
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic investigation of proton bonding at sub-kelvin temperatures†
América Y.
Torres-Boy
 a,
Martín I.
Taccone
a,
Katja
Ober
a,
Myles B.T.
Osenton
a,
Gerard
Meijer
a,
Gert
von Helden
*a and
Bruno
Martínez-Haya
*b
a,
Martín I.
Taccone
a,
Katja
Ober
a,
Myles B.T.
Osenton
a,
Gerard
Meijer
a,
Gert
von Helden
*a and
Bruno
Martínez-Haya
*b
aFritz Haber Institute of the Max Planck Society, 14195 Berlin, Germany. E-mail: helden@fhi-berlin.mpg.de
bCenter for Nanoscience and Sustainable Technologies (CNATS), Universidad Pablo de Olavide, 41013 Seville, Spain. E-mail: bmarhay@upo.es
First published on 25th December 2024
Abstract
The proton bond is a pivotal chemical motif in many areas of science and technology. Its quantum chemical description is remarkably challenged by nuclear and charge delocalization effects and the fluxional perturbation that it induces on molecular substrates. This work seeks insights into proton bonding at sub-kelvin temperatures. In this way, intrinsic features of the proton bond are exposed, essentially free from thermal fluctuations of the molecular frame. To this end, a proton is bound within the molecular ring cavity provided by the 12-crown-4 ether. The resulting ion is isolated in a He-droplet at ∼0.4 K, where it is interrogated by infrared laser spectroscopy. The recorded spectrum features narrow vibrational bands, consistent with a robust proton bond bridging ether sites across the cavity of the essentially frozen crown ether. The potential energy surface sustaining the proton bond is broad and markedly anharmonic. In consequence, common modeling methods within the harmonic approximation fail to capture the observed band positions, whose accurate description seems to be even beyond perturbative anharmonic approaches. Calculations show that at elevated temperatures, the crown ether backbone is highly fluxional and that the distance between the oxygen atoms fluctuates in time, modulating the potential that the proton or deuteron is exposed to, and yielding dynamic inhomogeneous broadening and blue shifts with respect to the cryogenic spectra. These observations call for novel computational developments, for which the vibrational signatures outlined in this work should provide a valuable benchmark.
1 Introduction
Proton bonding constitutes an archetypal chemical interaction that sustains supramolecular networks and commonly precedes proton and electron transfer reactions.1–7 The fundamental quantum chemical nature of the proton bond and its inherent charge delocalization are intriguing and pose considerable challenges to theoretical modeling.8 The electronic potential energy surface that sustains proton bonding is typically shallow and fosters a dynamic sharing of the proton between the electronegative moieties involved. Proton dynamics couples with the internal degrees of freedom of the molecular substrates, which can be monitored from distinct spectral signatures. Consistently, the first-principles rationalization of the structure of prototypical proton bonds has been pursued with various combinations of spectroscopic and computational approaches. Different studies have addressed the emergence of diffuse band fingerprints in the vibrational spectra of protonated moieties both at room temperature and at cryogenic temperatures.9–16 The application of ab initio Molecular Dynamics has pinpointed the role of the large-amplitude motion of the proton and of the fluctuations of the molecular substrate in the broad vibrational band features observed experimentally in benchmark systems.9,13–16Investigating proton bonding in rigid molecular frameworks is particularly appealing as a strategy to probe its intrinsic nuclear and electronic features and isolate them from broadening effects arising from fluxional backbone dynamics. The ring cavities of crown ether macrocycles constitute appropriate frameworks for this type of approach, as they provide semi-rigid molecular templates prone to host protons in a controlled environment. A previous investigation explored proton bonding in the 12-crown-4 (12c4), 15-crown-5 (15c5), and 18-crown-6 (18c6) ethers at room temperature, by means of infrared multiple-photon dissociation (IRMPD) ion spectroscopy.13 The 15-crown-5 and 18-crown-6 ethers displayed broad-band vibrational features that could be assigned to the active backbone dynamics and to the wandering of the proton inside the cavity, forming bonds with changing pairs of O-atom sites. The smaller 12-crown-4 has been chosen for the present study due to its reduced flexibility and simpler configurational landscape in comparison to the larger native crown ethers.
In 12c4, a stable proton bond is formed between opposing O atoms across the cavity. The three most stable conformations of protonated 12c4 calculated at MP2/aug-cc-pVTZ level of theory are represented in Fig. 1. Further conformations are provided in Fig. S3 of the ESI,† and details are given later. The ring backbone constraints the relative orientation of the lone pair orbitals of the O atoms and also imposes longer O–O distances relative to those achieved in proton-bonded dimers of free alkyl ethers.17Fig. 1 shows that this results in a shallow effective potential electronic energy surface for the proton, which may display a symmetric or asymmetric well, eventually with an internal barrier, depending on the 12c4 backbone conformation. The marked anharmonic topology of the potential energy surface can be expected to severely influence proton dynamics and the spectroscopic signatures of the system. Anharmonicity has been shown to contribute to band broadening observed in proton-bonded systems at low temperatures.10,11,15
 | ||
| Fig. 1 Top: Schematic representations of the three most stable configurations of protonated 12-crown-4 (H1, H2, H3). The corresponding point symmetries of the 12c4 backbone (proton excluded) and the O–H+ distances (Å) are indicated. R and r represent the largest and smallest O–H bond lengths of the nearest oxygens, respectively. The H-atoms of the methylene groups are not shown for a better appreciation of the backbone configurations. Relative energies of the conformers at different computational levels are indicated in Table S1 (ESI†). A larger set of conformations is provided in Fig. S3 (ESI†). Bottom: Relaxed electronic potential energy surfaces for the proton along the intramolecular bond in each conformer, computed at the B3LYP-D3(BJ) (lines) and MP2 (symbols) level (aug-cc-pVTZ basis set). | ||
The room temperature IRMPD spectrum of protonated 12c4 featured diffuse band structures that were reproduced with remarkable accuracy by BOMD computations.13 The computations suggested significant thermal fluctuations of the O atom positions and a marked delocalization of the proton along the intracavity bond. The present investigation provides insights into the nature of the proton bond in 12c4 at sub kelvin temperatures, within the ultracold cryogenic matrix provided by a superfluid helium droplet.18–23 In this scenario, backbone fluctuations are largely suppressed and fundamental questions arise, such as how sensitive the structural features of the proton bond are to the precise topology of the potential energy surface, or to what extent nuclear quantum effects, namely tunneling or zero-point energy, determine the nuclear and electronic structure of the proton bond.
2 Methods
2.1 Cryogenic gas-phase IR spectroscopy in superfluid helium droplets
Gas-phase fingerprint vibrational spectra of the 12c4-H+, 12c4-D+, and 12c4-Li+ complexes are obtained employing helium nanodroplet infrared action spectroscopy. This technique is based on the evaporation of superfluid helium droplets at photon wavelengths resonant with optical transitions of the dopant ion.18 The measurements were carried out with a custom-built instrument, extensively described in prior publications,19–23 only specific procedures and a brief overview of the instrument workflow are provided here.The protonated crown ether ions are produced via nanoelectrospray ionization (Pd/Pt-coated borosilicate capillary) of a solution of 200 μM 12c4 (>98%, Merck) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and acetonitrile (UHPLC). To produce the 12c4-Li+ complex (m/z = 183), 50 μM Lithium Chloride is added to the sample solution in water/acetonitrile. To substitute the exchangeable hydrogen with a deuterium atom, the source region is flooded with D2O-saturated nitrogen. The generated ions are mass-selected with a quadrupole mass filter, then orthogonally deflected by a quadrupole ion bender, and injected into a hexapole ion trap for storage and subsequent interaction with the He-droplet beam. Under these conditions, the electrospray process leads to comparable abundances of the two isotopologues 12c4-H+ (m/z = 177) and 12c4-D+ (m/z = 178). The 12c4H+ and 12c4D+ ions can be stored jointly in the trap by lowering the resolution of the quadrupole mass filter. This way, IR spectra of the two isotopologues can be recorded simultaneously. Separate measurements on the 12c4H+ ion alone were performed in order to monitor the contribution of its 13C-isotope in the m/z = 178 mass channel. Typical mass spectra are provided in Fig. S1 (ESI†).
