
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated diradical character assessment in large datasets of polybenzenoid hydrocarbons using xTB fractional occupation†
Alexandra Wahab a and
Renana Gershoni-Poranne*ab
a and
Renana Gershoni-Poranne*ab
aLaboratory for Organic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
bSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa 32000, Israel. E-mail: rporanne@technion.ac.il
First published on 29th November 2024
Abstract
Polybenzenoid hydrocarbons (PBHs) have garnered significant attention in the field of organic electronics due to their unique electronic properties. To facilitate the design and discovery of new functional organic materials based on these compounds, it is necessary to assess their diradical character. However, this usually requires expensive multireference calculations. In this study, we demonstrate rapid identification and quantification of open-shell character in PBHs using the fractional occupation number weighted electron density metric (NFOD) calculated with the semiempirical GFN2-xTB (xTB) method. We apply this approach to the entire chemical space of PBHs containing up to 10 rings, a total of over 19k molecules, and find that approximately 7% of the molecules are identified as having diradical character. Our findings reveal a strong correlation between xTB-calculated NFOD and the more computationally expensive Yamaguchi y and DFT-calculated NFOD, validating the use of this efficient method for large-scale screening. Additionally, we identify a linear relationship between size and NFOD value and implement a size-dependent threshold for open-shell character, which significantly improves the accuracy of diradical identification across the chemical space of PBHs. This size-aware approach reduces false positive identifications from 6.97% to 0.38% compared to using a single threshold value. Overall, this work demonstrates that xTB-calculated NFOD provides a rapid and cost-effective alternative for large-scale screening of open-shell character in PBHs.
Introduction
Organic compounds displaying diradical character have long fascinated chemists, as they provide unique platforms for investigating the nature of the chemical bond and other fundamental organic concepts. Classic examples include Tschitschibabin's hydrocarbon, bisphenalenyls, zethrenes, and indenofluorenes (Fig. 1), which have all been extensively investigated both computationally1–7 and experimentally.2–4,8–12 It is unsurprising that the majority of long-lived, synthetically accessible organic compounds with open-shell character are polycyclic aromatic hydrocarbons (PAHs). In such systems, termed “pro-aromatic” by Wu and coworkers,13 the closed-shell resonance structure (RS) is quinoidal whereas the open-shell RS contains additional disjoint 6-π electrons rings (compare the left and right RSs in Fig. 1; the diradical RSs contain additional Clar sextets). Hence, the increase in aromaticity gained by breaking the double bond stabilizes—and perhaps even drives—the adoption of diradical character.14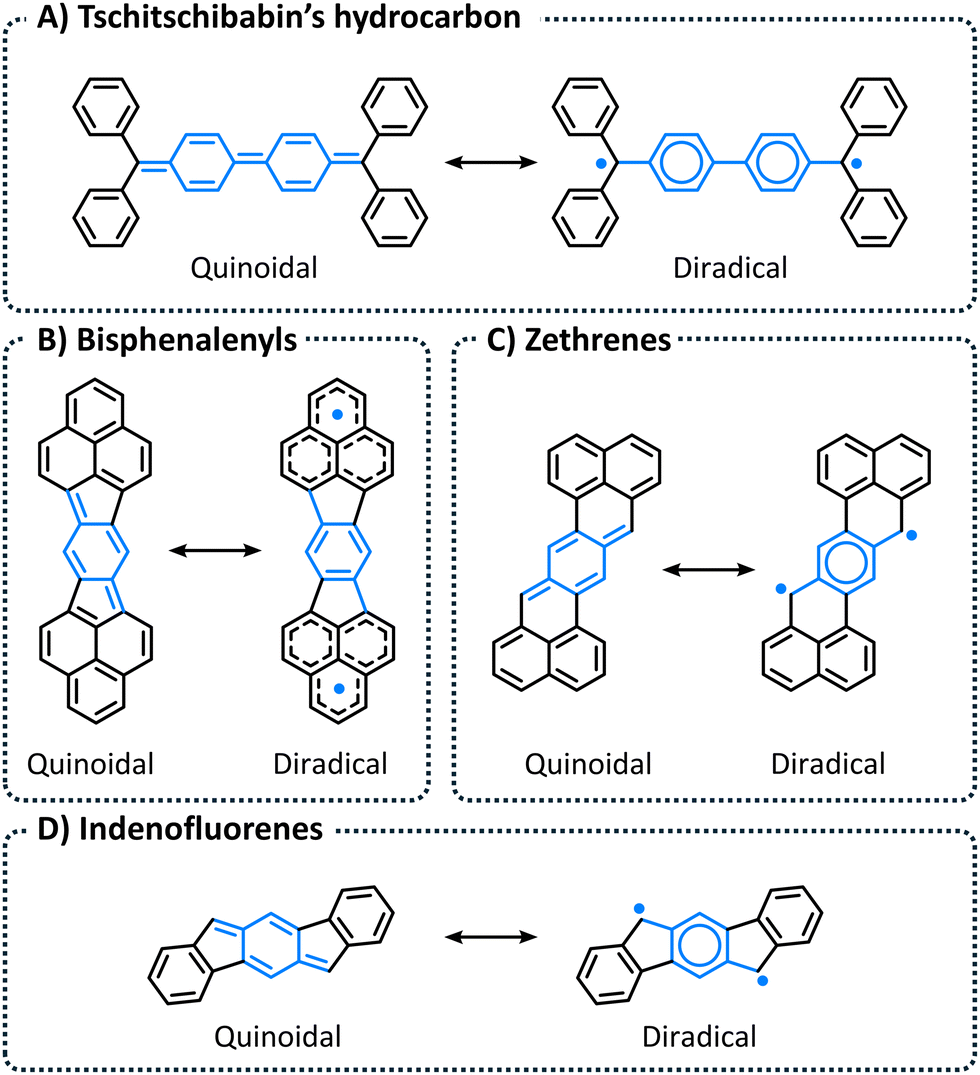 | ||
| Fig. 1 Examples of PAHs known to have appreciable diradical character: (A) Tschitschibabin's hydrocarbon; (B) Bisphenalenyls; (C) Zethrenes; and (D) Indenofluorenes. In all cases, left: quinoidal RS, right: diradical RS. | ||
Among such PAHs, polybenzenoid hydrocarbons (PBHs, cutouts of a graphene sheet) have emerged as especially promising for organic electronics. When endowed with significant open-shell character, PBHs demonstrate potential for a broad range of such uses, including light-emitting diodes, batteries, field-effect transistors, and photovoltaics.15 As the diradical character of a molecule is directly linked to its physical properties (optical, electronic, and magnetic), the identification and quantitative evaluation of this character is crucial to understanding and predicting molecular behavior, as well as to designing new functional compounds.
