
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lanthanide chloride clusters, LnxCl3x+1−, x = 1–6: an ion mobility and DFT study of isomeric structures and interconversion timescales†
Yuto
Nakajima
a,
Patrick
Weis
 *b,
Florian
Weigend
*bc,
Marcel
Lukanowski
c,
Fuminori
Misaizu
a and
Manfred M.
Kappes
*bd
*b,
Florian
Weigend
*bc,
Marcel
Lukanowski
c,
Fuminori
Misaizu
a and
Manfred M.
Kappes
*bd
aDepartment of Chemistry, Graduate School of Science, Tohoku University, Japan
bInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Germany. E-mail: patrick.weis@kit.edu; florian.weigend@kit.edu; manfred.kappes@kit.edu
cInstitute of Quantum Materials and Technologies, Karlsruhe Institute of Technology (KIT), Germany
dInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Germany
First published on 22nd November 2024
Abstract
Ion mobility spectrometry (IMS) (also including IMS–IMS measurements) as well as DFT calculations have been used to study isomer distributions and isomer interconversion in a range of electrospray-generated lanthanide chloride cluster anions, LnxCl3x+1− (where x = 1–6, and Ln corresponds to the 15 lanthanide elements (except for radioactive Pm)). Where measurement and structural rearrangement timescales allow, we obtain almost quantitative agreement between experiment and theory thus confirming isomer predictions and reproducing isomer intensity ratios. LnxCl3x+1− structures reflect strong ionic bonding with limited directionality. Ring and chain motifs dominate for smaller clusters while for larger clusters more compact three-dimensional structures become favourable. At cluster sizes with two or more closely lying isomers, the lanthanide contraction can lead to systematic variations in structure types across the series.
1. Introduction
Structural isomers and their thermally activated interconversion have been of chemical interest at least since Berzelius introduced the term isomerism almost 200 years ago.1 Since then, molecular isomerism has been extensively studied – often in liquid solution. Probing isomer distributions in the gas phase under high vacuum conditions helps to better address intrinsic aspects of their thermodynamics and dynamics because most environmental perturbations can be eliminated. Mass spectrometry (MS) based methods are well suited for this also because they can provide rigorous mass-selectivity which further reduces complexity. Ion mobility spectrometry (IMS) is a particularly powerful MS-based method with which to probe isomers. It allows us to measure the collision cross-sections (CCSs) and thus the structure of molecular ions. Recent technical developments have made CCS resolutions (CCS/delta CCS) approaching 1000 coupled with a mass resolution of 105 available in IMS–MS. Thus, two or more structural isomers of large molecular ions with CCS values differing only slightly can now be routinely distinguished which opens up increasingly larger regions of chemical space for gas-phase probes of molecular isomerism.Several different commercial IMS–MS platforms are available for measuring CCS values. All use some kind of reaction cell in which the ion of interest is collided with an inert collision gas and the transmission time through the cell is measured (similar to the retention time in chromatography). All such platforms have in common that higher CCS resolutions require longer measurement times. This characteristic time is typically on the order of hundreds of microseconds to hundreds of milliseconds, presently limited at the bottom end by the required CCS resolution to resolve the small structure changes of interest and at the top end by the instrumental duty cycle.
To gauge utility for structural characterization of isomers, this characteristic measurement time has to be related to the characteristic time for isomer interconversion. It is instructive to consider this for the simplest possible case of two thermalized isomers under high vacuum conditions which we will assume to be able to interconvert much more rapidly than any other competing reaction channels such as dissociation. If the interconversion rates are also much larger than the inverse measurement time for CCS determination, only one averaged CCS value is observable – even though two structurally distinct species are present during a large part of the measurement. By contrast, if both the forward and reverse unimolecular isomerization rates are significantly smaller than the inverse time required for CCS determination, the two isomers can be discriminated by IMS and their differing CCS values (and structures) determined with the resolution of the instrument.
The corresponding theory of the transport of internally reacting ions through a collision gas was first developed by Gatland in 1974 (for atomic ions undergoing reactions which “return…” them “to their former species”, i.e. electronic states).2 It was later reformulated and applied e.g. to measurements of isomer interconversion in cluster ions3,4 as well as more recently to interconverting organometallic complexes.5,6 The formalism can be used to fit arrival time distributions obtained under different experimental conditions, e.g. of varied transit times and temperatures, to obtain thermal rates for forward and reverse reactions and from them to gauge relative activation energies as well as Gibbs free energy differences between isomers.
50 years since Gatland's original derivation of the transport equations governing interconverting molecular isomers, IMS–MS studies have become routine in many areas of chemistry. It is therefore surprising that the IMS literature still contains comparatively few comprehensive experimental studies of isomer interconversion in an ensemble of isolated molecular ions. In part, this is due to the lack of suitable instrumentation with which to identify candidate systems – the setup should not only allow to determine CCS (distributions) at sufficient resolution but also to select, reinject/activate and probe specific isomers from within the isomer distribution (IMS–IMS) – ideally at variable vibrational temperatures. Recent advances in commercial IMS technology now allow routine IMS–IMS probing of ever larger and structurally more diverse molecules. Thus, new families of interconverting isomers are being uncovered which interconvert measurably while remaining stable towards dissociation.
In this study, we report such a system, lanthanide chloride LnnCl3n+1− clusters, and study it in detail. Lanthanide halide LnnX3n+1− clusters with their relatively high Ln atom counts also offer an ideal test system to probe the lanthanide contraction both experimentally in the gas phase and theoretically. Due to their insoluble nature, it is difficult to experimentally synthesize lanthanide fluoride clusters; therefore, we focus on chlorides. Specifically, isomer distributions and isomer interconversion have been characterized for a range of lanthanide chloride cluster anions, LnxCl3x+1−, x = 1–6, where Ln corresponds to each of the 15 lanthanide elements (except for radioactive Pm). From the pioneering work of Rutkowski et al.,7,8 it is known that such clusters can be readily formed from liquid solutions of lanthanide chlorides by electrospray ionization. They in fact combined gas-phase (electrospray-ionization mass spectrometry) and density functional theory computations to assign structures for several such species, LaxCl3x+1− and LuxCl3x+1− (x = 1–6). Apart from the consequences of x-dependent structural isomerism, the full range of LnxCl3x+1− offers an additional internal knob to influence properties: the lanthanide contraction. We will show below that this systematic change in Ln(III) ionic radius by more than 16% from Ln = La to Lu leads to corresponding changes in the relative stabilities of different common isomer types (at a given cluster size). This has consequences not only for thermodynamic properties but also for isomer interconversion. We have systematically probed these effects for LnxCl3x+1− (x = 1–6) held near room temperature using (i) a combination of high resolution IMS–MS (and IMS–IMS–MS), as implemented on a Waters select cyclic IMS platform, (ii) DFT calculations (using newly designed polarized effective core potential based triple zeta valence basis sets,9 lcecp-1-TZVP, developed for 4f-elements in anticipation of this study) to generate plausible structural models and (iii) trajectory method calculations to compare the structural models obtained by DFT with mobility measurements. We found isomer interconversion to be faster than our experimental timescale for x = 2 and at x = 4 and 5 for specific Ln elements. For x = 6 (and Ln = Sm–Lu), we are able to resolve two isomers which do not interconvert on the experimental timescale but which can be cleanly interconverted by moderate collisional excitation – at overall internal energies still significantly below that required for dissociation. The Ln-dependence of the isomer ratio observed for as-prepared hexamers correlates with the Ln-trend predicted on the basis of relative free energies from DFT calculations suggesting that near equilibrium conditions are being probed. On the basis of our measurements and calculations, we are able to assign isomeric structures for all sizes studied.
2. Experimental methods and results
2.1 ESI mass spectra
To generate the corresponding isolated cluster ions, solutions of ca. 0.05 mmol l−1 LnCl3 (Ln = La–Lu, except Pm) in isopropanol were electrosprayed into a Waters select series cyclic ion mobility mass spectrometer. Ref. 10 and 11 provide a detailed description of the setup, measurement principles, and multi-cycle calibration procedure. In negative mode, clusters with the composition LnnCl3n+1− (n ≤ 6) are formed. A typical mass spectrum (using LuCl3 as an example) is shown in Fig. 1.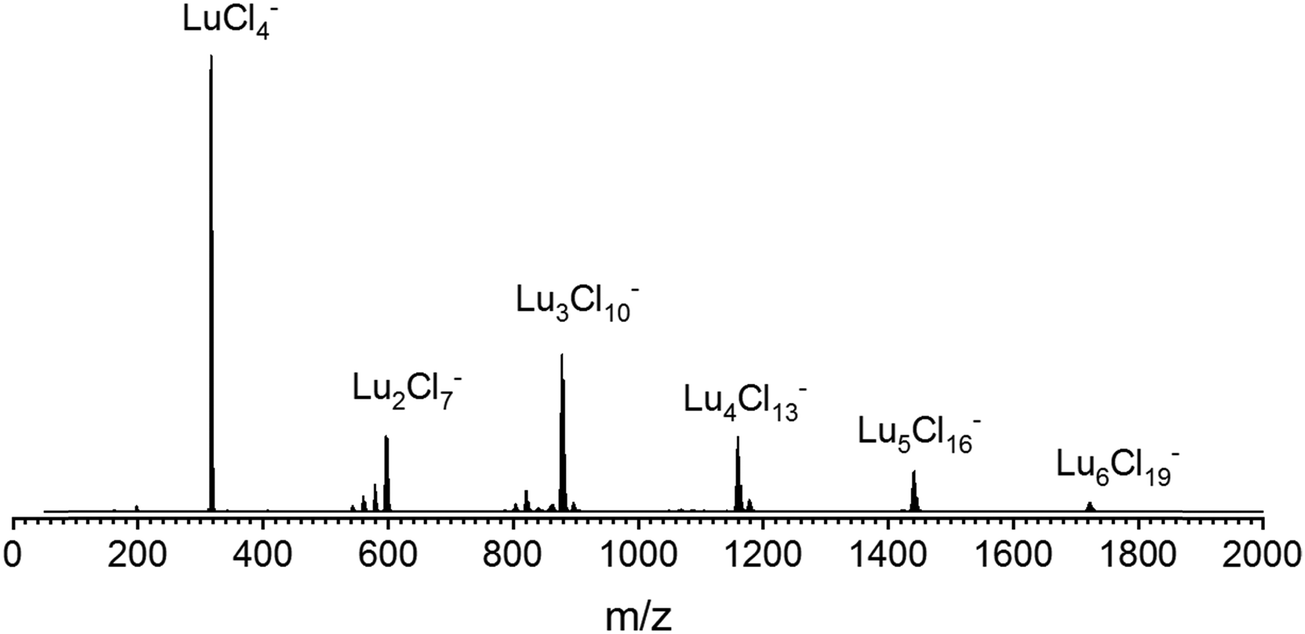 | ||
| Fig. 1 An overview ESI mass spectrum of a solution of 0.05 mmol l−1 LuCl3 in isopropanol, negative mode. The small peaks in front of the Lu2Cl7− and Lu3Cl10− signals correspond to Lu2Cl7−x(OH)x− (x = 1–3) and Lu3Cl10−x(OH)x− (x = 1–4), respectively. The small peaks after Lu3Cl10− and Lu4Cl13− correspond to water adducts, i.e. Lu3Cl10(H2O)− and Lu4Cl13(H2O)−, respectively. The other LnCl3 solutions showed analogous overview mass spectra. The IMS measurements reported below were performed under mass-selective conditions, i.e. results are specific to LnxCl3x+1−, x = 1–6, throughout (no contribution of –OH or H2O adducts). | ||
Solid LnCl3 samples were obtained mostly as hydrate salts from commercial sources and were used without further purification: LaCl3, HoCl3, LuCl3(H2O)6, and TmCl3(H2O)6 from Sigma Aldrich, NdCl3(H2O)x, SmCl3(H2O)x, TbCl3(H2O)x, and ErCl3(H2O)x from Chempur, CeCl3(H2O)7, DyCl3(H2O)6, and GdCl3(H2O)6 from Fluorchem, EuCl3(H2O)x from Alfa Aesar and PrCl3(H2O)x from Angene.
