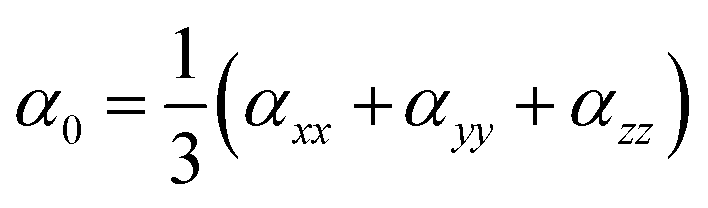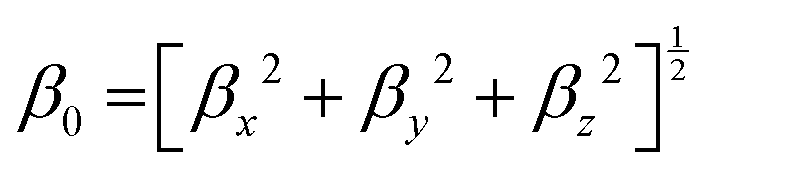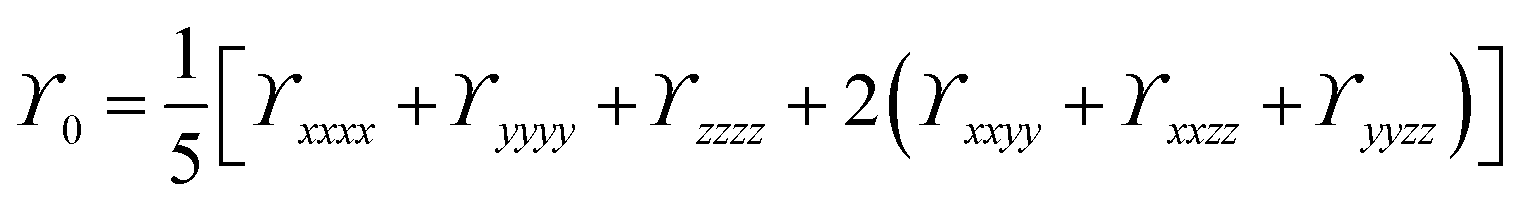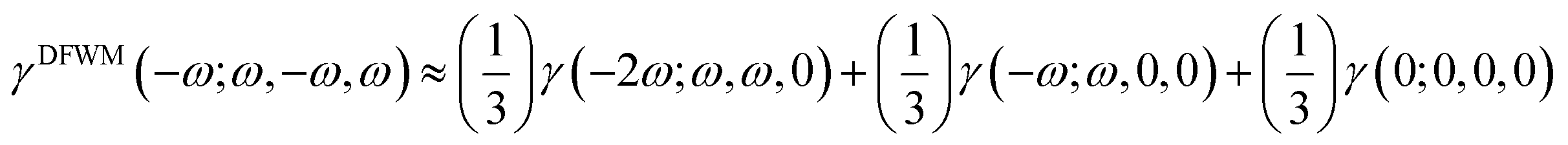Optimization of nonlinear properties of C6O6Li6-doped alkalides via group I/III doping for unprecedented charge transfer and advancements in optoelectronics†
Received
10th October 2024
, Accepted 22nd December 2024
First published on 23rd December 2024
Abstract
The design and synthesis of nonlinear optical (NLO) materials are rapidly growing fields in optoelectronics. Considering the high demand for newly designed materials with superior optoelectronic characteristics, we investigated the doping process of Group-IIIA elements (namely, B, Al and Ga) onto alkali metal (AM = Li, Na and K)-supported C6O6Li6 (AM@C6O6Li6) complexes to enhance their NLO response. The AM–C6O6Li6 complexes retained their structural features following interaction with the Group-IIIA elements. Interaction energies as high as −109 kcal mol−1 demonstrated the high thermodynamic stability of these complexes. An exceptional charge transfer behavior was predicted in these complexes, where the electronic density of the Group-III metals shifted toward the alkali metals, making these complexes behave as alkalides. The π conjugation of C6O6Li6 was found to withdraw excess electrons from the Group IIIA metals in these alkalides, which were subsequently transferred to the Group IA metals. The energy gap of the frontier molecular orbitals (FMOs) in the AM–C6O6Li6 complexes was notably reduced upon alkalide formation. UV-visible analysis explicitly showed a bathochromic shift in the alkalides. The first hyperpolarizability (β0) was calculated to confirm the NLO properties of these alkalides. B–C6O6Li6–K exhibited the highest β0 value of 1.75 × 105 au. The vibrational frequency-dependent first and second hyperpolarizability values illustrated an increase in hyperpolarizability at a frequency of 532 nm. A higher n2 value of 8.39 × 10−12 cm2 W−1 was obtained for B–C6O6Li6–Na at 532 nm. These results highlight the promising NLO response of the designed alkalides and their potential applications in the field of optics.
1. Introduction
Generation of excess electrons in a complex is a promising technique for enhancing the nonlinear optical (NLO) response of materials.1–8 NLO materials are on high demand due to their extensive applications in fields such as optics,7–9 electronics,10,11 bioimaging12,13 and memory storage devices.14,15 Various techniques are adopted to generate diffuse excess electrons, with the most widely known being the doping of electropositive metals onto a suitable substrate.16,17 The substrate holds the incoming diffuse excess electrons from the metals. After the diffusion of electrons, the geometric, electronic and optical properties are effectively tuned. To date, Group-IA18–21 metals, Group-IIA metals,22 transition metals23 and superalkalis24,25 have been used as sources for the generation of excess electrons. The metal-supported surfaces are further classified as electrides26,27 earthides,28 metalides29,30 and alkalides.31 In electrides, the excess electrons occupy the free space between the interacting metals and the surface, while in alkalides, the electrons shift to the alkali metals. Literature suggests that alkalides exhibit superior NLO responses. Extensive research has been performed on alkalides as effective NLO materials. For example, Sun et al. designed transition and alkali metal-doped adz alkalides, which exhibited a higher β0 value of 6.16 × 104 au. They further applied the same metal doping to TriPip222, where β0 effectively increased to 1.80 × 105 au.4 Li and coworkers reported alkaline earth metal-doped amine alkalides with β0 values up to 1.23 × 105 au.32 Li et al. designed alkali metal-doped graphene, graphdiyne and graphyne-based alkalides, with the alkali metal-doped graphdiyne exhibiting the highest β0 value of 3.93 × 105 au among these complexes.33
Recent literature reports on enhancement of NLO responses through the doping of metals on various surfaces is leading toward the development of new alkalides with better NLO properties. Chen et al. designed alkali metal-doped calix[4]pyrrole alkalides. A Li+@(calix[4]pyrrole)K− alkalide has a higher hyperpolarizability than that of the Li+@(calix[4]pyrrole)e− electride.34 Li and co-workers modelled alkali/superalkali-based calix[4]pyrrole alkalides. The β0 value of these alkalides was increased up to 3.47 × 103 au compared to pure calix[4]pyrrole.35 Li and colleagues suggested that an alkali metal-doped N3H3 complex may have enhanced NLO response because the hyperpolarizability of this complex is 6.27 × 104 au.36 Banerjee and Nandi designed alkalides based on alkali metal-doped calcium chain structures with extraordinary NLO properties with β0 ranging from 1.57 × 104 to 1.61 × 106 au.37 Ayub and co-workers explored judicious placement of alkali and alkaline earth metals on the hydrogen and fluorine faces of the F6C6H6 surface, respectively. A maximum β0 value of 2.91 × 104 au was notified in these complexes.28 Kang et al. observed the alkalide properties of alkali metal-doped open cage C50N5H5 fullerenes having good NLO response (β0 values up to 1.91 × 105 au).38 Wang et al. theoretically studied the NLO response of alkali and alkaline earth metal-doped Janus-type structures under an applied electric field and observed a high hyperpolarizability value in the range of 5.9 × 104 to 6.4 × 104 au.39 Sohaib et al. investigated the NLO properties of alkali metals doped with stacked Janus-type alkalides and reported a very large first hyperpolarizability of 5.13 × 107 au.40 Mahmod and colleagues worked on the alkali and alkaline earth metal-doped C6O6Li6 complexes where Mg–C6O6Li6–K has the highest β0 value of 1.75 × 105 au.41
Group-1A and group-IIA elements are effectively used as metal sources to acquire a high NLO response. Because these elements have low ionization potential and can easily donate excess electrons, which can enhance the NLO response of a system. However, the doping of Group-IIIA metals with Group-IA metals is not considered so far. Based on their individual doping influence on tuning the NLO response of different surfaces, we deemed to search their doping nature with both Group-I/III metals. We infer the withdrawal of diffuse excess electrons from Group IIA metals to alkali metals via the C6O6Li6 surface, which may give rise to the alkalide properties manifested in these investigated complexes. The alkalide properties of the Group-IA and Group-IIIA element-doped C6O6Li6 surface were evaluated. We hope that these investigated alkalides can be used as extraordinary NLO nanomaterials for the next-generation optics. C6O6Li6 is suggested to be a good supportive surface for alkali metals,42 alkaline earth metals,43 alkali and alkaline earth metals,41 transition metals,44 and super alkalis,45 which is used for optics,46 electronics,47 storage of hydrogen,48,49etc. Group-IIIA metals can also eject their electrons to generate diffuse excess electrons, and this system can be used as effective NLO materials. According to the literature, the ejection of electrons from boron atoms requires high-energy laser light, elevated temperatures, and UV photons.50–53 However, we propose that the current strategy of modeling novel alkalides through the doping of Group-IA and Group-IIIA metals on C6O6Li6 may offer a more energetically favorable approach. In this context, Group-IIIA metals can readily eject their electrons, which then accumulate on the alkali metals, thereby inducing alkalide properties in these complexes. The structural, electronic, and nonlinear optical (NLO) properties of these alkalides were investigated using density functional theory (DFT). Furthermore, we suggest that the NLO response of C6O6Li6 can be further enhanced through this doping strategy.
