
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolution of electronic structure and optical properties of naphthalenediimide dithienylvinylene (NDI-TVT) polymer as a function of reduction level: a density functional theory study†
Sushri Soumya
Jena
,
Mohit
Garg
 and
Sarbani
Ghosh
*
and
Sarbani
Ghosh
*
Department of Chemical Engineering, Birla Institute of Technology and Science, Pilani, Rajasthan 333031, India. E-mail: sarbani.ghosh@pilani.bits-pilani.ac.in
First published on 9th January 2025
Abstract
Naphthalenediimide (NDI)-based donor–acceptor co-polymers with tunable electronic, optical, mechanical, and transport properties have shown immense potential as n-type conducting polymers in organic (opto)electronics. During the operation, the polymers undergo reduction at different charged states, which alters their (opto)electronic properties mainly due to the formation of the quasiparticles, polaron/bipolaron. The theoretical study based on quantum mechanical calculations can provide us with a detailed understanding of their (opto)electronic properties, which is missing to a great extent. To date, a theoretical understanding of how these properties vary with reduction levels for NDI-based polymers is completely missing. Herein, the evolution of the electronic structure and optical properties of the naphthalenediimide dithienylvinylene (NDI-TVT) polymer with varying reduction levels (Cred) is studied using density functional theory and time-dependent density functional theory, respectively, in the gaseous phase and solvent phase. We have envisaged that at lower reduction levels, Cred ≤ 100% (i.e., up to one negative charge per NDI moiety), only radical anions, i.e., polarons, are formed. The bipolarons are observed to be formed only at higher reduction levels, Cred > 100%. We note the coexistence of polarons and bipolarons for the intermediate reduction levels (100% < Cred < 200%). Finally, at 200% reduction levels, the presence of two electrons per NDI unit leads to the completely spin-resolved bipolaronic state formation, where one bipolaron is localized at every NDI unit. This aforementioned evolution of polarons and bipolarons with varying reduction levels is also prominently reflected in the calculated UV-vis-NIR absorption spectra. The detailed theoretical insights gained from the evolution of the (opto)electronic properties of NDI-TVT with reduction levels due to the formation of polaronic/bipolaronic states can guide the systematic design of n-type NDI-TVT-based (opto)electronic devices and in their advancement.
Introduction
Conducting polymers are of great interest in organic electronics,1,2 mainly because of their unique and versatile properties, e.g., lightweight,3,4 flexible,5 tunable,6 and easy to fabricate via various large-area printing production protocols. The π-conjugated conducting polymers, due to their inherent conducting properties, which can be altered reversibly utilizing doping, i.e., oxidation or reduction,7 are useful in a wide variety of electronic devices,1 including organic light-emitting diodes,8 organic solar cells,9 organic thin-film transistors,10 sensors,11 batteries,12,13etc. The device's performance depends on the charge-carrier mobility, and the mobility depends on the charge-carrier distribution within the polymer chains and interchain distributions. Thus, it is possible to control the conductivity in one way via molecular engineering of the π-conjugated building blocks, which alters their electronic structures, and thereby, the optical properties. A better understanding of the electronic structure and optical properties at the molecular level is crucial to improving the charge carrier mobility and, thereby, the device's performance.Conducting polymers, depending on their charge carrier transport nature, are generally classified as p-type, i.e., hole-transporting and n-type, i.e., electron-transporting polymers.14,15 The hole-transporting polymers have been extensively studied in the past.16 On the other hand, the electron-transporting polymers are less studied mainly due to their stability issues in the air,7,17 and therefore, the performance of n-type devices lags behind that of p-type devices. Organic electronic devices such as complementary polymeric devices need both p-type and n-type polymers, and hence, recognition of a high-performance, stable, and solution-processed n-type polymer would lead us one step forward in the implementation of p-type/n-type complementary devices and the electron-transporting devices. Therefore, it is necessary to optimize the performance of the n-type polymers with good air stability and high electron affinity in developing polymer-based circuit technologies.
Among many electron-deficient building blocks, imide-based aromatic compounds, e.g., naphthalene diimide (NDI) and perylene diimide (PDI), have emerged as promising choices for advancing n-type organic semiconductors. The n-type polymers based on naphthalenediimide (NDI)15,18–20 have shown significant progress as a counterpart of thiophene-based p-type polymers, like poly(3-hexylthiophene) (P3HT)21,22 and poly(3,4-ethylenedioxythiophene) (PEDOT).23,24 The NDI building block has been proven to be an excellent electron transporting unit, with electron mobilities of 0.04 cm2 V−1 s−1 in a vacuum and 0.01 cm2 V−1 s−1 in the air.25–27 However, the polymerization of diimide derivatives, including NDI, possess a significant amount of steric hindrance with neighboring heteroarenes, leading to a higher degree of backbone distortion, which hampers the chain's coplanarity, and hence, the film morphology and conductivity. Therefore, the deteriorated crystallinity and consequent reduced charge carrier mobility limit the ability to improve the efficiency of the NDI polymer in many electronic devices. The structural modification of the NDI-based co-polymer by introducing different thiophene rings in between two NDI moieties decreases the steric hindrance on the monomers drastically, which in turn improves the planarity, crystallinity, and carrier mobility of the polymer significantly.28
NDI-based co-polymers are incredible due to their tunable large arrays of molecules with substantial electron transport characteristics of the NDI core, and hence, they show broad application versatility.29 The NDI-based polymers where the NDI unit is linked with the bithiophene unit, and dithienylvinylene/dithienylethane unit, as n-type donor–acceptor (D–A) co-polymers have gained substantial attention for organic electronic applications.30–33 For the NDI-thiophene-based D–A co-polymers, the lower-lying unoccupied molecular orbitals are spatially localized on the NDI unit, and the higher-lying occupied molecular orbitals are spatially localized on the thiophene unit. Erdmann et al., Zhao et al., and Ghosh et al. studied how the electronic structures, film's crystallinity, mechanical properties, and charge transport properties of the PNDI-TVTx (NDI-dithienylvinylene) co-polymer are affected by the change in π-conjugation lengths.34–36 n-Doping of the NDI-based co-polymers with molecular dopants e.g., 4-(2,3-dihydro-,3-dimethyl-1H-benzimidazol-2-yl)-N,N-dimethylbenzeneamine (N-DMBI), and tetrakis(dimethylamino)ethylene (TDAE), results in localization of polarons/bipolarons on the NDI-units, due to which their electronic structures and optical properties change. The changes in the (opto)electronic properties of the polymer result in altered conductivity37 and, eventually, affect the device's performance. The evolution of electronic structures and optical properties with the reduction level to have a clear insight into how the polaron(s)/bipolaron(s) form and how they change the (opto)electronic properties is very crucial for the deployment of NDI-based devices.
The theoretical study provides us with detailed information and a proper understanding of the evolution of electronic structures and optical properties, which is almost impossible to explore through experimental studies and is limited for PNDI-TVT to a great extent. The electronic, optical, morphological, mechanical, and charge transport properties of PNDI-TVTx co-polymers in the pristine form have been studied theoretically by Ghosh and coworkers.35,36 However, to date, to the best of our knowledge, a complete theoretical insight into the evolution of the (opto)electronic properties, viz., the polaron/bipolaron formation and how they affect the electronic transition in the excited-state of the NDI-TVT polymer upon reduction due to the delocalization of the excess charge carriers, is completely missing.
In this paper, using density functional theory (DFT), we have reported a complete theoretical understanding of the electronic structures, electron spin signal, and optical properties of the PNDI-TVT polymer in solvent and gas phases. We have reported the evolution of these properties with the reduction level by systematically increasing the total number of negative charges (Q) on the polymer chain ranging from Q = −1 to −6 and −10, corresponding to reduction levels (Cred) ranging from 10 up to 200%. We have found a consistently high absolute electron affinity value, i.e., ∼−3.8 eV for the PNDI-TVT copolymer for all the reduction levels in the solvent phase, with very minute variation with polaronic/bipolaronic states. This observation suggests the strong binding of the negatively added charges to the polymer, making PNDI-TVT a promising n-type conducting polymer that can be doped as high as up to 200% in the solvent. The bandgap of the polymer calculated from the difference between electron affinity and ionization energy shows excellent agreement with the experimental data. We have also investigated the UV-vis-NIR absorption peak for the PNDI-TVT polymer in both the solvent and gas phases for the pristine polymer and found a similar trend with the experimentally reported absorption spectra. We note that the UV-vis-NIR absorption spectra of the reduced polymer include peaks due to the electronic transitions from the polaronic/bipolaronic energy levels. The calculations provide a detailed understanding of the evolution of the (opto)electronic properties of the NDI-TVT polymer with the reduction levels in both the gas and solvent phase, which is completely missing to date and could be crucial to improve the performance of the polymer for the (opto)electronic devices.
Methods
For this study, we considered NDI5TVT4 as the model system, which is one oligomer chain of PNDI-TVT co-polymer consisting of five naphthalene diimide (NDI) units with a methyl group attached to each phenyl ring and four dithienylvinylene (TVT) units, i.e., two π-conjugated thiophene rings connected via ethene (with π-bond), see Fig. 4(a). We optimized the geometry of NDI5TVT4 at all the reduction levels using density functional theory (DFT) with the ωB97XD38 hybrid functional and 6-31+G(d) basis set39 in the Gaussian 1640 package. We have considered the lowest energy conformer of the NDI-TVT on the potential energy surface (PES) where the dihedral angle between NDI and TVT moieties θ ≈ 120°, having no imaginary frequencies, see Fig. S16 (discussed in detail in the ESI†). Note that, apart from the lowest energy conformer (θ ≈ 120°), the energies of three more conformers, viz., θ ≈ 60°, 240°, and 300°, lie in the range of 1–2 kJ mol−1 as of the minimum energy conformer, and therefore, all four conformers may coexist in the polymer chain and contribute to the experimental measurements. The functional, ωB97XD, overcomes the localization problem, capturing both short- and long-range interactions.41–43 It has also been observed that this functional quantitatively reproduces electronic structures for various conjugated polymers.43 We have tested the accuracy of the basis set 6-31+G(d) used in this study in predicting the structural, electronic, and optical properties of the NDI-TVT polymer. This was done by performing the calculations with lower and higher basis sets than 6-31+G(d), and considering a basis set that includes the diffuse function on all atoms, including hydrogen, see Fig. S17 and S18, discussed in the ESI.† We have optimized the charge-neutral/pristine (Q = 0) and the negatively-doped PNDI-TVT to obtain a minimum-energy structure at each reduction level, viz., Q = −1 (Cred = 20%), Q = −2 (Cred = 40%), Q = −3 (Cred = 60%), Q = −4 (Cred = 80%), Q = −5 (Cred = 100%), Q = −6 (Cred = 120%), and Q = −10 (Cred = 200%), and studied the effect of doping levels on the (opto)electronic properties. The solvent water was modelled implicitly by the polarizable continuum model, including the integral equation formalism variant (IEFPCM).44We used the corresponding DFT-optimized ground-state geometries of the lowest energy spin state for all the charged states to calculate the optical properties by analyzing the UV-VIS/NIR optical absorption spectra using time-dependent density functional theory (TD-DFT) calculations at the same level of theory, i.e., ωB97XD/6-31+G(d), and the same solvent model for the DFT calculations. To compare the calculated absorption spectra with the experimentally reported spectra of the pristine PNDI-TVT polymer, we calculated the UV-vis absorption spectra of the undoped/pristine state of the PNDI-TVT copolymer in chloroform solvent using TD-DFT at ωB97XD/6-31+G(d) with IEFPCM as the absorption spectra of the undoped NDI-TVT are primarily measured in chloroform.45–48 To obtain the absorption spectra of conducting polymers at different redox states through in situ spectroscopy, the film is electrochemically switched at different applied potentials in the presence of electrolytes, e.g., KCl and NaCl in water, water being the most prevalent solvent.3,47,49 Therefore, we have used water as the solvent to study the absorption spectra of reduced NDI-TVT chains (note that the experimental spectra of the reduced NDI-TVT are unavailable to date). More details on simulation methods are available in the ESI.†
We have also calculated the electronic properties of PNDI-TVT in the gaseous phase using the same basis set, 6-31+G(d), and a lower basis set, 6-31G(d) (without the diffuse basis function, +), see Section S2 and S3 in ESI.† We found that in the gas phase all the electronic properties, i.e., the lowest spin-multiplicity states (see Fig. S6, S10, and Table S1, ESI†), the charge distribution (see Fig. S3 and S7, ESI†), the electronic spin distribution (see Fig. S9 and S12, ESI†), and the total electronic spin (see Fig. S8, S12(h) and Table S1, ESI†), show qualitatively the same features for both the basis sets, 6-31G(d) and 6-31G+(d), but a significant difference in solvent phase.