:1 mixture of water and acetonitrile (UHPLC). To produce the 12c4-Li+ complex (m/z = 183), 50 μM Lithium Chloride is added to the sample solution in water/acetonitrile. To substitute the exchangeable hydrogen with a deuterium atom, the source region is flooded with D2O-saturated nitrogen. The generated ions are mass-selected with a quadrupole mass filter, then orthogonally deflected by a quadrupole ion bender, and injected into a hexapole ion trap for storage and subsequent interaction with the He-droplet beam. Under these conditions, the electrospray process leads to comparable abundances of the two isotopologues 12c4-H+ (m/z = 177) and 12c4-D+ (m/z = 178). The 12c4H+ and 12c4D+ ions can be stored jointly in the trap by lowering the resolution of the quadrupole mass filter. This way, IR spectra of the two isotopologues can be recorded simultaneously. Separate measurements on the 12c4H+ ion alone were performed in order to monitor the contribution of its 13C-isotope in the m/z = 178 mass channel. Typical mass spectra are provided in Fig. S1 (ESI†).
The housing of the hexapole ion trap was cooled to ∼90 K with a nitrogen gas flow. While the ions enter the trap, pre-cooled helium buffer gas is introduced to promote the collisional thermalization of the ions. For fluxional molecular systems, this cooling process can also reduce the kinetic trapping of high-energy conformers in the helium droplet. It will be shown that for the crown ether presently investigated, several low-energy conformers are nevertheless stabilized in the trap. After the removal of the buffer gas, a beam of helium nanodroplets traverses the ion trap. The helium nanodroplets are produced by expanding helium through a cryogenic nozzle (60 bar, 21 K) using a pulsed Even-Lavie valve with a repetition rate of 10 Hz. The droplets can capture the trapped ions, cool them down to the equilibrium temperature of ∼0.4 K, and carry them out of the trap. The capture of two or more ions by a single helium droplet is very unlikely due to the low density of ions inside the hexapole ion trap and further, if more than one ion would nonetheless be initially captured, Coulomb repulsion might render those droplets unstable.
The doped He nanodroplets are probed using the Fritz-Haber Institute infrared free-electron laser (FHI-FEL) in the 500–1800 cm−1 range. The laser provides IR light in the form of ∼10 μs-long macropulses at a repetition rate of 10 Hz. Each macropulse consists of a sequence of ∼10 ps-long micropulses, at a repetition rate of 1 GHz. The laser bandwidth amounts to ∼0.5% of the nominal frequency. When the photon energy is resonant with a vibrational transition, sequential absorption of multiple photons occurs. The energy of each photon is rapidly transferred within the molecular ion through intramolecular vibrational redistribution (IVR) and subsequently to the helium environment. This process results in the evaporation of helium atoms and the thermalization of the dopant ion back to the equilibrium temperature of ∼0.4 K. The relaxation and dissipation process is expected to be fast18 compared to the time lapse between micropulses, ensuring that all photon absorption events take place with cold ions in their vibrational ground state. Previous models report that the absorption of about 230 photons is required for complete evaporation of an average-sized droplet at 1500 cm−1 photon energy, whereas the smallest 10% of droplets require only 110 photons.22 The bare ion, released upon evaporation of the He droplet, is deflected by a second quadrupole bender and detected in a time-of-flight mass analyzer. Cold-ion infrared spectra are obtained by integrating the mass-to-charge selected signal of the bare ion(s) as a function of the photon wavenumber. No ion signal is produced off-resonance resulting in high signal-to-noise ratios. Earlier investigations employing the same action spectroscopy technique showed a non-linear correlation between the ion signal and the FEL energy.23 Nonetheless, as a first-order correction for variations in IR laser fluence, the intensity of the spectra is divided by the photon fluence. Each spectrum was recorded at least twice with the same operation parameters and then averaged. The spectra were measured in two different ranges: 500–900 cm−1 and 800–1800 cm−1. The relative intensity of the peaks from the different scan regions was estimated by calibrating to the intensity of the overlapping features in both scans.
2.2 Quantum chemical calculations
Density functional theory (DFT), Møller–Plesset perturbation theory (MP2) and Born–Oppenheimer Molecular Dynamics (BOMD) were employed to model the conformational structure and vibrational signatures of the ions. The configurational landscapes of lithiated and protonated 12-crown-4 were explored by means of simulated annealing. Conformational structures from previous investigations were also incorporated into the survey.13,24 Structure optimization was finally performed with the DFT and MP2 methods. Details are provided in the ESI.†For comparison with the experiment, computational spectra were generated from the MP2 and DFT calculations by convoluting the scaled harmonic stick spectra with Gaussian functions having a full-width at half-maximum (FWHM) of 0.4% of the transition frequency. An estimation of the anharmonic corrections to the calculated vibrational modes was attempted using generalized second-order vibrational perturbation theory (GVPT2) at the B3LYP-D3(BJ)/aug-cc-pVTZ level of theory.