2.2 Ion mobility measurements
For subsequent ion mobility analysis, the respective cluster sizes were first isolated in the quadrupole mass filter prior to the ion mobility cell in order to exclude any contamination of the mobilograms of the smaller cluster sizes by possible fragmentations of larger species during the ion mobility measurement. Furthermore, the mobility analysis is performed only for a narrow mass window (depending on the isotope pattern of the respective Ln) selected using instrument software (MassLynxV4.2). The ion mobility measurement is performed in a cyclic ion mobility cell (cIM) combined with a pre-storage device. The effective drift length and accordingly the IMS resolution can be modified by adjusting the number of cycles. The resolution (and measurement time) increases with the number of cycles. The arrival time distributions after 10 cycles are shown in Fig. 2 for all LnxCl3x+1− studied. As a general trend (at a given x), we observe the peak arrival time decreasing almost linearly from La to Lu in line with the lanthanide contraction. There are however some exceptions: for Ln4Cl13−, we observe a strong decrease in the arrival time from La to Nd while from Sm to Lu, the arrival time is almost constant. For Ln5Cl16−, the arrival time decreases from La to Ce, as expected, but increases from Ce to Nd, and therefore Nd has the largest arrival time along the series. For all LnxCl3x+1− (x ≤ 5), we found a single, sharp peak in the arrival time distribution with the peak width corresponding to the instrumental resolution. For Ln6Cl19−, the arrival time distribution is bimodal for Ln = Sm–Lu, with the relative intensity of the first peak decreasing from 94% (Sm) to 30% (Lu).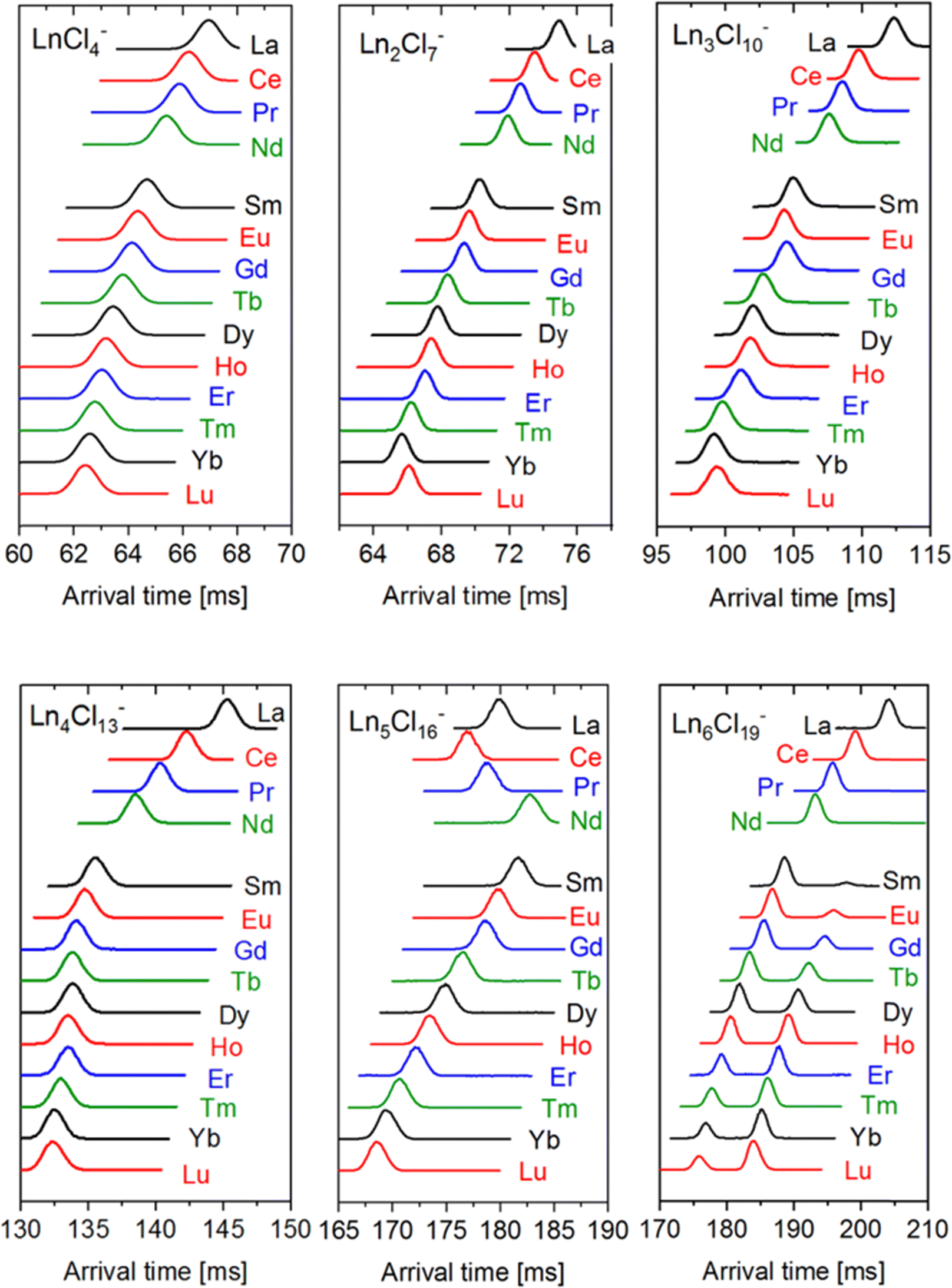 | ||
| Fig. 2 Typical arrival time distributions of LnxCl3x+1− anions after 10 cycles (for given x =1–6 as indicated). A travelling wave (TW) speed of 375 m s−1 was used in all cases. For x = 2–6, a TW height of 18 V was used. LnCl4− (x = 1) was measured with a TW height of 14 V (at 18 V the large mobility of this ion causes undesirable “surfing” on the TW rather than CCS dependent separation). | ||
Arrival times depend on many instrumental parameters which need to be taken into account before comparing with predictions from theory. Furthermore, the arrival time of an ion consists of the time it needs to pass the cIM and the (mass-dependent) transfer time to the detector.10 We eliminate transfer time contributions by measuring the arrival time after typically 1, 5 and 10 cycles and determining the time-per-cycle by a linear fit of the arrival time vs. the number of cycles. This time still depends on many instrumental parameters such as buffer gas (nitrogen) pressure, temperature, speed and height of the travelling wave. In order to obtain a device-independent collision cross-section (TWCCSN2), we calibrate against the set of “Agilent tunemix” ions using the CCS values of Stow et al.12 This calibration procedure is performed each day under exactly the same conditions as the mobility measurements of the lanthanide chloride clusters. All measurements were done at least in triplicate on different days. This workflow resulted in highly reproducible CCS values with statistical errors in most cases below 0.2%. The values are summarized in Table 1 and Fig. 3.