2. Computational methodology
The optimization and frequency analyses of individual C6O6Li6 and Group-IA & Group-IIIA metal-doped C6O6Li6 were carried out by the ωB97XD/6-31+G(d,p) method of DFT. All the simulations were executed using the Gaussian 09 package,54 whereas the results are analyzed using the GaussView 5.0 package.55 These analyses confirmed the energy minimization of all geometries with no imaginary frequency. Zero-point corrected vibrational energies were obtained from the analyses to calculate the interaction energy of all the complexes. The interaction energy of Group-IA & Group-IIIA metals with C6O6Li6 was obtained using eqn (1):| | | Eint = EG-IA/G-IIIAC6O6Li6 − (EC6O6Li6 + EG-IA/G-IIIA) | (1) |
Natural bond orbital (NBO) and electronic density difference (EDD) analyses were performed to estimate the charge transfer between the metals and C6O6Li6 in each alkalide. To understand the electronic properties, the energy difference between frontier molecular orbitals (EH–L) was calculated using eqn (2):The static second hyperpolarizability (γ0), the first hyperpolarizability (β0) and polarizability (α0) were calculated at ωB97XD/6-31+G(d,p) to estimate the NLO response. This ωB97XD density functional and 6-31+G(d,p) Pople basis set are important for the estimation of optical and nonlinear optical properties of complexes.56–59 Literature reveals the importance of density functionals in accurately describing the NLO properties of the metal-doped complexes based on the %HF exchange parameter in these functionals. Nowadays, computational chemists primarily apply ωB97XD to estimate the nonlinear optical properties of NLO nanomaterials. ωB97XD is a long-range corrected hybrid density functional with a dispersion correction density functional, which contains 100% HF exchange and performs exceptionally well for assessing NLO properties through the calculations of first hyperpolarizability. Among the Pople basis sets, especially medium-sized basis sets’ 6-31+G(d,p) gave better results and was suggested to be a suitable basis set for the estimation of the first hyperpolarizability.60 The α0, β0 and γ0 values of all complexes were obtained using the following equations:| | 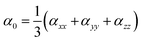 | (3) |
| | 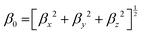 | (4) |
Here,
| βx = βxxx + βxyy + βxzz, βy = βyyy + βyzz + βyxx, βz = βzzz + βzxx + βzyy, |
| | 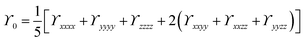 | (5) |
Frequency (ω)-dependent NLO properties of the complexes were also determined, which are important for experimentalists when they work on complexes in the lab work. For this purpose, the calculations were carried out at routinely used laser wavelengths of 532 nm and 1064 nm.61,62 Frequency-dependent hyperpolarizability contains the approximation of electro-optic Pockel's effect (EOPE) β(−ω; ω, 0) and second harmonic generation β(−2ω; ω, ω). Electric field-induced second harmonic generation (γ(−2ω; ω, ω, 0)), dc-Kerr effect γ(−ω; ω, 0, 0) and degenerate four-wave mixing (γDFWM(ω)) were calculated for the estimation of frequency-dependent second hyperpolarizability (γ(ω)). The mathematical equations for β(ω), SHG (βi), and EOPE (βi) were given using the following equations:| | | β(ω) = [βx2 + βy2 + βz2]1/2 | (6) |
| | | βi(SHG) = βiii(−2ω, ω, ω) + βijj(−2ω, ω, ω) + βikk(−2ω, ω, ω) | (7) |
| | | βi(EOPE) = βiii(−ω, ω, 0) + βijj(−ω, ω, 0) + βikk(−ω, ω, 0) | (8) |
The second hyperpolarizability coefficients including static (γ(0; 0, 0, 0)), ESHG (γ(−2ω; ω, ω, 0)) and dc-Kerr (γ(−ω; ω, ω, 0)) were also calculated. The degenerate four-wave mixing (γDFWM(ω)) equation was suggested by Tarazkar et al. in 2014,63 as given below.| | 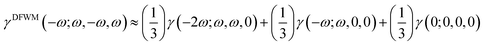 | (9) |
Additionally, the quadratic nonlinear refractive index (n2)64 of all complexes was obtained using eqn (10):| | | n2(cm2 W−1) = 8.28 × 10−23γDFWM | (10) |
We undergo a benchmark study of interaction energies and NLO properties using different methods to further ensure the accuracy of our method for the designed complexes. For this study, we also simulated all the complexes on LC-BLYP65 which is the long-range corrected density functional of the DFT. The simulations with LC-BLYP and ωB97XD are implemented with Dunning's aug-cc-PVDZ66 and aug-cc-PVTZ67 basis sets and Karlsruhe's def2-TZVP basis set.68,69 The results obtained at ωB97XD with 6-31+G(d,p) are more reliable, and these results are given in the main manuscript, while the results of the other basis sets are given in ESI† (ESI.† Tables S1 and S2). The aug-ccPVDZ and aug-cc-PVTZ basis sets show error when implemented on K containing complexes, as the atomic size of the K is out of reach of these dunning basis sets (aug-ccPVDZ and aug-cc-PVTZ basis sets). The total density of state spectra were generated through the GaussSam software to validate the energy states of molecular orbitals. Two-level model analysis was implemented to rationalize the internal factors responsible for the enhancement in NLO response. Furthermore, crucial excited states are analyzed using the TD-DFT method at the same level of theory.
3. Results and discussions
The pristine C6O6Li6 structure was evaluated first where six-membered rings of C, Li and O are linked together to form C6O6Li6 with a planar star-shaped structure. The point group symmetry of C6O6Li6 is D6h. The C–O, O–Li, and C–C bond lengths in C6O6Li6 are 1.38 Å, 1.79 Å, and 1.41 Å, respectively. Our calculated bond lengths are comparable to the one already reported in the literature.42
3.1. Structural and thermal stability of group-I and III metal-doped C6O6Li6 alkalides
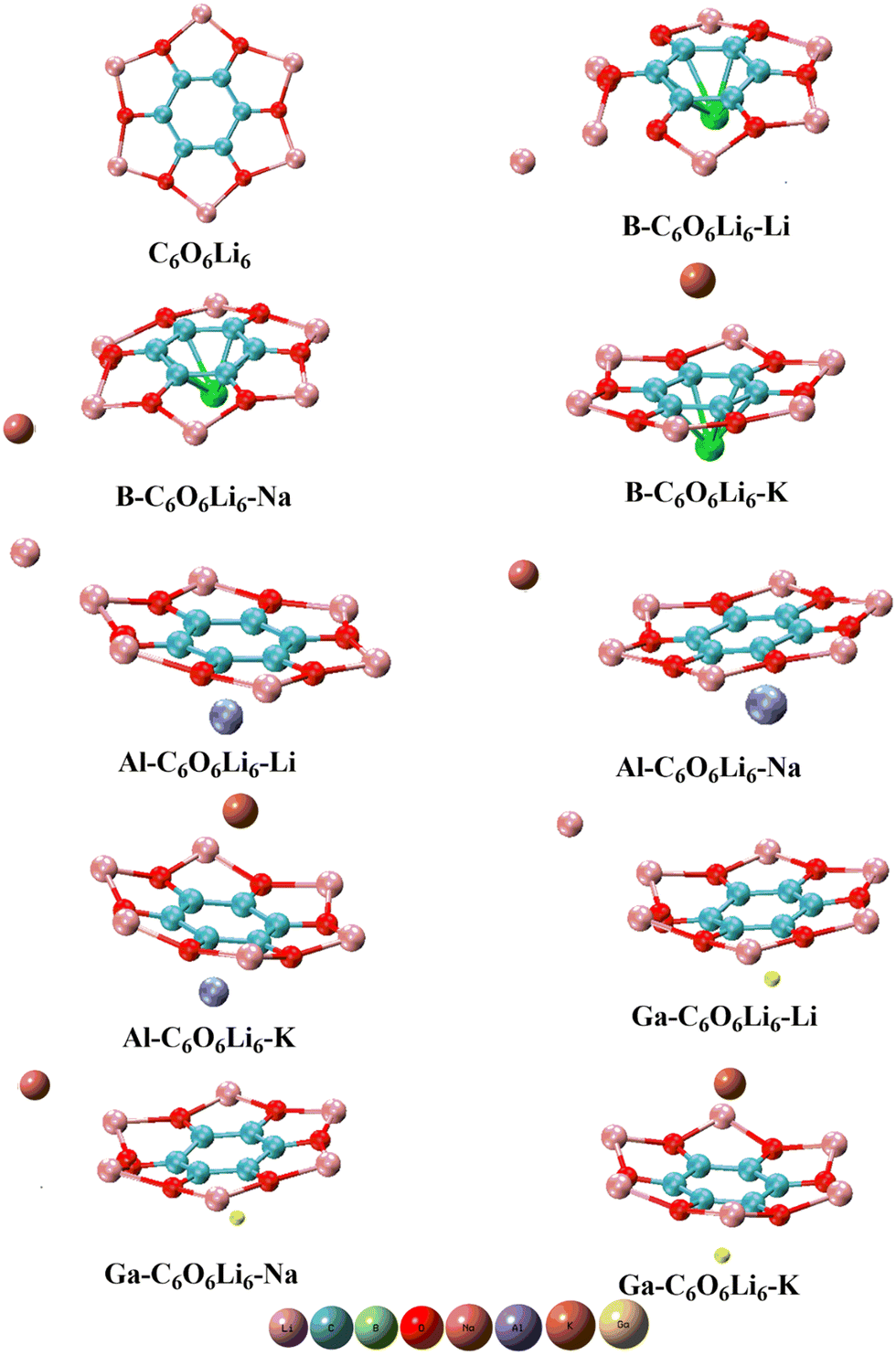
Nine complexes are designed by doping Group-I (K, Na, Li) and Group-III (Ga, Al, B) metals on the C6O6Li6 surface, and the structures of pristine and doped complexes are given in Fig. 1. Selected metals from Group-IA and Group-IIIA prefer to reside on the top of the central hexagonal ring of C6O6Li6 except Li and Na metal-doped Group-IIIA@C6O6Li6 alkalides. Li and Na metals have smaller atomic sizes than potassium and prefer to be adsorbed onto one side of C6O6Li6. The behavior is similar to alkali and alkaline earth metal-doped C6O6Li6 complexes.41 The interaction distances of Group-IIIA metals from the central position of C6O6Li6 are 1.60, 1.45, 1.46, 1.99, 2.04, 2.00, 2.11, 2.05, and 2.06 Å in B–C6O6Li6–K, B–C6O6Li6–Na, B–C6O6Li6–Li, Al–C6O6Li6–K, Al–C6O6Li6–Na, Al–C6O6Li6–Li, Ga–C6O6Li6–K, Ga–C6O6Li6–Na, and Ga–C6O6Li6–Li, respectively. Moving towards the adsorption sites of the alkali metals, it is noticed that the K metal holds the central position on C6O6Li6 (opposite Group-IIIA metals). The interaction distances of potassium from the central position of C6O6Li6 are 3.33, 3.24, and 3.15 Å in B–C6O6Li6–K, Al–C6O6Li6–K, and Ga–C6O6Li6–K, respectively. The average interaction distances of Li and Na metals from Li metals of the adsorption sides are 3.33, 3.45, 3.40, 3.50, 3.34 and 3.47 Å in B–C6O6Li6–Na, B–C6O6Li6–Li, Al–C6O6Li6–Na, Al–C6O6Li6–Li, Ga–C6O6Li6–Na, and Ga–C6O6Li6–Li, respectively. The interatomic distance of Group-IIIA metals is increased with the increase in atomic size. The outcomes are comparable to the research report on NLO response of alkali/alkaline earth metal-doped C6O6Li6 complexes, where the distance between both dopants and C6O6Li6 increases with the increase in the atomic size of alkali metals (from Li to K).41 However, the interatomic bond distance of alkali metals in respective complexes is decreased with the increase in atomic size. In the case of Group-IIIA metals (Ga, Al, and B), the lowest distance (1.45 Å) was calculated for B from the center of C6O6Li6 in B–C6O6Li6–Li. The highest interatomic distance (2.11 Å) was obtained for Ga from the center of C6O6Li6 in Ga–C6O6Li6–K. In the case of alkali metals (K, Na, and Li), the highest interatomic distance (6.23 Å) was obtained for Na from the center of C6O6Li6 in Al–C6O6Li6–Na, and the lowest interatomic distance (3.15 Å) was obtained for K from the center of C6O6Li6 in Ga–C6O6Li6–K (Table 1).