Results and discussion
Electronic structures
The band diagram of the undoped PNDI-TVT (NDI5TVT4) is plotted in Fig. 1, where the unoccupied electronic levels in the conduction band are depicted in red lines, and the occupied electronic levels in the valence band are depicted in blue lines. The highest occupied molecular orbital (HOMO) and a significant portion of the higher-lying occupied electronic states, HOMO−1, HOMO−2, and HOMO−3, have electron densities localized on the TVT moieties, see Fig. 1. On the other hand, the lowest unoccupied molecular orbital (LUMO) and the majority of the low-lying unoccupied electronic states, LUMO+1, LUMO+2, and LUMO+3, exhibit localized electron densities within the NDI units. As most of the lower-lying unoccupied states are localized on NDI moieties, upon reduction of the polymer, the NDI moieties will act as acceptor units to accommodate the added electrons. Interestingly, a mini-band formation appears in the undoped PNDI-TVT chain where the band of lower-lying states, both in the conduction band and valence band, is segregated by a mini-gap from the higher-lying states. To gain additional insight into the factors that contribute to the generation of the mini-band, we analyzed the molecular orbital plots of the LUMO to LUMO+4 and of the HOMO to HOMO−4 levels shown in Fig. 1.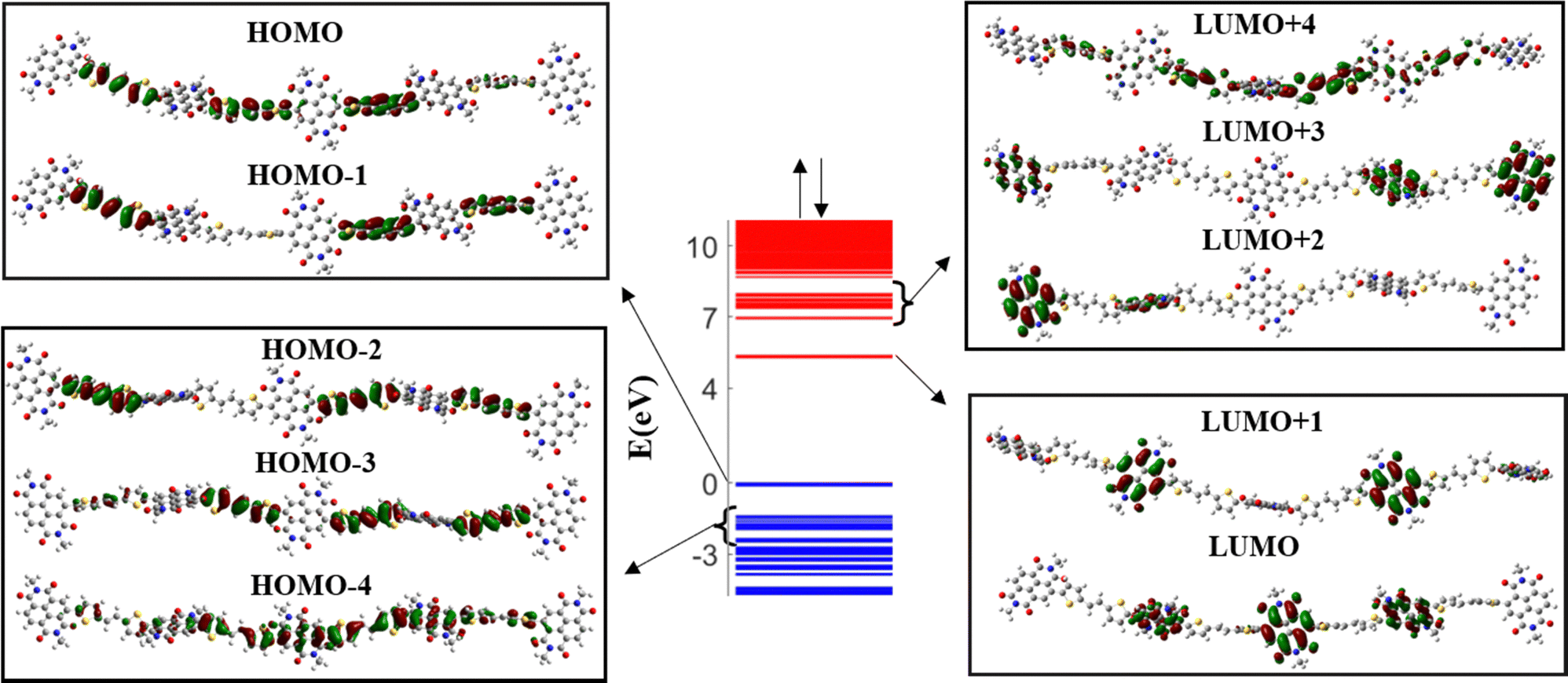 | ||
| Fig. 1 (center) Band diagram of the undoped (Q = 0, Cred = 0%) NDI-TVT chain indicating (left) the highest molecular orbital (HOMO) and some of the higher-lying occupied levels, HOMO−1, HOMO−2, HOMO−3, HOMO−4, and (right) the lowest molecular orbital (LUMO) and some of the lower-lying unoccupied levels, LUMO+1, LUMO+2, LUMO+3, LUMO+4. Empty electronic levels in the conduction band are visualized in red, and the occupied electronic levels in the valence bands are visualized in blue. | ||
We note that the molecular orbitals (MOs) of the lower-lying levels of the conduction band are localized on the NDI units. However, the MOs of the higher-lying levels (LUMO+4) extend towards the TVT units. Similarly, for the valence band, the MOs of the higher-lying levels are localized on the TVT units, and the MOs of the lower-lying levels (HOMO−4) extend towards the NDI units resulting in the formation of the mini-band. A similar picture is also noticed for the reduced (doped) polymers, see Fig. S2 (ESI†).
We have calculated the electron affinity (EA) of the NDI-TVT chain for the charged states ranging from Q = −1 to −6 (Cred = 20% to 120%) by deducting the energy of that system from the energy of the preceding system to have a complete picture of whether the added negative charges are tightly held to the polymer. The EA which defines the amount of energy released when an electron is added to a neutral system40 was calculated for all the charged states using the following formula,
| EAn = Enstate − E(n−1)state | (1) |
The variation of the electron affinity of the doped PNDI-TVT polymer chain with the addition of consecutive negative charges in the solvent phase (water) and the gaseous phase is shown in Fig. 2(a) and (b), respectively. The EA remains almost constant (ranging from −3.88 to −3.90 eV) with the successive addition of electrons up to 100% reduction level (Q = −5) in the solvent phase. We have compared the EA of PNDI-TVT in its 1st doping state with the reported EA of another widely studied n-type conducting polymer, BBL, and found that PNDI-TVT has a higher absolute value of electron affinity (EAabs) than BBL.50 However, on further addition of the electron at the 120% doping state, i.e., Q = −6, there is a slight but sudden decrease in the EAabs to −3.22 eV. The decrease in the EAabs is attributed to the first bipolaron formation in the chain. Therefore, we note that the electron affinity is higher in the case of the polaron formation, which decreases with the bipolaron formation. Earlier reports showed that the n-doped conjugated polymer with high EAabs is a benchmark property for high-performance (opto)electronic devices.51,52 In our work, we have observed consistently high EAabs of the NDI-TVT polymer in the solvent phase, even at very high reduction levels, making it a promising n-type conducting polymer for various applications in organic (opto)electronic devices.
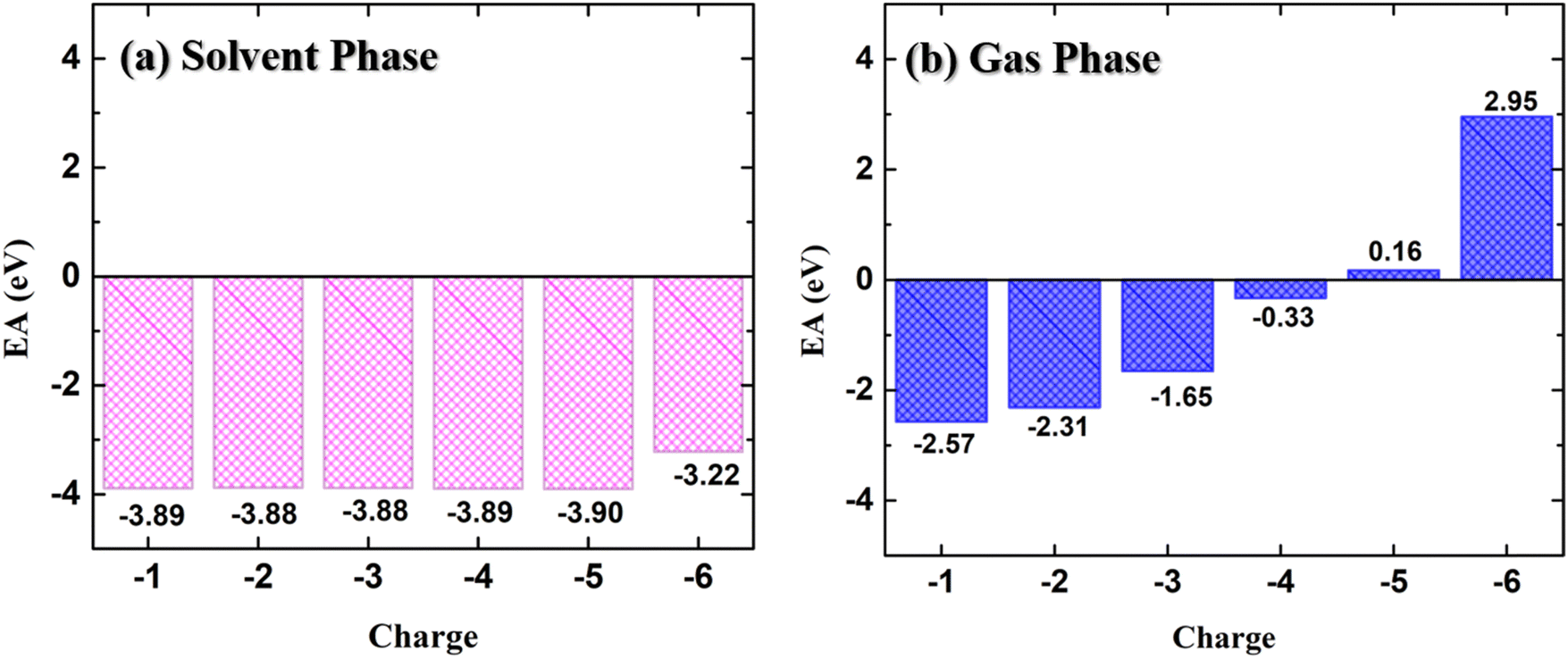 | ||
| Fig. 2 Electron affinity (EA) of NDI-TVT at different doped states from Q = −1 to Q = −6 (a) in the solvent phase and (b) in the gas phase. | ||
On the other hand, the electron affinity in the gas phase, shown in Fig. 2(b), indicates that with the increase in the reduction level, the EAabs decreases. Moreover, at a very highly reduced state, Q = −5 (Cred = 100%) onward, the EA becomes positive, suggesting that for Q = −5 and −6, the polymer is less likely to bind additional electrons in the gaseous phase. This may arise due to the increasing repulsion between the negatively charged regions within the multiple charged anions, which is well explained in the existing literature.41,42
We have compared the calculated electronic properties of NDI-TVT with the existing experimental results by calculating electron affinity (EA), ionization potential (IP), and bandgap (Eg). We calculated EA and IP by taking the energy difference between the neutral and the 1st reduced states and between the neutral and 1st oxidized states, respectively. The DFT calculated EA and the IP can be approximated as LUMO and HOMO, respectively, measured through the cyclic voltammetry (CV) by varying the applied potential, and the difference between them is considered as the bandgap.53 A schematic representation of the definition of IP, EA, and Eg used in this study is shown in Fig. S11(a) (ESI†). Experimentally, Kim et al.26 have calculated the HOMO of PNDI-TVT as −5.42 eV and the LUMO as −4.00 eV. The calculated IP, referred to as the HOMO, and the calculated EA, referred to as the LUMO, of PNDI-TVT in water are ∼−5.34 eV and ∼−3.89 eV, respectively, which are very close to the experimentally measured HOMO and LUMO values, as shown in Fig. 3. The calculated bandgap of PNDI-TVT in the solvent phase is ∼1.45 eV, which is very close to the experimentally estimated bandgap Eg of 1.42 eV.
