
A computational study of the ternary mixtures of NaPF6–EC and choline glycine ionic liquid
Eudes Eterno
Fileti
 a,
Iuliia V.
Voroshylova
*b,
M. Natália
D. S. Cordeiro
b and
Thaciana
Malaspina
*a
a,
Iuliia V.
Voroshylova
*b,
M. Natália
D. S. Cordeiro
b and
Thaciana
Malaspina
*a
aInstitute of Science and Technology, Federal University of São Paulo, 12247-014, São José dos Campos, São Paulo, Brazil. E-mail: thaciana.malaspina@unifesp.br
bREQUIMTE LAQV, Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007 Porto, Portugal. E-mail: voroshylova.iuliia@fc.up.pt
First published on 26th December 2024
Abstract
This study investigates the structural and dynamic properties of ternary mixtures composed of NaPF6, ethylene carbonate (EC), and the ionic liquid choline glycine (ChGly), with a focus on their potential as electrolytes for supercapacitors. The combination of NaPF6–EC, known for its high ionic conductivity, with the biodegradable and environmentally friendly ChGly offers a promising approach to enhancing electrolyte performance. Through molecular simulations, we analyze how the inclusion of small concentrations of ChGly affects key properties such as density, cohesive energy, and ion mobility. Our findings demonstrate that the NaPF6–EC–ChGly mixture exhibits a complex network of electrostatic interactions and hydrogen bonding, with the glycine anion significantly influencing the liquid structure. In mixtures with small additions of ChGly, we observed an optimal balance of diffusion and ionic mobility. These results highlight the potential of ChGly as a green additive to conventional electrolytes, paving the way for more sustainable and high-performance energy storage devices.
Introduction
A significant volume of research has focused on the development of electrolytes, as their properties ultimately determine the maximum operational voltage of electrochemical capacitors.1–7 Commercially used electrolytes can be classified into three main categories. The first category includes aqueous electrolytes, which exhibit good electrochemical properties, excellent performance in terms of power, and low cost.3–8 However, a notable disadvantage of water-based electrolytes is their narrow operational voltage window, constrained by the thermodynamic stability of water (approximately 1.23 V).5 The second category comprises organic electrolytes, characterized by a broad electrochemical stability of up to 3 V.5,9–11 These electrolytes, however, suffer from high volatility and flammability, elevated cost, sensitivity to moisture, and low conductivity.5 The third category refers to ionic liquid-based electrolytes, which offer advantages such as the ability to achieve high voltages of up to around 4 V, as well as being non-flammable and often environmentally friendly.5,12–14 Nonetheless, their high production cost and low ionic conductivity limit their application in high-power devices.4According to many researchers, an ideal electrolyte should be characterized by a wide potential window, low resistance, low viscosity, non-flammability, high electrochemical stability, and environmental friendliness.1,2 Consequently, the development of new electrolyte materials requires flexible strategies to optimize all these properties. In this regard, ternary mixtures of ionic liquids, consisting of three different components, can be employed to design electrolytes that combine the unique properties of ionic liquids with the characteristics of other components, resulting in solutions with adjustable and optimized properties for specific applications.15–17
Electrolytic solutions of NaPF6 (sodium hexafluorophosphate) in organic solvents such as ethylene carbonate (EC) are widely used in electrochemical energy storage devices due to their good solubility and ability to form electrolytes with high ionic conductivity.18–22 Ethylene carbonate efficiently dissolves NaPF6, allowing the dissociation of Na+ and PF6− ions and enhancing the ionic conductivity of the solution. In supercapacitors, this solution can increase capacitance through the adsorption of Na+ and PF6− ions at the electrode interfaces, contributing to energy storage.18–22
Ionic liquids based on choline and amino acids (AAILs), such as choline combined with glycine (ChGly), have been considered promising electrolytes for supercapacitors due to their unique physicochemical properties and potential advantages in terms of safety and performance compared to traditional ionic liquids.23–27 In addition to offering performance comparable to conventional electrolytes, ChGly and its aqueous mixtures can help reduce costs, increase ionic conductivity, and improve electrolyte performance.23–25,28
In this paper, we investigate the structural and dynamic properties of ternary mixtures of NaPF6–EC with the ionic liquid ChGly. The combination of NaPF6–EC, which provides high ionic conductivity, with ChGly, which possesses environmentally friendly properties, could result in an efficient, cost-effective, and less toxic electrolyte. The inclusion of ChGly, even in small concentrations, can expand the electrochemical window and improve the thermal stability of the mixture, making the system more robust under high-power conditions. Another advantage pertains to the issue of self-discharge in supercapacitors.29,30 This effect leads to voltage and energy loss due to charge redistribution, redox reactions, and ohmic leakage. One proposed solution to address this problem is to increase the electrolyte viscosity near the electrode.31 The alignment of particles by the electric field enhances viscosity, impeding ion diffusion, thereby reducing self-discharge and improving device efficiency.31 Given that ChGly is highly viscous, it may fulfill this role at the electrode–electrolyte interface.
While challenges such as component miscibility and viscosity control need to be overcome, these ternary mixtures of NaPF6–EC with ChGly could offer a high-performance and environmentally sustainable electrolytic solution, with the potential to optimize electrochemical energy storage.
Computational details
To investigate the effect of incorporating a NaPF6–EC solution into ChGly, the molar ratio of NaPF6 to EC in the 1 M solution was determined using experimental density data from the literature.32 The ratio was found to be 33 ion pairs of NaPF6 to 500 EC molecules. Then, the 1 M NaPF6–EC solution was added to ChGly at various concentrations to determine the optimal mixture composition, as outlined in Table 1.| x(NaPF6), % | x(ChGly), % | Number of molecules or ion pairs | Number of interaction sites | Box side length, nm | ||
|---|---|---|---|---|---|---|
| ChGly | EC | NaPF6 | ||||
| 0.0 | 100 | 500 | 0 | 0 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
5.03 |
| 0.6 | 89 | 450 | 50 | 3 | 14027 |
4.93 |
| 1.4 | 79 | 400 | 100 | 7 | 13053 |
4.85 |
| 2.0 | 69 | 350 | 150 | 10 | 12080 |
4.76 |
| 2.5 | 58 | 300 | 200 | 13 | 11107 |
4.68 |
| 3.3 | 48 | 250 | 250 | 17 | 10133 |
4.58 |
| 3.8 | 39 | 200 | 300 | 20 | 9160 | 4.49 |
| 4.4 | 29 | 150 | 350 | 23 | 8187 | 4.39 |
| 5.1 | 19 | 100 | 400 | 27 | 7213 | 4.30 |
| 5.7 | 9 | 50 | 450 | 30 | 6240 | 4.21 |
| 6.2 | 0 | 0 | 500 | 33 | 5267 | 4.09 |
Considering the number of interaction sites in all species involved in the simulations, and acknowledging that an increase in interaction sites prolongs simulation time, a balance between system size and computational feasibility was sought. As a result, the number of interaction sites in the systems ranged from ∼5300 for the 1 M NaPF6–EC solution (corresponding to an x(NaPF6) of 6.2%) to 15000 for the pure ChGly ionic liquid.
Cubic computational boxes were generated using PACKMOL33 by randomly distributing the species within a volume that approximately matches the desired density of the mixture. A representative configuration of these species and simulation boxes are shown in Fig. 1.