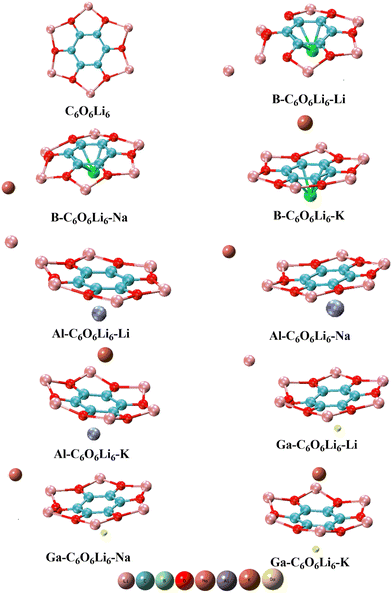 |
| | Fig. 1 Pristine C6O6Li6 and group-IA and group-IIIA metal-doped C6O6Li6 complexes. | |
Table 1 NBO charges on Group-IA metals (QG-IA in |e|), NBO charges on group-IIIA metals (QG-IIIA in |e|), interatomic distance of group-IIIA from the center of C6O6Li6 (dring-G-IIIA in Å, G-IIIA = Ga, Al, and B), interatomic distance of group-IA from the center of C6O6Li6 (dring-G-IA in Å, G-IA = K, Na, and Li), vertical ionization energy (VIE in eV) and interaction energies (Eint in kcal mol−1) of pristine C6O6Li6 and group-I and group-III metal-doped C6O6Li6–alkalides (group-IA = K, Na, and Li and group-IIIA = Ga, Al, and B)
| Pristine and group-I and group-III doping |
Q
G-IA
|
Q
G-IIIA
|
d
ring-G-IIIA
|
d
ring-G-IA
|
VIE |
E
int
|
| C6O6Li6 |
— |
— |
— |
— |
3.04 |
— |
| B–C6O6Li6–Li |
−0.29 |
0.11 |
1.46 |
3.33 |
3.75 |
−105.35 |
| B–C6O6Li6–Na |
−0.31 |
0.12 |
1.45 |
3.45 |
3.62 |
−104.66 |
| B–C6O6Li6–K |
−0.51 |
0.17 |
1.60 |
3.33 |
3.02 |
−88.13 |
| Al–C6O6Li6–Li |
−0.38 |
0.65 |
2.00 |
3.40 |
3.72 |
−97.41 |
| Al–C6O6Li6–Na |
−0.39 |
0.67 |
1.99 |
3.50 |
3.47 |
−97.12 |
| Al–C6O6Li6–K |
−0.47 |
0.69 |
2.04 |
3.24 |
3.14 |
−89.04 |
| Ga–C6O6Li6–Li |
−0.35 |
0.63 |
2.06 |
3.34 |
3.73 |
−109.13 |
| Ga–C6O6Li6–Na |
−0.37 |
0.63 |
2.05 |
3.47 |
3.57 |
−108.57 |
| Ga–C6O6Li6–K |
−0.46 |
0.68 |
2.11 |
3.15 |
3.12 |
−102.32 |
The C6O6Li6 structure retains its integrity during the doping of all elements, except for the small variations observed in B–C6O6Li6–Li and B–C6O6Li6–Na alkalides. The reason is the displacement of the Li atom of the C6O6Li6 surface towards the doped alkali metals. Li and co-workers observed a similar trend using their study on the interaction of Group-IIA metals with hexaammine.70 The point group symmetry of C6O6Li6 is changed to C1 symmetry after doping of Group-IA and IIIA metals. This change in symmetry was also observed by Kanis et al., and it plays an important role in the improvement of NLO response of a surface.71 This change in symmetry is common in almost all theoretical studies on NLO, specifically Wajid et al. observed such change during doping of Group-IIA43 and Group-IA42 metals on C6O6Li6.
The thermodynamic stability of a complex is inferred from the interaction energy (Eint). If a complex has high negative interaction energy, the complexation reaction is exothermic in nature and reflects the high thermodynamic stability of complexes. These properties confirmed the possibility of practical synthesis of these complexes.72 To estimate the thermal stability of a complex, we also calculated the interaction energy of all complexes (see Table 1).
Ga–C6O6Li6–Li, Ga–C6O6Li6–Na, Ga–C6O6Li6–K, Al–C6O6Li6–Li, Al–C6O6Li6–Na, Al–C6O6Li6–K, B–C6O6Li6–Li, B–C6O6Li6–Na, and B–C6O6Li6–K complexes have Eint of, −109.13, −108.57, −102.32, −97.41, −97.12, −89.04, −105.35, −104.66, and −88.13 kcal mol−1, respectively at ωB97XD by the 6-31+G(d,p) method. The negative Eint value of our designed complexes reflects the thermal stability and energetic favorability of complexation reactions. Pristine and doped complexes are also optimized by the LC-BLYP/aug-cc-PVDZ, ωB97XD/aug-cc-PVDZ, LC-BLYP/aug-cc-PVTZ, ωB97XD/aug-cc-PVTZ, LC-BLYP/def2-TZVP, and ωB97XD/def2-TZVP methods. A higher Eint value was obtained at the ωB97XD/6-31+G(d,p) level, which is given in main manuscript. The Eint values obtained by the other methods are given in the ESI† (ESI.† Table S1). The aug-ccPVDZ and aug-cc-PVTZ basis sets show error when implemented on K-containing complexes as the atomic size of K is out of reach of these dunning basis sets (aug-ccPVDZ and aug-cc-PVTZ basis sets).
Considering the interaction energies of both Group-IA metals, we observed a monotonic trend of decreasing interaction energy with the increase in the atomic number of Group-IA metals. The interaction energy decreases with the increase in the atomic size of the alkali metals, and subsequently their stability decreases. Among all metals, Li has the shorter interatomic distance from the adsorption side of the C6O6Li6 ring in each of Li–C6O6Li6@ Group-IIIA complexes, which is the reason for the strong interactions between Li and C6O6Li6@ Group-IIIA, and ultimately for the thermal stabilities of these Li-doped complexes. The trend of lowering interaction energy with the increase in atomic size is also observed during alkali and alkaline earth metal doping of the same C6O6Li6 surface.41 A nonmonotonic trend is seen among the Group-IIIA metals, and the interaction energy decreases from B to Al but increases in the case of Ga. The reason for the nonmonotonic behavior of the Group-IIIA metals is the smaller atomic size of B, which formed bond with C6O6Li6 that is validated from its shorter distance. Ga has a larger atomic size, and it can easily donate the electronic density and form stronger interactions with C6O6Li6, and these Ga–C6O6Li6–Alkali-IA alkalides have more interaction energy than others.
Overall, the Ga–C6O6Li6–Li complex is the most stable based on its highest interaction energy (−109.13 kcal mol−1). The atomic size of gallium is large, and it can easily donate electronic density to the C6O6Li6 surface. The surface becomes polarized and electron rich, and the interaction between two highly charged species is strong. This leads to a higher interaction energy in the case of Ga. However, smaller lithium metals interact on one side of the polarized C6O6Li6 surface to complete its outer shell. These stronger interactions are the reasons for the more chemical stability of the Ga–C6O6Li6–Li complex. Similar results are reported in the previous study of Group-IA73 and Group-IIA metal74-doped complexes. In alkali metal-doped gallium nitride nanocages, the stronger interaction is seen for lithium-doped complexes by Khurshid et al.75
3.2 NBO and EDD analyses of group-I/IIIA metal-doped C6O6Li6 alkalides
NBO analysis is used to understand the shifting of charge from the metal toward surface or vice versa. In all complexes, Group-IIIA metals have a positive charge that ranges from 0.11 to 0.69|e|. The highest charge (0.69|e|) for Al was obtained in Al–C6O6Li6–K and the lowest (0.11|e|) was obtained for Li in B–C6O6Li6–Li. Interestingly, the Group-IA metals have negative charges between −0.29 and −0.51|e|. The positive NBO values on Group-IIIA metals reflect that π conjugation of C6O6Li6 withdraws excess electrons from Group-IIIA metals in these alkalides. These excess electrons are then transferred to Group-IA metals and these metals have negative NBO values. This shift of excess electrons from Group-IIIA metals to Group-IA metals is based on electron push–pull mechanism. In the literature, it is mentioned that high energy laser light, high heat and UV-photons are required to eject electrons from the Boron atom,76 but our current strategy of metal doping with supportive C6O6Li6 makes it possible with high energetic and electronic feasibility. Both types of Group-IA metals get stable noble gas electronic configuration through this push–pull mechanism and form electronically stable alkalides. The negative charges on Group-IA metals justify their alkalide properties.
As we discussed above, doping of Group-I/IIIA metals causes variation in the dipole moment of C6O6Li6 which is zero in pristine form. When metals are doped on C6O6Li6, the shifting of charges occurs as a result of huge charge separation. This separation increases the dipole moment of the investigated alkalides, which also increases except B–C6O6Li6–K alkalides. Beside charge separation, the interatomic distance between Group-I/IIIA and surface (C6O6Li6) results in an increase in the dipole moment of alkalides compared to pristine C6O6Li6. The μ0 values of B–C6O6Li6–K, B–C6O6Li6–Na, B–C6O6Li6–Li, Al–C6O6Li6–K, Al–C6O6Li6–Na, Al–C6O6Li6–Li, Ga–C6O6Li6–K, Ga–C6O6Li6–Na and Ga–C6O6Li6–Li alkalides are 5.98, 16.46, 15.80, 5.00, 18.31, 16.83, 5.73, 16.61, and 15.58 D, respectively (Table 2). Al–C6O6Li6–Na has the highest μ0 value of 18.31 D among all alkalides because the charge separation in this alkalide is larger than that of other alkalides.
Table 2 First hyperpolarizability (β0 in au), polarizability (α0 in au), dipole moment (μ in Debye), HOMO–LUMO energy gap (EL–H in eV), energies of LUMO (ELUMO), and HOMO (EHOMO) of the pristine C6O6Li6 and group-I and group-III metal-doped C6O6Li6-alkalides (group-I = K, Na, and Li; group-III = Ga, Al, and B)
| Isolated and doped C6O6Li6 |
β
0
|
α
0
|
μ
|
E
L–H
|
E
LUMO
|
E
HOMO
|
| C6O6Li6 |
2.89 |
137 |
0.00 |
4.63 |
−0.24 |
−4.39 |
| B–C6O6Li6–Li |
1.11 × 104 |
410 |
15.80 |
3.49 |
−0.26 |
−3.75 |
| B–C6O6Li6–Na |
4.38 × 102 |
438 |
16.46 |
3.34 |
−0.28 |
−3.62 |
| B–C6O6Li6–K |
1.75 × 105 |
800 |
5.98 |
2.72 |
−0.29 |
−3.02 |
| Al–C6O6Li6–Li |
9.06 × 103 |
456 |
16.83 |
3.54 |
−0.18 |
−3.72 |
| Al–C6O6Li6–Na |
9.39 × 103 |
507 |
18.31 |
3.16 |
−0.30 |
−3.47 |
| Al–C6O6Li6–K |
7.50 × 104 |
690 |
5.00 |
2.88 |
−0.26 |
−3.14 |
| Ga–C6O6Li6–Li |
1.26 × 105 |
436 |
15.58 |
3.49 |
−0.23 |
−3.73 |
| Ga–C6O6Li6–Na |
7.49 × 103 |
629 |
16.61 |
3.31 |
−0.26 |
−3.57 |
| Ga–C6O6Li6–K |
7.17 × 104 |
231 |
5.73 |
2.88 |
−0.01 |
−3.12 |
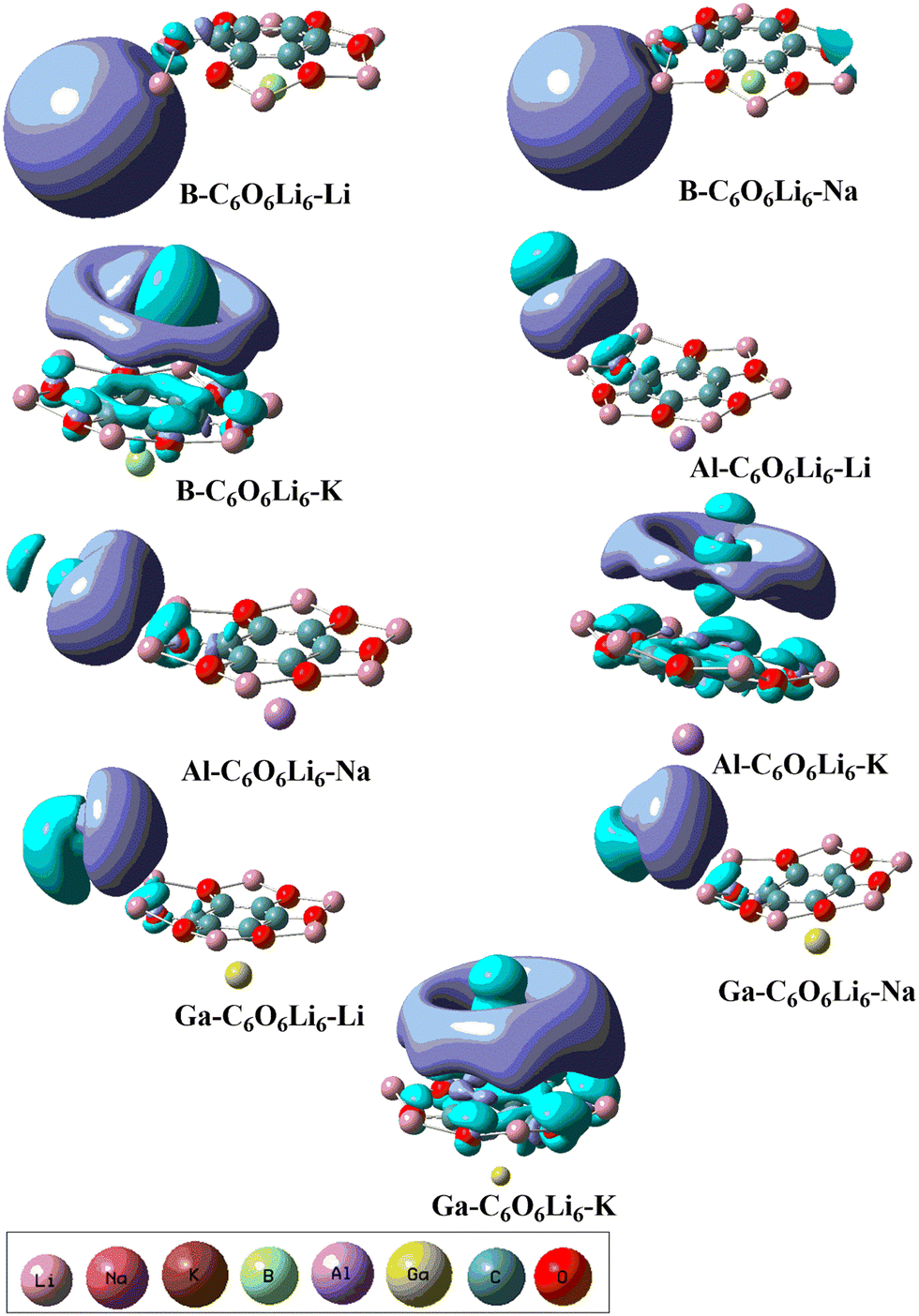
EDD analysis was also performed to understand the charge transfer in designed alkalides through qualitative pictorial representation (see Fig. 2). The two types of iso surfaces were generated on each of the complexes. Both are differentiated on the basis of purple color and blue color iso surfaces where purple represents the rich electron density region and cyan blue represents the poor electronic density region. The generation of iso surfaces between the interacting species in each of the complex represents the shifting of charge density after complexation. The cyan blue color iso surface is toward the Group-IA metals in each of the complexes, whereas the purple color iso surface has appeared towards C6O6Li6 that confirmed the charge transfer from G-IIIA-C6O6Li6 to the Group-IA metals.