Larger PBHs in general, and polyacenes (linearly annulated PBHs) in particular, are considered to have a tendency toward open-shell ground states.16,17 In contrast, the majority of small PBHs have closed-shell electron configurations. Nevertheless, open-shell configurations—i.e., radical or diradical character—can occur in three scenarios (Fig. 2): (a) an odd number of hydrogens/carbons (e.g., phenalenyl radical, C13H9, is a three-ring peri-condensed (pc-)PBH with a single unpaired electron); (b) non-Kekuléan structures, i.e., PBHs for which no classical closed-shell valence structure can be drawn18,19 (e.g., triangulene, C22H12, is a non-Kekuléan six-ring pc-PBH with two unpaired electrons in the ground state); and (c) molecules that possess a closed-shell resonance structure, but have appreciable diradical character (e.g., longer polyacenes, zethrenes, periacenes).19
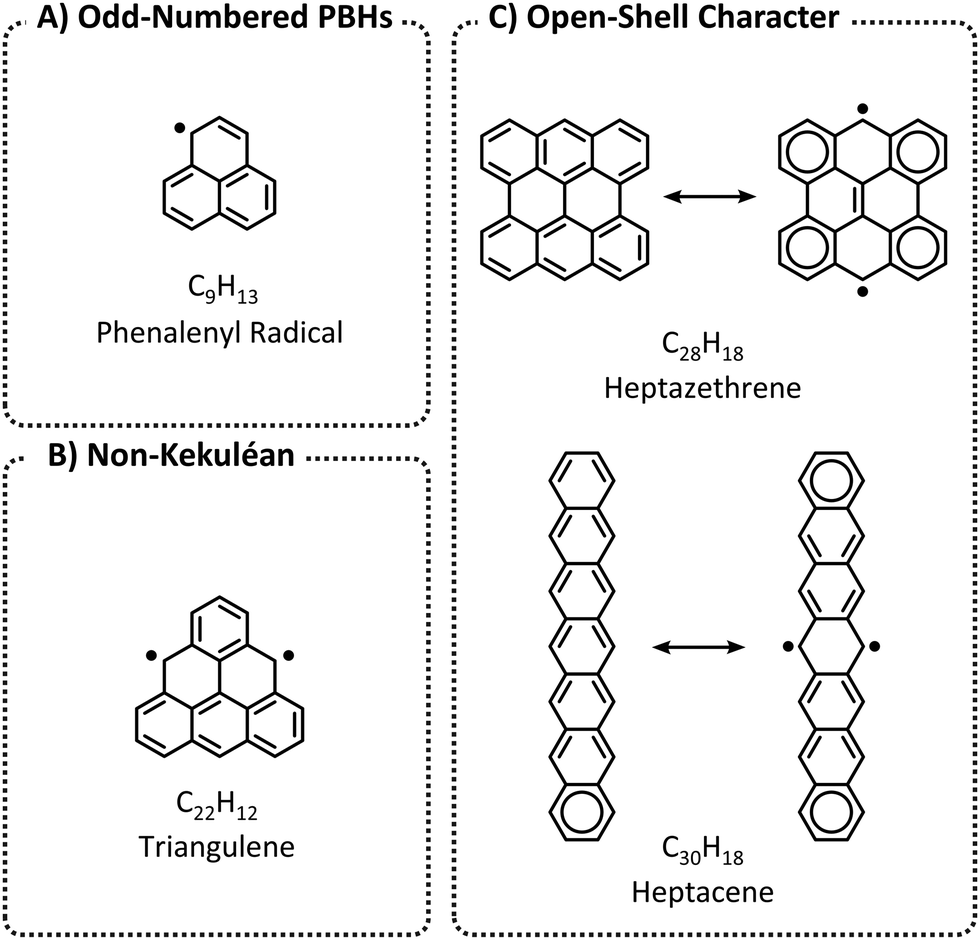 | ||
| Fig. 2 Three possible scenarios for PBHs to obtain diradical character: (A) Odd-numbered PBHs; (B) Non-Kekuléan PBHs; and (C) PBHs with open-shell character. | ||
The first two cases are relatively easily identified. It is the third scenario that is most challenging, as the open-shell character is not implied by either the molecular or the structural formula. Thus, reliable methods for identification of open-shell character are required for these molecules.
However, such assessment is not trivial because open-shell PAHs often have multireference (MR) character, meaning they are described by two or more dominant contributing electronic configurations. Hence, to adequately describe the wavefunction of the molecule, MR methods are needed.17,20–24 In addition to their high computational cost and their molecular-size limitations, such methods usually require a high level of user expertise and are not easily accessible to non-specialists. Therefore, density functional theory (DFT) remains the most prevalent method, despite the inherent difficulty of choosing the most appropriate functional. To simplify the evaluation of radical and diradicaloid character, Lischka and coworkers recently benchmarked various DFT-derived descriptors and showed that the number of “hot” electrons, NFOD, obtained from the fractional occupation number weighted electron density (FOD), provides a fast and reliable assessment of different classes of PAHs, without the need to perform MR calculations.25 In their report, the authors used thermally-assisted DFT to obtain the NFOD values for a test set of 29 polycyclic aromatic hydrocarbons, which they compared to the numbers of unpaired electrons (NU) obtained from MR-averaged quadratic coupled-cluster (MR-AQCC). Additional systems were investigated in a subsequent report, published very recently.26 Their work highlights both the ongoing interest in PAHs and the need for inexpensive and user-friendly methods for evaluating diradical character. Indeed, they conclude their report with the statement “This finding opens the possibility of large scale and reliable screening of PAH biradical properties, which is expected to have a significant impact on the PAH research field.”‡
Herein, we build upon the work of Lischka and coworkers in two directions. First, we continue the process of simplification by demonstrating that NFOD values obtained from a semiempirical method (namely, GFN2-xTB; referred to as xTB in this text for conciseness) can be used for this evaluation, rather than DFT. We note that a very recent report26 by the same group also demonstrated the use of xTB NFOD for various PAHs in the context of bond dissociation energies. Second, using this computationally efficient tool, we embark on the large-scale screening they envisioned by assessing the diradical character of all isomers in the chemical space of PBHs containing up to 10 rings.
The work described in this manuscript is detailed in the following sections: (a) the data used in the study, (b) the open-shell character descriptors employed, (c) the benchmarking procedure from which a threshold value was identified and verified, (d) the process developed to make the approach transferable across molecules of various sizes, and (e) comparison of xTB- and DFT-calculated NFOD.
Data and descriptors
Data
Polybenzenoid hydrocarbons (PBHs) are made up of only one type of ring—benzene. Their structural diversity, therefore, stems from variations in the number of fused rings and in the patterns of fusion that are possible. In the context of diradical character, the most extensively studied systems are the extended polyacenes, zethrenes, and periacenes. Several computational investigations have focused on these types of scaffolds, using a variety of methods, including unrestricted DFT,1,29 CASSCF,21,22 restricted active space configuration interaction,20,24 spin–flip techniques,20,30 and MR-AQCC.17,23 In this work, we sought to extend beyond these recognized structures and explore the chemical space of PBHs more broadly, therefore we investigate the entire chemical space of PBHs comprising up to 10 rings (i.e., all isomers containing nrings ≤ 10).For the purpose of this study, we divided this chemical space into various groups, as depicted in Fig. 3. First, we distinguished between cata-condensed (cc-PBHs) and peri-condensed (pc-PBHs). In cc-PBHs, a single carbon may only participate in up to two rings; in pc-PBHs, at least one carbon participates in three rings. Second, we further divided each of the subgroups into two groups: cc-PBHs—Groups A and B, pc-PBHs—Groups C and D. For the cc-PBHs, the division was performed using a structural descriptor: the longest linear stretch within the molecule, nLL. Bendikov et al.16 and Yang et al.29 independently identified hexacene as the smallest polyacene to show indications of multireference character. This was corroborated by the experimental observations of instability of the acenes larger than pentacene.31–34 Accordingly, we divided our data such that Group A contained molecules with nLL < 6, which were expected to be closed-shell, and Group B contained molecules with nLL ≥ 6, which were expected to be diradical. For the pc-PBHs, the division was performed using an NxTBFOD-based threshold, which we identified in the course of this study (NxTBFOD = 1.2, vide infra). For this subset of data, Group C contained the molecules with NxTBFOD < 1.2 and Group D contained the molecules with NxTBFOD ≥ 1.2.
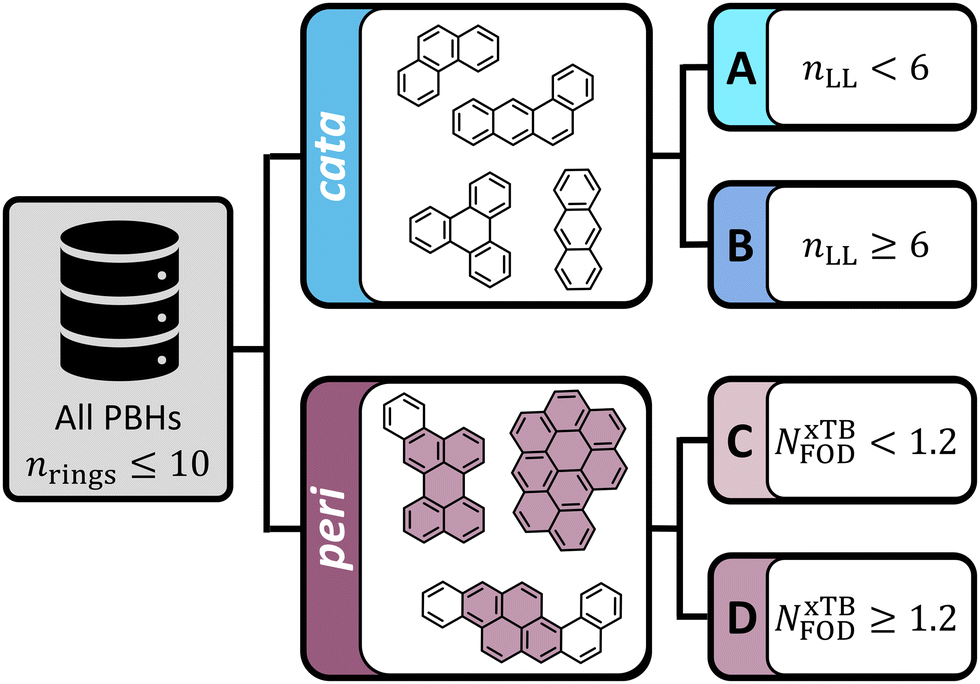 | ||
| Fig. 3 Overview of the data. All isomers of nrings ≤ 10 PBHs were included, divided into cata- and peri-condensed subsets (representative examples are shown). The cc-PBHs were divided into Groups A and B according to the structural parameter nLL. The pc-PBHs molecules were divided into Groups C and D according to an NxTBFOD threshold. | ||
As stated above, in this work we explored the entire chemical space of PBHs with nrings ≤ 10. The majority of molecular geometries were extracted from the COMPAS (COMputational database of Polycyclic Aromatic Systems) datasets.35,36 The COMPAS datasets were designed to focus on closed-shell molecules and, accordingly, COMPAS-1D closely corresponds to our Group A and COMPAS-3D closely corresponds to our Group C. All additional molecules were calculated in the course of the current investigation to ensure coverage of the entire chemical space.