| LnCl4− | Ln2Cl7− | Ln3Cl10− | Ln4Cl13− | Ln5Cl16− | Ln6Cl19− | ||
|---|---|---|---|---|---|---|---|
| La | 116.8 | 150.3 | 176.5 | 196.6 | 216.0 | 228.7 (100%) | |
| Ce | 116.4 | 149.1 | 174.8 | 194.9 | 214.3 | 226.2 (100%) | |
| Pr | 116.0 | 148.3 | 173.8 | 193.6 | 215.3 | 224.4 (100%) | |
| Nd | 115.7 | 147.8 | 173.2 | 192.5 | 217.4 | 223.0 (100%) | |
| Sm | 115.1 | 146.3 | 171.5 | 190.7 | 216.8 | 220.5 (94%) | 225.5 (6%) |
| Eu | 114.8 | 145.8 | 170.9 | 190.2 | 215.7 | 219.5 (89%) | 224.3 (11%) |
| Gd | 114.6 | 145.5 | 171.0 | 189.7 | 215.1 | 218.8 (80%) | 223.6 (20%) |
| Tb | 114.2 | 144.7 | 169.8 | 189.6 | 214.0 | 217.6 (69%) | 222.4 (31%) |
| Dy | 114.0 | 144.1 | 169.2 | 189.6 | 213.0 | 216.7 (55%) | 221.4 (45%) |
| Ho | 113.9 | 143.8 | 169.0 | 189.4 | 212.3 | 215.9 (47%) | 220.6 (53%) |
| Er | 113.7 | 143.6 | 168.8 | 189.5 | 211.7 | 215.5 (42%) | 220.1 (58%) |
| Tm | 113.5 | 142.8 | 167.7 | 189.0 | 210.7 | 214.4 (38%) | 219.0 (62%) |
| Yb | 113.3 | 142.4 | 167.4 | 188.8 | 210.2 | 214.1 (33%) | 218.7 (67%) |
| Lu | 113.1 | 142.7 | 167.5 | 188.8 | 209.5 | 213.4 (30%) | 217.8 (70%) |
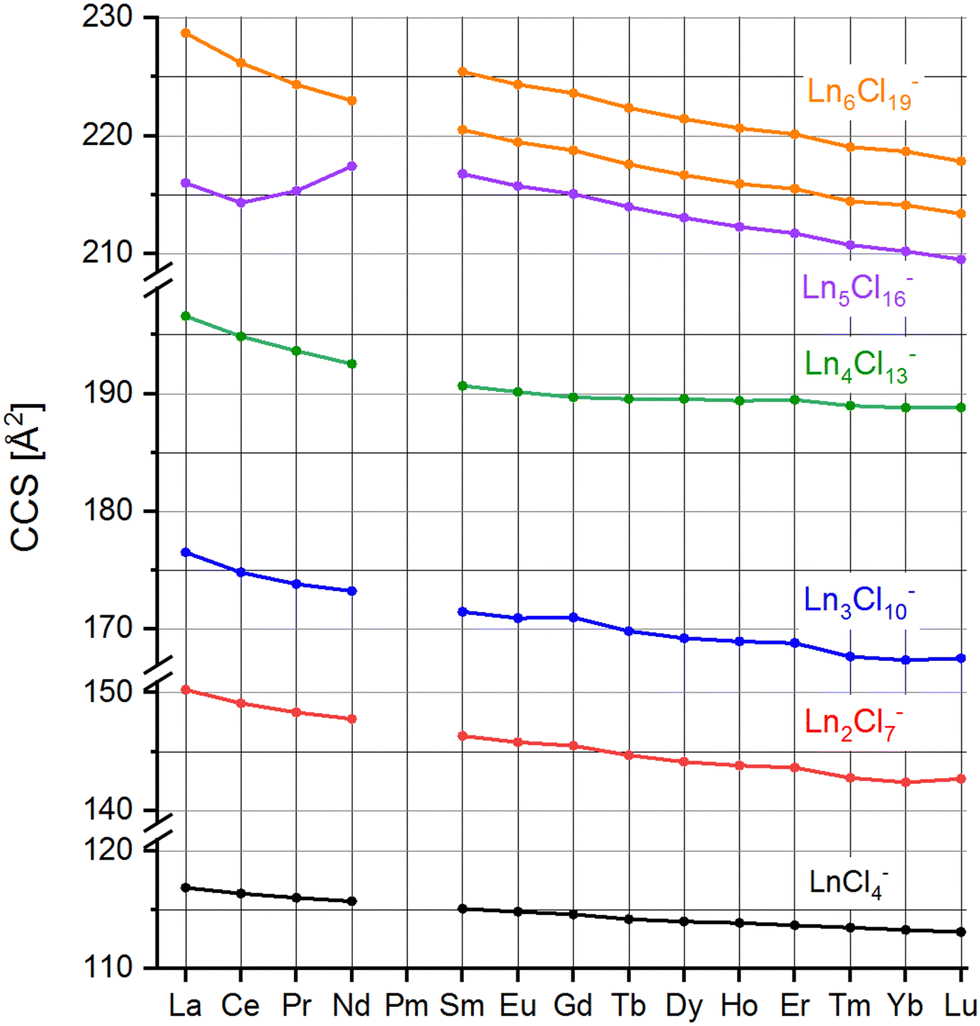 | ||
| Fig. 3 Collision cross-sections TWCCSN2 of LnxCl3x+1−, x = 1–6, Ln = La–Lu except Pm (see Table 1). | ||
2.3 IMS–IMS experiment
The Ln6Cl19− arrival times show a bimodal distribution for Sm–Lu, with the relative intensity of the second (larger CCS) peak increasing smoothly from 6% for Sm6Cl19− to 70% for Lu6Cl19−. The question that arises is whether this is merely a consequence of the ionisation process (solvent, concentration, and source voltages), i.e. some kinetic trapping of the solution phase composition, or whether it reflects the thermodynamic stability of the two isomers in the gas phase. In a first step, we widely varied the source conditions (needle voltage: 2.5–4 kV and cone voltage: 40–80 V) but could not find a significant change in isomer compositions (within 5% relative intensity). A unique feature of the Waters cyclic instrument (among commercial systems) is the capability to perform IMS–IMS experiments with a collisional activation step between the first and second IMS step. The workflow for it is as follows: for the mass selected Ln6Cl19−, we first perform a five-cycle separation sequence (5 cycles are enough to baseline separate the two peaks). In a second step, we remove the ions corresponding to either the first or the second peak by switching the cIM at the appropriate time. The remaining ion packet is transferred into the pre-store (an ion trap located prior to the cIM).10 Third, these ions are reinjected into the cIM with an adjustable kinetic energy (0–100 eV, lab frame (against N2 collision gas)) and cycled for another 5 cycles. The results are shown in Fig. 4a for Ho6Cl19− as an example. When the first peak “isomer 1” is selected, then transferred into pre-store, reinjected with nominally 0 eV and separated for another 5 cycles, only the unchanged peak at 199 ms is observed, corresponding to the unchanged arrival time of isomer 1. When the injection energy is increased, a second peak at 203 ms appears that corresponds to isomer 2. Finally, at 60 eV, this second peak dominates. If the second peak (“isomer 2”, Fig. 4b) is isolated first and reinjected with 0 eV, only the peak at 203 ms corresponding to isomer 2 is observed. With increasing injection energy, the peak at 199 ms appears, corresponding to isomer 1. At 60 eV and above, we see the same intensity ratio, 35% of isomer 1 and 65% of isomer 2, independent of which isomer is selected in the first place (Fig. 4c). This is a clear indication that during the annealing step associated with reinjection, (i) ions become sufficiently excited to rapidly interconvert multiple times before (ii) cooling back down to room temperature on a timescale faster than one cIM cycle, i.e. there is no further interconversion in the cIM (otherwise we would observe a single peak at the weighted average of the two isomers, or at least a significant filling of the baseline in between the two peaks).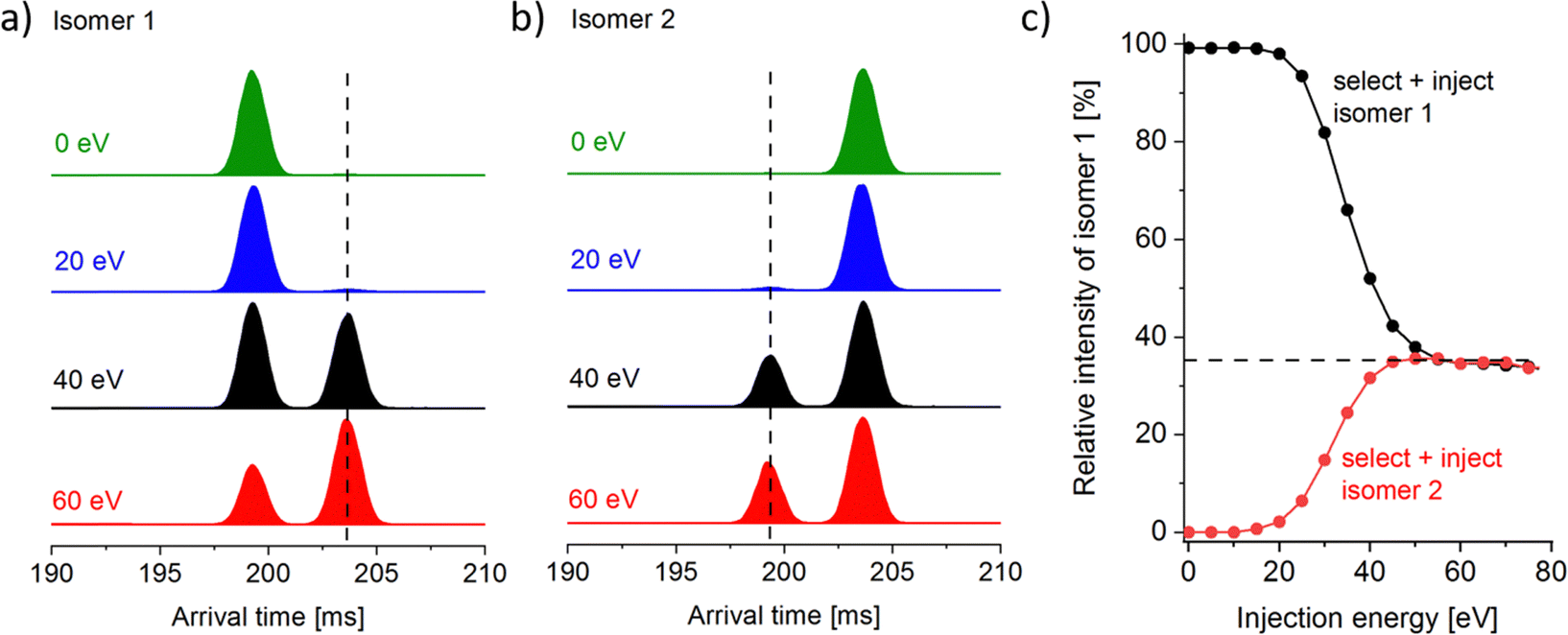 | ||
| Fig. 4 IMS–IMS experiment for Ho6Cl19−. (a) The first peak of the arrival time distribution (isomer 1 shown after 5 cycle separation) is selected, transferred into the pre-store, reinjected with variable kinetic energy and separated for another 5 cycles. (b) The same workflow with the second peak selected (isomer 2). (c) The isomer distribution as a function of injection energy. It should be noted that this is the same for either prehistory (a) or (b). | ||
For the other lanthanides (Sm–Lu), different equilibrium isomer ratios are observed, see Fig. 5. In all cases, the isomer ratio does not depend on the isomer that is selected and reinjected (Fig. 5, black and red data points). Across the lanthanide series, there is a clear trend: the relative intensity of isomer 1 decreases strongly from 70% for Sm to 21% for Lu. This parallels the observed intensity ratios observed by electrospraying the respective lanthanide chloride solution (and by measuring the “as-prepared” isomer composition with minimal injection energy (Fig. 5, green data points)).
