
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wavelength-dependent intersystem crossing dynamics of phenolic carbonyls in wildfire emissions†
Rashid R.
Valiev
 *a,
Yiheng
He
b,
Tate
Weltzin
b,
Allen
Zhu
b,
Dong
Lee
b,
Ellery
Moore
b,
Aidan
Gee
b,
Greg
Drozd
b and
Theo
Kurten
a
*a,
Yiheng
He
b,
Tate
Weltzin
b,
Allen
Zhu
b,
Dong
Lee
b,
Ellery
Moore
b,
Aidan
Gee
b,
Greg
Drozd
b and
Theo
Kurten
a
aDepartment of Chemistry, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: valievrashid@gmail.com
bDepartment of Chemistry, Colby College, Waterville, Maine, USA
First published on 11th December 2024
Abstract
Quantum chemical calculations were employed to construct Jablonski diagrams for a series of phenolic carbonyls, including vanillin, iso-vanillin, 4-hydroxybenzaldehyde, syringaldehyde, and coniferyl aldehyde. These molecules can enter the Earth's atmosphere from forest fire emissions and participate in photochemical reactions within the atmospheric condensed phase, including cloud and fog droplets and aqueous aerosol particles. This photochemistry alters the composition of light-absorbing organic content, or brown carbon, in droplets and particles through the formation and destruction of key chromophores. This study demonstrates that following photon absorption, phenolic carbonyls efficiently transition to triplet states via intersystem crossings (ISC), with rate coefficients ranging from 109 to 1010 s−1. Despite the presence of multiple potential ISC pathways due to several lower-lying triplet states, a single channel is found to dominate for each system. We further investigated the dependence of the ISC rate constant (kISC) on the vibrational excitation energy of the first accessible (ππ*) singlet excited state (S1 or S2, depending on the molecule), and compared it with the measured wavelength dependence of the photochemical quantum yield (Φloss). Although our model only accounts for intramolecular nonradiative electronic transitions, it successfully captures the overall trends. All studied molecules, except coniferyl aldehyde, exhibit saturation in the dependence of both kISC and Φloss on the wavelength (or vibrational excitation energy). In contrast, coniferyl aldehyde displays a single maximum, followed by a monotonic decrease as the excitation energy increases (wavelength decreases). This distinct behavior in coniferyl aldehyde may be attributed to the presence of a double-bonded substituent, which enhances π-electron conjugation, and reduces the exchange energy and thus the adiabatic energy gap between the S1(ππ*) state and the target triplet state. For small energy gaps, the classical acceptor modes of the ISC process are less effective, leading to a low effective density of final states. Larger gaps enhance the effective density of states, making the wavelength dependence of the ISC more pronounced. Our calculations show that while all the studied phenolic carbonyls have similar acceptor modes, coniferyl aldehyde has a substantially smaller adiabatic gap (1700 cm−1) than the other molecules. The magnitude of the adiabatic energy gap is identified as the primary factor determining the energy/wavelength dependence of the ISC rate and thus Φloss.
1. Introduction
Phenolic carbonyls are released into the atmosphere during biomass burning, including both forest fires and domestic heating.1–4 Once in the atmosphere, these compounds become important components of organic aerosols, specifically brown carbon (BrC).5–11 BrC plays a significant role in climate processes due to its ability to absorb light, which directly influences radiative forcing.5–12 Estimates indicate that BrC may have a global warming potential approximately 20% of that of black carbon.5–12In water droplets, such as fog and aqueous aerosol particles, phenolic carbonyls undergo photochemical reactions under sunlight exposure.12–15 During these reactions, they can transition into triplet excited states, which are a crucial step in their photochemical activity.12–15 These triplet states facilitate unique oxidation pathways for organic aerosol components.12–16 For example, the interaction between triplet excited state vanillin and ground state vanillin leads to concentration-dependent rates of photochemical loss, significantly impacting the transformation and chemical activity of organic aerosols.8,17,18 The scheme of loss of phenolic carbonyls is shown in ESI.†
