
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of radicals in the reaction of oxygen difluoride with monohydrogenated silicon†
Henry
Thake
 and
Stephen J.
Jenkins
*
and
Stephen J.
Jenkins
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: sjj24@cam.ac.uk
First published on 27th November 2024
Abstract
We present first-principles molecular dynamics simulations of oxygen difluoride impinging upon the monohydrogenated Si{001}(2 × 1) surface. Adsorption occurred in fewer than 10% of our computed trajectories, but in each reactive case the initial step involved partial dissociation to yield an adsorbed fluorine atom and a free oxygen monofluoride radical. In one trajectory, the adsorbed fluorine atom displaced a hydrogen atom into an unusual Si–H–Si bridge position, consistent with three-centre two-electron bonding. In another, a Si–Si–F motif was created, consistent with three-centre four-electron bonding. Depending upon its recoil direction, the ubiquitous monofluoride species either migrated across the surface before itself reacting to form a Si–O–Si bridge and a second adsorbed fluorine atom, or desorbed intact as a gas-phase radical.
1 Introduction
Some years ago, our group reported first-principles molecular dynamic simulations of the adsorption of ozone on hydrogen-passivated silicon that revealed an unusual mechanism proceeding via the formation of an intermediate radical.1,2 Specifically, we found that an incoming ozone molecule was typically repelled from a monohydrogenated Si{001} surface but could nevertheless, in a minority of trajectories, actually abstract a hydrogen atom to form a hydrotrioxy (HO3) radical and a silicon dangling bond. The unstable free radical persisted for no more than about 200 fs before dissociating into triplet O2 (which departed from the surface) and a hydroxy radical (OH) that rapidly saturated the dangling bond to create a Si–OH surface moiety. Despite its short lifetime, the intermediate radical was found to be crucial for adsorption to take place at all.More recently, in studies into the reaction of molecular fluorine with the same monohydrogenated silicon surface, we found (amongst other less exotic mechanisms) a small number of trajectories in which abstraction of hydrogen led to the creation of a silicon dangling bond and a very short-lived hydrodifluoro (HF2) species of either radical or anionic character.3 Dissociation of this moiety, again after no more than 200 fs, resulted in saturation of the dangling bond with a single fluorine atom, leaving a closed-shell HF molecule to one of two possible fates: in some trajectories, it simply desorbed intact, while in another it scavenged a second hydrogen atom from the surface resulting eventually in an additional Si–F surface moiety and a desorbing H2 molecule. In this system, the unstable (and possibly radical) intermediate was not essential for adsorption, but on the occasions when it was implicated the surface reaction took a strikingly different course to occasions when it was not.
In search of other similar radical-mediated reactions, we note that a common feature between ozone and molecular fluorine is their highly oxidising nature. This is doubtless central to their ability to abstract hydrogen from any given surface. We therefore set out to identify alternative species that might be equally interesting in their adsorption at hydrogenated silicon surfaces, settling upon oxygen difluoride (OF2) as a likely candidate. In its ground-state configuration, this fascinating molecule4,5 strongly resembles ozone, adopting a bent geometry with O–F bond lengths6 of 1.41 Å (cf. 1.27 Å for the O–O bond lengths7 in ozone) and an O–F–O bond angle6 of 103° (cf. 117° for the O–O–O bond angle7 in ozone). Where the electronic structure of ozone is rather complicated, however, having singlet biradical character,8 oxygen difluoride is simply a bog-standard closed-shell species. Nevertheless, the standard reduction potentials for the reactions
| O2 + 4H+ + 4e− ⇌ 2H2O |
| O3 + 2H+ + 2e− ⇌ O2 + H2O |
| OF2 + 2H+ + 4e− ⇌ H2O + 2F− |
| F2 + 2H+ + 2e− ⇌ 2HF |
Turning from similarities to differences, however, the manifestation of that power is likely to be dominated by the nature of the lowest unoccupied molecular orbital (LUMO) in each case, because we expect strong oxidising agents to be electron acceptors. In ozone, the LUMO has π-antibonding character (Fig. 1a) that suggests greatest reactivity when approached along a direction normal to the molecular plane. This surmise was, indeed, borne out by our prior simulations of ozone adsorption on clean and monohydrogenated silicon surfaces.1,2 By virtue of two additional valence electrons, however, this state becomes the highest occupied molecular orbital of oxygen difluoride (HOMO) while the LUMO now has σ-antibonding character (Fig. 1b) and suggests greatest reactivity when approached along the axis of one or other O–F bond. We anticipate that these differing steric constraints may give rise to qualitatively dissimilar adsorption modes for the latter molecule with respect to the former.
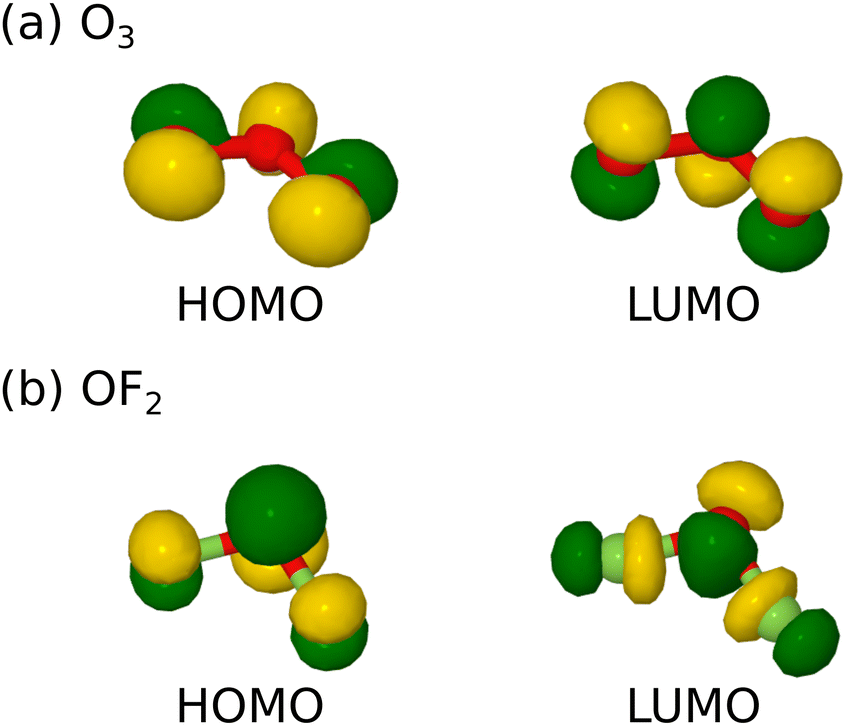 | ||
| Fig. 1 HOMO and LUMO for (a) ozone, and (b) oxygen difluoride. Oxygen and fluorine atoms are shown as red and green spheres respectively, while green and gold isosurfaces indicate opposing phases of the wavefunction in each case. | ||
In the present work, we investigate the adsorption of oxygen difluoride on monohydrogenated Si{001} by means of first-principles molecular dynamics. This assumption-free and rather general technique has been used by ourselves and others in studies of surface adsorption, diffusion, reaction and desorption,1–3,10–26 where it permits one to follow detailed trajectories for individual events and hence to extract valuable mechanistic insight not amenable to static (geometry optimisation) or quasistatic (transition state search) calculations. The computational cost per trajectory can be significant, however, limiting the number that may be calculated and restricting practical application to systems with fairly high sticking probability. It is advisable, therefore, to adopt a systematic approach to initialising and analysing a meaningful set of trajectories, based on knowledge not only of the adsorbate but also of the substrate.
To this end, we shall briefly recap some essential aspects of our chosen target, which is arguably the single most important surface in the context of the commodity semiconductor industry. Modern integrated circuits are lithographically fabricated on high-purity single-crystal wafers with well-defined crystallographic orientation. In the case of silicon, the exposed surface facet is usually of {001}, {111} or {110} type, with the first of these being preferred for most (although not all) electronic applications. During processing, the silicon surface is generally passivated with hydrogen, to avoid build-up of contaminants, and this layer may later need to be removed if other surface coatings are to be substituted in its place. These considerations motivate our interest in the reactivity of this particular surface and we shall begin by highlighting the role of its most characteristic and ubiquitous structural motif – the silicon dimer.27,28 These geometric features are an inevitable consequence of the fact that top-layer atoms in the ideal Si{001} surface each possess two semi-occupied dangling bonds. Lateral pairing of top-layer atoms (dimerisation) can saturate half of these dangling bonds, resulting in a doubling of the surface repeat spacing in one direction – an example of a Peierls distortion, where lowering of translational symmetry, in this case from (1 × 1) to (2 × 1) periodicity, goes hand-in-glove with increased stability. This still leaves one remaining semi-occupied dangling bond on each dimer atom, however, which on the clean surface gives rise to a Jahn–Teller distortion, where lowering of local point symmetry is associated with a further increase in stability. The necessary asymmetry is manifest through buckling of the dimers, coincident with charge transfer between their dangling bonds, and may or may not alter the periodicity depending upon the relative buckling of adjacent dimers. The higher-lying dimer atoms end up with fully occupied dangling bonds, while their lower-lying partners host empty dangling bonds.
