
Unlocking the potential of Ni-rich LiNi0.9Co0.1O2 cathodes: a DFT investigation of performance-limiting factors†
Temitayo Ojuetimi
Ikuerowo
 a,
Olusegun
Tomomewo
a and
Salawu Omotayo
Akande
*b
a,
Olusegun
Tomomewo
a and
Salawu Omotayo
Akande
*b
aDepartment of Energy Studies, College of Engineering and Mines, University of North Dakota, North Dakota, USA
bDepartment of Chemical and Biological Engineering, Illinois Institute of Technology, Chicago, Illinois, USA. E-mail: omotayo.salawu@kaust.edu.sa
First published on 19th December 2024
Abstract
Ni-rich layered oxides, particularly LiNi0.9Co0.1O2, have garnered significant attention in the realm of high-capacity cathodes for lithium-ion batteries. Despite their promise, their commercialization is hindered by challenges related to structural instability and defect formation. This study utilizes density functional theory (DFT) to unravel the intricate structural, defect formation, and transport properties of LiNi0.9Co0.1O2, thereby providing insights into the performance-limiting factors. Our findings reveal that a 10% cobalt doping while enhancing lithium mobility, is insufficient to significantly mitigate antisite defects and oxygen vacancy formation. These defects are critical in influencing the electrochemical performance and durability of the material. The study further delves into the implications of defect formation on the electrochemical characteristics, emphasizing the need for a higher concentration of cobalt doping to effectively stabilize the Ni-rich cathode. This theoretical investigation contributes to the understanding of defect behaviors in Ni-rich cathodes and paves the way for optimized material design in future high-energy-density battery technologies.
1. Introduction
In response to the escalating global demand for clean energy, the development of efficient and sustainable energy storage technologies has become a critical focus. Among various solutions, electrochemical batteries, particularly lithium-ion batteries (LIBs), have emerged as pivotal in revolutionizing the energy sector.1–3 Renowned for their high specific power, energy densities, and reliability, LIBs have been instrumental in diverse applications, including renewable energy integration and automotive advancements.4–6Despite their widespread applications in diverse fields such as renewable energy integration, the mobile electronics market, and the automotive industry,4,7–10 the potential of LIB technologies faces significant limitations related to cost, safety, and degradational issues.5 To advance renewable energy integration, and consumer electronics, and extend the driving ranges of electric vehicles, there's a pressing need for more cost-effective, higher-capacity, and environmentally friendly materials for LIB applications.11–13 In the pursuit of addressing limitations hampering LIBs, substantial research efforts over the last few decades have zeroed in on the cathode component of LIBs. The cathode significantly influences the power and energy density of a battery system and constitutes over 20% of the total cost.12,14–17
Layered transition metal oxides, such as LiMO2 (where M = Co, Ni, Mn, etc.), stand out as promising cathode materials due to their high theoretical capacities and operating voltages.18 Initially introduced in 1980 with the development of LiCoO2 (LCO) cathodes by Goodenough's team,19 LCO chemistry has been predominant in the electronics industry. However, LCO cathodes face limitations in delivering only about half of their theoretical capacity (approximately 140 mA h g−1) and contain cobalt, a toxic and expensive component.14,15,20 Research exploring LiNiO2 (LNO) chemistries as alternatives to LCOs has gained attention due to their increased specific energy (>200 mA h g−1), reduced toxicity, and a more economically viable raw material supply.21 Yet, challenges persist, primarily stemming from the structural instability inherent in LNO compounds, a well-recognized issue in the literature, ultimately impeding their electrochemical performance.22–24 The structural instability in Ni-rich cathodes occur in the form of irreversible phase transformations and formation of new active sites which deter the electrochemical properties of the material. This instability can be attributed to nickel migration due to the spontaneous change in oxidation states of Ni ions causing them to occupy Li sites.25,26 The inherent difficulty in synthesizing a pure LNO compound results from the lower stability of Ni3+ in layered structures compared to Co3+, which prompts the formation and migration of Ni2+ ions, affecting the electrochemical performance of the material.20,27,28 Recent advancements in Co-doping have shown a potential to address these issues by enhancing the structural integrity and electrochemical properties of LNO cathodes.26,28–37 Notably, despite earlier experimental findings indicating insufficiency in mitigating cation mixing at low concentrations of Co dopants (<20% of Ni site), studies have shown promise in improving structural and electrochemical performance.38–41 Specifically, Ni-rich LiNi1−xCoxO2 (NC) cathodes have demonstrated the potential to meet the energy requirements for next-generation electric vehicles, albeit with accelerated capacity losses after repeated cycling.28,42–44 However, there exists a gap in the comprehensive theoretical understanding of these improvements, particularly at low Co doping levels. Specifically, a comprehensive understanding of defect behavior within these materials remains lacking. Computational tools, notably density functional theory (DFT), offer a promising avenue to explore material defects and establish structure–property relationships.9,45,46 While previous studies have used DFT to examine the stability and electronic properties of Ni-rich cathodes, comprehensive theoretical calculations specifically elucidating the effects of low-level Co doping on LNO are scarce to the best of our knowledge. With the global shift towards low-cobalt cathodes, there remains a significant gap in understanding how reduced levels of cobalt impact the performance and stability of Ni-rich cathodes. Our research uniquely addresses this gap by providing a comprehensive analysis of the intrinsic effects of lower Co-doping levels on structural and defect properties of LNO.
In recent years, Ni-rich layered oxides, such as LiNi0.9Co0.1O2 (NC91), have garnered attention due to their high energy densities, as highlighted in a recent study by Susai et al.47 Their investigation underscored the degradational issues arising from 180° Ni–O–Ni bonds that facilitate antisite defects, limiting the performance of NC91 cathodes. While Susai et al.47 demonstrated the stabilization potential of dual Mo and B doping, our work focuses on the effects of 10% Co doping in mitigating these interactions. Although this doping level fails to entirely prevent antisite formation, consistent with their findings, we uniquely reveal its positive impact on lithium-ion mobility, offering an explanation for the enhanced energy densities of NC91. This complementary perspective bridges the gap between experimental observations and theoretical insights, contributing to the broader understanding of performance-limiting factors in Ni-rich cathodes.
This study aims to leverage first-principles DFT simulations to investigate the impact of Co doping in Ni-rich LiNi0.9Co0.1O2 (NC91). The research systematically probes the effects of Co-doping on the material's structural and defect properties, examining antisite and vacancy defect formation energies and lithium migration behavior in comparison to pristine LNO and LCO. By providing a theoretical foundation, this work seeks to complement experimental findings and facilitate a deeper understanding of Ni-rich cathodes, aiming to address their structural limitations for broader application. The novelty of our approach lies in the detailed theoretical exploration of low-level Co-doping effects, which have been less addressed in existing literature. We hypothesize that this investigation will reveal critical insights into improving the performance and stability of Ni-rich cathodes, potentially leading to more efficient, safer, and cost-effective LIBs. The broader implications of our study extend beyond the technical enhancements of cathode materials. By contributing to the development of improved LIBs, this research aligns with the global push towards sustainable energy solutions, potentially impacting future battery technologies and supporting the transition to a more sustainable energy landscape.
