combinations. As such, the host compound (0.05 g, 0.14 mmol) was dissolved in equimolar binary, ternary and quaternary guest mixtures, where the combined guest quantity was 5 mmol. The sealed glass vials were stored at 4 °C to facilitate crystallization. Once crystals formed, they were treated as per the single-solvent crystallization experiments. The overall H:G ratios were quantified using 1H-NMR spectroscopy, while guest-to-guest (G:G) ratios were determined through GC analyses. To ensure reproducibility, all experiments were conducted in duplicate, with percentage estimated standard deviations (% e.s.d.s) thus also reported here.
2.5 Binary guest competition experiments with varied guest concentrations
These host crystallization experiments were undertaken to systematically assess the selective binding behaviour of H in binary guest mixtures of guest A (GA) and guest B (GB). Thus, the host compound (0.05 g, 0.14 mmol) was dissolved in GA:GB molar solutions (5 mmol combined amount) prepared at defined molar ratios of 20:80, 40:60, 60:40 and 80:20. The sealed glass vials were stored at 4 °C to promote crystallization. Following crystal growth, the solids were isolated and treated according to the method in the equimolar guest experiments. Guest compositions within the mixed complexes were quantified by means of GC analyses. Selectivity profiles were then constructed based on the selectivity model of Pivovar et al., where the amount of GA or GB in the crystalline phase (ZA or ZB) was plotted against the corresponding molar fractions of the same guest in the original solution (XA or XB).41 The selectivity coefficient (K), reflecting the preferential affinity of H for a specific guest, was determined in accordance with the equation KA:B = ZA/ZB × XB/XA, where XA + XB = 1. A theoretical reference line corresponding to KA:B = 1 was included in these plots, representing a scenario in which the host compound exhibits no preferential behaviour for either guest solvent present. The experimentally determined selectivity coefficients were evaluated against this benchmark to ascertain deviations indicative of host selectivity. In addition, according to the criteria set by Nassimbeni and coworkers, a K-value of 10 or higher signifies a highly efficient host compound, ensuring it to be an excellent candidate for separations on a practical setting42 (note that the results from the equimolar binary experiments (Table 2) were also inserted into these selectivity plots).
2.6 Software
Crystallographic analysis was carried out using program Mercury,43 which facilitated the generation of all crystal structure representations, including unit cell, host–guest packing, host–guest interaction and void diagrams. In the latter case, guest molecules were excluded from the packing calculations, and the resultant void spaces were quantitatively examined using a spherical probe with a 1.2 Å radius. Moreover, Crystal Explorer (version 21.5)44 was employed to perform Hirshfeld surface analysis, a powerful tool for quantifying guest⋯host (G⋯H) interactions within supramolecular complexes. Three-dimensional Hirshfeld surfaces were generated around each guest molecule and from these surfaces, two-dimensional fingerprint plots were derived, providing visual representations and quantification of each interaction contribution.453. Results and discussion
3.1 Single-solvent host crystallization experiments
Crystallization of H from the four pyridyl guest solvents afforded inclusion complexes with each of PYR, 3MP and 4MP (Table 1), and the H:G ratios remained consistently 1:1. Notably, the H/2MP experiment did not crystallize and a gel resulted after repeated attempts (the relevant 1H-NMR spectra are provided in the ESI,† Fig. S1a–c).