Despite the stability of such a buckled-dimer reconstruction relative to an unreconstructed surface, the dangling bonds nevertheless present reactive sites for incoming adsorbates to attack. Broadly, we might expect electrophilic adsorbates to be drawn toward the higher-lying dimer atoms, where there is a concentration of electronic charge, and nucleophilic ones to be drawn toward the lower-lying dimer atoms, where there is a relative lack. Alternatively, however, we might consider the passivation of Si{001} by exposure to hydrogen.27,28 At modest coverages (the monohydride regime) hydrogen reverses the Jahn–Teller distortion, saturating one dangling bond per top-layer atom to form Si–H surface moieties and allowing the dimers to adopt a symmetric (unbuckled) geometry of (2 × 1) periodicity. At higher coverages (the dihydride regime) even the Peierls distortion may be reversed too, breaking the dimers apart and forming H–Si–H surface moieties with (1 × 1) periodicity. We shall focus on the monohydride regime, so our surface of interest is characterised by symmetric silicon dimers, each end of each dimer being decorated with a single hydrogen atom. Our interest lies in whether an intermediate radical species is created upon interaction with oxygen difluoride (either by abstraction of hydrogen or otherwise) and in what role any such species might play in determining the endpoint of adsorption.
2 Computational method
All calculations were performed subject to periodic boundary conditions using the CASTEP computer code (Version 18.1)29 with most key parameters identical to those employed in our group's previous investigation into the dynamics of fluorine adsorption on silicon.3 Specifically, electronic wavefunctions were expanded in a basis set of plane waves up to a kinetic energy cutoff at 350 eV, exchange–correlation interactions between valence electrons were included via the Perdew–Burke–Ernzerhof functional,30 and electron–ion interactions were included through use of ultrasoft pseudopotentials.31 Fractional eigenstate occupancies were allowed and spin was unconstrained, both in anticipation of semi-occupied dangling bonds and other radical species.Although the initial geometry of our surface displays (2 × 1) periodicity, we adopted a supercell of lateral dimensions consistent with a c(4 × 4) surface unit cell to minimise interactions between adsorbates and their periodic images; the supercell length was chosen equivalent to 16 layers of silicon in the [001] crystallographic direction, and Brillouin-zone sampling was accomplished by means of a 2 × 2 × 1 Monkhorst–Pack mesh.32 The surface itself was represented by a slab comprising eight silicon layers, with one side initialised in monohydrogenated and dimerised form and the other in ideal and dihydrogenated form. The back three layers of silicon were held fixed at their bulk positions and all other atoms were permitted to relax according to calculated forces.
For subsequent dynamic simulations, a time-step of 0.5 fs was adopted within the NVE ensemble (i.e. atom numbers, supercell volume, and total energy were all held constant) and electronic convergence at each step was judged against tolerances of 10−5 eV per atom for the total energy and 10−6 eV for individual eigenvalues. This ensemble is necessary to account correctly for the complementary variation in potential and kinetic energies implied by a chemical reaction; were a thermostat to be applied (as in the NVT ensemble) the kinetic energy of our system would be unphysically quenched. Once again, the back three silicon layers were held fixed at their bulk positions, but this time the hydrogen atoms saturating the back surface were also held fixed (at the relaxed positions determined during the previous geometry optimisation procedure). The incoming molecule was initialised with its centre of mass at a height of a little over 6.0 Å above the top-layer silicon atoms of the relaxed surface, travelling vertically along the inward surface normal at a speed of 295 m s−1 (kinetic energy 0.0245 eV) toward one of five sites (A–E) that uniformly sample the surface unit cell. Our chosen translational energy corresponds to the most probable speed for gaseous OF2 at 283 K, according to kinetic theory, while the molecules are both rotational and vibrationally cold. Substrate atoms were also initially cold, starting with zero velocity.
Initial orientations were chosen with reference to the molecule's principal axes of inertia, specifying how these align with high-symmetry directions of the surface. For the latter, we define the α direction to lie along the dimer rows, the β direction to lie across the dimer rows, and the γ direction to be the outward surface normal (Fig. 2). In descending order of their moments of inertia, the principal axes of the molecule may be taken to lie along
| I1||(r1 × r2) |
| I2||(r1 + r2) |
| I3||(r2 − r1) | (1) |
![[small gamma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c4.gif) α orientation, on the other hand, aligns the I1 axis along the inward surface normal (the overline signifying negation) and the I2 axis along the dimer-row direction. In this way, we find that there are eight symmetrically distinct orientations when aiming the molecule at one of the high-symmetry sites (A–D) but twelve when aiming at the low-symmetry site (E). Taking sites and orientations together, therefore, we investigate a total of 44 trajectories, listed in Table 1 alongside their symmetry equivalents.
α orientation, on the other hand, aligns the I1 axis along the inward surface normal (the overline signifying negation) and the I2 axis along the dimer-row direction. In this way, we find that there are eight symmetrically distinct orientations when aiming the molecule at one of the high-symmetry sites (A–D) but twelve when aiming at the low-symmetry site (E). Taking sites and orientations together, therefore, we investigate a total of 44 trajectories, listed in Table 1 alongside their symmetry equivalents.
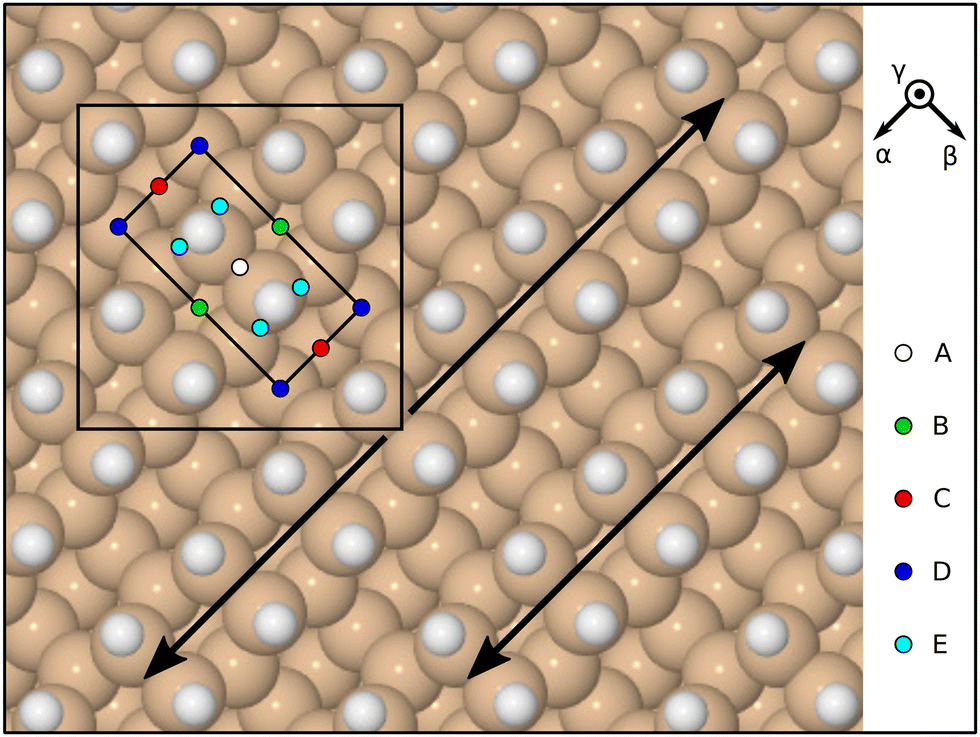 | ||
| Fig. 2 Schematic top-down view of the monohydrogenated Si{001} surface, highlighting two adjacent dimer rows with double-headed arrows. The surface exhibits a (2 × 1) reconstruction, whose primitive cell is marked with the inner rectangle, but calculations were performed within a c(4 × 4) unit cell, marked with the large square. Incoming molecules were aimed at sites A–E, shown here in multiple instances to emphasise the uniformity of their distribution. Reference axis orientations are indicated by vectors labelled α (along the dimer rows) and β (across the dimer rows). A third orientation, γ, corresponds to the outward surface normal. | ||
| Site | Primary | Redundant | Reflection |
|---|---|---|---|
| A–D | αβ |
![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) β β
|
α![[small beta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c3.gif) , , |
| αγ |
γ
|
— | |
|
α
|
|
— | |
| βα |
α
|
β, |
|
| βγ |
γ
|
— | |
|
β
|
|
— | |
| γα |
α
|
γ, |
|
| γβ |
β
|
γ, |
|
| E | αβ |
β
|
— |
| αγ |
γ
|
— | |
|
α
|
|
— | |
| βα |
α
|
— | |
| βγ |
γ
|
— | |
|
β
|
|
— | |
| γα |
α
|
— | |
| γβ |
β
|
— | |
|
α
|
|
— | |
|
β
|
|
— | |
|
γ
|
|
— | |
|
γ
|
|
— |
In order to analyse our computed results in a somewhat quantitative manner, we plot selected interatomic separations as a function of time, noting in particular those occasions where distances deviate significantly from the equilibrium bond lengths summarised in Table 2. Such plots are also convenient when estimating vibrational frequencies (quoted to the nearest multiple of 5 cm−1) from the periodic spacing of successive minima (averaging over at least five) in the relevant traces. Finally, we are aided in identifying instances of bond making or breaking by consulting plots of two different spin measures. The first of these is the integrated net spin, defined as
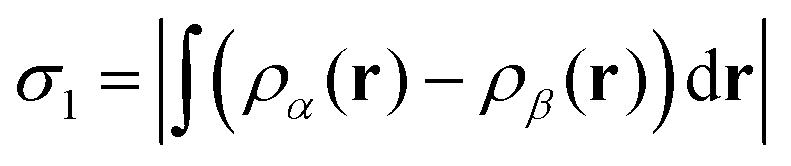 | (2) |
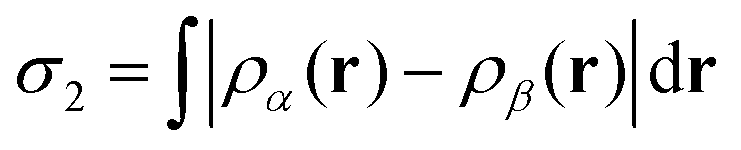 | (3) |
| Bond | Notes | Length (Å) |
|---|---|---|
| F–O | (Gas-phase OF2) | 1.42 |
| F–O | (Gas-phase OF radical) | 1.39 |
| F–Si | (F attached to Si dimer) | 1.60 |
| Si–Si | (Typical single bond) | 2.35 |
The significance of these spin measures may best be understood by considering a few idealised cases, starting with the fully spin-compensated situation where both σ1 and σ2 are zero. Clearly this can only occur when all electrons are paired, so that both substrate and adsorbate may be considered to be closed-shell sub-systems. In the event that a single unpaired spin is localised somewhere within the system, on the other hand, one would expect both σ1 and σ2 to take values close to 1μB (being the spin attributable to a single isolated electron in free space). And if there were to be two unpaired spins within the system, localised in different spatial regions, we must expect σ2 to approach 2μB while σ1 may either do the same (in the case of a triplet state) or vanish (in the case of a singlet state). That is to say, the moments in the two regions of spin localisation may either lie parallel or anti-parallel with respect to one another. In the present case, our system begins in a fully spin-compensated configuration, but may pass through (and indeed perhaps settle upon) various of the above-mentioned spinful states in the course of executing our computed trajectories. These conditions, if present, indicate the transient or permanent existence of radical molecular species and/or semi-occupied surface-related dangling bonds. This, we believe, is often a more pertinent factor than electron transfer when seeking to understand radical-mediated reactions, so we do not focus upon charge to nearly the same degree as spin. Moreover, standard methods of attributing charge to individual atoms, such as the Mulliken, Hirshfeld, or Bader approaches,33–35 are notoriously fickle and routinely underestimate the degree of charge separation in extended systems. As we shall see, however, significant charge transfer may be inferred from the sudden absence of spin where it might ordinarily be expected, and this we have highlighted where appropriate.
