
Enhancing the stability and performance of Ni-rich cathode materials through Ta doping: a combined theoretical and experimental study†
Frederike
Monsees
 *ab,
Casimir
Misiewicz
c,
Mert
Dalkilic
b,
Diddo
Diddens
d and
Andreas
Heuer
a
*ab,
Casimir
Misiewicz
c,
Mert
Dalkilic
b,
Diddo
Diddens
d and
Andreas
Heuer
a
aInstitute for Physical Chemistry, University of Münster, Münster, Germany
bPowerCo, Salzgitter, Germany. E-mail: frederike.monsees@powerco.de
cDepartment of Chemistry - Ångström Laboratory, Uppsala University, Uppsala, Sweden
dHelmholtz Institute Münster (IMD-4), Forschungszentrum Jülich GmbH, Münster, Germany
First published on 11th December 2024
Abstract
As the demand for high-energy batteries to power electric vehicles continues to grow, Ni-rich cathode materials have emerged as promising candidates due to their high capacity. However, these materials are prone to rapid degradation under increased voltages, posing significant challenges for their long-term stability and safety. In this study, we investigate the effects of tantalum (Ta) doping on the performance and stability of LiNi0.80Mn0.1Co0.1O2 (NMC811) cathode materials. Using a combined theoretical and experimental approach, we employ density functional theory (DFT) and cluster expansion models to analyze the electronic structure and oxygen vacancy formation enthalpy in Ta-doped NMC811. Experimental validation is conducted using cycling and gas measurements via on-line electrochemical mass spectrometry (OEMS) on in-house synthesized cathode active materials. Both theoretical and experimental approaches show an improvement in oxygen binding due to tantalum doping, with the DFT results highlighting the impact of Ni4+ concentration on the proximity of the vacancy. Our results suggest that Ta doping inhibits the formation of oxygen vacancy-induced side phases, reducing cracking and enhancing the longevity and safety of Ni-rich cathodes.
1 Introduction
As we strive for a more sustainable and long-lasting transport sector, the development of robust batteries is crucial. New regulations mandate batteries to last over 10 years while retaining at least 80% of their capacity.1 However, a significant challenge arises: higher volumetric capacity in batteries often leads to faster degradation.2 This issue is particularly noticeable with Ni-rich cathode materials. As the nickel content in the active material increases, so does the capacity, but this also makes the material significantly more prone to aging.3 Common aging mechanisms for Ni-rich cathode active materials (CAM) include gassing and cracking, both of which heavily impact battery safety and capacity.4,5 Among these, oxygen release is particularly concerning as it is the primary driver for safety hazards such as thermal runaway.6 Oxygen release typically occurs at high voltages, which are applied at the end of charge.7 The interface between different phases in cathode materials also plays a critical role in determining their performance and longevity. Ni-rich materials, in particular, suffer from the formation of secondary phases such as spinel and rock salt at the interface between cathode and electrolyte.8 These phases can disrupt the material's structure and degrade its electrochemical performance. Moreover, the presence of Ni4+ at the interface is problematic as it can act as a catalytic site for undesirable side reactions, leading to increased oxygen release and degradation.9 Mitigating oxygen release is a key focus in cathode design, with doping and coating being typical countermeasures.10–13Both can stabilize the interface by altering the local electronic structure and reducing the formation of Ni4+ ions. Similarly, both may induce a core–shell structure that can stabilize the Ni-rich phase at the surface, thus enhancing the overall stability of the cathode.14,15
An examination of the total amounts of gas released between different doped and undoped Ni-rich cathodes reveals that the identity and concentration of the dopant significantly influence gas evolution. Data suggest that dopants such as Mn and Al are effective at suppressing gas release, particularly when present in higher concentrations. Comparatively, Mn substitution for Ni exhibits a greater influence on reducing gas evolution than Co, highlighting the nuanced effects that different elements have on cathode performance.16
A recent trend has emerged in which high-valence elements such as tantalum, tungsten, molybdenum, and niobium are used as dopants to enhance the lifespan of CAMs. Initially, these elements were thought to be too large and positively charged to integrate into the material structure, but recent studies indicate otherwise.17,18 How and where the dopant is incorporated into the material is significantly affected by the synthesis routine and calcination temperature.19 Interestingly, these dopants do not only influence the crystal's host structure but also modify grain boundaries and particle growth.18 One reason why these relatively large dopants have a positive impact might be found in their high oxygen binding energy compared to Ni, Mn, or Co.20
This work builds on our previously published study (ref. 17), which focused on the synthesis and characterization of Ta-doped and pristine LiNi0.80Mn0.1Co0.1O2 (NMC811). While that study identified 0.5% Ta doping as optimal for enhancing material performance, the present work offers a detailed evaluation of how Ta doping affects oxygen-binding behavior.
Despite extensive gassing analyses on Ni-rich CAMs,16,21 no studies to date have explored the gassing behavior of Ta-doped NMCs, making this investigation novel. To assess the impact of Ta on oxygen binding, we combine ab initio methods with experimental validation.
Density functional theory (DFT) was used to analyze the electronic structure, while a cluster expansion algorithm enabled the creation of model systems with accurately distributed components and various states of charge (SOCs). These computational methods allowed us to generate meaningful configurations of Ta-doped NMC for atomistic-scale investigations of oxygen binding. Complementing this theoretical approach, we conducted on-line electrochemical gas spectrometry on the previously synthesized NMC811 and NMC811 with 0.5% Ta, validating our theoretical results against experimental gas measurements.
2 Methods
2.1 Computational methods
All simulations were performed using the Vienna Ab initio Simulation Package (VASP), a widely used first-principles simulation tool based on density functional theory (DFT) with the projector-augmented-wave (PAW) method22–25 in the MedeA software environment.26Spin polarization was considered in all calculations. All structures underwent a stepwise relaxation with a cutoff energy of 520 eV and a k-point spacing of 0.5 Å−1 until the maximum force on each atom was lower than 0.02 eV Å−1 using the optB86b-vdW functional.27–29 To calculate the electronic structure and to ensure an accurate binding behavior, single point calculations were performed using the SCAN functional with vdW correction, proven reliable for layered cathode active materials.30–35 Here the numerically more efficient version R2SCAN with vdW correction is applied.36
Cluster expansion (CE) calculations were carried out applying the universal cluster expansion (UNCLE) method.37 This method performs a CE using a genetic algorithm to generate a training set based on one input structure with defined occupancies for the targeted sublattice. The application of a genetic algorithm aims to efficiently explore the configurational space and identify the most relevant structures that contribute to the model's accuracy. In the application of the genetic algorithm, the fitness of randomly generated structures is determined by the total energy of the configuration. The higher the stability of the structure, the more weighting it gets in the generation of new populations. By this iterative process a convex hull is created, representing the set of lowest-energy configurations (ground states) at various compositions. It serves as a reference to identify stable and metastable phases. In order to obtain a LiNi0.80Mn0.1Co0.1O2 (NMC811) structure for doping and gassing investigation two cluster expansion (CE). Models were set up. The first one starting from the primitive R3/m LiNiO2 (LNO) cell, investigating different nickel, cobalt and manganese distributions on the 3b Wyckoff position. The second one starting from a P2/m NMC811 cell obtained from the first CE to generate thermodynamically stable structures at 0–90% lithium occupancy (Wyckoff position 2i, 2j, 1b and 1d). The training data for the CE model was generated using the optB86b-vdW functional. This choice balances computational cost and efficiency, as optB86b-vdW, while not as precise in predicting electronic structure, provides sufficiently accurate forces for effective model training.