2. Computational details
The computational methods employed in this study utilized first-principles density functional theory (DFT) calculations conducted with the Vienna Ab initio Simulation Package (VASP)48 and the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.49 Core–electron interactions were described by employing the projector-augmented wave (PAW) approach;50 2s1 (Li), 3d8 4s2 (Ni), 3d8 4s1 (Co) and 2s2 2p4 (O) are treated as valence electrons. To provide an accurate representation of localized states, a Hubbard U correction (Dudarev approach) of 6 eV was applied to the transition metal d orbitals.47,51–53 Preliminary analysis indicates that our chosen value of U = 6.0 eV yields results consistent with experimental data and represents a good compromise between structural accuracy and electronic properties. Additionally, the vacancy formation energies remain qualitatively consistent across different U values. More information can be found in the ESI† (Tables S1 and S2).The initial structural relaxations for the LiNiO2 (LNO) and LiCoO2 (LCO) cathode materials were carried out using 12-atom unit cells and an 8 × 8 × 2 k-point mesh. Atomic positions were optimized until the forces reached a convergence criterion of 0.001 eV Å−1. For ionic relaxation, a convergence criterion of 0.05 eV Å−1 was applied, while electronic relaxation used a convergence criterion of 10−5 eV. A plane wave cutoff energy of 520 eV was chosen in order to adequately describe the electronic wavefunctions of the oxides.54,55 Symmetry constraints were applied during the geometry optimization calculations to preserve crystal symmetry while optimizing computational resources.
In extending the structural relaxations to study defects, we adopted a 120-atom supercell approach with dimensions of 5 × 2 × 1 based on convergence calculations (Fig. S1, ESI†). The selected supercell size effectively simulates defect formation energies, approximating defect interactions in a manner that closely resembles the real system's behavior while preserving periodicity. To construct the LiNi0.9Co0.1O2 (NC91) supercell, we randomly substituted 10% of the Ni sites with Co atoms using the special quasi-random structure (SQS) approach.56,57 The SQS approach gives more accurate descriptions of the essential correlations than methods such as coherent potential approximation (CPA) methods. Therefore, features such as near-neighbor pair distributions and multisite correlations found in random alloys are well-mimicked resulting in enhanced reliability of material property predictions.58,59 The configuration with the lowest energy served as the basis for further investigations, including defect calculations. We investigated the effect of London dispersion and found that the effect is minimal both on the structural and energetic properties discussed here (see Tables S3 and S4, ESI†).
Two major kinds of defects were formed: antisites and vacancies. The defect formation energies of the defective supercells were obtained by evaluating the energy difference between the defective and pristine system using eqn (1) below:60
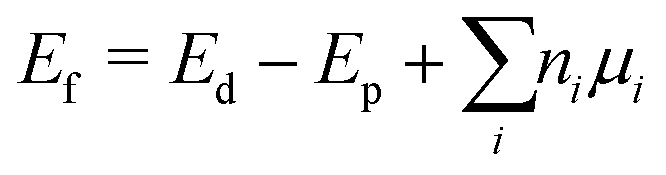 | (1) |
| Ef = Ed − Ep | (2) |
The concentration of antisite and vacancy defects (Cd) under thermal equilibrium can be calculated using the expression below:61
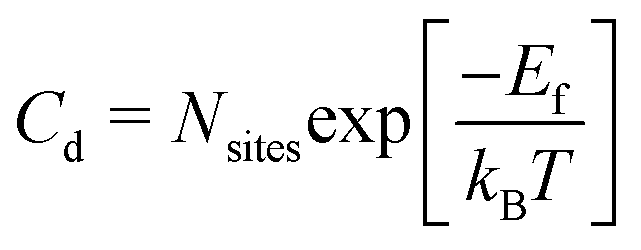 | (3) |
To determine the migration barriers for lithium vacancies, we employed the nudged elastic band (NEB) method.62 We utilized a linear interpolation involving eight images between the initial and final states to ascertain the migration pathway and energy barriers. Finally, all atomic structures and visualizations were created using VESTA.63
3. Results and discussion
3.1. Structural properties
The optimized lattice parameters and cell volumes for the pristine LiMO2 (M = Ni, Co, Ni0.9Co0.1) cathode materials are listed in Table 1 and are in good agreement with experimental values. The 3.5% deviation observed with the lattice parameter a of NC91 likely stems from the complex electronic interactions of a mixed Ni/Co environment which may be difficult to capture perfectly with DFT+U. The materials adopt the α-NaFeO2 layered structure in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group36,64 as shown in Fig. 1. The cell volumes follow the trend of NC91 > LNO > LCO, with NC91 having the largest volume of 102.66 Å3, then LNO with 100.71 Å3 and LCO has the smallest volume of 98.11 Å3 consistent with the relative ionic radii of Ni2+ and Co3+. It is important to state that some studies may show slightly different volume trends, but this can be attributed to the different synthesis or characterization methods used. The calculated c/a ratio exhibits only minor variations across the materials, ranging from 4.978 for LNO to 5.020 for LCO. The O–M–O slab thickness and M–O bond lengths also show incremental differences, with values increasing as Ni content rises, as seen in Table 2. Overall, the structures contain alternating Li and transition metal (TM) layers separated by close-packed oxygen layers in the expected layered motif. The minor variations in lattice parameters, c/a ratio, O–M–O slab thickness, and M–O bond lengths can be attributed to the different ionic radii of Ni2+ and Co3+. The stoichiometric cation concentrations were confirmed from the optimized NC91 structure, which contained 9 Ni, 1 Co, and 10 Li atoms as expected. The Co dopant is coordinated octahedrally within the transition metal slab. The introduction of 10% Co thus does not significantly alter the basal lattice parameters or layered structural motif. Moreover, the overall structural properties of the LiMO2 cathode materials are consistent with the expected layered motif, suggesting that Co doping is a viable strategy for tuning the electrochemical properties of LNO without compromising its structural stability.
m space group36,64 as shown in Fig. 1. The cell volumes follow the trend of NC91 > LNO > LCO, with NC91 having the largest volume of 102.66 Å3, then LNO with 100.71 Å3 and LCO has the smallest volume of 98.11 Å3 consistent with the relative ionic radii of Ni2+ and Co3+. It is important to state that some studies may show slightly different volume trends, but this can be attributed to the different synthesis or characterization methods used. The calculated c/a ratio exhibits only minor variations across the materials, ranging from 4.978 for LNO to 5.020 for LCO. The O–M–O slab thickness and M–O bond lengths also show incremental differences, with values increasing as Ni content rises, as seen in Table 2. Overall, the structures contain alternating Li and transition metal (TM) layers separated by close-packed oxygen layers in the expected layered motif. The minor variations in lattice parameters, c/a ratio, O–M–O slab thickness, and M–O bond lengths can be attributed to the different ionic radii of Ni2+ and Co3+. The stoichiometric cation concentrations were confirmed from the optimized NC91 structure, which contained 9 Ni, 1 Co, and 10 Li atoms as expected. The Co dopant is coordinated octahedrally within the transition metal slab. The introduction of 10% Co thus does not significantly alter the basal lattice parameters or layered structural motif. Moreover, the overall structural properties of the LiMO2 cathode materials are consistent with the expected layered motif, suggesting that Co doping is a viable strategy for tuning the electrochemical properties of LNO without compromising its structural stability.