Studies show that the quantum yield for the photochemical loss of vanillin depends on the wavelength of sunlight.17–19 Utilizing this wavelength dependence allows more accurate application of laboratory experiments to actual atmospheric conditions. Previously, our quantum chemical studies demonstrated that the intersystem crossing (ISC) rate constant for vanillin exhibits a wavelength-dependent behavior that closely mirrors the quantum yield for its photochemical loss.18 As the absorption wavelength decreased, the intersystem crossing rate constants from the singlet excited state to the accessible triplet states initially rose, and then reached a saturation point. This behavior was attributed to the specific energy dependence of the overlap of the nuclear wave functions of these electronic excited states, known as the Franck–Condon (FC) factors. The FC factors are influenced by molecular structure, including the presence or absence of single or double bonds.20 Consequently, it is anticipated that other molecules in the phenolic carbonyl series may exhibit entirely different dependencies. In this study, we address this issue, both theoretically and experimentally, developing the theory and applying it to a range of phenolic carbonyl molecules: iso-vanillin, 4-hydroxybenzaldehyde, syringaldehyde, and coniferyl aldehyde. The studied molecules are shown in Fig. 1. This study demonstrates that different phenolic carbonyls exhibit varying behavior in terms of the dependence of the ISC rate on the wavelength of radiation absorbed. This variation is expected to influence also their atmospheric chemistry.
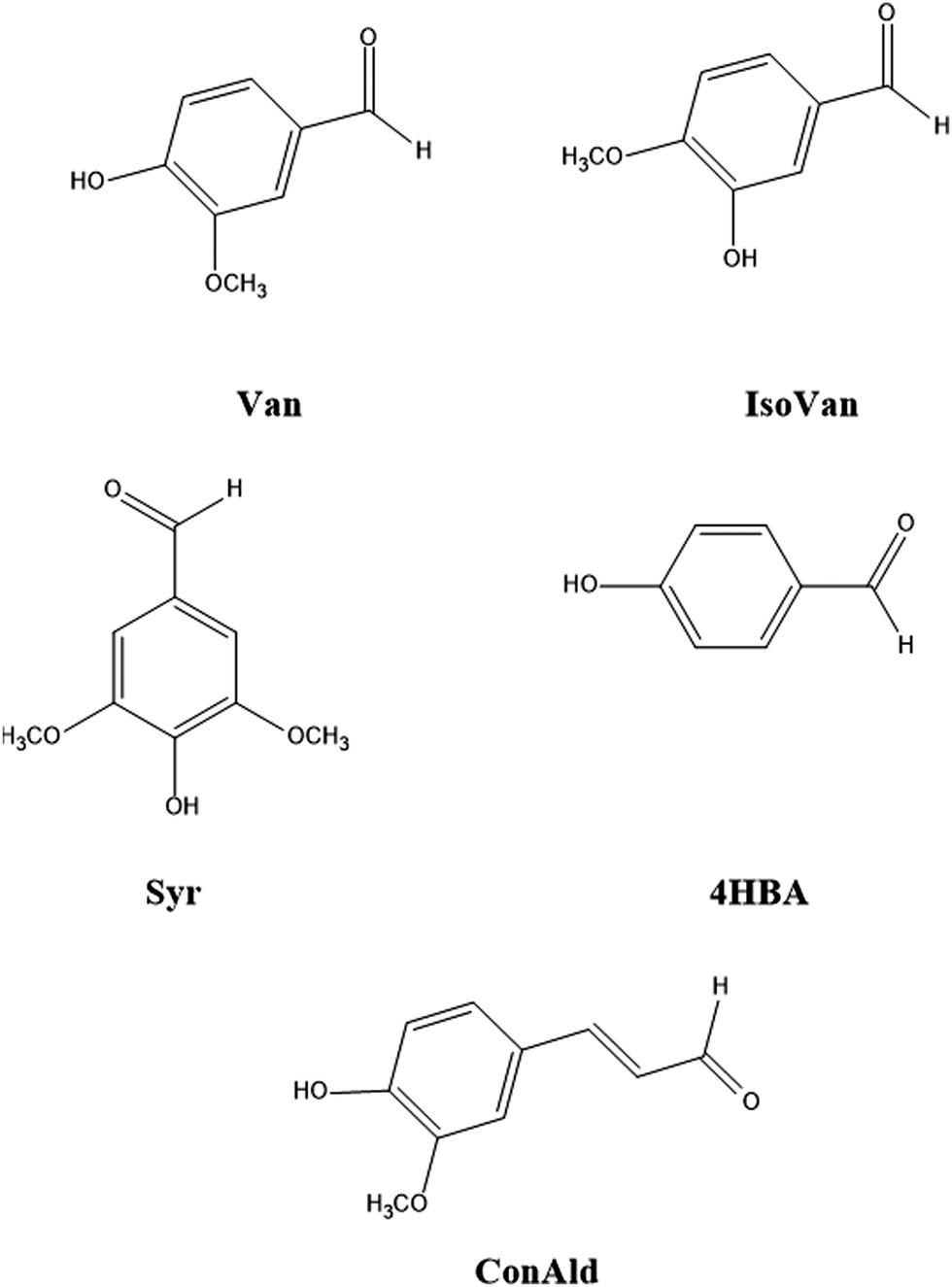 | ||
| Fig. 1 The molecular structure of the studied phenolic carbonyls: vanillin (Van), iso-vanillin (IsoVan), 4-hydroxybenzaldehyde (4HBA), syringaldehyde (Syr), and coniferyl aldehyde (ConAld). | ||
2. Theory and computation
2.1. Photophysical processes
As previously noted, the triplet state of phenolic carbonyls plays a critical role in their photochemical activity.12–15 Phenolic carbonyls absorb light in the 295–400 nm range, transitioning into the first ππ* excited singlet state. This can be either the lowest excited singlet state (S1) or the second-lowest excited singlet state (S2), as the lowest excited singlet state S1 may in some cases be a dark nπ* state.18,21 For example, vanillin has an nπ* state as its first singlet electronic state, with almost zero probability of an electronic transition from the ground state (S0). After absorption and vibrational relaxation, the electronic excited state energy can be dissipated through three intramolecular pathways: photon emission back to the ground singlet state (S0), internal conversion (IC) to S0, and intersystem crossing (ISC) to a lower-energy triplet state (Tn).18,21 When the S1 state is a nπ* dark state, an IC from S2 to S1 is possible as well. It is well known that when the first excited singlet state is a ππ* state, and when the state is more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (less than 500 nm) above the ground state, its main deactivation channel is the ISC process.20,22 The energy gap criterion is fulfilled for phenolic carbonyls.18,21 The reason for the dominance of the ISC process is the difference in configuration between the ππ* excited singlet state, and a lower-energy triplet state (Tn, where n can be 1, 2 or 3) of type nπ*. This leads to large spin–orbit coupling matrix elements (SOCME) between these states, resulting in large intersystem crossing rate constants (kISC), according to El-Sayed's rule.23,24 We have previously proven this conclusion for vanillin using quantum chemical calculations.18
000 cm−1 (less than 500 nm) above the ground state, its main deactivation channel is the ISC process.20,22 The energy gap criterion is fulfilled for phenolic carbonyls.18,21 The reason for the dominance of the ISC process is the difference in configuration between the ππ* excited singlet state, and a lower-energy triplet state (Tn, where n can be 1, 2 or 3) of type nπ*. This leads to large spin–orbit coupling matrix elements (SOCME) between these states, resulting in large intersystem crossing rate constants (kISC), according to El-Sayed's rule.23,24 We have previously proven this conclusion for vanillin using quantum chemical calculations.18
2.2. ISC calculation details
Within the Franck–Condon approximation, and assuming a linear coupled model (i.e. the Hessians of the initial (Si) and final electronic (Tj) states are equal), kISC(Si → Tj) can be calculated as18,20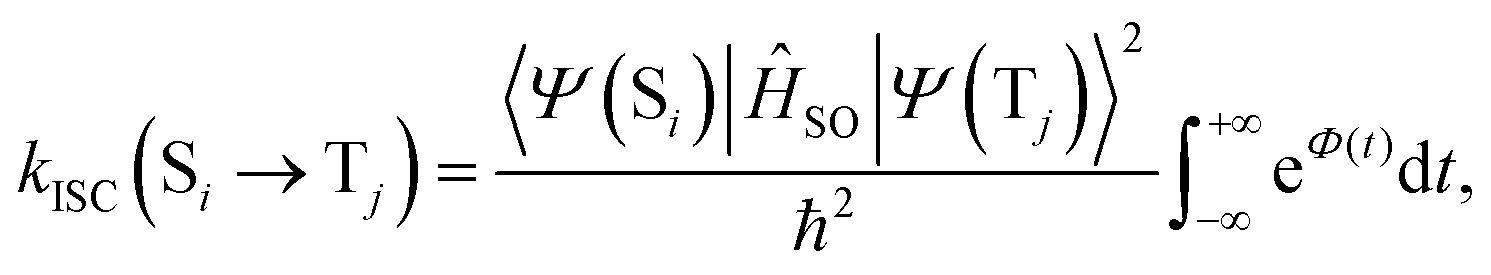 | (1) |
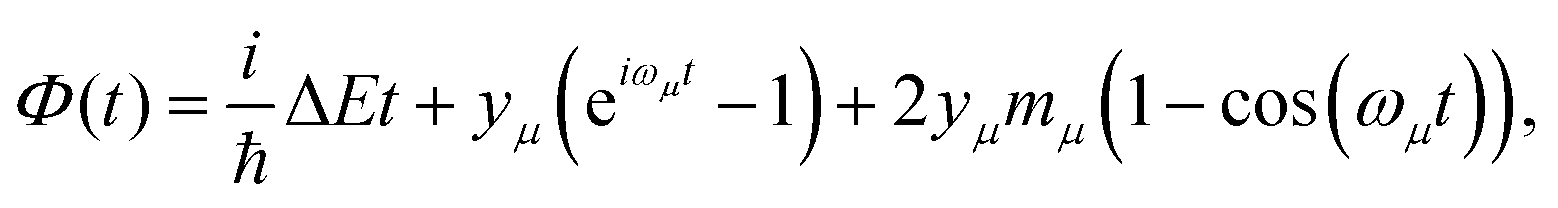 | (2) |
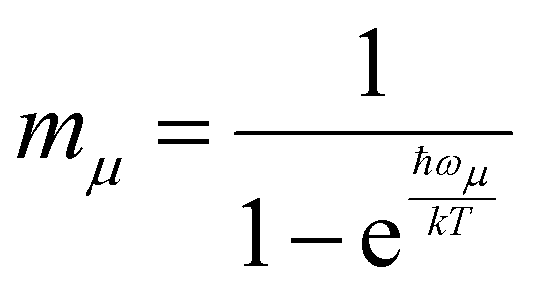 is Boltzmann population of the μ-th mode vibrational level with frequency ωμ. eΦ(t) is a correlation function, while yμ is the Huang–Rhys’ factor related to the equilibrium position displacement of the μ-th mode during the Si → Tj transition, and ΔE is the a diabatic energy gap of the transition.
is Boltzmann population of the μ-th mode vibrational level with frequency ωμ. eΦ(t) is a correlation function, while yμ is the Huang–Rhys’ factor related to the equilibrium position displacement of the μ-th mode during the Si → Tj transition, and ΔE is the a diabatic energy gap of the transition.