The maximum cross validation score (CVS) over all subsets was below 65 meV per atom for the Ni–Mn–Co sublattice and below 10 meV for the Li-Vac sublattice in the NMC811 system. A higher CVS in systems with more components has also been observed by Yang et al.38 Reflecting the complexity of the ternary CE model, the training set for the transition metal sublattice contains 172 structures (see Table S5, ESI†), while the training set for the Li-vacancy sublattice includes 39 structures.
All presented atomic structures have been rendered using VESTA®39 and OVITO®.40
2.2 Electrode and cell preparation
Two cathode active materials, NMC811 and Ta-doped NMC811 (synthesised and characterised in previous studies by Dalkilic et al.17) were made into slurries and subsequent electrodes according to the same procedure. Each cathode powder was mixed with conductive carbon (Super P Li, Imerys) and polyvinylidene difluoride (PVDF, Kynar HSV 900, Arkema) dissolved in N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich) using a stand disperser (PT 2500 E equipped with a PT-D4 0.7 aggregate, Kinematica) such that a 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3 ratio of NMC, conductive carbon and PVDF was achieved. The slurries were mixed between 15k–12k rpm ramping 2k rpm every 2 min for 12 min followed by 5 min at 30k rpm. They were then coated onto stainless steel mesh (212/90 μm, Bopp AG) using a 150 μm doctor blade, dried in a vacuum oven at 100 °C for 4 hours before being punched into ∅ 13 mm disks. The Ta-doped and pristine NMC811 electrodes had an active mass loading of 24.9 ± 1.3 mg cm−2 and 29.9 ± 1.9 mg cm−2 respectively. Before cell preparation the electrodes were transferred into an Ar-atmosphere glovebox (CO2 content <0.1 ppm, H2O content 0.3 ppm) and dried in a vacuum oven at 120 °C for 12 hours. Cells were assembled in our in-house dedicated OEMS cell-housings (described in previous studies) with ∅ 16 mm Li metal disk as the counter electrode, ∅ 16 mm 2325 Celgard separator, ∅ 13 mm cathode as the working electrode and soaked in 100 μL LP40 (1 M LiPF6 EC:DEC, 1:1).
:3:3 ratio of NMC, conductive carbon and PVDF was achieved. The slurries were mixed between 15k–12k rpm ramping 2k rpm every 2 min for 12 min followed by 5 min at 30k rpm. They were then coated onto stainless steel mesh (212/90 μm, Bopp AG) using a 150 μm doctor blade, dried in a vacuum oven at 100 °C for 4 hours before being punched into ∅ 13 mm disks. The Ta-doped and pristine NMC811 electrodes had an active mass loading of 24.9 ± 1.3 mg cm−2 and 29.9 ± 1.9 mg cm−2 respectively. Before cell preparation the electrodes were transferred into an Ar-atmosphere glovebox (CO2 content <0.1 ppm, H2O content 0.3 ppm) and dried in a vacuum oven at 120 °C for 12 hours. Cells were assembled in our in-house dedicated OEMS cell-housings (described in previous studies) with ∅ 16 mm Li metal disk as the counter electrode, ∅ 16 mm 2325 Celgard separator, ∅ 13 mm cathode as the working electrode and soaked in 100 μL LP40 (1 M LiPF6 EC:DEC, 1:1).
2.3 Online electrochemical mass spectrometry