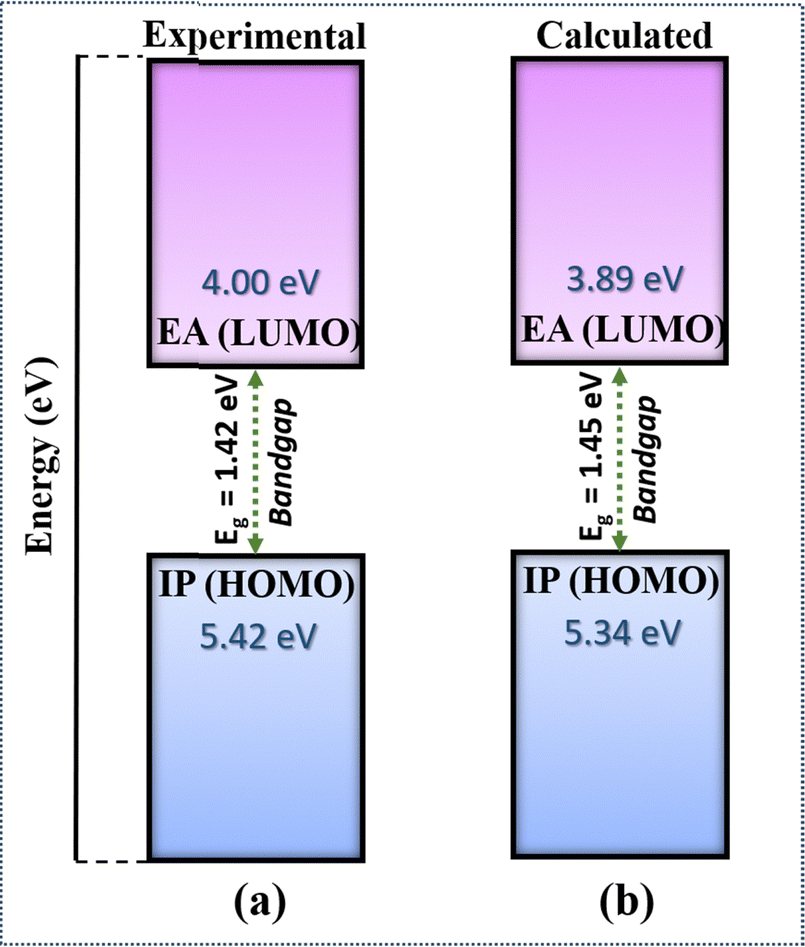 | ||
| Fig. 3 (a) Electron affinity (EA), ionization potential (IP) and bandgap of the NDI-TVT polymer taken from experimental data of Kim et al.26 (b) Calculated IP, EA and bandgap of the NDI-TVT polymer in the solvent phase. | ||
Electronic structures of conducting polymers change with doping (oxidation/reduction) levels due to the formation of the quasiparticle polaron and/or spin-resolved bipolaron (bound pair of two polarons). Precisely, the bipolaron is a spinless pair of polarons (denoted as “BP”) having opposite spins (spin-up and spin-down), sharing the same energy levels and wave functions. To understand the evolution of the electronic structures of NDI-TVT, we have optimized the structure of NDI-TVT at each charged state (Q = 0, −1, −2, −3, −4, −5, −6, and −10) for all the possible spin states (S) corresponding to the multiplicity M (= 2S + 1), and calculated the ground-state energy for all the cases, as shown in Fig. 4(b). The distributions of electron spin on individual NDI units within the PNDI-TVT chain in the gas and solvent phases at all the reduction levels are plotted in Fig. S12 (ESI†) and Fig. 5(a)–(g), respectively. The charge distribution over all the NDI units for the gas and solvent phase is shown in the ESI,† see Fig. S3 and S15, respectively. The only possible spin state for the neutral system (Q = 0) is the singlet. For the case of one added electron, i.e., a single charge per chain (Q = −1, Cred = 20%), there is one unpaired electron, and the only practicable state is a doublet, M = 2. The one added electron to the PNDI-TVT polymer produces one polaron of spin S = 1/2, and the negative charge density of the polaron is accumulated on the 5th NDI moiety, NDI5, see Fig. 5(a). Therefore, the spin of NDI5 is 0.5, whereas the spin of the rest of the NDI units is zero, see Fig. 5(a). For Q = −2 (Cred = 40%), there are two added electrons in the chain, which can result in the formation of either a spin-resolved bipolaron (S = 0, M = 1) or a pair of two distinct polarons (S = 1, M = 3). Our calculation shows that, for the case of two added electrons, the ground state energy of the state with a triplet multiplicity is the lowest, indicating the formation of two distinct polarons rather than a single bipolaron (discussed in the following sections in detail) and as a result, the negative charges are distributed in two different NDI units, i.e., NDI1 and NDI5, see Fig. 5(b). Note, the predicted electronic structure using the modern DFT technique of two added charge states forming two distinct polarons differs from the pre-DFT predictions,54 which typically claim that the presence of two excess charges forms spinless bipolaron states (S = 0, M = 1) and also differs from the reported electronic structure of n-doped polymers where the potential existence of a triplet ground-state has not been examined.55 This is further confirmed by performing open-shell broken-symmetry singlet-state (U-singlet) calculation, which shows that both closed-shell singlet-state (R-singlet) and U-singlet converge at higher energy compared to the open-shell triplet state calculation (S = 1) and the degeneracy of the energy levels is intact in U-single as it is in the R-single, see Fig. S19 (ESI†). However, our present study also lines up with the earlier investigation on the n-doped BBL and PEDOT polymers using the modern DFT approach, where the addition of two electrons exhibits the same nature of forming the two distinct polarons having a triplet ground state instead of forming a bipolaron having the single spin state.56 We have observed precisely the same pattern of the charge density and spin distribution on the NDI moieties in the gaseous phase to the solvent phase for Q = −1 (Cred = 20%) and Q = −2 (Cred = 40%), see Fig. S3(a), (b), S12(a), (b) and S15(a), (b) (ESI†).
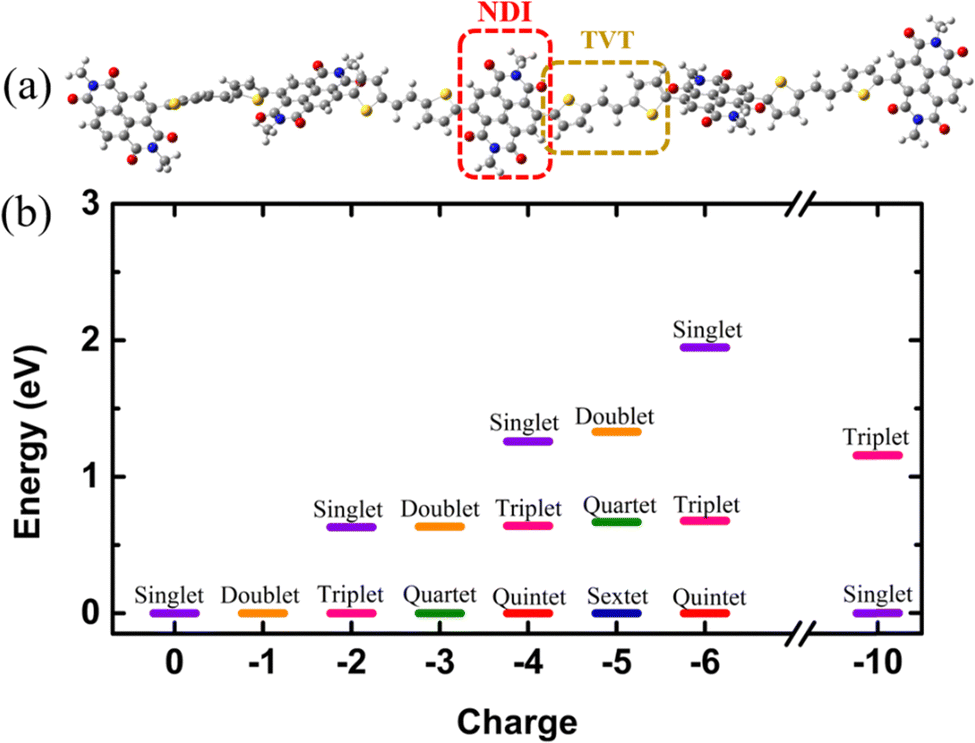 | ||
| Fig. 4 (a) The PNDI-TVT (NDI5TVT4) chain, where an NDI moiety is highlighted in the red box, and one TVT moiety is highlighted in the yellow box. (b) The total energy of the NDI-TVT polymer chain at different spin states of neutral and reduced at different charged states from 0 to −6 and −10 in the solvent phase, where all the ground-state energies are set to zero for convenience. | ||
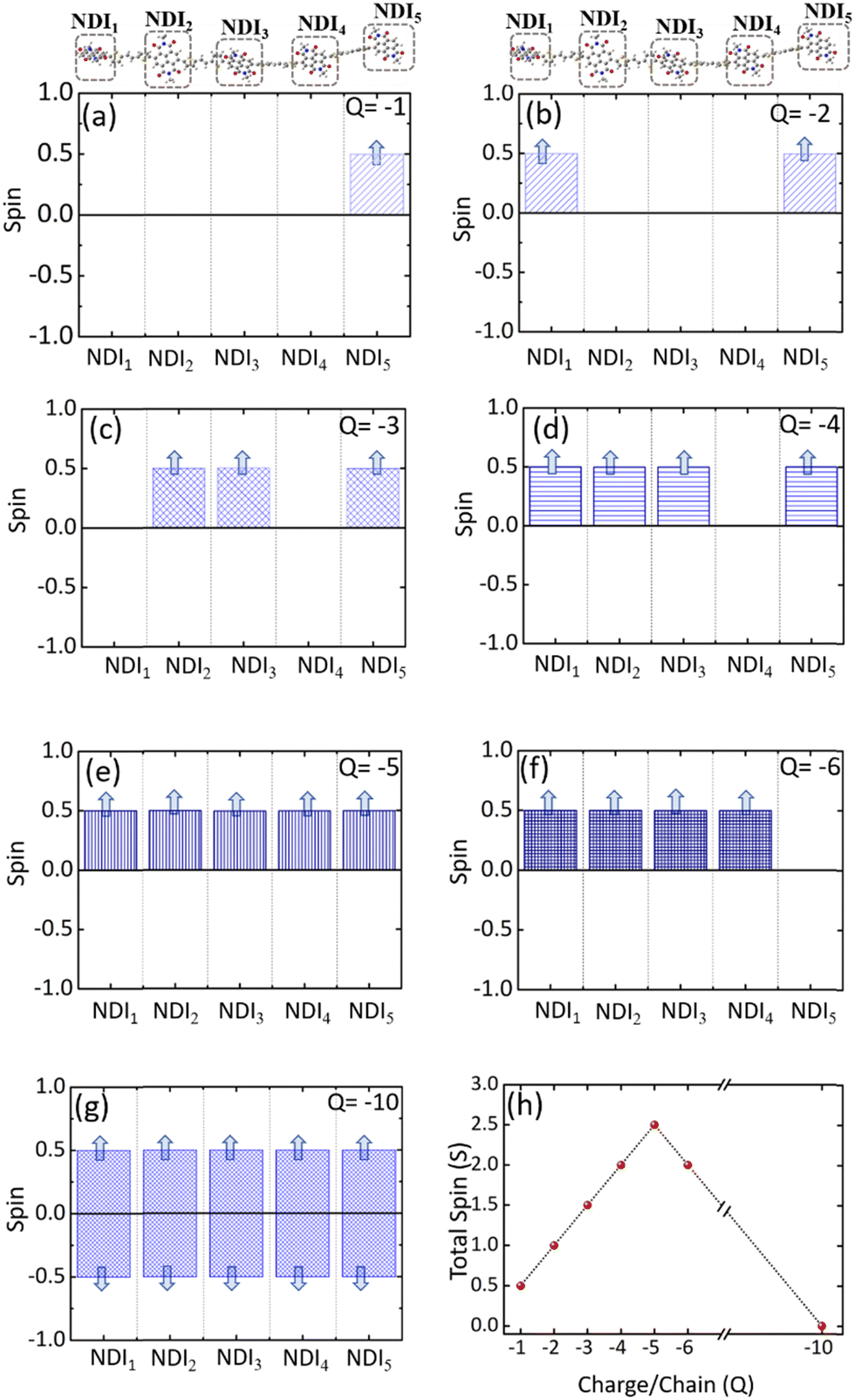 | ||
| Fig. 5 The electronic spin distribution on individual NDI-units within the NDI-TVT polymer chain showing distinct spin states at all the reduction levels in the solvent phase (water), (a) Q = −1, (b) Q = −2, (c) Q = −3, (d) Q = −4, (e) Q = −5, (f) Q = −6, and (g) Q = −10. (h) The electronic total spin (S) of the PNDI-TVT chain at different charged states in the solvent phase (water). | ||
For Q = −3, −5, the system with an odd number of charges, the state could either be a doublet (M = 2), a quartet (M = 4) or a sextet (M = 6). For Q = −3, i.e., the addition of three electrons, we found a quartet as the lowest ground state, see Fig. 4(b), and three NDI units, viz., NDI2, NDI3, and NDI5 moieties have a spin of 0.5 each, see Fig. 5(c). As a result, the total spin of the chain for Q = −3 (Cred = 60%) increases to S = 3/2, see Fig. 5(h). For the systems with an even number of charges (Q = −2, −4, −6), the plausible states could be quintet (M = 5), triplet (M = 3), or singlet (M = 1). The chain with four added electrons Q = −4 (Cred = 80%) has a quintet ground state (M = 5). The NDI1, NDI2, NDI3, and NDI5 have spins of 0.5 each, see Fig. 5(d), and the total spin again increases to 2 (S = 2), as shown in Fig. 5(h). The accumulation of the negative charges on NDI1, NDI2, NDI3, and NDI5 can be seen in Fig. S15(d) (ESI†). Further inclusion of an electron dopes the NDI-TVT polymer to 100%, i.e., Q = −5 (Cred = 100%), and the lowest ground state is a sextet. At this stage, all the NDIs, i.e., NDI1, NDI2, NDI3, NDI4, and NDI5 moieties, have a spin of 0.5 containing a single polaron by each of them. These electron spins added up effectively, yielding a total spin S = 5/2 (M = 6), see Fig. 5(e).