The integral (2) cannot be calculated because it diverges.18 Typically, to ensure convergence, a physical regularization is introduced by adding a damping term such as exp(−γ|t|) for Lorentzian damping or exp(−γt2) for Gaussian damping.18,25,26 Generally, the Gaussian term significantly underestimates the rate constant, while the Lorentzian term overestimates it.18,25
One of the effective methods for evaluating this integral is the method of the first saddle point,18 which provides the following analytical solution:18
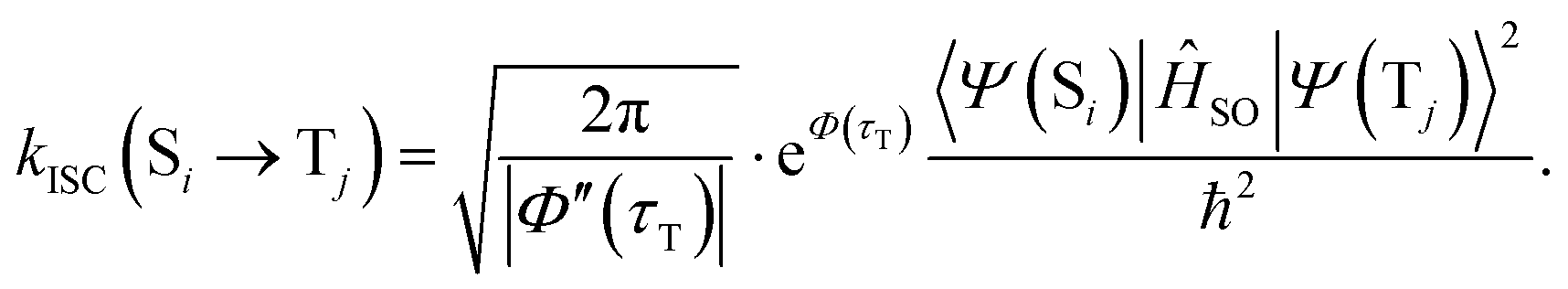 | (3) |
| Φ′′(t) = −yμωμ2(eiωμt + 2mμch(ωμt)). | (4) |
| ħωμyμ(eiωμτT + 2mμsh(ωμτT)) = ΔE. | (5) |
Previously, we applied this method to vanillin (Van). Here, we apply it to iso-vanillin (IsoVan), 4-hydroxybenzaldehyde (4HBA), syringaldehyde (Syr), and coniferyl aldehyde (ConAld). Additionally, we calculated the internal conversion (kIC) and radiative (kr) rate constants from the lowest energy excited singlet state of type π–π* (S1 or S2, depending on the molecule) to the ground state (S0) to estimate the fluorescence quantum yield (φfl) using the method described in details in ref. 27–31. This calculation requires the oscillator strength (f), energy (E), configuration interaction coefficients (CI), and molecular orbital basis set (BF) coefficients involved in the electronic transition.
2.3. Computational details
The molecular geometries of all studied systems were optimized in the first excited singlet state of type ππ*, referred to for simplicity as S(ππ*) in the following discussion. For Van, IsoVan and 4HBA S(ππ*) corresponds to state S2 (as these molecules have a lower-lying dark state of type nπ*), while for Syr and ConAld S(ππ*) corresponds to S1. For all studied systems, we explicitly included one H-bonded water molecule in the structure.In our preliminary SOCME calculations based on the S(ππ*) optimized geometries, one electronic transition from S(ππ*) to Tn (where n is 1, 2 or 3 depending on the system) was found to dominate over others. The reason is that these specific transition have a ππ* → nπ* character, while the other ISC are ππ* → ππ*. The dominant transition is into T2 for Van, IsoVan and 4HBA, and into T3 for Syr and ConAld. For simplicity, we refer to this as the target triplet T(nπ*) state. To examine the ISC dependence on the adiabatic energy gap (ΔE), the molecular geometries were optimized also in the target triplet state. Hessians for the initial S(ππ*) and target T(nπ*) states were calculated using time-dependent density functional theory (TDDFT).32 The calculations employed the CAM-B3LYP33 exchange–correlation functional, a 6-31G**34 basis set, and the polarizable continuum model35 to account for solvation beyond the first explicit water molecule. We stress that it is important to explicitly include the interaction of the molecules under investigation with one water molecule to accurately account for solvent effects in the electronic excited state calculations.18,21 The choice of CAM-B3LYP is due to its ability to account for the charge-transfer character of electronic transitions, which can be relevant for carbonyls with larger substituents. We note that B3LYP is unable to properly treat these systems. As will be shown later, the adiabatic energy gap ΔE is a key parameter in the studied process, and it can vary with the functional used. Our calculations on vanillin show that for this system, B3LYP and CAM-B3LYP yield similar values, with a difference of only 0.04 eV. We performed further test calculations with the ωb97X-D functional on ConAld (lowest-energy conformer), Van, and 4HBA to assess the sensitivity of ΔE to the used functional for different absolute values of ΔE magnitudes.
Due to the presence of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, the ConAld molecule has both cis and trans isomers, each with multiple conformers. These were all explicitly studied. For the other molecules, the lowest-energy conformer found in the initial optimizations was used. We note that the results presented here for Van differ slightly from those in our earlier study due to the use of the CAM-B3LYP functional rather than B3LYP.18
C double bond, the ConAld molecule has both cis and trans isomers, each with multiple conformers. These were all explicitly studied. For the other molecules, the lowest-energy conformer found in the initial optimizations was used. We note that the results presented here for Van differ slightly from those in our earlier study due to the use of the CAM-B3LYP functional rather than B3LYP.18
The SOCME between the electronic states of different multiplicities were calculated using MOLSOC software36 at the TDDFT level of theory with CAM-B3LYP/6-31G**. BF, CI, f and E, needed for the kr and kIC calculation, were also all computed using TDDFT/CAM-B3LYP/6-31G*.
All quantum chemical calculations were performed using Gaussian 1637 software on the PUHTI CSC supercomputer.38
3. Experimental details
3.1. Methods
3.2. Illumination source characterization
6 UV-LED (Violumas) with nominal wavelengths of 300, 318, 327, 342, 370, and 382 nm were used to illuminate aqueous phenolic carbonyl samples. The LEDs (light emitted diodes) were connected to a Tekpower TP1803D variable DC power supply with current controlled under 1 A. The LED spectral profiles are shown in Fig. S1 (ESI†). The photon flux of each LED was determined using 2-nitrobenzaldehyde (NBA) as an actinometer which has a known and constant quantum yield (0.43) between 300–400 nm. The conversion of NBA to 2-nitrosobenzoic acid was monitored with an Agilent Cary 60 UV-Vis spectrometer over a period of 2–3 lifetimes. Typical volume averaged fluxes for LED varied between 2.0 × 1015–3 × 1017 photons cm−3 s−1, with the highest fluxes for longer wavelengths where absorption is weakest.3.3. Photolysis experiments