To remain consistent with the COMPAS datasets, we calculated Groups B and D at the same level of theory. Thus, all geometry optimizations were performed with ORCA version 5.0.3,37,38 at the (U)CAM-B3LYP39–43/def2-SVP44 level, using Grimme's D3 dispersion correction45 with Becke–Johnson damping.46,47 Groups A and C were optimized with restricted Kohn–Sham DFT (RKS), while Groups B and D were optimized with unrestricted KS DFT (UKS).
Descriptors
We benchmarked the NFOD values against two widely used methods for assessment of open-shell character: the DFT-derived NFOD and the unrestricted Hartree–Fock (UHF)-derived Yamaguchi y value. Both are described below.
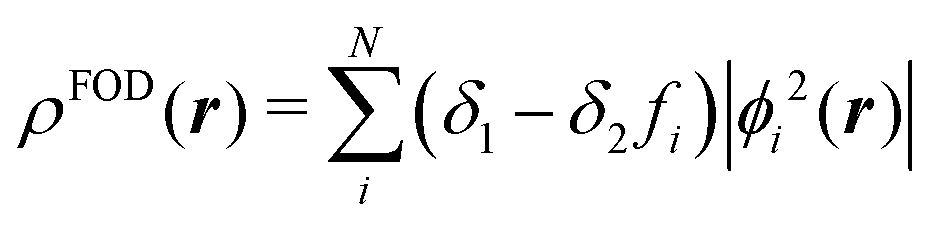 | (1) |
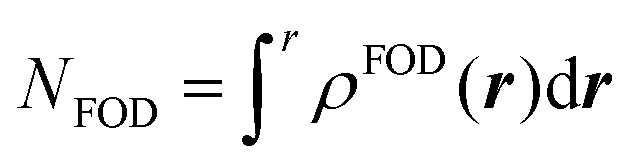 | (2) |
The main advantage of NFOD, as highlighted in Lischka and coworkers’ recent report,25 is its low cost, which carries an even more substantial benefit when working with large datasets. This advantage can be further exploited by calculating the NFOD value with xTB, a semiempirical method, thereby accelerating calculations by three orders of magnitude. An additional advantage has been pointed out by Chan, who showed that NxTBFOD can be used to identify potentially problematic components in a larger system, whereas many MT diagnostics are formulated for small systems and do not provide accurate characterization of large systems.49 However, NFOD also has two main disadvantages. The first is that, similarly to most MR diagnostics, there is no well-defined threshold for identifying open-shell character unambiguously. Nevertheless, previous work by Grimme and coworkers50 did indeed find a correlation between NFOD and y values on a small test set, demonstrating the possibility to use NFOD when it is benchmarked against a less ambiguous diagnostic. The second disadvantage is that NFOD is size-extensive, which makes it difficult, if not impossible, to define a universal threshold applicable to molecules of varying sizes. In this work, we shed light on the size-dependency of NFOD and provide a framework for alleviating this problem in the context of PBHs.
In this work, we calculated the NFOD values with both DFT (NDFTFOD) and xTB (NxTBFOD). For the DFT calculations, we used the B3LYP functional with the def2-SVP basis set and set the temperature to 9000 K, as recommended by Grimme and coworkers,51 based on the empirical formula Tel = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K × ax + 5000 K, where ax is the Fock exchange percentage of the functional (ax = 20% for B3LYP). For the xTB calculations, we set the temperature to 5000 K, as recommended by Grimme and coworkers.51 We note that ref. 26, which was published after completion of this work, recommends a temperature of 5200 K for xTB calculations. See Section S2 of the ESI† for a comparison of the results obtained with the two temperatures.
000 K × ax + 5000 K, where ax is the Fock exchange percentage of the functional (ax = 20% for B3LYP). For the xTB calculations, we set the temperature to 5000 K, as recommended by Grimme and coworkers.51 We note that ref. 26, which was published after completion of this work, recommends a temperature of 5200 K for xTB calculations. See Section S2 of the ESI† for a comparison of the results obtained with the two temperatures.
 | (3) |
In this work, we used only the y0 values (see Section S7 of the ESI† for additional discussion on the y1 values). Hence, for conciseness, we use the simple symbol y throughout.
Although the y value also does not have a well-defined threshold for identifying open-shell character, it is one of the most commonly used diagnostics in the organic chemistry community This is likely due to the fact that it uses a [0,1] scale. For a ‘perfect’ closed-shell system, the occupation of the HONO is 2 and the occupation of the LUNO is 0; accordingly, y0 = 0. For a ‘perfect’ diradical system, the occupation of both the HONO and the LUNO is 1, resulting in y0 = 1. The 0%-to-100% scale makes it very intuitive and straightforward to interpret. Because it is most often applied using the UHF orbital approach, this metric is rather inexpensive. Nevertheless, it requires broken symmetry calculations, which may become prohibitively expensive when applied to large molecules and/or large datasets. For context, in this investigation, we calculated the y values of 19875 molecules, with a median size of 62 atoms (median of 40 carbon atoms), using the UHF method and the def2-SVP basis set. The cumulative CPU time for these calculations was ∼178650 hours. In comparison, NxTBFOD calculations required ∼5 CPU hours and NDFTFOD calculations required ∼7810 CPU hours.
Results and discussion
Benchmarking
| nrings | Polyacenes | Polyphenacenes | ||||
|---|---|---|---|---|---|---|
| NxTBFOD | NDFTFOD | y (%) | NxTBFOD | NDFTFOD | y (%) | |
| 1 | 0.03 | 0.11 | 1 | — | — | — |
| 2 | 0.12 | 0.27 | 4 | — | — | — |
| 3 | 0.32 | 0.53 | 13 | 0.18 | 0.40 | 5 |
| 4 | 0.58 | 0.87 | 26 | 0.27 | 0.54 | 7 |
| 5 | 0.89 | 1.25 | 39 | 0.34 | 0.68 | 7 |
| 6 | 1.22 | 1.65 | 51 | 0.42 | 0.82 | 9 |
| 7 | 1.56 | 2.07 | 61 | 0.50 | 0.96 | 9 |
| 8 | 1.91 | 2.48 | 69 | 0.57 | 1.11 | 9 |
| 9 | 2.25 | 2.90 | 76 | 0.65 | 1.25 | 9 |
| 10 | 2.60 | 3.32 | 82 | 0.73 | 1.39 | 10 |
As expected, for every nrings, the acene isomer showed a higher value than the corresponding phenacene, for all three metrics. Additionally, although both families of compounds showed an increase in open-shell character as nrings increases, this behavior was much more pronounced for the polyacenes. For the polyacenes, open-shell character became dominant (i.e., y > 50%) from nrings = 6 (specifically, y = 51% for hexacene). In contrast, all polyphenacenes had negligible y values (≤10%). For both families of compounds, the results were in agreement with experimental data,55,56 which validated our choice of the y value as a benchmarking metric for the NFOD value.
We set y = 50% as the transition point between predominant closed-shell and predominant open-shell character, which made hexacene the first in the series to cross that threshold, as expected. The NFOD values for hexacene were NxTBFOD = 1.22 and NDFTFOD = 1.65. Therefore, we set the NxTBFOD threshold at 1.2, meaning that molecules with NxTBFOD ≥ 1.2 were considered to be predominantly open-shell.