Experimental analysis was carried out on the cells using our online electrochemical mass spectrometry (OEMS) setup (described and validated by Lundström et al. in previous works) in order to monitor the evolution rate of select gaseous species during cycling (data given in mol min−1 g−1). Gas is extracted from the cell (10% of cell volume) every 10 minutes while fresh argon carrier gas (99.9999%, Ari liquid) is used to maintain a constant internal cell pressure. The instrument is calibrated for H2, CH4, C2H4, O2 and CO2 (m/z channels: 2, 26, 28, 32, 44 and 85) using 2000 ppm H2 and 4000 ppm CO2 in Ar, 2000 ppm CH4 and C2H4 in Ar and 2000 ppm CO2 in Ar (Air liquide). All other unknown gasses are calibrated against the total pressure evolution which is determined by a pressure transducer. All cells were cycled galvanostatically on an astrol potentiostat (Bat-small) with the same overcharge protocol (4 × 0.1C; with charge step CC; between 3.0–4.6 V vs. Li+/Li redox couple). Particular attention was placed in monitoring the oxygen and carbon dioxide gas evolution rates (on m/z channels 32 and 44 respectively).3 Results
3.1 Ternary cluster expansion model for obtaining NMC811
A ternary CE model was employed to identify potential stable structures for NMC811, within the constraint imposed by the size of the supercell used as a ref. 41. Here, the dataset comprised R3/m supercells (see Fig. 1a) with up to 10 transition metal sites, corresponding to concentrations ranging from 10 to 100% for nickel, cobalt, and manganese, respectively. Fig. 2 visualizes this with each colored hexagon corresponding to a micro-state of the ternary mixture at 0 K. The formation energy for a given composition is determined by using its total energy along with the energies of the reference compounds, LiCO2, LiMnO2, and LiNiO2:| Ef(x, y, z) = E(LiNixMnyCozO2) − xE(LiNiO2) − yE(LiMnO2) − zE(LiCoO2) | (1) |
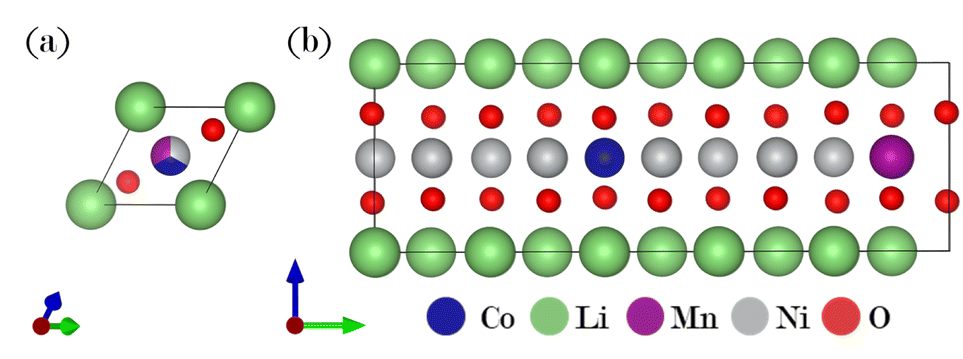 | ||
| Fig. 1 (a) Starting structure for the ternary cluster expansion (CE) model. (b) Thermodynamically stable NMC811 structure resulting from the CE. | ||
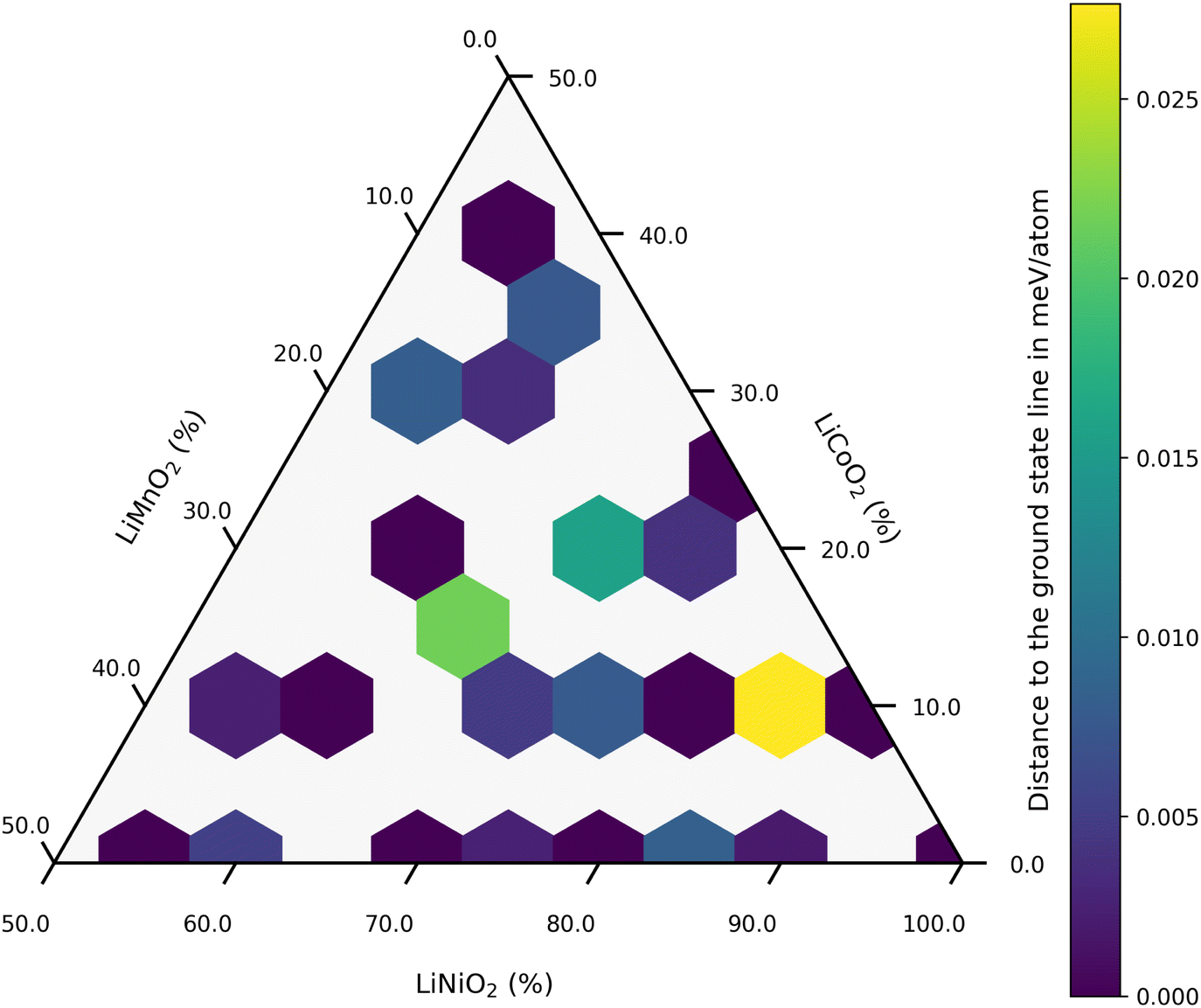 | ||
| Fig. 2 Composition and stability of training set structures for the CE Model with a Ni content >50%. | ||
The color gradient from yellow to blue represents the distance to the ground state line; only the structures lying on the hull are considered absolutely stable. Dark blue regions in Fig. 2 indicate that these compositions are on the convex hull. Within the dataset, a thermodynamically stable NMC811 structure was produced and chosen as a reference for NMC811 in further calculations (see Fig. 1b). The model structure for NMC811 closely resembles the experimental and theoretical structures found in literature.44,45 Direct conclusions about phase stability cannot be drawn from the convex hull at 0 Kelvin, which does not account for entropy contributions or temperature effects. Especially in ternary systems, configurational entropy can play a significant role. Phases that appear unstable at 0 K might be stabilized by entropy at higher temperatures. By employing a CE in combination with Monte Carlo sampling methods, configurational entropy and temperature effects can be incorporated, allowing the construction of phase diagrams.34,46 However, this was not pursued as it is beyond the scope of this study. Additionally, mesoscale effects such as the segregation of Co or Mn-rich regions cannot be observed due to the size of the chosen supercell.