1.0 mL of phenolic carbonyl solution was added to a quartz vial (Technical Glass Products, 15 mm dia) with UV LED illumination from below and a pathlength of 0.88 cm. To minimize internal screening, solution absorbance was kept below 0.2 (Section S1, ESI†). For shorter experiments fresh solution was used for each time point, and for longer experiments aliquots were removed, never reducing total solution volume by more than 10%. Vials were tightly sealed with a Teflon-lined screw cap to prevent evaporation. The laboratory temperature was 23 ± 2 °C. All samples had pH = 2, as atmospheric particle pH is generally observed to be between 0 and 4, with an approximate range recently considered to span −1 to 5.39–42 Cloud or fog droplets generally have higher pH, approximately 2–7.433.4. HPLC analysis
Eluents were ultrapure water (eluent A), and acetonitrile (eluent B) both with 0.1% formic acid. The solvent flow rate was 0.5 ml min−1. The mobile phase gradient was: starting with 0% B, isocratic for 2 min, followed by a gradient to 100% B in 5 minutes, a hold at 100% B for 1 minute, and a gradient to 0% B in 1 minute followed by a 3 minute hold at 0% B. The HPLC system was connected to the ToF-MS via a diode array detector (DAD) equipped with a 500 nL flow cell, 10 mm pathlength, using a reference wavelength of 500 nm. The electrospray ion source of the MS was operated using the following setup: nebulizer pressure 2400 mbar, drying gas flow 8 l min−1, dry gas temperature 325 °C, and spray voltage 3500 V. Quantification of each phenolic carbonyl was determined using calibration from the UV chromatograms of known standards.4. Results and discussion
4.1. Conformer analysis of ConAld and Van, IsoVan
Due to the non-aromatic double bond, ConAld can exist in both cis and trans isomeric forms, which do not interconvert at atmospheric temperatures. Each isomer then has multiple possible conformers, which do interconvert. Fig. 2 shows all the six conformers of ConAld discovered in our sampling. The structures of Van and IsoVan are also shown for comparison.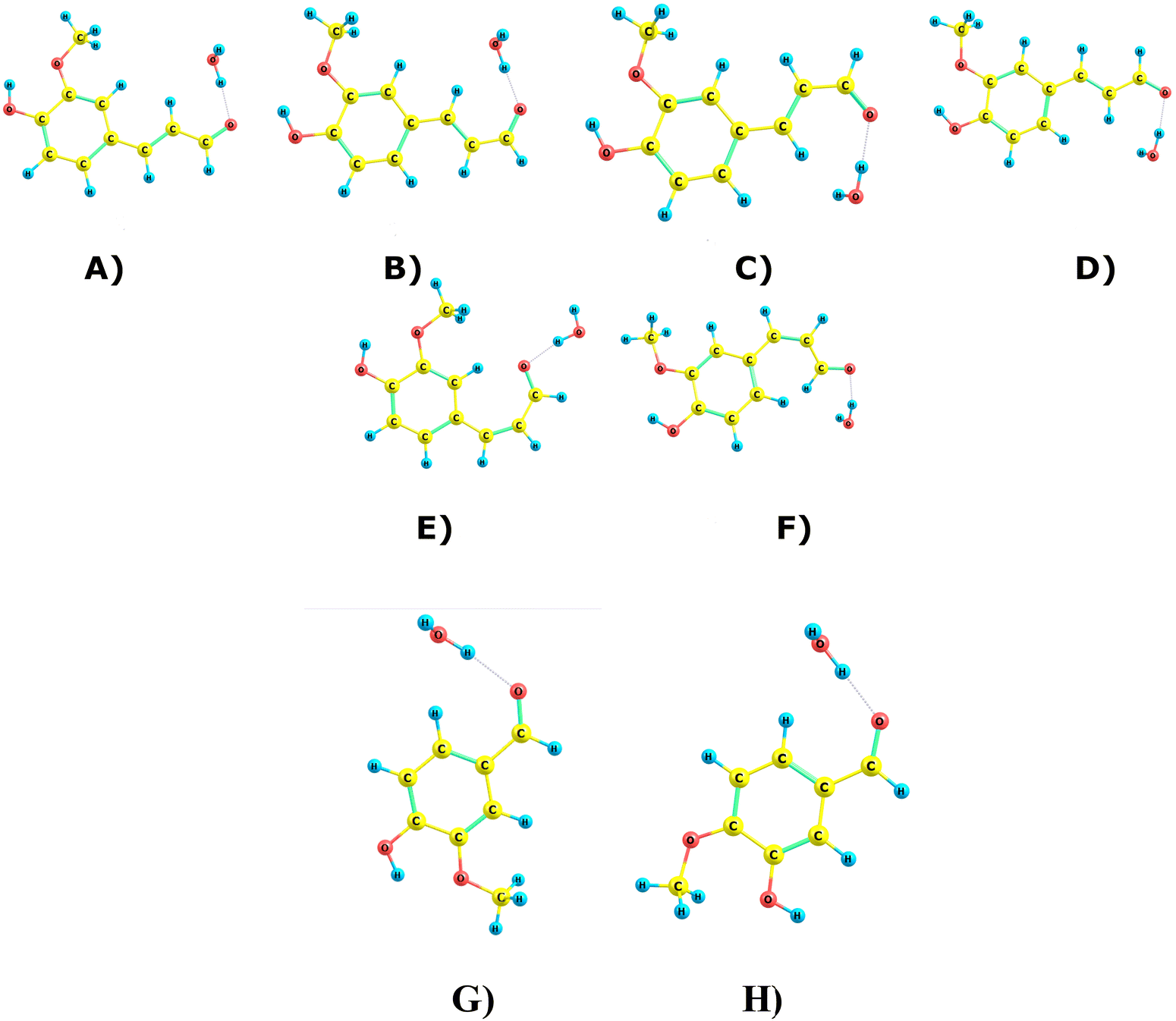 | ||
| Fig. 2 All discovered conformers of the trans- and cis-isomers of ConAld. The respective energies are 0.00, 1.72, 1.41, 0.38, 6.29 and 5.75 kcal mol−1 at the PCM/CAM-B3LYP/6-31G(d,p) level for (A) TRANS-I, (B) TRANS-II, (C) TRANS-III, (D) TRANS-IV, (E) CIS-I, (F) CIS-II, respectively. (G) Van (H) IsoVan have relative energies of 0.00 kcal mol−1 and 6.00 kcal mol−1, respectively. | ||
We found 4 trans-conformers and 2 cis-conformers of ConAld. The lowest-energy conformer of the cis-isomer is over 5 kcal mol−1 higher in energy than the lowest-energy conformer of the trans-isomer. At equilibrium, the trans structures would thus dominate. However, we caution that due to the high barrier for interconversion, the actual isomer distribution depends on the reactions forming the ConAld molecules in the first place. Note that our computed energies are both quantitatively and qualitatively in good agreement with the results reported in ref. 44, where it was shown that the four trans-isomers are within 1.7 kcal mol−1 of each other at the B3LYP/6-31G(d,p) level of theory. In our case, using CAM-B3LYP, we obtained a similar energy range of 0–1.72 kcal mol−1.
We further note that vanillin (Van) and isovanillin (IsoVan) are structural isomers, with the latter 6 kcal mol−1 higher in energy. However, as the interconversion reaction has an extremely high barrier, this energy difference is not relevant to their atmospheric chemistry.
4.2. Photophysical properties
The SOCME values and all other intramolecular rate constants for the phenolic carbonyls under investigation are presented in Table 1. For ConAld, the calculation was done for the lowest energy conformer TRANS-I. As expected, the main deactivation pathway of the lowest energy singlet state is intersystem crossing (ISC) to the triplet state with a nπ* configuration, for which kISC = 108–1010 s−1. Only for the 4HBA, the kIC(S2 → S1) prevails over kISC(S2 → T2). The fluorescence quantum yield is negligible due to the large kISC and kIC.| Molecule | Van | IsoVan | 4HBA | Syr | ConAld |
|---|---|---|---|---|---|
| 〈Si|HSO|T1〉, cm−1 | 0.3 | 1.0 | 26.90 | 0.32 | 0.05 |
| 〈Si|HSO|T2〉, cm−1 | 12.0 | 10.00 | 2.70 | 0.22 | 0.08 |
| 〈Si|HSO|T3〉, cm−1 | 0.6 | 0.4 | — | 12.8 | 14.4 |
| k ISC(Si → T1), s−1 | 1.0 × 101 | 1.0 × 101 | 5 × 108 | 1 × 10−1 | 1 × 10−3 |
| k ISC(Si → T2), s−1 | 2.2 × 109 | 2.0 × 109 | 1.0 × 107 | 3.0 × 103 | 7.0 × 105 |
| k ISC(Si → T3), s−1 | 3.0 × 107 | 3.0 × 107 | — | 4.6 × 109 | 1.0 × 1010 |
| k r(Si → S0), s−1 | 7.0 × 107 | 7.0 × 107 | 1.0 × 108 | 2.0 × 108 | 2.0 × 108 |
| k IC(Si → S0), s−1 | 1.0 × 106 | 5.0 × 106 | 1.0 × 104 | 5.0 × 104 | 5.0 × 104 |
| k IC(S2 → S1), s−1 | 1.0 × 109 | 1.0 × 109 | 1.0 × 109 | — | — |
| ϕ fl(Si → S0), s−1 | 0.01 | 0.01 | 0.07 | 0.04 | 0.04 |
It should be noted that the considered molecules have multiple low-energy triplet states. For most molecules, three triplet states lower in energy than the lowest excited singlet state of type π–π* are observed. The exception is 4HBA, which exhibits only two triplet states below S2. Among these triplet states, only one has an ISC channel that significantly contributes to the total intersystem crossing (ISC) rate constant. Specifically, the kISC(S2 → T2) pathway serves as the main deactivation channel for Van, IsoVan and 4HBA. In contrast, the kISC(S1 → T3) pathway is predominant for ConAld and Syr. The latter result is attributed to the T3(nπ*) state being close in energy to S1, along with a substantial SOCME for ConAld. Consequently, the kISC(S1 → T3) rate for ConAld exhibits the highest value among all the studied rate constants. Further, we note that there is a reasonably good agreement between the experimental and theoretical fluorescence quantum yields, as the values are within one order of magnitude from each other. For Van, the experimental φfl(S1 → S0) is 0.01221 while the theoretical value is 0.01. For Syr, the corresponding numbers are 0.00821 and 0.04, respectively.