| LCO | LNO | NC91 | ||||
|---|---|---|---|---|---|---|
| Calculated | Expt.65 | Calculated | Expt.66 | Calculated | Expt.67 | |
| a (Å) | 2.825 | 2.818 | 2.858 | 2.873 | 2.970 | 2.877 |
| b (Å) | 2.825 | 2.818 | 2.858 | 2.873 | 2.970 | 2.877 |
| c (Å) | 14.190 | 14.064 | 14.227 | 14.189 | 14.199 | 14.169 |
| V (Å3) | 98.11 | 96.74 | 100.71 | 101.49 | 102.66 | 101.58 |
| α (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| γ (°) | 120 | 120 | 120 | 120 | 120 | 120 |
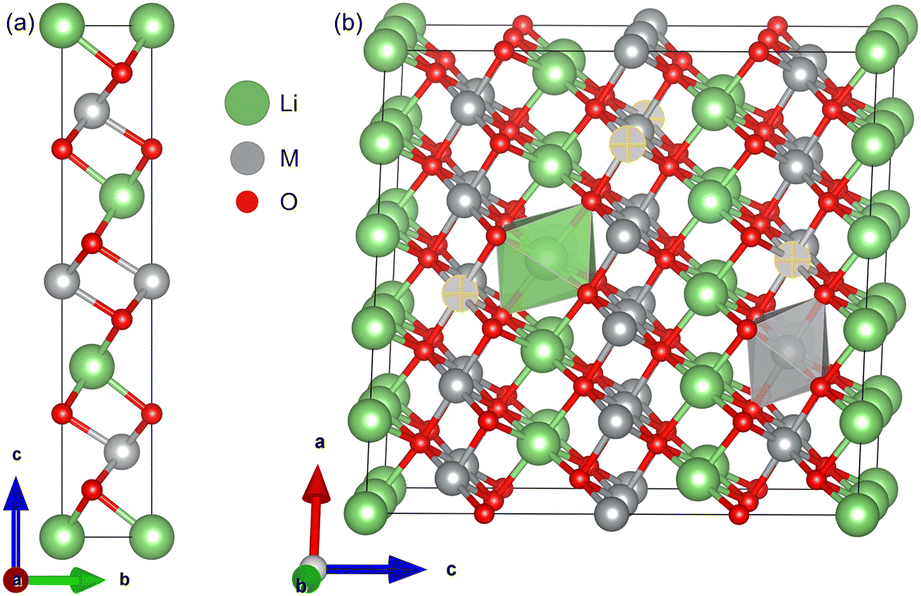 | ||
| Fig. 1 (a) Unit cell and (b) supercell of LMO, where M = Co or Ni. Selected atoms show dopant positions in NC91. | ||
3.2. Vacancy defect formation
The presence of vacancy defects in lithium-ion batteries significantly influences their electrochemical performance. The ease of lithium vacancy formation is crucial for good electrochemical performance, while the presence of oxygen vacancies could deter the performance of the battery.24,69 In order to calculate the energy associated with the formation of vacancy defects, it is crucial to define a valid range of chemical potentials. These chemical potentials are influenced by both equilibrium conditions and constraints preventing the emergence of undesirable secondary or rival phases. The energies of the formation of secondary phases used in this work were acquired from published peer-reviewed articles.70 To maintain the stability of the LiMO2 phases in an equilibrium state, it is necessary for the chemical potentials of the components to meet the following criteria:| ΔμLi + ΔμCo + 2ΔμO = ΔμEf(LCO), | (4) |
| ΔμLi + ΔμNi+ 2ΔμO = ΔμEf(LNO). | (5) |
| Δμi = μi − μoi | (6) |
In the above equations, ΔμLi, ΔμCo, ΔμNi and ΔμO are the relative chemical potentials of Li, Co, Ni and O, respectively. μoi represents the atomic chemical potential of element i in solid state form. The formation energy of the desired phase, LiMO2, must be less than that of rival phases to prevent their formation. By factoring these conditions (see eqn (S1)–(S15), ESI†), the stability boundaries for each system were computed and are shown in Fig. 2. A–B, C–D, E–F, G–H, I–J–K, represent the stability lines for Li2O, Li2O2CoO, Co3O4 and Li6CoO4 phases respectively in LiCoO2. A–B, C–D, E–F–G represent the stability lines of NiO, Li2NiO2 and Li2NiO3 phases in LiNiO2. The points, A–J in Fig. 2(a) and points A–G in Fig. 2(b) signify the positions at which the secondary phases will not precipitate. The Li-, Co/Ni-, and O-rich positions are indicated by the key. Eqn (1) was then used to compute the formation energies of the considered defect types employing the relative chemical potential values for the respective positions.
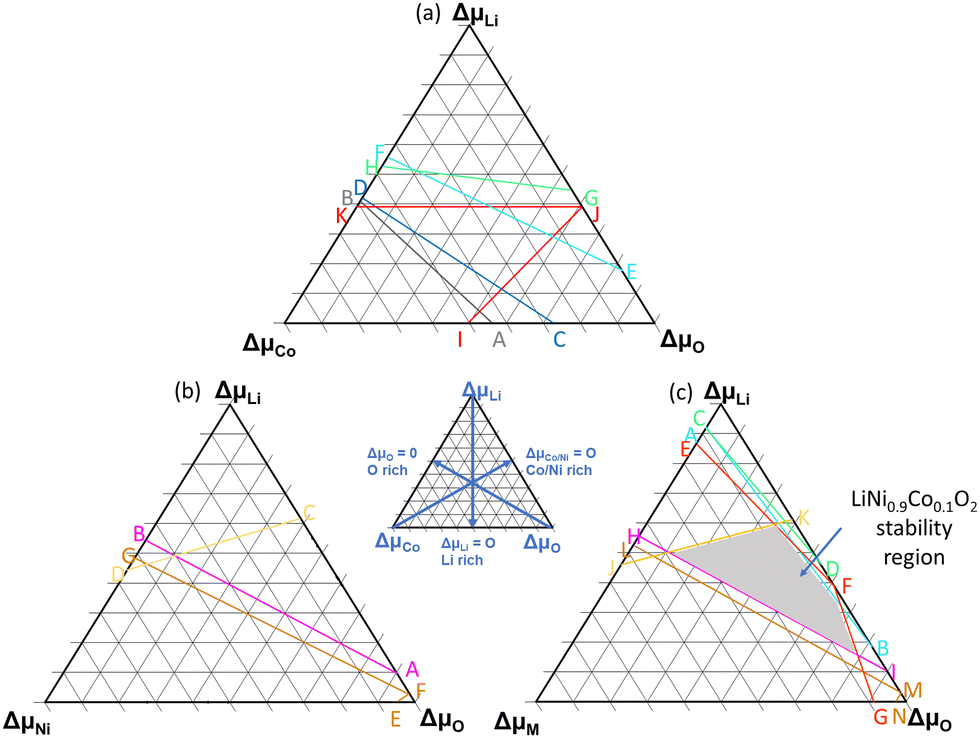 | ||
| Fig. 2 Stability diagram and chemical potential analysis for (a) LCO, (b) LNO, (c) NC91. More information on formalism used to derive stability limit is presented in the ESI.† | ||
Fig. 3 displays the Li(VLi), Co/Ni(VCo/Ni) and O(VO) vacancy formation energies in LNO, LCO and NC91. It can be observed that the vacancy formation energy is largely dependent on the chemical potentials. For Li-rich positions, I, A, and C, in Fig. 2(a), VLi is at its highest. Li-rich point E in Fig. 2(b) also appears to have the highest Li-vacancy formation energy in LNO and NC91. This signifies that the formation of lithium vacancies is least favorable in Li-rich conditions. Oxygen vacancy formation, VO, appears to be averagely higher in LCO than in LNO. It can also be observed that on average, the vacancy formation energies in NC91 system are slightly less than the LNO system.