Relaxed electronic potential energy surfaces (PES) were computed at the MP2 and B3LYP levels to aid in the rationalization of the proton and backbone dynamics. These computations scan a specified coordinate, in the present study the position of the proton along the intramolecular bond, while all other degrees of freedom are allowed to relax to the most stable configuration of the molecular ion. A set of potential energy curves was additionally generated with the crown ether frozen at its equilibrium structure or, alternatively, at relaxed configurations with fixed O–O distances in the proton bond. In this latter case, the proton was displaced along the coordinates corresponding to the stretching mode between the two nearest oxygens, and the orthogonal motion of the proton to the bending in and out of the cavity plane. The resulting one-dimensional potential energy curves were employed to estimate the frequency of oscillation of each of the three modes within the discrete variable representation (DVR) scheme.25
BOMD computations were performed with the main aim of monitoring the fluctuations of the crown ether backbone at cryogenic temperatures compared to room temperature. The B3LYP-D3(BJ) functional and the double-ζ DZVP basis set were employed, as implemented in the CP2K package.26 Technical details are provided in the ESI.†
3 Results
3.1 He-droplet spectrum of 12c4-Li+
The alkali metal complex 12c4-Li+ was included in the study as reference for the rationalization of the protonated crown ether. The stability of 12c4-Li+ relies on electrostatic interactions, favored by the high charge density of Li+ compared to the heavier alkali cations. The optimum docking of the cation results from a balanced interaction with the four O atoms of the crown ether ring. The four conformations of lowest energy found in our survey denoted Li1–Li4 (point group symmetries C4, CS, C1 and C2V, respectively), are depicted in Fig. 2. A more detailed representation of the conformers is provided in Fig. S4 of the ESI,† which also includes two further higher-energy conformations. The conformers are ordered according to their relative zero-point corrected electronic energies at the MP2/aug-cc-pVTZ level, which updates previous studies of the low-energy configurational landscape of this system.27–30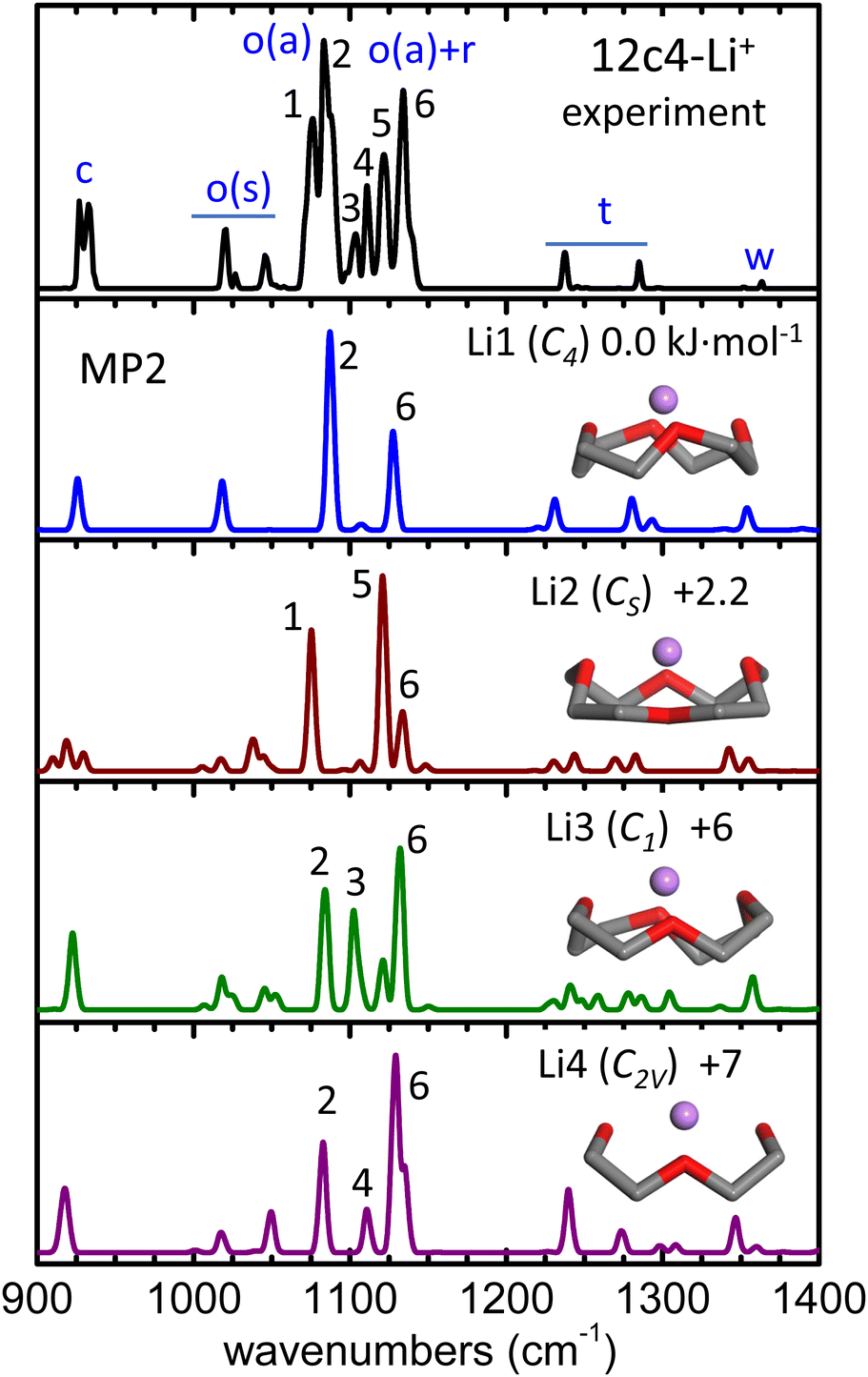 | ||
| Fig. 2 He-droplet IR spectrum of the 12c4-Li+ complex (top black trace) and MP2/aug-cc-pVTZ harmonic spectra (frequencies scaled by factor 0.97) for the four conformers of lowest energy, Li1–Li4. Side views of the conformers are inserted (H atoms of the methylene groups obviated) and the corresponding point group symmetries and relative zero-point corrected energies (kJ mol−1) are indicated. A qualitative assignment of the vibrational bands is outlined as follows: C–C stretching (c); symmetric/asymmetric COC stretching (o(s), o(a)); methylene rocking, twisting, and wagging (r, t, w). The peak numbering is only meant to guide the visualization of the correlation between the experimental and computational bands. Computed spectra at other levels of theory are included in Fig. S6 (ESI†). | ||
In the Li1–Li4 conformers, the Li+ cation interacts with the O atoms at distances within 1.95–2.05 Å and at angles 120°–130° with respect to the COC plane. In comparison, in the analogous complex with dimethoxyethane, which can be viewed as one half of the 12c4 cycle, these parameters are of 1.85 Å and 160°, respectively.27 Moreover, in the complex with diethyl ether, the O–Li+ distance is 1.82 Å, and the location of the cation is coplanar with the COC bonds so that both lone pairs of the O atom contribute evenly to the interaction. It becomes apparent that host–guest interactions in the 12c4-Li+ complex are intrinsically constrained by the limited flexibility of the macrocyclic backbone and by the compromise of an optimization of the orientation of the lone pairs of the four O atoms.
Spectroscopic investigations of the 12c4-Li+ complex are scarce to date. Infrared and Raman spectra have been reported for equimolar solutions of 12c4 and LiCl in methanol and for the crystalline solid formed upon solvent evaporation.28 The comparison to B3LYP and MP2 computations found the recorded spectra to be consistent with a predominance of the Li1 (C4) conformer. In an investigation more closely related to the present one, infrared spectroscopy of the isolated 12c4-Li+ ion was performed under low-temperature conditions with the Ar-tagging method.30 The band structures observed in the C–H stretching region were in this case rationalized in terms of the Li2 (CS) conformer.
The He-droplet spectrum of the 12c4-Li+ complex produced in our experiments is shown in Fig. 2, where it is compared to the computational MP2 spectra of conformers Li1–Li4. The comparison to the predictions from quantum chemistry calculations help to associate the observed spectral features to different vibrational modes of the crown ether backbone. Bands at wavenumbers above 1200 cm−1 arise from CH2 bending motions, whereas at lower wavenumbers, vibrational modes are dominated by C–C and C–O stretching motions. The experimetnal spectrum displays a sequence of narrow bands, particularly congested in the 1000–1150 cm−1 region. To ease the discussion, the peaks observed in this spectral region are labeled 1 through 6 in Fig. 2. These transitions are assigned to the symmetric and asymmetric stretching modes of the C–O–C moieties, with a varying degree of mixing with the rocking motions of the methylene groups. The computations predict vibrational transitions in this region that vary significantly among conformers, suggesting that the modes involved are particularly susceptible to the precise structure of the crown ether backbone and to its interactions with the Li+ cation. The computational bands are numbered according to the experimental band that they most closely match. The comparison suggests that the highly structured vibrational signatures observed experimentally are unlikely to arise from a single supramolecular arrangement and are rather the result of joint contributions from an ensemble of conformers of the complex. Indeed, the sum of the spectral features of the most stable conformers, Li1 and Li2, reproduces the most salient bands of the He-droplet spectrum (e.g., peaks 1, 2, 5, 6). The incorporation of conformers Li3 and Li4 might serve to account for the weaker bands observed within 1100–1115 cm−1 (peaks 3 and 4) while, however, the relative intensity of peak 4 in Li4 differs from what is observed in the experiment.