4.3. Intersystem crossing wavelength dependence
We investigated the dependence of the main kISC(S(ππ*) → T(nπ*)) process on the excitation wavelength. By increasing the frequency of electromagnetic radiation from that corresponding to the energy of the pure electronic transition S(ππ*) → S0, we can excite the vibrational modes of S(ππ*). This has two consequences. First, it leads to increase in energy (ΔE + ħmiωi) of the S(ππ*) state with respect to the ground vibrational state of the target triplet electronic state T(nπ*). Here, mi and ωi are vibrational quantum numbers and frequencies, respectively, of the vibrational modes of S(ππ*). This, in turn, alters the Franck–Condon factor between the S(ππ*) and T(nπ*) states, which finally determines the rate constant kISC(S(ππ*) → T(nπ*)) as defined by eqn (1)–(5). Consequently, there emerges a dependence of kISC(S(ππ*) → T(nπ*)) on the wavelength of the electromagnetic radiation.The results for all molecules, including all isomers, are presented in Fig. 3. The figure plots the ISC rate as a function of the excess excitation energy (the energy in excess of the S(ππ*)–S0 energy gap), in wavenumbers. Additionally, the values of the adiabatic energy gap and the main acceptive modes with the y (Huang–Rhys factors) are given in Table 2. As observed from Table 2, the adiabatic energy gap of the main deactivation channel (ISC) decreases from 4HBA to ConAld, reaching a minimum value of 1630 cm−1. In contrast, the principal accepting modes and their Huang–Rhys factors are similar across the entire series of molecules. These modes are primarily associated with the breathing vibrations of the benzene ring. The wavenumber region 1200–1700 cm−1, with Huang–Rhys factors y ranging from 0.1 to 0.6, corresponds to the typical accepting modes of the ISC process in organic molecules.21,22,27–29 This region is characterized by an average wavenumber of approximately 1400 cm−1, and an average Huang–Rhys factor of y ≈ 0.3.21,22,27
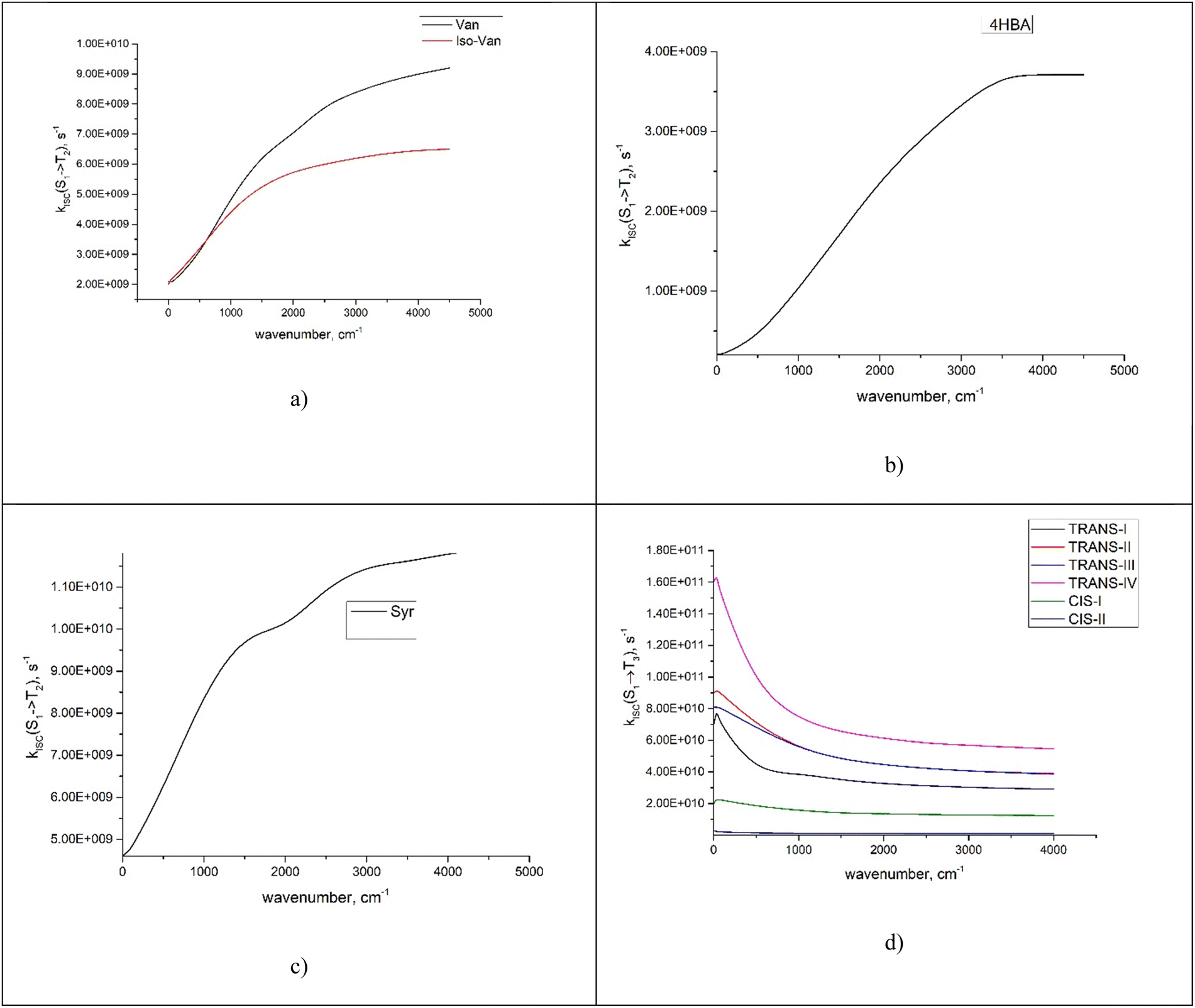 | ||
| Fig. 3 The dependence of kISC(S2 → T2) (a)–(c) for Van, IsoVan, 4HBA, Syr and kISC(S1 → T3) (d) for ConAld and Syr on the vibrational energy of the initial state. | ||
| Molecule | Adiabatic energy gap (cm−1) | Main acceptors ω(y) in cm−1 |
|---|---|---|
| Van | ΔE(S2–T2) = 5800 | 1320(0.08), 1573(0.09) |
| IsoVan | ΔE(S2–T2) = 5860 | 1386(0.09), 1595(0.22) |
| 4HBA | ΔE(S2–T2) = 9500 | 1386(0.65), 1594(0.21), 1695(0.12) |
| Syr | ΔE(S1–T3) = 5100 | 1442(0.15), 1485(0.16), 1607(0.16), 1657(0.14) |
| ConAld | ΔE(S1–T3) = 1630 | 1252(0.16), 1410(0,40), 1532(0.12), 1571(0.10), 1650(0.11) |
From Fig. 3, it can be observed that the dependence of kISC on vibrational excitation is notably different only for ConAld. In this case, kISC rapidly reaches a maximum and then decreases, whereas for the other molecules, it increases and then reaches saturation. We previously reported this saturation behavior for Van.18 Now, we observe that it occurs for all studied molecules with an adiabatic energy gap larger than that of ConAld. It is important to note that all cis-isomers of ConAld exhibit significantly lower kISC values, with a much weaker dependence on vibrational excitation energy compared to the trans-isomers.
Van and IsoVan have similar vibrational excitation energy dependences for the region 0–1000 cm−1, but the value of kISC is larger (and depends more strongly on the wavelength) for Van than for IsoVan at larger energies. In any case, both reach saturation at around 5000 cm−1.