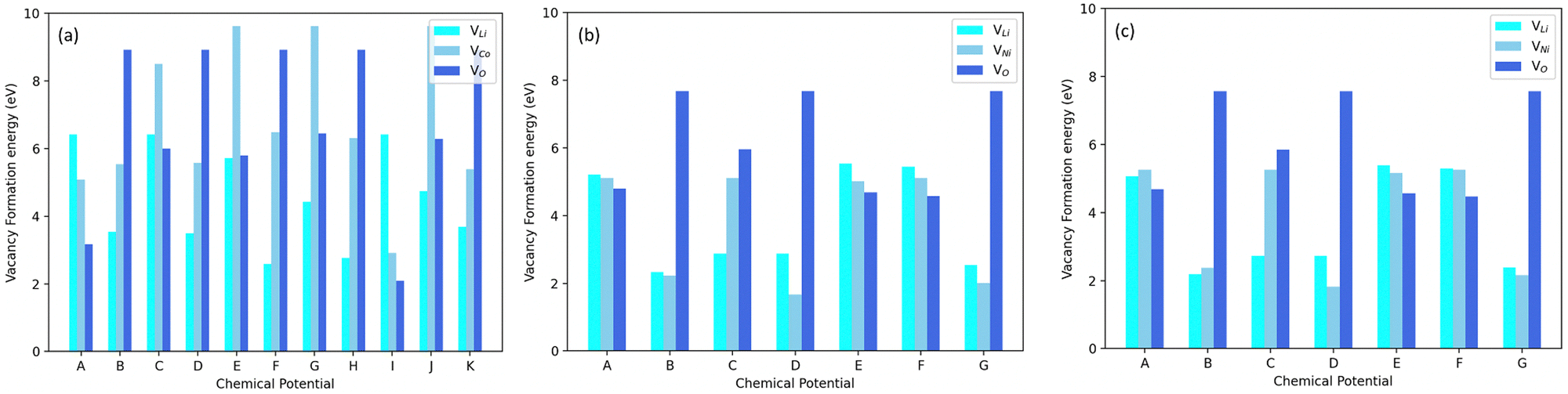 | ||
| Fig. 3 Calculated vacancy formation energies in (a) LCO, (b) LNO, (c) NC91 as a function of the atomic chemical potential positions shown in the figure (for NC91 vacancy formation diagram, the chemical potential positions for LNO in (b) we used). All vacancy formation energies can be observed to be higher in LCO than LNO and NC91. Data revels lower VLi energies in NC91 than LNO which connotes easier lithium deintercalation in NC91. | ||
The reversible nature of the vacancy formation process, largely dependent on chemical potentials, holds significant implications for battery materials. By controlling these chemical potentials, we can manipulate the creation or elimination of vacancies. This has the potential to enhance battery performance, making materials more resistant to capacity degradation caused by vacancy formation. Additionally, the slightly lower vacancy formation energies in the NC91 system compared to the LNO system indicate that Co doping may be a promising strategy to enhance the energy capacity of LNO-based batteries. Vacancies can trap Li ions, reducing battery capacity. Co doping can lower vacancy formation energy, improving Li-ion retention and overall battery capacity.
Moreover, the higher oxygen vacancy formation in LCO compared to LNO aligns with experimental findings,24,69 suggesting that LCO is more prone to oxygen loss during cycling. This implies that Co doping may also contribute to stabilizing LNO-based batteries and reducing the risk of oxygen loss during operation. Furthermore, considering that lithium vacancy formation is the fundamental process during lithium-ion battery charging,61 Co doping could potentially enhance the charging performance of LNO-based batteries. Lowering the vacancy formation energy through Co doping can facilitate the insertion of Li ions into the battery material during charging.
3.3. Antisite defect formation
In an ordered compound, antisite defects occur when there is a switch in the atomic positions of different types of atoms in a crystal structure.53 This phenomenon may occur during synthesis and after repeated cycling;71 during Li/Ni intermixing, Ni ions in the transition metal layer migrate to the Li layer and these have been reported to have both favourable and detrimental consequences.71 Studies have shown that excessive Li/Ni intermixing compromises the interlayer spacing of the Li slab and hinders Li migration due to electrostatic repulsion.72 On the other hand, a study performed by Li et al. demonstrated that a 5.7% Li/Ni antisite improved the electrochemical properties of Ni-rich and Co-poor (<20%) layered transition metal cathode.73 By studying antisite defects, we believe more insight may be obtained on the impacts of Co-doping LNO on antisite formation.This study explored and analyzed Li/transition-metal (TM) intermixing by swapping the positions of Co/Ni and Li atoms in pristine supercells of LCO, LNO and NC91. Fig. 4 shows the antisite positions (I–IV) sampled in NC91. The antisite defect formation energies were calculated using eqn (2) and the most stable antisites were noted. The antisite energies follow the trend LCO > LNO > NC91, with LCO being the least stable for antisite formation. This agrees with findings in literature; stoichiometric LNO is known to be more unstable and forms antisite defects more easily than LCO.61 The antisite formation energy of LNO decreases after 10% Co doping, with 1.18 eV as the energy of the most stable antisite configuration (position I). It was additionally observed that of the four antisite configurations studied in NC91, the antisite Ni atom at position I, which is the farthest from the Co atom, was the most stable antisite formation. This indicates that the antisite nickel ions are more energetically favored to be positioned farther from the Co ion. Corroborating this claim, the closest Ni antisite (position II) was the least stable for antisite formation. This would connote that although 10% Co doping does not sufficiently inhibit Li/Ni mixing, slightly higher concentrations of Co (>10%) would improve the material's ability to withstand antisite formation; this is corroborated by existing literature.74 Furthermore, A study by Susai et al.47 investigated the electronic structure of NC91 and revealed that Ni2+ ions dominate the valence band, which explains the limitation of 10% Co in mitigating antisite formation. In addition, the inclusion of additional cationic dopants, such as Al and Mo has also proved to be a favorable method of further stabilizing low-Co NC cathodes.75,76 It is also worthy of note that the average volumetric change of the supercell in all four antisite configurations was less than 1%.