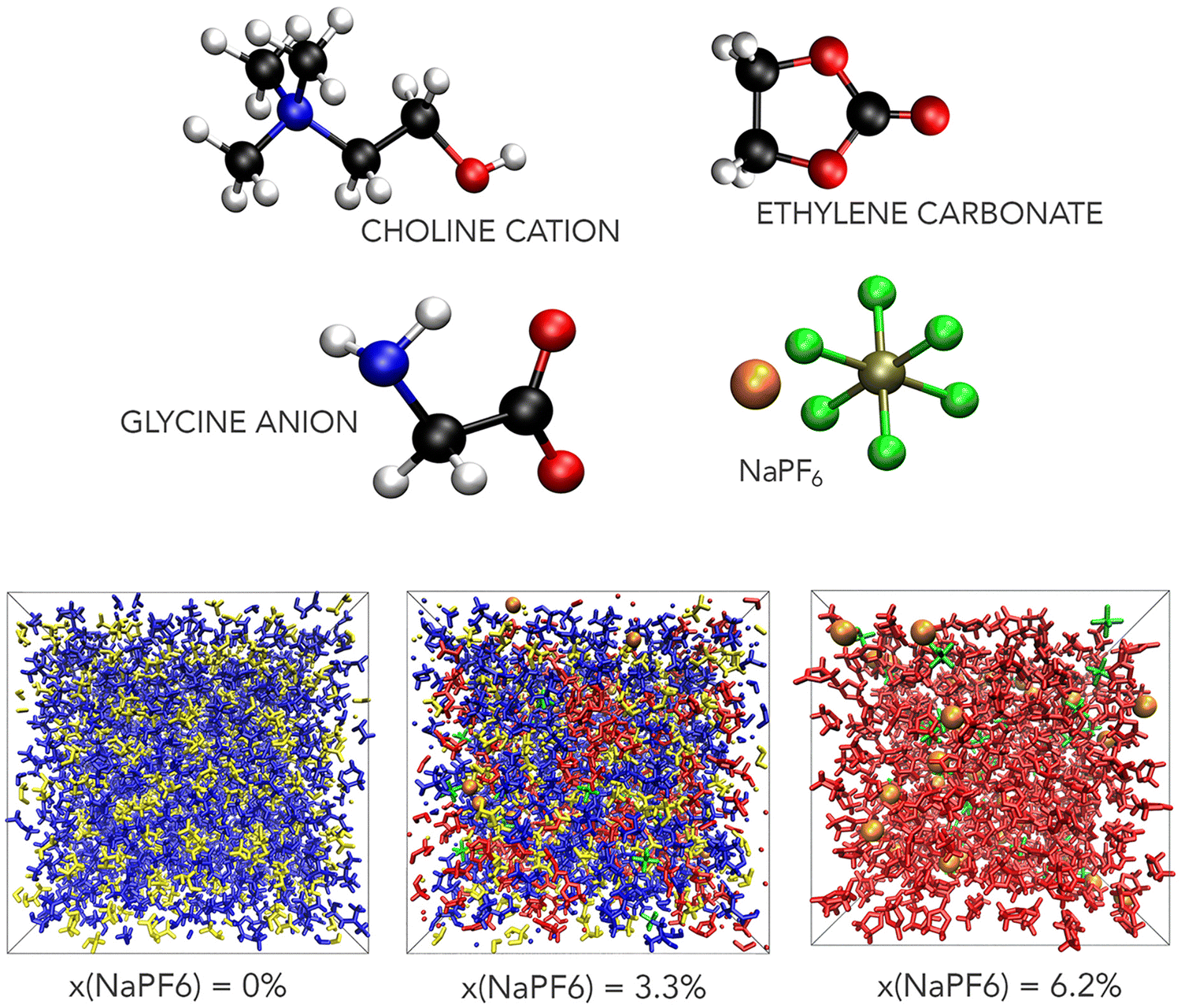 | ||
| Fig. 1 The molecular components of the ChGly and NaPF6–EC solutions (top) and typical simulation boxes for these systems at three different NaPF6 concentrations (bottom). In the molecules representation, hydrogen, carbon, oxygen, nitrogen, sodium, fluorine and phosphorus atoms are shown with white, black, red, blue, orange, green, and tan, respectively; in the snapshots choline, glycine, ethylene carbonate, sodium and hexafluorophosphate structures are shown in blue, yellow, red, orange and green colors, respectively. | ||
The intermolecular interaction model for all species is based on the OPLS-AA force field,34 which has demonstrated good performance in similar systems.11,35–37 For the ChGly system, its charges were quantum mechanically determined using the CHelpG method38 as implemented in Gaussian 09 program.39 Each simulation box was energy minimized and then pre-equilibrated for approximately 5 ns in the NVT ensemble. Subsequently, two different calculations were performed: one generated a 10 ns NPT trajectory, which was used to obtain cohesive energy and density, and another longer trajectory (100 ns) in the NVT ensemble, to determine dynamic properties. In all cases, an integration time step of 2 fs was used, with frames collected every 5 ps for the NPT simulations and every 0.02 ps for the NVT simulations. The systems were subjected to temperature and pressure coupling using the Nose–Hoover40,41 and Parrinello–Rahman42 schemes, with coupling constants of 2.0 and 1.0 ps, respectively. Electrostatic interactions were simulated using the direct Coulomb law up to 1.2 nm of separation between each pair of interaction sites. Beyond 1.2 nm, electrostatic interactions were accounted for by the computationally efficient Particle–Mesh–Ewald (PME) method.43 The Lennard-Jones 12-6 interactions were smoothly tapered to zero from 1.1 to 1.2 nm using the classical shifted force technique. All simulations were performed using the GROMACS 2020.4 program44 using a NaRiBaS scripting framework to automate the repetitive tasks and avoid human errors.45
Results and discussion
Structural properties
An initial characterization of all ternary solutions of ChGly, NaPF6, and EC can be described in terms of the variation in their density and cohesive energy as a function of NaPF6 concentration. As illustrated in Fig. 2, the density of the mixtures shows an approximately monotonic variation, ranging from 1160 kg m−3 for the pure ChGly liquid to 1208 kg m−3 for the pure NaPF6–EC mixture. Fig. 2 also shows that the cohesive energy density of the solutions, which is directly related to the strength of intermolecular interactions, decreases monotonically from the pure ChGly liquid to the NaPF6–EC mixture. Here, we observe that at low concentrations of NaPF6, the cohesive energy is high, indicating stronger intermolecular interactions between the species that comprise the solution. In other words, the presence of higher amounts of NaPF6 reduces the stability of the mixture, which directly impacts its structural and dynamic properties.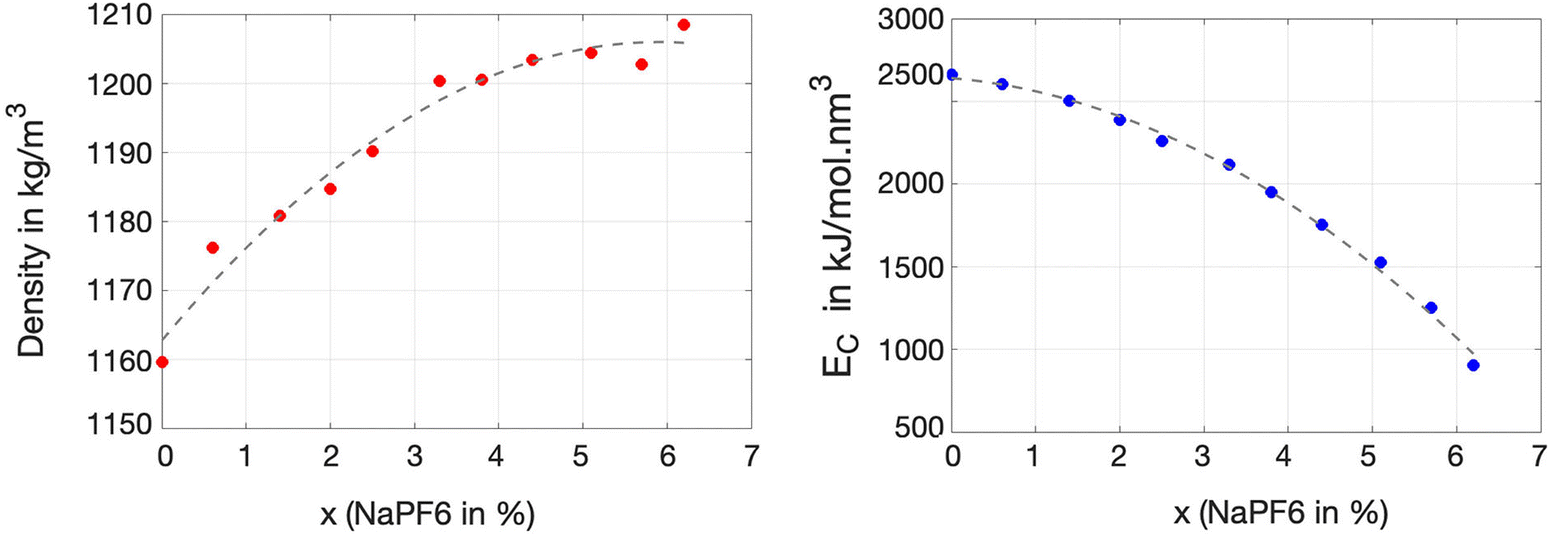 | ||
| Fig. 2 Density (in kg m−3) and cohesive energy density (in kJ mol−1 nm−3) as a function of NaPF6 concentration. The cohesive energy was calculated according to the equation Ec = (Epotmix − N1 × EChGlygas − N2 × ENaPF6gas − N3 × EECgas)/Vmix. In this equation Ec is the cohesive energy, Epotmix and Vmix are the potential energy and volume of the ternary mixture, N1, N2 and N3 are the numbers of ChGly, NaPF6, and EC molecules, respectively, and EChGlygas, ENaPF6gas and EECgas are the energies of the corresponding gas phases (isolated system). The magnitude of the standard deviations for all densities and energies was below 0.56 kg m−3 and 50 kJ mol−1 nm−3, respectively. The dashed lines represent a second-order polynomial fit of the data points. | ||
The structure of all ternary solutions of ChGly and NaPF6–EC was analyzed by calculating the radial distribution functions (RDFs) of selected sites for each molar fraction, as shown in Fig. 3. The selected pairs involve atoms with the highest charges, with one site carrying a positive charge and the other a negative charge. For all these pairwise interactions, we observed that the glycine anion, in particular, played a significant role in structuring the liquid, which explains why nearly all the most pronounced RDFs involve this anion. For instance, the RDFs in Fig. 3(a) and (b) describe the hydrogen bonding between the polar sites in the glycine anion, which exhibit the highest opposite partial charges within the anion. In the hydrogen bond characterized in Fig. 3(b), HGLY⋯OGLY, the involved sites have partial charges of +0.33e and −0.85e, respectively. In both cases, there is a first peak around 0.2 nm, corresponding to the interaction between the donated proton and acceptor atoms of the anion. This peak gradually increases in intensity as the NaPF6–EC concentration rises (and consequently the choline glycine concentration decreases), indicating that the interaction between glycine anions