3 Results
In common with the interaction of ozone and molecular fluorine with the monohydrogenated Si{001} surface,1–3 the majority of trajectories for incoming oxygen difluoride (41 out of 44) resulted in repulsion rather than reaction. We shall only briefly summarise these rather uninteresting results before turning our attention in much more detail to the few reactive trajectories (B/βα, D/βα, D/αβ). The first two of these are somewhat similar to one another (see snapshots in Fig. 3) but the third is of an altogether different and more surprising character. What all three have in common, however, is that they start with the molecule in an orientation that can readily be steered to point one of its fluorine atoms directly toward one or more of the silicon atoms. Such a scenario maximises coupling of the LUMO with surface orbitals, destabilising the involved O–F bond and prompting dissociation.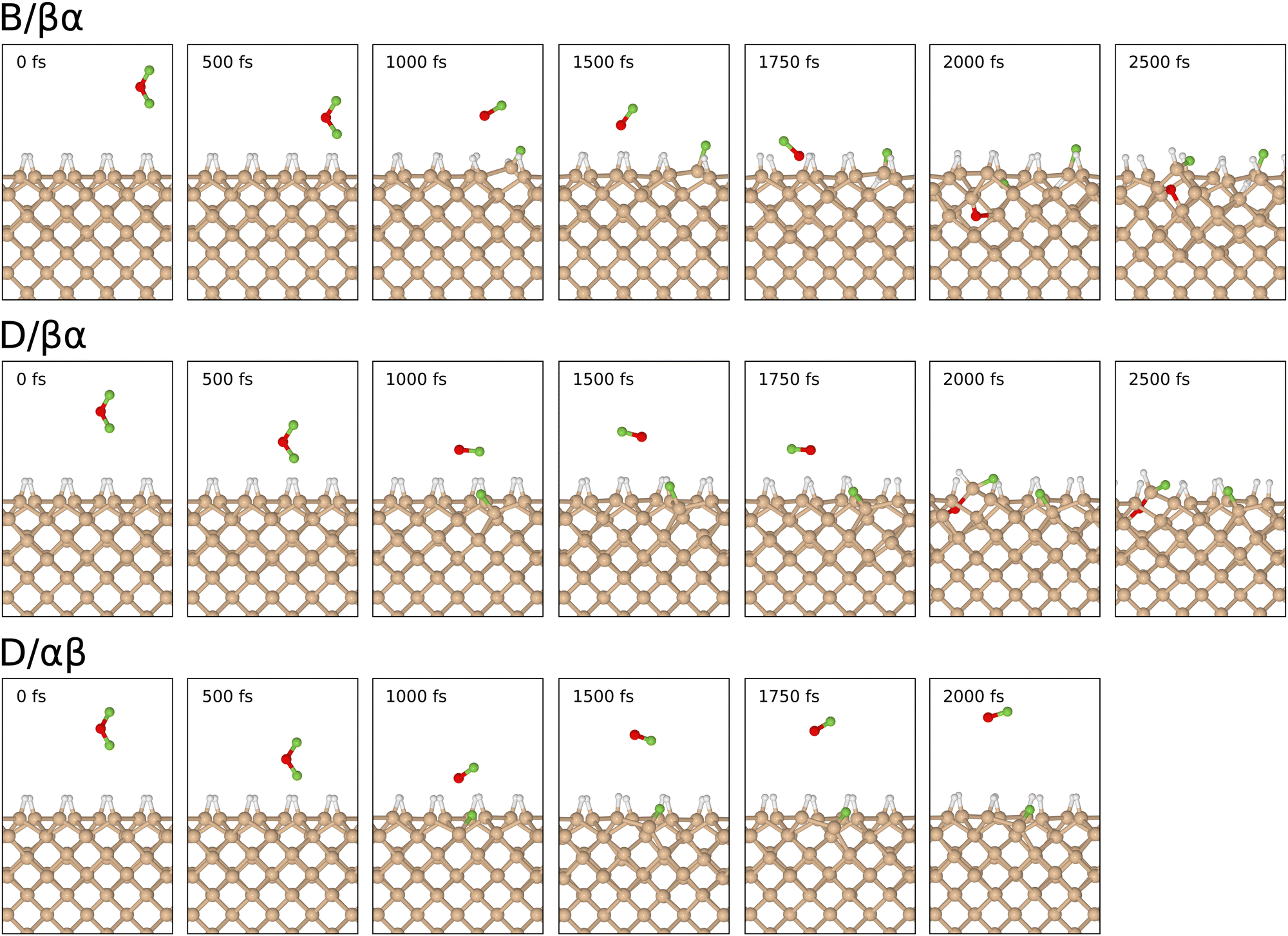 | ||
| Fig. 3 Snapshots of the reactive trajectories. Note uneven timestamps (denser time-sampling may be examined in the ESI†) and that the third trajectory has been halted early, before interaction of the OF radical with the periodic image of the slab's back surface becomes an issue. | ||
3.1 Non-reactive trajectories
Typical non-reactive trajectories began with very slight acceleration of the molecule toward the surface, but this swiftly gave way to repulsion. In all cases, the smallest interatomic separation achieved between the molecule and the surface involved a hydrogen atom, most often with one of the fluorine atoms but sometimes with the oxygen atom. In a handful of cases, this closest approach occurred at a distance in the 1.75–2.00 Å range, but more generally in the 2.00–2.50 Å range and sometimes yet more distant. These are to be compared with typical O–H and F–H equilibrium bond lengths, which should both be in the ballpark of 1.00 Å or less. There is thus no evidence for any significant affinity between the approaching molecule and pre-adsorbed hydrogen. Simulated trajectories were terminated once clear signs of repulsion were observed, and it is conceivable of course that some of these instances may possibly have led to chemisorption if continued indefinitely, perhaps via some long-lived physisorbed state. Undoubtedly, however, such a process would first require adoption of an orientation and impact site similar to those described below.3.2 B/βα Trajectory
In order to describe trajectories in detail, we shall focus upon plots of atomic separation versus elapsed time, as exemplified by Fig. 4. Here, the two O–F separations are depicted in the first (uppermost) panel with orange and cyan traces (for those involving the initially lower-lying and higher-lying fluorine atoms respectively). The separation between the lower-lying fluorine atom and the silicon atom with which it eventually bonds is shown in the same panel with a red trace, while a blue trace shows the separation between the higher-lying fluorine atom and the (different) silicon atom with which it will eventually bond.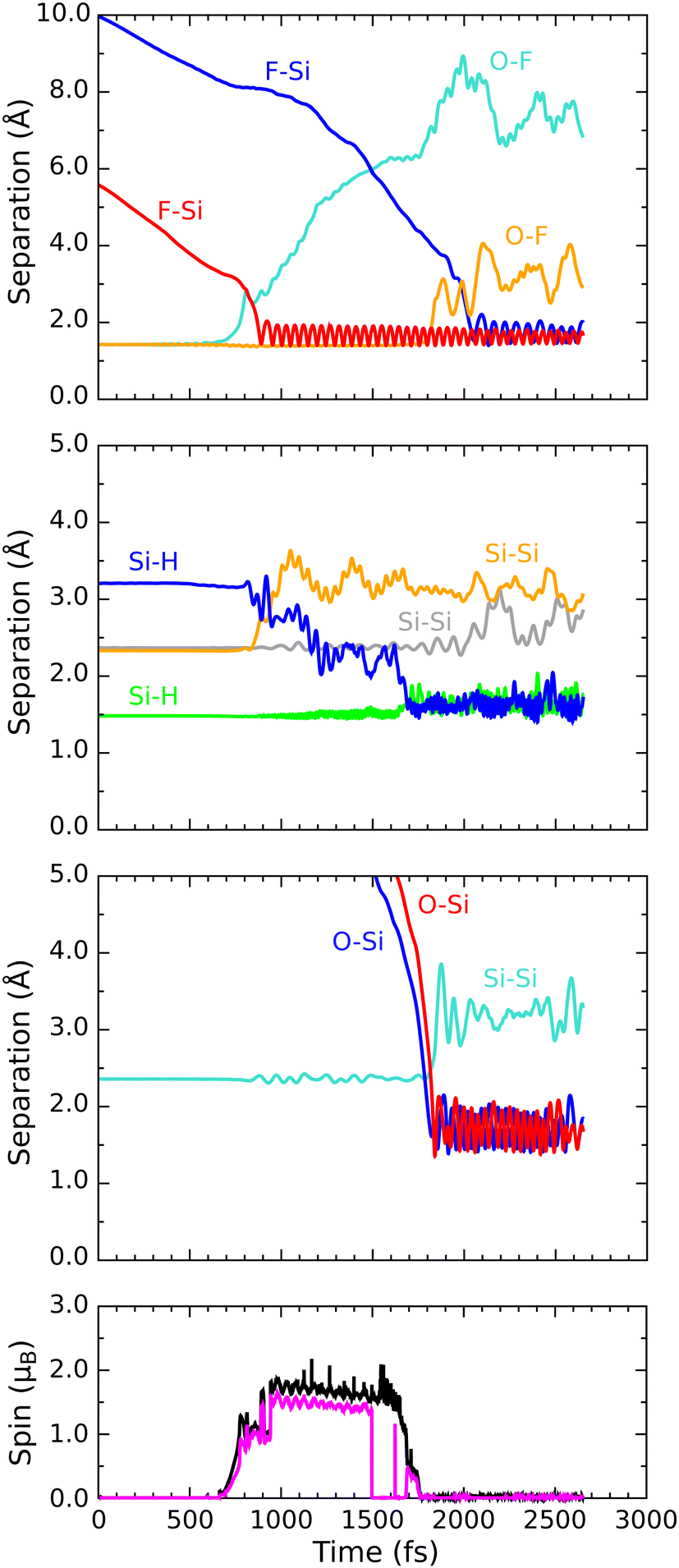 | ||
| Fig. 4 Evolution of the B/βα trajectory. The three upper panels show interatomic separations, coded as follows: bond-making is depicted with red or blue traces; bond-breaking is depicted with cyan or orange traces; and separations having equivocal character with respect to these criteria are depicted with green or grey traces. The lower panel shows the integrated net spin in magenta and the integrated spin modulus in black. | ||
During the first 700 fs of the simulation, the molecule drifts toward the surface with little sign of any significant interaction. Shortly thereafter, however, rapid acceleration becomes evident and the lower-lying fluorine atom moves decisively toward one of the silicon dimer atoms, reaching its closest approach (1.42 Å) at the 810 fs mark. The O–F separation involving this fluorine atom rises rapidly at much the same time (first panel, cyan trace) while the other O–F separation remains almost unaltered from the gas-phase bond length of 1.42 Å (orange trace). Simultaneously, the integrated net spin and integrated spin modulus (magenta and black traces in the fourth panel) increase to around 1μB. In effect, the molecule has dissociated leaving an oxygen monofluoride (OF) radical above the surface (hence the unpaired spin) alongside a nascent H–Si–F moiety.