From the above considerations, it can be inferred that the Li1 and Li2 conformers are stabilized in the ion trap along with additional higher energy conformers, presumably Li3 and Li4. Nevertheless, it must be pointed out that the precise rationalization of the He-droplet spectrum is challenged by computational accuracy. For instance, Fig. S6 of the ESI† shows that peak assignments are partially altered when the spectral analysis is based on the B3LYP-D3(BJ) or the ωB97xD-D3(BJ) DFT computations, due to sizeable changes in the predicted band positions and intensities with respect to MP2. While the overall conclusions about the relevant 12c4-Li+ conformations do not change, it is not straightforward to assess which computational method is more accurate to describe the individual vibrational signatures observed at the cryogenic temperatures presently employed.
3.2 He-droplet spectra of 12c4-H+ and 12c4-D+
 | ||
| Fig. 3 He-droplet IR spectra of the two isotopologues 12c4-H+ (top) and 12c4-D+ (bottom), recorded at m/z 177 and 178, respectively. The shaded curve along the 12c4-D+ spectrum corresponds to the signal of the 13C isotope of 12c4-H+ on the m/z 178 mass channel. Greek letters indicate peaks presumably associated with neat vibrational modes of the proton bond. The right-hand panels magnify the central part of the spectra for better visualization of partially resolved d/d′ band components. The corresponding room temperature IRMPD spectra from a previous work13 are shown for comparison (broader-band gray traces). | ||
The spectra of 12c4-H+ and 12c4-D+ display a series of narrow bands, some of which partially overlap leading to congested band envelopes. A particularly prominent group of barely resolved transitions is observed within 1150–1200 cm−1, as shown in close-up representations in Fig. 3. It should be noted that the helium droplet method produces spectra where the intensities are nonlinear with laser fluence, making it difficult to quantitatively interpret relative intensities. In the spectra presented in Fig. 3, the intensities of the stronger bands such as, for example, bands d and g, suffer from saturation and when measurements are performed with lower FEL macropulse energies (see Fig. S2, ESI†), band d is significantly more prominent than the others. Under those conditions, however, many weaker bands are barely visible.
The peak patterns have qualitative similarities in the case of the two isotopologues in Fig. 3 but are markedly different from the pattern observed in the Li+ complex. For instance, the spectrum of the lithiated crown ether has most of the intense features in the 1000–1150 cm−1 region. In contrast, the spectra of 12c4-H+ and 12c4-D+ exhibit intense transitions over a much wider range. This already suggests that the incorporation of the proton induces a perturbation of the bond strengths along the crown ether backbone that significantly differs from that associated with the electrostatic binding of alkali ions.
Fig. 3 also includes the room temperature IRMPD spectra for 12c4-H+ and 12c4-D+ from a previous investigation.13 For most bands, their positions in the He-droplet spectra match nicely with the central parts of the much broader bands in the IRMPD spectra. In the IRMPD spectrum of 12c4-D+, all bands appear red-shifted compared to the cryogenic spectra. For the IRMPD spectrum of 12c4-H+, most bands appear red shifted as well. Exceptions are the broad band (A) between ≈ 1430 and ≈ 1700 cm−1 and the asymmetric band (M) between 700 and 800 cm−1 which both are clearly blue shifted, compared to positions of bands in the helium droplet spectrum. The band B at 1385 cm−1 does not exhibit a clear blue or red shift.
The drastic narrowing of the vibrational bands under cryogenic conditions can be partly the result of the nature of the two experiments where during the IRMPD excitation process, the ion is being heated by the IR light and (cross)-anharmonicities lead to red shifts and broadening. The observation of apparent blue shifts is therefore unusual. The helium droplet spectra, on the other hand, result from cold ions during the entire excitation process.
The overall similarity of the 12c4-H+ and 12c4-D+ spectra can be used to guide the assignment of some of the bands. For instance, all computations indicate that the vibrational motions of the heavy deuteron bond (O–D+⋯O) have a negligible contribution to the vibrational modes of the 12c4-D+ system at wavenumbers above 1200 cm−1. Based on this prediction, bands denoted a′, b′, and c′ in Fig. 3 are traced back to predominant CH2 twisting, wagging, and scissoring bending modes, respectively. In the 12c4-H+ system, the vibrational motions of the lighter proton bond potentially overlap and couple with the CH2 bending modes, which lays out a more complex scenario in this spectral region. In fact, whereas bands c and c′ at ∼1300 cm−1 have positions and intensities coincident in the two isotopologues, the spectrum of 12c4-H+ displays two pronounced bands (denoted α and β), apparently on top of weaker methylene bending a and b bands. At least the presence of the a band is apparent in the form of a shoulder in the blue flank of the intense α band.
The assignment of bands α and β to vibrational signatures of the proton bond in 12c4-H+ is appealing. The significant strength of the two transitions implies large oscillations of the dipole moment in the modes involved. The lower band intensities recorded for 12c4-D+ in this region indicate that CH2 bending modes alone are unlike to produce such strong bands.
Further strong bands in the 12c4-H+ spectrum are observed at ∼1180, 1046, 993, and 900 cm−1 (bands d, g, h, and j, respectively). These bands would be associated to C–C and C–O stretching motions that also drag stretching motions in the proton bond, leading to enhanced transition moments. A similar coupling of proton motions to bending motions along the [–C–C–O–] backbone gives rise to the strong transitions observed towards the low-frequency flank of the spectrum, within 500–800 cm−1 (peaks l, m, n, o).
The assignment of bands involving deuteron motions in the spectrum of the 12c4-D+ isotopologue may be attempted in a similar way. Salient bands, in terms of intensity, are found at 1180, 1045, 986 and 883 cm−1 (bands d′, g′, h′ and j′, respectively). In the vibrational motions associated with these modes, deuteron motions couple to the C–C and C–O stretching modes. Such couplings also complicate the identification of bands associated with dominant motions of the deuteron bond (analogous to bands α and β for 12c4-H+). Bands h′ and j′ in the 12c4-D+ spectrum at 986 and 883 cm−1, respectively, likely correspond to C–C/C–O stretching modes shifted with respect to the analogous bands at observed for 12c4-H+ (bands h and j). Such shifts would be related to the participation of proton/deuteron motions in the vibrational modes. The inspection of the 12c4-D+ spectrum also reveals the presence of a salient band at 1029 cm−1 that is absent in 12c4-H+. This characteristic band may tentatively be ascribed to a deuterated analog to band α at 1500 cm−1, based on reduced mass considerations (1500/1029 ∼ √2) and it is therefore labeled α′. Its low intensity is however indicative of an extensive mixing with backbone modes that would partly cancel changes in the dipole moment along the corresponding normal coordinates. Finally, the presence of only one prominent band in the 500–800 cm−1 range (band m′) for the 12c4-D+ complex is noticeable, given the strong transitions observed in this region for 12c4-H+. We tentatively assume that band m′, the only spectral signature clearly observed in this region for 12c4-D+, would be analogous to band m at 695 cm−1 in 12c4-H+, and that further neighboring bands are not observed in the deuterated complex due to the low transition moments. The computations indeed predict a significant lowering of the transition moments in the 12c4-D+ complex compared to 12c4-H+.
Further insights into the interpretation of the He-droplet spectra are developed below, in the light of computational insights into the structural and dynamic features of the low-energy conformational landscape of the protonated crown ether, and into the vibrational signatures of the individual conformers that may contribute to the experiment.
3.3 Computational analysis of 12c4-H+ and 12c4-D+
Modeling of the structure and vibrational signatures of the 12c4-H+ and 12c4-D+ complexes was carried out with the MP2 and DFT methods. These same methods were employed to investigate the potential energy surface driving the intracavity proton bond. Born–Oppenheimer Molecular Dynamics was applied to assess the magnitude of backbone fluctuations and their effect on the proton bond.In Fig. S5 (ESI†), we compare the lowest-energy conformations and respective IR spectra of protonated crown ether with neutral, bare crown ether. The computations suggest that the neutral crown ether adopts a bowl-like conformation of S4 symmetry in which the repulsion between the lone pairs of the O atoms is minimized. Hence, their structures as well as IR spectra are very different and a comparison does not help in assigning spectral features.