:G ratiosa of complexes formed with H when crystallized from the pyridyl solvents
3.2 Equimolar guest competition experiments
Table 2 presents the results after GC analyses of the complexes formed when H was crystallized from the various equimolar mixtures of PYR, 2MP, 3MP and 4MP (Fig. S2–S12 in the ESI†). Preferred guest solvents in each experiment are denoted in bold text. Each experiment was carried out in duplicate, and so percentage estimated standard deviations (% e.s.d.s) are also provided here (in parentheses) to determine the repeatability of these results.:G and overall H:G ratios of complexes formed from H crystallization experiments in equimolar mixed pyridinesa
| PYR | 2MP | 3MP | 4MP | Guest ratios (% e.s.d.s) | Overall H:G ratio |
|---|---|---|---|---|---|
| a G:G and overall H:G ratios were obtained using GC and 1H-NMR spectroscopy, respectively. |
|||||
| X | X | 75.1:24.9 (2.2) |
1:1 |
||
| X | X | 8.4:91.6 (1.0) |
1:1 |
||
| X | X | 5.4:94.6 (3.3) |
1:1 |
||
| X | X | 1.7:98.3 (0.2) |
1:1 |
||
| X | X | 0.7:99.3 (0.4) |
1:1 |
||
| X | X | 0:100 (0.0) |
1:1 |
||
| X | X | X | 8.2:2.4:89.4 (0.7)(0.6)(1.3) |
1:1 |
|
| X | X | X | 68.1:31.4:0.5 (0.7)(0.7)(0.0) |
1:1 |
|
| X | X | X | 6.5:93.5:0.0 (0.2)(0.2)(0.0) |
1:1 |
|
| X | X | X | 2.3:0.0:97.7 (0.3)(0.0)(0.3) |
1:1 |
|
| X | X | X | X | 11.2:1.9:86.9:0.0 (1.1)(0.2)(0.9)(0.0) |
1:1 |
From Table 2, it is clear that H, in the binary solvent experiments, possessed an overwhelming affinity for 4MP. The resulting complexes contained 94.6%, 99.3%, and 100% 4MP when H was crystallized from PYR/4MP, 2MP/4MP and 3MP/4MP mixtures, respectively. In the absence of 4MP, 3MP emerged as the preferred guest species, yielding complexes containing 91.6% and 98.3% 3MP in PYR/3MP and 2MP/3MP mixtures, respectively. When neither 3MP nor 4MP was present, PYR was then preferentially accommodated, and the crystals contained 75.1% PYR in the PYR/2MP experiment. The latter result is not unexpected given that H did not crystallise with 2MP in the single-solvent experiment (Table 1).
From the ternary experiments, 3MP was most favoured in the PYR/2MP/3MP and PYR/3MP/4MP solutions, the resultant crystals having 89.4% and 93.5% 3MP, respectively. This latter observation is remarkable in that the crystals contained no 4MP whatsoever, despite 4MP being preferred in the equimolar binary solutions. When all three MPs were combined, the host affinity was then again overwhelmingly in favour of 4MP (97.7%), while 3MP was completely excluded (0.0%) from the complex. A possible trend subsequently emerged: 4MP was favoured in the presence of 3MP but only in the absence of PYR. Conversely, when PYR was present, 3MP was preferred over 4MP, indicating that PYR plays a significant role in directing the selectivity behaviour of the host compound in these guest mixtures.
In quaternary guest solutions, this observed trend persisted and, since PYR was present, 3MP was preferentially incorporated over 4MP (86.9%) (remarkably, once more, no 4MP was measured in the complex).
These experiments, therefore, revealed that 3MP and 4MP were, more usually, the favoured guest solvents, whereas 2MP and PYR were less preferred by the host compound. In fact, 2MP was never preferentially selected by H, in accordance with the fact that H did not crystallise from this solvent (Table 1). These results are particularly promising as H demonstrated remarkable selectivities in many of these guest mixtures, and the low % e.s.d.s emphasise the reliability of these results.
The overall H:G ratios remained 1:1 in all of these experiments, which is congruent with the consistent 1:1 ratio observed in the single-solvent crystallization experiments (Table 1).
3.3 Binary guest competition experiments with varied guest concentrations
The selectivity profiles obtained from the crystallization experiments of H from the binary guest mixtures with varied concentrations, as outlined in the experimental section, are provided in Fig. 1a–f (relevant GC traces are presented in Fig. S13–S18 of the ESI†). | ||
| Fig. 1 Selectivity profiles of H when crystallized from (a) PYR/2MP, (b) 3MP/PYR, (c) 4MP/PYR, (d) 3MP/2MP, (e) 4MP/2MP and (f) 4MP/3MP binary systems, where the straight diagonal lines denote a host compound that exhibits no selectivity. | ||
The selectivity profile for H when crystallized from mixtures of PYR/2MP (Fig. 1a) revealed a constant preference for PYR over 2MP across the concentration range. However, the overall selectively was low, with all K-values below 10. The highest value, 4.7, was recorded in a mixture containing 60% PYR. These results suggest that H lacks the requisite selectivity to serve as an effective host compound for the separation of PYR/2MP binary mixtures.
H exhibited a preference for 3MP over PYR in all 3MP/PYR mixtures (Fig. 1b). The 20, 40, 50, 60 and 80% 3MP solutions furnished crystals with remarkable amounts of 3MP, that is, 69.0, 87.0, 91.6, 95.1 and 99.7%, respectively. Only when 20% 3MP was present in the solution was the K-value below 10 (8.9), while in the remaining mixtures (40, 50, 60 and 80% 3MP), these values were 10.1, 10.9, 12.9 and 84.8, alluding to the feasibility of employing H effectively for these four separations.