To further clarify the dependence of kISC on vibrational excitation, we constructed the relationship between the integral 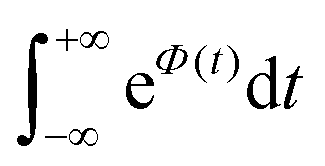 , which can be associated with the effective density of the final T(nπ*) state, occupancies of different energy levels of the vibrational modes in S(ππ*), and the adiabatic energy gap. This is shown in Fig. 4, with different colors associated with energy gaps of 500–5000 cm−1. Here, we used the characteristics of accepting modes (ω(y)) corresponding to Van. It can be seen from Fig. 4 that if the adiabatic energy gap is larger than 2500 cm−1, the effective density first increases as a function of vibrational energy and then reaches saturation. This behavior can be explained by the existence of good and bad acceptors for kISC.20 In the ISC process, the energy ΔE + ħmiωi of the S(ππ*) state should be transferred into the excitation of vibrational modes of the T(nπ*) state, ħniωi (where ni and ωi are the vibrational quantum numbers and frequencies, respectively, of the T(nπ*) state). There are good and bad acceptor modes, corresponding to large and small yi values, respectively. The good acceptors make large contributions to the
, which can be associated with the effective density of the final T(nπ*) state, occupancies of different energy levels of the vibrational modes in S(ππ*), and the adiabatic energy gap. This is shown in Fig. 4, with different colors associated with energy gaps of 500–5000 cm−1. Here, we used the characteristics of accepting modes (ω(y)) corresponding to Van. It can be seen from Fig. 4 that if the adiabatic energy gap is larger than 2500 cm−1, the effective density first increases as a function of vibrational energy and then reaches saturation. This behavior can be explained by the existence of good and bad acceptors for kISC.20 In the ISC process, the energy ΔE + ħmiωi of the S(ππ*) state should be transferred into the excitation of vibrational modes of the T(nπ*) state, ħniωi (where ni and ωi are the vibrational quantum numbers and frequencies, respectively, of the T(nπ*) state). There are good and bad acceptor modes, corresponding to large and small yi values, respectively. The good acceptors make large contributions to the 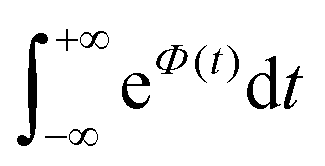 integral. When the adiabatic energy gap is less than 2500 cm−1, the accepting modes (1200–1700 cm−1) are not efficient in accepting the additional vibrational energy from the S(ππ*) state. Consequently, we tend to observe an initial maximum, followed by a decrease in the kISC value. However, the detailed dependence of
integral. When the adiabatic energy gap is less than 2500 cm−1, the accepting modes (1200–1700 cm−1) are not efficient in accepting the additional vibrational energy from the S(ππ*) state. Consequently, we tend to observe an initial maximum, followed by a decrease in the kISC value. However, the detailed dependence of 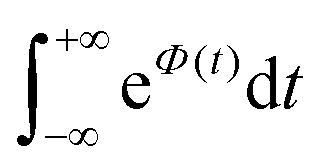 on the yi and ΔE is more complicated, and can even have a saddle point, as seen in Fig. 3c. Additionally, it is important to note that the absolute value of the effective density at a given vibrational energy decreases with an increasing adiabatic energy gap, which is in complete agreement with the energy gap law for the radiationless rate constant of electronic transitions.20
on the yi and ΔE is more complicated, and can even have a saddle point, as seen in Fig. 3c. Additionally, it is important to note that the absolute value of the effective density at a given vibrational energy decreases with an increasing adiabatic energy gap, which is in complete agreement with the energy gap law for the radiationless rate constant of electronic transitions.20
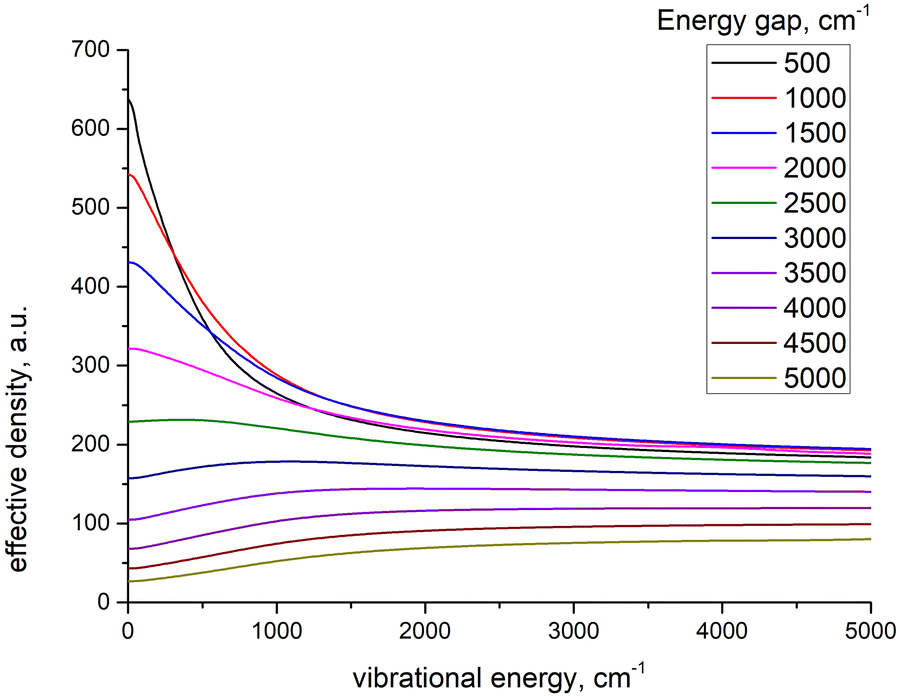 | ||
| Fig. 4 The model dependence of effective density of final state on the vibrational energy of initial state at different energy gaps. | ||
Thus, the wavelength dependence of kISC depends primarily on the adiabatic energy gap between the electronic states, with 2500 cm−1 as a key threshold (with different behavior for gaps larger and smaller than this value). As ΔE is the key parameter for the studied process, we carried out additional calculation of ΔE using the wb97xD to test how sensitive our results are to the choice of method. The result were ΔE(S1–T3) = 1700 cm−1 and 〈S1|HSO|T3〉 = 140 cm−1 for ConfAld, ΔE(S2–T2) = 5551 cm−1 and 〈S2|HSO|T2〉 = 10 cm−1 for Van, and ΔE(S2–T2) = 8700 cm−1 and 〈S2|HSO|T2〉 = 20 cm−1 for 4HBA. These results are close to the values calculated by CAM-B3LYP (see e.g.Table 2).
Also, we can speculate that the presence of a double bond increases the π-electronic conjugation in ConAld. Usually, this reduces the exchange energy, and finally the energy gap between the singlet and triplet states.20–24 Thus, the presence of a double bond in the structure of ConAld can cause (among the molecules studied here) a unique wavelength dependence for the ISC process, and ultimately for its photochemistry.
4.4. The wavelength dependence on the experimental quantum yield for loss
Experimental results for quantum yields of phenolic carbonyl loss, Φloss, are shown in Fig. 5. For most structures, Φloss consistently decreases with increasing wavelength. Syr exhibits an approximately constant Φloss in the range from 325–340 nm, with a continued decrease at longer wavelengths. Negligible loss for 4HBA was observed for wavelengths above 320 nm, likely due to a decrease in Φloss and minimal photon absorption, so Φloss for 4HBA is not reported above 320 nm. Φloss for ConAld shows a single, well-defined peak near 325 nm.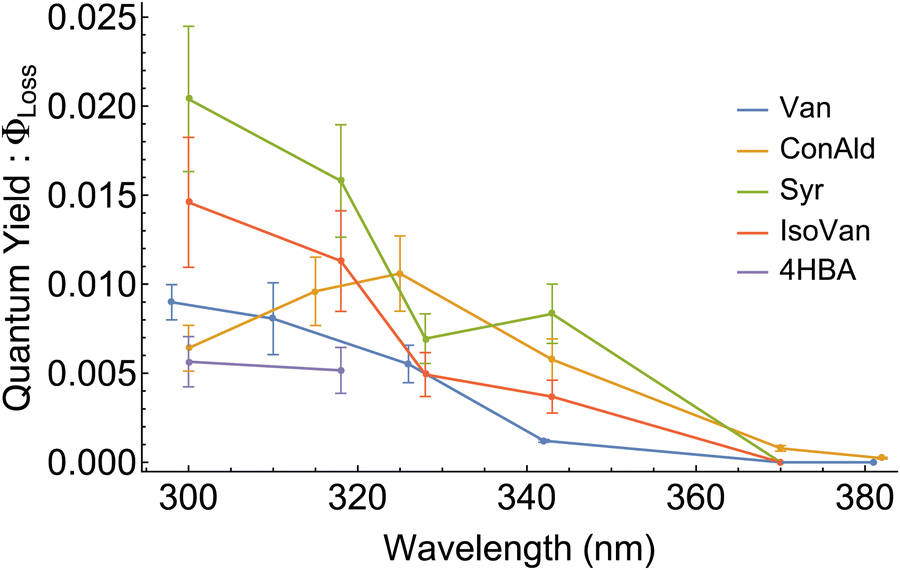 | ||
| Fig. 5 Quantum yield for loss, Φloss, measurements for the 5 structural variations of phenolic carbonyls studied. | ||
We can now qualitatively compare the experimental measurements with the theoretical results. The comparison is shown in Fig. 6. If we assume that loss processes are dominated by triplet state formation, then the kISC should be correlated with Φloss for all molecules. Indeed, only ConAld has a clear maximum in the Φloss dependence. kISC(S1–T3) for ConAld also has a maximum in its dependence on the vibrational energy of the initial state for all its conformers. The Φloss dependences of the other molecules do not show a peak, and tend to saturate at short wavelengths, similar to their kISC. The only deviation occurs for Syr near 325nm, where Φloss is relatively constant or possibly decreases slightly, but moving to shorter wavelengths Φloss for Syr shows continued increase and subsequent saturation. Analogously, the kISC vibrational excitation energy dependence for Syr has a saddle point at 2000 cm−1, with a plateau between 2000 cm−1 and 3000 cm−1 as seen from Fig. 3c. At higher energies, further increase followed by saturation are observed.