The fluxional character of the protonated crown ether suggests that relaxation during the higher temperature stages of ion production and storage is likely to be more efficient than for the more rigid 12c4-Li+ complex. Hence, despite the uncertainty about the abundance of different conformers that are stabilized in the He droplets, we will attempt to rationalize the recorded spectra in terms of the two conformers of lowest energy, H1 and H2. We will see that these two conformers already lay out a highly complex scenario for the modeling of the spectral signatures of the 12c4-H+ and 12c4-D+ isotopologues.
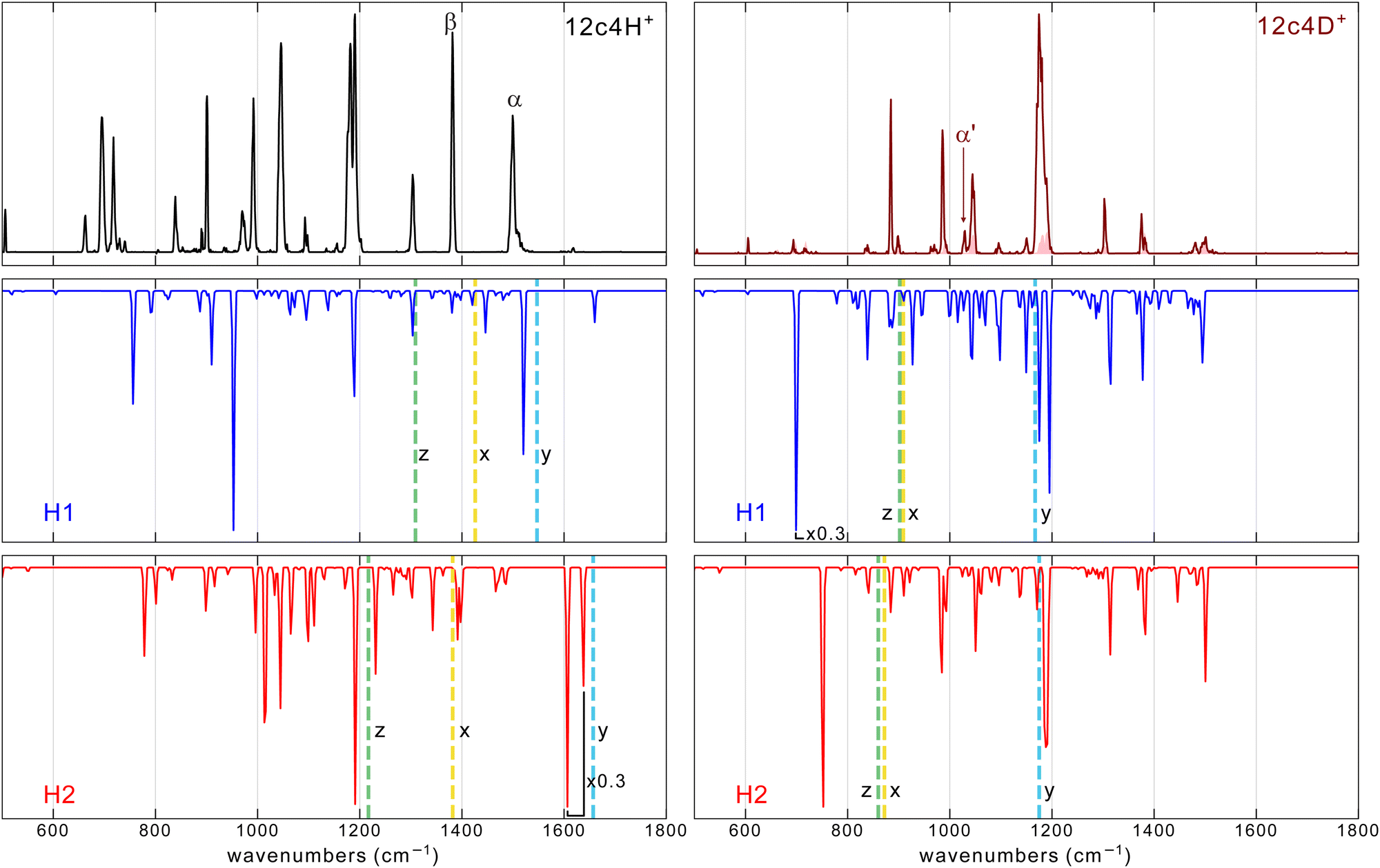 | ||
| Fig. 4 Comparison of the He-droplet IR spectra of the 12c4-H+ and 12c4-D+ complexes (top traces) with the MP2/aug-cc-pVTZ spectra (frequencies scaled by factor 0.975) for the two conformers of lowest energy, H1 and H2 (see Fig. 1). The vertical dashed lines correspond to the estimated frequencies computed by one-dimension DVR of three vibrational modes of H+ (D+): the O–H stretching mode along the two nearest oxygen (x), the proton motion orthogonal to the stretching in the plane (y) and out of the plane (z) of the backbone. More details are provided in the (ESI†). | ||
The MP2 method provides a fair approximation to the vibrational band features observed experimentally for the 12c4-D+ system. In particular, the MP2 spectrum for the H2 conformer of 12c4-D+ accounts for the most salient features of the He droplet measurement. While the analogous spectrum for the H1 conformer compares less satisfactorily, its incorporation helps to reproduce the structured envelope of the most intense and congested band in the 12c4-D+ spectrum at around 1180 cm−1.
For the 12c4-H+ system, the differences between the computed MP2 spectra and the He-droplet spectrum are quite significant, despite a partial agreement for some of the main bands in the 1100–1500 cm−1 range. The mismatch of the experimental bands with the MP2 predictions becomes particularly evident in the low-frequency region (below 1100 cm−1), where the experiment displays a sequence of prominent bands. A detailed discussion of the computed spectra is given in the ESI.†
The apparent limitations of the harmonic MP2 or DFT computations to reproduce the vibrational signatures of the protonated crown ethers at cryogenic temperatures were not completely unexpected. Fig. 1 shows that for the semi-rigid crown ether backbone, the concerted interaction of the two O atoms with the proton across the cavity leads to a markedly non-harmonic potential energy surface, and this poses fundamental difficulties to the prediction of spectral observables. In an attempt to capture the effects of anharmonicities, we included them using a perturbation treatment at the GVPT2/B3LYP-D3(BJ)/aug-cc-pVTZ level of theory. In those calculations, some modes got lowered by more than 1000 cm−1 with respect to their harmonic values, which clearly shows that for this system, the inclusion of anharmonicities as a mere perturbation of harmonic normal modes is insufficient and invalid. Alternative strategies seem to be required to describe the vibrational dynamics in the here presented protonated crown ethers.
The BOMD trajectories computed sample the actual potential electronic energy surface of the system (at a given computational level) and intrinsically incorporate anharmonic vibrational features from the Fourier Transform deconvolution of the time evolution of dipole moments of the ion. This approach leads in fact to an excellent agreement with the room-temperature IRMPD spectra of the protonated 12c4, 15c5, and 18c6 crown ethers.13 Fig. S8 (ESI†) shows that BOMD, as applied here with the B3LYP functional, does not improve the agreement with the experiment, with respect to DFT. It must be noted that BOMD trajectories must be interpreted with caution at cryogenic temperatures as the description of proton motions and the associated mode couplings and vibrational signatures can be expected to be severely affected by the lack of nuclear quantum effects (e.g., zero-point energy and tunneling) within the BOMD approach.