The plot obtained for the 4MP/PYR experiments (Fig. 1c) was characterised by an S-shape, indicating that the selectivity behaviour of H was guest concentration dependent. At high concentrations of 4MP (50, 60 and 80%), 4MP was overwhelmingly favoured (94.6, 98.7 and 99.0% of 4MP were measured in the crystals) and the K-values were significant, 17.5, 49.2, and 25.5. However, at high PYR concentrations (60 and 80%), PYR was then preferred (the crystals contained 98.6 and 98.7% PYR), with substantial K-values of 48.2 and 18.4, respectively, being calculated. Thus, H would serve as an effective separatory tool in all 4MP/PYR mixtures, displaying overwhelming selectivities for one or the other guest solvent, dependent upon their relative amounts.
In the binary mixture comprising 3MP/2MP (Fig. 1d), H consistently preferred 3MP. For solutions with 20, 40, 50, 60 and 80% 3MP, the mixed complexes contained 76.8, 94.7, 98.3, 99.4 and 99.0% 3MP. Moreover, all the K-values exceeded 10 (13.3, 26.7, 57.9, 117.7 and 23.6) and so these results indicate that H is more than capable of effecting all of these 3MP/2MP separations.
The host compound showed an overwhelming affinity towards 4MP in all of the 4MP/2MP mixtures (Fig. 1e), specifically when the solutions contained 40% or more 4MP (40, 50, 60 and 80% mixtures produced crystals with as much as 98.8, 99.3, 99.5 and 99.3% 4MP). Unsurprisingly, therefore, from these experiments were calculated extremely high K-values, 126.7, 134.0, 122.1 and 33.7, indicating that H would be exceptionally effective for these separations. However, this was not the case at a lower concentration of 4MP (20%), where 41.4% of 4MP was measured in the crystals, and a low K-value of 2.8 was obtained.
Finally, in binary mixtures comprising 4MP/3MP (Fig. 1f), H exhibited a clear preference for 4MP throughout. Remarkably, all of the solutions, 20, 40, 50, 60 and 80% 4MP, resulted in complexes with 100.0% of this guest species, and the K-values were, consequently, infinite in each instance. Hence, H is an excellent candidate to use for all 4MP/3MP mixture separations. This is an extremely important result given the nearly identical boiling points of 3MP and 4MP (144 and 145 °C), ensuring enormously challenging separations through fractional distillations; supramolecular chemistry strategies with H as the host compound is certainly a more attractive prospect for these separations.
Table 3 summarises the K-values that were calculated in each of the binary guest mixtures. Preferred guests are in parentheses and K-values that were 10 or greater are provided in bold text.
| Guest percentage in original solution | K-Values (in favour of) | |||||
|---|---|---|---|---|---|---|
| PYR/2MP | 3MP/PYR | 4MP/PYR | 3MP/2MP | 4MP/2MP | 4MP/3MP | |
| 20 | 3.4 (PYR) | 8.9 (3MP) | 18.4 (PYR) | 13.3 (3MP) | 2.8 (4MP) | ∞ (4MP) |
| 40 | 3.1 (PYR) | 10.1 (3MP) | 48.2 (PYR) | 26.7 (3MP) | 126.7 (4MP) | ∞ (4MP) |
| 50 | 3.0 (PYR) | 10.9 (3MP) | 17.5 (4MP) | 57.9 (3MP) | 134.0 (4MP) | ∞ (4MP) |
| 60 | 4.7 (PYR) | 12.9 (3MP) | 49.2 (4MP) | 117.7 (3MP) | 122.1 (4MP) | ∞ (4MP) |
| 80 | 3.1 (PYR) | 84.8 (3MP) | 25.5 (4MP) | 23.6 (3MP) | 33.7 (4MP) | ∞ (4MP) |
The summary presented in Table 3 thus demonstrates that H is a highly effective host compound for the separation of many of these binary pyridyl mixtures. In particular, solutions of 3MP/PYR (excluding the 20% 3MP experiment), 4MP/PYR (all), 3MP/2MP (all) and 4MP/3MP (all), as well as 4MP/2MP (once more, excluding the 20% 4MP experiment) may all be separated in this fashion since K-values exceeded 10 in all of these cases, indicating high host selectivity for one guest solvent rather than another. Only in the PYR/2MP experiments were low host affinities observed and no separations are feasible in these particular mixtures.