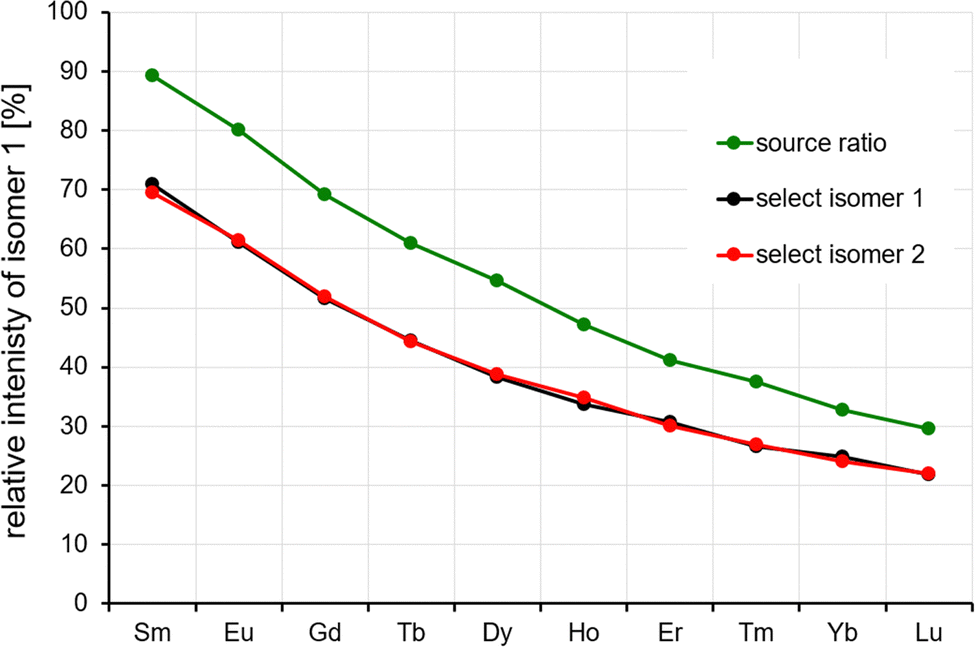 | ||
| Fig. 5 IMS–IMS experiment for Ln6Cl19−, Ln = Sm–Lu. The observed relative intensities at 50 eV reinjection energy are independent of the choice of the isomer stored and reinjected (red and black data points). For comparison, the isomer ratios observed directly from the source are shown as green data points. See the text. | ||
However, the ratios are slightly different quantitatively, probably due to the fact that the two different experimental workflows are associated with different excitation, cooling and detection histories.
3. Theoretical methods and results
3.1 Quantum chemical calculations
The energy surfaces of LnxCl3x+1−, Ln = La–Lu and x = 2–6, were investigated with TURBOMOLE13 in the following way. First, for each x and each Ln, a genetic algorithm procedure14 was carried out, scanning in total more than 5000 isomers for each Ln at the level PBE/lcecp-1-SVP, i.e. employing the PBE functional15 with def2-SVP bases16 for Cl; for lanthanides, large-core fn−1 effective core potentials17,18 were employed (covering, e.g. for Pr, the inner shells plus two f electrons) together with newly designed error-balanced polarized double zeta basis sets9 lcecp-1-SVP. For a given x, the structure lowest in energy and all energetically following, up to ca. 100 kJ mol−1, were collected for all Ln, and then redundant structures were removed. In this way, typically about three isomers for each size were obtained. These sets of structures were optimized for all Ln at the level PBE/lcecp-1-TZVP9 with fine grids (grid size 519) and tight convergence (energy to 10−9Eh). The optimized structures were symmetrized to the highest symmetry, which in the case of imaginary frequencies was again lowered until their disappearance. For estimating the consistency with other methods, the resulting structures were additionally re-optimized at the level TPSS20/lcecp-1-TZVP and for x = 2 additionally PBE021/lcecp-1-TZVP and PBE0/lcecp-1-QZVP,9 and further with the all-electron scalar relativistic X2C method22,23 with the PBE0 functional and x2c-TZVPall24 as well as x2c-QZVPall25 basis sets. Structure parameters for all these methods were compared for the monomer. Furthermore, for each species, vibrational spectra were calculated, which yielded real frequencies throughout and served for the free energy calculation within the harmonic oscillator rigid rotor model at 0, 300 and 600 K. Finally, transition pathways between the lowest and the second-lowest isomer were optimized with a generalized Newton-method26 at the level PBE/lcecp-1-TZVP and the identified maxima were fine-optimized employing the corresponding tool in TURBOMOLE with default options.3.2 Comparison of experimental and calculated CCS
The experimental TWCCSN2 can be used to rule out or confirm the quantum chemically determined candidate structures by comparison with calculated theoCCSN2. This is done by trajectory method calculations as implemented in IMoS1.09.27,28 The interaction potential between the drifting ion and a nitrogen buffer gas molecule is modelled (atom by atom) by a combination of Lennard-Jones, ion-induced dipole and ion-quadrupole interactions with the following assumptions: the Lennard-Jones parameters εl and σl are treated as element specific, the ion-induced dipole is modelled with the polarizability of nitrogen (αN2) and the partial charges ql (from ESP-fit) on each atom. For the ion-quadrupole interaction, the quadrupole moment of nitrogen is modelled by 3 partial charges qi located at appropriate distances.29While for H, C, O, N and F the respective Lennard-Jones parameters have been optimized in IMoS using a test-set of small (covalently bound, organic) molecules;30 for other elements, only the default values of ε = 2.6 meV and σ = 3.5 Å are implemented in IMoS. With these values, we obtain theoCCSN2 that are ca. 7% too small; therefore, a further calibration process is needed. In test calculations, it turns out that the influence of σLn on the calculated CCS is ca. ten-fold smaller than σCl (see Fig. 6). We therefore opt to use the same values for all Ln while optimizing the parameters for Cl. εCl and σCl are correlated, i.e. several combinations thereof give similar CCS. In order to stick as close as possible to the IMoS default parameters, we have kept ε constant at its default value of 2.6 meV and have only adjusted σCl.
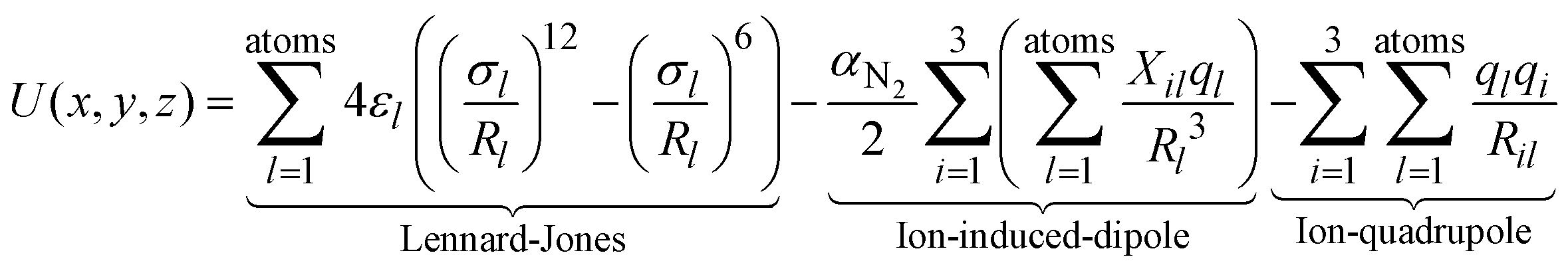
 | ||
| Fig. 6 Experimental (black, with error bars; TWCCSN2) and calculated CCS (red, blue, green; theoCCSN2) for LnCl4−. Red: IMoS-parameters σCl = 3.85 Å and (constant) σLn = 3.00 Å, blue: σCl = 3.95 Å and (constant) σLn = 3.00 Å, green: σCl = 3.95 Å and σLn linearly decreasing from 3.00 Å for La to 2.8 Å for Lu. It should be noted that σLn has comparatively little influence on theoCCSN2 of the monomers because the collision gas scatters from the enclosing chloride shell. | ||
In the following, we have used our TWCCSN2 values for LaCl4−, La3Cl10−, La4Cl13− and La6Cl19− as calibration points to iteratively find an optimal value for σCl because, based on the DFT calculations (see Section 3.1 and also below), there is little doubt about the respective energetically favored structures: LaCl4− is a tetrahedron, La3Cl10− and La4Cl13− are ring structures and La6C19− is an octahedron; other isomers are energetically significantly less favorable. The tabulated ionic radii of Cl−, La3+ and Lu3+ (in coordination number 6) are 1.81 Å, 1.032 Å and 0.861 Å,31 respectively, i.e. on average the Ln3+ ion is expected to be 0.8–0.9 Å smaller than the Cl− ion. Keeping this difference in mind we find for σCl = 3.87 Å and σLa = 3.00 Å a close fit (within 1%) between the experimental (196.6 Å2) and calculated (195.0 Å2) CCS of La4Cl13−. If we calculate theoCCSN2 of monomer LaCl4− with these parameters, we find that it is 1.9% below the experimental value (114.5 Å2vs. 116.8 Å2). For the trimer La3Cl10−, the deviation is −1.4%. For the hexamer La6Cl19−, the order is reversed, and the theoretical CCS is 2.1% larger than the experimental value (233.4 Å2vs. 228.7 Å2). While a deviation of 2–3% is not uncommon for CCS calculations based on DFT-optimized structures, a better fit for the four calibration points would be desirable. However, with just element-specific parameters, we clearly cannot achieve a perfect fit for all four calibration points simultaneously: if we adjust parameters to perfectly match the theoretical and experimental CCS of La4Cl13−, LaCl4− is always slightly (ca. 2%) too small while La6Cl19− is slightly (ca. 2%) too large (see Fig. S1, ESI† for plots of theoCCSN2 as a function of σCl). The problem most likely arises from the use of element specific, but charge-independent L-J parameters in IMoS. This is more critical in highly polar systems like the clusters considered here: the overall charge per atom is larger in the smaller ions like LaCl4− and therefore their effective van der Waals radii are larger. Rather than taking the somewhat questionable step of also adjusting this parameter for each cluster size we decided to keep the σCl constant at 3.87 Å (a value which minimizes the overall error) unless otherwise noted. Therefore, for the following, we have to keep in mind that with this parameter choice, the theoCCSN2 of the smaller cluster sizes will be slightly underestimated and the larger cluster sizes will be slightly overestimated.