is favored at lower choline glycine concentrations. Fig. 3(c) presents the structuring of ethylene carbonate concerning the carbon of the glycine anion. Unlike the hydrogen bonds discussed earlier, this interaction shows a reduced peak as the NaPF6–EC concentration increases. This represents the only modest contribution of ethylene carbonate to the liquid structure, as all other RDFs involving EC display less pronounced peaks and lower coordination with other species in the solution. The interaction between Ch and Gly ions, which have a profound impact on the structure of the mixtures, is described in terms of their hydrogen bonds in Fig. 3(d) and (e), where we clearly observe strong primary peaks that become even more intense as the NaPF6–EC concentration increases. These peaks beyond the first maximum do not represent long-range hydrogen bond order but rather longer-range correlations between the corresponding atoms. Specifically, they reflect the order in the relative alignment of choline and glycine ions, resulting in preferred distances of their non-hydrogen-bonding H–O and H–N pairs. This indicates that the interactions between the OCH⋯HGLY and NCH⋯HGLY pairs persist over longer distances, confirming that these species in the investigated solutions are indeed highly interactive. Additionally, we have the interaction between the HCH⋯OGLY sites, which, besides occurring at longer distances, results in intense coordination of the choline and glycine ions, as evidenced by very high first peaks. We will further discuss the dynamics of this interaction in greater detail later. The most relevant interaction involving the PF6 anion occurs between the phosphorus atom and the oxygen of the choline cation. Despite both sites carrying significant charges of opposite signs, the P⋯OCH electrostatic interaction is relatively weak, likely due to the positive charge of the PF6 anion being shielded by the six negatively charged fluorine atoms. RDFs in Fig. 3(g) and (h) show that the glycine anion strongly coordinates with the Na cation. For both interactions, Na⋯NGLY and Na⋯OGLY, the first maximum and minimum occur at 2.5 and 2.9 nm, respectively, defining a clear solvation shell of glycine around the cation. Overall, the analyzed RDFs allow us to assign two distinct characteristics for the investigated solutions. The first is typical of molecular liquids, which display smoother distributions and weaker electrostatic interactions (Fig. 3(a)–(d) and (f)). The second characteristic, typical of ionic liquids, discloses a much more structured RDF, with more evident charge distributions and a solvation shell defined by a strong maximum and a well-defined minimum (Fig. 3(e), (g) and (h)).
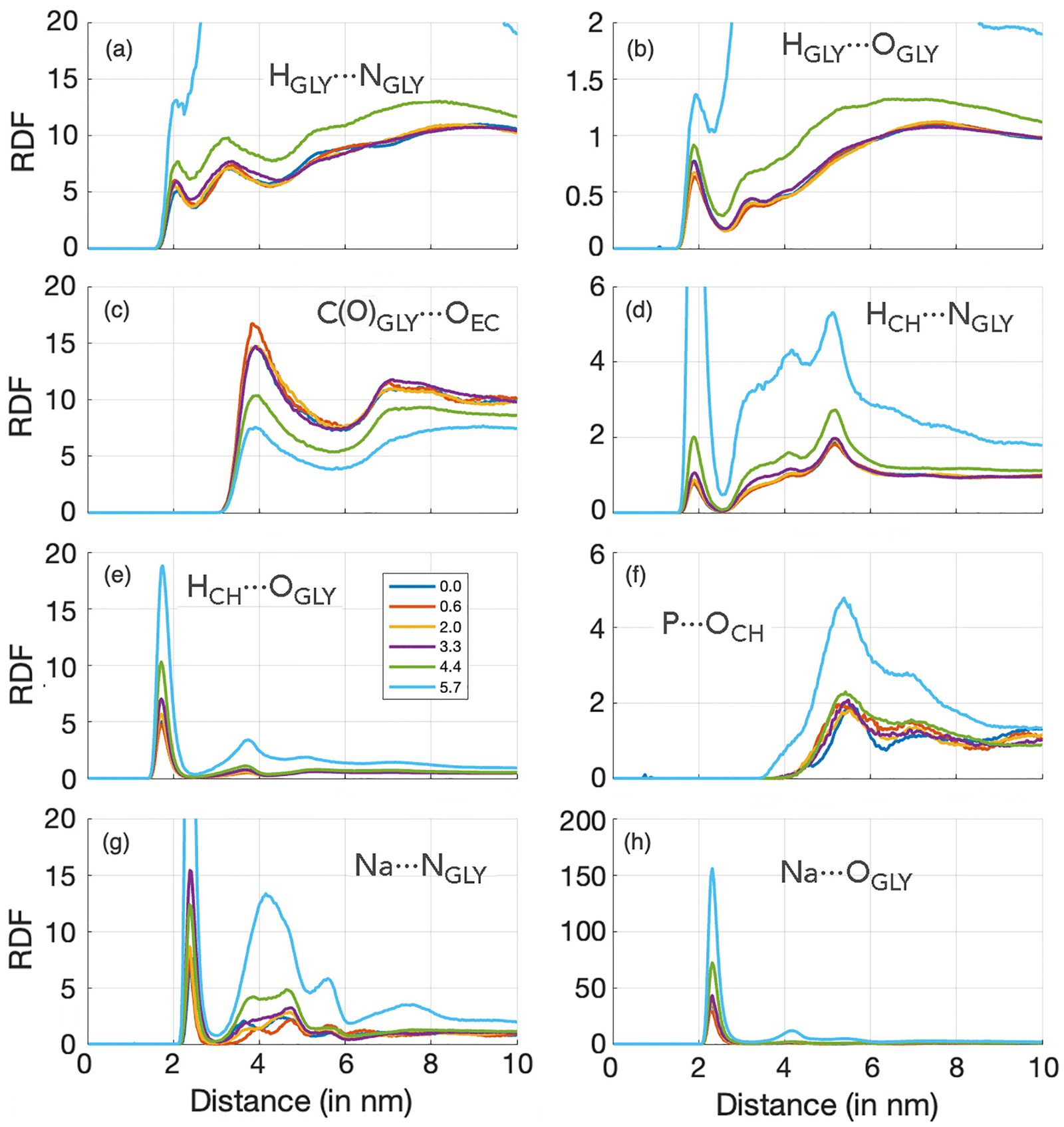 | ||
| Fig. 3 Radial distribution function (RDF) for selected ion pairs across the studied molar fractions of NaPF6. The pairs were chosen based on those exhibiting the highest absolute charge values. Since all relevant ion pairs involve either choline or glycine, the RDF for the 6.2% molar fraction is not displayed. RDFs for intermediate concentrations were omitted for clarity, as their behavior is similar to the other fractions. The H atoms in panels a, b, d, and e correspond to the donated proton. | ||
As it can be seen from the analysis of RDFs, the systems investigated here are characterized by complex interactions: two anions (one with two polar sites) interacting with two cations, all solvated by an organic solvent. In our previous study on ChGly,23 we observed that the RDF peaks for HCH⋯OGLY and HCH⋯NGLY appear at similar distances, with the HCH⋯OGLY interaction being more intense, which may suggest either a preference for the OGLY site by cation or simply reflect the higher concentration of OGLY atoms in the system. These RDFs indicate a competition between the anion's active sites for coordination with Ch+ cations, typical for complex ion-molecular systems.46–48 When diluted with water, the peak positions remain constant, but the intensity of HCH⋯OGLY decreases and that of HCH⋯NGLY increases, suggesting that water forms hydrogen bonds with OGLY, thereby favoring HCH⋯NGLY interactions.23
In this study, we find that in pure ChGly and systems with low NaPF6–EC content, there is no clear preference for Ch+ to coordinate with either the O or N site of Gly−, as both RDF peaks appear at similar distances. Still, the HCH⋯OGLY interaction maximum appears at slightly shorter distances (∼0.17 nm) and is more intense than HCH⋯NGLY (with ∼0.19 nm peak), indicating that Ch+ primarily interacts with OGLY. Only at 5.7% of NaPF6–EC do HCH⋯NGLY interactions occur at the same distance as HCH⋯OGLY, highlighting that dilution promotes competition between OGLY and NGLY sites, allowing for HCH⋯NGLY close contacts. This is consistent with the coordination numbers (CN): for the 0.6% system, CN is ∼1 for HCH⋯OGLY and ∼0.2 for HCH⋯NGLY, while for the 5.7% system, CN shifts to ∼0.2 for HCH⋯OGLY and ∼0.7 for HCH⋯NGLY. These findings align with our previous observations for aqueous ChGly, where dilution favored HCH⋯NGLY interactions.23