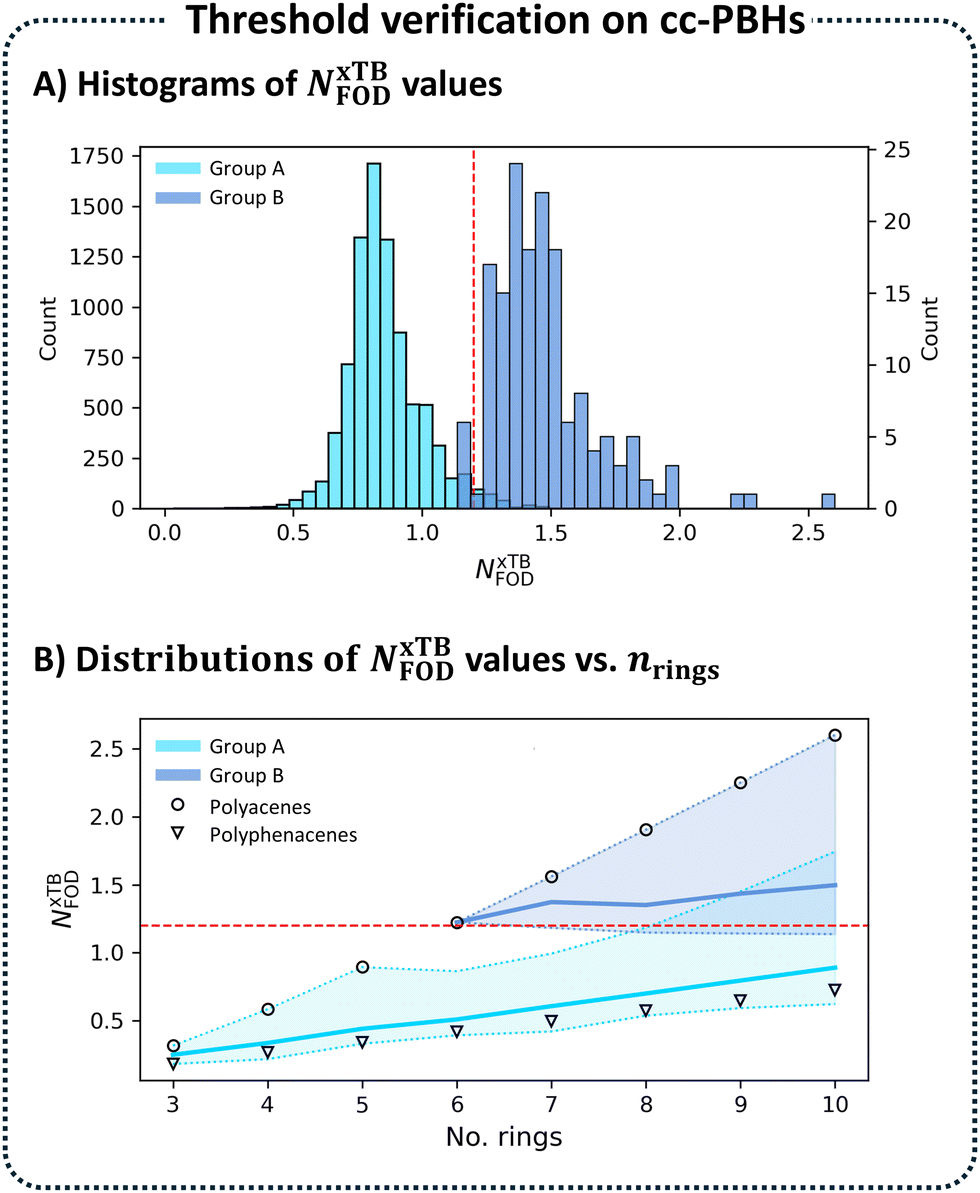 | ||
| Fig. 4 Threshold verification on cc-PBH data. (A) Histograms of NxTBFOD for the entire chemical space of cc-PBHs, divided into Group A (nLL < 6, 8556 molecules, light blue) and Group B (nLL ≥ 6, 161 molecules, dark blue). Each histogram is normalized such that ∑Area = 1. (B) Distributions of Groups A and B versus nrings. The colored areas show the ranges of the NxTBFOD values (min to max). The bold lines denote the respective mean values. The values for the polyacenes (circles) and polyphenacenes (triangles) are marked. | ||
We then plotted distributions of the same NxTBFOD values and the same groupings (Fig. 4B; Group A in light blue, Group B in dark blue), this time against nrings. For each nrings, we denoted the values for the respective acene (circle) and phenacene (triangle). Interestingly, we observed that these two were the ‘bookends’ of the distributions, with the acene consistently marking the highest NxTBFOD value and the phenacene marking the lowest. The colored areas between these min and max values denoted the range of NxTBFOD values for each nrings and the bold lines within the areas denoted the mean values. This plot clearly showed that the diradical character increases with molecular size. It also provided additional validation that our choice of benchmarking molecules was appropriate. Moreover, it is yet further indication that the linear stretches are the source of diradical character (further discussion and analysis of the role of the linear stretches is provided in Section S4 of the ESI†).
As a final evaluation, we plotted a histogram of the y values for Group A. As expected, Fig. 5A showed that all compounds in this group had y ≤ 0.5, which served as our ground truth threshold value.
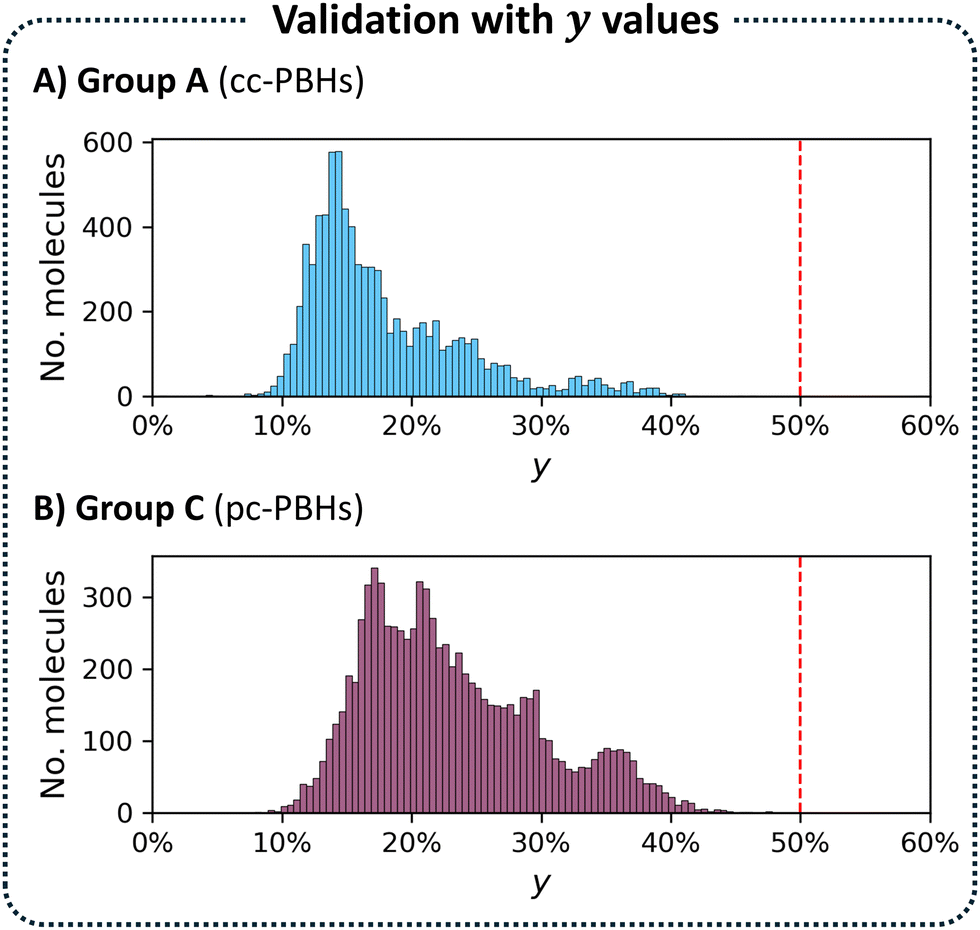 | ||
| Fig. 5 Histograms of the y values for: (A) Group A (8556 molecules); (B) Group C (8701 molecules). | ||
Overall, these analyses affirmed our choice of the NxTBFOD = 1.2 threshold value and provided insight into the structure–property relationship governing the open-shell character of cc-PBHs.
Transferability of the approach
Having confirmed the validity of our approach and the NxTBFOD threshold value for the cc-PBHs, we next turned to testing their transferability by applying them to the pc-PBH (nrings ≤ 10) dataset. Analogously to the cc-PBH division, we divided the pc-PBH dataset into two groups, as well, this time using the threshold value we had identified for cc-PBHs, rather than a structural parameter. Accordingly, Group C comprised pc-PBHs with NxTBFOD < 1.2 and Group D comprised pc-PBHs with NxTBFOD ≥ 1.2. Based on this division, Group C molecules were expected to be closed-shell. To test this, we calculated the y values for all 8701 molecules in this group. As seen in the resulting histogram (Fig. 5B), the values were all below 0.5, corroborating that these were indeed closed-shell molecules.Thus, the application to the pc-PBHs suggested that our NxTBFOD threshold successfully filtered out molecules with appreciable open-shell character, for both types of PBHs. However, it did not indicate whether this threshold was too conservative. In other words, perhaps by setting the threshold too low, molecules with y < 0.5 had been mistakenly identified as predominantly open-shell. Conversely, if the threshold were too high, some molecules had been mistakenly identified as closed-shell. To obtain a more comprehensive evaluation of the transferability of the method, we needed to account for four different scenarios (assuming the y values as the ground truth):
1. False negatives (FN): molecules that NxTBFOD identifies as not open-shell, but which are diradical according to y.
2. True negatives (TN): molecules that both NxTBFOD and y identify as closed-shell.
3. True positives (TP): molecules that both NxTBFOD and y identify as open-shell.
4. False positives (FP): molecules that NxTBFOD identifies as open-shell, but which are closed-shell according to y.
Plotting y versus NxTBFOD for the entire pc-PBH chemical space (nrings ≤ 10) allowed us to see each of these scenarios. As shown in Fig. 6A, the majority of the data were identified correctly as TP (quadrant 1) and TN (quadrant 3). There were no cases of FNs (quadrant 2), however a certain percentage of the molecules (6.97%) were falsely identified as open-shell, i.e., FP (quadrant 4). Meaning, our NxTBFOD threshold of 1.2 overestimated the open-shell character of these molecules. For context, the percentage of TP molecules in the entire chemical space is 7.23%; hence, the identification of 6.97% of the molecules as FP almost doubled the number of molecules identified as open-shell according to NxTBFOD.