Very quickly, however, this latter feature itself decomposes, with the lower-lying fluorine atom hopping across to bond with a silicon atom from the neighbouring dimer within the next 80 fs. After this moment, the new Si–F bond (first panel, red trace) oscillates around a mean length of 1.68 Å at a frequency of 730 cm−1 – the latter figure being practically identical to that reported in a similar situation after the adsorption of molecular fluorine.3 Interestingly, the hydrogen atom attached to this complex is eventually displaced (in a process beginning around the 800 fs mark and completed some 900 fs later) into a bridging position between first-layer and second-layer silicon atoms. This may be seen in the second panel of Fig. 4, where the original Si–H bond (green trace) remains intact while the relevant Si–Si bond (orange trace) breaks and a new Si–H bond (blue trace) forms. The two constituent Si–H bonds of the newly created bridge oscillate in antiphase with one another (green and blue traces) at a joint frequency of 1700 cm−1 and both average around 1.68 Å in length. We note in passing that a similar Si–H–Si motif has been reported experimentally in the context of organosilicon chemistry from time to time,36,37 and theoretically in the context of silicon carbide surface chemistry.38
As for the OF radical, it remains intact and vibrationally cold, rotating gently at some distance from the surface, for at least the first 500 fs after its formation. Only after a total simulated time of 1400 fs, when it has drifted over the trough between dimer rows, does significant interaction with the surface become evident. Over the following 400 fs, the radical rapidly accelerates into the trough (oxygen atom downward) after which it abruptly dissociates (first panel, orange trace). By the 1850 fs mark, the oxygen atom has inserted into the bond between a second-layer silicon atom and one of its third-layer neighbours (third panel, cyan trace) forming a siloxane (Si–O–Si) unit. The two constituent Si–O bonds (third panel, red and blue traces) oscillate in antiphase with one another, at a joint frequency of 845 cm−1 and with identical average lengths of 1.72 Å.
Concerning the integrated net spin and integrated spin modulus (fourth panel, magenta and black traces) these had both jumped to over 1.5μB when the first fluorine atom attached to its eventual bonding partner, subsequently declining rather slowly during formation of the Si–H–Si bridge (see Fig. 5 for the spin distribution at 1250 fs). Only as the two constituent Si–H bonds finally start to oscillate around a common length (1.66 Å) does the spin in our system abruptly vanish (at about the 1750 fs mark). This likely coincides with the Si–H–Si bridge attaining the status of a true three-centre two-electron bond, implying that there must now be a spare electron in need of accommodation elsewhere. The Si–O–Si bridge, formed at the same moment, is not a likely recipient of this electron, implying that it is the isolated fluorine atom that gains anionic character (consistent with a Mulliken charge of −0.70|e| at the 1875 fs mark) in its final moments before bonding with the surface. This it does after about the 2000 fs mark. The Si–F bond thus created (Fig. 4, first panel, blue trace) oscillates around a mean length of 1.74 Å at 560 cm−1 – quite a bit different from the parameters of the first-formed Si–F bond (which itself had shortened to a mean length of 1.63 Å and blue-shifted to 780 cm−1 upon completion of the Si–H–Si bridge) but this is perhaps unsurprising in light of its different local environment. Most importantly, the formation of this second Si–F bond coincides with cleavage of the Si–Si bond belonging to the dimer to which it attaches, so that the ultimate home of the excess electron is to be found in a fully occupied dangling bond on the other dimer atom. Mulliken analysis indicates that the net charge on this atom switches from +0.11|e| at the 650 fs mark to −0.06|e| at the 2500 fs mark. The F–Si–H–Si complex, comprising the bridge and its attendant fluorine atom, acquires a somewhat complementary net charge of +0.21|e| by the same time. As mentioned in our methodology section, one ought not to seek quantitative accuracy in such values (nor in that cited above for the isolated fluorine anion) but the trend is certainly meaningful and consistent with our interpretation.