4. Discussion
Next, we discuss the inferences which can be made by comparing TWCCSN2 with DFT derived model structures (following the procedure described in the previous section) in order of increasing oligomer size.4.1 Calculated structures and CCS
 | ||
| Fig. 7 Left: Experimental TWCCSN2 (black) with error bars and calculated theoCCSN2 (red, blue) for the two lowest energy isomers of Ln2Cl7−. IMoS-parameters σCl = 3.85 Å and σLn = 3.00 Å. Right: Calculated energy of isomer B with respect to isomer A (ΔE, no ZPE correction). | ||
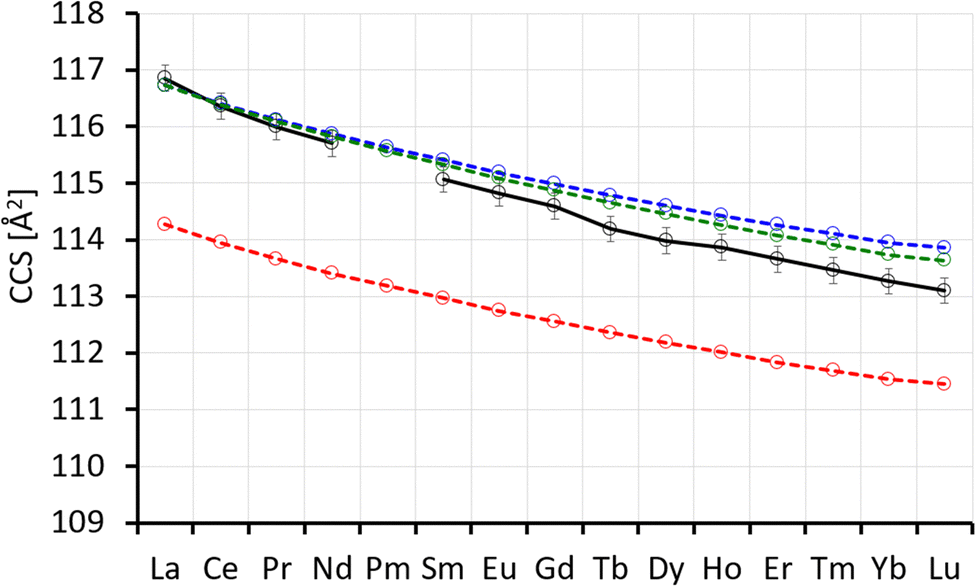
The theoCCSN2 value for isomer A is in the range of 174.2–164.0 Å2, i.e. 1.0–2.1% below the experimental value and that for isomer B is 2.9–4.9% above, see Fig. 8. The linear chain, isomer C, is between 1.0% and 2.1% above the experimental value. Among the three possibilities, isomer A is in best agreement with the experiment, especially as we expect the CCS calculated for smaller clusters like Ln3Cl10− to be slightly below the experimental value (based on our calibration procedure, see above). Isomer interconversion on the experimental timescale does not need to be invoked here. The closest higher lying isomers are uniformly 15–18 kJ mol−1 less favorable and would therefore not be expected to contribute significantly to a room temperature equilibrium distribution.
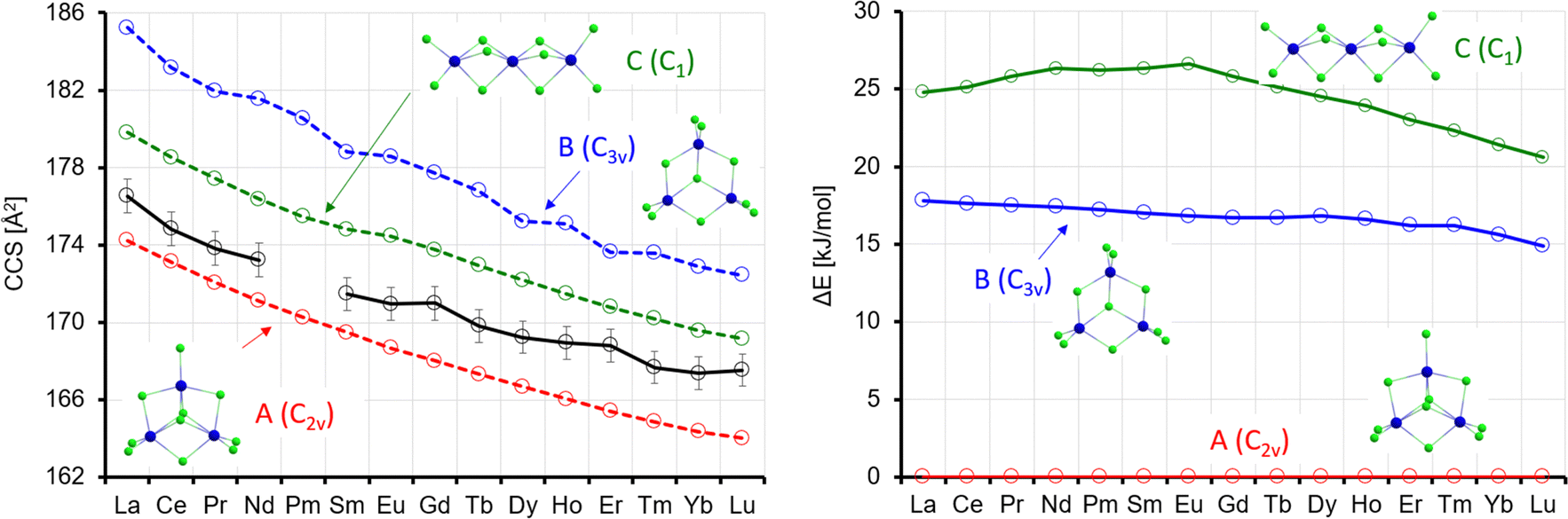 | ||
| Fig. 8 Left: Experimental (black) with error bars and calculated CCS (red, blue, green) for Ln3Cl10−. Right: Calculated energy differences (with respect to isomer A) (ΔE, no ZPE correction). | ||
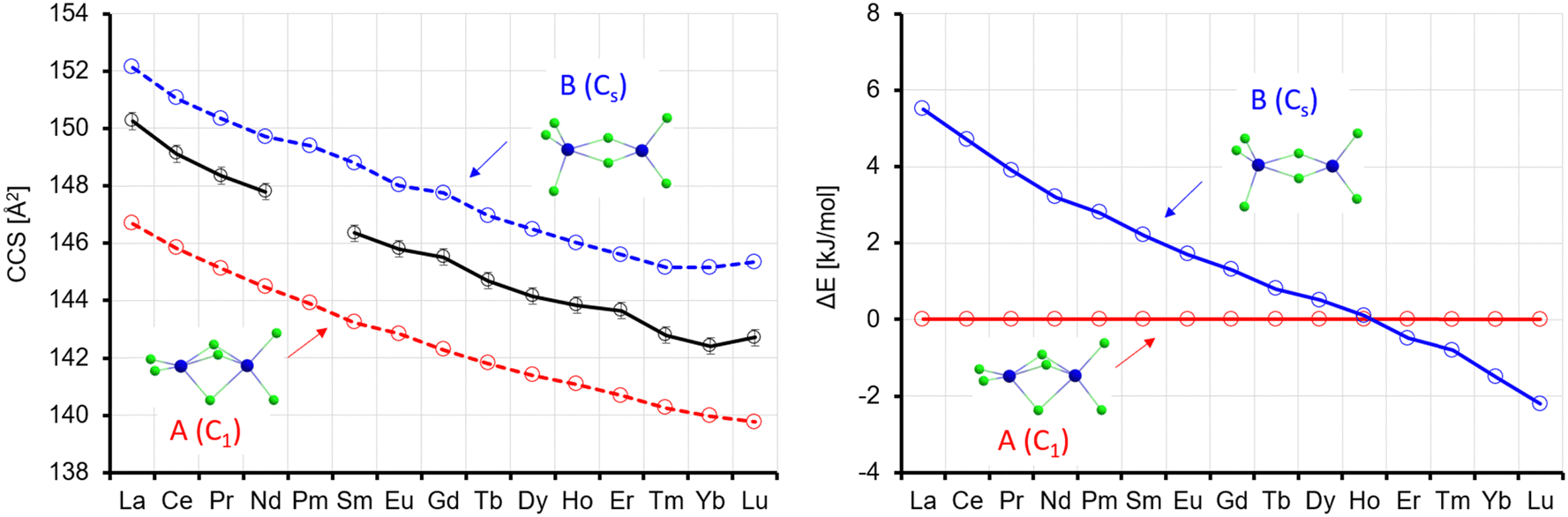
In the lowest energy isomer A for La4Cl13−, the four Ln atoms form a square with two bridging Cl atoms on each edge. Each Ln atom has a terminal Cl atom, and the last Cl atom is in the centre. For La, this isomer is preferred by 16 kJ mol−1 over B, where the Ln atoms form a butterfly structure, with edges and planes bridged by Cl, and additionally two terminal Cl each at two of the Ln atoms and one each at the two others. The preference for A continuously decreases, and from the element Tb onwards isomer B is preferred (by up to 12 kJ mol−1 for Lu, see Fig. 9). It should be noted that Rutkowski et al. found isomer A for both La4Cl13− and Lu4Cl13−. Other isomers are more than 50 kJ mol−1 higher in energy and can be ruled out (see Table S5 and Fig. S7, ESI†). The theoCCSN2-curve for A (Fig. 9, red curve) has a basically constant slope and closely follows the experimental CCS curve from La to Gd. The theoCCSN2 curve of B (Fig. 9, left, blue curve) parallels the curve of A (but shifted to larger CCS by 5–7 Å2). It meets the experimental data in the range from Tm to Lu. This suggests that a structural transition from isomer A to isomer B occurs along the series, which is perfectly in line with the predicted relative energies: the energy difference between isomer B and isomer A decreases from La to Gd and for Tb–Lu isomer B becomes favored (Fig. 9, right).