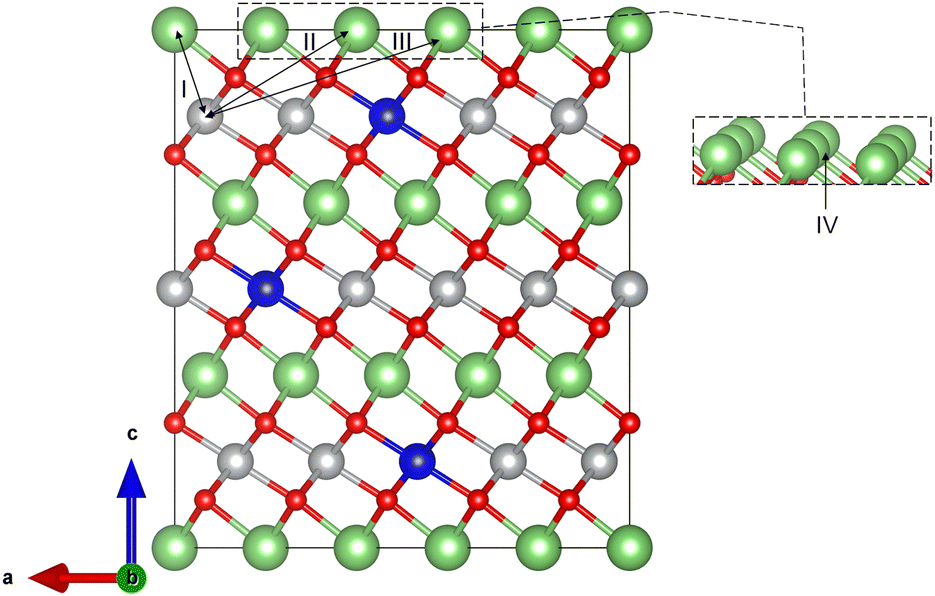 | ||
| Fig. 4 Illustration of sampled inequivalent Li/Ni antisite defect positions (I–IV) in NC91. Position I, farthest from Co, shows the lowest antisite formation energy due to strong 180° Ni–O–Ni superexchange interactions. Position II, shows highest antisite formation energy due to close proximity to Co. | ||
Further study of the antisite formation energies reveals that there is a direct relationship between the antisite formation energies and the strength of superexchange interactions. Ni2+ and Ni3+ ions are known to have partially filled doubly degenerate (eg) orbitals which causes them to have strong superexchange interactions when they form 180° Ni–O–Ni bonds. On the other hand, Co3+ and Co4+ ions have weak superexchange interactions due to their fully occupied eg states. The defect formation energies reveal the effects of Ni–O–Ni and Ni–O–Co bonds formation upon antisite formation in NC91 system.
Looking closely at the results and structure of NC91, upon the formation of antisite defects in position I, only 180° Ni–O–Ni bonds are formed. The strong superexchange interactions due to the 180° Ni–O–Ni bonds stabilize antisite defect formation, which is evident in the fact that this position exhibits the lowest defect formation energy. Position III, which gave an antisite defect formation energy of 1.8024 eV formed a 180° Ni–O–Ni bond and a 180° Ni–O–Co. The presence of the 180° Ni–O–Co bond is able to suppress the Li/Ni mixing making that position less energetically favored for antisite defects. Next, in position IV, we observed a formation energy of 2.0482 eV with both 180° Ni–O–Ni and 180° Ni–O–Co bonds formed. Similar to postion 3, the weak 180° Ni–O–Co superexchange interactions are able to screen Li/Ni mixing thereby mitigating antisite defect formation. The higher antisite formation energy in position IV than in postion III can be attributed to the proximity of the antisite Ni atom to a Co atom. The highest defect formation energy was observed in position II with a value of 3.0246 eV. Although, Ni–O–Ni bonds are formed, the proximity of Co to the antisite Ni disfavors mobilization of Ni to the postion II. This further affirms our claim that antisite nickel atoms closest to the Co atom possess higher resistance to antisite formation. In corroboration with our findings, You et al. performed X-ray diffraction Rietveld refinements to investigate the quantity of cation-disordering in Co-doped LNO samples. They discovered that low Co-dopant levels were able to effectively reduce the level of cation disordering which they attributed to the existence of 180° Ni–O–Co superexchange interactions.77 Susai et al.47 also attributed antisite formation to 180° Ni–O–Ni bonds.
This study also considered the effects of the presence of antisites on vacancy formation. Li vacancies were created in the most stable antisite configurations of the three systems. It was discovered in all three structures, that the ground state energies increased with the addition of vacancies, which corroborates experimental findings that antisite formation impedes Li-ion release during cycling.78
The variation in antisite formation energy among different cathode materials, with the lowest in LNO and the highest in LCO, aligns with experimental observations, indicating that LNO is more prone to antisite defect formation compared to LCO. This observation is crucial in understanding the inherent stability of these materials. Moreover, the discovery that Co doping reduces the antisite formation energy in LNO is a promising development, as it implies that Co doping could serve as a viable strategy for enhancing the stability of LNO cathode materials. This has significant implications for the advancement of battery technology. Interestingly, the observation that the antisite Ni atom is energetically more stable when located farther from the Co ion in NC91, hints at a possible influence of the differing ionic radii of Ni and Co. Additionally, the presence of Co screens antisite formation due to the weak superexchange interactions of Ni–O–Co bonds. This insight provides a deeper understanding of the dynamics at play within these materials. Furthermore, the fact that the average volumetric change in all four antisite configurations remains below 1% suggests that antisite defects have a minimal impact on cell volume. This observation is particularly noteworthy, as it implies that Co doping can effectively reduce antisite defect formation without causing significant structural alterations in the LNO cathode material. This balance between defect reduction and material integrity is a crucial consideration for the practical application of Co doping in battery technology.
This study delved into investigating vacancy and antisite defect concentrations in three phases, as illustrated in Fig. 5. The calculations employed atomic chemical potentials aligned with Li-rich synthesis conditions, incorporating an adjusted term for the oxygen atomic potential. The predominant defects identified across all phases were Li/TM intermixing (TMLi), referring to Ni in LNO and NC91, and Co in LCO, along with oxygen vacancy formation. Our findings reveal lower defect concentrations in LCO compared to LNO for both oxygen vacancy formation (VO) and antisite formation, affirming a higher predisposition for antisite formation in LNO than in LCO.61,81 Specifically, the oxygen vacancy (VO) concentration in LCO is notably lower, 2 orders of magnitude less than in LNO (108vs. 1010 defects per cm3 at 1300 C). Additionally, the NiLi antisite concentration in LNO is approximately three times higher than the CoLi concentration in LCO (1015vs. 1012 defects per cm3 at 1300). These trends align with the previously observed higher antisite defect formation energy and structural stability of pristine LCO. The higher synthesis temperature decreases the activation energy for defects, but the stronger Co–O interactions and stable layered motif of LCO hinder defect formation.