3.4 Single crystal X-ray diffractometry
The pertinent crystallographic data for the crystal structures of the PYR-, 3MP- and 4MP-containing inclusion complexes of H are summarized in Table 4 (their respective ORTEP diagrams are provided in Fig. S19a–c in the ESI†). No disorder was observed in any of these complexes. The H·PYR and H·3MP inclusion compounds crystallized in the monoclinic crystal system and space group P2/n, while the 4MP-containing complex crystallized in the orthorhombic crystal system and space group P212121.| H·PYR | H·3MP | H·4MP | |
|---|---|---|---|
| Chemical formula | C26H18O2·C5H5N | C26H18O2·C6H7N | C26H18O2·C6H7N |
| Formula weight | 441.50 | 455.53 | 455.53 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P2/n | P2/n | P212121 |
| μ (Mo-Kα)/mm−1 | 0.077 | 0.077 | 0.077 |
| a/Å | 20.3320(8) | 20.6545(7) | 10.4369(5) |
| b/Å | 9.1554(4) | 9.0895(3) | 10.7155(4) |
| c/Å | 26.9029(11) | 27.2828(10) | 21.6126(9) |
| Alpha/° | 90 | 90 | 90 |
| Beta/° | 108.812(1) | 109.272(1) | 90 |
| Gamma/° | 90 | 90 | 90 |
| V/Å3 | 4740.4(3) | 4835.0(3) | 2417.08(18) |
| Z | 8 | 8 | 4 |
| F(000) | 1856 | 1920 | 960 |
| Temp./K | 200 | 200 | 200 |
| Restraints | 0 | 0 | 0 |
| Nref | 11766 |
12019 |
5532 |
| Npar | 622 | 642 | 322 |
| R | 0.0453 | 0.0483 | 0.0310 |
| wR2 | 0.1151 | 0.1216 | 0.0817 |
| S | 1.03 | 1.08 | 1.06 |
| ϑ min–max/° | 2.1, 28.3 | 2.1, 28.3 | 2.1, 27.5 |
| Tot. data | 198861 |
164342 |
74747 |
| Unique data | 11766 |
12019 |
5532 |
| Observed data [I > 2.0 sigma(I)] | 10130 |
10384 |
5265 |
| Rint | 0.051 | 0.043 | 0.052 |
| Completeness | 0.999 | 0.999 | 0.998 |
| Min. resd. dens./e Å−3 | −0.25 | −0.22 | −0.16 |
| Max. resd. dens./e Å−3 | 0.41 | 0.36 | 0.19 |
| Density/g cm−3 | 1.237 | 1.252 | 1.252 |
The unit cell dimensions of complexes H·PYR and H·3MP were comparable (Table 4). As such, their powder XRD patterns were calculated using Mercury (after guest deletion), and Fig. S20a and b in the ESI† illustrate these. Clearly, the two patterns resemble one another closely and it may be concluded that the host packing in these two complexes is isostructural.
Fig. 2a–c display the unit cells and the host–guest packing arrangements (left), where the guest molecules are in spacefilling representation while the host framework is depicted in capped-stick notation. The corresponding void diagrams are presented on the right. In each case, the guest species were located in wide-open, continuous and unidirectional channels within the complex (the isostructural host packing motif in H·PYR and H·3MP is also obvious here when considering Fig. 2a and b).
 | ||
| Fig. 2 Unit cells illustrating the host–guest packing (left) and corresponding void diagrams (right) for the complexes (a) H·PYR, (b) H·3MP and (c) H·4MP; host molecules are in capped stick and guests in spacefill representation. | ||
Each pyridyl guest molecule engaged in classical (host)O–H⋯N(guest) hydrogen bonding with the host compound. The appropriate parameters for these close contacts are summarised in Table 5: H⋯N and O⋯N distances measured between 1.87 and 1.88 Å, and 2.7025(15) and 2.7144(19) Å, while the bond angles ranged between 168 and 174°.
| Complex | H⋯N/Å | O⋯N/Å | O–H⋯N/° |
|---|---|---|---|
| H·PYR | 1.87 | 2.7025(15) | 168 |
| H·3MP | 1.87 | 2.7084(16) | 174 |
| H·4MP | 1.88 | 2.7144(19) | 174 |
While less preferred PYR experienced a statistically significantly shorter hydrogen bond with H than favoured 4MP, these bond distances were similar when comparing PYR and 3MP, and 3MP and 4MP. However, notably, preferred guests 3MP and 4MP were involved in short contacts of this type in which the O–H⋯N angles were significantly closer to linearity (174 vs. 168°). This observation may explain the affinity behaviour of H in the guest competition experiments.