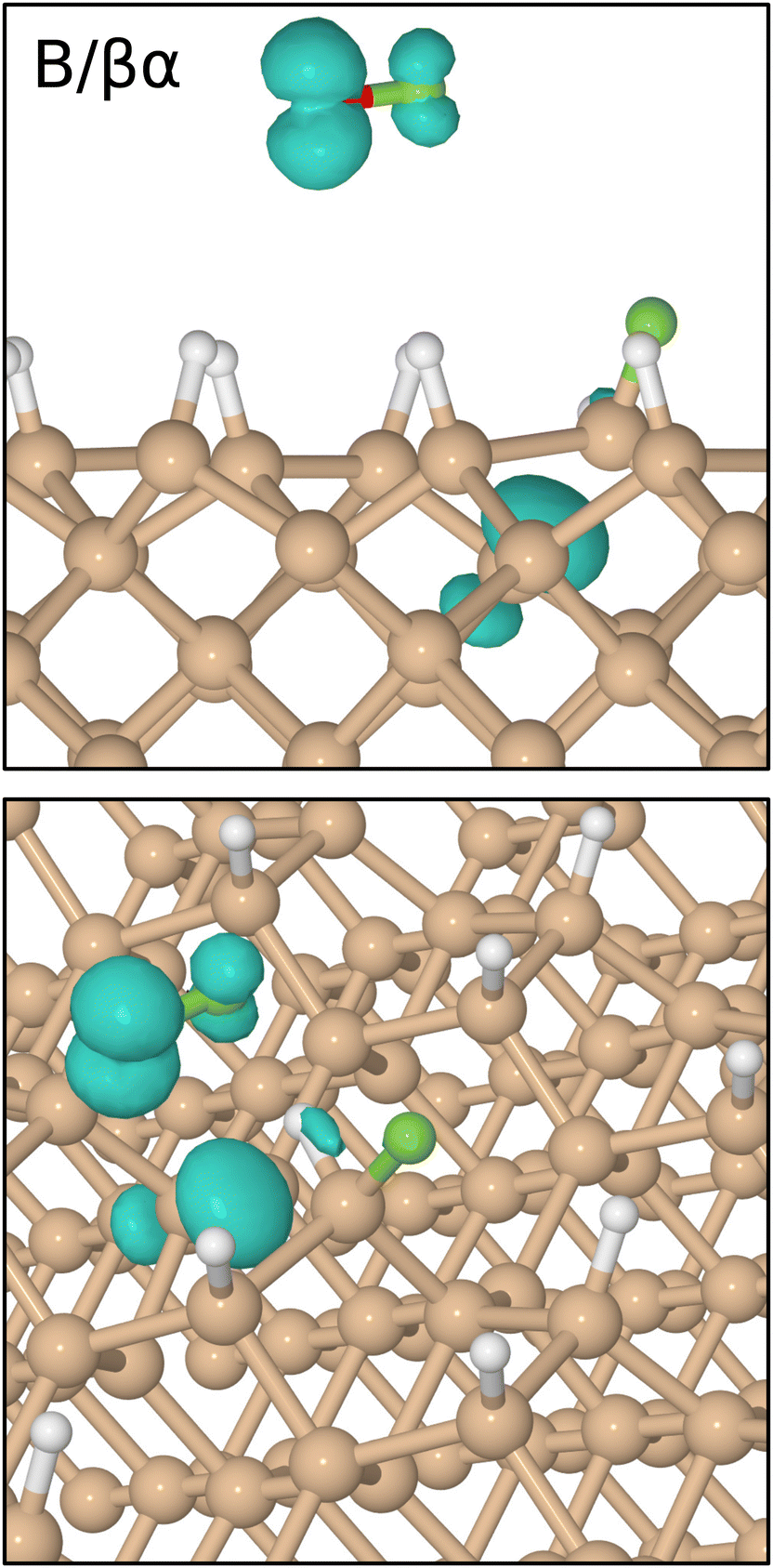 | ||
| Fig. 5 Two views of net spin density at 1250 fs in the B/βα trajectory, the isosurfaces being drawn at 0.05μB Å−3. Note the regions of unpaired spin in the vicinity of the transient radical (above the surface) and the nascent Si–H–Si bridge (within the surface). | ||
Taking stock, we see that a radical species is indeed created during adsorption, but not by abstraction of hydrogen. Instead, it is a byproduct of depositing a single fluorine atom onto one end of a silicon dimer – an event that in fact displaces a hydrogen atom into an unusual Si–H–Si bridge with three-centre two-electron bonding. The OF radical species persists above the surface for a much greater duration than either of the two previously discussed species (HO3 and HF2) but eventually decomposes to form a Si–O–Si bridge and anionic F. The latter then attaches to a second silicon dimer, breaking that bond in the process.
3.3 D/βα Trajectory
Progress along our second reactive trajectory is not entirely dissimilar to our first for the initial 700 fs or so, during which time the molecule coasts along the inward surface normal with only very gradual acceleration (Fig. 6). When significant interaction does become apparent, however, the lower-lying fluorine atom darts toward not one of the top-layer silicon dimer atoms, but instead one of the second-layer silicon atoms, making its initial closest approach at a distance of 1.46 Å after 885 fs of simulated time (first panel, red trace). After then stretching to a distance of 2.27 Å, the newly formed Si–F bond subsequently oscillates around a mean length of 1.66 Å, at a frequency of 680 cm−1, for the next 1000 fs. Needless to say, the corresponding O–F bond breaks (first panel, cyan trace) and an OF radical is formed. The integrated net spin and integrated spin modulus (fourth panel, magenta and black traces) both rise to around 1μB at this time, as one might expect in the circumstances.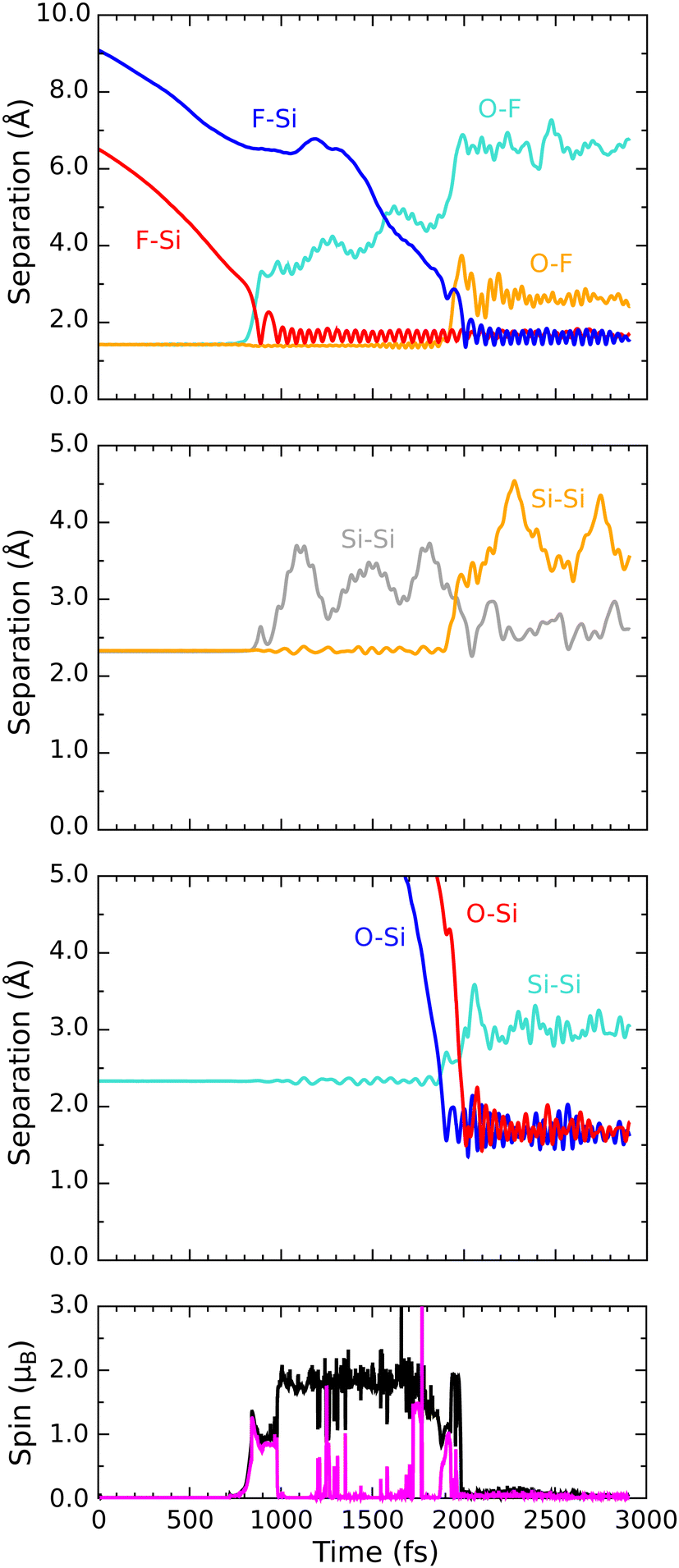 | ||
| Fig. 6 Evolution of the D/βα trajectory. The three upper panels show interatomic separations (colours as per Fig. 4) while the lower panel shows integrated net spin in magenta and integrated spin modulus in black. | ||
As a result of gaining a bond with the first fluorine atom, the second-layer atom mentioned above finds itself over-coordinated and so moves away from the third-layer silicon atom that lies under the adjacent dimer row. This Si–Si bond breaks (second panel, grey trace) and again remains broken for around 1000 fs, its nominal length not falling below 2.74 Å within this period. Notably, this bond cleavage event is accompanied by a collapse to zero of the integrated net spin and an increase to around 2μB of the integrated spin modulus (fourth panel, magenta and black traces) that implies a singlet state with around 1μB localised on the OF radical and equal but opposite spin localised at the dangling bond created on the now under-coordinated third-layer silicon atom (see Fig. 7 for the spin distribution at 1250 fs).