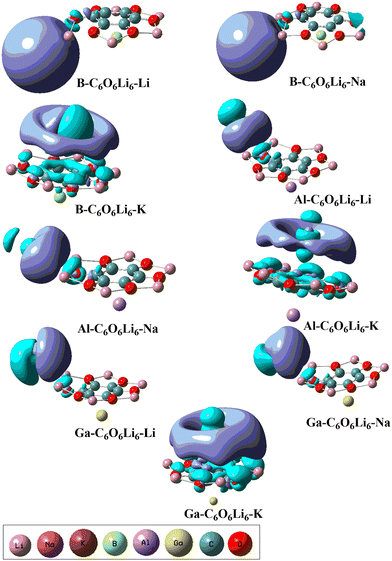 |
| | Fig. 2 EDD analysis for group-IA and group-IIIA metal-doped C6O6Li6 alkalides representing electron-rich and electron-poor iso surface regions at an isovalue of 0.04 au. | |
3.3. FMO analysis and alkalide properties of group-I/III metal-doped C6O6Li6
FMO analysis was performed to understand the electronic stability and conductivity of pristine C6O6Li6 and Group-I/IIIA metal-doped complexes (see Table 2). Pristine C6O6Li6 has an energy gap (EH–L) of 4.63 eV. When Group-I/IIIA metals are doped on pristine C6O6Li6, the EH–L value is reduced to be in the range of 2.72 eV to 3.54 eV. The lowest gap of 2.72 eV is seen for B–C6O6Li6–K. For the B–C6O6Li6–K complex, the EHOMO, ELUMO and EH–L values are −3.02, −0.29, and 2.72 eV, respectively. The decreased H–L gap is attributed to the increase in the energy of new occupied orbitals and the decrease in the energy of new unoccupied orbitals. The decrease in the energy gap confirmed the semiconductive properties of these alkalides with sufficient thermal stability. Li and coworkers in their work on the NLO response of N3H3 with alkali metals reported a similar type of behavior.77
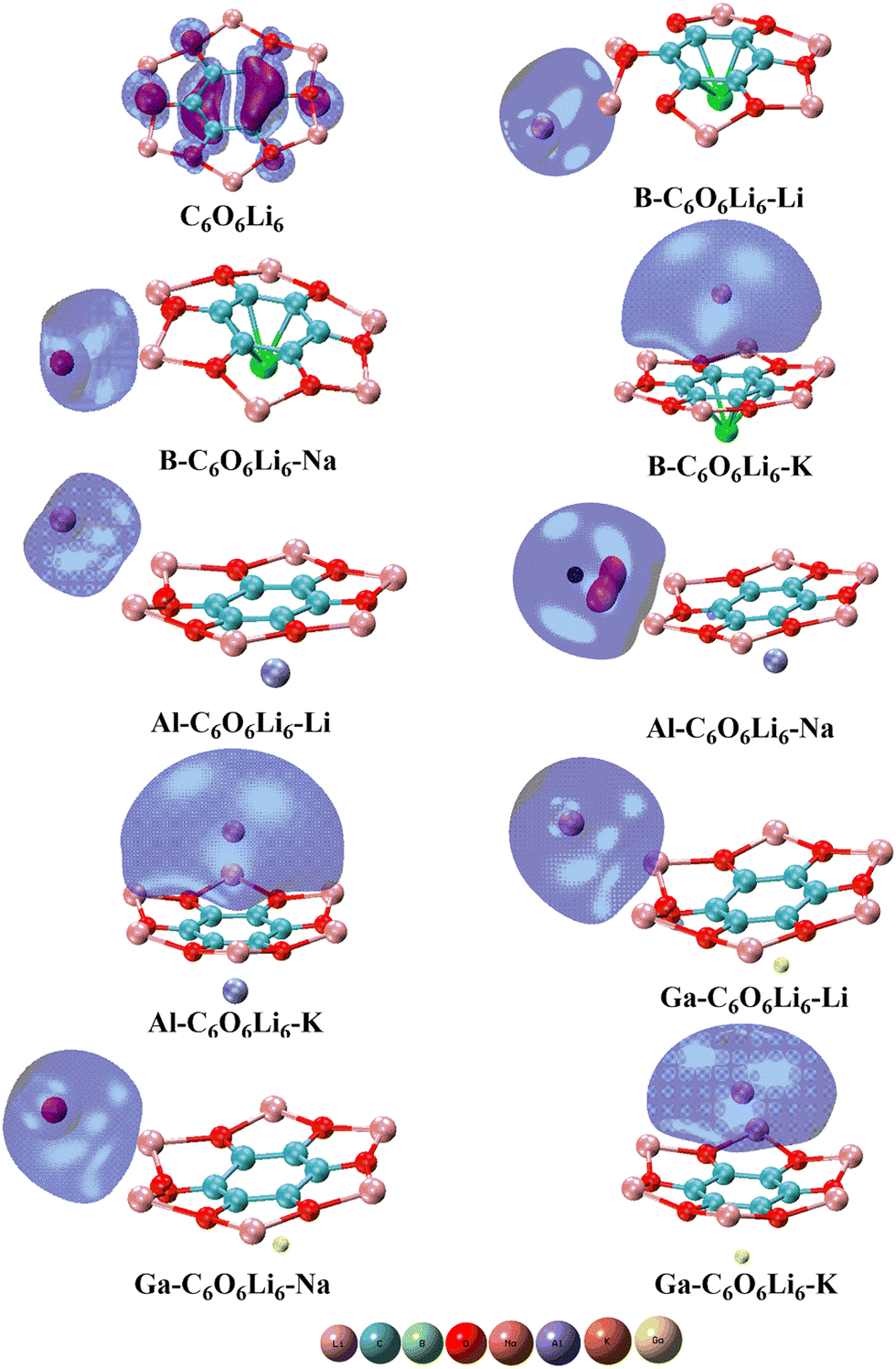
The isodensities of the highest occupied and the lowest unoccupied frontier orbitals are also studied, and their graphics are given in Fig. 3. It is observed that HOMO isodensities are present on Group-IA metals and LUMO isodensities are located on the Group-IIIA metals. The anionic alkali metals with HOMO densities reflect the alkalide behavior of the investigated complexes. The presence of HOMOs densities on metals justifies alkalide characteristics. Here, the electron push–pull mechanism is operated in each complex where the π conjugation of C6O6Li6 withdraws valence shell electrons from the Group-IIIA metals and these electrons act as excess electrons. Meanwhile, these excess electrons are shifted to the alkali metals doped on the other side of C6O6Li6. The outcomes are more comparable to the research work on alkalide properties of diamantanes with alkali metals.78
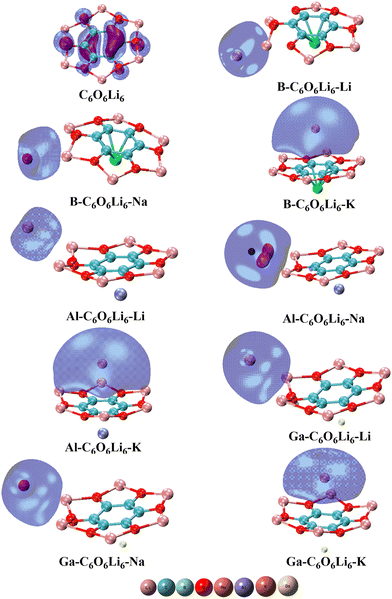 |
| | Fig. 3 Pristine C6O6Li6 and group-IA and group-IIIA metal-doped C6O6Li6 alkalides with their highest occupied molecular orbitals. | |
The stabilization of excess electrons on negatively charged alkali metals is a very crucial factor, which depends on the VIE. These alkalides have VIEs ranging from 3.02 to 3.75 eV to justify their electronic stability. As the VIE increases, it means that the alkalides are stable enough for further synthesis.79–81 Group-IA metals exhibit a monotonic decreasing trend in VIEs, whereas Group-IIIA metals display a nonmonotonic trend, with the VIE values decreasing from B to Al before increasing again as one moves towards Ga.