Upon the addition of one more electron to the polymer chain, i.e., Q = −6 (Cred = 120%), the lowest ground state is a quintet. NDI1, NDI2, NDI3, and NDI4 have a spin of 0.5 each, and NDI5 has a spin equal to zero; hence, the overall electronic spin of the chain becomes 2. The zero spins on NDI5 indicate the formation of the first bipolaron, a spatially localized pair of two polarons, at a reduction level beyond 100%. It has been found in the literature that the NDI-based polymers, including PNDI-TVT polymer, can be doped up to 200% reduction levels.56 Therefore, we added ten negative charges in the chain to study the 200% reduction state. For Q = −10 (Cred = 200%), the presence of ten electrons in a PNDI-TVT chain results in the formation of five distinct bipolarons localized on every NDI moiety, and the ground state is a singlet (S = 0, M = 1), see Fig. 4(b). At this stage, all the NDI units are filled with one bipolaron containing one spin-up and one spin-down electron, and as all the added electrons are spin-resolved bipolarons, the total electronic spin of the chain is 0.
The variation of the total electronic spin of the NDI-TVT chain as a function of added charges (Q) is plotted in Fig. 5(h). The total electronic spin of the chain continuously increases with doping and reaches its peak at Cred = 100%. However, from Cred = 120% onwards, the spin starts decreasing gradually, and at Cred = 200%, it becomes zero. We note that the addition of electrons follows a ‘Λ’-shape pattern, which says a continuous increase of electronic spin up to 100% reduction level where each acceptor unit, i.e., an NDI moiety, has one electron and then the electronic spin of the chain starts to decrease until it becomes zero at 200% reduction level where all the acceptor moieties have two electrons each, as seen in Fig. 5(h). This pattern supports the first single-filling of the electron to the available acceptor units, and upon the unavailability of available space for single-filing, doubly occupied electronic states are formed. Notably, Ghosh et al.56 reported a similar spin pattern in n-doped polymer BBL, which exhibits an increase in the spin signal and decreases to zero beyond 100% reduction levels in the gaseous phase. On the contrary, we observed a zig-zag pattern of the total spin of the PNDI-TVT chain in the gaseous phase due to a different arrangement of the polarons in spin-up and spin-down states within the chain from Q = −3, Cred = 60% onwards, see Fig. S12(h) (ESI†). In the case of the gaseous phase for Q = −3, NDI3 and NDI5 acquired a spin of 0.5 each, and NDI1 has a spin of −0.5, as shown in Fig. S12(c) (ESI†), which is in contrast with the solvent phase where all the polarons are formed in spin-up states resulting in an increase in total spin. Therefore, the total spin of the chain in the gas phase decreases to 0.5. For Q = −4, in the gaseous phase, the spin distribution of the chain shows that the NDI1, NDI3, and NDI5 have a spin of 0.5 each, and NDI4 has a spin of −0.5, resulting in a total spin of 1 (S = 1), see Fig. S12(d) (ESI†). The corresponding distribution of the negative charges is accumulated at NDI1, NDI3, NDI4, and NDI5, see Fig. S3(d) (ESI†), while the negative charges are accumulated at NDI1, NDI2, NDI3 and NDI5 in the solvent phase, see Fig. S15(d) (ESI†). For Q = −6, the NDI1 is localized with one spin-up and one spin-down electron, and the rest of the NDI units have a spin of ∓0.5. Specifically, NDI1, NDI2, NDI3, NDI4, and NDI5 have a spin of 0, 0.5, 0.5, −0.5, and 0.5, respectively, and the total electronic spin of the chain becomes 1. The zigzag pattern of the evolution of total electronic spin of the PNDI-TVT chain from undoped to the fully reduced state in the gas phase, along with the positive EA value Cred = 100% onward, suggests that the reduction of the polymer at the higher doping levels is unfavorable in the gas phase. However, past experimental study says that the polymer can be electrochemically doped to its fully reduced state, i.e., 200% reduction levels.56 For Q = −10, Cred = 200%, in both the solvent phase and gaseous phase, all the NDI units are filled with spin-resolved bipolarons, the total spin of the chain being zero. In our calculation, electron affinity in the solvent phase (water) is negative even beyond 100% reduction level of the polymer. This observation implies that the negatively added charges are tightly bound to the polymer in the solvent phase, and the polymer can be reduced up to 200% reduction level, making PNDI-TVT a promising n-type conducting polymer. Note, the obtained lowest energy spin-state corresponding to each reduction level has been considered in all the follow-up calculations.
The electronic structure of the PNDI-TVT chain obtained from the ground state DFT calculations of undoped, Q = 0 (Cred = 0%), and negatively-doped, Q = −1, −2, −3, −4, −5, −6 and −10 (Cred = 20%, 40%, 60%, 80%, 100%, 120%, and 200%) are shown in Fig. 6(e)–(l), which depict the occupied electronic levels in the valence band as blue lines and the unoccupied electronic levels in the conduction band as red lines. The up (↑) and down arrows (↓) signify the spin-up and spin-down orbitals, respectively. The ground state of the pristine NDI chain has a singlet spin multiplicity; hence, the spin degeneracy is intact for all the levels. For the charge Q = −1 (Cred = 20%), the spin degeneracy is lifted due to the formation of a polaronic state in the spin-up state (shown in a cyan dashed line); see Fig. 6(f). Due to the formation of a new occupied energy level between the valence band and the conduction band, the gap between the occupied and unoccupied energy levels significantly decreases as compared to the undoped chain, which in turn would reduce the energy barrier of an electron to jump from the polaronic state to the conduction band. Henceforward, to differentiate the (bi)polaronic levels from the other occupied levels present in the valence band, formed due to the added electrons in the gap between the valence and conduction band, they are denoted with dashed cyan lines. Note, the observed electronic structure of the polaronic state (for Q = −1) aligns with the electronic structure of other corresponding n-doped conducting polymers56–58 as documented in previous research. Due to the formation of a single polaronic state on the addition of one electron to the PNDI-TVT chain, we observed the localization of corresponding MOs on only one of the acceptor (NDI) units, viz., the 5th NDI unit (NDI5), see Fig. 6(a). This result is in line with the individual spin calculation of each NDI, which also shows that the polaron is localized on NDI5, the spin of the NDI5 is ∼0.49 (nearly equal to ½), and the spins of the other NDI units remain 0, see Fig. 5(a). It is well known that (bi)polarons alter the molecular geometry in the conjugated polymers as benzoid-type rings convert into quinoid types, changing the bond lengths and bond angles.58 To quantify the structural changes due to the formation of (bi)polarons, we calculated the change in the bond length of all the bonds present in the NDI units, neglecting the C–H bond as defined in Fig. S1 (ESI†). We observed that for Q = −1 (one polaron localized on the 5th NDI unit), the 5th NDI unit is structurally altered, whereas the remaining NDI units are left structurally unchanged, see Fig. 6(a). In the case of 2 added electrons to the PNDI-TVT (Q = −2, Cred = 40%), the ground state is a triplet, and the corresponding electronic structure is shown in Fig. 6(g). We observed two newly added occupied electronic levels in the spin-up state. The corresponding electronic distributions of the two polaronic states are observed to be localized on two spatially different NDI units, namely on NDI1 and NDI5, see Fig. 6(b). This further confirms that the two added electrons (Cred = 40%) result in two polarons that are spatially isolated from each other, as clearly seen in Fig. 6(b). Furthermore, this is completely in line with the observed change in the bond length and the non-zero spin of NDI1 and NDI5, see Fig. 5(b) and 6(b). For Q = −1 and Q = −2, in the gaseous phase, we have observed the same trend of localization of polarons with the structural alteration on the same NDI units as in the solvent phase, i.e., NDI5 for Q = −1 and NDI1 and NDI5 for Q = −2, see Fig. S13(a) and (b) (ESI†).