Regarding competition between Gly− and PF6− for interaction with Na+, the RDFs show that Na+ prefers Gly− over the PF6− anion, approaching Gly− more closely at higher NaPF6 concentrations (e.g., OGLY⋯Na peaks at ∼0.22 nm in 0.6% NaPF6 and ∼0.23 nm in 5.7%). Na–FPF6 contacts occur at longer distances, and no interaction is observed at lower NaPF6–EC concentrations, confirming that Gly− is more attractive to both Ch+ and Na+ cations.
The solutions investigated here exhibit an intricate network of electrostatic interactions, not only consisting of hydrogen bonds but also of intense and diverse electrostatic linkages. Among the hydrogen bonds, those formed between glycine anions and between glycine and choline stand out, as discussed earlier. Key intense electrostatic interactions manifest in the solvation structures surrounding sodium ions. One way to visually capture this complex network of interactions is through the contact matrix displayed in Fig. 4. In this matrix, the rows and columns correspond to all the most relevant and non-equivalent interaction sites (with the highest absolute charges). The determination of each matrix element was based on the calculation of the radial distribution function, which identified the distance and height of the first maximum. As a result, the matrix was constructed in a way that, if there is no contact between a specific atom pair, the corresponding matrix element is marked with a cross, whereas if a contact exists, the matrix element is colored based on a two-dimensional color scale shown in Fig. 4. Cells with colors tending towards red represent stronger electrostatic interactions, characterized by very short distances and high intensities at the RDF's first peak.
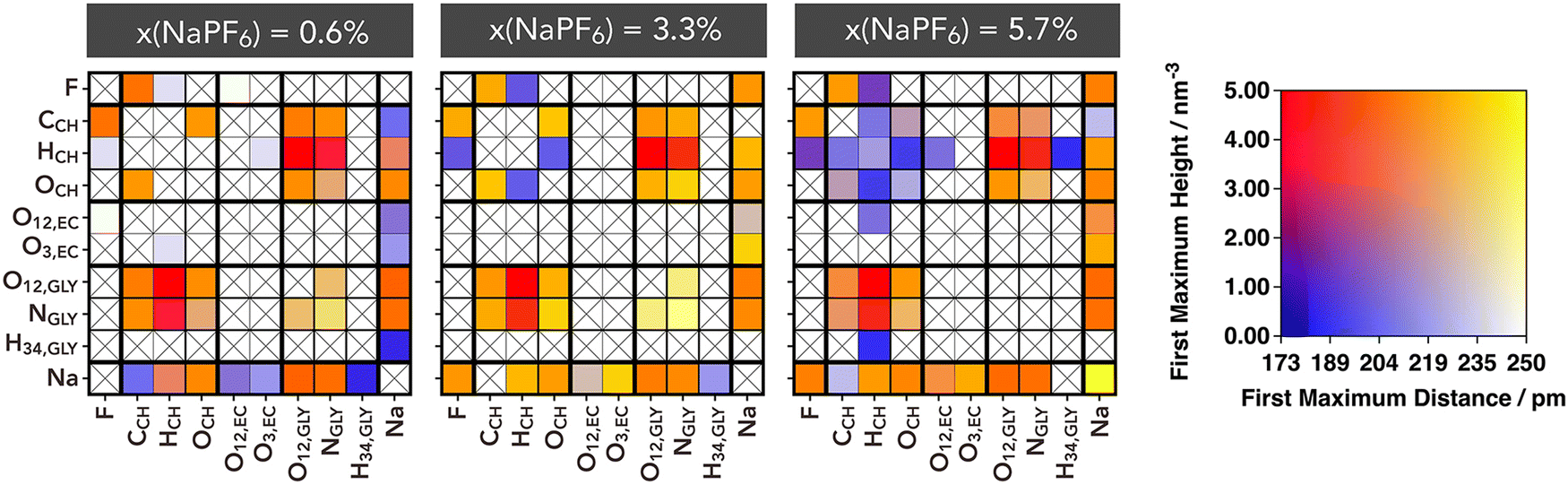 | ||
| Fig. 4 Connection matrices between specific sites of the electrolyte components for three selected molar fractions. The colors represent the intensity and distance of the first maximum in the corresponding RDF as defined in the 2D colormap. | ||
Similarly to the above analysis of RDFs, the competition between ions is clearly reflected in the connection matrixes (Fig. 4). The distances and intensities of HCH⋯OGLY and HCH⋯NGLY interactions are nearly indistinguishable, indicating no strong preference between the two. At low NaPF6–EC content, there is no detectable interaction between Na+ and PF6−; this interaction begins to emerge at intermediate concentrations and reaches its peak at higher NaPF6–EC concentrations. Throughout, the Na⋯OGLY/NGLY interactions remains consistently significant, highlighting strong affinity of Gly− anion for Na+ regardless of the PF6− presence.
A more detailed and specific approach to investigating the interactions within ChGly mixtures is through the use of Sankey diagrams.49 In these, the positively charged atomic sites (including proton-donating hydrogen atoms) are depicted on the left side of the circular diagram, while the negatively charged atomic sites (such as proton-accepting oxygen atoms) are displayed on the right. Due to the ternary nature of the mixtures studied here, these diagrams are more complex than those typically analyzed for binary mixtures.23,50
In this context, the width of each band corresponding to a positively charged group is proportional to the number of bonds formed with the respective negatively charged group. For the three molar fractions presented, we observe that the interactions predominantly involve the sodium cation, which interacts frequently with nearly all negative sites (with an interaction count ranging from 4.7 to 5.8), particularly the polar sites of the glycine anion, as already noted in the RDF analyses. This characteristic highlights and quantifies the significant role of the sodium ion in structuring the mixtures investigated here. The interaction involving the carbon of the glycine anion (CGLY), which predominantly interacts with the oxygen of the choline cation, is also noteworthy. Being non-directional in nature, this interaction appears crucial for the stability of the mixture's structure. Another strong electrostatic interaction involves the phosphorus atom of the PF6− anion. At low NaPF6–EC concentrations (0.6%), this site preferentially interacts with the oxygen of the choline cation, but at higher concentrations, these interactions shift towards those with the oxygen or nitrogen atoms of the glycine anion. The Sankey diagrams clearly show that hydrogen bonds are mainly formed between the choline and glycine ions, with the most numerous bonds being those between the hydrogen of choline and the oxygen of glycine (HCH⋯OGLY). This fact still corroborates our discussion about ions competition, since here we observe that the HCH, forms relatively more contacts with the oxygen accepting anion glycine than with its nitrogen, which can be observed comparing the correspondents of Sankey diagrams for 0.6% and 5.7% concentrations. Overall, we also observe that hydrogen bonds are far less numerous than the intense electrostatic interactions, indicating that the mixtures studied here are primarily dominated by electrostatic forces.