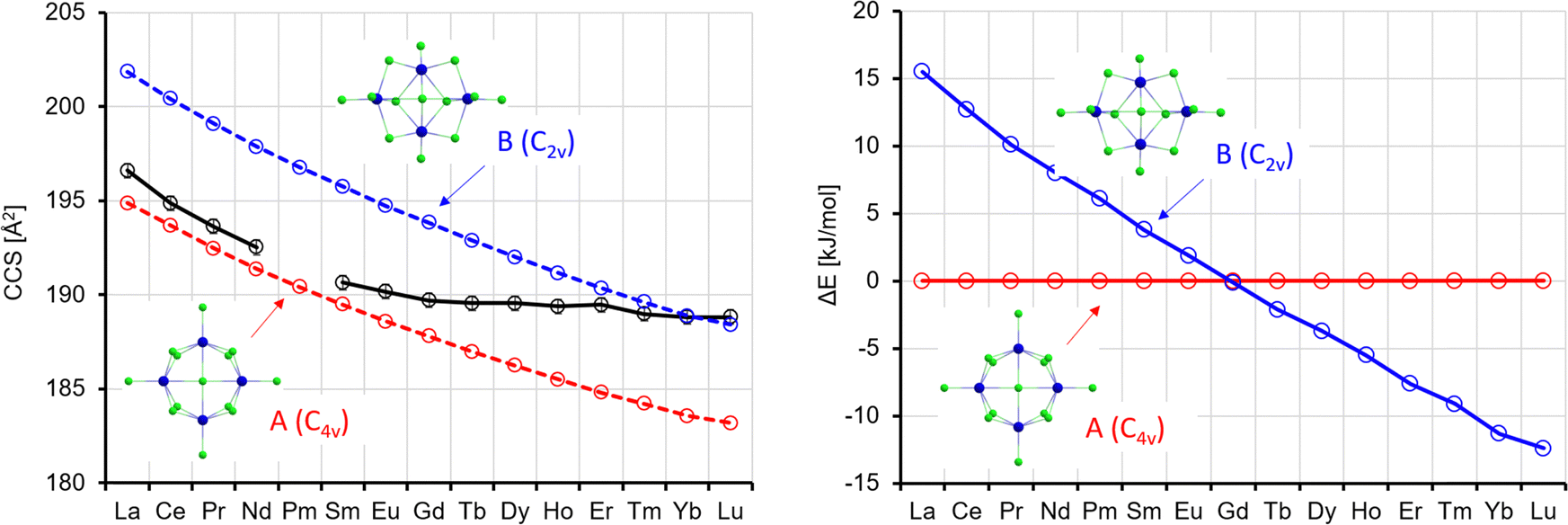 | ||
| Fig. 9 Left: Experimental (black) with error bars and calculated CCS (red, blue) for Ln4Cl13−. Right: Calculated energy differences (with respect to isomer 1); ΔE, no ZPE correction. | ||
It is interesting to note that we always observe only one sharp peak in the arrival time distribution (see Fig. 2), even in the intermediate region from Eu to Tm. This can most likely be explained by a quick interconversion between the two isomers in this region (the barrier height is ca. 60 kJ mol−1, see Table S8, ESI†). This leads to an averaged collision cross-section which is weighted by the relative amounts of isomers A and B in the Ln-dependent equilibrium distributions (in turn reflecting free energy differences which correlate with ΔE's as shown in Fig. 9).32
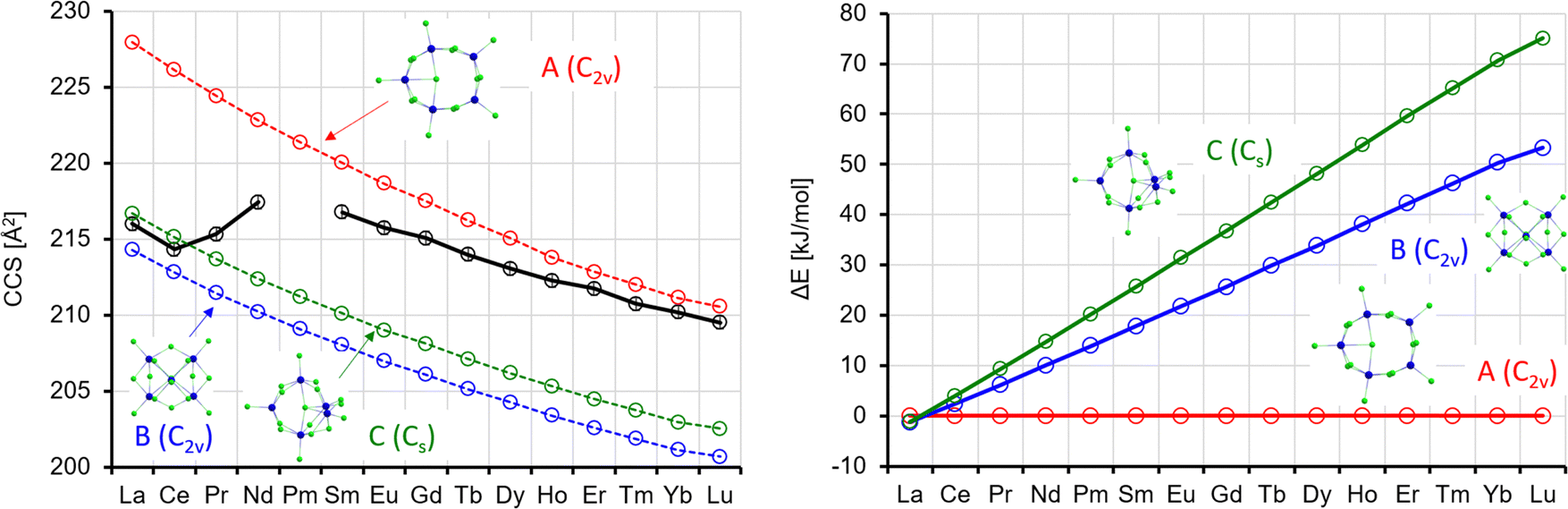 | ||
| Fig. 10 Left: Experimental (black) with error bars and calculated CCS (red, blue, green) for Ln5Cl16−. Right: Calculated energy differences (with respect to isomer 1) (ΔE, no ZPE correction). | ||
According to the calculations, the lowest energy isomer A shows C5v symmetry, from Ho to Lu and thus is a straightforward structural extension of the most stable isomer for x = 4. For the lighter lanthanides, it is slightly distorted to C2v (in-line with the calculations of Rutkowski et al.,8 who considered this isomer only for La and Lu). Also, isomer C is a ring-type isomer, but here only three atoms reside in the plane. At the last corner of an imaginary square, there is one Ln atom above and one below the plane. For the remainder, the Cl bridges are similar to isomer A, and, like there, one Cl is in the middle of the ring. Isomer B may be regarded as the next step in this structural development. One corner of an imaginary triangle is occupied with one Ln atom, the two others each with one Ln atom above and one below the plane. In total, there are ten bridging Cl atoms, further one terminal Cl per Ln, and one Cl in the middle. Whereas for La, isomers A–C are practically degenerate, B and C become progressively less favourable across the Ln series to reach 53(71) kJ mol−1 for B(C) at Lu (see also Table S6 and Fig. S8, ESI†).
The theoCCSN2 for isomer A closely matches the experimental TWCCSN2 for Sm–Lu (see Fig. 10). For La and Ce, however it is much (>5%) larger than the experimental values and can therefore be ruled out in the experiment. On the other hand, the quasi-degenerate isomers B and C match the experimental CCS almost perfectly (within 1%) in this region (see Fig. 10, left). So, from the experimental point of view, it is clear that isomers B and/or C are dominant for La (unlike the prediction by Rutkowski et al.8) and Ce, while isomer A dominates for Sm–Lu. This is in line with the relative energies: for the early lanthanides, all three isomers are close in energy (within 10 kJ mol−1 or lower); for the late lanthanides, isomer A is strongly favored (by >30 kJ mol−1, see Fig. 10, right). For the intermediate region (Pr and Nd), the situation is less clear: the experimental CCS are in between the calculations for isomers A, B, and C. On the other hand, the arrival time distributions for Pr and Nd are not significantly broader than the curves for the other lanthanides (Fig. 2) – we observe only one narrow peak. Therefore, we conclude that we observe a dynamical interconversion in this region, similar to what we have inferred for the intermediate region of Ln4Cl13− (the barrier height is again ca. 60 kJ mol−1, see Table S9, ESI†).
 | ||
| Fig. 11 Left: Experimental (black) with error bars and calculated CCS (red, blue, green) for Ln6Cl19−. Right: Calculated energy differences (with respect to isomer 1) (ΔE, no ZPE correction). | ||
In the Oh-symmetric isomer A, the Ln atoms form an octahedron, whose edges are bridged by two Cl atoms each. Each Ln has one terminal Cl, and one Cl resides in the centre. In C3v-symmetric B, all corners of an imaginary triangle are occupied each with one Ln atom above and one below the plane; it is thus closely related to isomer B for x = 5, also concerning the positions of the Cl atoms. C is a Cl double-bridged six-membered Ln ring with one more terminal Cl at five of the Ln atoms. At the sixth Ln atom, the ring is bent inwards. There is no terminal Cl here; instead, there are two Cl inside the ring. For La, B(C) is higher in energy by 21(34) kJ mol−1 (see Table S7 and Fig. S9, ESI†). Whereas the energetic disfavouring of C increases somewhat towards Lu, it decreases for B. If we compare the calculated CCS for isomer A (La: 233.9 Å2, using the “standard” σCl value of 3.85 Å, see above), it is the isomer that fits best (within ca. 2%) to the experiment (228.7 Å2) for La. Isomers B and C overestimate the CCS by ca. 4 and 11%, respectively, and it is clear that the single peak in the arrival time distribution of La6Cl19− corresponds to isomer A. For the other lanthanides, isomer A parallels the experimental curve, and it is always ca. 2–3% too large (see Fig. 11, left, red and lower black curve). Isomer B corresponds to the second peak that we observe in the arrival time distribution of Ln6Cl19− for Ln = Sm–Lu (see Fig. 2). Again, its CCS is ca. 2% larger than the experiment (Fig. 11, left, blue and upper black curve). Furthermore, the intensity increase of the second peak is in line with the decrease of energetic disfavoring of B (vs. A). Isomer C can be clearly ruled out both on the basis of relative energy and CCS. Rutkowski et al.8 predicted for La6Cl19− a ring of 5 La atoms with a La atoms in the center and for Lu6Cl19− a Lu-6-ring with a central Cl atom. These structures are more than 50 kJ mol−1 above isomer A and can be ruled out. Note that the calculated barrier height between A and B is in the range of 100 kJ mol−1 (Sm) to 130 kJ mol−1 (Lu), see Table S8 (ESI†).
At this point, we should remind the reader that our calculations are performed at 0 K and do not include zero-point energy (ZPE), while the experimental arrival time distributions are determined in a measurement that is performed on cluster anions that are thermalized to near room temperature. Since we experimentally observe two base-line separated peaks in this measurement, it is clear that there is no significant interconversion on the experimental time-scale (10–100 ms) of the CCS determination. Therefore, the relative intensities of the two peaks as determined for the as-prepared clusters must reflect an elevated temperature, that is accessed somewhere upstream from the cIM for long enough to approach an equilibrium ratio (perhaps in the ESI† source or in the Stepwave ion guide). Clusters at this elevated temperature then quickly cool at latest after injection into the cIM cell.