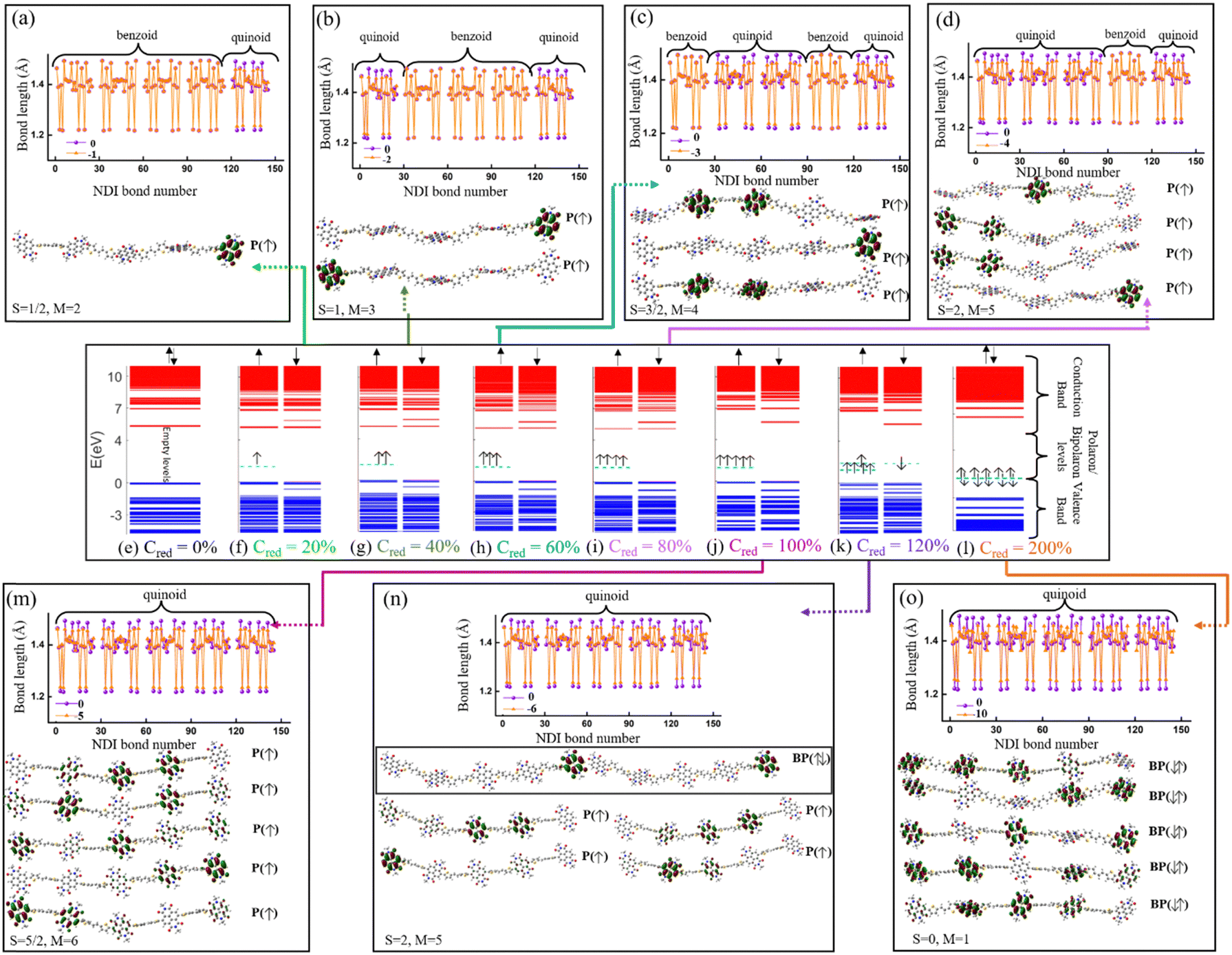 | ||
| Fig. 6 Molecular orbitals of polaron(P)/bipolaron(BP) states along with bond-length alteration of NDI moieties for all the charged states (a) Q = −1 (Cred = 20%), (b) Q = −2 (Cred = 40%), (c) Q = −3 (Cred = 60%), (d) Q = −4 (Cred = 80%), (m) Q = −5 (Cred = 100%), (n) Q = −6 (Cred = 120%), (o) Q = −10 (Cred = 200%). Band diagrams of (e) the undoped PNDI-TVT chain (Q = 0) and (f)–(l) the doped PNDI-TVT chain in the solvent phase. In the band diagrams, the empty electronic levels in the conduction band are plotted as red lines, occupied electronic levels in the valence band are plotted as blue lines, and occupied polarons/bipolarons are plotted as cyan dashed lines. Up and down -arrows indicate the spin-up and spin-down state, respectively. S indicates the total electronic spin of the chain, and M indicates the corresponding spin multiplicity. | ||
For the case of three added electrons (Q = −3, Cred = 60%), we observed the localization of three individual polarons, all formed in the same spin-up state on three different NDI units (NDI2, NDI3, NDI5). The corresponding bond-length alteration is observed on these three NDI units, see Fig. 6(c). Similarly, in the gas phase, three polarons are formed, out of which two are with spin-up state and the third polaron is with spin-down state, see Fig. S12(c) (ESI†). For Q = −4, Cred = 80%, the energy levels of the 4 individual polarons, having the same spin state (spin-up) are shown in Fig. 6(i). The electron and spin distributions of the polarons are mainly localized on these four NDI units, NDI1, NDI2, NDI3 and NDI5, and the corresponding bond length alteration is observed on the same NDI moieties, see Fig. 6(d). On the contrary, in the gaseous phase for Q = −4, Cred = 80%, three of the four individual polarons are formed in the same spin state (spin-up), and the other is formed in the opposite spin state (spin-down). The electron and spin distributions are localized only on these four NDI units, namely, NDI1, NDI3, NDI4, and NDI5, see Fig. S13(d) and (i) (ESI†). 100% reduction level (Cred = 100%, Q = −5 i.e., one added electron per monomer) results in the formation of five individual polarons. The electronic structure shows that newly formed polarons are in the spin-up state, making the total spin S = 5/2 (M = 6). Moreover, we observed that the electron distribution of the polarons is localized on all the NDI units, according to Hund's rule of exclusive packing of the electrons to the available states. As a result, the bond length alternation plot also shows that all the NDI units are structurally altered, see Fig. 6(m). However, the trend is entirely different in the gaseous phase. Among the five individual polarons, three polarons are in the spin-up state, and two are in the spin-down state, making the total spin S = ½ (M = 2), which is not very clear from the bond length alternation plot as it shows that all the NDI units are structurally altered, see Fig. S13(m) (ESI†). Beyond this reduction level, when all the NDI units are occupied by one polaron each, the addition of one more electron to the chain (Q = −6, Cred = 120%) shows an interesting behavior of the electronic structure in PNDI-TVT, which is the presence of both polarons and bipolarons in the same chain. The polaron with spin-up is localized on four distinct NDI units (NDI1, NDI2, NDI3, and NDI4), and the bipolaron is localized on one NDI unit, viz., NDI5 moiety, see Fig. 6(k), resulting in the total electronic spin of PNDI-TVT as S = 2 (counting only the spin of the four polarons). The structural change is more pronounced on NDI5 than on the remaining NDI units due to the presence of bipolarons, see Fig. 6(n). It is quite interesting that the presence of both polarons and bipolarons is also observed in the gas phase at a 120% reduction level with the bipolaron being localized on the NDI1 unit, and therefore, structural change is more noticeable on the same NDI unit, see Fig. S13(n) (ESI†). From the above analysis, we have observed that the bipolarons are only formed when the PNDI-TVT polymer is reduced beyond 100%.
At a 200% reduction level (Q = −10), it is noteworthy that the MOs of a single bipolaron is distributed among more than one NDI moiety, which is not the case in the gaseous phase where we observed localization of the MO of a single bipolaron on a single NDI moiety, see Fig. S13(o) (ESI†). It is also seen from the electronic structure that five spin-degenerate bipolarons are formed with no well-separated energy levels, as we observed that there are overlaps of electronic density distributions among the bipolarons as a single bipolaron is delocalized over three NDI units. At this stage, all the NDI units are occupied with bipolarons, and the prominent bond-length alternation and the corresponding electron distribution also support this fact, see Fig. 6(o). In the gas phase at a 200% reduction level (Q = −10), two excess electrons are present per NDI moiety, and five spin-degenerate spatially distinct bipolarons are formed, one per NDI unit, see Fig. S13(l) (ESI†). Although the polarons/bipolarons are predominantly localized on the NDI moieties, note, at the higher reduction levels (Cred >100%), there is a minute spatial extension of the electronic densities to the neighboring thiophene rings of the TVT units to accommodate a large number of excess charges.
The evolution of the electronic structures of PNDI-TVT is completely different in the solvent phase as compared to the gaseous phase, see Table S1 (ESI†). Electronic structure analysis reveals that although the presence of solvent does not affect the number of polaron(s) and bipolaron(s) formed, a significant effect is noticed in the total energy of the system, the lowest energy state, total spin, electron affinity (EA), HOMO, LUMO, and bandgap of the PNDI-TVT polymer system. The aforementioned calculated parameters in the solvent phase are in good agreement with experimental data compared to those in the gas phase. It is noteworthy that the observed spin multiplicity of the ground states, the charge distribution patterns, and the total electronic spin at all the charged states remain qualitatively the same for the DFT calculations with the diffuse basis function (+), 6-31+G(d) and without the diffuse basis function, 6-31G(d), as shown in Table S1 (ESI†). Although the distribution of the added charge(s)/electron(s) over the individual NDI moieties might differ for different calculations, the overall picture of charge/electron distribution considering the whole chain as a function of reduction level is qualitatively the same. Our detailed electronic structure analysis reveals that up to 100% reduction levels, only polarons are formed, in the intermediate reduction levels, 100% > Cred < 200%, an admixture of polaronic and bipolaronic states are formed, and upon 200% reduction, complete bipolaronic states are observed in both solvent and gas phases causing a ‘Λ’-shape spin pattern (see Fig. 5(h)) irrespective of the basis sets and the diffuse function and this is also independent of the polymer's chain length, see Fig. S21 (ESI†).