3.2 Cluster expansion model for the Li-vacancy sublattice of NMC811
To simulate a structure at high SOC, a second cluster expansion is performed with a lithium-vacancy-sublattice spanning concentrations from 0–100% on the previously selected NMC811 structure, with 10 different lattice positions within the Li sublattice.| Ef(x) = E(LixNi0.8Mn0.2Co0.2O2) − xE(LiNi0.8Mn0.2Co0.2O2) − (1 − x)E(Ni0.8Mn0.2Co0.2O2) | (2) |
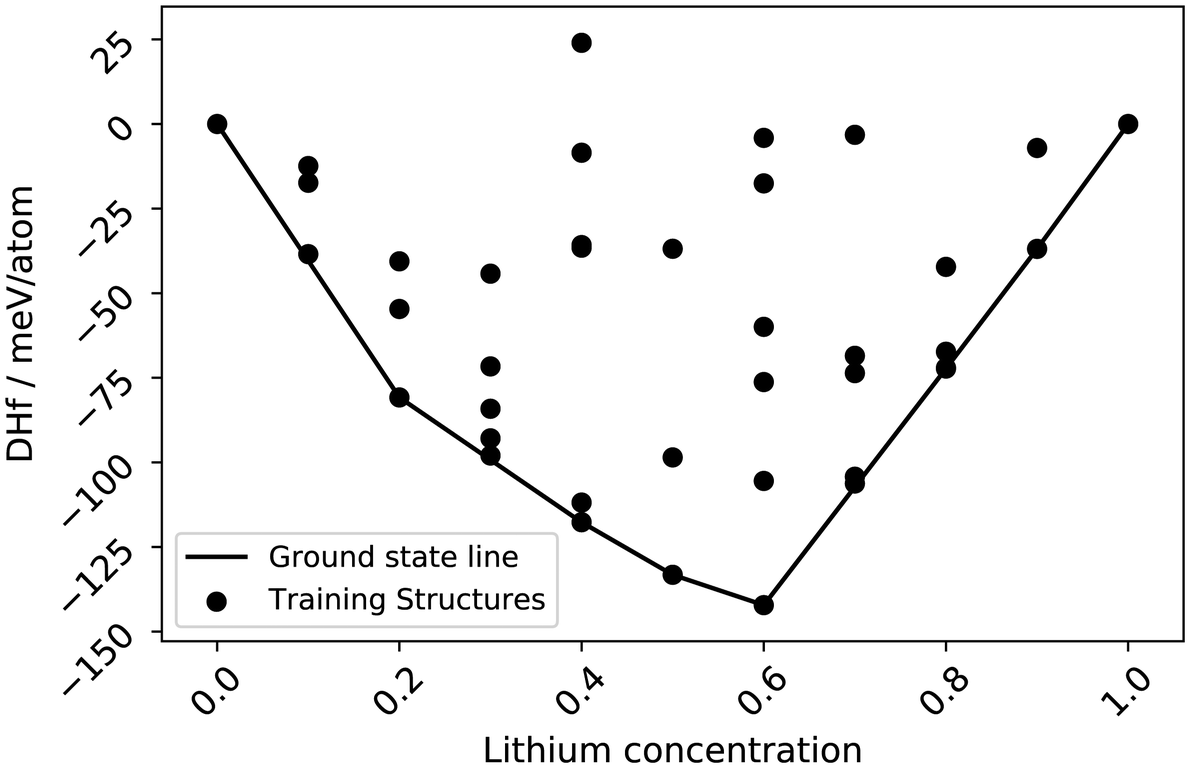 | ||
| Fig. 3 Convex hull and training structures of the Li-vacancy CE model. | ||
3.3 Dopant position analysis of Ta in NMC811
Before studying the impact of Ta doping on oxygen release, we investigate the Ta substitution energy. Substitution was performed on a fully lithiated supercell at all geometrically invariant transition metal positions. Li and O positions were not considered.15,17,49 The selection of reservoirs for calculating substitution energies is not fixed and can vary based on the study's context, making the choice somewhat arbitrary. There are multiple sensible options for reservoirs depending on the specific needs of the calculation. One practical approach is to use native oxides as reservoirs for transition metals. This method reflects the natural oxidation states of the metals involved and the phases that form during calcination, providing a relevant and chemically intuitive baseline for evaluating the thermodynamics of substitution within a compound. Also this approach aligns with the chosen synthesis process of Ta-doped NMC17 and previous reports of doping induced side phases.3The formulation refers to the reaction:
| Ecell + EMO → Edoped + EXO + EO2 | (3) |
| ΔEsub = (Edoped + EXO + EO2) − (Ecell + EMO) | (4) |
For symmetry reasons, there is one value for each Co and Mn, but four for Ni substitution (see Fig. 1). The mean energy value for Ni substitution is displayed in Table 1, as the substitution energies for the four Ni sites range from 0.86 eV to 1.41 eV. This variation arises from differences in the local chemical environment, particularly the influence of neighboring atoms such as Mn and Co. In future calculations, we will focus on the thermodynamically most preferable Ni site, which aligns well with the previously observed trends.48 The increased disorder introduced by substituting the most abundant component, Ni, contributes to this preference. While DFT does not explicitly account for configurational entropy, its influence is crucial in understanding the thermodynamic behavior during the doping process.50 By considering the oxides and taking into account meaningful side phases rather then the elements, the substitution energies are endothermic. This reflects on the actual process of doping high-valence elements, which involves temperatures around 800 °C.17
| Dopant | Ni site (eV) | Co site (eV) | Mn site (eV) |
|---|---|---|---|
| Ta | 1.02 | 2.17 | 1.44 |
3.4 Oxygen release energy via first-principles calculations
Combining the results from the substitution analysis and the cluster expansion models, two P2/m 2 × 1 × 2 supercells corresponding to the compositions (Li2Ni8CoMnO20)4 and Li8TaNi31Mn4Co4O80 (Fig. 4) were created. We aimed for a tantalum concentration of 2.5%, considering computational cost and the desire for a realistic model. The shortest distance between two Ta positions is approximately 10 Å, determined by the size of the supercell. We hereby intend to minimize the self-interaction of the doping element, allowing for a more distinct analysis of their impact on the overall system. | ||
| Fig. 4 Structural model for Ta-doped NMC811 at 20% Li. | ||
To evaluate the impact of Ta on oxygen binding behavior, an analysis of geometrically invariant oxygen positions yielded 40 and 6 different positions for doped and pristine structures, respectively. Wandt et al.51 report the onset of singlet oxygen release above 4.55 V, corresponding to a lithiation degree of 10–20%.21 Following the reaction path in 5, the oxygen vacancy formation enthalpy can be estimated as shown in eqn (6).
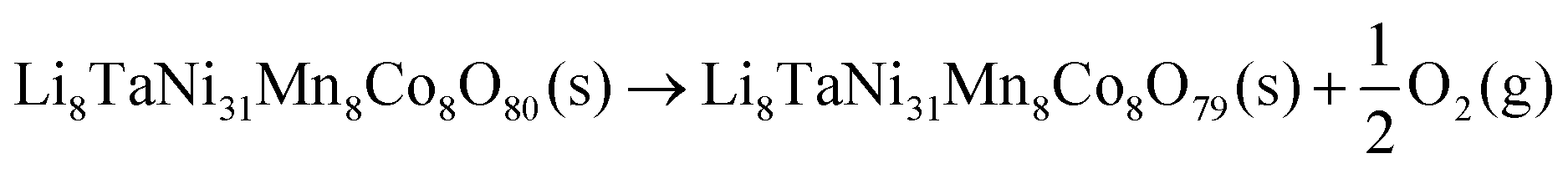 | (5) |
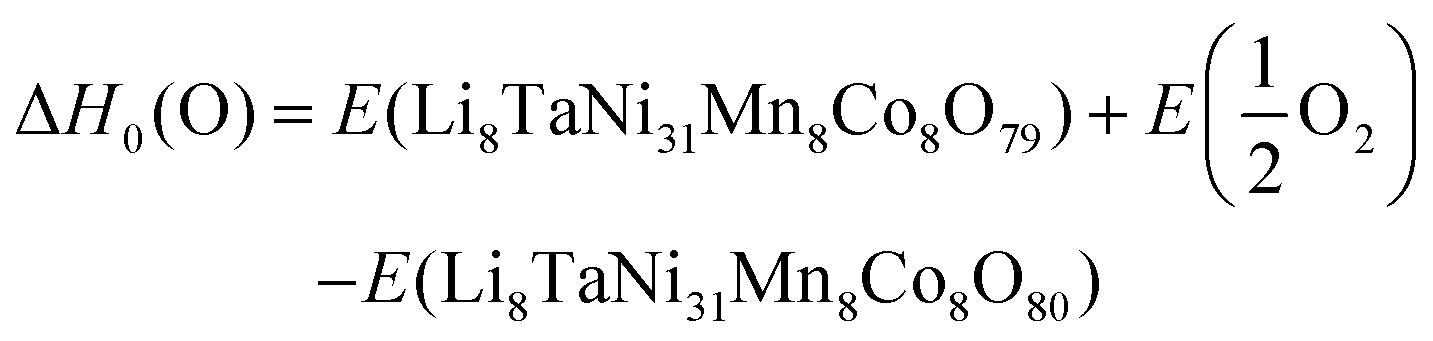 | (6) |
As a reference, molecular oxygen in a triplet state is used to ensure consistency with the ground state properties and standard thermodynamic conditions.52 In contrast, experimental conditions may lead to the formation of singlet oxygen due to additional energy inputs.53 When comparing experimental results with theoretical calculations, it is important to acknowledge that ab initio methods might not fully capture the complete thermodynamics and, particularly, the kinetics of reaction pathways. However, when evaluating the impact of doping on a system, these limitations have minimal influence on our analysis. Our primary focus is to identify trends on the effect of oxygen binding—a thermodynamic property—which can be effectively evaluated using theoretical approaches. By comparing the oxygen vacancy formation energy in two different crystal systems, we can therefore predict how doping alters oxygen bond strength.