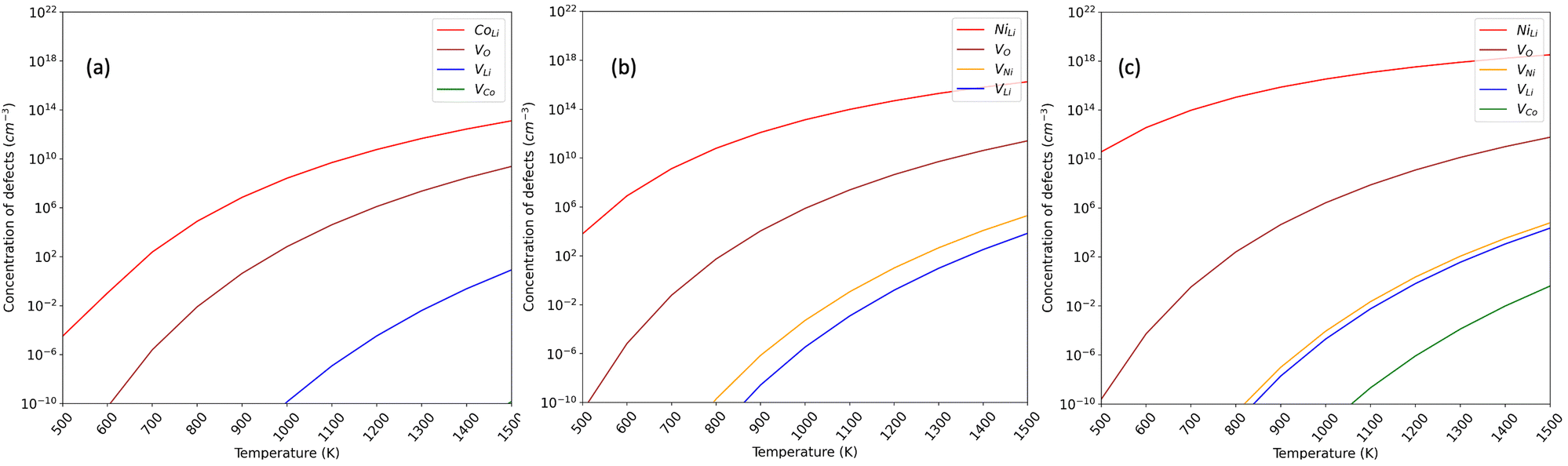 | ||
| Fig. 5 Equilibrium defect concentrations per volume (cm−3) under high-temperature conditions in (a) LCO; (b) LNO (c) NC91. | ||
For the 10% Co-doped NC91 material, no substantial improvement in Vo and NiLi defects was observed when compared to LNO. The NiLi concentration increases from 1015 defects per cm3 in LNO to 1018 defects per cm3 in NC91, indicating no stabilizing effect due to the 10% Co substitution. However, the difference is marginal, corroborating the earlier finding that this level of Co substitution doesn't effectively minimize antisite mixing. Similarly, the oxygen vacancy concentration is not improved with a value of 1010 and 1011 defects per cm3 observed, suggesting that higher concentrations of cobalt are required for maintaining oxygen stoichiometry.
3.4. Transport properties
The lithium-ion migration barriers were calculated using the climbing image nudged elastic band (NEB) method to determine the energy profile along the migration pathway between adjacent lithium sites in the cathode materials. One of the pathways that has been identified for lithium migration in layered transition metal oxides is tetrahedral site hopping (TSH). Here, lithium migrates from an octahedral site to another octahedral site via a neighboring tetrahedral site.82 As depicted in Fig. 6, the migration barrier exhibits a decreasing trend in the order of LCO > LNO > NC91.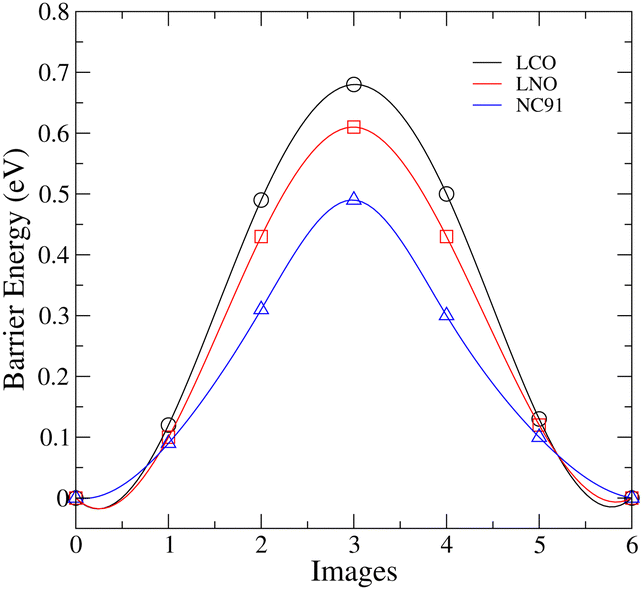 | ||
| Fig. 6 Lithium-ion migration barriers in LCO, LNO, and NC91 cathodes. | ||
The LCO possesses the highest migration barrier of 0.69 eV, indicating facile lithium diffusion will be most difficult in this Co-based cathode. This aligns with the observed high Li vacancy and antisite defect formation energies and the structural stability of undoped LCO. The strong Co–O bonding interactions and stable layered structure likely impede Li mobility. In contrast, the Ni-rich LNO exhibits a lower migration barrier (0.6 eV). The weaker Ni–O interactions, lower Li vacancy formation energy and inherent structural instabilities of LNO facilitate greater Li ion diffusivity. However, the extent of cation mixing and oxygen loss risks in LNO warrant caution. Intriguingly, the 10% Co-doped NC91 material displays the lowest Li migration barrier of only 0.49 eV, signifying enhanced lithium ion mobility compared to pure LNO and LCO. The incorporation of Co achieves a balance between stabilizing the layered structure sufficiently while still allowing Li conduction. This corroborates the findings that minor Co doping reduces the prevalence of performance-limiting antisite defects and structural changes in LNO.
The improved Li diffusivity and migration kinetics are extremely advantageous for cathode charging–discharging processes in lithium-ion batteries. Therefore, the combined results strongly indicate 10% Co doping unlocks the potential of Ni-rich cathodes by lowering the migration barrier to enable fast Li mobility while mitigating structural instabilities. This explains the observed high energy densities observed in NC91-based chemistries such as reported by Susai et al.47 Further work is warranted to investigate if slightly higher Co fractions can optimize the trade-off between stability and Li conductivity. Overall, these insights provide a compelling rationale for the enhancements in electrochemical performance achieved via minor Co substitution in Ni-rich layered oxide cathodes.