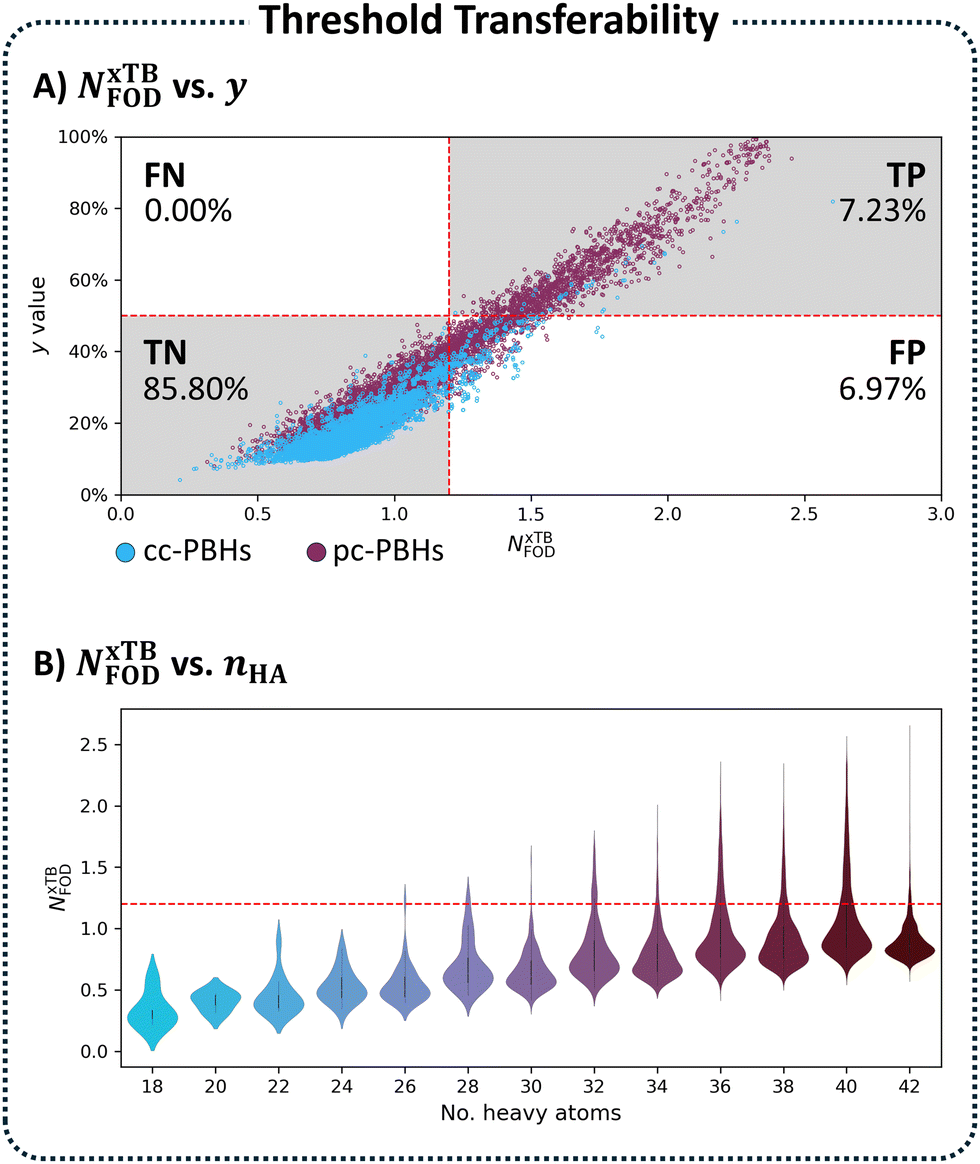 | ||
| Fig. 6 (A) Scatter plot of y vs. NxTBFOD values for all PBHs (cc: blue, pc: burgundy; n ≤ 10 rings). (B) Violin plots of the KDE distributions of all PBHs (nrings ≤ 10 rings) grouped by nHA. | ||
Thus, we concluded that although the approach had merit, it was not trivially transferable between different classes of molecules, even ones as similar as cc-PBHs and pc-PBHs! We hypothesized that the source of this issue may be the size-extensive nature of the NFOD metric, as observed in both our benchmarking set and in the analysis on the cc-PBH chemical space. Therefore, we envisioned it may be possible to provide a solution by taking the molecular size into account.
The effect of size
To solve the transferability problem, we began by elucidating the relationship between molecular size and the NxTBFOD metric. To this end, we plotted kernel density estimates (KDE) distributions of the NxTBFOD values for all molecules (Groups A–D combined), grouped by the number of heavy atoms, nHA (the numbers of molecules in each group are detailed in Table S3 in Section S5 of the ESI†). We observed a clear upward trend, whereby larger molecules had higher NxTBFOD values (Fig. 6B), and further noted that the majority of the data were below the chosen threshold of NxTBFOD = 1.2.Based on this, we concluded that a different threshold should be identified for each heavy-atom group, i.e., a size-aware threshold. To identify the most suitable threshold for each group, we plotted y against NxTBFOD and fit individual regression lines for the data within each respective group (Fig. 7A; panel B shows a zoomed-in section of the plot for additional clarity). Groups containing fewer than 3 molecules were omitted (namely, the groups of nHA = 6, 10, 14, and 16, which account for 5 molecules altogether). Using the respective fitting equations (see Fig. S6 in Section S5 of the ESI,† for the individual fitting equations), we calculated the NxTBFOD value corresponding to y = 0.5 for each group (shown pictorially in Fig. 7A and B as the intercepts of the various regression lines with the horizontal y = 0.5 line). We plotted the calculated thresholds against nHA (Fig. 7C) and found an excellent linear fitting, confirming the suspected size effect. This indicated that the size effect should not be neglected, in particular when characterizing increasingly larger molecules.