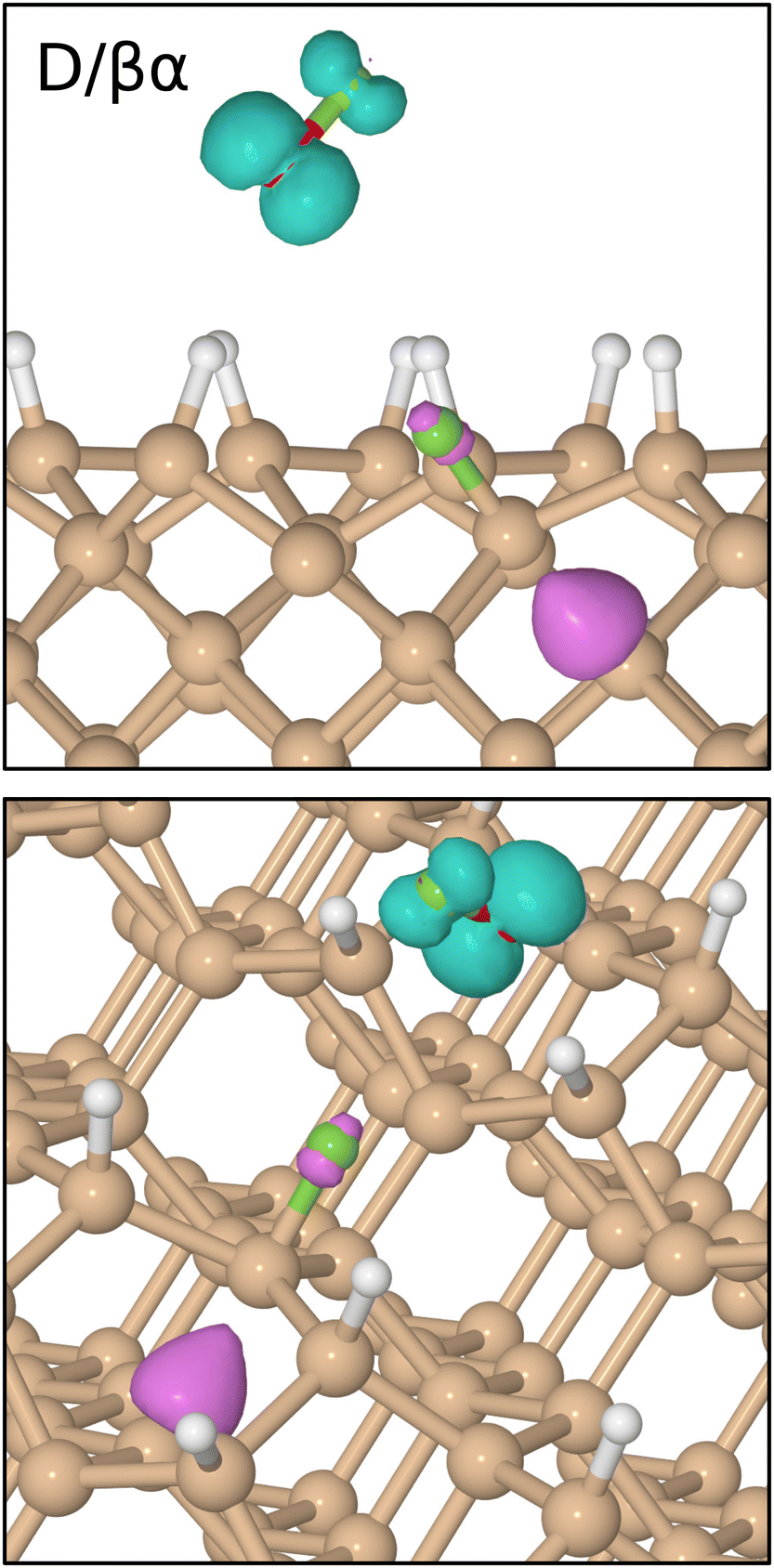 | ||
| Fig. 7 Two views of net spin density at 1250 fs in the D/βα trajectory, the isosurfaces being drawn in contrasting colours at ± 0.05μB Å−3. Note the regions of unpaired spin in the vicinity of the transient radical (above the surface) and on a third-layer dangling bond (within the surface). | ||
After formation at around the 900 fs mark, the OF radical drifts across the surface without a great deal of evidence for interaction until at least the 1600 fs mark. Beyond this point, however, the oxygen atom starts to accelerate toward one of the top-layer silicon atoms of the nearest dimer row, initially approaching to a distance of 1.55 Å after 1900 fs of simulated time (Fig. 6, third panel, red trace). Thereafter, the oxygen atom inserts into the bond made between this top-layer silicon atom and one of its second-layer neighbours (third panel, cyan trace). The resulting Si–O–Si bridge is somewhat similar to that formed in the previously discussed trajectory, with its two constituent Si–O bonds (third panel, red and blue traces) oscillating in antiphase with one another at a joint frequency of 770 cm−1 and with identical average lengths of 1.71 Å.
In the process of forming the Si–O–Si bridge, of course, the second fluorine atom is liberated from the oxygen atom and at the precise instant (1985 fs) that the relevant O–F separation (first panel, orange trace) reaches its maximum value (3.74 Å) the spin of the system abruptly vanishes, never to return. As in the previous trajectory, this implies that the isolated fluorine atom may exist transiently in anionic form at this moment. By now, however, it lies only 2.30 Å from the uppermost atom of the Si–O–Si bridge and is already heading rapidly toward it. This Si–F separation reaches its minimum value of 1.38 Å just after the 2000 fs mark, after which the new bond oscillates around a mean length of 1.62 Å at a frequency of 765 cm−1. Compared with the original Si–F bond, this second one seems to be a little stronger, as evidenced by its shorter bond length and higher stretch frequency.
Interestingly, the formation of the second Si–F bond coincides with a distinct change in the character of the first; specifically, its mean bond length extends slightly to around 1.69 Å and its vibrational frequency drops dramatically to 580 cm−1. These changes are accompanied by an almost complete reversal of the Si–Si bond cleavage event (second panel, grey trace) that had accompanied the first Si–F bond creation, bringing the central silicon atom into a trigonal bipyramidal geometry that makes sense only if the axial Si–Si–F component forms a three-centre four-electron bond. This implies that an extra electron must be gleaned from elsewhere within the surface, but fortunately there is a ready source for this. Formation of the second Si–F bond is accompanied, it turns out, by cleavage of the bond between the involved top-layer silicon atom and its only remaining second-layer neighbour after formation of the Si–O–Si bridge (second panel, orange trace). That second-layer atom then adopts a trigonal planar geometry with respect to its remaining neighbours, stabilised by donation of an electron (its Mulliken charge switching from −0.04|e| at the 650 fs mark to +0.23|e| at the 2500 fs mark) so as to leave the inevitable dangling bond entirely empty. Transfer of this electron to the hypervalent Si–Si–F complex (which gains a net Mulliken charge of −0.21|e| in the process) then ensures a spin-compensated surface.
In common with the first-described trajectory, therefore, we once again observe the crucial role of a radical intermediate in the present mechanism, but not one that involves abstraction of a hydrogen atom. This time, the first step is deposition of a single fluorine atom onto a second-layer silicon atom, as opposed to a top-layer dimer atom, and this results in cleavage of a Si–Si bond rather than formation of a Si–H–Si bridge. The OF radical byproduct is slightly longer-lived in this instance (700 fs compared with 500 fs) but this is likely a contingent finding, rather than anything significant. Notably, however, this moiety's eventual fate is (more-or-less as before) to insert its oxygen atom into a Si–O–Si bridge, leaving anionic F to bind with a top-layer silicon atom. Instead of breaking a dimer bond in this last step, the formation of an empty second-layer dangling bond provides an electron that stabilises a three-centre four-electron bond in the vicinity of the original fluorine adsorption site.