4. Conclusion
In summary, this work utilized first-principles DFT calculations to provide valuable theoretical insights into the impacts of low-level Co doping on the properties of Ni-rich LiNi0.9Co0.1O2 (NC91) cathode material. The structural characterization reveals 10% Co incorporation preserves the layered α-NaFeO2 motif without significantly altering the lattice parameters or slab thickness compared to undoped LNO and LCO. Examination of point defects shows Co addition moderately decreases the formation energies for lithium vacancies compared to LNO, suggesting possible enhancements in capacity. Analysis of lithium migration barriers reveals a significant lowering with Co doping, inferring improved Li mobility in NC91 over LNO and LCO. This combination of mitigated antisite defects and enhanced Li conductivity offers a compelling rationale for the electrochemical improvements achieved through minor Co incorporation. The high-temperature defect calculations provide further evidence that more than 10% Co is required to substantially stabilize the Ni-rich cathode against structural changes. Overall, these fundamental insights expand the theoretical understanding of strategies to unlock the potential of Ni-rich Li-transition metal–oxide cathodes. This work helps elucidate the mechanisms by which minor Co additions enhance the performance of Ni-rich layered oxide cathodes, paving the way for advanced Li-ion battery materials.Author contributions
Temitayo Ojuetimi Ikuerowo: investigation, methodology, writing, data curation, visualization, discussion of results Olusegun Tomomewo: fund acquisition, manuscript editing Salawu Omotayo Akande: conceptualization, methodology, resources, project administration, supervision, writing, discussion of results.Data availability
The data that support the findings of this study are available upon reasonable request from the corresponding author. A supplementary page containing additional details and relevant data is also included with this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The advanced computing facility of Texas A & M University at Qatar is used for all calculations.References
- M. Armand, P. Axmann, D. Bresser, M. Copley, K. Edström, C. Ekberg, D. Guyomard, B. Lestriez, P. Novák and M. Petranikova, et al. , J. Power Sources, 2020, 479, 228708 Search PubMed.
- M. M. Rahman, A. O. Oni, E. Gemechu and A. Kumar, Energy Convers. Manage., 2020, 223, 113295 Search PubMed.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 Search PubMed.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 Search PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. Chen, Materials, 2013, 6, 156–183 CrossRef PubMed.
- M. D. Bhatt and C. O'Dwyer, Phys. Chem. Chem. Phys., 2015, 17, 4799–4844 Search PubMed.
- L. Qiu, M. Zhang, Y. Song, Y. Xiao, Z. Wu, W. Xiang, Y. Liu, G. Wang, Y. Sun and J. Zhang, et al. , EcoMat, 2021, 3, e12141 CrossRef CAS.
- M. Tuccillo, O. Palumbo, M. Pavone, A. B. Muñoz-Garca, A. Paolone and S. Brutti, Crystals, 2020, 10, 526 CrossRef CAS.
- S. Mahmud, M. Rahman, M. Kamruzzaman, M. O. Ali, M. S. A. Emon, H. Khatun and M. R. Ali, Results Eng., 2022, 15, 100472 Search PubMed.
- M. S. Whittingham, MRS Bull., 2008, 33, 411–419 CrossRef CAS.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., 2012, 73, 51–65 Search PubMed.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS.
- B. E. Murdock, K. E. Toghill and N. Tapia-Ruiz, Adv. Energy Mater., 2021, 11, 2102028 CrossRef CAS.
- J. U. Choi, N. Voronina, Y.-K. Sun and S.-T. Myung, Adv. Energy Mater., 2020, 10, 2002027 Search PubMed.
- C. Pillot, Proceedings of the 33rd Annual International Battery Seminar & Exhibit, Fort Lauderdale, FL, USA, 2017.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- J.-M. Kim, X. Zhang, J.-G. Zhang, A. Manthiram, Y. S. Meng and W. Xu, Mater. Today, 2021, 46, 155–182 CrossRef CAS.
- K. Mizushima, P. Jones, P. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- Y. Ding, D. Mu, B. Wu, R. Wang, Z. Zhao and F. Wu, Appl. Energy, 2017, 195, 586–599 Search PubMed.
- J. Välikangas, P. Laine, M. Hietaniemi, T. Hu, P. Tynjälä and U. Lassi, Appl. Sci., 2020, 10, 8988 Search PubMed.
- J. Li, G. Liang, W. Zheng, S. Zhang, K. Davey, W. K. Pang and Z. Guo, Nano Mater. Sci., 2023, 5(4), 404–420 Search PubMed.
- L. Fang, M. Wang, Q. Zhou, H. Xu, W. Hu and H. Li, Colloids Surf., A, 2020, 600, 124940 CrossRef CAS.
- Y. Kim, D. Kim and S. Kang, Chem. Mater., 2011, 23, 5388–5397 CrossRef CAS.
- Z. Ahaliabadeh, X. Kong, E. Fedorovskaya and T. Kallio, J. Power Sources, 2022, 540, 231633 CrossRef CAS.
- T. Xu, F. Du, L. Wu, Z. Fan, L. Shen and J. Zheng, Electrochim. Acta, 2022, 417, 140345 CrossRef CAS.
- V. Bianchi, D. Caurant, N. Baffier, C. Belhomme, E. Chappel, G. Chouteau, S. Bach, J. Pereira-Ramos, A. Sulpice and P. Wilmann, Solid State Ionics, 2001, 140, 1–17 Search PubMed.
- A. Iqbal and D. Li, Chem. Phys. Lett., 2019, 720, 97–106 Search PubMed.
- H.-H. Ryu, G.-T. Park, C. S. Yoon and Y.-K. Sun, J. Mater. Chem. A, 2019, 7, 18580–18588 RSC.
- R. Sathiyamoorthi, P. Shakkthivel, S. Ramalakshmi and Y.-G. Shul, J. Power Sources, 2007, 171, 922–927 CrossRef CAS.
- C. S. Yoon, M.-J. Choi, D.-W. Jun, Q. Zhang, P. Kaghazchi, K.-H. Kim and Y.-K. Sun, Chem. Mater., 2018, 30, 1808–1814 CrossRef CAS.
- Q. Hao, F. Du, T. Xu, Q. Zhou, H. Cao, Z. Fan, C. Mei and J. Zheng, J. Electroanal. Chem., 2022, 907, 116034 Search PubMed.
- C. S. Yoon, U.-H. Kim, G.-T. Park, S. J. Kim, K.-H. Kim, J. Kim and Y.-K. Sun, ACS Energy Lett., 2018, 3, 1634–1639 Search PubMed.
- H. Cao, F. Du, J. Adkins, Q. Zhou, H. Dai, P. Sun, D. Hu and J. Zheng, Ceram. Int., 2020, 46, 20050–20060 CrossRef CAS.
- X. Kong, D. Li, E. O. Fedorovskaya, T. Kallio and X. Ren, Int. J. Energy Res., 2021, 45, 10489–10499 CrossRef CAS.