We observe a decrease in the relative intensity of the first isomer in the arrival time distribution (experimental: isomer 1, calculation: isomer A) decreasing from 90 to 70% for Sm to 20–30% for Lu (depending on whether the ion intensities are measured directly by IMS or in IMS-selection-activation-IMS mode, see above, Fig. 5). In order to compare experiment and theory, we determined the equilibrium constants and relative ion intensities as a function of temperature (i.e. from relative free energies) expected for DFT-calculated relative energies, rotational constants and vibrational frequencies. The results are shown in Table 2 and Fig. 12. It turns out that isomer B is favored entropically for all Ln6Cl19− and should therefore dominate the distribution at room temperature for Ho–Lu, which is very close to our experimental findings. It should be noted, however, that the predicted intensities are very sensitive to the parameters used in the calculation such as temperature, vibrational frequencies and relative energies, so the agreement might be fortuitous. For comparison, we include in the ESI† the relative energies obtained with TPSS and PBE0 functionals. The values are similar but they differ by 2–3 kJ mol−1 with respect to the PBE values – which is enough to shift the intensity ratio significantly.
| Temperature [K] | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| 200 | 100 | 100 | 100 | 100 | 100 | 99 | 99 | 98 | 97 | 95 | 93 | 89 | 84 | 75 | 66 |
| 300 | 94 | 91 | 90 | 89 | 87 | 83 | 79 | 72 | 65 | 60 | 53 | 45 | 40 | 31 | 26 |
| 400 | 65 | 57 | 56 | 55 | 53 | 49 | 43 | 37 | 32 | 29 | 25 | 21 | 19 | 15 | 13 |
| 500 | 34 | 28 | 28 | 29 | 28 | 26 | 23 | 20 | 17 | 16 | 14 | 12 | 11 | 9 | 8 |
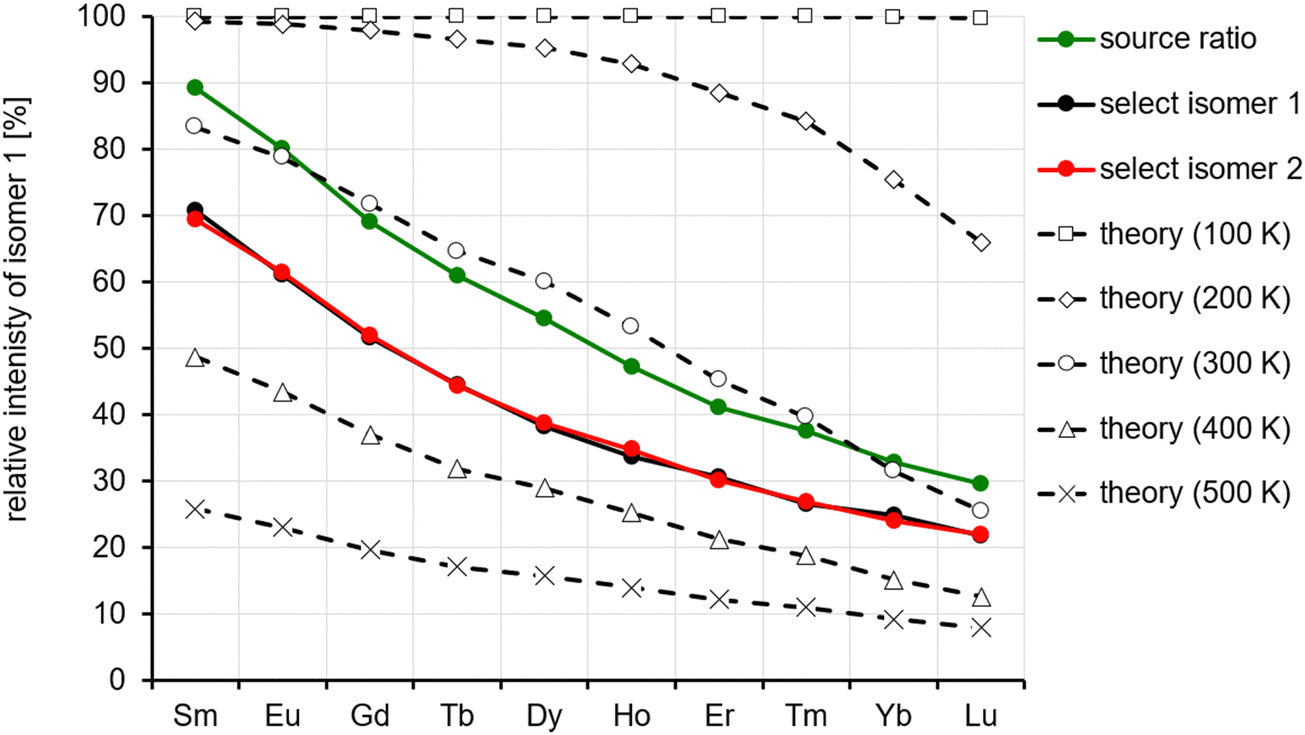 | ||
| Fig. 12 Comparison of observed ion intensities of the first isomer in the arrival time distribution (assigned as isomer A) with predictions based on DFT calculated relative energies, rotational constants and vibrational frequencies. | ||
Conclusions
We have characterized the structures and isomer distributions of isolated LnxCl3x+1−, x = 1–6, for all Ln = La–Lu (except Pm). Clusters were generated by ESI from isopropanol solutions. Based on observed concentration dependencies, they are likely being formed during the spray process itself rather than being present in solution. To probe Ln-dependent structural characteristics of these clusters, we have used cyclic IMS which can determine CCS to high precision (better than 0.2%) and resolution (significantly better than 200) by probing near room temperature ion ensembles during measurement times which range from 10 to 100 ms in length. In some cases, we have also used IMS–IMS to study isomer interconversion. Comparison of experimental results to comprehensive DFT calculations across the complete Ln series together with trajectory method modelling of the observed collision cross sections allows the studied anions to be categorized into two groups:(a) those showing interconversion between two or more isomeric structures on a much faster than the experimental timescale (Ln2Cl7−, Ln4Cl13− (Gd–Tm) and Ln5Cl16− (Pr and Nd)) thus yielding an averaged CCS value and
(b) structurally rigid species which do not show significant isomeric interconversion during the measurement. Of special interest in this regard is the Ln6Cl19− system. For Sm–Lu, two baseline separable isomers are observed whose relative ratios are strongly dependent on Ln. While they are rigid on a 100 ms timescale, each of these two isomers can be converted to the other structure by moderate collisional excitation – at energies still well below that required for dissociation. The Ln-dependent isomer ratios observed correlate with the equilibrium ratios predicted from DFT based relative free energies calculated for the two lowest energy Ln6Cl19− structures.
For all LnxCl3x+1− studied, measurements are consistent with the lowest energy structures predicted in DFT calculations. In fact, where measurement and structural rearrangement timescales allow, we obtain an almost quantitative agreement between experiment and theory thus confirming isomer predictions and reproducing isomer intensity ratios.
Overall, the structures of LnxCl3x+1− reflect strong ionic bonding with limited directionality. They comprise Ln(III) centres bridged (and sometimes terminated) by multiple chlorides. This gives rise to ring and chain motifs for smaller clusters while for larger clusters more compact three-dimensional structures become favourable. At cluster sizes with two or more close lying isomers, the lanthanide contraction can lead to changes in structure types across the series. Interestingly, there is a condensed phase analogue to the Oh isomer A of Ln6Cl19−: W. P. Kretschmer et al. have reported the crystal structure of [Cp6Yb6Cl13]− with 6 terminating Cp rings instead of chlorides as in our case (and [Cp3Yb3Cl5(thf)3]+ counterions).33 It is also instructive to consider how the gas-phase cluster structures can be related to each other in terms of a hypothetical assembly sequence involving sequential addition of LnCl3− units (bearing in mind that our experiments are consistent with the formation of thermodynamically favored isomeric structures even though the clusters must be growing in the presence of solvent molecules). This is shown in Fig. 13 which also summarizes the results from theory (structures and relative energies) for all species/cluster sizes.