Fig. 3a–c are stereoviews that were prepared in order to clearly illustrate these hydrogen bonds between the guest and host molecules. In addition to the intermolecular interactions, host⋯host intramolecular contacts of this type were also observed, and H⋯O and O⋯O distances measured 2.24, 2.51 and 2.52 Å, and 2.6840(17), 3.408(2) and 3.2673(15) Å, respectively. The associated bond angles ranged from 113 to 135°. The only significant non-classical C–H⋯O intramolecular hydrogen bonding interaction was observed in the H·PYR complex, with distances of 2.57 Å and 3.0431(16) Å, and a bond angle of 111°.
 | ||
| Fig. 3 Stereoviews of the hydrogen bonds (classical and non-classical) in (a) H·PYR, (b) H·3MP and (c) H·4MP; intermolecular bonds are shown in green and intramolecular bonds in magenta. | ||
Interestingly, no meaningful π⋯π interactions were identified between or within any of the molecules in the three complexes.46 However, notable (guest)C–H⋯π(host) close contacts were observed in both H·PYR (PYR being a disfavoured guest species) and H·3MP (containing a preferred guest solvent), as detailed in Table 6 and illustrated in Fig. 4. In direct accordance with observations in the guest competition experiments, this contact was significantly shorter in the 3MP-containing complex (2.72, 3.551(2) Å) compared with that containing PYR (2.96, 3.805(2) Å), explaining the host preference for 3MP relative to PYR (the bond angles were comparable, 147 and 149°). However, the guest molecules in H·4MP (4MP also a favoured guest) did not interact with the host molecule in this manner.
| Complex | H⋯π/Å | X⋯π/Å | X–H⋯π/° |
|---|---|---|---|
| H·PYR | 2.96 | 3.805(2) | 149 |
| H·3MP | 2.72 | 3.551(2) | 147 |
 | ||
| Fig. 4 The close (guest)C–H⋯ π(host) interactions in (a) H·PYR and (b) H·3MP. | ||
Furthermore, and notably, the favoured guest species 3MP and 4MP formed complexes with greater crystal densities (both 1.252 g cm−1) compared with H·PYR (1.237 g cm−1) with the less preferred guest solvent (Table 4), alluding to a tighter and more thermodynamically stable packing arrangement in these two complexes.
3.5 Hirshfeld surface analyses
Hirshfeld surface analysis was subsequently employed in order to quantify the guest⋯host (G⋯H) interactions within the three complexes.44,45 Three-dimensional Hirshfeld surfaces were generated around each guest molecule using program Crystal Explorer 21.5, and these were then converted into two-dimensional fingerprint plots. In these plots, di signifies the shortest distance to atoms located within the surface, while de represents the shortest distance to atoms external to this surface. Fig. 5a–c (left) depict the three-dimensional Hirshfeld surfaces enveloping each pyridyl guest molecule, with red regions indicating areas of pronounced hydrogen bonding interactions. On the right, the corresponding fingerprint plots illustrate the distribution of all of the intermolecular G⋯H interactions, while the bar graph in Fig. 6 summarises the percentage of these interactions. | ||
| Fig. 5 Three-dimensional Hirshfeld surfaces (left) and their corresponding fingerprint plots (right) for (a) H·PYR, (b) H·3MP and (c) H·4MP. | ||
 | ||
| Fig. 6 Quantification of the various G⋯H interactions in the three complexes. | ||
From Fig. 6, it is noteworthy that the percentage of hydrogen atom contacts between the host and guest species was greatest in the complex with 4MP (56.1%), followed by 3MP (43.6%), while in the PYR-containing inclusion compound, only 38.7% were interactions of this type. While this aligns with the fact that 3MP and 4MP were preferred in the guest competition experiments, it must be borne in mind that these favoured guest species do possess methyl groups while PYR does not, and therefore it is expected that 3MP and 4MP would experience the greater percentage of these interactions.
3.6 Thermal analysis
To evaluate the relative thermal stabilities of the three complexes prepared in this work, thermal analyses were performed on each one. The resultant thermogravimetric (TG), its derivative (DTG) and differential scanning calorimetric (DSC) traces are thus illustrated in Fig. S21a–c in the ESI.† These were obtained after heating each sample at a rate of 10 °C min−1 over a temperature range of approximately 50 to 280 °C. The key thermal events, summarised in Table 7, include the onset temperature (Ton) marking the commencement of the guest release process and which serves as a measure of the relative thermal stability of the complex, and the peak temperature (Tp) of the endotherm representing the melting of the host compound, together with the calculated and observed mass losses.47| Complex | Ton/°C | Tp/°C | Measured mass loss/% | Expected mass loss/% |
|---|---|---|---|---|
| a H did not crystallize in 2MP and so no thermal traces could be provided in this instance.b Ton is the onset temperature for the guest release event and Tp is the peak temperature for the host melt endotherm. | ||||
| H·PYR | 71.2 | 190.1 | 17.4 | 17.9 |
| H·3MP | 79.3 | 187.9 | 23.5 | 20.4 |
| H·4MP | 80.5 | 190.6 | 22.3 | 20.4 |
The mass losses observed during these experiments for the H·PYR, H·3MP and H·4MP complexes, 17.4, 23.5 and 22.3%, respectively, agreed reasonably well with those that were calculated, 17.9, 20.4, and 20.4% (Table 7).