3.4 D/αβ Trajectory
As hinted at above, our third reactive trajectory ultimately displays mechanistic detail that differs considerably from the previous two, but this is not at all apparent during the first 800 fs of the simulation. The molecule initially drifts along the inward surface normal at fairly constant speed, as may be seen, for example, in two Si–F separations plotted in Fig. 8 (first panel, grey and red traces). After the onset of rapid acceleration and cleavage of its O–F bond (first panel, cyan trace) the lower-lying fluorine atom makes its closest approach (1.42 Å) to one of the second-layer silicon atoms at the 930 fs mark (first panel, grey trace) before rebounding across the inter-row trench to approach a second such silicon atom almost equally closely (1.47 Å) at the 1015 fs mark. It is, indeed, this second second-layer atom with which it forms a lasting Si–F bond, oscillating about a mean length of 1.71 Å at a frequency of 635 cm−1 for the remainder of the simulation (first panel, red trace). The original contact with the first second-layer silicon atom coincides with a rise in integrated net spin and integrated spin modulus (fourth panel, magenta and black traces) to around 1μB, but both measures rise further to around 2μB as the bond with the second silicon atom is formed. This implies a triplet state, with an unpaired spin localised on the OF radical matched by an equal spin somewhere on the surface.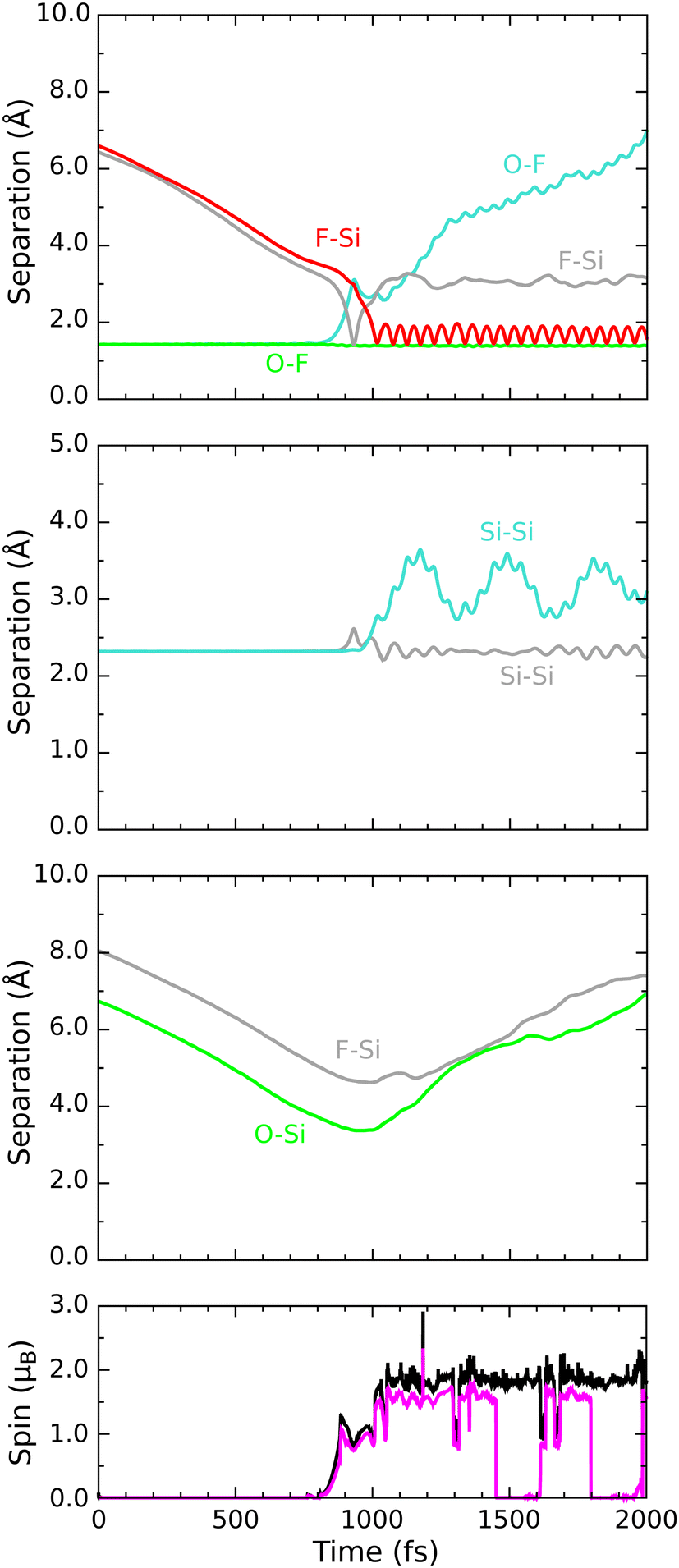 | ||
| Fig. 8 Evolution of the D/αβ trajectory. The three upper panels show interatomic separations (colours as per Fig. 4) while the lower panel shows integrated net spin in magenta and integrated spin modulus in black. | ||
The location of the surface spin component is not hard to deduce (see Fig. 9 for the spin distribution at 1250 fs) since the bonding of fluorine to a second-layer atom induces cleavage of the bond between the involved silicon atom and its third-layer neighbour from beneath the nearest dimer row (Fig. 8, second panel, cyan trace). A dangling bond is clearly located on the third-layer atom, for which the Mulliken spin at 1250 fs is 0.47μB and the corresponding charge just −0.02|e|; the latter measure is unaltered from its pre-reaction value, indicating that no significant charge transfer to or from the dangling bond has taken place. We note, in passing, that the direction of the surface-localised spin oscillates as the distance between the two cleaved silicon atoms varies, so that the overall spin of the system switches between singlet and triplet configurations on the same timescale. By way of comparison, we also show the corresponding bond between second- and third-layer silicon atoms that involves the atom first struck by fluorine (second panel, grey trace) and see that it is only slightly excited by the collision.
 | ||
| Fig. 9 Two views of net spin density at 1250 fs in the D/αβ trajectory, the isosurfaces being drawn at 0.05μB Å−3. Note the regions of unpaired spin in the vicinity of the transient radical (above the surface) and on a third-layer dangling bond (within the surface). | ||
Finally, we turn to the OF radical, whose mean bond length drops only very slightly from 1.42 Å at the start of the simulation to around 1.39 Å at the end (first panel, green trace). Compared with the same species in our two previous trajectories, its velocity here is aligned much more closely with the outward surface normal, raising the prospect of departure from the surface. This may be seen most clearly in Fig. 10, where we show the vertical position of the relevant O–F bond's centre of mass in all three trajectories. By the 1500 fs mark, both the B/βα and D/βα trajectories already show turning points in the OF vertical position, whereas the D/αβ trajectory shows this species travelling rapidly away from the surface with little sign of further deceleration. We have analysed the energetic characteristics of desorbing OF, along the lines described in detail previously for HF and H2 desorption after decomposition of F2 on monohydrogenated silicon,3 but here find translational kinetic energy of only around 0.04 eV (cf. 0.31 & 0.90 eV for those other species) and rovibrational energy around 0.01 eV (cf. 2.23 & 0.94 eV before) at the 2000 fs mark. It is debatable, therefore, whether the outgoing molecule possesses sufficient energy to overcome van der Waals interactions (not formally included in our calculations) and hence actually to escape from the surface in reality. The present result nevertheless suggests at least the possibility that adsorption of OF2 on Si{001} may generate a modest outward flux of OF radicals. Needless to say, this is an unusual and unexpected finding.
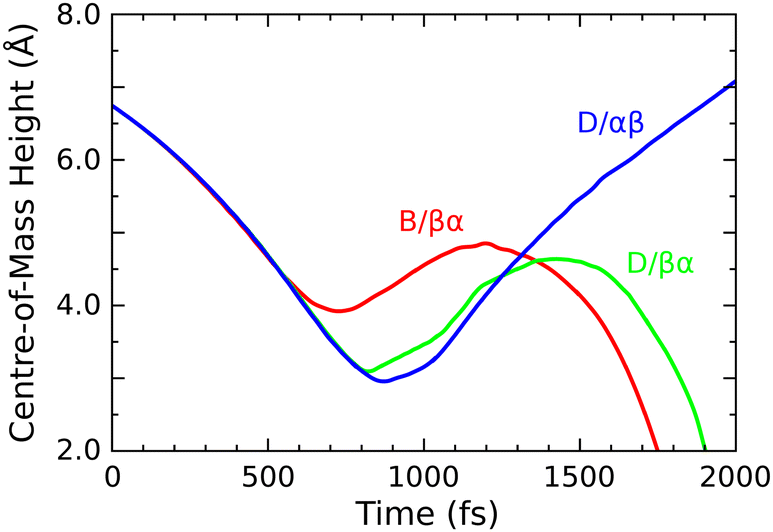 | ||
| Fig. 10 Evolution of the centre-of-mass height (measured relative to the initial height of top-layer silicon atoms) of the O–F moiety that forms a radical intermediate or product in trajectories B/βα, D/βα and D/αβ (red, green and blue traces). | ||
4 Conclusions
Although we have described each of our three reactive trajectories in some detail, the fact remains that these represent only a rather sparse sampling of the phase space available to an incoming molecule. It is important, therefore, to consider what broad inferences may be drawn from their totality, rather than dwelling unduly upon their particulars. In this spirit, we shall start with the observation that only three of our computed trajectories resulted in adsorption. This amounts to 3/44 of the symmetrically distinct trajectories actually calculated, or 6/60 of the total trajectories that these ultimately represent. We thus expect the sticking probability to fall broadly into the 1–10% range, although a little higher or lower would not be much of a surprise, given the uncertainties and limitations of the present work.It is also fairly certain from our results that there exists no particular tendency for the incoming molecule to abstract hydrogen from the surface, contrasting with the cases of ozone or fluorine adsorption.1–3 Instead, it is fluorine that is abstracted from the molecule by the surface, doubtless facilitated by incoming orientations in which one such atom lies closer to the surface than the other. This is consistent with the LUMO favouring interactions along the O–F bond directions, rather than perpendicular to the plane of the molecule as in ozone. Partial population of the LUMO then destabilises the molecule, leading to dissociation and subsequent adsorption of an adatom at either a surface dimer site or at a second-layer site. In the latter situation, a compensating dangling bond is instantly created on a third-layer atom, while in the former a Si–H–Si bridge starts to take shape but is not immediately completed. Although variations upon these scenarios are to be expected, however, the underlying theme of unpaired surface spin following initial adatom adsorption is likely to be fundamental. The fate of the OF radical, meanwhile, depends critically upon the velocity imparted to it during the initial fluorine abstraction process.
In two of our three reactive trajectories, the OF radical gains relatively little velocity along the surface normal and simply traverses a short distance across the surface before reacting to form a Si–O–Si bridge and a further fluorine adatom (bound to a silicon dimer atom in both instances studied here). The resulting electronic redistribution permits the Si–H–Si bridge to adopt a spin-free three-centre two-electron bond in one scenario, while a Si–Si–F motif manifests a spin-free three-centre four-electron bond in the other. These differing end-points are inextricably linked to the mechanistic detail that follows from the initial approach of the adsorbate and are unlikely to have been predicted on the basis of static calculations, underlining the merits of a molecular dynamics approach. In both cases, the Si–O–Si bridge comprises two ordinary bonds and also carries no spin, while any remaining dangling bonds within the surface are found to be either completely filled or entirely empty. Mulliken analysis confirms substantial transfer of electronic charge from the Si–H–Si bridge to a dangling bond located on a top-layer silicon atom in the former case, and from a dangling bond located on a second-layer silicon atom to the Si–Si–F motif in the latter, although the magnitude of charges calculated in this way should be treated with caution. In the end, the surface as a whole is spin-compensated, as it was in the beginning.