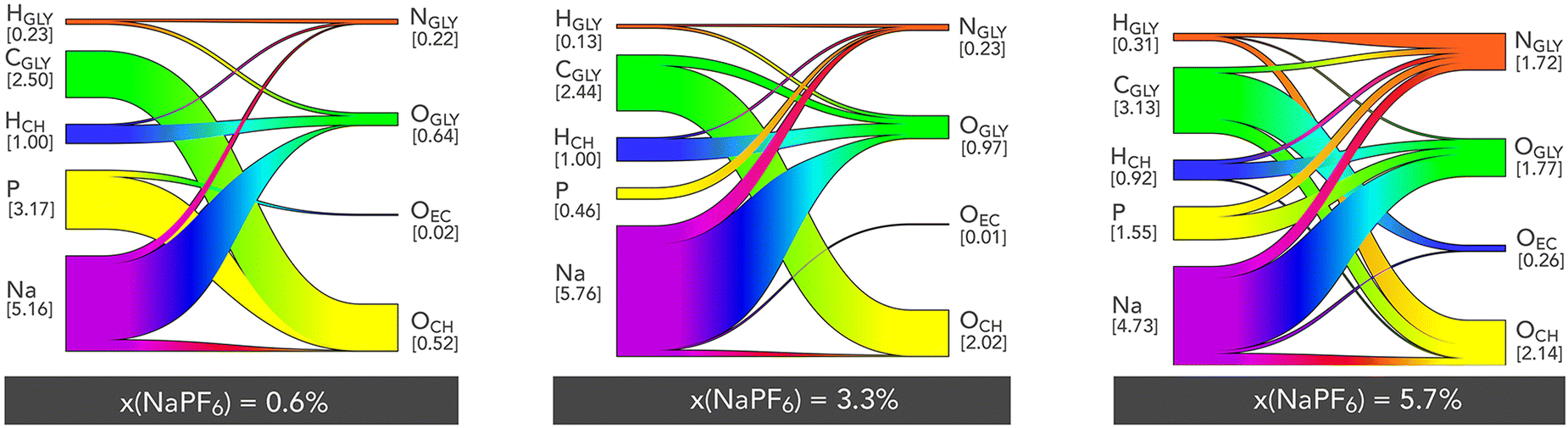 | ||
| Fig. 5 Sankey diagrams depicting the topology of hydrogen bonds and electrostatic interactions for three different molar fractions. The numbers in square brackets represent the average number of interactions per positive/negative site, as defined by the first minimum of the corresponding radial distribution function. | ||
Dynamic properties
The determination of dynamic properties is essential for elucidating the transport mechanisms in these systems. These properties not only offer valuable insights into ion and molecular mobility, solvation efficiency, and the structural stability of the mixtures, but also have a direct impact on their performance as electrolytes in electrochemical energy storage applications. Determining dynamic properties through molecular dynamics simulations is often challenging, particularly when it comes to viscosity.36,51 This is especially true for highly viscous liquids, such as pure ChGly, where the calculation becomes even more complex. The accuracy of these calculations is contingent upon several factors, including the simulation time, sample size, the force field model employed, and critically, the specific methodology applied, such as the Green–Kubo or Einstein approaches. For viscosity, in particular, rigorous protocols have been proposed due to the fact that small variations in simulation conditions or data processing can lead to significant discrepancies in the viscosity results.51 Therefore, obtaining precise results for the dynamic properties of the solutions investigated here is virtually impossible. Consequently, the results presented at Fig. 6 should be considered as qualitative indicators of the dynamics of NaPF6–EC solutions in ChGly.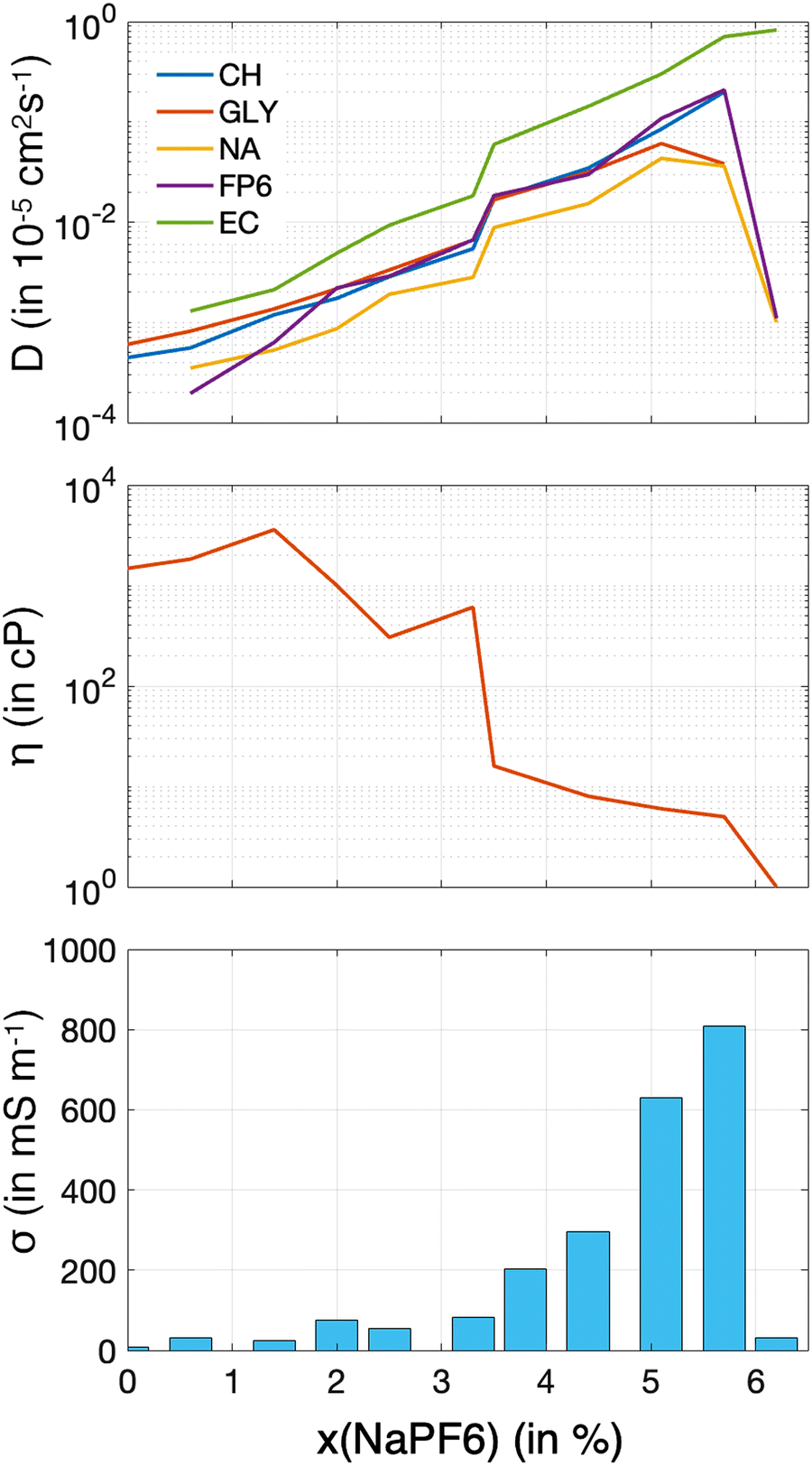 | ||
| Fig. 6 Dynamic properties as a function of NaPF6 concentration in the solutions. (a) Logarithm of the diffusion coefficient, (b) logarithm of viscosity, and (c) ionic conductivity. | ||
Fig. 6(a) presents the diffusion coefficient for each component of the mixtures and shows that, at lower concentrations of ChGly, ionic mobility is significantly enhanced. However, the diffusion coefficient reaches a peak at a concentration of 5.7% and then decreases back to the value for the pure NaPF6–EC solution (without ChGly). Fig. 6(b) illustrates the variation in viscosity as a function of NaPF6–EC concentration. For this property, the data is less precise due to the fact that our methodology involved a single trajectory, which was relatively short (only 50 ns). Nonetheless, the qualitative behavior of viscosity, which shows an approximately exponential reduction between the pure extremes of ChGly and NaPF6–EC, can be observed. Finally, the results for ionic conductivity (Fig. 6(c)) reveal that, across the concentration range, the ionic conductivity of the mixtures is relatively low but increases significantly with concentration, peaking at 5.7%.