The onset temperatures for the guest release events for complexes of H with 3MP and 4MP were higher (Ton 79.3 and 80.5 °C), indicative of greater relative thermal stabilities, compared with H·PYR, which commenced degradation at a lower 71.2 °C (Table 7). This observation aligns with the results of the guest competition experiments in which PYR was not favoured, while 3MP and 4MP were both preferred guest solvents of the host compound. According to Weber et al.,40 the pure host species melted between 190 and 192 °C, which was in accordance with observations in the present investigation, where the endotherm peak temperatures representing the melting of the host compound was between 187.9 and 190.6 °C.
4. Conclusions
Crystallization of 9,9′-bifluorenyl-9,9′-diol (H) from each of PYR, 2MP, 3MP and 4MP afforded complexes with each one with the exception of 2MP, in which crystallization did not occur (and a gel resulted). Successfully formed complexes had 1:1 H:G ratios. In the equimolar binary guest competition experiments, H demonstrated a higher affinity for 4MP, while in the ternary and quaternary guest experiments, 3MP was the most favoured guest solvent. Interestingly, the presence of PYR affected the host selectivity behaviour, steering it towards enclathrating more of 3MP than 4MP. For the experiments involving binary guest mixtures in various concentrations, H was demonstrated to possess the ability to effectively separate very many of these mixtures through supramolecular chemistry strategies since significant K-values were oftentimes calculated. SCXRD analyses revealed that during classical hydrogen bonding with H, preferred 3MP and 4MP adopted more favourable hydrogen bonding geometries, with bond angles notably closer to linearity compared with this bond in H·PYR. The crystal densities were also higher in the complexes with preferred 3MP and 4MP. These findings align with those obtained from Hirshfeld surface analyses, whereby H·3MP and H·4MP presented a greater percentage of hydrogen atom interactions. Thermal analyses further demonstrated that the H·4MP complex exhibited the highest stability, followed by H·3MP, while H·PYR was the least stable. Collectively, these results support the observations from the mixed guest experiments. In summary, H is a highly effective host candidate for very many of the guest mixtures employed in this work.
Data availability
The crystal structures of complexes H·PYR, H·3PYR and H·4PYR were deposited at the Cambridge Crystallographic Data Centre, and their respective CCDC numbers are 2409400, 2409401 and 2409402.Author contributions
Jaime-lee Groenewaldt: investigation; methodology; validation; writing original draft. Benita Barton: conceptualization; methodology; funding acquisition; project administration; resources; supervision; visualization; editorial assistance for the original draft. Eric C. Hosten: data curation; formal analysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support is acknowledged from the Nelson Mandela University and the National Research Foundation (NRF) of South Africa.References
- S. Shimizu, N. Watanabe, T. Kataoka, T. Shoji, N. Abe, S. Morishita and H. Ichimura, Pyridine and Pyridine Derivatives, Ullmann's Encyclopedia of Industrial Chemistry, 2000, pp. 557–586 Search PubMed.
- Y. Higashio and T. Shoji, Heterocyclic compounds such as pyrrole, pyridines, pyrrolidine, piperidine, indole, imidazol and pyrazines, Appl. Catal., A, 2004, 260, 251–259 CrossRef CAS.
- D. Sahu, P. S. R. Sreekanth, P. K. Behera, M. K. Pradhan, A. Patnaik, S. Salunkhe and R. Cep, Advances in synthesis, medicinal properties and biomedical applications of pyridine derivatives: A comprehensive review, Eur. J. Med. Chem. Rep., 2024, 12, 100210 CAS.
- R. J. S. Lewis, Hawley's Condensed Chemical Dictionary, John Wiley & Sons, Inc, New York, NY, 15th edn, 2007 Search PubMed.
- G. D. Henry, De novo synthesis of substituted pyridines, Tetrahedron, 2004, 60, 6043–6061 CrossRef CAS.
- S. E. Golunski and D. Jackson, Heterogeneous conversion of acyclic compounds to pyridine bases - a review, Appl. Catal., 1986, 23, 1–14 CrossRef CAS.