Optical properties
Using the TD-DFT approach, we have investigated the optical absorption spectra of the NDI-TVT polymer in both its pristine/undoped states (Q = 0) in water as a solvent, see Fig. 7(a), and the doped states (Q = −1 to −10), see Fig. 7(b)–(h). The absorption spectra of the neutral PNDI-TVT show two distinct, prominent peaks, a unique feature that differs from various linear polymers, see Fig. 7(a). The peak at ∼460 nm is due to the electronic transition from the HOMO energy level to the LUMO energy level, called ICT i.e., the intramolecular charge transfer band caused by the donor and acceptor configuration of the polymer structure. The peak at the lower wavelength, i.e., at ∼343 nm, called π–π* transition, has a major contribution from three electronic transitions, viz., (1) transition from the HOMO energy level to the higher-lying unoccupied energy level, LUMO+7, (2) transition from the lower-lying occupied energy level, HOMO−2 to the higher-lying unoccupied energy level, LUMO+5, and (3) transition from the further lower-lying occupied energy level, HOMO−12 to the higher-lying unoccupied energy level LUMO+2, as shown in Fig. S20 (ESI†). Notably, the estimated spectra for the neutral PNDI-TVT are in line with experimentally recorded spectra reported by Erdmann et al.34 and TD-DFT calculated spectra reported by Ghosh et al.,35 which says that the peak at ∼343 nm corresponds to the transition from the valence band to the mini-band within the conduction band and the peak at ∼460 nm is due to the HOMO–LUMO transition.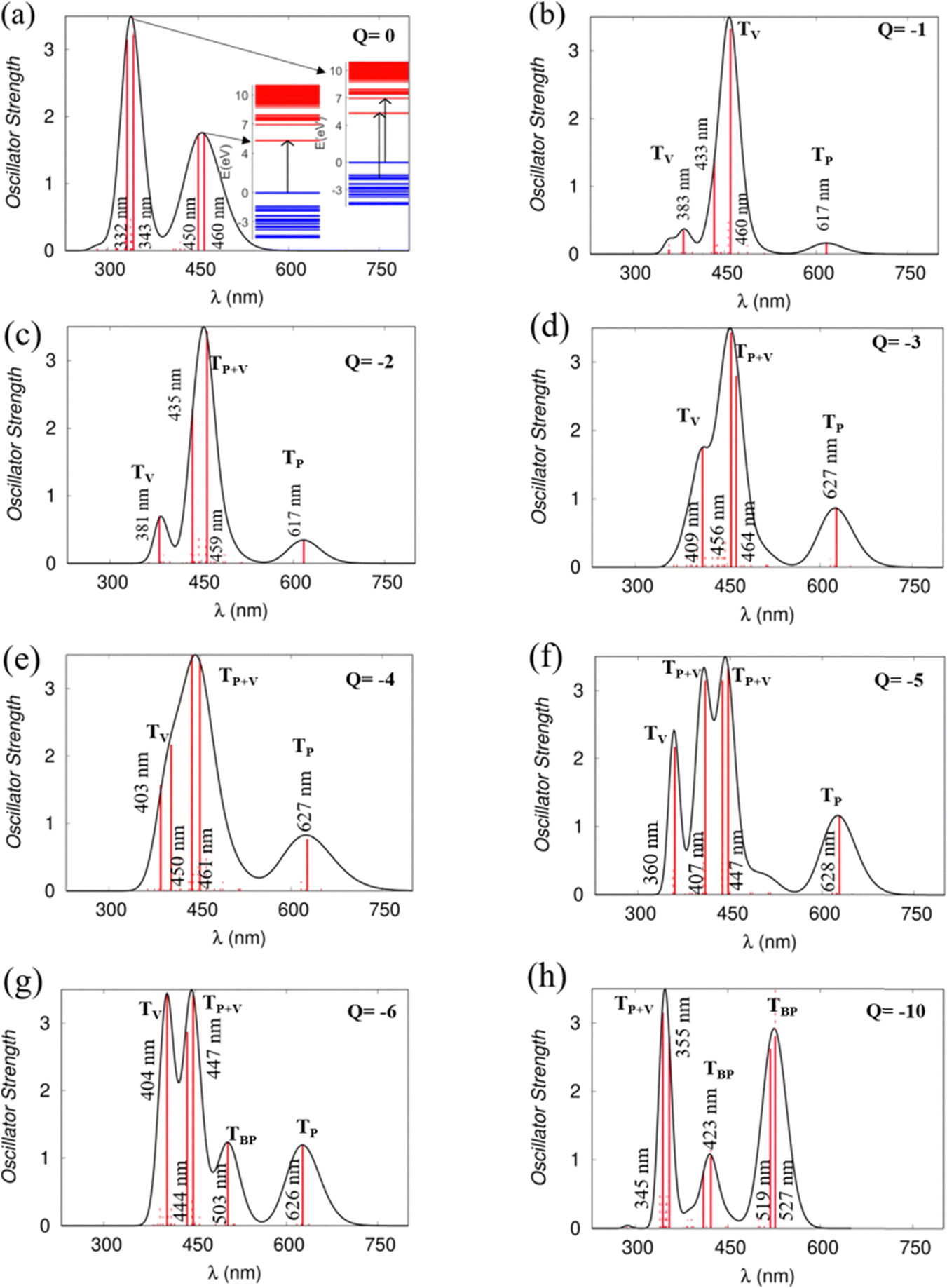 | ||
| Fig. 7 UV vis/NIR absorption spectra of (a) the charge neutral PNDI-TVT chain and (b)–(h) the doped PNDI-TVT chain at various charged states from Q = −1 to −6 and −10 indicating the corresponding electronic transitions calculated in the solvent phase. | ||
We have also compared the TD-DFT calculated spectra of undoped PNDI-TVT in both gaseous and solvent phases with the available experimental measured spectra taken from the study of Kim et al.,26 where they measured them both in chloroform solution and as a solid film, see Fig. 8. Precisely, we have compared the experimentally measured spectra of PNDI-TVT in chloroform with TD-DFT calculated spectra in the solvent phase, chloroform, and the experimentally measured spectra of the dry polymeric film with the TD-DFT calculated spectra in the gaseous phase, to thus establish a comprehensive comparison between theory and experiment, shown in Fig. 8(a) and (b). In the experimental spectra, two distinct absorption peaks are observed both in the solution and dry phase, see Fig. 8(b). Similarly, the TD-DFT calculated spectra also show two major absorption peaks in both the solvent conditions, chloroform and water, and in the gaseous phase, see Fig. 7(a) and 8(a). The peak at a higher wavelength that corresponds to the HOMO–LUMO transition, i.e., the ICT peak, shows a noticeable red shift for the case of the TD-DFT calculated spectra in the gas phase and the experimentally measured spectra of the dry film, compared to the ICT peak present in both experimental and calculated absorption spectra in solvent. The observed blue/hypsochromic shift, i.e., shifting towards a shorter wavelength, and the observed hypsochromic effect i.e., shorter absorbance intensity of the ICT peak in the solvent phase spectra are caused by the solvent's polarity, which increases the bandgap between the HOMO–LUMO level and shifts the peak towards a lower wavelength by stabilizing the ground state more than the excited states.59 We found that the calculated absorption peaks of the neutral PNDI-TVT in its pristine state in the solvent and gas phase are in good agreement with the experimental absorption spectra across different environments, which shows the accuracy of the computational models. One should note that the spectra measured from in situ spectroscopy show peaks at higher wavelength values than the TD-DFT calculated spectra, with the difference being very prominent for the 2nd peak, i.e., the absorption peak at the longer wavelength. This is also in line with the previously reported TD-DFT studies of the n-doped polymers60,61 which might be attributed to the fact that the experiments are performed for longer polymeric chains, whereas, in TD-DFT calculations, oligomers (dimer, trimer, tetramers, etc.) are used.61 Future improvement in the TDDFT approach in predicting the doped-state absorption spectra of n-type conducting polymers may result in better alignment between the calculated and experimental peak positions. However, we note that the qualitative agreement between the calculated and experimental spectra can be used to demonstrate the qualitative nature of the evolution of the optical properties with doping.
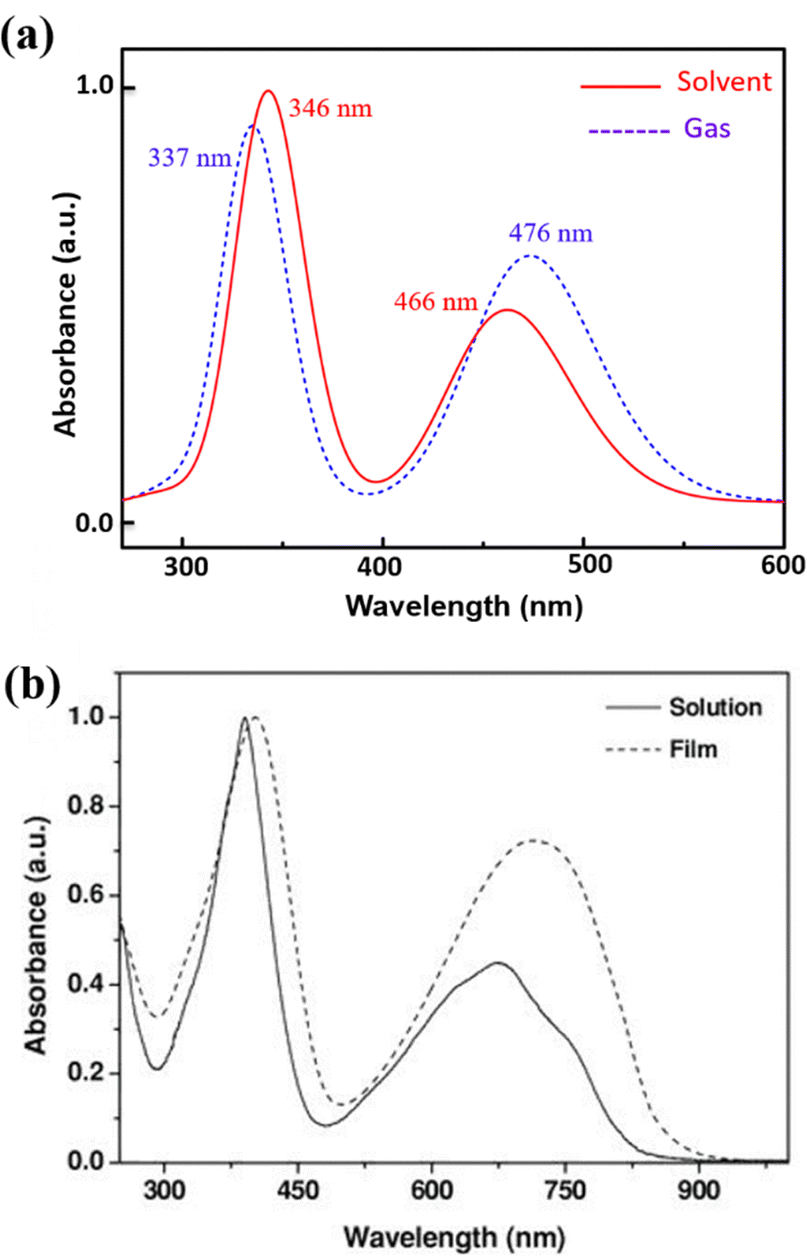 | ||
| Fig. 8 (a) TD-DFT calculated UV-vis absorption spectra of PNDI-TVT in chloroform solution and the gas phase. (b) Experimental data of UV-vis absorption spectra of PNDI-TVT in chloroform solution and thin film taken from the work of Kim et al.26 Reproduced with permission from WILEY. | ||
The absorption spectra of the doped PNDI-TVT are depicted in Fig. 7(b)–(h). The peaks in the spectra are classified in four ways according to the dominant electronic transitions occurring from the occupied energy levels to the unoccupied levels with the highest configuration interaction factors, as defined in Table S2 (ESI†), namely, (1) the transitions from polaronic energy levels to the energy levels in the conduction band (TP), (2) the transitions from bipolaronic energy levels to the energy levels in the conduction band (TBP), (3) the transitions between the energy levels of the valence and the conduction band and also the transitions from both polaronic/bipolaronic levels (TP+V), and, (4) the energy levels in the valence band to the energy levels in the conduction band (TV). The schematic definition of all the four peaks is shown in Fig. 9.
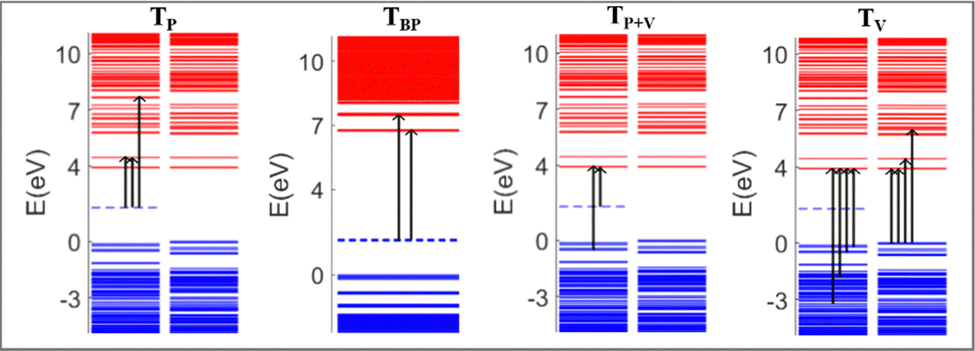 | ||
| Fig. 9 Schematic definition of four different transitions, where TP represents the transition from polaronic states to the energy levels in the conduction band, TBP represents the transition from bipolaronic energy levels to higher energy levels in the conduction band, TP+V represents the electronic transitions both from the polaronic/bipolaronic states and any other states in the valence band to the energy levels in the conduction band, and TV represents the electronic transition from the energy levels present in the valence band to the energy levels present in the conduction band. | ||
Upon single-charge (Q = −1, Cred = 20%) and double-charge (Q = −2, Cred = 40%) doping, in the absorption spectra of PNDI-TVT in the solvent phase, we observed the formation of a new prominent peak at a higher wavelength around ∼617 nm compared to the spectra of the neutral chain, see Fig. 7(a)–(c). The new peak has the contribution of electronic transition solely from the polaronic energy level to the conduction band marked as TP. Whereas the other two peaks at ∼380 nm and ∼430 nm represent the electronic transitions from the valence band to the conduction band, they are marked as TV, see Table S2 (ESI†). For the charged state Q = −3 (Cred = 60%), i.e., in the case of three polarons, the contribution from the polaronic energy levels to the absorption spectra increases, and the contribution from the energy levels in the valence band decreases significantly. Precisely, we observed a prominent peak at a higher wavelength of ∼627 nm compared to Q = −1, −2 attributed to the electronic transition from the polaron to the conduction band. Besides, the spectra show a sharp peak at ∼456 nm due to the transition from the polaron and valence band to the conduction band, and we observed a shoulder peak at ∼409 nm due to the electronic transition from the valence band to the conduction band. For the reduction levels from Q = −1 (Cred = 20%) to Q = −6 (Cred = 120%), all the spectra show a distinct peak at approximately 600 nm due to the electronic transitions from the polaronic energy levels, and as the doping level increases, we observed the evolution towards a sharper peak in the absorption spectra from the contribution of the electronic transitions from the polaronic energy levels, see Fig. 7(b)–(g). Similarly, in the gas phase for reduction levels from Q = −1 (Cred = 20%) to Q = −4 (Cred = 80%), all the spectra show a distinct peak at a comparatively higher wavelength ∼1000 nm attributed to electronic transitions from the polaronic energy levels, and as the doping level increases, we observed more contribution of the electronic transition from the polaronic energy levels to the overall absorption spectra. At Q = −5 (Cred = 100%), in the gaseous phase, we observed that most of the peaks in the spectra are in the range of 400–800 nm, and there is one TP peak at ∼3665 nm, shown in the inset plot of Fig. S14(f) (ESI†). In the solvent phase, up to the 100% reduction level, we note that all the spectra exhibit an overall red shift towards a longer wavelength and a distinct polaronic absorption peak, TP, at ∼600 nm. Besides, among all the spectra of all reduction levels in the solvation phase, we didn’t observe any peak beyond ∼650 nm, but in the gas phase, we observed peaks at a much higher wavelength of ∼3665 nm in the IR region.