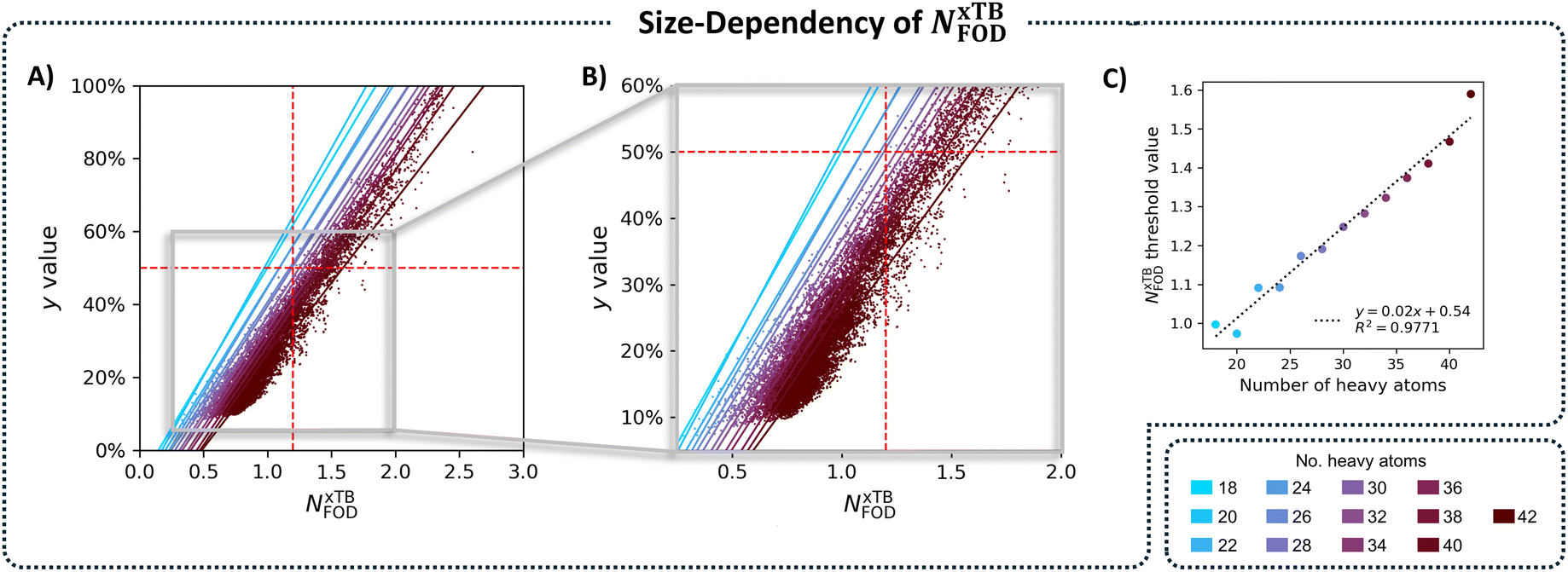 | ||
| Fig. 7 (A) Scatter plot of NxTBFOD against y, colored by the number of heavy atoms, nHA. Color-coded regression lines are shown for each grouping. (B) A zoomed-in portion of the scatter plot in panel (A), corresponding to the area in the gray rectangle. (C) Scatter plot of the individual size-aware thresholds for each grouping plotted against nHA, color-coded with the same colors as panels (A) and (B). | ||
With these results in hand, we repeated our previous evaluation, identifying the TP, TN, FN, FP percentages for each nHA group using its respective size-aware threshold. Fig. 8 displays the confusion matrices obtained with the initial single threshold approach of NFOD = 1.2 (panel A) and with the size-aware approach (panel B). Using the size-aware method resulted in an 18-fold decrease in the number of FP identifications (from 6.97% to 0.38%). We also noted an increase in the percentage of FN cases (from 0.00% to 0.51%), which suggested that the new thresholds were slightly more permissive (allowing diradical molecules into the so-called closed-shell group). Considering that the previous threshold of 1.2 was overly conservative (especially for the larger molecules), this was not surprising. Nevertheless, we emphasize that, cumulatively, FN and FP accounted for fewer than 1% of the molecules when using the size-aware approach. Overall, the results indicated that incorporating molecular size allows for identification of appropriate thresholds and renders the approach transferable across the chemical space of PBHs.
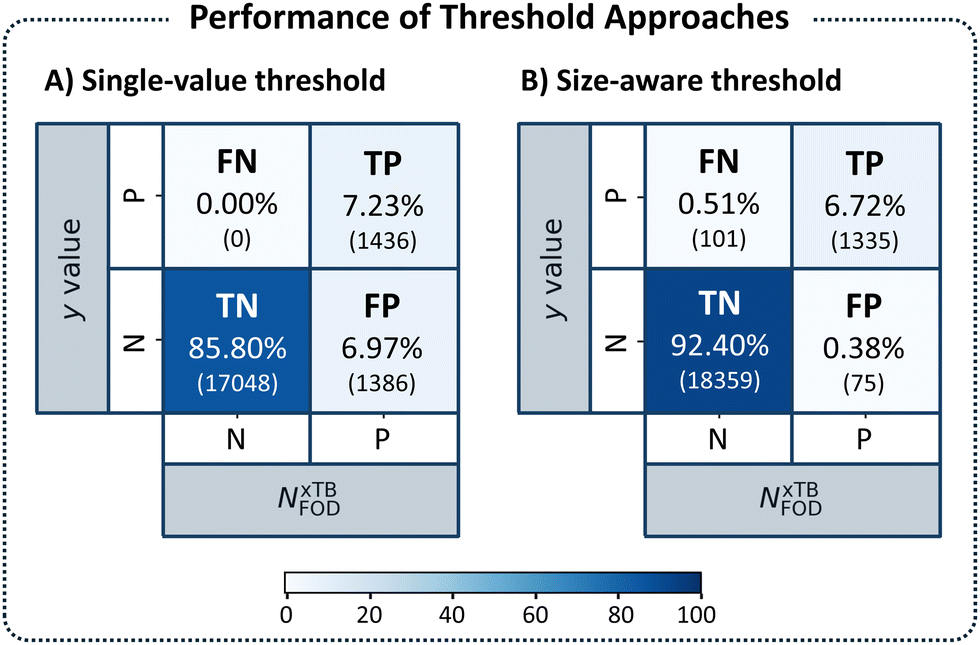 | ||
| Fig. 8 Confusion matrices assessing the performance of the threshold approaches: (A) the single-value threshold approach and (B) the size-aware threshold approach. Each quadrant identifies the type of identification (TP, FN, FP, and TN, respectively) and details the percentage and number (in parentheses) of molecules identified. The color corresponds to the percentage. The five molecules in groups with nHA < 18 were omitted. | ||
Two important comments should be made at this point. First, we note that in the original publication of the GFN2-xTB method, Grimme and co-workers suggested normalization of the NFOD value using the number of electrons.48 For a comparison between the two approaches, we refer the reader to Section S6 of the ESI.† Second, as we mentioned above, the Yamaguchi y value used in this work only measures diradical character, whereas the NFOD value measures total open-shell character. Therefore, the two quantities are not directly comparable and the latter may be biased by partial tetraradical character. The good linear agreement shown in Fig. 6A suggests that this is not the case for the molecules studied herein. Nevertheless, to ensure this, we investigated the y1 values, as well. These are presented along with additional discussion in Section S7 of the ESI.†
xTB vs. DFT
As we mentioned in the introduction, Lischka and coworkers25 originally demonstrated that NFOD values enable accurate estimation of the diradical character of PAHs at a fraction of the computational cost of MR-AQCC. In this work, we demonstrated even more cost-effective characterization by using xTB-generated NFOD, instead of DFT, for a large dataset. As a final aspect of this investigation, we were interested in comparing the xTB and DFT NFOD values.We generated a scatter plot of the two NFOD metrics (Fig. 9A) and observed a rather good correlation between the two methods (R2 = 0.93). Coloring the individual data points according to nHA further revealed a series of parallel correlations, which shift with the molecular size; the larger the molecules, the farther away from the x = y line (Fig. 9B shows a zoomed-in portion of the total plot, color-coded by nHA). Similarly to NxTBFOD, we extracted the individual threshold values for NDFTFOD, to compare the size-dependency of the two metrics. Plotting the individual size-aware thresholds obtained for the two NFOD metrics against nHA (Fig. 9C) showed that NDFTFOD has a much steeper dependence on size than NxTBFOD.