The average value of all geometrically invariant positions is calculated for the oxygen release energy and printed in Table 2. Tantalum doping shows a positive impact, increasing the endothermic reaction energy by approximately 35%. Fig. 5 visualizes the oxygen vacancy formation energy for each oxygen atom in the doped system at a high SOC. The intensity of the red color correlates with the oxygen vacancy formation energy, with higher energy indicating a stronger binding (darker color). Particularly, oxygen atoms in direct proximity to Ta are strongly bound. Lower binding can be observed in Ni-rich and Li-depleted areas.
| Doped (eV) | Pristine (eV) |
|---|---|
| 2.10 | 1.56 |
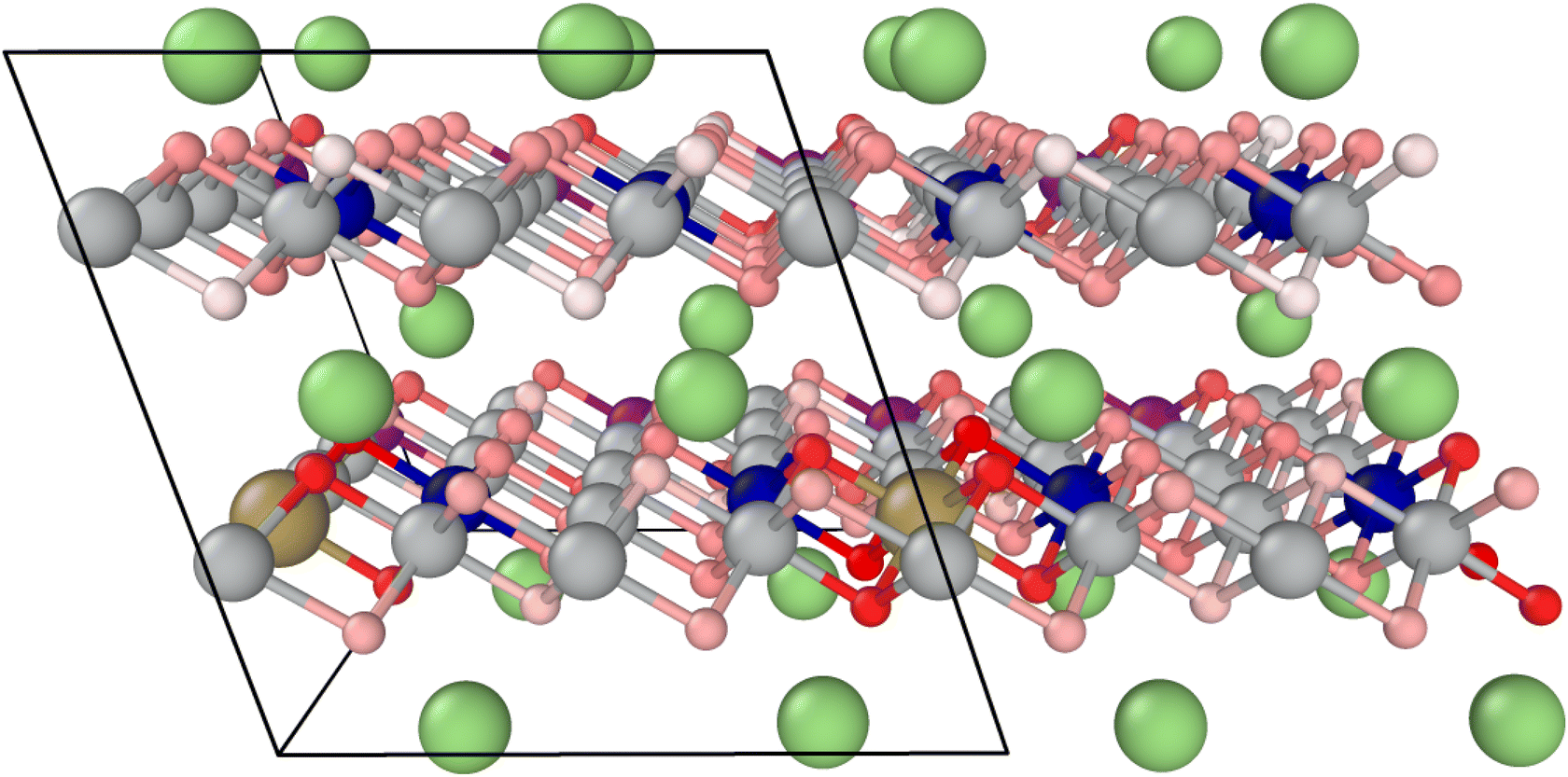 | ||
| Fig. 5 Structural model for Ta-doped NMC811 at 20% Li: oxygen atoms (shades of red) are color-coded according to their vacancy formation energy. | ||
To further investigate the factors influencing oxygen binding, we conducted a correlation analysis on environmental properties surrounding an oxygen vacancy: distance to the closest Ta, Li, O, and Ni atoms, and the Bader charge of the oxygen atom prior to removal (Fig. S7 and Table S3, ESI†). The most pronounced correlation with increased oxygen release energy (0.66) was observed for the distance between the removed oxygen and its nearest neighboring Ni atom. A shorter Ni–O bond suggests electron-depleted bonding, which weakens the oxygen's lattice interaction and facilitates its release. In NMC811, manganese has an oxidation state of 4+, therefore only Ni and Co are redox-active.54 The stronger hybridization of the Ni–O bond at high SOCs drives oxygen oxidation, directly linking it to oxygen release.16 The second strongest correlation (0.58) involved the distance between the removed oxygen and the nearest remaining oxygen atom. A shorter O–O distance reduces Coulomb repulsion and indicates lower electron density, which can precede oxidation and the formation of peroxides.52 The third most significant correlation (0.52) related to the Bader charge of the oxygen atom being removed, where a higher charge indicates stronger bonding and reduced oxygen release tendency. These correlations provide additional insights into oxygen stability mechanisms in the doped material. While the Bader charge provides a useful descriptor, it is not the primary determinant of oxygen binding strength. Instead, the Ni–O bond distance and O–O proximity emerge as critical parameters, with Ta doping stabilizing oxygen by modifying the local electronic and structural environment.