At the sub-Kelvin temperature of the present experiments, the crown ether backbone can be considered frozen, in its zero-point energy state. In contrast, at room temperature, thermal atomic motion becomes significant and induces continuous changes in the distance across the cavity between the proton-bonded O atoms, and therefore, the PES that drives proton bonding fluctuates in time. This scenario is illustrated in Fig. 5, which represents the distribution of O–O distances in protonated 12c4 from BOMD computations at temperature 350 K and 1 K. At the higher temperature, the crown ether adopts O–O distances over a comparably wide range, within 2.3–2.7 Å. Fig. 5 shows that a change in the O–O distance of only ±0.1 Å induces dramatic changes in the PES for the proton; at shorter O–O distances the PES becomes single-welled, while at longer distances a significant barrier emerges between the O atoms. Therefore, at room temperature, the PES felt by the proton is highly dynamic. As a consequence, the frequencies of the vibrational modes more directly associated with the proton-bonded moiety are likely to be spread over a wide spectral range. Similar considerations would apply to the coupling of the proton bond with the vibrational modes of the crown ether backbone.
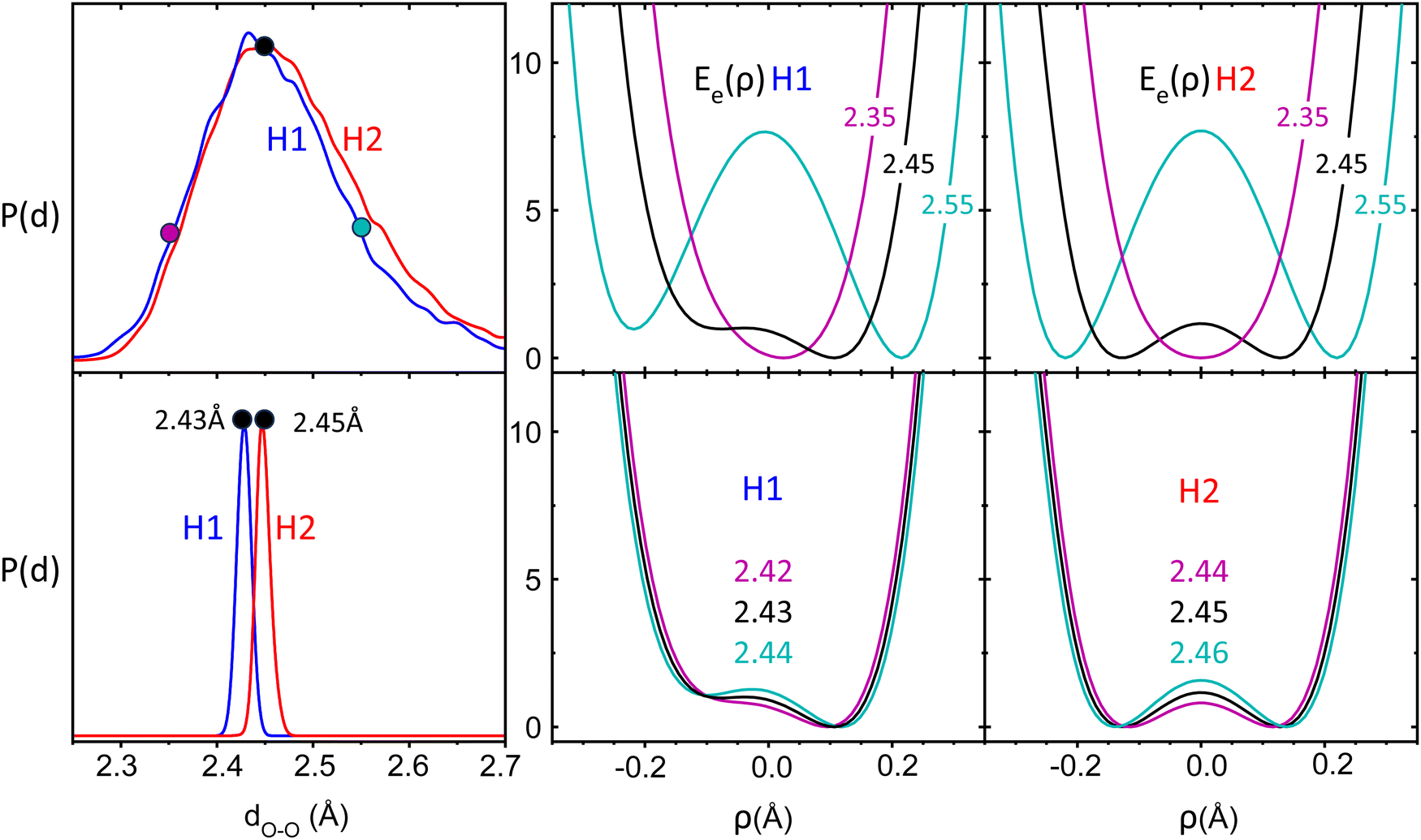 | ||
| Fig. 5 Distribution of distances between the proton-bonded O atoms, P(d), from BOMD computations of 12c4-H+ in the H1 and H2 conformations at temperatures 350 K (top) and 1 K (bottom). Middle and right: Relaxed potential energy curves (kJ mol−1) for the proton in the intramolecular bond at fixed O–O distances representative of each temperature (coordinate ρ defined as (R − r)/2). | ||
At cryogenic temperatures, represented by the BOMD computation at 1 K, the fluctuations in the O–O distances are dramatically reduced down to the ±0.01 Å range. The changes in the potential energy curve for the proton are correspondingly small, falling below the 1 kJ mol−1 range. This freezing of the crown ether backbone gives rise to a virtually static PES for the proton in the intramolecular bond. Valuable insights into the vibrational signatures of the cryogenic proton bond may be then derived from considering the effective potential energy surface for the bound proton. To this end, we have employed the discrete variable representation (DVR) scheme31 to determine the vibrational eigenstates of the proton in one-dimensional cuts through the PES. In particular, we use DVR to solve for the energy levels associated with the stretching of the O–H+⋯O proton bond and with the orthogonal bending motions, in-plane and out-of-plane (Fig. S9, ESI†). For each mode, the potential energy curve is computed along the corresponding coordinate, while keeping the crown ether core frozen. We employ an iterative scheme, wherein the first iteration, the B3LYP crown ether core is used. Next, the ground state DVR wave function and the expectation value for the proton location are calculated. Then, the structure is re-optimized at the B3LYP level with the O–H bond distance fixed to the DVR expectation value. This process is converged to a stable proton location after two iterations. Depending on the structure and isotope, this process caused the O–O distance to contract by 0.02–0.05 Å. (details are provided in the ESI†).
The resulting potential energy curves for the stretching mode of the proton bond along the nearest oxygen atoms as well as eigenvalues and wavefunctions for the n = 0 and 1 vibrational levels for 12c4H+ and 12c4D+ are shown in Fig. 6 (see Table S3, ESI† for numerical values). For the H2 conformer, this curve does not contain a barrier as in Fig. 5. This is due to the fact that for the latter, a relaxed scan is performed where at each point, the crown ether backbone is re-optimized and only the O–H distance is kept fixed, while it is completely frozen in the DVR calculations.