In the third of our reactive trajectories, however, the momentum of the OF radical is directed almost perfectly parallel to the surface normal and desorption becomes a real prospect, depending upon the possible influence of dispersion forces. Here, we find that the surface retains an unpaired spin, associated with a third-layer dangling bond. That radical desorption may happen in the real world remains merely a tantalising proposition at this stage, but one that may nevertheless be open to experimental verification. Regardless, the role of radical intermediates in the adsorption of oxygen difluoride on monohydrogenated silicon is clearly demonstrated by the present work, adding to the rather limited number of radical-mediated adsorption mechanisms identified to date.
Data availability
The trajectories analysed in this study are openly available in the University of Cambridge Data Repository at DOI: 10.17863/CAM.113772.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computational resources were provided by the Cambridge Service for Data-Driven Discovery (CSD3). For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.References
- C. K. Fink and S. J. Jenkins, Radical-Mediated Adsorption: Ozone Oxidation of Passivated Silicon, Surf. Sci., 2008, 602, L100 CrossRef CAS.
- C. K. Fink, K. Nakamura, S. Ichimura and S. J. Jenkins, Silicon Oxidation by Ozone, J. Phys.: Condens. Matter, 2009, 21, 183001 CrossRef.
- I. Y. H. Wu and S. J. Jenkins, First Principles Dynamics of Fluorine Adsorption on Clean and Monohydrogenated Si{001}, Langmuir, 2022, 38, 7256–7271 CrossRef CAS.
- N. V. Sidgwick, The Chemical Elements and Their Compounds, Oxford University Press, vol. II, 1950 Search PubMed.
- J. E. House and K. A. House, Descriptive Inorganic Chemistry, Academic Press, 3rd edn, 2016 Search PubMed.
- L. Pierce, N. Di Cianni and R. H. Jackson, Centrifugal Distortion effects in asymmetric Rotor Molecules. I. Quadratic Potential Constants and Average Structure of Oxygen Difluoride from the Ground-State Rotational Spectrum, J. Chem. Phys., 1963, 38, 730–739 CrossRef.
- T. Tanaka and Y. Morino, Coriolis Interaction and Anharmonic Potential Function of Ozone from the Microwave Spectra in the Excited Vibrational States, J. Mol. Spectrosc., 1970, 33, 538–551 CrossRef CAS.
- F. K. Sheong, J.-X. Zhang and Z. Lin, Revitalizing Spin Natural Orbital analysis: Electronic Structures of Mixed-Valence Compounds, Singlet Biradicals, and Antiferromagnetically Coupled systems, J. Comput. Chem., 2019, 40, 1172–1184 CrossRef CAS.
- ed. Haynes, W. M., CRC Handbook of Chemistry and Physics, 95th edn, CRC Press, 2014 Search PubMed.
- C. K. Fink and S. J. Jenkins, First-Principles Molecular Dynamics of the Initial Oxidation of Si{001} by Ozone, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195407 CrossRef.
- M. Sacchi, D. J. Wales and S. J. Jenkins, Mode-Specific Chemisorption of CH4 on Pt{110}-(1× 2) Explored by First-Principles Molecular Dynamics, J. Phys. Chem. C, 2011, 115, 21832 CrossRef CAS.
- M. Sacchi, D. J. Wales and S. J. Jenkins, Bond-Selective Energy Redistribution in the Chemisorption of CH3 D and CD3 H on Pt{110}-(1× 2): A First-Principles Molecular Dynamics Study, Comput. Theor. Chem., 2012, 990, 144 CrossRef CAS.
- M. Sacchi, D. J. Wales and S. J. Jenkins, Mode-Specificity and Transition State-Specific Energy Redistribution in the Chemisorption of CH4 on Ni{100}, Phys. Chem. Chem. Phys., 2012, 14, 15879 RSC.
- A. Chatterjee, F. Cheng, L. Leung, M. Luo, Z. Ning and J. C. Polanyi, Molecular Dynamics of the Electron-Induced Reaction of Diiodomethane on Cu(110), J. Phys. Chem. C, 2014, 118, 25525–25533 CrossRef CAS.
- D. G. Sangiovanni, D. Edström, L. Hultman, I. Petrov, J. E. Greene and V. Chirita, Ab Initio and Classical Molecular Dynamics Simulations of N2 Desorption from TiN(001) Surfaces, Surf. Sci., 2014, 624, 25–31 CrossRef CAS.
- D. G. Sangiovanni, B. Alling, P. Steneteg, L. Hultman and A. Abrikosov, Nitrogen Vacancy, Self-Interstitial Diffusion, and Frenkel-Pair Formation/Dissociation in B1 TiN Studied by Ab Initio and Classical Molecular Dynamics with Optimised Potentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 054301 CrossRef.
- W. Y. Guo, S. J. Jenkins, W. Ji, J. C. Polanyi, M. Sacchi and G.-C. Wang, Repulsion-Induced Surface Migration by Ballistics and Bounce, Chem. Lett., 2015, 6, 4093–4098 Search PubMed.
- K. Huang, O. MacLean, S. Y. Guo, I. R. McNab, Z. Ning, G.-C. Wang, W. Ji and J. C. Polanyi, Dynamics of Surface Migration: Electron-Induced Reaction of 1,2-Dihaloethanes on Si(100), Surf. Sci., 2016, 652, 312–321 CrossRef CAS.
- F. Nattino, O. Galparsoro, F. Costanzo, R. Díez Muiñno, M. Alducin and G.-J. Kroes, Modelling Surface Motion Effects in N2 Dissociation on W(110): Ab Initio Molecular Dynamics Calculations and Generalized Langevin Oscillator Model, J. Chem. Phys., 2016, 144, 244708 CrossRef PubMed.
- D. G. Sangiovanni, A. B. Mei, L. Hultman, V. Chirita, I. Petrov and J. E. Greene, Ab Initio Molecular Dynamics Simulations of Nitrogen/VN(001) Surface Reactions: Vacancy-Catalyzed N2 Dissociative Chemisorption, N Adatom Migration, and N2 Desorption, J. Phys. Chem. C, 2016, 120, 12503–12516 CrossRef CAS.
- D. G. Sangiovanni, G. K. Gueorguiev and A. Kakanakova-Georgieva, Ab Initio Molecular Dynamics of Atomic-Scale Surface Reactions: Insights into Metal Organic Chemical Vapor Deposition of AlN on Graphene, Phys. Chem. Chem. Phys., 2018, 20, 17751–17761 RSC.
- S. C. Matysik, D. J. Wales and S. J. Jenkins, Surface Chirality Influences Molecular Rotation upon Desorption, Phys. Rev. Lett., 2021, 126, 126101 CrossRef.
- S. C. Matysik, D. J. Wales and S. J. Jenkins, Rotational Dynamics of Desorption: Methane and Ethane at Stepped and Kinked Platinum Surfaces, J. Phys. Chem. C, 2021, 125, 27938 CrossRef CAS.
- S. C. Matysik, D. J. Wales and S. J. Jenkins, Dynamic Diastereomerism on Chiral Surfaces, J. Phys. Chem. C, 2023, 127, 229–233 CrossRef CAS PubMed.
- D. G. Sangiovanni, R. Faccio, G. K. Gueorguiev and A. Kakanakova-Georgieva, Discovering Atomistic Pathways for Supply of Metal Atoms from Methyl-Based Precursors to Graphene Surface, Phys. Chem. Chem. Phys., 2023, 25, 829–837 RSC.
- H. Thake and S. J. Jenkins, First-Principles Dynamics of the Surface Fluorination of Diamond. (submitted).
- G. P. Srivastava, Theory of Semiconductor Surface Reconstruction, Rep. Prog. Phys., 1997, 60, 561–613 CrossRef CAS.
- S. J. Jenkins, Foundations of Surface Science, Oxford University Press, 2023 Search PubMed.
- S. J. Clarke, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, First Principles Methods using CASTEP, Z. Kristallogr., 2005, 220, 567–570 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- D. Vanderbilt, Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin Zone Integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- R. S. Mulliken, Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- F. L. Hirshfeld, Bonded-Atom Fragments for Describing Molecular Charge Densities, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- T. Müller, A Silyl Cation with a Three-Center Si–H–Si Bond, Angew. Chem., Int. Ed., 2001, 40, 3033–3036 CrossRef.
- J. P. Nimoth and T. Müller, Hydrogen-Bridged Oligosilanylsilyl Mono- and Oligosilanylsilyl Dications, Chem. – Eur. J., 2022, 28, e202104318 CrossRef CAS PubMed.
- H. Chang, J. Wu, B.-L. Gu, F. Liu and W. Duan, Physical Origin of Hydrogen-Adsorption-Induced Metallization of the SiC Surface: n-Type Doping via Formation of Hydrogen Bridge Bond, Phys. Rev. Lett., 2005, 95, 196803 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03375b |
| This journal is © the Owner Societies 2025 |