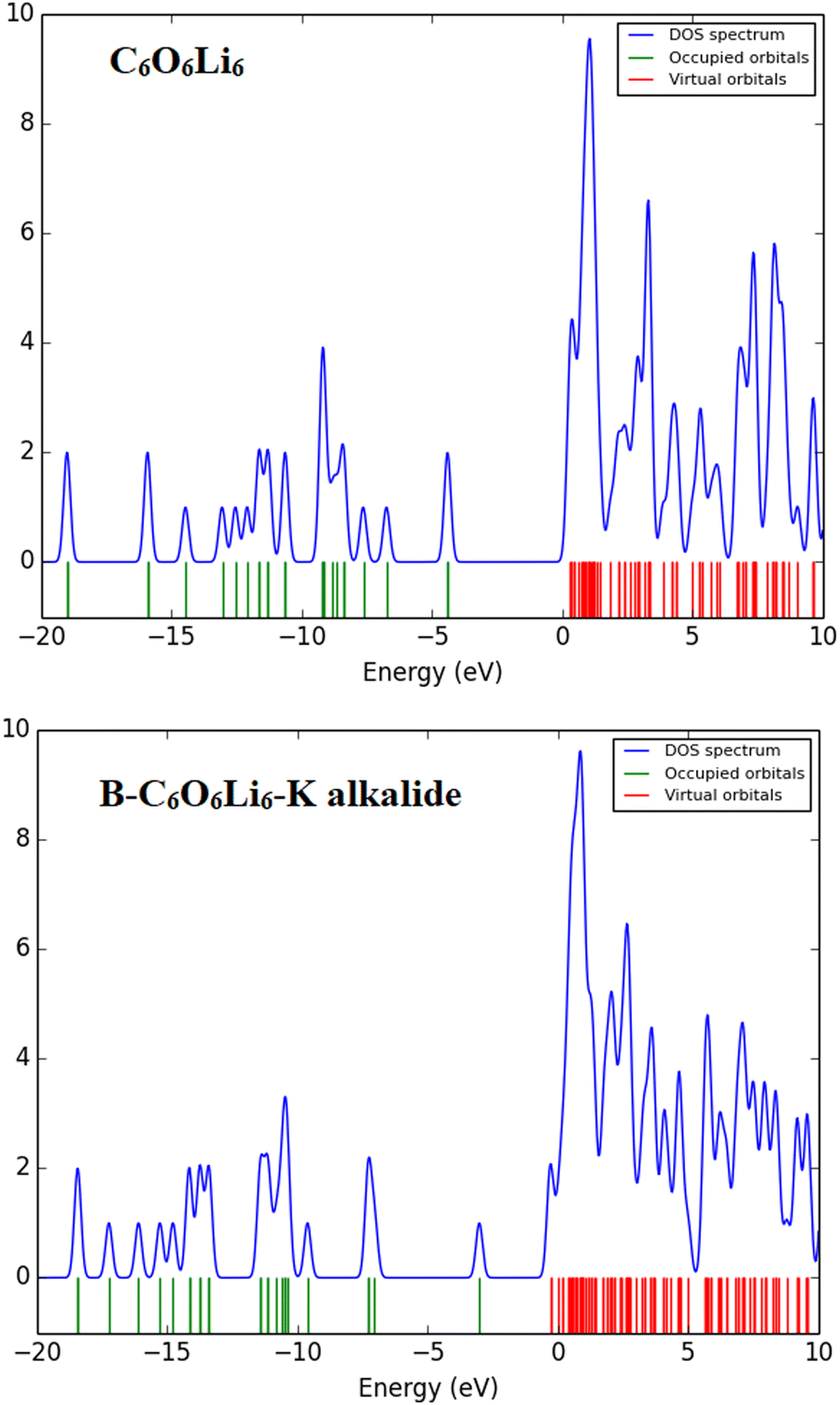
The TDOS spectra give information about the occupied and unoccupied orbitals with energy states and electronic interactions in complexes. The total density of states (TDOS) spectra of the designed complexes were studied. These spectra reconfirmed the change in the energy states after complexation compared to individual C6O6Li6. New energy states are generated, and energy gaps are reduced upon complexation, as discussed vide supra in FMO analysis. The intensity of peaks also increases after complexation, which justifies the electronic contribution in each complex, where the overlapping of peaks shows stronger interactions between the Group-IA and Group-IIIA metals and C6O6Li6 (see Fig. 4 and ESI.† Fig. S1).
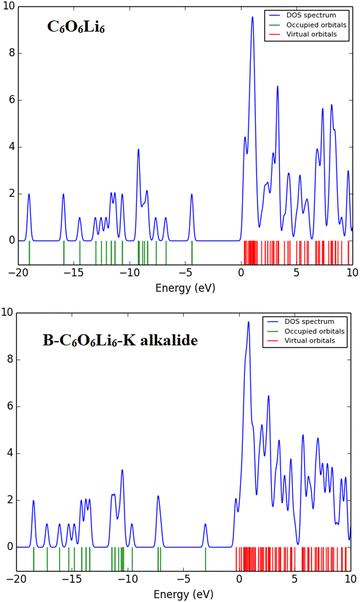 |
| | Fig. 4 Individual C6O6Li6 and B–C6O6Li6–K alkalides with total density of state spectra. | |
3.4. NLO analysis of group-IA/IIIA-doped C6O6Li6 alkalides
The geometrical and electronic properties of Group-IA/IIIA metal-doped C6O6Li6 alkalides validate the strong interaction between the selected metals and C6O6Li6. A significant amount of charge transfer occurred. These excess electrons are responsible for the extraordinary NLO response of these alkalides. We also calculated the important NLO parameters to estimate the NLO response of these alkalides such as polarizability (α0) and hyperpolarizability (β0). These parameters provide insights into the NLO response. The α0 and β0 values were calculated to estimate the NLO response of these alkalides, and the data are given in Table 2. The α0 value of Groups-IA/IIIA@C6O6Li6 alkalides is in the range of 231 to 800 au, which is remarkably higher than that of pristine C6O6Li6 (137 au). The B–C6O6Li6–K alkalide has the highest α0 (800 au) and the lowest α0 (231 au) values found for Ga–C6O6Li6–K. A large amount of charges are transferred (−0.51|e|) from B–C6O6Li6 to the K metal in B–C6O6Li6–K alkalides, which is the crucial factor responsible for the increase in polarizability.18 In the ongoing work, the α0 values reflect significant changes in the polarizability of each alkalide because of the interactions between metals and C6O6Li6. The polarizability increases with the increase in the atomic size of Group-IA metals in each alkalide. Nisar et al. also observed changes in polarizability values during their work on supramolecular assemblies of azobenzene and alkoxystilbazole molecules.82
Besides polarizability, when Groups-IA/IIIA metals are adsorbed on C6O6Li6, the β0 value is considerably increased compared to β0 (2.89 au) of pristine C6O6Li6. The increase in β0 notifies enhancement in the NLO response of the designed complexes. The β0 value is in the range of 4.38 × 102–1.75 × 105 au for all alkalides by the ωB97XD/6-31+G(d,p) method. The B–C6O6Li6–K alkalide has the highest β0 value of 1.75 × 105 au and B–C6O6Li6–Na has the lowest β0 value of 4.38 × 102 au. It is observed that the β0 value increases with the increase in the atomic size of alkali metals. The β0 results are similar to our previous work on NLO response of alkali metal-doped C6O6Li6 complexes, where the K@C6O6Li6 complex has the highest β0 value of 2.9 × 105 au compared to other complexes.42 We see in our present results that K-doped complexes have a higher β0 value. The second factor is the inverse relationship of energy gap with a β0 value, and the β0 value increases with the decrease in energy gap, as observed by Nouman and coworkers during working on alkali metal-doped 26 adamanzane.83 In our study, the B–C6O6Li6–K alkalide has the lowest energy gap of 2.72 eV, but, it has the highest β0 value of 1.75 × 105 au. Iqbal and coworkers depict from their results on alkaline earth metal-doped adamanzane that the large amount of charge transfer causes an increase in the polarizability of the alkali metal-doped complexes, which causes an increase in hyperpolarizability.84 We also observed that a large amount of charge (−0.51|e|) is transferred to the K metal in B–C6O6Li6–K alkalides, which increases the polarizability and hyperpolarizability of this complex. In the literature, the inverse relationship between the VIE and β0 values has been reported. VIE of a complex is inversely proportional to hyperpolarizability as observed by Asif et al. upon doping of superalkli with aminated graphdiyne.85 VIE is also inversely proportional to β0, and B–C6O6Li6–K has the lowest VIE of 3.02 eV and has the highest β0. Nisar et al. also observed a change in β0 values during their work on supramolecular assemblies of azobenzene and alkoxystilbazole molecules.86 We compared the results of our designed B–C6O6Li6–K complex with a number of designed metal-doped complexes reported in the recent literature (see Table 3). This comparative study justifies the better hyperpolarizability result of the B–C6O6Li6–K complex and identifies it to be a good NLO response-generating nanomaterial.
Table 3 Comparative analysis of the hyperpolarizability values of the reported complexes with our best designed B–C6O6Li6–K complex
| S. no. |
Complexes |
Hyperpolarizability (β0 in au) |
Ref. |
| 1. |
Superalkali-doped B38 |
3.5 × 104 |
87
|
| 2. |
Na@B12N12 |
1.89 × 104 |
88
|
| 3. |
Li3O@C32H15 graphene |
1.40 × 105 |
89
|
| 4. |
M1(26 adz)M2 where (M1 = M2 = Be, Mg and Ca) |
8.19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × 103 × 103 |
84
|
| 5. |
K@bicorannulenyl (C40H38) |
2.68 × 106 |
16
|
| 6. |
K@B12N11 nanocage |
1.3 × 104 |
90
|
| 7. |
Alkali metal@all-cis-1,2,3,4,5,6-hexafluorocyclohexane F6C6H6 |
2.91 × 104 |
28
|
| 8. |
Na@2N-atoms functionalized corannulene (C18N2H10) |
4.84 × 104 |
91
|
| 9. |
Ca2@C20 nanocage |
5.86 × 104 |
92
|
| 10. |
K@Boron phosphide |
4.41 × 105 |
17
|
| 11 |
B–C6O6Li6–K |
1.75 × 105 |
Current work |
The doped complexes are also analyzed by the LC-BLYP/aug-cc-PVDZ, ωB97XD/aug-cc-PVDZ, LC-BLYP/aug-cc-PVTZ, ωB97XD/aug-cc-PVTZ, LC-BLYP/def2-TZVP, and ωB97XD/def2-TZVP methods. The higher β0 values are obtained at the ωB97XD/6-31+G(d,p) level, which is given in main manuscript. The NLO parameters including β0, α0 and μ values of other methods are given in the ESI† (ESI.† Table S2). The μ values are between 14.43 and 17.51 Debye, and the α0 values are up to 507 au and the β0 values range from 8.89 × 102 to 1.90 × 104 au at ωB97XD/aug-cc-PVDZ. The μ values are up to 17.01 Debye, the α0 values are between 413 and 523 au and the β0 values range from 8.93 × 102 to 2.01 × 104 au at LC-BLYP/aug-cc-PVDZ. The μ values are between 15.99 and 18.31 Debye, the α0 values are up to 521 au and the β0 values range from 5.37 × 103 to 1.65 × 104 au at ωB97XD/aug-cc-PVTZ. The μ values are up to 18.12 Debye, the α0 values are between 388 and 503 au and the β0 values range from 4.04 × 103 to 1.76 × 104 au at LC-BLYP/aug-cc-PVTZ. The μ values are between 5.11 and 17.56 Debye, the α0 values are up to 762 au and the β0 values range from 1.16 × 103 to 1.46 × 105 au at ωB97XD/def2-TZVP. The μ values are up to 17.91 Debye, the α0 values are between 434 and 810 au and the β0 values range from 2.89 × 103 to 1.29 × 105 au at LC-BLYP/def2-TZVP. The aug-ccPVDZ and aug-cc-PVTZ basis sets show error when implemented on K-containing complexes as the atomic size of K is out of reach of these dunning basis sets (aug-ccPVDZ and aug-cc-PVTZ basis sets).
3.5. Two-level model of group-IA/IIIA metal-doped C6O6Li6 alkalides
A two-level model (βTLM) was applied to calculate the internal parameters, which are responsible for the variations in hyperpolarizability (β0) and ultimately can cause the variation in NLO response (Table 4). These internal parameters are variational dipole moment (Δμ), oscillation strength (f0) and crucial excitation energies. The equation for βTLM is given as follows:These changes in dipole moment between the ground state and the crucial excited state dipole moment (Δμ) and oscillation strength (f0) are directly correlated with βTLM and crucial excitation energies to this βTLM.93 The Δμ values lie between −0.08 and 1.71 Debye, whereas f0 ranges from 0.38 to 0.44. The trend of increase in f0 is opposite to β0. The trend of change in dipole moment is similar to β0 in each complex. In B–C6O6Li6, K-doped B–C6O6Li6 has a higher Δμ value (0.04 Debye) and it also has large β0 (1.75 × 105 au). Similarly in Al–C6O6Li6, K-doped Al–C6O6Li6 has a higher Δμ value (0.04 Debye) and it also has large β0 (7.50 × 104 au). Then in Ga–C6O6Li6, Li-doped Ga–C6O6Li6 has a higher Δμ value (1.76 Debye) and it also has large β0 (1.26 × 105 au).