- G.-X. Huang, R.-H. Wang, X.-Y. Lv, J. Su, Y.-F. Long, Z.-Z. Qin and Y.-X. Wen, J. Electrochem. Soc., 2022, 169, 040533 CrossRef CAS.
- C. Geng, D. Rathore, D. Heino, N. Zhang, I. Hamam, N. Zaker, G. A. Botton, R. Omessi, N. Phattharasupakun and T. Bond, et al. , Adv. Energy Mater., 2022, 12, 2103067 CrossRef CAS.
- H. Yu, Y. Li, Y. Hu, H. Jiang and C. Li, Ind. Eng. Chem. Res., 2019, 58, 4108–4115 CrossRef CAS.
- J. Liang, Y. Lu, J. Wang, X. Liu, K. Chen, W. Ji, Y. Zhu and D. Wang, J. Energy Chem., 2020, 47, 188–195 CrossRef.
- X. Li, L. Cheng, L. Chen, B. Huang, J. Yang, Y. Li and W. Li, Solid State Ionics, 2022, 386, 116028 CrossRef CAS.
- I. Saadoune and C. Delmas, J. Mater. Chem., 1996, 6, 193–199 Search PubMed.
- H. W. Park, J. U. Hwang, J. S. Im and J. D. Lee, J. Solid State Electrochem., 2022, 26, 1567–1576 Search PubMed.
- J. Li, W. Li, Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1801957 CrossRef.
- C. Chen, J. Liu, M. Stoll, G. Henriksen, D. Vissers and K. Amine, J. Power Sources, 2004, 128, 278–285 CrossRef CAS.
- W. Shin, J. C. Garcia, A. Vu, X. Ji, H. Iddir and F. Dogan, J. Phys. Chem. C, 2022, 126, 4276–4285 Search PubMed.
- Y. Kim, J. Mater. Sci., 2012, 47, 7558–7563 Search PubMed.
- F. A. Susai, A. Bano, S. Maiti, J. Grinblat, A. Chakraborty, H. Sclar, T. Kravchuk, A. Kondrakov, M. Tkachev and M. Talianker, et al. , J. Mater. Chem. A, 2023, 11(24), 12958–12972 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- K. Saritas, E. R. Fadel, B. Kozinsky and J. C. Grossman, J. Phys. Chem. C, 2020, 124, 5893–5901 Search PubMed.
- M. Tuccillo, A. Costantini, A. Celeste, A. B. M. Garca, M. Pavone, A. Paolone, O. Palumbo and S. Brutti, Crystals, 2022, 12, 723 CrossRef CAS.
- S. Kim, V. I. Hegde, Z. Yao, Z. Lu, M. Amsler, J. He, S. Hao, J. R. Croy, E. Lee and M. M. Thackeray, et al. , ACS Appl. Mater. Interfaces, 2018, 10, 13479–13490 Search PubMed.
- D.-H. Seo, A. Urban and G. Ceder, et al. , Phys. Rev. B, 2015, 92, 115118 CrossRef.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- S. Yang, J. Peng, H. Huang, Z. Li, H. Dong and F. Wu, Mater. Sci. Semicond. Process., 2022, 144, 106552 CrossRef CAS.
- J. Zhang, Y.-P. Zhang and C.-M. Su, Calphad, 2020, 71, 102007 CrossRef CAS.
- C. Jiang, Acta Mater., 2009, 57, 4716–4726 CrossRef CAS.
- S. O. Akande and D. Çakr, Phys. Rev. Mater., 2023, 7, 053610 CrossRef CAS.
- Y. Koyama, H. Arai, I. Tanaka, Y. Uchimoto and Z. Ogumi, Chem. Mater., 2012, 24, 3886–3894 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Wang, N. Wang, W. Nan, C. Wang, X. Chen, X. Qi, S. Yan and S. Dai, Int. J. Electrochem. Sci., 2020, 15, 9282–9293 CrossRef.
- S. Hao, Y. Li, S. Wang, J. Yang, Z. Tan, X. Li, X. Shen, X. Xi, J. Zheng and Z. He, Mater. Today Energy, 2022, 25, 100980 CrossRef CAS.
- A. Mesnier and A. Manthiram, ACS Appl. Mater. Interfaces, 2020, 12, 52826–52835 CrossRef CAS PubMed.
- X. Shi, C. Wang, X. Ma and J. Sun, Mater. Chem. Phys., 2009, 113, 780–783 CrossRef CAS.
- S. Laubach, S. Laubach, P. C. Schmidt, D. Ensling, S. Schmid, W. Jaegermann, A. Thißen, K. Nikolowski and H. Ehrenberg, Phys. Chem. Chem. Phys., 2009, 11, 3278–3289 RSC.
- W. Hu, H. Wang, W. Luo, B. Xu and C. Ouyang, Solid State Ionics, 2020, 347, 115257 CrossRef CAS.
- K. Hoang and M. D. Johannes, J. Mater. Chem. A, 2014, 2, 5224–5235 RSC.
- H.-X. Wei, L.-B. Tang, Z.-Y. Wang, Y.-H. Luo, Z.-J. He, C. Yan, J. Mao, K.-H. Dai and J.-C. Zheng, et al. , Mater. Today, 2021, 51, 365–392 CrossRef CAS.
- R. Clément, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345–373 RSC.
- C. Li, W. H. Kan, H. Xie, Y. Jiang, Z. Zhao, C. Zhu, Y. Xia, J. Zhang, K. Xu and D. Mu, et al. , Adv. Sci., 2019, 6, 1801406 CrossRef PubMed.
- E. Lee, H. Iddir and R. Benedek, Electrochemical Society Meeting Abstracts, 2016, pp. 847–847 Search PubMed.
- H. Zhu, H. Yu, H. Jiang, Y. Hu, H. Jiang and C. Li, Chem. Eng. Sci., 2020, 217, 115518 CrossRef CAS.
- K. Zhou, Q. Xie, B. Li and A. Manthiram, Energy Storage Mater., 2021, 34, 229–240 CrossRef.
- B. You, Z. Wang, D. Wang, G. Luo, D. Deng, G. Yan, H. Guo, X. Li, W. Peng and M. Dong, et al. , Prog. Nat. Sci.: Mater. Int., 2023, 33, 425–432 CrossRef CAS.
- J. Hu, Q. Wang, B. Wu, S. Tan, Z. Shadike, Y. Bi, M. S. Whittingham, J. Xiao, X.-Q. Yang and E. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 2622–2629 CrossRef CAS PubMed.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- R. Koksbang, J. Barker, H. Shi and M. Saidi, Solid State Ionics, 1996, 84, 1–21 CrossRef CAS.
- Y. Kim, Phys. Chem. Chem. Phys., 2019, 21, 24139–24146 RSC.
- C. Wang, R. Wang, Z. Huang, M. Chu, W. Ji, Z. Chen, T. Zhang, J. Zhai, H. Lu and S. Deng, et al. , Energy Storage Mater., 2022, 44, 1–9 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03475a |
| This journal is © the Owner Societies 2025 |