The enhancement of ion self-diffusion and electric conductivity in systems with high NaPF6–EC content is expected. In concentrated ionic liquid systems like pure ChGly or those with low NaPF6–EC content, the ions form a dense network of electrostatic and hydrogen bonding interactions, as seen in Fig. 4 and 5, resulting in high viscosity, restricted ion mobility, and low diffusion and conductivity. As NaPF6 and the EC solvent are added, this network disrupts, reducing cation–anion interactions and increasing ion and solvent mobility. NaPF6 introduces more ions, enhancing charge transport, while EC lowers viscosity by solvating the ions. According to ref. 13 and 52, at around 10–20 molar percent of ionic liquid, molecular solvents stabilize ion pairs while disrupting larger ion clusters, as suggested by analysis of clusters on the same systems.53 The system with 5.7% NaPF6, corresponds to nearly 10% of ChGly, supporting this behavior. In contrast, in the NaPF6–EC system without ChGly, weaker ion–ion interactions and less efficient ion pairing in EC lead to reduced ion mobility, explaining the drop in conductivity and diffusion. Therefore, the enhanced behavior with dilution is due to the balanced interactions between NaPF6, ChGly, and EC, promoting better ion transport, while the absence of ChGly weakens the system's overall performance.
The RDF can be combined with temporal correlation functions to investigate how local interactions evolve over time, which is crucial for understanding mobility and diffusion processes in liquid mixtures. In this context, the Van Hove correlation function (VHCF) extends the structural analysis of the RDF by incorporating time dependence.54 Thus, the VHCF describes the probability of finding a site at a given distance from another site at different time intervals.
In Fig. 7, we present the Van Hove correlation function for the HCH⋯OGLY distance in the ternary mixtures investigated here. The choice of this particular distance is justified by the impact of the interactions between choline and glycine ions on the mixture structures. In each frame of Fig. 7, the correlation time, which measures the time lag between the positions of the two sites from different trajectory frames, is shown as a function of the distance between the sites. At τ = 0, we reproduce the RDF from Fig. 3(e), but as the correlation time increases, the RDF features quickly fade. We observe that the first peak of the functions, between 150 and 200 pm, is very prominent for t close to zero and gradually but rapidly diminishes as t progresses. Under high NaPF6–EC concentrations, the deterioration of the RDF characteristics becomes more pronounced, which is especially evident at concentrations of 5.1% and 5.7%.
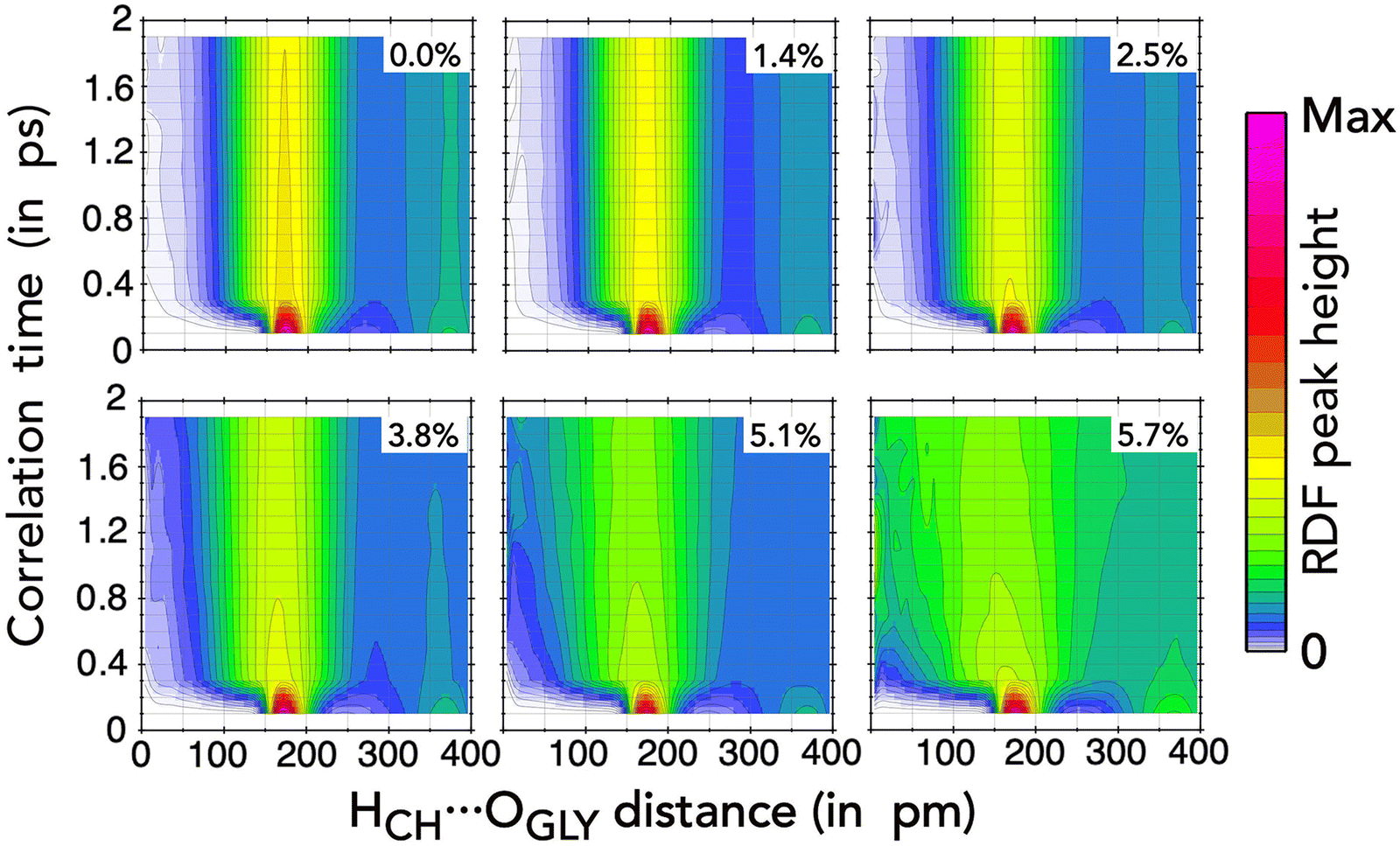 | ||
| Fig. 7 Van Hove correlation function (VHCF) for the HCH⋯OGLY distance in ChGly and NaPF6–EC mixtures. VHCFs for intermediate concentrations were omitted for clarity, as their behavior closely resembles that of the other fractions. | ||
Conclusions
In this study, we investigated the structural and dynamic properties of NaPF6–EC–ChGly ternary mixtures as potential electrolytes for supercapacitors through molecular simulations. Overall, the analyzed solutions exhibit a complex network of electrostatic interactions. Structural analysis, via radial distribution functions, revealed that the glycine anion plays a crucial role in structuring the liquid, showing significant interaction peaks at lower NaPF6–EC concentrations. Additionally, the hydrogen bonding between the glycine anion sites is strong, while the contribution of ethylene carbonate is modest. Interactions between choline and glycine ions intensify with increasing NaPF6–EC concentration, indicating a highly interactive behavior. The preference of the Na+ cation to coordinate with the glycine anion over PF6− highlights the attractiveness of glycine, which may have significant implications for the development of advanced electrolytes and electrochemical systems. Both contact matrices and Sankey diagrams further support the complexity of these mixtures, emphasizing the importance of hydrogen bonds between choline and glycine.The dynamic properties, essential for understanding the transport mechanisms in the studied mixtures, were analyzed qualitatively. We observed that the diffusion coefficient, at lower ChGly concentrations, indicates an increase in ionic mobility, peaking at a concentration of 5.7% before decreasing again. Viscosity, while less precisely measured, exhibits an exponential reduction between the extremes of ChGly and NaPF6–EC, while ionic conductivity significantly increases with concentration. The addition of NaPF6 and EC disrupts the dense network of electrostatic interactions and hydrogen bonds in ChGly, enhancing ionic mobility. The Van Hove correlation function, analyzed for one of the key observed interactions (HCH⋯OGLY), reveals that the structure of the mixtures deteriorates rapidly over time, particularly at higher NaPF6–EC concentrations, indicating a decline in RDF characteristics.