Table 4 Maximum wavelength (λmax in nm), oscillating strength (f0), change in excitation energy (ΔE in eV), variation in dipole moment between ground and crucial excited states (Δμ in Debye), hyperpolarizability in au in the two-level model (βTLM in au) and hyperpolarizability (β0 in au) of pristine C6O6Li6 and Group-I and Group-III metal-doped C6O6Li6-alkalides (group-I = K, Na, and Li; group-III = Ga, Al, and B)
| Parameters |
λ
max
|
f
0
|
ΔE |
Δμ |
β
TLM
|
β
0
|
| C6O6Li6 |
515 |
0.03 |
2.42 |
0.01 |
0.000 |
2.89 |
| B–C6O6Li6–Li |
580 |
0.43 |
2.14 |
−0.14 |
−0.006 |
1.11 × 104 |
| B–C6O6Li6–Na |
606 |
0.38 |
2.05 |
−0.08 |
−0.004 |
4.38 × 102 |
| B–C6O6Li6–K |
1556 |
0.41 |
0.80 |
0.04 |
0.032 |
1.75 × 105 |
| Al–C6O6Li6–Li |
629 |
0.41 |
1.97 |
−0.1 |
−0.005 |
9.06 × 103 |
| Al–C6O6Li6–Na |
654 |
0.38 |
1.90 |
−0.09 |
−0.005 |
9.39 × 103 |
| Al–C6O6Li6–K |
1393 |
0.42 |
0.89 |
0.04 |
0.024 |
7.50 × 104 |
| Ga–C6O6Li6–Li |
602 |
0.40 |
0.83 |
1.71 |
0.076 |
1.26 × 105 |
| Ga–C6O6Li6–Na |
625 |
0.39 |
1.98 |
1.69 |
0.085 |
7.49 × 103 |
| Ga–C6O6Li6–K |
1375 |
0.44 |
0.90 |
1.06 |
0.640 |
7.17 × 104 |
The trend of decrease in excitation energy values is comparable to β0 in each complex. In B–C6O6Li6, K-doped B–C6O6Li6 has the lowest ΔE value (0.80 eV) and has the largest β0 (1.75 × 105 au). Similarly in Al–C6O6Li6, K-doped Al–C6O6Li6 has a lower ΔE value (0.89 eV) and it also has large β0 (7.50 × 104 au). Exceptional behavior is seen in Ga–C6O6Li6 where Li-doped Ga–C6O6Li6 has a lower ΔE value (0.83 eV) and a larger β0 value (1.26 × 105 au). The excitation energy plays a key role in increasing the hyperpolarizability of alkalides. The β0 value of B–C6O6Li6–K is 1.75 × 105 au, which has the lowest excitation energy of 0.80 eV among the investigated alkalides, which is in accordance with the two-level model. These results indicated that the excitation energy is responsible for the enhancement of NLO response. Overall, the increasing trend of βTLM is comparable to β0, and βTLM outcomes support our results. Similar results have also been previously reported for superalkali-doped graphdiyne by Kosar et al. in the literature.94
3.6. Vibrational frequency-dependent first hyperpolarizability of group-I/IIIA metal-doped C6O6Li6 alkalides
We also analyzed the frequency-dependent first hyperpolarizability to illustrate the vibrational frequency-dependent behavior of wave function on NLO response, as previously reported by Kirtman et al. in the literature.95 Two important coefficients including second-harmonic generation (SHG represented β(−2ω; ω, ω)) and electro optic Pockel effect (EOPE represented −β(−ω; ω, 0)) coefficients were obtained. These coefficients were calculated at two frequencies mostly used in laser technology,96,97 and their values are given in Table 5.
Table 5 Coefficients of vibrational frequency-dependent first hyperpolarizability (β(ω)), i.e., electro-optic Pockel's effect β0(−ω; ω, 0) and second harmonic generation β0(−2ω; ω, ω) with static first hyperpolarizability β0(0; 0, 0) in au calculated at ωB97XD/6-31+G(d,p) of pristine C6O6Li6 and group-I and group-III metal-doped C6O6Li6-alkalides (group-I = K, Na, and Li; group-III = Ga, Al, and B)
| Pristine and doped complexes |
Frequency |
β
0(0; 0, 0) |
β
0(−ω; ω, 0) |
β
0(−2ω; ω, ω) |
| C6O6Li6 |
0 |
2.89 × 100 |
|
|
| 532 |
|
9.20 × 102 |
3.63 × 101 |
| 1064 |
|
3.06 × 100 |
2.02 × 101 |
|
|
| B–C6O6Li6–Li |
0 |
1.11 × 104 |
|
|
| 532 |
|
2.41 × 107 |
8.62 × 105 |
| 1064 |
|
4.37 × 103 |
4.76 × 105 |
|
|
| B–C6O6Li6–Na |
0 |
4.38 × 102 |
|
|
| 532 |
|
2.02 × 106 |
9.12 × 104 |
| 1064 |
|
1.36 × 104 |
7.02 × 105 |
|
|
| B–C6O6Li6–K |
0 |
1.75 × 105 |
|
|
| 532 |
|
1.78 × 108 |
1.71 × 107 |
| 1064 |
|
4.98 × 106 |
8.84 × 106 |
|
|
| Al–C6O6Li6–Li |
0 |
9.06 × 103 |
|
|
| 532 |
|
2.28 × 107 |
7.45 × 104 |
| 1064 |
|
2.29 × 104 |
1.36 × 105 |
|
|
| Al–C6O6Li6–Na |
0 |
9.39 × 103 |
|
|
| 532 |
|
1.82 × 106 |
1.69 × 106 |
| 1064 |
|
3.87 × 104 |
5.42 × 105 |
|
|
| Al–C6O6Li6–K |
0 |
7.50 × 104 |
|
|
| 532 |
|
5.33 × 105 |
4.11 × 104 |
| 1064 |
|
5.67 × 105 |
3.15 × 105 |
|
|
| Ga–C6O6Li6–Li |
0 |
1.26 × 105 |
|
|
| 532 |
|
6.48 × 105 |
7.07 × 104 |
| 1064 |
|
1.39 × 104 |
4.21 × 105 |
|
|
| Ga–C6O6Li6–Na |
0 |
7.49 × 103 |
|
|
| 532 |
|
5.97 × 106 |
9.24 × 107 |
| 1064 |
|
1.02 × 104 |
3.05 × 105 |
|
|
| Ga–C6O6Li6–K |
0 |
7.17 × 104 |
|
|
| 532 |
|
6.94 × 105 |
7.31 × 104 |
| 1064 |
|
5.51 × 105 |
5.17 × 105 |
This Table illustrates that the change in frequency caused a prominent increase in first hyperpolarizability, which described a significant enhancement in NLO response at both frequencies (532 and 1064 nm). Static hyperpolarizability ranges from 4.38 × 102 to 1.75 × 105 au. The SHG values for all designed nine complexes are between 4.11 × 104 and 9.24 × 107 au and the EOPE values for all the designed nine complexes are between 5.33 × 105 and 1.78 × 108 au at 532 nm. The SHG values for all the designed nine complexes are between 1.36 × 105 and 8.84 × 106 au and the EOPE values for all the designed nine complexes are between 4.37 × 103 and 4.98 × 106 au at 1064 nm. The higher EOPE (9.24 × 107 au) and SHG (1.78 × 108 au) values were obtained for B–C6O6Li6–K and B–C6O6Li6–K complexes. These results described the prominent EOPE and SHG effects at a lower frequency of 532 nm.
3.7. Static and vibrational frequency-dependent second hyperpolarizability of group-I/IIIA metal-doped C6O6Li6 alkalides
We further elaborate our study by analyzing the static (γ(0)) and frequency-dependent second hyperpolarizability (γ(ω)). Two important coefficients including electric field-induced second-harmonic generation (γ(−2ω; ω, ω, 0)) represented by ESHG, and the dc Kerr effect (γ(−ω; ω, ω, 0)) coefficients were calculated at 532 and 1064 nm frequencies. The values of these coefficients are given in Table 6.
Table 6 Coefficients of vibrational frequency-dependent second hyperpolarizability (γ(ω)), i.e., electric field-induced second-harmonic generation (SHG) with β(−2ω; ω, ω, 0) term, the electro-optical Karr effect (EOKE) with (β(−ω; ω, 0, 0)) term with static second hyperpolarizability (γ(0)) in au, and nonlinear refractive index (n2 in cm W−1) calculated at ωB97XD/6-31+G(d,p) of pristine C6O6Li6 and group-I and group-III metal-doped C6O6Li6-alkalides (group-I = K, Na, and Li; group-III = Ga, Al, and B)
| Pristine and doped complexes |
Frequency |
γ
0(0; 0, 0, 0) |
γ
0(−ω; ω, ω, 0) |
γ
0(−2ω; ω, ω) |
n
2
|
| C6O6Li6 |
0 |
5.21 × 106 |
|
|
|
| 532 |
|
3.69 × 108 |
9.52 × 108 |
3.66 × 10−14 |
| 1064 |
|
3.12 × 107 |
4.04 × 107 |
2.12 × 10−15 |
|
|
| B–C6O6Li6–Li |
0 |
4.11 × 106 |
|
|
|
| 532 |
|
1.95 × 108 |
9.33 × 108 |
3.12 × 10−14 |
| 1064 |
|
2.28 × 107 |
4.44 × 107 |
1.97 × 10−15 |
|
|
| B–C6O6Li6–Na |
0 |
5.31 × 107 |
|
|
|
| 532 |
|
2.90 × 1011 |
1.39 × 1010 |
8.39 × 10−12 |
| 1064 |
|
1.26 × 109 |
1.13 × 1010 |
3.48 × 10−13 |
|
|
| B–C6O6Li6–K |
0 |
4.43 × 106 |
|
|
|
| 532 |
|
7.18 × 108 |
9.57 × 107 |
2.26 × 10−14 |
| 1064 |
|
2.35 × 107 |
1.02 × 108 |
3.59 × 10−15 |
|
|
| Al–C6O6Li6–Li |
0 |
6.25 × 106 |
|
|
|
| 532 |
|
1.15 × 109 |
1.99 × 1010 |
5.81 × 10−13 |
| 1064 |
|
4.69 × 107 |
2.77 × 108 |
9.11 × 10−15 |
|
|
| Al–C6O6Li6–Na |
0 |
3.00 × 107 |
|
|
|
| 532 |
|
1.47 × 109 |
2.09 × 109 |
9.91 × 10−14 |
| 1064 |
|
1.48 × 108 |
3.14 × 108 |
1.36 × 10−14 |
|
|
| Al–C6O6Li6–K |
0 |
4.55 × 106 |
|
|
|
| 532 |
|
8.00 × 108 |
1.23 × 109 |
5.62 × 10−14 |
| 1064 |
|
2.37 × 107 |
9.76 × 107 |
3.47 × 10−15 |
|
|
| Ga–C6O6Li6–Li |
0 |
6.41 × 106 |
|
|
|
| 532 |
|
6.46 × 108 |
9.27 × 108 |
4.36 × 10−14 |
| 1064 |
|
4.81 × 107 |
3.40 × 108 |
1.09 × 10−14 |
|
|
| Ga–C6O6Li6–Na |
0 |
2.97 × 107 |
|
|
|
| 532 |
|
4.05 × 108 |
1.94 × 108 |
1.74 × 10−14 |
| 1064 |
|
1.56 × 108 |
3.24 × 108 |
1.41 × 10−14 |
|
|
| Ga–C6O6Li6–K |
0 |
1.45 × 105 |
|
|
|
| 532 |
|
1.33 × 108 |
2.67 × 108 |
1.10 × 10−14 |
| 1064 |
|
3.08 × 105 |
8.80 × 106 |
2.55 × 10−16 |