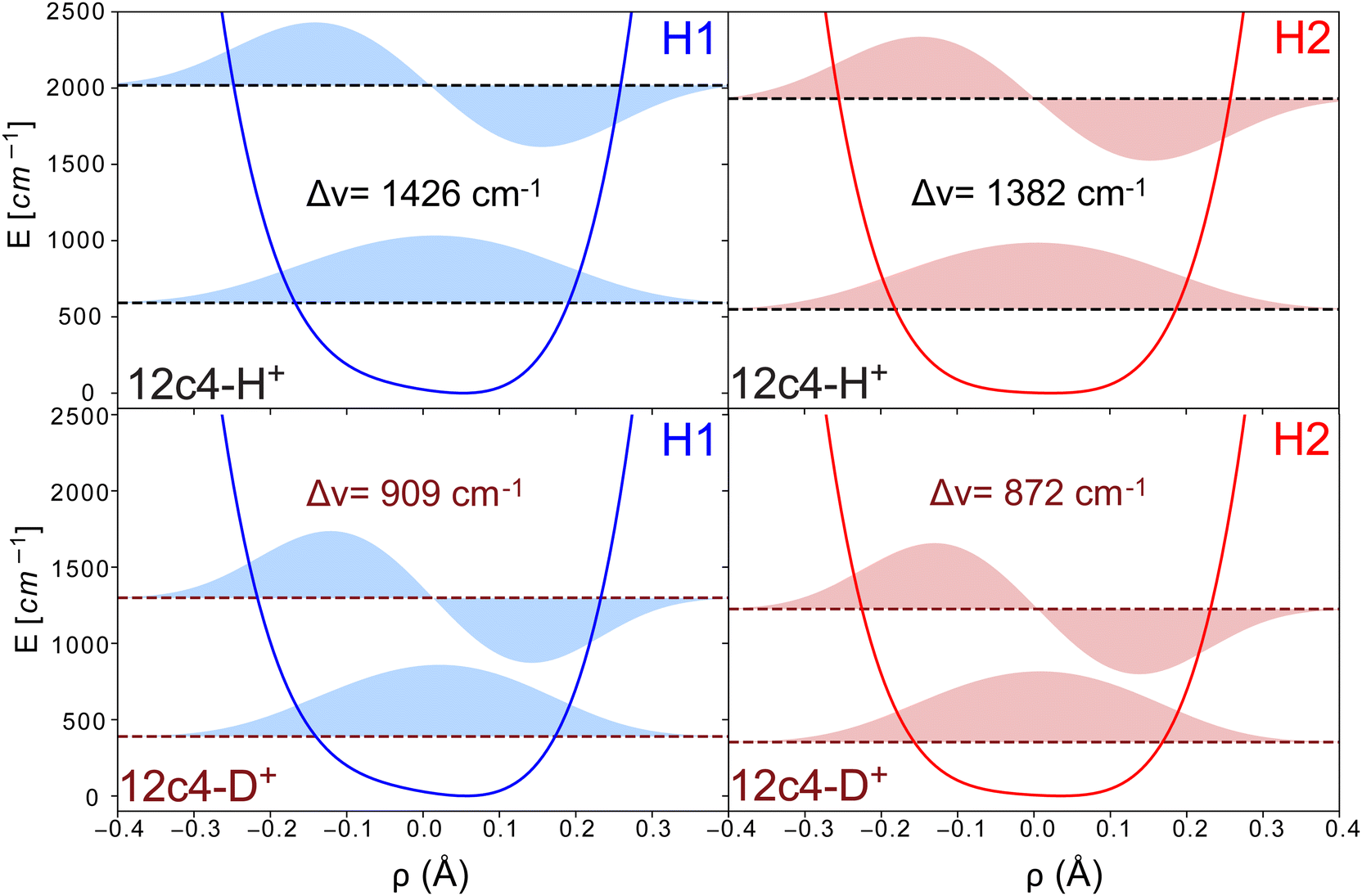 | ||
| Fig. 6 Schematic representation of the potentials for the O–H+⋯O stretching coordinate in the H1 and H2 structures, along with their corresponding eigenvectors and eigenvalues for the first two vibrational states, calculated using DVR computations (coordinate ρ defined as (R − r)/2). The different panels depict the results for both conformers and H/D isotopologues. | ||
The transition frequencies for the stretching modes derived from the DVR calculations are 1426 and 1382 cm−1 (909 and 872 cm−1) for the H1 and H2 conformers of 12c4H+ (values in brackets for 12c4D+). The corresponding frequencies for the in-plane bending motion are higher, at 1547 and 1657 cm−1 (1167 and 1175 cm−1) and for the out-of-plane bending motion, the DVR transition frequencies are found at 1309 and 1217 cm−1 (902 and 860 cm−1). While the treatment of anharmonicity provided by the one-dimensional DVR approach is certainly simplistic, it provides support to a scenario where the proton bond modes are expected to be found above 1200 cm−1 for 12c4H+ and below 1200 cm−1 for 12c4D+. For 12c4H+, the main vibrational signatures of the bound proton would be near the bending modes of the –CH2 groups of the crown ether. For 12c4D+, they would fall in the range of the main stretching and modes of the [–COC–]4 crown ether ring.
In addition, we calculated the transition strengths from the changes of the dipole moments along the DVR coordinates. In those calculations, the stretching modes showed much larger intensities than those of the in-plane and out-of-plane perpendicular modes. However, experimentally, we expect many modes to be coupled with the proton motion, making it difficult to make reliable predictions of the relative intensities.
Based on the DVR results it seems tempting to assign the experimental bands β and α in the spectra of 12c4H+ to the stretching and orthogonal in plane mode of the proton, respectively. Alternatively, bands β and α could stem from the simultaneous presence of H1 and H2. The similarity of the intensities of the two peaks would support this latter option. Nevertheless, based on the calculations, it seems difficult to make a firm statement on which of the two scenarios is giving rise to those bands.
It is well known that bands involving hydrogen or proton bonding are often observed to be broad,32–35 usually attributed to homogeneous broadening due to anhamonicities and short excited state lifetimes. In the experimental spectrum of 12c4-H+ in helium droplets (Fig. 3), a narrow band is observed at 1500 cm−1, denoted as α, with a width (FWHM) of ≈ 10 cm−1. In contrast, the same feature in the IRMPD spectrum appears slightly blue-shifted and spans over more than 200 cm−1. Since this spectral region involves bands associated with the mobile proton, it suggests that anhamonicities and short excited state lifetimes are not the primary causes for the spectral broadening observed for this band. A more likely explanation is that the broadening is caused by the dynamics of the backbone which modulates in time the potential energy surface (PES) experienced by the proton. These backbone fluctuations occur on a timescale slower than the timescale of the photon absorption, leading to fluctuations in transition frequencies and resulting in a broad envelope over the corresponding bands. Broadening for thermally fluctuating molecules can occur when either an ensemble of molecules is probed in a single photon experiment and/or when a single molecule is probed in a multiple photon excitation process. The spectral broadening here can be described as a dynamic inhomogeneous broadening, being only inhomogeneous because of the difference in timescale between the dynamics of the backbone fluctuations and the photon absorption process.
4 Summary and conclusions
The mid-IR spectra of 12-crown-4 complexed with Li+, H+ and D+ have been measured at cryogenic temperatures in liquid helium droplets. Together with results from theoretical calculations, it can be concluded that Li+ adopts a position where it interacts with similar strength with all oxygen atoms and the experimental spectra show evidence for the presence of multiple conformers, differing in the puckering in the crown ether backbone. The experimental spectrum is fairly well described by ab initio calculated spectra at the harmonic approximation.Compared to the spectrum of 12c4-Li+, the cryogenic spectra of 12c4-H+ and 12c4-D+ show the presence of bands over a much wider range. The comparison to ab initio and BOMD calculations show that the proton or deuteron prefers to be bound to one oxygen atom or to be shared by two. For those systems, ab initio calculated spectra do not reproduce the experimental spectra well, both at the harmonic approximation and when including anharmonic effects in a perturbative scheme.