These findings highlight the potential application of this innovative mixture in next-generation supercapacitors, particularly in contexts requiring long-term stability, improved energy storage capacity, and environmentally responsible solutions. Future research could focus on experimentally validating these results and further exploring the role of other ionic liquids in enhancing electrolyte properties for energy storage systems.
Data availability
All data generated in this study, including simulation trajectories totaling approximately 1 TB, are available from the corresponding author upon request. Due to the large size of the data files, direct public sharing is not feasible. However, the configurations of equilibrated simulation cells and associated itp-files are available on GitHub: https://github.com/voroshylova/ChGly-NaPF6-EC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from FCT/MCTES (UIDP/50006/2020 DOI 10.54499/UIDP/50006/2020) through Portuguese national funds. This work was supported by research grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (TM 2023/12286-8). Support from FCT/MCTES through the Portuguese national funds (LA/P/0008/2020 DOI 10.54499/LA/P/0008/2020, UIDP/50006/2020 DOI 10.54499/UIDP/50006/2020 and UIDB/50006/2020 DOI 10.54499/UIDB/50006/2020, REQUIMTE LAQV) are also acknowledged. The authors acknowledge the National Laboratory for Scientific Computing (SDumont supercomputer, LNCC/MCTI, Brazil) for providing computational resources for the calculations reported in this paper.References
- M. Saha, A. Kumar, R. Kanaoujiya, K. Behera and S. Trivedi, A Comprehensive Review of Novel Emerging Electrolytes for Supercapacitors: Aqueous and Organic Electrolytes Versus Ionic Liquid-Based Electrolytes, Energy Fuels, 2024, 38, 8528–8552, DOI:10.1021/acs.energyfuels.4c00685.
- T. Zhou, C. Gui, L. Sun, Y. Hu, H. Lyu, Z. Wang, Z. Song and G. Yu, Energy Applications of Ionic Liquids: Recent Developments and Future Prospects, Chem. Rev., 2023, 123, 12170–12253, DOI:10.1021/acs.chemrev.3c00391.
- W. Raza, F. Ali, N. Raza, Y. Luo, K.-H. Kim, J. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Recent advancements in supercapacitor technology, Nano Energy, 2018, 52, 441–473, DOI:10.1016/j.nanoen.2018.08.013.
- M. Salanne, Ionic Liquids for Supercapacitor Applications, Top. Curr. Chem., 2017, 375, 63, DOI:10.1007/s41061-017-0150-7.
- L. Xia, L. Yu, D. Hu and G. Z. Chen, Electrolytes for electrochemical energy storage, Mater. Chem. Front., 2017, 1, 584–618, 10.1039/C6QM00169F.
- C. Zhan, C. Lian, Y. Zhang, M. W. Thompson, Y. Xie, J. Wu, P. R. C. Kent, P. T. Cummings, D. Jiang and D. J. Wesolowski, Computational Insights into Materials and Interfaces for Capacitive Energy Storage, Adv. Sci., 2017, 4, 1700059, DOI:10.1002/advs.201700059.
- P. Simon and Y. Gogotsi, Materials for electrochemical capacitors, Nat. Mater., 2008, 7, 845–854, DOI:10.1038/nmat2297.
- V. V. Chaban and N. A. Andreeva, Aqueous electrolytes at the charged graphene Surface: Electrode-Electrolyte coupling, J. Mol. Liq., 2023, 387, 122724, DOI:10.1016/j.molliq.2023.122724.
- I. V. Voroshylova, S. R. Smaga, E. V. Lukinova, V. V. Chaban and O. N. Kalugin, Conductivity and association of imidazolium and pyridinium based ionic liquids in methanol, J. Mol. Liq., 2015, 203, 7–15, DOI:10.1016/j.molliq.2014.12.028.
- V. V. Chaban and N. A. Andreeva, Shorter-chained trialkylsulfonium cations are preferable as admixtures to lithium-ion and sodium-ion electrolytes in acetonitrile, J. Mol. Liq., 2023, 385, 122399, DOI:10.1016/j.molliq.2023.122399.
- N. M. Figueiredo, I. V. Voroshylova, V. A. Koverga, E. S. C. Ferreira and M. N. D. S. Cordeiro, Influence of alcohols on the inter-ion interactions in ionic liquids: A molecular dynamics study, J. Mol. Liq., 2019, 111538, DOI:10.1016/j.molliq.2019.111538.
- V. V. Chaban and N. A. Andreeva, Magnesium-based electrolytes with ionic liquids chloride additives: Quantum chemical stationary point analysis, J. Mol. Liq., 2024, 402, 124804, DOI:10.1016/j.molliq.2024.124804.
- O. N. Kalugin, A. V. Riabchunova, I. V. Voroshylova, V. V. Chaban, B. A. Marekha, V. A. Koverga and A. Idrissi, Transport Properties and Ion Aggregation in Mixtures of Room Temperature Ionic Liquids with Aprotic Dipolar Solvents, Mod. Probl. Mol. Phys., Springer, Cham, 2018, pp. 67–109 DOI:10.1007/978-3-319-61109-9_5.
- I. V. Voroshylova, E. S. C. Ferreira, V. A. Koverga, C. M. Pereira and M. N. D. S. Cordeiro, Chapter 4 – Structure and noncovalent interactions in ionic liquids mixtures and deep eutectic solvents, in Theor. Comput. Approaches Predict. Ion. Liq. Prop., ed. A. Joseph and S. Mathew, Elsevier, 2021, pp. 105–157 DOI:10.1016/B978-0-12-820280-7.00013-9.
- E. Karpierz, L. Niedzicki, T. Trzeciak, M. Zawadzki, M. Dranka, J. Zachara, G. Z. Żukowska, A. Bitner-Michalska and W. Wieczorek, Ternary mixtures of ionic liquids for better salt solubility, conductivity and cation transference number improvement, Sci. Rep., 2016, 6, 35587, DOI:10.1038/srep35587.
- P. Martínez-Crespo, M. Otero-Lema, O. Cabeza, H. Montes-Campos and L. M. Varela, Structure, dynamics and ionic conductivities of ternary ionic liquid/lithium salt/DMSO mixtures, J. Mol. Liq., 2022, 359, 119188, DOI:10.1016/j.molliq.2022.119188.
- P. Vallet, S. Bouzón-Capelo, T. Méndez-Morales, V. Gómez-González, Y. Arosa, R. de la Fuente, E. López-Lago, J. R. Rodríguez, L. J. Gallego, J. J. Parajó, J. Salgado, M. Turmine, L. Segade, O. Cabeza and L. M. Varela, On the physical properties of mixtures of nitrate salts and protic ionic liquids, J. Mol. Liq., 2022, 350, 118483, DOI:10.1016/j.molliq.2022.118483.
- D. Qin, F. Cheng, M. Cao, F. Yan, Q. Wang, C. Fang and J. Han, Unraveling the incompatibility mechanism of ethylene carbonate-based electrolytes in sodium metal anodes, J. Energy Chem., 2024, 94, 560–567, DOI:10.1016/j.jechem.2024.02.012.
- S. Sarkar, M. J. Lefler, B. S. Vishnugopi, R. B. Nuwayhid, C. T. Love, R. Carter and P. P. Mukherjee, Fluorinated ethylene carbonate as additive to glyme electrolytes for robust sodium solid electrolyte interface, Cell Rep. Phys. Sci., 2023, 4, 101356, DOI:10.1016/j.xcrp.2023.101356.
- M. Dahbi, T. Nakano, N. Yabuuchi, S. Fujimura, K. Chihara, K. Kubota, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Effect of Hexafluorophosphate and Fluoroethylene Carbonate on Electrochemical Performance and the Surface Layer of Hard Carbon for Sodium-Ion Batteries, ChemElectroChem, 2016, 3, 1856–1867, DOI:10.1002/celc.201600365.