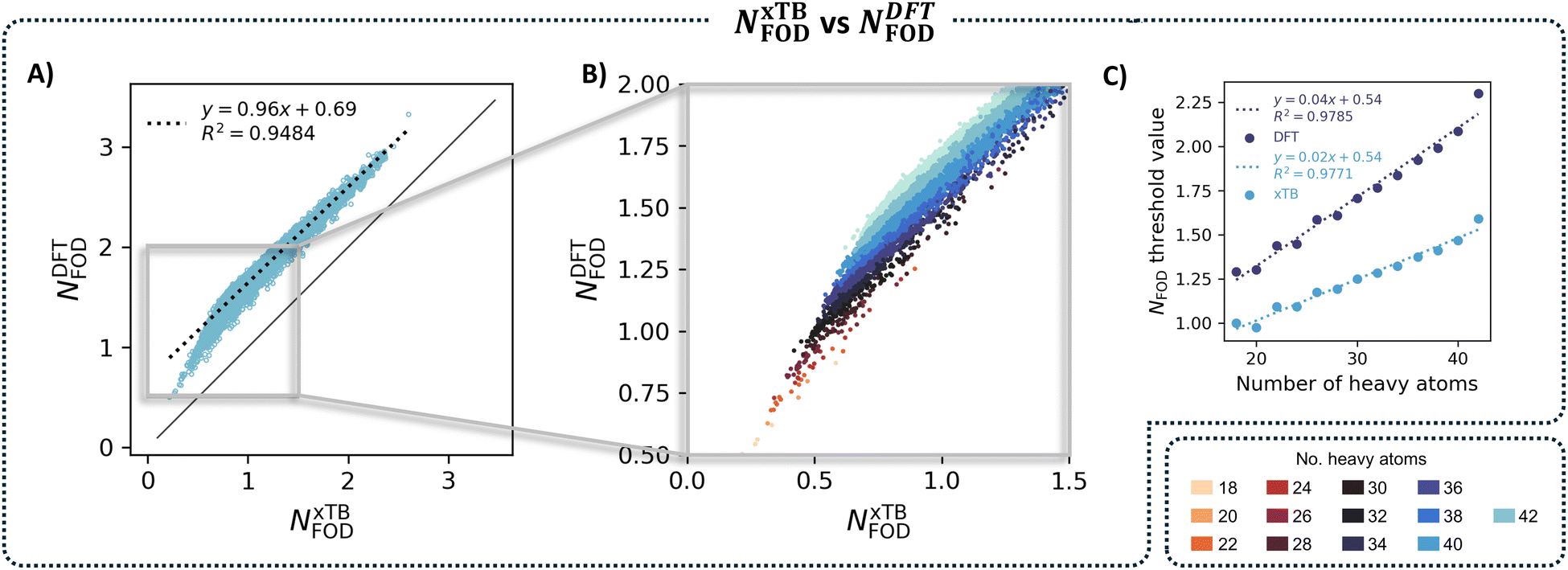 | ||
| Fig. 9 (A) Scatter plot of NxTBFOD against NDFTFOD with trendline. (B) A zoomed-in portion of the scatter plot in panel (A), corresponding to the area in the gray rectangle. The data are colored by nHA. (C) Scatter plots of the individual size-aware thresholds for NxTBFOD (light blue) and NDFTFOD (dark blue) plotted against nHA. | ||
Conclusions
In this study, we demonstrated the use of xTB-calculated NFOD values as a computationally efficient approach for identifying open-shell character in polybenzenoid hydrocarbons (PBHs). We computed the Yamaguchi y and NxTBFOD values for the entire chemical space of PBHs containing nrings ≤ 10 and identified the value of NxTBFOD = 1.2 as a suitable threshold for distinguishing between closed- and open-shell systems. In general, NxTBFOD values show good correlation with the more computationally expensive y values, validating their use as a predictor of open-shell character. Based on this analysis, we found that 6.72% of this chemical space (1335 molecules) are identified as open-shell. However, our findings also highlighted a size-dependency that makes transferability of the approach non-trivial. We elucidated the relationship between NxTBFOD and molecular size, finding a linear correlation that allows for easy adjustment of thresholds for molecules of varying sizes. Applying the individual size-aware thresholds for different sized molecules reduced false positive identifications from 6.97% to 0.38%, compared to using a single threshold value. Finally, comparison between xTB- and DFT-calculated NFOD values showed that the former has a much less pronounced size dependency than the latter. As high-throughput calculations with xTB and DFT methods are becoming more widely used for data-driven research, these findings have important implications for the field of organic electronics and materials science. In this context, we wish to add a note of caution regarding the choice of computational method. To retain consistency between all methods, the NxTBFOD values included in this report were obtained for DFT-optimized geometries. However, we also performed a similar analysis using xTB-optimized geometries. We found that the general trends and conclusions remain the same, but the single-value threshold increases to 1.3 and the size-aware thresholds increase accordingly. Furthermore, when using xTB-optimized geometries, a subset of molecules (mostly zethrene derivatives) appeared as outliers on the y vs. NxTBFOD scatter plot, whereas well-behaved correlations were obtained when using DFT-optimized geometries. This indicates that the NxTBFOD value is sensitive to small geometric changes, and that xTB does not provide accurate geometries for some of the molecules in our dataset (see Section S8 of the ESI† for further details).In conclusion, we believe that the contribution of the current study is twofold. From a practical perspective, it demonstrates rapid and accurate assessment of open-shell character in large datasets of PBHs, striking a balance between computational efficiency and accuracy. As such, it facilitates the design and discovery of new functional organic materials with tailored electronic properties and can be a valuable tool for researchers in the field of organic chemistry and materials chemistry. From a conceptual perspective, we have shed light on the behavior of the NFOD metric. We have shown that there is a linear relationship between size and NFOD in PBHs.
Data and availability
The data underlying this study are openly available on Gitlab at https://gitlab.com/porannegroup/nfod_xTB and https://gitlab.com/porannegroup/compas. The datasets are provided as .csv files. Further description of the data structure is provided on the GitLab repositories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Dr Peter Chen for his ongoing support, and Dr Eno Paenurk and Dr Alexandra Tsybizova for helpful commentary on the manuscript. The anonymous reviewers are gratefully acknowledged for their helpful comments and suggestions, including the addition of the y1 values and the analysis based on number of electrons. The authors also express their deep appreciation to the Branco Weiss Fellowship for supporting this research as part of a Society in Science grant and to the Israel Science Foundation for financial support (Grant No. 1745/23). R. G. P. is a Branco Weiss Fellow, a Horev Fellow, and an Alon Scholarship recipient.References
- S. Markovic, S. Radenkovic, Z. Markovic and I. Gutman, DFT study on singlet diradical character of zethrenes, Russ. J. Phys. Chem., 2011, 85, 2368–2372, DOI:10.1134/S0036024411130127.
- Z. Sun, Z. Zeng and J. Wu, Zethrenes, Extended p-Quinodimethanes, and Periacenes with a Singlet Biradical Ground State, Acc. Chem. Res., 2014, 47, 2582–2591, DOI:10.1021/ar5001692.
- G. Gryn'ova, M. L. Coote and C. Corminboeuf, Theory and practice of uncommon molecular electronic configurations, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 440–459, DOI:10.1002/wcms.1233.
- P. Hu and J. Wu, Modern zethrene chemistry, Can. J. Chem., 2017, 95, 223–233, DOI:10.1139/cjc-2016-0568.
- A. Das, M. Pinheiro Jr., F. B. C. Machado, A. J. A. Aquino and H. Lischka, Tuning the Biradicaloid Nature of Polycyclic Aromatic Hydrocarbons: The Effect of Graphitic Nitrogen Doping in Zethrenes, ChemPhysChem, 2018, 19, 2492–2499, DOI:10.1002/cphc.201800650.
- J. Casado, in Physical Organic Chemistry of Quinodimethanes, ed. Y. Tobe and T. Kubo, Springer International Publishing, Cham, 2018, pp. 209–248 DOI:10.1007/978-3-319-93302-3_5.
- G. Kundu, S. R. Dash, R. Kumar, K. Vanka, A. Ghosh and S. S. Sen, Enhancing Diradical Character of Chichibabin’s Hydrocarbon through Fluoride Substitution, ChemPlusChem, 2023, 88, e202300273 CrossRef CAS.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, The molecular structures of Thiele's and Chichibabin's hydrocarbons, J. Am. Chem. Soc., 1986, 108, 6004–6011, DOI:10.1021/ja00279a056.
- A. Shimizu, S. Nobusue, H. Miyoshi and Y. Tobe, Indenofluorene congeners: biradicaloids and beyond, Pure Appl. Chem., 2014, 86, 517–528, DOI:10.1515/pac-2014-5043.
- Z. X. Chen, Y. Li and F. Huang, Persistent and Stable Organic Radicals: Design, Synthesis, and Applications, Chem, 2021, 7, 288–332, DOI:10.1016/j.chempr.2020.09.024.
- W. Zeng and J. Wu, Open-Shell Graphene Fragments, Chem, 2021, 7, 358–386, DOI:10.1016/j.chempr.2020.10.009.
- I. Pozo and L. Bogani, A perspective on radicaloid conjugated polycyclic hydrocarbons, Treds Chem., 2024, 6, 581–595, DOI:10.1016/j.trechm.2024.08.005.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Pro-aromatic and anti-aromatic p-conjugated molecules: an irresistible wish to be diradicals, Chem. Soc. Rev., 2015, 44, 6578–6596, 10.1039/C5CS00051C.
- T. Ishida and J.-I. Aihara, Aromaticity of neutral and doubly charged polyacenes, Phys. Chem. Chem. Phys., 2009, 11, 7197–7201, 10.1039/B903815A.
- T. Minami and M. Nakano, Diradical Character View of Singlet Fission, J. Phys. Chem. Lett., 2012, 3, 145–150, DOI:10.1021/jz2015346.
- M. Bendikov, H. M. Duong, K. Starkey, K. N. Houk, E. A. Carter and F. Wudl, Oligoacenes: Theoretical Prediction of Open-Shell Singlet Diradical Ground States, J. Am. Chem. Soc., 2004, 126, 7416–7417, DOI:10.1021/ja048919w.
- F. Plasser, H. Pašalic, M. H. Gerzabek, F. Libisch, R. Reiter, J. Burgdörfer, T. Müller, R. Shepard and H. Lischka, The Multiradical Character of One- and Two-Dimensional Graphene Nanoribbons, Angew. Chem., Int. Ed., 2013, 52, 2581–2584, DOI:10.1002/anie.201207671.
- S. J. Cyvins and I. Gutman, Topological properties of benzenoid hydrocarbons: part XLIV. Obvious and concealed non-Kekuléan benzenoids, J. Mol. Struct.: THEOCHEM, 1987, 150, 157–169, DOI:10.1016/0166-1280(87)80035-0.