This table illustrates that the change in frequency caused a valuable increase in second hyperpolarizability, which described a significant enhancement in NLO response at both frequencies (532 and 1064 nm). The static, dc Karr and EFSHG values for pristine C6O6Li6 are 5.21 × 106, 3.69 × 108 and 3.12 × 107 au, respectively. After complexations, the static hyperpolarizability values range from 4.11 × 106 to 5.31 × 107 au. The EFSHG values for all designed nine complexes are between 9.57 × 107 and 2.09 × 1010 au and the dc Karr effect values for all designed nine complexes are between 1.33 × 108 and 2.90 × 1011 au at 532 nm. The SHG values for all the designed nine complexes are between 8.80 × 106 and 1.13 × 1010 au and the EOPE values for all the designed nine complexes are between 3.08 × 105 and 1.26 × 109 au at 1064 nm. The higher dc Karr effect (2.90 × 1011 au) and EFSHG (2.09 × 1010 au) values were obtained for B–C6O6Li6–Na and Al–C6O6Li6–Na complexes. These results depict the prominent EOPE and EFSHG effects at a lower frequency of 532 nm. Similar results were obtained by Mahmood and coworkers, which also show the higher vibrational frequency-dependent first and second hyperpolarizability values at 532 nm.98
The nonlinear refractive index (n2) of Group IA/IIIA-doped C6O6Li6 range from 8.39 × 10−12 to 2.55 × 10−16 cm2 W−1. The higher n2 value (8.39 × 10−12 cm2 W−1) was obtained for B–C6O6Li6–Na at 532 nm and the lower n2 value (2.55 × 10−16 cm2 W−1) was obtained for Ga–C6O6Li6–K at 1064 nm. The charge transfer from Group-IIIA to Group-IA metals occurs through C6O6Li6 in a complex. The excess charge transfer within this complex causes polarization, which can possibly be responsible for the increase in nonlinear refractive index and enhancement of NLO response, as previously reported in the literature.99–101
3.8. TD-DFT calculation of group-I/IIIA metal-doped C6O6Li6 alkalides
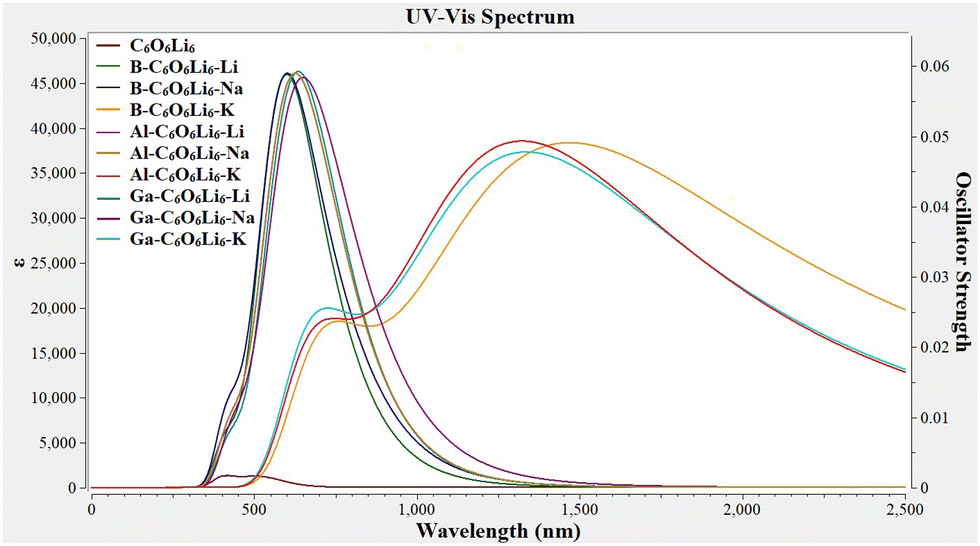
The absorption maxima (λmax) of Group-IIIA-C6O6Li6-Group-IA range from 580 to 1556 nm, where the highest value (1556 nm) is observed for B–C6O6Li6–K and the lowest value (580 nm) is observed for B–C6O6Li6–Li alkalides (see Fig. 5). All these values are higher than those of pristine C6O6Li6 (λmax = 515 nm). K-Doped Group-IIIA-C6O6Li6 has a higher wavelength than that of Na and Li doped alkalides. The outcomes of UV-vis analysis are comparable to the research report on NLO response of alkali metal-doped C6O6Li6 complexes, where K–C6O6Li6–Mg complex has the highest λmax value among all selected alkali metal-doped C6O6Li6 complexes.41 These results show bathochromic shifts in all alkalides. Comparable red shift behavior of Group I/IIIA metal-doped complexes92,95 reported in the literature justifies our results.
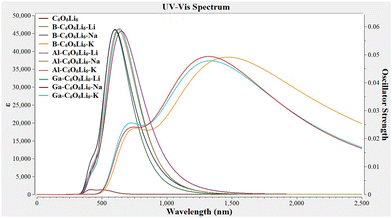 |
| | Fig. 5 Ultra-violet-visible (UV-vis) spectra of pristine C6O6Li6 and group-I and group-III metal-doped C6O6Li6-alkalides (group-I = K, Na, and Li; group-III = Ga, Al, and B). | |
4. Conclusion
Metals act as sources of excess electrons that enhance the nonlinear optical (NLO) response of complexes. The NLO response of C6O6Li6 was evaluated with Group IA (K, Na, and Li) and Group IIIA (Ga, Al, and B) metals. In these alkalides, π conjugation of C6O6Li6 withdraws excess electrons from Group IIIA metals, which are transferred to Group IA metals. All alkalides are thermodynamically stable, with the internal energy (Eint) increasing to −109.13 kcal mol−1 after complexation. The vertical ionization energies (VIEs) of 3.75 eV and 3.02 eV indicate electronic stability. Charge analysis shows positive charges on Group IIIA and negative charges on Group IA metals, with HOMO densities on anionic Group IA metals confirming alkalide properties. The semiconducting behavior is evident from HOMO–LUMO energy gaps compared to pristine C6O6Li6. The NLO response, characterized by hyperpolarizability, is highest in the B–C6O6Li6–K alkalide, with the highest β0 of 1.75 × 105 au. A two-level model elucidates changes in the first hyperpolarizability due to excitation energy and dipole moment variations. The vibrational frequency-dependent first and second hyperpolarizability values were also calculated, which depicts the increase in hyperpolarizability values at a frequency of 532 nm. The higher n2 value (8.39 × 10−12 cm2 W−1) was obtained for B–C6O6Li6–Na at 532 nm. The UV-vis analysis confirms a bathochromic shift in all alkalides, marking them as promising candidates for future electronics with significant NLO responses.
Data availability
The data supporting the findings of this study are available from the corresponding author mahmood@cuiatd.edu.pk (T. M) upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the Higher Education Commission (NRPU project; 20-16279/NRPU/HEC/2021-2020) of Pakistan. M. I express appreciation to the Deanship of Scientific Research at King Khalid University Saudi Arabia through the research groups program under Grant Number RGP1/86/44. The experiments presented in this paper were carried out using the facilities of the Benefit Advanced AI and Computing Lab at the University of Bahrain—see https://ailab.uob.edu.bh with support from Benefit Bahrain Company—see https://benefit.bh.
References
- H. Ai, Y. Bu, P. Li and L. Sun, J. Phys. Chem. A, 2004, 108, 4156–4162 CrossRef CAS.
- R.-L. Zhong, H.-L. Xu, Z.-R. Li and Z.-M. Su, J. Phys. Chem. Lett., 2015, 6, 612–619 CrossRef CAS PubMed.
- Y. Bai, Z.-J. Zhou, J.-J. Wang, Y. Li, D. Wu, W. Chen, Z.-R. Li and C.-C. Sun, J. Phys. Chem. A, 2013, 117, 2835–2843 CrossRef CAS PubMed.
- W.-M. Sun, X.-H. Li, J. Wu, J.-M. Lan, C.-Y. Li, D. Wu, Y. Li and Z.-R. Li, Inorg. Chem., 2017, 56, 4594–4600 CrossRef CAS PubMed.
- A. Ahsin and K. Ayub, Mater. Sci. Semicond. Process., 2022, 138, 106254 CrossRef CAS.
- R. Zheng, B. Zhang, C. Wang and J. Hou, New J. Chem., 2022, 46, 15334–15343 RSC.
- Y. Y. Liang, B. Li, X. Xu, F. Long Gu and C. Zhu, J. Comput. Chem., 2019, 40, 971–979 CrossRef CAS PubMed.
- N. Maqsood, A. Asif, K. Ayub, J. Iqbal, A. Y. Elnaggar, G. A. M. Mersal, M. M. Ibrahim and S. M. El-Bahy, RSC Adv., 2022, 12, 16029–16045 RSC.
- P. Khan, T. Mahmood, K. Ayub, S. Tabassum and M. Amjad Gilani, Opt. Laser Technol., 2021, 142, 107231 CrossRef CAS.
- L. Dalton, M. Lauermann and C. Koos, NLO: Electro-Optic Applications, 2016, 369–396 CAS.
- S. Di Bella, I. Fragalà, I. Ledoux, M. A. Diaz-Garcia and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 9550–9557 CrossRef CAS.
- V. Parodi, E. Jacchetti, R. Osellame, G. Cerullo, D. Polli and M. T. Raimondi, Front. Bioeng. Biotechnol., 2020, 8, 585363 CrossRef PubMed.
- T. Tian, Y. Fang, W. Wang, M. Yang, Y. Tan, C. Xu, S. Zhang, Y. Chen, M. Xu, B. Cai and W.-Q. Wu, Nat. Commun., 2023, 14, 4429 CrossRef CAS PubMed.
- N. Li, J. Lu, H. Li and E.-T. Kang, Dyes Pigm., 2011, 88, 18–24 CrossRef CAS.
- R. Kaur, K. P. Singh and S. K. Tripathi, J. Alloys Compd., 2022, 905, 164103 CrossRef CAS.
- A. Parveen, J. Yaqoob, S. Ijaz, R. Baloach, M. U. Khan, R. Hussain, H. M. Abo-Dief, A. K. Alanazi and Z. M. El-Bahy, ChemistrySelect, 2023, 8(21), e202300843 CrossRef CAS.
- M. Rashid, J. Yaqoob, N. Khalil, R. Jamil, M. U. Khan and M. A. Gilani, Mater. Sci. Semicond. Process., 2022, 151, 107007 CrossRef CAS.
- N. Kosar, H. Tahir, K. Ayub, M. A. Gilani, M. Imran and T. Mahmood, Mater. Sci. Semicond. Process., 2022, 138, 106269 CrossRef CAS.
- W. Jin, C. Xie, X. Hou, M. Cheng, E. Tikhonov, M. Wu, S. Pan and Z. Yang, Chem. Mater., 2023, 35, 5281–5290 CrossRef CAS.
- N. Hou and X.-H. Fang, Inorg. Chem., 2022, 61, 10756–10767 CrossRef CAS PubMed.
- X. Li, J. Mater. Chem. C, 2018, 6, 7576–7583 RSC.
- F. Ullah, N. Kosar, A. Ali, Maria, T. Mahmood and K. Ayub, Phys. E, 2020, 118, 113906 CrossRef CAS.
- S. Taboukhat, N. Kichou, J.-L. Fillaut, O. Alévêque, K. Waszkowska, A. Zawadzka, A. El-Ghayoury, A. Migalska-Zalas and B. Sahraoui, Sci. Rep., 2020, 10, 15292 CrossRef CAS PubMed.
- N. Kosar, T. Mahmood, K. Ayub, S. Tabassum, M. Arshad and M. A. Gilani, Opt. Laser Technol., 2019, 120, 105753 CrossRef CAS.
- N. Kosar, K. Shehzadi, K. Ayub and T. Mahmood, Optik, 2020, 218, 165033 CrossRef CAS.
- Y.-F. Wang, J. Huang, L. Jia and G. Zhou, J. Mol. Graphics Model., 2014, 47, 77–82 CrossRef PubMed.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723 RSC.
- A. Ahsin, A. Ali and K. Ayub, J. Mol. Graphics Model., 2020, 101, 107759 CrossRef CAS PubMed.
- M. A. Alkhalifah, N. S. Sheikh, Y. S. S. Al-Faiyz, I. Bayach, R. Ludwig and K. Ayub, Materials, 2023, 16, 3447 CrossRef CAS PubMed.
- A. Ahsin, A. Ali and K. Ayub, Mater. Sci. Semicond. Process., 2023, 162, 107506 CrossRef CAS.