- K. Suresh Kumar Reddy, C. Srinivasakannan and K. V. Raghavan, Catalytic Vapor Phase Pyridine Synthesis: A Process Review, Catal. Surv. Asia, 2012, 16, 28–35 CrossRef CAS.
- A. E. Chichibabin and M. P. Oparina, Über die Synthese des Pyridins aus Aldehyden und Ammoniak, J. Prakt. Chem., 1924, 107, 154–158 CrossRef CAS.
- R. L. Frank and R. P. Seven, Pyridines. IV. A Study of the Chichibabin Synthesis, J. Am. Chem. Soc., 1949, 71, 2629–2635 CrossRef CAS.
- R. Bicker, H. Deger, W. Herzog, K. Rieser, H. Pulm, G. Hohlneicher and H. Freund, X-ray photoelectron spectroscopy study of silica-alumina catalysts used for a new pyridine synthesis, J. Catal., 1985, 94, 69–78 CrossRef CAS.
- N. G. Grigor'eva, A. N. Filippova, M. I. Tselyutina and B. I. Kutepov, Synthesis of pyridine and methylpyridines over zeolite catalysts, Appl. Petrochem. Res., 2014, 5, 99–104 CrossRef.
- S. Shimizu, N. Abe, A. Iguchi, M. Dohba, H. Sato and K. I. Hirose, Synthesis of pyridine bases on zeolite catalyst, Microporous Mesoporous Mater., 1998, 21, 447–451 CrossRef CAS.
- S. Shimizu, N. Abe, A. Iguchi and H. Sato, Synthesis of pyridine bases: General methods and recent advances in gas phase synthesis over ZSM-5 zeolite, Catal. Surv. Jpn., 1998, 2, 71–76 CrossRef CAS.
- U. Kameswari, C. S. Swamy and C. N. Pillai, Methylation of pyridine over zeolites, Stud. Surf. Sci. Catal., 1994, 84, 1959–1964 CrossRef CAS.
- F. J. Van der Gaag, F. Louter and J. C. Oudejans, Reaction of ethanol and ammonia to pyridines over zeolite ZSM-5, Appl. Catal., 1986, 26, 191–201 CrossRef CAS.
- J. Vymetal, Separation of pyridine bases by combining simple rectification and azeotropic distillation with water - Part II, Chem. Prum., 1987, 37, 307–311 CAS.
- Y. Zeng, C. Liang, X. Lu, L. Zhao, F. Wu, T. Hou, A. Zhao, M. Lv, Z. Tao and Q. Li, Perfect separation of pyridine and 3-methylpyridine by cucurbit[6]uril, Chin. Chem. Lett., 2024, 110807 CrossRef.
- Y. Zhang, G. Zhang, X. Xiao, Q. Li and Z. Tao, Cucurbit[n]uril-Based supramolecular separation materials, Coord. Chem. Rev., 2024, 514, 215889 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry Concepts and Perspectives, VCH Verlagsgesellschaft mbH, Weinheim (Bundesrepublik Deutschland), 1st edn, 1995 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, 3rd edn, 2022 Search PubMed.
- J. Bacsa, M. R. Caira, A. Jacobs, L. R. Nassimbeni and F. Toda, Complexation with diol host compounds. Part 33. Inclusion and separation of pyridines by a diol host compound, Cryst. Eng., 2000, 3, 251–261 CrossRef CAS.
- S. A. Bourne, K. C. Corin, L. R. Nassimbeni and F. Toda, Selective enclathration of picolines, Cryst. Growth Des., 2005, 5, 379–382 CrossRef CAS.
- B. Barton, M. R. Caira, E. C. Hosten and C. W. McCleland, A computational, X-ray crystallographic and thermal stability analysis of TETROL and its pyridine and methylpyridine inclusion complexes, Tetrahedron, 2013, 69, 8713–8723 CrossRef CAS.
- B. Barton, E. C. Hosten and D. V. Jooste, Comparative investigation of the inclusion preferences of optically pure versus racemic TADDOL hosts for pyridine and isomeric methylpyridine guests, Tetrahedron, 2017, 73, 2662–2673 CrossRef CAS.
- D. L. Recchia, B. Barton and E. C. Hosten, Extremely effective separations of pyridine/picoline mixtures through supramolecular chemistry strategies employing (4R,5R)-bis(diphenylhydroxymethyl)-2-spiro-1′-cyclohexane-1,3-dioxolane as the host compound, CrystEngComm, 2025, 27, 1960–1976 RSC.