- S. Das and J. Wu, Polycyclic Hydrocarbons with an Open-Shell Ground State, Phys. Sci. Rev., 2017, 2, 20160109, DOI:10.1515/psr-2016-0109.
- D. Casanova and M. Head-Gordon, Restricted active space spin–flip configuration interaction approach: theory, implementation and examples, Phys. Chem. Chem. Phys., 2009, 11, 9779–9790 RSC.
- S. Chattopadhyay, R. K. Chaudhuri and U. Sinha Mahapatra, Ab Initio Multireference Investigation of Disjoint Diradicals: Singlet versus Triplet Ground States, ChemPhysChem, 2011, 12, 2791–2797, DOI:10.1002/cphc.201100430.
- A. E. Torres, P. Guadarrama and S. Fomine, Multiconfigurational character of the ground states of polycyclic aromatic hydrocarbons. A systematic study, J. Mol. Model., 2014, 20, 2208, DOI:10.1007/s00894-014-2208-6.
- A. Das, T. Müller, F. Plasser and H. Lischka, Polyradical Character of Triangular Non-Kekulé Structures, Zethrenes, p-Quinodimethane-Linked Bisphenalenyl, and the Clar Goblet in Comparison: An Extended Multireference Study, J. Phys. Chem. A, 2016, 120, 1625–1636, DOI:10.1021/acs.jpca.5b12393.
- A. Das, T. Müller, F. Plasser, D. B. Krisiloff, E. A. Carter and H. Lischka, Local Electron Correlation Treatment in Extended Multireference Calculations: Effect of Acceptor–Donor Substituents on the Biradical Character of the Polycyclic Aromatic Hydrocarbon Heptazethrene, J. Chem. Theory Comput., 2017, 13, 2612–2622, DOI:10.1021/acs.jctc.7b00156.
- R. Nieman, J. R. Carvalho, B. Jayee, A. Hansen, A. J. A. Aquino, M. Kertesz and H. Lischka, Polyradical character assessment using multireference calculations and comparison with density-functional derived fractional occupation number weighted density analysis, Phys. Chem. Chem. Phys., 2023, 25, 27380–27393, 10.1039/D3CP03734G.
- J. R. Carvalho, R. Nieman, M. Kertesz, A. J. A. Aquino, A. Hansen and H. Lischka, Multireference calculations on bond dissociation and biradical polycyclic aromatic hydrocarbons as guidance for fractional occupation number weighted density analysis in DFT calculations, Theor. Chem. Acc., 2024, 143, 69, DOI:10.1007/s00214-024-03143-8.
- in The IUPAC Compendium of Chemical Terminology: The Gold Book, ed. V. Gold, International Union of Pure and Applied Chemistry (IUPAC), 4th edn, 2019 DOI:10.1351/goldbook.
- M. Abe, Diradicals, Chem. Rev., 2013, 113, 7011–7088, DOI:10.1021/cr400056a.
- Y. Yang, E. R. Davidson and W. Yang, Nature of ground and electronic excited states of higher acenes, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5098–E5107 CAS.
- C. U. Ibeji and D. Ghosh, Singlet–triplet gaps in polyacenes: a delicate balance between dynamic and static correlations investigated by spin–flip methods, Phys. Chem. Chem. Phys., 2015, 17, 9849–9856, 10.1039/C5CP00214A.
- M. M. Payne, S. R. Parkin and J. E. Anthony, Functionalized Higher Acenes: Hexacene and Heptacene, J. Am. Chem. Soc., 2005, 127, 8028–8029, DOI:10.1021/ja051798v.
- R. Mondal, R. M. Adhikari, B. K. Shah and D. C. Neckers, Revisiting the Stability of Hexacenes, Org. Lett., 2007, 9, 2505–2508, DOI:10.1021/ol0709376.
- R. Mondal, C. Tönshoff, D. Khon, D. C. Neckers and H. F. Bettinger, Synthesis, Stability, and Photochemistry of Pentacene, Hexacene, and Heptacene: A Matrix Isolation Study, J. Am. Chem. Soc., 2009, 131, 14281–14289, DOI:10.1021/ja901841c.
- B. Purushothaman, S. R. Parkin and J. E. Anthony, Synthesis and Stability of Soluble Hexacenes, Org. Lett., 2010, 12, 2060–2063, DOI:10.1021/ol100178s.
- A. Wahab, L. Pfuderer, E. Paenurk and R. Gershoni-Poranne, The COMPAS Project: A Computational Database of Polycyclic Aromatic Systems. Phase 1: cata-Condensed Polybenzenoid Hydrocarbons, J. Chem. Inf. Model., 2022, 62, 3704–3713, DOI:10.1021/acs.jcim.2c00503.
- A. Wahab and R. Gershoni-Poranne, COMPAS-3: a dataset of peri-condensed polybenzenoid hydrocarbons, Phys. Chem. Chem. Phys., 2024, 26, 15344–15357, 10.1039/D4CP01027B.
- F. Neese, The ORCA Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78, DOI:10.1002/wcms.81.
- F. Neese, Software update: the ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606, DOI:10.1002/wcms.1606.
- A. D. Becke, Density-functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-energy Formula Into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157, 200–206, DOI:10.1016/0009-2614(89)87234-3.
- R. H. Hertwig and W. Koch, On the Parameterization of the Local Correlation Functional. What is Becke-3-LYP?, Chem. Phys. Lett., 1997, 268, 345–351, DOI:10.1016/S0009-2614(97)00207-8.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57, DOI:10.1016/j.cplett.2004.06.011.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297, 10.1039/b508541a.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- E. R. Johnson and A. D. Becke, A post-Hartree–Fock Model of Intermolecular Interactions, J. Chem. Phys., 2005, 123, 024101, DOI:10.1063/1.1949201.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- S. Grimme and A. Hansen, A Practicable Real-Space Measure and Visualization of Static Electron-Correlation Effects, Angew. Chem., Int. Ed., 2015, 54, 12308–12313, DOI:10.1002/anie.201501887.
- B. Chan, The Paradox of Global Multireference Diagnostics, J. Phys. Chem. A, 2024, 128, 9829–9836, DOI:10.1021/acs.jpca.4c06148.
- C. A. Bauer, A. Hansen and S. Grimme, The Fractional Occupation Number Weighted Density as a Versatile Analysis Tool for Molecules with a Complicated Electronic Structure, Chem. – Eur. J., 2017, 23, 6150–6164, DOI:10.1002/chem.201604682.
- S. Grimme, Towards First Principles Calculation of Electron Impact Mass Spectra of Molecules, Angew. Chem., Int. Ed., 2013, 52, 6306–6312, DOI:10.1002/anie.201300158.
- K. Yamaguchi, The electronic structures of biradicals in the unrestricted Hartree–Fock approximation, Chem. Phys. Lett., 1975, 33, 330–335, DOI:10.1016/0009-2614(75)80169-2.
- M. Nakano, Open-Shell-Character-Based Molecular Design Principles: Applications to Nonlinear Optics and Singlet Fission, Chem. Rec., 2017, 17, 27–62, DOI:10.1002/tcr.201600094.
- M. Nakano, Excitation Energies and Properties of Open-Shell Singlet Molecules, SpringerBriefs in Molecular Science; Springer International Publishing, 2014 DOI:10.1007/978-3-319-08120-5.
- T. J. Chow, Hexacene: Synthesis, Properties and Future Perspectives, Chem. Rec., 2015, 15, 1137–1139, DOI:10.1002/tcr.201510003.
- R. Einholz, T. Fang, R. Berger, P. Grüninger, A. Früh, T. Chassé, R. F. Fink and H. F. Bettinger, Heptacene: Characterization in Solution, in the Solid State, and in Films, J. Am. Chem. Soc., 2017, 139, 4435–4442, DOI:10.1021/jacs.6b13212.
Footnotes |
| † Electronic supplementary information (ESI) available: General computational details, further analysis of the effect of temperature on the NxTBFOD value, examples of mistaken classification, the effect of the longest linear stretch in cc-PBHs, the effect of size, comparison of normalization approaches (number of heavy atoms versus number of electrons), investigation of y1 values, and the effect of geometry; tables and figures for the identification of the size-aware thresholds, and analysis on the effect of geometry on NxTBFOD. See DOI: https://doi.org/10.1039/d4cp04059g |
| ‡ For clarity, please note that we use the terminology introduced in the IUPAC Gold-Book27 and detailed by Abe.28 Accordingly, ‘biradical’ refers to molecules in which the two electrons act independently or nearly independently, whereas ‘diradical’ refers to species in which the magnitude of the dipole–dipole interaction is large enough to produce two spin states, namely, singlet (S, S = 0, spin multiplicity = 1) and triplet (T, S = 1, spin multiplicity = 3). For PBHs, the latter term is more suitable. |
| This journal is © the Owner Societies 2025 |