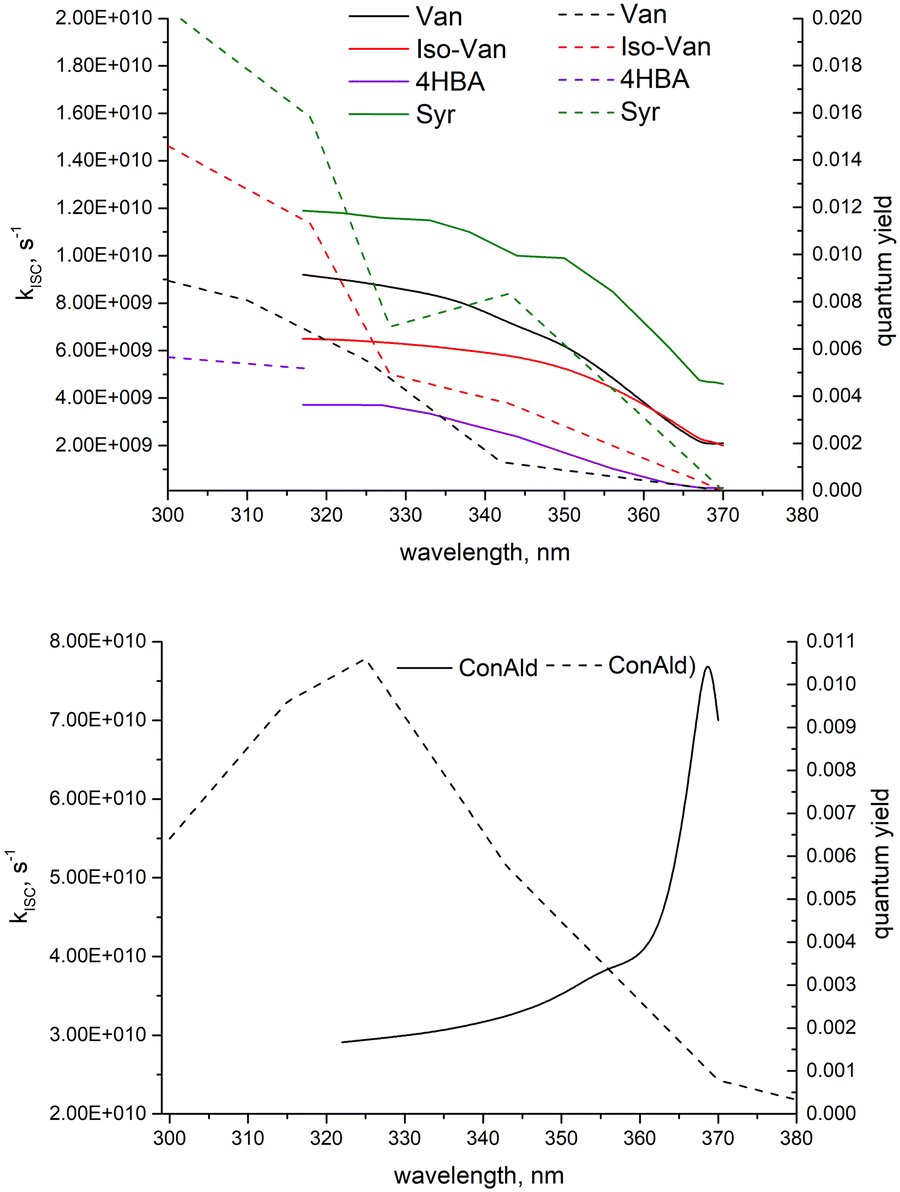 | ||
| Fig. 6 The comparison between the measured dependence of quantum yields of phenolic carbonyl loss and calculated dependence of kISC on wavelength. The solid lines correspond to the calculated values and the dashed lines correspond to the measured ones. | ||
It should be noted that for the lowest-energy conformer of ConAld, kISC reaches its maximum very rapidly, at an energy of only 100 cm−1. In contrast, Φloss increases from 380 nm to 325 nm, corresponding to an energy difference of approximately 4450 cm−1. This additional broadening, compared to the wavelength dependence of kISC, may be due to the aqueous environment of the molecule, which cannot be fully accounted for in our theoretical model. Nevertheless, the model successfully reproduces the overall qualitative trend in the dependence of Φloss, characterized by a single maximum, which is not observed for other phenolic carbonyl molecules.
Thus, the comparison between the measured dependence of quantum yields of phenolic carbonyl loss and calculated dependence of kISC on wavelength supports the hypothesis that transitions to the triplet state indeed dictate their photochemical loss.
5. Summary and conclusion
Quantum chemical calculations were employed to construct Jablonski diagrams for several phenolic carbonyls: vanillin, iso-vanillin, 4-hydroxybenzaldehyde, syringaldehyde, and coniferyl aldehyde. These molecules can be present in Earth's atmosphere due to forest fires and participate in photochemical reactions within water droplets and aqueous aerosol particles, leading to the formation of brown carbon which influences the climate. It has been shown that, following photon absorption to the first accessible (ππ* type) singlet state, denoted here S(ππ*), these molecules efficiently transition to triplet states via the intersystem crossing (ISC) mechanism, at rates of 109–1010 s−1. Although the ISC process can in principle proceed through multiple ISC channels due to the presence of several lower-lying triplet states, a single channel dominates for each molecule. For vanillin, iso-vanillin and 4-hydroxybenzaldehyde, this dominant channel is S1 → T2, whereas for coniferyl aldehyde and syringaldehyde, it is S1 → T3.For the dominant ISC channel, the dependence of kISC on the vibrational excitation energy of S(ππ*) was computed, and compared with the wavelength dependence of photochemical loss. Although a relatively simple model was used, accounting only for intramolecular nonradiative electronic transitions, the overall trend of both dependencies is similar. All but one of the molecules investigated exhibit saturation in the dependence of kISC and Φloss on the photon wavenumber or the vibrational excitation energy of the S1 state. The exception is coniferyl aldehyde, where a single maximum and a subsequent monotonic decrease are observed with increasing wavenumber. This behavior may be attributed to the fact that addition of a substituent with a double bond in the phenolic carbonyl structure, such as in coniferyl aldehyde, increases π-electron conjugation, thereby reducing the exchange energy and the adiabatic energy gap between S1 and the target triplet state. For small energy gaps, the classical acceptor modes of the ISC process, with an average frequency of 1400 cm−1 and a Huang–Rhys factor ∼0.3, may be ineffective, resulting in a low effective density of final states.20 Conversely, at larger energy gaps, the excitation of these modes leads to the formation of a highly effective density of states, and the dependence becomes more pronounced. Indeed, quantum chemical calculations clearly demonstrate that the adiabatic gap in coniferyl aldehyde is significantly smaller (1700 cm−1) compared to the other investigated phenolic carbonyls; the set of acceptor modes is nearly the same as in these molecules, with characteristic frequencies of 1200–1600 cm−1 and y = 0.1–0.3. Thus, the primary parameter distinguishing the different types of ISC dependencies and, consequently, Φloss, is the magnitude of the adiabatic gap. We have shown that for adiabatic gaps greater than 2500 cm−1, growth and saturation are observed, while for smaller gaps, a maximum followed by a decrease with increasing photon wavenumber is observed.