- B. Barton, L. de Jager and E. C. Hosten, Host proficiency of N,N′-bis(9-phenyl-9-thioxanthenyl)ethylenediamine for pyridine and the methylpyridine guests–a competition study, Supramol. Chem., 2018, 30, 61–71 CrossRef CAS.
- B. Barton, M. R. Caira, D. V. Jooste and E. C. Hosten, Investigation of the separation potential of xanthenyl- and thioxanthenyl-based host compounds for pyridine and isomeric picoline mixtures, J. Inclusion Phenom. Macrocyclic Chem., 2020, 98, 223–235 CrossRef CAS.
- B. Barton, M. R. Caira, D. V. Jooste and E. C. Hosten, Alternative purification protocols of mixed pyridines in the presence of trans-N,N′-bis(9-phenyl-9-xanthenyl)cyclohexane-1,4-diamine, J. Inclusion Phenom. Macrocyclic Chem., 2021, 99, 235–243 CrossRef CAS.
- B. Barton, L. de Jager, U. Senekal, E. Ferg and E. C. Hosten, Potential facile separation strategy for mixtures of 3- and 4-methylpyridine by employing N,N′-bis(9-phenyl-9-xanthenyl)ethylenediamine as an alternative host compound, J. Inclusion Phenom. Macrocyclic Chem., 2021, 100, 233–241 CrossRef CAS.
- B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, Selectivity considerations of host compound trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid when presented with pyridine and picoline mixtures: charge-assisted versus classical hydrogen bonding, CrystEngComm, 2022, 24, 4573–4583 RSC.
- B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, Complementary host behaviour of three anthracenyl-derived roof-shaped compounds in mixed pyridines, CrystEngComm, 2023, 25, 1740–1754 RSC.
- B. Barton, J. Vorgers and E. C. Hosten, Behavior of the Wheel-and-Axle Host Compound 1,4-Bis(diphenylhydroxymethyl)benzene in Mixed Pyridyl Guest Solvents, Cryst. Growth Des., 2023, 23, 6641–6650 CrossRef CAS.
- D. L. Recchia, B. Barton and E. C. Hosten, Exploring Supramolecular Chemistry as an Innovative Strategy for Pyridine Guest Separations When Employing a TADDOL Derivative as the Host Compound, Cryst. Growth Des., 2024, 24, 6438–6449 CrossRef CAS.
- A. Bruker, APEX2, SADABS and SAINT, Bruker AXS Inc., Madison (WI), USA, 2010 Search PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- Bruker, APEX4, SADABS and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- L. J. Farrugia, WinGX and ORTEP for Windows: An update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- E. Weber, S. Nitsche, A. Wierig and I. Csöregh, Inclusion compounds of diol hosts featuring two 9-hydroxy-9-fluorenyl or analogous groups attached to linear spacer units, Eur. J. Org. Chem., 2002, 856–872 CrossRef CAS.
- A. M. Pivovar, K. T. Holman and M. D. Ward, Shape-selective separation of molecular isomers with tunable hydrogen-bonded host frameworks, Chem. Mater., 2001, 13, 3018–3031 CrossRef CAS.
- L. Nassimbeni, Useful Techniques in Host-Guest Chemistry, Supramol. Chem., 2000, 12, 161–167 CrossRef CAS.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: From visualization to analysis, design and prediction, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32 RSC.
- V. Sivasakthi, P. Anitha, K. M. Kumar, S. Bag, P. Senthilvel, P. Lavanya, R. Swetha, A. Anbarasu and S. Ramaiah, Aromatic-aromatic interactions: analysis of π-π interactions in interleukins and TNF proteins, Bioinformation, 2013, 9, 432 CrossRef PubMed.
- S. A. Bourne, J. Charles, M. Luigi, E. Weber, K. Skobridis and F. Toda, Complexation with Hydroxy Host Compounds. Part. 4. Structures and Thermal Stabilities of Inclusion Compounds with Dioxane as the Guest, J. Chem. Soc., Perkin Trans. 2, 1991, 1707–1713 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The crystal structures of complexes H·PYR, H·3MP and H·4MP were deposited at the Cambridge Crystallographic Data Centre (CCDC) and their CCDC numbers are 2409400, 2409401 and 2409402, respectively. Crystallographic data for these structures are freely accessible at http://www.ccdc.cam.ac.uk/structures/. The relevant 1H-NMR spectra and GC traces may be found in the ESI together with the ORTEP diagrams and computed powder X-ray diffraction patterns. Additionally, the thermograms have also been placed in the ESI. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00432b |
| This journal is © The Royal Society of Chemistry 2025 |