While our study provides insights into the bulk properties of Ta-doped NMC811 through first-principles calculations, its direct applicability to surface effects remains limited. However, recent studies15,55,56 report a significant impact of Ta doping on grain boundaries. Line scans of Ta-doped LNO reveal that dopant concentration is particularly high near the surface and grain boundaries.18 This enrichment often results in the formation of Li–X–O compounds during calcination, such as LiTaO3.57 The bulk calculations of oxygen vacancy formation enthalpies provide valuable insights into how Ta strengthens oxygen–metal bonds. These findings can be extended to understand the role of Ta at grain boundaries, where it may similarly stabilize oxygen atoms. The enrichment of Ta and the formation of Li–Ta–O surface layers likely enhance oxygen binding at grain boundaries, helping to prevent oxygen release and contributing to structural stability. These surface effects complement our bulk-based conclusions, suggesting a protective mechanism against oxygen loss that aligns with the role of Ta in stabilizing oxygen in the bulk.
3.5 OEMS measurement results for gas release
Fig. 6 shows the gas evolution rate, n in nmol min−1 g−1 of O2 and CO2 from a pristine NMC811 and a Ta-doped NMC811 half-cell. Only the first two of four cycles are shown in order to be able to better compare with the DFT calculation results (for characterization and long term cycling please see Dalkilic et al.17 or ESI,† Fig. S10 and S11). Examination of the electrochemical performance reveals that both cells exhibit comparable discharge/charge capacities during the first cycle, with the pristine cell demonstrating a slightly higher coulombic efficiency (CE, 89.1%) compared to the Ta-doped cell (87.8%). Previous works on aluminum-doped NMCs show similar trends.58 In subsequent cycles, the Ta-doped cells seem to perform marginally better than the pristine cells with a CE of 99.9% and 99.0% respectively. This difference implies that Ta-doped cells contain fewer parasitic side reactions, such as oxygen release and/or electrolyte oxidation processes, from the second cycle on. It is important to note here that since the powders were synthesised for this study, no performance enhancing modifications were applied. Most commercial NMC materials on the market today have been altered significantly with additives, coatings and a myriad of additional dopants. As such, all processes observed in this study are due to the NMC material in a relatively “pristine” state, with and without a single modification (Ta dopant).17 | ||
| Fig. 6 Gas evolution rate of O2 and CO2 for pristine and Ta-doped NMC811 cycled galvanostatically against lithium metal at 0.1C between 3.0–4.6 V vs. Li+/Li. | ||
The dominant observable gas is CO2 and is seen to start evolving at 3.8 V vs. Li+/Li in relatively minor amounts compared to a spike in evolution at >4.3 V vs. Li+/Li. The process at 3.8 V is attributed to the oxidation of a variety of impurities in the electrolyte and on the electrode surfaces (resulting in CO2, protons, and organic products).59,60 This is corroborated by the fact that the amount of CO2 observed at 3.8 V on the second and subsequent cycles is significantly lower, implying that the impurities have predominantly already reacted. The profiles of the CO2 evolution are almost identical between the pristine and Ta-doped cells, varying only in the total amount observed, with ∼42 μmol g−1 and ∼29 μmol g−1 in the first and second cycle for Ta-doped and ∼80 μmol g−1 and 37 μmol g−1 in the first and second cycles for the pristine cell respectively. This means that when Ta is present, there is an 88%/29% decrease in CO2 evolution in the first and second cycles. We know from extensive previous publications that Ni-rich layered cathode materials release reactive singlet oxygen at potentials above 4.2 V vs. Li+/Li which in turn reacts with carbonate electrolytes to form CO2, which we clearly observe too.51,53 The fact that the Ta-doped material seemingly releases significantly less CO2 at these high potentials (where oxygen release is the dominant mechanism for electrolyte degradation) implies two potential Ta-induced mechanisms: (a) Ta does in fact mitigate the loss of reactive oxygen, or (b) it alters the NMC particle surface such that fewer electrochemical oxidation processes occur. Further analysis is required to be able to differentiate between the two.
Unexpectedly, we are able to see oxygen evolution during cycling of both the pristine and Ta-doped electrodes. This is a rather uncommon occurrence in OEMS setups, since the singlet oxygen preferentially reacts with the electrolyte before combining to gaseous oxygen (in the triplet state). As mentioned, most NMC cathode materials have been pre-treated with coatings that limit oxygen release; however, since we have uncoated NMC electrodes and relatively high mass loadings (30 mg cm−2), oxygen gas is detectable, albeit roughly 6 times less than the O2 evolution. Gasteiger et al. have additionally shown that triplet oxygen is more likely to form in more porous materials, i.e., with less cracking, suggesting we have electrodes with a higher number of closed pores.21 A unique double peak evolution profile is observed with the expected peak at >4.3 V on charge followed by a second peak at 4.2 V vs. Li+/Li on discharge. Both the Ta-doped and pristine electrodes produce roughly the same amount of total O2 (∼8 μmol g−1 in the first cycle for both and ∼2.6/2.3 μmol g−1 for the pristine/doped electrodes on the second cycle). However, the onset for oxygen release/detection varies considerably between the two electrodes. While the pristine electrode produces more O2 during the charge step than that of the Ta-doped, the opposite is true during the event at 4.2 V on discharge. Although oxygen evolution during discharge has been discussed in prior studies, particularly for LNO,61 the mechanisms and specific conditions under which it occurs remain less explored compared to the charge step. However, since its onset potential corresponds exactly to the NMC structural relaxation occurring after expansion during the H3-to-H2 phase transition, prior studies suggest that oxygen that had been previously trapped within pores/layers while the system was above 4.2 V is now able to be released.62–64
The differences between the two materials in this study are likely attributed to variations in surface area and particle size, which impact the electrode's tortuosity, thereby affecting the rate and timing of gas release. As shown in our sister paper, Ta doping leads to smaller, needle-like primary particles17 (Fig. S9, ESI†). Building on previous discussions of oxygen release, we propose that the presence of Ta in the NMC structure reduces cracking (due to the stronger Ta–O bond and changed morphology65), maintains pore integrity, and traps singlet oxygen. This oxygen can subsequently convert to triplet oxygen and be released (as observed via OEMS) during the H3-to-H2 phase transition in NMC.16,21,66,67 Taking into consideration the aforementioned observations, we can conclude that Ta-doping within the NMC cathode material has a marked effect on the gas evolution profile, specifically the amount of CO2 evolved (and therefore the amount of singlet oxygen released from the layered structure). Ta seems to be able to reduce the amount of reactive oxygen species released at high potentials as well as impact its onset (the charge or discharge step). Further experimental work is necessary however, in order to more closely understand how the presence of Ta is impacting the retention of the reactive oxygen species in the Ni-rich layered structure.