At Q = −6, one prominent and intense new peak appears at ∼500 nm; this is due to the formation of the first bipolaron at a 120% reduction level. For the case of Q = −10 (Cred = 200%), each bipolaron is shared by three NDI units, see Fig. 6(o), and the corresponding absorption spectra in the range of ∼350–600 nm show three distinguishable peaks, at ∼350 nm, at ∼430 nm and at ∼ 530 nm. The peak at ∼350 nm is due to the electronic transitions from the polaronic energy level to the conduction band along the transition from the valence band to the conduction band. The spectra range from ∼380 to ∼600 nm due to the electronic transitions from the bipolaronic energy levels to the conduction band. We note that the formation of the bipolarons diminishes the mini-gap in the electronic structure, increasing the energy gap between the occupied and unoccupied energy levels compared to the cases where polarons are formed, see Fig. 6(l).
Conclusions
In the present work, we provided a complete theoretical understanding in terms of electronic structures, electronic spin signal, and optical properties of the PNDI-TVT polymer at different doping levels ranging from 0–200% studied using density functional theory and the time-dependent density functional theory calculations in both solvent and gaseous phases. The electronic structure calculation of the undoped NDI-TVT polymer chain shows that the occupied levels are localized on the imide group, i.e., NDI moieties, and the unoccupied levels are localized at the TVT moieties and hence, upon reduction, the added electrons will accumulate on the NDI moieties. We found a strong alignment of the electronic structures of the pristine polymer with the existing experimental data, which we compared by calculating the electron affinity, ionization potential, and bandgap of PNDI-TVT. The polymer exhibits consistently high electron affinity up to 200% reduction level, indicating the potential of PNDI-TVT to be used as an n-type conducting polymer in the solvent phase (water). However, we note from the gaseous phase electron affinity calculation that the polymer is unfavorable to reduce beyond 100% without solvent. The comparison of the UV-vis-NIR absorption spectra of the neutral/pristine polymer in the solvent and gas phase shows a red shift in the gas phase, which aligns with the experimentally measured absorption spectra of the dry PNDI-TVT film compared to that in solvent. The evolution of the electronic spin signal with the reduction level exhibits a very specific ‘Λ’-shaped pattern. Initially, the spin signal increases as the doping level reaches its peak at 100% and eventually becomes zero at the 200% reduction level as all the states of the excess electrons are spin-resolved bipolarons. The DFT calculations predict that upon doping, the bipolarons are rarely formed for PNDI-TVT at the lower reduction levels, and they exist only at the higher reduction level Cred > 100%. At 200% reduction levels, the presence of two electrons per NDI unit leads to the formation of spin-resolved bipolarons, where one bipolaron is delocalized over three NDI units. At the intermediate reduction levels (100% < Cred < 200%) both the polarons and bipolarons coexist in the same PNDI-TVT. The evolution of UV-vis-NIR spectra with reduction levels shows four distinguished electronic transitions that are accountable for the absorption peak in the spectra, namely, TP, TBP, TV, and TP+V. Specifically, peak TP denotes the transitions from the polaronic levels to the conduction band, peak TBP denotes the transitions from the bipolaronic levels to the conduction band, peak TV corresponds to the hopping from the valence band to the conduction band, and peak TP+V indicates a combination of transitions from the energy levels of the polaronic/bipolaronic and valence band. With the increase in doping level, from Q = −1 (Cred = 20%) to Q = −6 (Cred = 120%), we observed the evolution of a sharper and distinct absorption peak, TP, ranging from ∼615 to ∼630 nm, originating from the electronic transition due to the polaronic energy levels. Beyond 100% doping level, it is observed that the peak position at the maximum wavelength slightly shifts towards a shorter wavelength but remains longer than that of the undoped PNDI-TVT chain. At a 200% reduction level, we observed a left shift of the maximum spectra, attributed to the bipolaron formation, which diminishes the mini-band in the electronic structure and the consequent increases in the energy gap between occupied and unoccupied energy levels, which is hardly ever explained in the past study. This evolution of polarons and bipolarons observed in the electronic structures is also reflected in the calculated UV-vis-NIR absorption spectra, which show the presence of only polaronic peaks (i.e., no bipolaronic peak) in the reduced polymer up to Cred = 100%, a mixture of polaronic and bipolaronic peaks at the intermediate reduction levels (100% < Cred < 200%), and only bipolaronic peaks (i.e., no polaronic peak) at the 200% reduction level. The detailed theoretical insights gained from this comparative study in both solvent and gas phases addressing the electronic and optical properties of PNDI-TVT at different reduction levels can be useful to guide the design of future NDI-TVT-based (opto)electronic devices.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the SERB Start-up Research Grant, file no. SRG/2021/002169. The authors thank the Department of Chemical Engineering, BITS Pilani. MG thanks the support of SERB Start-up Research Grant, file no. SRG/2022/001617. The computations were performed on the SERB-funded HPC facility and BITS Pilani HPC facility.References
- V. Saxena and B. D. Malhotra, Prospects of Conducting Polymers in Molecular Electronics, Curr. Appl. Phys., 2003, 3(2–3), 293–305 Search PubMed.
- C. W. Lee, O. Y. Kim and J. Y. Lee, Organic Materials for Organic Electronic Devices, J. Ind. Eng. Chem., 2014, 20(4), 1198–1208 Search PubMed.
- K. Namsheer and C. S. Rout, Conducting Polymers: A Comprehensive Review on Recent Advances in Synthesis, Properties and Applications, RSC Adv., 2021, 11(10), 5659–5697 RSC.
- H. Yao, Z. Fan, H. Cheng, X. Guan, C. Wang, K. Sun and J. Ouyang, Recent Development of Thermoelectric Polymers and Composites, Macromol. Rapid Commun., 2018, 39(6), 1700727 CrossRef PubMed.
- X. Wu, W. Fu and H. Chen, Conductive Polymers for Flexible and Stretchable Organic Optoelectronic Applications, ACS Appl. Polym. Mater., 2022, 4(7), 4609–4623 CrossRef CAS.
- X. Jia, Y. Ge, L. Shao, C. Wang and G. G. Wallace, Tunable Conducting Polymers: Toward Sustainable and Versatile Batteries, ACS Sustainable Chem. Eng., 2019, 7(17), 14321–14340 CrossRef CAS.
- S. Griggs, A. Marks, H. Bristow and I. McCulloch, N-Type Organic Semiconducting Polymers: Stability Limitations, Design Considerations and Applications, J. Mater. Chem. C, 2021, 9(26), 8099–8128 RSC.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Light-Emitting Diodes Based on Conjugated Polymers, Nature, 1990, 347(6293), 539–541 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Recent Advances in Bulk Heterojunction Polymer Solar Cells, Chem. Rev., 2015, 115(23), 12666–12731 Search PubMed.
- A. Tsumura, H. Koezuka and T. Ando, Macromolecular Electronic Device: Field-effect Transistor with a Polythiophene Thin Film, Appl. Phys. Lett., 1986, 49(18), 1210–1212 CrossRef CAS.
- J. Janata and M. Josowicz, Conducting Polymers in Electronic Chemical Sensors, Nat. Mater., 2003, 2(1), 19–24 CrossRef CAS PubMed.
- C. P. Grey and J. M. Tarascon, Sustainability and in Situ Monitoring in Battery Development, Nat. Mater., 2017, 16(1), 45–56 CrossRef PubMed.
- D. O. Akinyele and R. K. Rayudu, Review of Energy Storage Technologies for Sustainable Power Networks, Sustainable Energy Technol. Assess., 2014, 8, 74–91 CrossRef.
- Y. Yamashita, Organic Semiconductors for Organic Field-Effect Transistors, Sci. Technol. Adv. Mater., 2009, 10(2), 24313 CrossRef PubMed.
- S. M. Sze, Y. Li and K. K. NgPhysics of Semiconductor Devices, John Wiley & Sons, 2021 Search PubMed.
- D. M. De Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Stability of N-Type Doped Conducting Polymers and Consequences for Polymeric Microelectronic Devices, Synth. Met., 1997, 87(1), 53–59 CrossRef CAS.
- A. Facchetti, π-Conjugated Polymers for Organic Electronics and Photovoltaic Cell Applications, Chem. Mater., 2011, 23(3), 733–758 CrossRef CAS.
- X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, A High-Mobility Electron-Transport Polymer with Broad Absorption and Its Use in Field-Effect Transistors and All-Polymer Solar Cells, J. Am. Chem. Soc., 2007, 129(23), 7246–7247 CrossRef CAS PubMed.
- L. Zhang, Z. Wang, C. Duan, Z. Wang, Y. Deng, J. Xu, F. Huang and Y. Cao, Conjugated Polymers Based on Thiazole Flanked Naphthalene Diimide for Unipolar N-Type Organic Field-Effect Transistors, Chem. Mater., 2018, 30(22), 8343–8351 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, A High-Mobility Electron-Transporting Polymer for Printed Transistors, Nature, 2009, 457(7230), 679–686 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang and N. Gasparini, High-Efficiency and Air-Stable P3HT-Based Polymer Solar Cells with a New Non-Fullerene Acceptor, Nat. Commun., 2016, 7(1), 11585 CrossRef CAS PubMed.
- N. P. Holmes, S. Ulum, P. Sista, K. B. Burke, M. G. Wilson, M. C. Stefan, X. Zhou, P. C. Dastoor and W. J. Belcher, The Effect of Polymer Molecular Weight on P3HT: PCBM Nanoparticulate Organic Photovoltaic Device Performance, Sol. Energy Mater. Sol. Cells, 2014, 128, 369–377 CrossRef CAS.
- W. F. Quirós-Solano, N. Gaio, C. Silvestri, G. Pandraud and P. M. Sarro, PEDOT: PSS: A Conductive and Flexible Polymer for Sensor Integration in Organ-on-Chip Platforms, Procedia Eng., 2016, 168, 1184–1187 CrossRef.
- Y. Yang, H. Deng and Q. Fu, Recent Progress on PEDOT: PSS Based Polymer Blends and Composites for Flexible Electronics and Thermoelectric Devices, Mater. Chem. Front., 2020, 4(11), 3130–3152 RSC.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, Naphthalenedicarboximide-vs. Perylenedicarboximide-Based Copolymers. Synthesis and Semiconducting Properties in Bottom-Gate n-Channel Organic Transistors, J. Am. Chem. Soc., 2009, 131(1), 8–9 CrossRef CAS PubMed.