- G. Serdaroğlu, N. Uludag, P. Sugumar and P. Rajkumar, J. Mol. Struct., 2021, 1244, 130978 CrossRef.
- W.-M. Sun, D. Wu, Y. Li, J.-Y. Liu, H.-M. He and Z.-R. Li, Phys. Chem. Chem. Phys., 2015, 17, 4524–4532 RSC.
- X. Li and S. Li, J. Mater. Chem. C, 2019, 7, 1630–1640 RSC.
- W. Chen, Z. R. Li, D. Wu, Y. Li, C. C. Sun, F. L. Gu and Y. Aoki, J. Am. Chem. Soc., 2006, 128, 1072–1073 CrossRef CAS PubMed.
- W.-M. Sun, L.-T. Fan, Y. Li, J.-Y. Liu, D. Wu and Z.-R. Li, Inorg. Chem., 2014, 53, 6170–6178 CrossRef CAS PubMed.
- W.-M. Liang, Z.-X. Zhao, D. Wu, W.-M. Sun, Y. Li and Z.-R. Li, J. Mol. Model., 2015, 21, 311 CrossRef PubMed.
- P. Banerjee and P. K. Nandi, Struct. Chem., 2018, 29, 859–870 CrossRef CAS.
- D. Kang, L. Zhao, Z.-M. Su and H.-L. Xu, J. Mol. Liq., 2023, 385, 122400 CrossRef CAS.
- Y.-F. Wang, J. Li, J. Huang, T. Qin, Y.-M. Liu, F. Zhong, W. Zhang and Z.-R. Li, J. Phys. Chem. C, 2019, 123, 23610–23619 CrossRef CAS.
- M. Sohaib, H. Sajid, S. Sarfaraz, M. H. S. A. Hamid, M. A. Gilani, M. Ans, T. Mahmood, S. Muhammad, M. A. Alkhalifah, N. S. Sheikh and K. Ayub, Heliyon, 2023, 9, e19325 CrossRef CAS PubMed.
- N. Kosar, S. Wajid, K. Ayub, M. A. Gilani, N. H. Binti Zainal Arfan, M. H. Sheikh Abdul Hamid, M. Imran, N. S. Sheikh and T. Mahmood, Heliyon, 2023, 9, e18264 CrossRef CAS PubMed.
- S. Wajid, N. Kosar, F. Ullah, M. A. Gilani, K. Ayub, S. Muhammad and T. Mahmood, ACS Omega, 2021, 6, 29852–29861 CrossRef CAS PubMed.
- N. Kosar, S. Wajid, K. Ayub, M. A. Gilani and T. Mahmood, Chem. Phys., 2023, 570, 111894 CrossRef CAS.
- N. Kosar, S. Wajid, K. Ayub and T. Mahmood, Optik, 2023, 276, 170660 CrossRef CAS.
- N. Kosar, L. Zari, K. Ayub, M. A. Gilani and T. Mahmood, Phys. Scr., 2023, 98, 065909 CrossRef CAS.
- D. Gounden, N. Nombona and W. E. van Zyl, Coord. Chem. Rev., 2020, 420, 213359 CrossRef CAS.
- R. Sreedharan, S. Ravi, K. R. Raghi, T. K. M. Kumar and K. Naseema, SN Appl. Sci., 2020, 2, 578 CrossRef CAS.
- M. Aetizaz, F. Ullah, T. Mahmood and K. Ayub, Comput. Theor. Chem., 2024, 1232, 114469 CrossRef CAS.
- S. Kaviani, I. Piyanzina, O. V. Nedopekin and D. A. Tayurskii, Int. J. Hydrogen Energy, 2023, 48, 30069–30084 CrossRef CAS.
- M. A. Adebisi, C.-D. Chen, M. Maaza, C. Ronning, E. Manikandan, D. E. Motaung, P. S. Christopher, K. Bharuth-Ram, N. S. Aliyu, M. N. Pillay and M. K. Moodley, Phys. Status Solidi, 2023, 220(1), 2200464 CrossRef CAS.
- Y. Deng and J. Jiang, IEEE Sens. J., 2022, 22, 13811–13834 CAS.
-
X. Zhang and J. Meng, Ultra-Wide Bandgap Semiconductor Materials, Elsevier, 2019, pp. 347–419 Search PubMed.
- H. Hora, J. Energy Power Eng., 2020, 14, 156–177 CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. K. J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. C. N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. J. R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. D. J. F. Cioslowski, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
R. I. Dennington, T. Keith and J. Millam, GaussView version 5.0.8, semichem, Inc., Shawnee Mission, KS, 2008 Search PubMed.
- S. Tariq, A. R. Raza, M. Khalid, S. L. Rubab, M. U. Khan, A. Ali, M. N. Tahir and A. A. C. Braga, J. Mol. Struct., 2020, 1203, 127438 CrossRef CAS.
- N. Islam and A. H. Pandith, J. Mol. Model., 2014, 20, 2535 CrossRef PubMed.
- D. R. Mohbiya and N. Sekar, ChemistrySelect, 2018, 3, 1635–1644 CrossRef CAS.
- M. Sherafati, A. Shokuhi Rad, M. Ardjmand, A. Heydarinasab, M. Peyravi and M. Mirzaei, Curr. Appl. Phys., 2018, 18, 1059–1065 CrossRef.
- N. Hou, Y.-Y. Wu and J.-Y. Liu, Int. J. Quantum Chem., 2016, 116, 1296–1302 CrossRef CAS.
- Z. Liu, J. Sun, C. Yan, Z. Xie, G. Zhang, X. Shao, D. Zhang and S. Zhou, J. Mater. Chem. C, 2020, 8, 12993–13000 RSC.
- P. S. Halasyamani and J. M. Rondinelli, Nat. Commun., 2018, 9, 2972 CrossRef PubMed.
- M. Tarazkar, D. A. Romanov and R. J. Levis, J. Phys. B: At., Mol. Opt. Phys., 2015, 48, 094019 CrossRef.
- C. Bree, A. Demircan and G. Steinmeyer, IEEE J. Quantum Electron., 2010, 46, 433–437 CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- Maria, J. Iqbal, R. Ludwig and K. Ayub, Mater. Res. Bull., 2017, 92, 113–122 CrossRef CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- X. Zhang, G. Liu, K. Meiwes-Broer, G. Ganteför and K. Bowen, Angew. Chemie, 2016, 128, 9796–9799 CrossRef.
- A. Ahsan and K. Ayub, J. Mol. Liq., 2020, 297, 111899 CrossRef CAS.
- K. Srinivasu and S. K. Ghosh, J. Phys. Chem. C, 2012, 116, 5951–5956 CrossRef CAS.
- Y. Arshad, S. Khan, M. A. Hashmi and K. Ayub, New J. Chem., 2018, 42, 6976–6989 RSC.
- J. Kotakoski, C. H. Jin, O. Lehtinen, K. Suenaga and A. V. Krasheninnikov, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 113404 CrossRef.
- W.-M. Liang, Z.-X. Zhao, D. Wu, W.-M. Sun, Y. Li and Z.-R. Li, J. Mol. Model., 2015, 21, 311 CrossRef PubMed.
- P. Khan, T. Mahmood, K. Ayub, S. Tabassum and M. Amjad Gilani, Opt. Laser Technol., 2021, 142, 107231 CrossRef CAS.
- V. Galasso, B. Kovač, A. Modelli, M. F. Ottaviani and F. Pichierri, J. Phys. Chem. A, 2008, 112, 2331–2338 CrossRef CAS PubMed.
- M.-S. Liao, J. D. Watts and M.-J. Huang, J. Phys. Chem. C, 2014, 118, 21911–21927 CrossRef CAS.
- D. W. Boukhvalov, A. N. Rudenko, D. A. Prishchenko, V. G. Mazurenko and M. I. Katsnelson, Phys. Chem. Chem. Phys., 2015, 17, 15209–15217 RSC.
- A. Nisar, S. Tabassum, K. Ayub, T. Mahmood, H. AlMohamadi, A. L. Khan, M. Yasin, R. Nawaz and M. A. Gilani, Phys. Chem. Chem. Phys., 2023, 25, 20430–20450 RSC.
- R. Akbar, A. Asif, N. Maqsood and M. Nouman, Struct. Chem., 2024, 35, 1943–1962 CrossRef CAS.
- A. Rafique, H. Maqbool, R. A. Shehzad, I. A. Bhatti, K. Ayub, A. Elmushyakhi, A. M. Shawky and J. Iqbal, Int. J. Quantum Chem., 2023, 123(6), e27060 CrossRef CAS.
- M. Asif, H. Sajid, K. Ayub, M. A. Gilani and T. Mahmood, Polyhedron, 2022, 215, 115695 CrossRef CAS.
- A. Nisar, S. Tabassum, K. Ayub, T. Mahmood, H. AlMohamadi, A. L. Khan, M. Yasin, R. Nawaz and M. A. Gilani, Phys. Chem. Chem. Phys., 2023, 25, 20430–20450 RSC.
- M. U. Khan, M. R. S. A. Janjua, J. Yaqoob, R. Hussain, M. Khalid, A. Syed, A. M. Elgorban and N. S. S. Zaghloul, J. Photochem. Photobiol., A, 2023, 440, 114667 CrossRef CAS.
- N. Hou, Y.-Y. Wu and J.-Y. Liu, Int. J. Quantum Chem., 2016, 116, 1296–1302 CrossRef CAS.
- N. Kosar, K. Ayub and T. Mahmood, J. Mol. Graphics Model., 2021, 102, 107794 CrossRef CAS PubMed.
- Maria, J. Iqbal and K. Ayub, J. Alloys Compd., 2016, 687, 976–983 CrossRef CAS.
- R. Nazir, J. Yaqoob, M. U. Khan, M. A. Gilani, R. Hussain, M. U. Alvi, M. Rashid, M. A. Assiri and M. Imran, Phys. B, 2022, 640, 414041 CrossRef CAS.
- N. Kosar, H. Tahir, K. Ayub, M. A. Gilani, M. Arshad and T. Mahmood, Comput. Theor. Chem., 2021, 1204, 113386 CrossRef CAS.
- Z.-G. Shao and Z.-L. Sun, Phys. E, 2015, 74, 438–442 CrossRef CAS.
- N. Kosar, K. Shehzadi, K. Ayub and T. Mahmood, J. Mol. Graphics Model., 2020, 97, 107573 CrossRef CAS PubMed.
- N. Kosar, S. Gul, K. Ayub, A. Bahader, M. A. Gilani, M. Arshad and T. Mahmood, Mater. Chem. Phys., 2020, 242, 122507 CrossRef CAS.
- S. Muhammad, A. G. Al-Sehemi, Z. Su, H. Xu, A. Irfan and A. R. Chaudhry, J. Mol. Graphics Model., 2017, 72, 58–69 CrossRef CAS PubMed.
- A. Plaquet, B. Champagne and F. Castet, Molecules, 2014, 19, 10574–10586 CrossRef CAS PubMed.
- N. Kosar, S. Kanwal, H. Sajid, K. Ayub, M. A. Gilani, K. Elfaki Ibrahim, M. K. Gatasheh, Y. S. Mary and T. Mahmood, J. Mol. Graphics Model., 2024, 126, 108646 CrossRef CAS PubMed.
- N. Kosar, S. Kanwal, H. Sajid, K. Ayub, M. A. Gilani, K. Elfaki Ibrahim, M. K. Gatasheh, Y. S. Mary and T. Mahmood, J. Mol. Graphics Model., 2024, 126, 108646 CrossRef CAS PubMed.
- J. Yaqoob, T. Mahmood, K. Ayub, S. Tabassum, A. F. Khan, S. Perveen, J. Yang and M. A. Gilani, Eur. Phys. J. Plus, 2022, 137, 233 CrossRef CAS.
- X. Zhou, C. Zhao, G. Wu, J. Chen and Y. Li, Appl. Surf. Sci., 2018, 459, 354–362 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Abdulaziz A.
Al-Saadi
b,
Abdulaziz A.
Al-Saadi