Author contributions
All authors have discussed the results and contributed to the final manuscript.Data availability
Quantum chemical software utilized: (1) the optimized molecular structures, excitation energies, and matrix elements of spin–orbital coupled interaction operators were computed using GAUSSIAN 16 [https://gaussian.com/gaussian16/] and MOLSOC [https://github.com/gaox-qd/pysoc]. The results of these calculations are compiled and discussed in detail in the main text of the article. The molecular structures are given in zip-file. (2) The intramolecular rate constants were determined using proprietary code developed by Valiev and colleagues: [https://rsc.66557.net/en/content/articlelanding/2020/cp/d0cp03231j]. (3) The working expressions for intramolecular rate constants, along with the initial parameters, are provided and discussed within the main text. (4) All measured photophysical values are reported in the main text of the article, with detailed measurement procedures described.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Academy of Finland through project 340582 and 346369. We thank CSC – IT Center for Science, Finland for computer time.References
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. De Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS.
- C. G. Nolte, J. J. Schauer, G. R. Cass and B. R. T. Simoneit, Environ. Sci. Technol., 2001, 35, 1912–1919 CrossRef CAS PubMed.
- M. V. Misovich, A. Priyadarshani Silva Hettiyadura, W. Jiang, Q. Zhang and A. Laskin, ACS Earth Space Chem., 2021, 5, 1983–1996 CrossRef CAS.
- R. F. Hems, E. G. Schnitzler, C. Liu-Kang, C. D. Cappa and J. P. D. Abbatt, ACS Earth Space Chem., 2021, 5, 722–748 CrossRef CAS.
- Y. L. Sun, Q. Zhang, C. Anastasio and J. Sun, Atmos. Chem. Phys., 2010, 10, 4809–4822 CrossRef CAS.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Atmos. Chem. Phys., 2014, 14, 13801–13816 CrossRef.
- L. Yu, J. Smith, A. Laskin, K. M. George, C. Anastasio, J. Laskin, A. M. Dillner and Q. Zhang, Atmos. Chem. Phys., 2016, 16, 4511–4527 CrossRef CAS.
- D. Vione, A. Albinet, F. Barsotti, M. Mekic, B. Jiang, C. Minero, M. Brigante and S. Gligorovski, Atmos. Environ., 2019, 206, 197–207 CrossRef CAS.
- L. Ma, C. Guzman, C. Niedek, T. Tran, Q. Zhang and C. Anastasio, Environ. Sci. Technol., 2021, 55, 5772–5781 CrossRef CAS.
- W. Jiang, M. V. Misovich, A. P. S. Hettiyadura, A. Laskin, A. S. McFall, C. Anastasio and Q. Zhang, Environ. Sci. Technol., 2021, 55, 5199–5211 CrossRef CAS.
- T. Felber, T. Schaefer, L. He and H. Herrmann, J. Phys. Chem. A, 2021, 125, 5078–5095 CrossRef CAS PubMed.
- S. Rossignol, K. Z. Aregahegn, L. Tinel, L. Fine, B. Nozière and C. George, Environ. Sci. Technol., 2014, 48, 3218–3227 CrossRef CAS PubMed.
- C. Anastasio, B. C. Faust and C. Janakiram Rao, Environ. Sci. Technol., 1997, 31, 218–232 CrossRef CAS.
- J. D. Smith, H. Kinney and C. Anastasio, Atmos. Environ., 2016, 126, 36–44 CrossRef CAS.
- Ó. Guzmán-Méndez, M. M. Reza, B. Meza, J. Jara-Cortés and J. Peón, J. Phys. Chem. B, 2023, 127, 5655–5667 CrossRef.
- R. Ossola, R. Gruseck, J. Houska, A. Manfrin, M. Vallieres and K. McNeill, Environ. Sci. Technol., 2022, 56, 13449–13460 CrossRef CAS PubMed.
- T. A. Gawargy, B. Wang and J. C. Scaiano, Photochem. Photobiol., 2022, 98, 429–433 CrossRef CAS PubMed.
- G. T. Drozd, T. Weltzin, S. Skiffington, D. Lee, R. Valiev, T. Kurtén, L. R. Madison, Y. He and L. Gargano, Environ. Sci., 2024, 4, 509–518 CAS.
- R. Ossola, R. Gruseck, J. Houska, A. Manfrin, M. Vallieres and K. McNeill, Environ. Sci. Technol., 2022, 56, 13449–13460 CrossRef CAS PubMed.
- E. S. Medvedev and V. I. Osherov, Radiationless Transitions in Polyatomic Molecules, Springer-Verlag, Berlin, 1995 Search PubMed.
- O. K. Bazyl’, V. Ya. Artyukhov, G. V. Mayer, P. P. Pershukevich, M. V. Bel’kov, O. I. Shadyro and S. N. Samovich, Opt. Spectrosc., 2019, 127, 242–250 CrossRef.
- V. G. Plotnikov, Int. J. Quant. Chem., 1979, 16, 527–554 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- R. R. Valiev, B. S. Merzlikin, R. T. Nasibullin, A. Kurtzevitch, V. N. Cherepanov, R. R. Ramazanov, D. Sundholm and T. Kurtén, Phys. Chem. Chem. Phys., 2023, 25, 6406–6415 RSC.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118(15), 6975–7025 CrossRef CAS.
- R. R. Valiev, V. N. Cherepanov, G. V. Baryshnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 6121–6133 RSC.
- R. R. Valiev, V. N. Cherepanov, R. T. Nasibullin, D. Sundholm and T. Kurtén, Phys. Chem. Chem. Phys., 2019, 21, 18495–18500 RSC.
- R. R. Valiev, R. T. Nasibullin, V. N. Cherepanov, G. V. Baryshnikov, D. Sundholm, H. Ågren, B. F. Minaev and T. Kurtén, Phys. Chem. Chem. Phys., 2020, 22, 22314–22323 RSC.
- R. R. Valiev, R. T. Nasibullin, V. N. Cherepanov, A. Kurtsevich, D. Sundholm and T. Kurtén, Phys. Chem. Chem. Phys., 2021, 23, 6344–6348 RSC.
- R. R. Valiev, B. S. Merzlikin, R. T. Nasibullin, V. N. Cherepanov, D. Sundholm and T. Kurtén, Phys. Chem. Chem. Phys., 2024, 26, 4151–4158 RSC.
- M. Casida, Recent Advances in Density Functional Methods, Part I, World Scientific, Singapore, 1999, pp. 155–192 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- S. G. Chiodo and M. Leopoldini, Comput. Phys. Commun., 2014, 185, 676–683 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- https://www.csc.fi .
- R. J. Weber, H. Guo, A. G. Russell and A. Nenes, Nat. Geosci., 2016, 9, 282–286 CrossRef CAS.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I. T. Ku, V. Faye McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, Atmos. Chem. Phys., 2020, 20, 4809–4888 CrossRef CAS PubMed.
- K. J. Angle, D. R. Crocker, R. M. C. Simpson, K. J. Mayer, L. A. Garofalo, A. N. Moore, S. L. Mora Garcia, V. W. Or, S. Srinivasan, M. Farhan, J. S. Sauer, C. Lee, M. A. Pothier, D. K. Farmer, T. R. Martz, T. H. Bertram, C. D. Cappa, K. A. Prather and V. H. Grassian, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2018397118 CrossRef PubMed.
- G. Zheng, H. Su, S. Wang, M. O. Andreae, U. Pöschl and Y. Cheng, Science, 2020, 369, 1374–1377 CrossRef CAS PubMed.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I. T. Ku, V. Faye McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, Atmos. Chem. Phys., 2020, 20, 4809–4888 CrossRef CAS PubMed.
- C. T. Calvert and E. G. Schnitzler, ACS Earth Space Chem., 2023, 7, 490–500 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03501a |
| This journal is © the Owner Societies 2025 |