- P. Wróbel, P. Kubisiak and A. Eilmes, MeTFSI (Me = Li, Na) Solvation in Ethylene Carbonate and Fluorinated Ethylene Carbonate: A Molecular Dynamics Study, J. Phys. Chem. B, 2021, 125, 1248–1258, DOI:10.1021/acs.jpcb.0c10622.
- D. Morales, L. G. Chagas, D. Paterno, S. Greenbaum, S. Passerini and S. Suarez, Transport studies of NaPF6 carbonate solvents-based sodium ion electrolytes, Electrochim. Acta, 2021, 377, 138062, DOI:10.1016/j.electacta.2021.138062.
- T. Malaspina, I. V. Voroshylova, M. N. D. S. Cordeiro and E. E. Fileti, Probing the local structures of Choline-Glycine Electrolytes: Insights from ab initio simulations, J. Mol. Liq., 2023, 390, 122946, DOI:10.1016/j.molliq.2023.122946.
- H. de Araujo Chagas, E. E. Fileti and G. Colherinhas, Comparing supercapacitors with graphene/graphyne electrodes and [Bmim][PF6], [Emim][BF4], [Ch][Gly] and [Pyr][Tfsi] ionic liquids using molecular dynamics, J. Mol. Liq., 2023, 379, 121703, DOI:10.1016/j.molliq.2023.121703.
- H. de Araujo Chagas, E. E. Fileti and G. Colherinhas, A molecular dynamics study of graphyne-based electrode and biocompatible ionic liquid for supercapacitor applications, J. Mol. Liq., 2022, 360, 119494, DOI:10.1016/j.molliq.2022.119494.
- Y. Li, F. Yang, Y. Li, M. Cai, H. Li, X. Fan and M. Zhu, Choline amino acid ionic Liquids: A novel green potential lubricant, J. Mol. Liq., 2022, 360, 119539, DOI:10.1016/j.molliq.2022.119539.
- V. Alizadeh, F. Malberg, A. A. H. Pádua and B. Kirchner, Are There Magic Compositions in Deep Eutectic Solvents? Effects of Composition and Water Content in Choline Chloride/Ethylene Glycol from Ab Initio Molecular Dynamics, J. Phys. Chem. B, 2020, 124, 7433–7443, DOI:10.1021/acs.jpcb.0c04844.
- G. Colherinhas, T. Malaspina and E. E. Fileti, Storing Energy in Biodegradable Electrochemical Supercapacitors, ACS Omega, 2018, 3, 13869–13875, DOI:10.1021/acsomega.8b01980.
- X. Yan, Y. He, X. Liu, S. Jing, J. Guan, W. Gao, S. Ray, Y. Xiong, T. Li and X. Ge, Deterministic Effect of the Solid-State Diffusion Energy Barrier for a Charge Carrier on the Self-Discharge of Supercapacitors, ACS Energy Lett., 2023, 8, 2376–2384, DOI:10.1021/acsenergylett.3c00453.
- C. Zhao, X. Sun, W. Li, M. Shi, K. Ren and X. Lu, Reduced Self-Discharge of Supercapacitors Using Piezoelectric Separators, ACS Appl. Energy Mater., 2021, 4, 8070–8075, DOI:10.1021/acsaem.1c01373.
- P. Galek, A. Slesinski, K. Fic and J. Menzel, Peculiar role of the electrolyte viscosity in the electrochemical capacitor performance, J. Mater. Chem. A, 2021, 9, 8644–8654, 10.1039/D0TA11230E.
- K. Chayambuka, R. Cardinaels, K. L. Gering, L. Raijmakers, G. Mulder, D. L. Danilov and P. H. L. Notten, An experimental and modeling study of sodium-ion battery electrolytes, J. Power Sources, 2021, 516, 230658, DOI:10.1016/j.jpowsour.2021.230658.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations, J. Comput. Chem., 2009, 30, 2157–2164, DOI:10.1002/jcc.21224.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236, DOI:10.1021/ja9621760.
- E. S. C. Ferreira, I. V. Voroshylova, C. M. Pereira and M. N. D. S. Cordeiro, Improved Force Field Model for the Deep Eutectic Solvent Ethaline: Reliable Physicochemical Properties, J. Phys. Chem. B, 2016, 120, 10124–10137, DOI:10.1021/acs.jpcb.6b07233.
- E. S. C. Ferreira, I. V. Voroshylova, N. M. Figueiredo and M. N. D. S. Cordeiro, Molecular dynamic study of alcohol-based deep eutectic solvents, J. Chem. Phys., 2021, 155, 064506, DOI:10.1063/5.0058561.
- E. S. C. Ferreira, I. V. Voroshylova, N. M. Figueiredo, C. M. Pereira and M. N. D. S. Cordeiro, Computational and experimental study of propeline: a choline chloride based deep eutectic solvent, J. Mol. Liq., 2020, 298, 111978 CrossRef.
- C. M. Breneman and K. B. Wiberg, Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis, J. Comput. Chem., 1990, 11, 361–373, DOI:10.1002/jcc.540110311.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. MontgomeryJr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Revision A.02, 2009 Search PubMed.
- S. Nosé, A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys., 1984, 52, 255–268, DOI:10.1080/00268978400101201.
- W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697, DOI:10.1103/PhysRevA.31.1695.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182–7190, DOI:10.1063/1.328693.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092, DOI:10.1063/1.464397.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25, DOI:10.1016/j.softx.2015.06.001.
- E. Roos Nerut, K. Karu, I. V. Voroshylova, K. Kirchner, T. Kirchner, M. V. Fedorov and V. B. Ivaništšev, NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab, Computation, 2018, 6, 57, DOI:10.3390/computation6040057.
- I. V. Voroshylova, E. S. C. Ferreira, M. Malček, R. Costa, C. M. Pereira and M. N. D. S. Cordeiro, Influence of the anion on the properties of ionic liquid mixtures: a molecular dynamics study, Phys. Chem. Chem. Phys., 2018, 20, 14899–14918, 10.1039/C8CP01541D.
- M. Brüssel, M. Brehm, T. Voigt and B. Kirchner, Ab initio molecular dynamics simulations of a binary system of ionic liquids, Phys. Chem. Chem. Phys., 2011, 13, 13617–13620, 10.1039/C1CP21550G.
- S. Cha and D. Kim, Anion exchange in ionic liquid mixtures, Phys. Chem. Chem. Phys., 2015, 17, 29786–29792, 10.1039/C5CP04276C.
- M. Brehm, M. Thomas, S. Gehrke and B. Kirchner, TRAVIS-A free analyzer for trajectories from molecular simulation, J. Chem. Phys., 2020, 152, 164105, DOI:10.1063/5.0005078.
- T. Malaspina, G. Colherinhas, S. E. Weitzner, B. C. Wood and E. Eterno Fileti, Unraveling local structures of Salt-in-Water and Water-in-Salt electrolytes via ab initio molecular dynamics, J. Mol. Liq., 2023, 383, 122097, DOI:10.1016/j.molliq.2023.122097.
- E. J. Maginn, R. A. Messerly, D. J. Carlson, D. R. Roe and J. R. Elliot, Best Practices for Computing Transport Properties 1. Self-Diffusivity and Viscosity from Equilibrium Molecular Dynamics [Article v1.0], Living J. Comput. Mol. Sci., 2019, 1, 6324, DOI:10.33011/livecoms.1.1.6324.
- O. N. Kalugin, I. V. Voroshylova, A. V. Riabchunova, E. V. Lukinova and V. V. Chaban, Conductometric study of binary systems based on ionic liquids and acetonitrile in a wide concentration range, Electrochim. Acta, 2013, 105, 188–199, DOI:10.1016/j.electacta.2013.04.140.
- V. V. Chaban, I. V. Voroshylova, O. N. Kalugin and O. V. Prezhdo, Acetonitrile Boosts Conductivity of Imidazolium Ionic Liquids, J. Phys. Chem. B, 2012, 116, 7719–7727, DOI:10.1021/jp3034825.
- L. Van Hove, Correlations in Space and Time and Born Approximation Scattering in Systems of Interacting Particles, Phys. Rev., 1954, 95, 249–262, DOI:10.1103/PhysRev.95.249.
| This journal is © the Owner Societies 2025 |