- R. Kim, P. S. K. Amegadze, I. Kang, H. J. Yun, Y. Y. Noh, S. K. Kwon and Y. H. Kim, High-Mobility Air-Stable Naphthalene Diimide-Based Copolymer Containing Extended π-Conjugation for n-Channel Organic Field Effect Transistors, Adv. Funct. Mater., 2013, 23(46), 5719–5727 CrossRef CAS.
- S. Shao, Z. Chen, H.-H. Fang, G. H. Ten Brink, D. Bartesaghi, S. Adjokatse, L. J. A. Koster, B. J. Kooi, A. Facchetti and M. A. Loi, N-Type Polymers as Electron Extraction Layers in Hybrid Perovskite Solar Cells with Improved Ambient Stability, J. Mater. Chem. A, 2016, 4(7), 2419–2426 RSC.
- Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, Naphthodithiophenediimide (NDTI): Synthesis, Structure, and Applications, J. Am. Chem. Soc., 2013, 135(31), 11445–11448 CrossRef CAS PubMed.
- Z. Yuan, Y. Ma, T. Geßner, M. Li, L. Chen, M. Eustachi, R. T. Weitz, C. Li and K. Müllen, Core-Fluorinated Naphthalene Diimides: Synthesis, Characterization, and Application in n-Type Organic Field-Effect Transistors, Org. Lett., 2016, 18(3), 456–459 CrossRef CAS PubMed.
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. A. Sengrian and P. C. T. Souza, Enhancing Molecular N-type Doping of Donor–Acceptor Copolymers by Tailoring Side Chains, Adv. Mater., 2018, 30(7), 1704630 CrossRef PubMed.
- J. Liu, G. Ye, B. Zee, J. van der; Dong, X. Qiu, Y. Liu, G. Portale, R. C. Chiechi and L. J. A. Koster, N-Type Organic Thermoelectrics of Donor–Acceptor Copolymers: Improved Power Factor by Molecular Tailoring of the Density of States, Adv. Mater., 2018, 30(44), 1804290 CrossRef PubMed.
- S. Wang, H. Sun, T. Erdmann, G. Wang, D. Fazzi, U. Lappan, Y. Puttisong, Z. Chen, M. Berggren and X. Crispin, A Chemically Doped Naphthalenediimide-bithiazole Polymer for N-type Organic Thermoelectrics, Adv. Mater., 2018, 30(31), 1801898 CrossRef PubMed.
- Y. M. Gross, D. Trefz, C. Dingler, D. Bauer, V. Vijayakumar, V. Untilova, L. Biniek, M. Brinkmann and S. Ludwigs, From Isotropic to Anisotropic Conductivities in P (NDI2OD-T2) by (Electro-) Chemical Doping Strategies, Chem. Mater., 2019, 31(9), 3542–3555 CrossRef CAS.
- T. Erdmann, S. Fabiano, B. Milián-Medina, D. Hanifi, Z. Chen, M. Berggren, J. Gierschner, A. Salleo, A. Kiriy and B. Voit, Naphthalenediimide Polymers with Finely Tuned In-Chain Π-Conjugation: Electronic Structure, Film Microstructure, and Charge Transport Properties, Adv. Mater., 2016, 28(41), 9169–9174 CrossRef CAS PubMed.
- S. Ghosh, N. Rolland and I. Zozoulenko, Electronic Structure, Optical Properties, Morphology and Charge Transport in Naphthalenediimide (NDI)-Based n-Type Copolymer with Altered π-Conjugation: A Theoretical Perspective, Appl. Phys. Lett., 2021, 118(22), 223302 CrossRef CAS.
- D. Zhao, D. Kim, S. Ghosh, G. Wang, W. Huang, Z. Zhu, T. J. Marks, I. Zozoulenko and A. Facchetti, Mechanical, Morphological, and Charge Transport Properties of NDI Polymers with Variable Built-in Π-Conjugation Lengths Probed by Simulation and Experiment, Adv. Funct. Mater., 2024, 34(4), 2310071 CrossRef CAS.
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, Heavily N-Dopable π-Conjugated Redox Polymers with Ultrafast Energy Storage Capability, J. Am. Chem. Soc., 2015, 137(15), 4956–4959 CrossRef CAS PubMed.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections, J. Chem. Theory Comput., 2013, 9(1), 263–272 CrossRef CAS PubMed.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, 6-31G* Basis Set for Third-row Atoms, J. Comput. Chem., 2001, 22(9), 976–984 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. NakatsujiGaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Systematic Optimization of Long-Range Corrected Hybrid Density Functionals, J. Chem. Phys., 2008, 128(8), 084106 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- U. Salzner and A. Aydin, Improved Prediction of Properties of π-Conjugated Oligomers with Range-Separated Hybrid Density Functionals, J. Chem. Theory Comput., 2011, 7(8), 2568–2583 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General Formalism, J. Chem. Phys., 2010, 132(11), 114110 CrossRef PubMed.
- B. Kang, R. Kim, S. B. Lee, S.-K. Kwon, Y.-H. Kim and K. Cho, Side-Chain-Induced Rigid Backbone Organization of Polymer Semiconductors through Semifluoroalkyl Side Chains, J. Am. Chem. Soc., 2016, 138(11), 3679–3686 CrossRef CAS PubMed.
- R. Kim, P. S. K. Amegadze, I. Kang, H. Yun, Y. Noh, S. Kwon and Y. Kim, High-mobility Air-stable Naphthalene Diimide-based Copolymer Containing Extended Π-conjugation for N-channel Organic Field Effect Transistors, Adv. Funct. Mater., 2013, 23(46), 5719–5727 CrossRef CAS.
- Q. Li, J.-D. Huang, T. Liu, T. P. A. van der Pol, Q. Zhang, S. Y. Jeong, M.-A. Stoeckel, H.-Y. Wu, S. Zhang, X. Liu, H. Y. Woo, M. Fahlman, C.-Y. Yang and S. Fabiano, A Highly Conductive N-Type Conjugated Polymer Synthesized in Water, J. Am. Chem. Soc., 2024, 146(23), 15860–15868 CrossRef CAS PubMed.
- C. Beaumont, J. Turgeon, M. Idir, D. Neusser, R. Lapointe, S. Caron, W. Dupont, D. D’Astous, S. Shamsuddin, S. Hamza, É. Landry, S. Ludwigs and M. Leclerc, Water-Processable Self-Doped Conducting Polymers via Direct (Hetero)Arylation Polymerization, Macromolecules, 2021, 54(12), 5464–5472 CrossRef CAS.
- V. Sethumadhavan, K. Zuber, C. Bassell, P. R. Teasdale and D. Evans, Hydrolysis of Doped Conducting Polymers, Commun. Chem., 2020, 3(1), 153 CrossRef CAS PubMed.
- S. Ghosh, M. Berggren and I. Zozoulenko, Electronic Structures and Optical Properties of P-Type/n-Type Polymer Blends: Density Functional Theory Study, J. Phys. Chem. C, 2020, 124(17), 9203–9214 CrossRef CAS.
- M. Vagin, V. Gueskine, E. Mitraka, S. Wang, A. Singh, I. Zozoulenko, M. Berggren, S. Fabiano and X. Crispin, Negatively-Doped Conducting Polymers for Oxygen Reduction Reaction, Adv. Energy Mater., 2021, 11(3), 2002664 CrossRef CAS.
- S. C. Moratti, R. Cervini, A. B. Holmes, D. R. Baigent, R. H. Friend, N. C. Greenham, J. Grüner and P. J. Hamer, High Electron Affinity Polymers for LEDs, Synth. Met., 1995, 71(1–3), 2117–2120 CrossRef CAS.
- A. Dittmer, R. Izsak, F. Neese and D. Maganas, Accurate Band Gap Predictions of Semiconductors in the Framework of the Similarity Transformed Equation of Motion Coupled Cluster Theory, Inorg. Chem., 2019, 58(14), 9303–9315 CrossRef CAS PubMed.
- W. R. Salaneck, R. H. Friend and J. L. Brédas, Electronic Structure of Conjugated Polymers: Consequences of Electron–Lattice Coupling, Phys. Rep., 1999, 319(6), 231–251 CrossRef CAS.
- C. Bruno, F. Paolucci, M. Marcaccio, R. Benassi, C. Fontanesi, A. Mucci, F. Parenti, L. Preti, L. Schenetti and D. Vanossi, Experimental and Theoretical Study of the P-and n-Doped States of Alkylsulfanyl Octithiophenes, J. Phys. Chem. B, 2010, 114(26), 8585–8592 CrossRef CAS PubMed.
- S. Ghosh, V. Gueskine, M. Berggren and I. V. Zozoulenko, Electronic Structures and Optical Absorption of N-Type Conducting Polymers at Different Doping Levels, J. Phys. Chem. C, 2019, 123(25), 15467–15476 CrossRef CAS.
- F. Alkan and U. Salzner, Theoretical Investigation of Excited States of Oligothiophene Anions, J. Phys. Chem. A, 2008, 112(27), 6053–6058 CrossRef CAS PubMed.
- W. S. R. Jayasundara and G. Schreckenbach, Theoretical Study of P-and n-Doping of Polythiophene-and Polypyrrole-Based Conjugated Polymers, J. Phys. Chem. C, 2020, 124(32), 17528–17537 CrossRef CAS.
- M. F. Nicol, Solvent Effects on Electronic Spectra, Appl. Spectrosc. Rev., 1974, 8(2), 183–227 CrossRef CAS.
- D. Trefz, A. Ruff, R. Tkachov, M. Wieland, M. Goll, A. Kiriy and S. Ludwigs, Electrochemical Investigations of the N-Type Semiconducting Polymer P (NDI2OD-T2) and Its Monomer: New Insights in the Reduction Behavior, J. Phys. Chem. C, 2015, 119(40), 22760–22771 CrossRef CAS.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Design and Evaluation of Conjugated Polymers with Polar Side Chains as Electrode Materials for Electrochemical Energy Storage in Aqueous Electrolytes, Energy Environ. Sci., 2019, 12(4), 1349–1357 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Section-1: Model and methods: additional simulation details are provided. Section-2: Additional properties calculated at ωB97XD/6-31+G(d) in the gas phase: band diagram and MOs of the HOMO and some lower-lying HOMOs and LUMO and some higher-lying LUMOs of reduced NDI-TVT (Q = −1); the charge distribution on different NDI-units within the NDI-TVT polymer chain. Section-3: Effect of diffuse function: comparison table of spin multiplicity (M) and total spin (S) of the NDI-TVT oligomeric chain using both basis set 6-31+G and 6-31G for pristine and different charged states from Q = −1 to −6 and −10 in the gas and solvent phase (electronic structures calculated at a lower basis set 6-31G(d) without diffuse basis functions ‘+’ in the gas phase), band diagrams and MOs of the HOMO and some lower lying HOMOs and the LUMO and some higher lying LUMOs of doped NDI-TVT; total energy of the NDI-TVT chain of different spin states; charge and spin distribution on each NDI, and total spin (S) of the NDI-TVT chain at all the reduction levels. Electronic structure and optical properties of the NDI-TVT polymer in the gas phase calculated using the 6-31+G(d) total energy of the NDI-TVT chain of different spin states; schematic representation of the definition of the ionization potential (IP) and the electron affinity (EA) and IE and EA of NDI-TVT polymer in the gas phase; the charge and spin distribution on individual NDI-units and total electronic spin at all the reduction levels; electron densities of polaron/bipolaron states along with bond-length alteration of NDI moieties for all the doped states; UV vis/NIR absorption spectra of neutral and doped NDI-TVT. Section-4: Additional properties of NDI-TVT polymer calculated using the optimized functional/basis-set, ωB97XD/6-31+G(d) in the solvent phase, wavelengths, coefficients and the respective transition calculated for NDI-TVT in the solvent phase, water for pristine and different doped states; the charge distribution on different NDI-units in the solvent phase within the NDI-TVT polymer chain showing distinct distribution of charge at various doped levels. See DOI: https://doi.org/10.1039/d4cp02770a |
| This journal is © the Owner Societies 2025 |