The cryogenic spectra of 12c4-H+ and 12c4-D+ are also compared to IRMPD spectra, which show much broader and shifted bands. For 12c4-H+, the band associated with the motion of the proton of the IRMPD spectrum shows an unusual blue-shift with respect to its spectrum recorded in liquid helium droplets. BOMD calculations show that at elevated temperatures, the crown ether backbone is highly fluxional and that the distance between the oxygen atoms flanking the proton or deuteron distance fluctuates in time. Calculating one dimensional potentials for the proton in the crown ether cavity shows that those coordinates are highly non harmonic. Using DVR, energy levels are calculated, and derived transition frequencies show fair qualitative agreement with experimental bands. It is concluded that at elevated temperatures, the thermal backbone fluctuations induce a modulation of this potential, resulting in dynamic inhomogeneous broadening and a blue shift of the corresponding IRMPD band.
The vibrational signatures of protonated 12-crown-4 at cryogenic temperatures emerge as a valuable benchmark for quantum chemical methods. The crown ether ring provides a semi-rigid template to investigate fundamental features of proton bonding, whose description seems to be elusive within the framework of a harmonic or perturbative anharmonic description.
Data availability
The data within the article or its ESI† are available and can be accessed at [https://doi.org/10.1039/d4cp04058a]. Researchers interested in accessing the data should contact helden@fhi-berlin.mpg.de. Any additional information regarding the data can be provided upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Sandy Gewinner, Marco de Pas, and Wieland Schöllkopf for providing high-energy FEL radiation. A. Y. T. B. acknowledges support by the IMPRS for Elementary Processes in Physical Chemistry. M. I. T. gratefully acknowledges the support of the Alexander von Humboldt Foundation. K. O. acknowledges funding from the Max Planck-Radboud University Center for Infrared Free Electron Laser Spectroscopy. B. M. H. receives support from the Government of Spain (project TED2021-130683b-C21). We are grateful to the BIO-MS laboratory and to the C3UPO High Performance Computing Center of Universidad Pablo de Olavide for technical support. Additionally, we thank Priyansh Agarwal for helping with the implementation of the DVR code in Python. Open Access funding provided by the Max Planck Society.Notes and references
-
Hydrogen Transfer Reactions, ed. J. T. Hynes, J. P. Klinman, H. H. Limbach and R. L. Schowen, Wiley-VCH, Weinheim, 2007 Search PubMed
.
- G. Zundel, Adv. Chem. Phys., 2007, 111, 1–217 CrossRef
- M. Meot-Ner, Chem. Rev., 2012, 112, PR22–PR103 CrossRef CAS PubMed
- H. Ishikita and K. Saito, J. R. Soc., Interface, 2014, 11, 20130518 CrossRef PubMed
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. Nibbering, Science, 2005, 310, 83–86 CrossRef CAS PubMed
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed
- H. Knorke, H. Li, J. Warneke, Z.-F. Liu and K. R. Asmis, Phys. Chem. Chem. Phys., 2020, 22, 27732–27745 RSC
- J. R. Roscioli, L. R. McCunn and M. A. Johnson, Science, 2007, 316, 249–254 CrossRef CAS PubMed
- X. Li, D. T. Moore and S. S. Iyengar, J. Chem. Phys., 2008, 128, 184308 CrossRef PubMed
- C. Leavitt, A. DeBlase, C. Johnson, M. van Stipdonk, A. McCoy and M. Johnson, J. Phys. Chem. Lett., 2013, 4, 3450–3457 CrossRef CAS
- S. M. Craig, F. S. Menges, C. H. Duong, J. K. Denton, L. R. Madison, A. B. McCoy and M. A. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4706–E4713 CAS
- J. Oomens, G. Berden, J. Martens and T. H. Morton, Int. J. Mass Spectrom., 2017, 418, 188–192 CrossRef CAS
- F. Gámez, J. R. Avilés-Moreno, G. Berden, J. Oomens and B. Martínez-Haya, Phys. Chem. Chem. Phys., 2021, 23, 21532–21543 RSC
- B. Martínez-Haya, J. R. Avilés-Moreno, F. Gámez, J. Martens, J. Oomens and G. Berden, J. Phys. Chem. Lett., 2023, 14, 1294–1300 CrossRef PubMed
- L. Chen, E. L. Sibert III and J. A. Fournier, J. Phys. Chem. A, 2023, 127, 3362–3371 CrossRef CAS PubMed
- F. Gámez, J. R. Avilés-Moreno, J. Martens, G. Berden, J. Oomens and B. Martínez-Haya, J. Chem. Phys., 2024, 160, 094311 CrossRef PubMed
- D. T. Moore, J. Oomens, L. van der Meer, G. von Helden, G. Meijer, J. Valle, A. G. Marshall and J. R. Eyler, ChemPhysChem, 2004, 5, 740–743 CrossRef CAS PubMed
- J. Toennies and A. Vilesov, Angew. Chem., Int. Ed., 2004, 43, 2622–2648 CrossRef CAS PubMed
- A. I. G. Flórez, D. S. Ahn, S. Gewinner, W. Schöllkopf and G. von Helden, Phys. Chem. Chem. Phys., 2015, 17, 21902–21911 RSC
- D. A. Thomas, M. Marianski, E. Mucha, G. Meijer, M. A. Johnson and G. von Helden, Angew. Chem., Int. Ed., 2018, 57, 10615–10619 CrossRef CAS PubMed
- M. Taccone, W. Schöllkopf, D. Thomas, G. Meijer, K. Ober, S. Gewinner and G. von Helden, Phys. Chem. Chem. Phys., 2023, 25, 10907 RSC
- A. Y. Torres-Boy, M. I. Taccone, C. Kirschbaum, K. Ober, T. Stein, G. Meijer and G. von Helden, J. Phys. Chem. A, 2024, 128, 4456–4466 CrossRef CAS PubMed
-
E. Mucha, D. Thomas, M. Lettow, G. Meijer, K. Pagel and G. von Helden, Spectroscopy of small and large biomolecular ions in helium-nanodroplets, Springer International Publishing Cham, 2022, pp. 241–280 Search PubMed
- A. El-Azhary and A. Al-Kahtani, J. Phys. Chem. A, 2005, 109, 8041–8048 CrossRef CAS PubMed
- J. Light and T. Carrington, Adv. Chem. Phys., 2000, 114, 263–310 CrossRef
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt and F. Schiffmann,
et al.
, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed
- D. Ray, D. Feller, M. More, E. Glendening and P. Armentrout, J. Phys. Chem., 1996, 100, 16116–16125 CrossRef CAS
- S. Al-Rusaese, A. Al-Kahtani and A. El-Azhary, J. Phys. Chem. A, 2006, 110, 8676–8687 CrossRef CAS PubMed
- S. De, A. Boda and S. Ali, J. Mol. Struct., 2010, 941, 90–101 CrossRef CAS
- J. Rodriguez, D. Kim, P. Tarakeshwar and J. Lisy, J. Phys. Chem. A, 2010, 114, 1514–1520 CrossRef CAS PubMed
-
J. C. Light and T. Carrington, Advances in Chemical Physics, John Wiley & Sons, Inc., 2000, pp. 263–310 Search PubMed
- L. Al-Adhami and D. Millen, Nature, 1966, 211, 1291 CrossRef CAS
- J. B. Asbury, T. Steinel and M. Fayer, J. Phys. Chem. B, 2004, 108, 6544–6554 CrossRef CAS
- T. Yamashita and K. Takatsuka, J. Chem. Phys., 2007, 126, 074304 CrossRef PubMed
- E. T. Nibbering and T. Elsaesser, Chem. Rev., 2004, 104, 1887–1914 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Including additional cryogenic spectra, further theoretical conformations and spectra calculated at different levels of theory, and details of the DVR analysis. See DOI: https://doi.org/10.1039/d4cp04058a |
| This journal is © the Owner Societies 2025 |