4 Conclusions
In this study, we aimed to enhance the stability and performance of Ni-rich cathode materials by investigating the effects of Ta doping on in-house synthesized NMC811. We approached this problem using both theoretical (DFT calculations) and experimental (OEMS) analyses.Our main theoretical findings reveal that Ta dopants preferentially occupy Ni-rich regions within the material, resulting in a decrease in Ni4+ ions and an increase in Ni2+ and Ni3+ ions due to charge compensation. Additionally, our calculations suggest that oxygen binds more strongly to Ta than to other transition metals, implying that Ta doping could effectively mitigate oxygen release. These theoretical predictions were confirmed by OEMS measurements, which showed that Ta doping reduces oxygen release and subsequent CO2 gassing during the first few cycles by approximately 50% compared to the pristine material. Moreover, the coulombic efficiency increased from 99.00% to 99.9% in the presence of Ta from the second cycle onward, approaching commercial standards—likely due to the reduction of side reactions. We propose that the stronger Ta–O bond inhibits oxygen vacancy-induced side phase formation, thereby reducing cracking in overcharged regions. Also the decrease in Ni oxidation state by Ta reduces the catalytic effect of Ni4+ on surface reactions.
Overall, our findings suggest that Ta doping primarily influences the material's surface, inhibiting oxygen release and improving the stability of Ni-rich cathode materials. Thus, Ta doping emerges as a promising strategy to enhance the longevity and safety of high-capacity batteries. This study also highlights the value of integrating experimental methods with first-principles calculations to achieve a deeper understanding of doping effects at the atomic level. The combined approach can be extended to investigate other dopants, ultimately contributing to the development of more durable and efficient batteries for sustainable transportation.
Data availability
All data supporting the findings of this study are available within the manuscript and its ESI.† Additional data, including raw simulation outputs and experimental measurements, are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Sabrina Sicolo for her valuable guidance and insights during the course of this work. This work was supported by PowerCo SE. All practical work on syntheses related to this work was conducted at Zentrum für Sonnenenergie- und Wasserstoff-Forschung in Ulm, Germany. Analytical work was carried out at the Ångström Laboratory, Sweden.Notes and references
- European Parliament, Regulation (EU) 2023/1542 of the European Parliament and of the Council of 12 July 2023 concerning batteries and waste batteries, amending Directive 2008/98/EC and Regulation (EU) 2019/1020 and repealing Directive 2006/66/EC, Official Journal of the European Union, 2023, L191, 1–104 Search PubMed . Available at: https://eur-lex.europa.eu/eli/reg/2023/1542/oj.
- Z. Xiao, P. Liu, L. Song, Z. Cao, J. Du, C. Zhou and P. Jiang, Ionics, 2021, 27, 3207–3217 CrossRef CAS.
- U. H. Kim, D. W. Jun, K. J. Park, Q. Zhang, P. Kaghazchi, D. Aurbach, D. T. Major, G. Goobes, M. Dixit, N. Leifer, C. M. Wang, P. Yan, D. Ahn, K. H. Kim, C. S. Yoon and Y. K. Sun, Energy Environ. Sci., 2018, 11, 1271–1279 RSC.
- H. H. Sun and A. Manthiram, Chem. Mater., 2017, 29, 8486–8493 CrossRef CAS.
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 CrossRef CAS.
- X. Y. Hou, Y. Kimura, Y. Tamenori, K. Nitta, H. Yamagishi, K. Amezawa and T. Nakamura, ACS Energy Lett., 2022, 1687–1693 CrossRef CAS.
- A. T. Freiberg, M. K. Roos, J. Wandt, R. De Vivie-Riedle and H. A. Gasteiger, J. Phys. Chem. A, 2018, 122, 8828–8839 CrossRef CAS PubMed.
- F. Wu, N. Liu, L. Chen, N. Li, J. Dong, Y. Lu, G. Tan, M. Xu, D. Cao, Y. Liu, Y. Chen and Y. Su, J. Energy Chem., 2021, 62, 351–358 CrossRef CAS.
- Y. Y. Wang, Y. Y. Wang, S. Liu, G. R. Li, Z. Zhou, N. Xu, M. T. Wu and X. P. Gao, Energy Environ. Mater., 2022, 5, 1260–1269 CrossRef CAS.
- T. Li, X. Zi, Y. Lei, Z. Datong, S. Kaiyuan and S. Christina, Degradation Mechanisms and Mitigation Strategies of Nickel-Rich NMC-Based Lithium-Ion Batteries, Springer, Singapore, 2020, vol. 2018, pp. 43–80 Search PubMed.
- J. Cheng, B. Ouyang and K. A. Persson, ACS Energy Lett., 2023, 8, 2401–2407 CrossRef CAS.
- G. Ko, S. Jeong, S. Park, J. Lee, S. Kim, Y. Shin, W. Kim and K. Kwon, Energy Storage Mater., 2023, 60, 102840 CrossRef.
- H. Qian, H. Ren, Y. Zhang, X. He, W. Li, J. Wang, J. Hu, H. Yang, H. M. K. Sari, Y. Chen and X. Li, Surface Doping vs. Bulk Doping of Cathode Materials for Lithium-Ion Batteries: A Review, Springer Nature, Singapore, 2023, vol. 6, pp. 1–32 Search PubMed.
- P. Hou, H. Zhang, Z. Zi, L. Zhang and X. Xu, J. Mater. Chem. A, 2017, 5, 4254–4279 RSC.
- N. Y. Park, G. Cho, S. B. Kim and Y. K. Sun, Adv. Energy Mater., 2023, 13, 1–10 Search PubMed.
- R. Sim, Z. Cui and A. Manthiram, ACS Energy Lett., 2023, 5143–5148 CrossRef CAS.
- M. Dalkilic, A. Schmidt, T. D. Schladt, P. Axmann, M. Wohlfahrt-Mehrens and M. Lindén, J. Electrochem. Soc., 2022, 169, 090504 CrossRef CAS.
- N. Y. Park, S. B. Kim, M. C. Kim, S. M. Han, D. H. Kim, M. S. Kim and Y. K. Sun, Adv. Energy Mater., 2023, 13, 1–9 Search PubMed.
- S. L. Dreyer, P. Kurzhals, S. B. Seiffert, P. Müller, A. Kondrakov, T. Brezesinski and J. Janek, J. Electrochem. Soc., 2023, 170, 060530 CrossRef CAS.
- Y.-R. Luo and J. Alistair Kerr, Bond dissociation energies, CRC handbook of chemistry and physics, 89 edn, 2012, pp. 65–98 Search PubMed.
- S. Oswald and H. A. Gasteiger, J. Electrochem. Soc., 2023 DOI:10.1149/1945-7111/acbf80.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- MedeA 3.6 (Materials Design, San Diego, USA). MedeA is a registered trademark of Materials Design, Inc.
- M. Sadowski, L. Koch, K. Albe and S. Sicolo, Chem. Mater., 2022, 35, 584–594 CrossRef.
- S. Sicolo, M. Sadowski, K. Vettori, M. Bianchini, J. Janek and K. Albe, Chem. Mater., 2023, 36, 492–500 CrossRef.