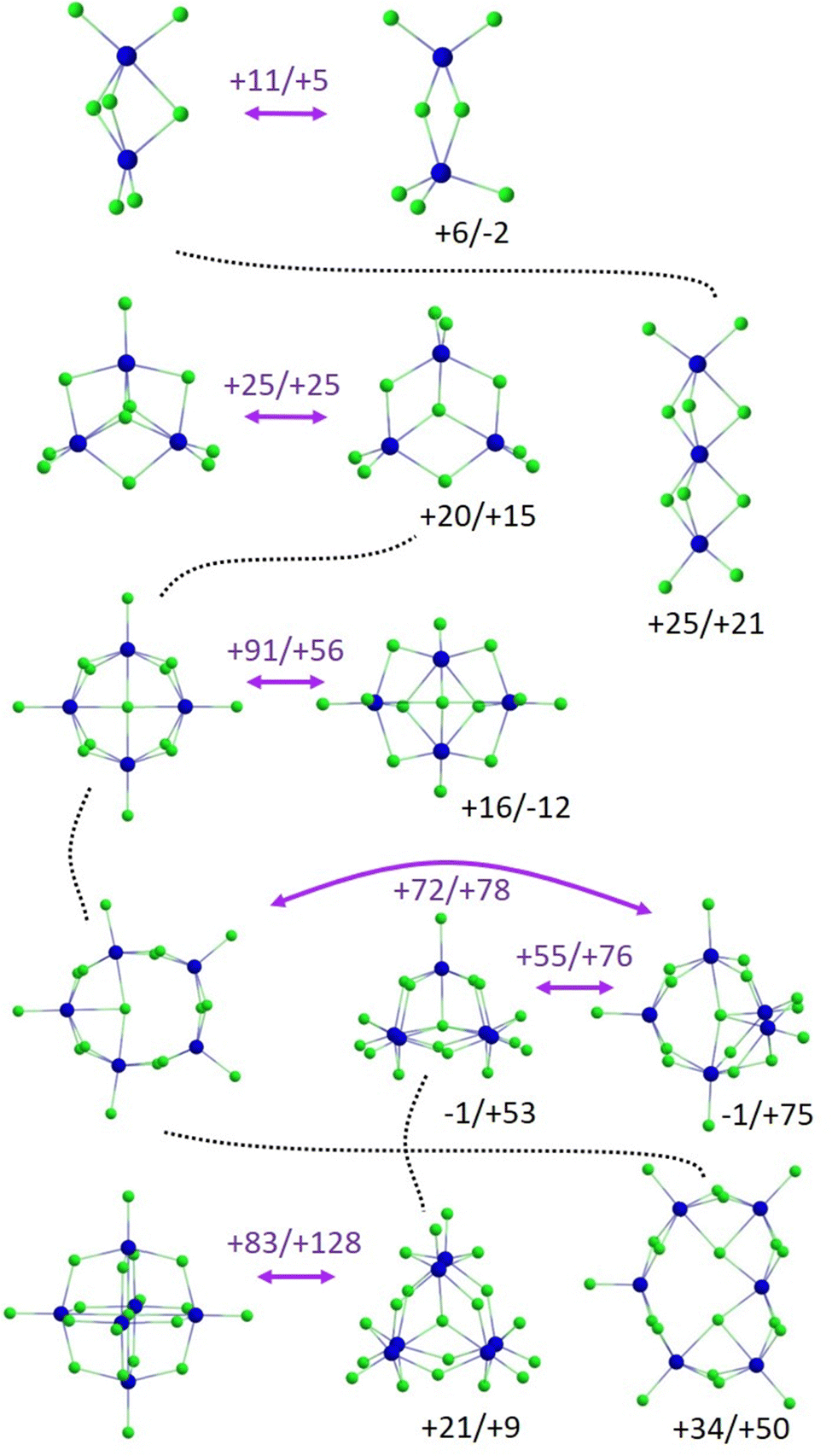 | ||
| Fig. 13 Energetically most favourable isomers of LnxCl3x+1− for each x and energy differences relative to isomer A (left) for La/Lu in kJ mol−1. Straight purple arrows show calculated transitions within a given x and the energies for the optimized transition states for La/Lu relative to isomer A. Dotted lines indicate structural relationships between clusters of sizes x and x + 1 (via addition/insertion of LnCl3− units). The ESI† contains the top and side views of all structures shown. | ||
We have begun to computationally address the daunting problem of multidimensional barrier surfaces and associated interconversion pathways (see Fig. S10–S13, ESI†). It would clearly also be interesting to experimentally study the T-dependence of isomer interconversion (on a different instrumental platform), e.g. to make interconversion slower by cooling the ion ensemble down sufficiently and thus to hopefully resolve the contributing isomers. Similar experiments with ESI sprayed ions in a T-variable drift cell have recently been performed by one of the groups contributing to this study.6 Along the same lines, we expect that interconversion of Ln6Cl19− isomers A and B should become measurable by moderate heating.
A further direction for future work will be to extend this study to other lanthanide halides. For example, preliminary measurements on the Ln6Br19− system indicates analogous isomerism but interestingly the interconversion becomes fast enough to cause broadening along the lines of the ATD's observed in ref. 4. Such data would allow fits of the interconversion kinetics to yield forward and reverse reaction rates (perhaps even as a function of Ln). This could be modelled by statistical rate theory to obtain effective activation energies which could be compared with DFT prediction.
Data availability
Most of the experimental data have been reported in the main text. Details of the collision cross-section and quantum chemical calculations including employed basis sets as well as coordinates and vibrational frequencies of the relevant species have been included as part of the ESI† (as separate files).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
MK, FW, ML and PW gratefully acknowledge support from the German Science Foundation (DFG) as administered by the Collaborative Research Center 1573 “4f-for-future” in projects A2, C3 and Q. MK and PW are also grateful to DFG and KIT for the funding of a Cyclic IMS–MS instrument under Art. 91b GG. YN acknowledges funding of a six-month research stay in Karlsruhe by “GP-Chem” the International Joint Program in Integrated Chemistry of Tohoku University.References
- J. J. Berzelius, Om sammansättningen af vinsyra och drufsyra (John's säure aus den Voghesen), om blyoxidens atomvigt, samt allmänna anmärkningar om sådana kroppar som hafva lika sammansättning, men skiljaktiga egenskaper, Kungliga Svenska vetenskapsacademiens Handling (Transactions of the Royal Swedish Science Academy), 1840, vol. 49 Search PubMed.
- I. Gatland, Case Stud. At. Phys., 1974, 4, 369–437 CAS.
- R. R. Hudgins, P. Dugourd, J. M. Tenenbaum and M. F. Jarrold, Structural transitions in sodium chloride nanocrystals, Phys. Rev. Lett., 1997, 78, 4213–4216 CrossRef CAS.
- P. Weis, T. Bierweiler, E. Vollmer and M. M. Kappes, Au9+: Rapid isomerization reactions at 140 K, J. Chem. Phys., 2002, 117, 9293–9297 CrossRef CAS.
- S. Poyer, C. Comby-Zerbino, C. M. Choi, L. MacAleese, C. Deo, N. Bogliotti, J. Xie, J. Y. Salpin, P. Dugourd and F. Chirot, Conformational Dynamics in Ion Mobility Data, Anal. Chem., 2017, 89, 4230–4237 CrossRef CAS.
- R. Ito, X. He, K. Ohshimo and F. Misaizu, Large Conformational Change in the Isomerization of Flexible Crown Ether Observed at Low Temperature, J. Phys. Chem. A, 2022, 126, 4359–4366 CrossRef CAS PubMed.
- P. X. Rutkowski, M. C. Michelini, T. H. Bray, N. Russo, J. Marçalo and J. K. Gibson, Hydration of gas-phase ytterbium ion complexes studied by experiment and theory, Theor. Chem. Acc., 2011, 129, 575–592 Search PubMed.
- P. X. Rutkowski, M. C. Michelini and J. K. Gibson, Gas-phase lanthanide chloride clusters: relationships among ESI abundances and DFT structures and energetics, Phys. Chem. Chem. Phys., 2012, 14, 1965–1977 RSC.
- M. Lukanowski and F. Weigend, manuscript in preparation; the lcecp-1-TZVP bases are provided within the ESI† as separate file.
- K. Giles, J. Ujma, J. Wildgoose, S. Pringle, K. Richardson, D. Langridge and M. Green, A Cyclic Ion Mobility-Mass Spectrometry System, Anal. Chem., 2019, 91, 8564–8573 CrossRef CAS PubMed.
- F. Hennrich, S. Ito, P. Weis, M. Neumaier, S. Takano, T. Tsukuda and M. M. Kappes, Cyclic ion mobility of doped [MAu24L18]2− superatoms and their fragments (M = Ni, Pd and Pt; L = alkynyl), Phys. Chem. Chem. Phys., 2024, 26, 8408–8418 RSC.
- S. M. Stow, T. J. Causon, X. Y. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. E. Rennie, E. S. Baker, R. D. Smith, J. A. McLean, S. Hann and J. C. Fjeldsted, An Interlaboratory Evaluation of Drift Tube Ion Mobility-Mass Spectrometry Collision Cross Section Measurements, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS.
- Turbomole, Version 7.8 2023; a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH 1989–2007, Turbomole GmbH, since 2007; available viahttps://www.turbomole.org.
- M. Sierka, J. Döbler, J. Sauer, G. Santambrogio, M. Brümmer, L. Wöste, E. Janssens, G. Meijer and K. R. Asmis, Unexpected structures of aluminum oxide clusters in the gas phase, Angew. Chem., Int. Ed., 2007, 46, 3372–3375 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Energy-adjusted pseudopotentials for the rare-earth elements, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, A combination of quasi-relativistic pseudopotential and ligand-field calculations for lanthanoid compounds, Theor. Chim. Acta, 1993, 85, 441–450 CrossRef CAS.
- O. Treutler and R. Ahlrichs, Efficient molecular numerical-integration schemes, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for mixing exact exchange with density functional approximations, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- D. L. Peng, N. Middendorf, F. Weigend and M. Reiher, An efficient implementation of two-component relativistic exact-decoupling methods for large molecules, J. Chem. Phys., 2013, 138, 14 Search PubMed.
- Y. J. Franzke, N. Middendorf and F. Weigend, Efficient implementation of one- and two-component analytical energy gradients in exact two-component theory, J. Chem. Phys., 2018, 148, 104110 CrossRef PubMed.
- P. Pollak and F. Weigend, Segmented Contracted Error-Consistent Basis Sets of Double- and Triple-ζ Valence Quality for One- and Two-Component Relativistic All-Electron Calculations, J. Chem. Theory Comput., 2017, 13, 3696–3705 CrossRef CAS PubMed.
- Y. J. Franzke, L. Spiske, P. Pollak and F. Weigend, Segmented Contracted Error-Consistent Basis Sets of Quadruple-ζ Valence Quality for One- and Two-Component Relativistic All-Electron Calculations, J. Chem. Theory Comput., 2020, 16, 5658–5674 CrossRef CAS PubMed.
- P. Plessow, Reaction Path Optimization without NEB Springs or Interpolation Algorithms, J. Chem. Theory Comput., 2013, 9, 1305–1310 CrossRef CAS PubMed.
- C. Larriba and C. J. Hogan, Ion Mobilities in Diatomic Gases: Measurement versus Prediction with Non-Specular Scattering Models, J. Phys. Chem. A, 2013, 117, 3887–3901 CrossRef CAS PubMed.
- C. Larriba and C. J. Hogan, Free molecular collision cross section calculation methods for nanoparticles and complex ions with energy accommodation, J. Comput. Phys., 2013, 251, 344–363 CrossRef CAS.
- V. Shrivastav, M. Nahin, C. J. Hogan and C. Larriba-Andaluz, Benchmark Comparison for a Multi-Processing Ion Mobility Calculator in the Free Molecular Regime, J. Am. Soc. Mass Spectrom., 2017, 28, 1540–1551 CrossRef CAS PubMed.
- T. Y. Wu, J. Derrick, M. Nahin, X. Chen and C. Larriba-Andaluz, Optimization of long range potential interaction parameters in ion mobility spectrometry, J. Chem. Phys., 2018, 148, 074102 CrossRef PubMed.
- R. D. Shannon, Revised Effective Ionic-radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- Y. Nakajima, P. Weis, M. Kappes, F. Wiegend and F. Misaizu, to be published Search PubMed.
- W. P. Kretschmer, J. H. Teuben and S. I. Troyanov, Novel, highly symmetrical halogen-centered polynuclear lanthanide complexes: [Cp6Yb6Cl13]− and [Cp12Sm12Cl24], Angew. Chem., Int. Ed., 1998, 37, 88–90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04057k |
| This journal is © the Owner Societies 2025 |