- J. L. Kaufman, J. Vinckevičicutė, S. Krishna Kolli, J. Gabriel Goiri and A. Van Der Ven, Philos. Trans. R. Soc., A, 2019, 377, 20190020 CrossRef CAS.
- S. Sicolo, M. Mock, M. Bianchini and K. Albe, Chem. Mater., 2020, 32, 10096–10103 CrossRef CAS.
- M. M. E. Cormier, N. Zhang, A. Liu, H. Li, J. Inglis and J. R. Dahn, J. Electrochem. Soc., 2019, 166, A2826–A2833 CrossRef CAS.
- X. Li, Q. Wang, H. Guo, N. Artrith and A. Urban, ACS Appl. Energy Mater., 2022, 5, 5730–5741 CrossRef CAS.
- N. Artrith, J. A. Garrido Torres, A. Urban and M. S. Hybertsen, Phys. Rev. Mater., 2022, 6, 1–13 Search PubMed.
- M. Mock, M. Bianchini, F. Fauth, K. Albe and S. Sicolo, J. Mater. Chem. A, 2021, 9, 14928–14940 RSC.
- A. Chakraborty, S. Kunnikuruvan, S. Kumar, B. Markovsky, D. Aurbach, M. Dixit and D. T. Major, Chem. Mater., 2020, 32, 915–952 CrossRef CAS.
- J. Ning, M. Kothakonda, J. W. Furness, A. D. Kaplan, S. Ehlert, J. G. Brandenburg, J. P. Perdew and J. Sun, Phys. Rev. B, 2022, 106, 1–17 Search PubMed.
- D. Lerch, O. Wieckhorst, G. L. Hart, R. W. Forcade and S. Müller, Modell. Simul. Mater. Sci. Eng., 2009, 17, 055003 CrossRef.
- J. H. Yang, T. Chen, L. Barroso-Luque, Z. Jadidi and G. Ceder, npj Comput. Mater., 2022, 8, 1–11 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2009, 015012 Search PubMed.
- P. A. Olsson and J. Blomqvist, Comput. Mater. Sci., 2017, 139, 368–378 CrossRef CAS.
- X. Yu, W. A. Yu and A. Manthiram, Small Methods, 2021, 2001196, 1–30 Search PubMed.
- G. Liang, V. K. Peterson, K. W. See, Z. Guo and W. K. Pang, J. Mater. Chem. A, 2020, 8, 15373–15398 RSC.
- J. P. Peres, F. Weill and C. Delmas, Solid State Ionics, 1999, 116, 19–27 CrossRef CAS.
- V. N. Kothalawala, A. A. Sasikala Devi, J. Nokelainen, M. Alatalo, B. Barbiellini, T. Hu, U. Lassi, K. Suzuki, H. Sakurai and A. Bansil, Condens. Matter, 2022, 7, 1–9 Search PubMed.
- H. Das, A. Urban, W. Huang and G. Ceder, Chem. Mater., 2017, 29, 7840–7851 CrossRef CAS.
- J. M. Lim, H. Kim, K. Cho and M. Cho, Extreme Mech. Lett., 2018, 22, 98–105 CrossRef.
- J. Lin, M. Li, Z. Lv, J. Luo, B. Wu and R. Hong, J. Electron. Mater., 2023, 52, 7833–7841 CrossRef CAS.
- Y.-G. Zou, H. Mao, X.-H. Meng, Y.-H. Du, H. Sheng, X. Yu, J.-L. Shi and Y.-G. Guo, Angew. Chem., Int. Ed., 2021, 60, 26535–26539 CrossRef CAS.
- B. Ouyang and Y. Zeng, Nat. Commun., 2024, 15, 1–5 Search PubMed.
- J. Wandt, A. T. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS.
- A. R. Genreith-Schriever, H. Banerjee, A. S. Menon, E. N. Bassey, L. F. Piper, C. P. Grey and A. J. Morris, Joule, 2023, 7, 1623–1640 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- H. J. Shin, M. Kim, N. Kim, C. Jung, J. G. Chung, K. H. Kim and W. S. Jeon, J. Phys. Chem. Solids, 2021, 148, 109732 CrossRef CAS.
- Y. B. Hendri, L. Y. Kuo, M. Seenivasan, Y. S. Wu, S. H. Wu, J. K. Chang, R. Jose, M. Ihrig, P. Kaghazchi and C. C. Yang, J. Colloid Interface Sci., 2024, 661, 289–306 CrossRef CAS.
- H. H. Sun, U. H. Kim, J. H. Park, S. W. Park, D. H. Seo, A. Heller, C. B. Mullins, C. S. Yoon and Y. K. Sun, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- X. Li, W. Ge, K. Zhang, G. Peng, Y. Fu and X. Ma, Electrochim. Acta, 2022, 403, 139653 CrossRef CAS.
- M. Leißing, F. Horsthemke, S. Wiemers-Meyer, M. Winter, P. Niehoff and S. Nowak, Batteries Supercaps, 2021, 4, 1344–1350 CrossRef.
- M. Metzger, P. Walke, S. Solchenbach, G. Salitra, D. Aurbach and H. A. Gasteiger, J. Electrochem. Soc., 2020, 167, 160522 CrossRef CAS.
- B. L. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS PubMed.
- L. de Biasi, A. Schiele, M. Roca-Ayats, G. Garcia, T. Brezesinski, P. Hartmann and J. Janek, ChemSusChem, 2019, 12, 2240–2250 CrossRef CAS PubMed.
- K. Zhou, Q. Xie, B. Li and A. Manthiram, Energy Storage Mater., 2021, 34, 229–240 CrossRef.
- M. Juelsholt, J. Chen, M. A. Pérez-Osorio, G. J. Rees, S. De Sousa Coutinho, H. E. Maynard-Casely, J. Liu, M. Everett, S. Agrestini, M. Garcia-Fernandez, K. J. Zhou, R. A. House and P. G. Bruce, Energy Environ. Sci., 2024, 17, 2530–2540 RSC.
- J. J. Marie, R. A. House, G. J. Rees, A. W. Robertson, M. Jenkins, J. Chen, S. Agrestini, M. Garcia-Fernandez, K. J. Zhou and P. G. Bruce, Nat. Mater., 2024, 23, 818–825 CrossRef CAS PubMed.
- U. H. Kim, G. T. Park, B. K. Son, G. W. Nam, J. Liu, L. Y. Kuo, P. Kaghazchi, C. S. Yoon and Y. K. Sun, Nat. Energy, 2020, 5, 860–869 CrossRef CAS.
- J. Schmitt, B. Kraft, J. P. Schmidt, B. Meir, K. Elian, D. Ensling, G. Keser and A. Jossen, J. Power Sources, 2020, 478, 228661 CrossRef CAS.
- C. Misiewicz, R. Lundström, I. Ahmed, M. J. Lacey, W. R. Brant and E. J. Berg, J. Power Sources, 2023, 554, 23231 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03911d |
| This journal is © the Owner Societies 2025 |