
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene-based single-atom catalysts for electrochemical CO2 reduction: unraveling the roles of metals and dopants in tuning activity†
Colin
Gallagher
,
Manish
Kothakonda
and
Qing
Zhao
 *
*
Department of Chemical Engineering, Northeastern University, Boston, Massachusetts 02115, USA. E-mail: q.zhao@northeastern.edu
First published on 7th January 2025
Abstract
Discovering electrocatalysts that can efficiently convert carbon dioxide (CO2) to valuable fuels and feedstocks using excess renewable electricity is an emergent carbon-neutral technology. A single metal atom embedded in doped graphene, i.e., single-atom catalyst (SAC), possesses high activity and selectivity for electrochemical CO2 reduction (CO2R) to CO, yet further reduction to hydrocarbons is challenging. Here, using density functional theory calculations, we investigate stability and reactivity of a broad SAC chemical space with various metal centers (3d transition metals) and dopants (2p dopants of B, N, O; 3p dopants of P, S) as electrocatalysts for CO2R to methane and methanol. We observe that the rigidities of these SACs depend on the type of dopants, with 3p-coordinating SACs exhibiting more severe out-of-plane distortion than 2p-coordinating SACs. Using CO adsorption energy as a descriptor for CO2R reactivity, we narrow down the candidates and identify seven SACs with near-optimal CO binding strength. We then elucidate full reaction mechanisms towards methane and methanol generation on these identified candidates and observe highly dopant-dependent activity and rate-limiting steps, divergent from conventional mechanistic understanding on metallic surfaces, calling into question whether previous design principles established on metals are directly transferrable to SACs. Consequently, we find that zinc embedded in boron-doped graphene (Zn–B–C) is a highly active catalyst for electrochemical CO2R to C1 hydrocarbons. Our work reveals the opportunities of tuning SAC reactivity via engineering dopants and metals and highlights the importance of re-elucidating CO2R reaction mechanisms on SACs towards unearthing new design principles for SAC chemistry.
I. Introduction
Consumption of fossil fuels and other CO2-emitting energy resources contributes significantly to global warming.1,2 As energy demands are projected to increase substantially,3,4 mitigation of CO2 concentrations in the air will be crucial to avoiding climate-related ecological and humanitarian problems.5 One of the most promising solutions is the conversion of atmospheric CO2 to valuable chemical feedstocks via electrochemical CO2 reduction (CO2R) using excess renewable electricity, such as solar and wind.6–8 Various materials, including metals, metal alloys, and metal oxides, have been used to catalyze CO2R.9–13 Currently, copper (Cu)-based materials remain the best metal electrocatalysts for CO2R towards generating hydrocarbon products.14–20 However, these catalysts require high overpotentials to achieve reasonable reaction rates and exhibit poor selectivity towards valuable products, which is impractical for commercializing this technology,9,21 motivating rational design of more efficient CO2R catalysts with improved activity and selectivity.Beyond conventional metal surfaces, single-atom catalysts (SACs) consisting of an isolated metal site in doped graphene (typically with nitrogen or N dopants) have become emergent catalysts for CO2R in the past decade.22,23 Dispersing isolated metal atoms on graphene maximizes atom utilization and suppresses competitive hydrogen evolution reaction (HER).24–27 In addition, dopants, such as N, can stabilize active metal centers embedded on graphene, enabling proton-coupled electron transfer (PCET) to enhance CO2R activity.28–30 Carbon monoxide (CO) has been experimentally identified as the major product of electrochemical CO2R for SACs on N-doped graphene. Zheng et al. synthesized nickel (Ni) SAC on N-doped graphene (Ni–N–C) and reported a nearly 100% faradaic efficiency for electrochemical CO2 to CO conversion at an applied potential of −0.68 V vs. the reversible hydrogen electrode (RHE).31 Zheng et al. confirmed the superior catalytic performance of Ni–N–C in reducing CO2 to CO with a faradaic efficiency of 98.5% and observed that Ni–N–C outperforms other SACs, including Fe–N–C, Cu–N–C, and Co–N–C, in generating CO.32 Wang et al. noted that the poor selectivity of Co–N–C for CO production comes from the undercoordinated N species in the synthesized materials and removing those undercoordinated N atoms results in a highly improved CO faradaic efficiency of 99.4% at an applied potential of −0.60 V vs. the RHE.33 In addition to CO, hydrocarbons have also been identified as major products of CO2R for SACs on N-doped graphene. Yang et al. reported a faradaic efficiency of 44% for methanol generation using Cu–N–C at −0.9 V vs. the RHE,34 while Han et al. observed a high faradaic efficiency of 85% for methane production using Zn–N–C at −1.8 V vs. the saturated calomel electrode.35 Beyond C1 products, SACs on N-doped graphene can also generate multi-carbon hydrocarbons with notable faradaic efficiencies. Guan et al. observed production of both methane and ethylene on Cu–N–C and noted that a high Cu loading favors ethylene generation, while a low Cu loading prefers producing methane.36 To improve activity and selectivity of SACs towards generating hydrocarbons, SACs incorporating dopants other than N or heteroatom dopants have been synthesized. Cai et al. achieved a high faradaic efficiency of 78% for methane production at −1.44 V vs. the RHE when mixing oxygen (O) dopants with N in Cu–N2O2–C SAC.37 Mixing boron (B) dopants with N can also boost the catalytic performance for CO2R to methane using Cu SACs.38 In addition to SACs with 2p dopants, SACs with 3p dopants have also been successfully synthesized. For example, Co SACs with both N and sulfur (S) dopants (Co–N2S2–C) have been used for CO2R to CO, outperforming Co–N–C at all overpotentials.39
Despite these achievements, discovering practical SACs towards electrochemically reducing CO2 to hydrocarbons remains a grand challenge due to the lack of in situ spectroscopy to detect atomic-scale, short-lived reaction intermediates and the existence of a large SAC chemical space that cannot be fully enumerated by experimental approaches.40 Instead, computational modeling, typically with density functional theory (DFT), plays an important role in understanding reaction mechanisms at the electronic structure level and assessing hypothetical catalysts prior to experimental synthesis.41–43 For example, Zhao and Liu used ab initio molecular dynamics simulations and enhanced sampling to investigate the CO2R to CO reaction pathways via an adsorbed COOH intermediate (*COOH; * refers to an adsorption site) on Ni–Nx–C SACs (x = 0–4).44 They found that an appropriate explicit description of solvents and surface charge can lead to the experimentally observed CO2 chemisorption of forming *CO2 prior to protonation and proposed that a hybrid coordination environment with one nitrogen and three carbon atoms is the most active and selective Ni–N–C SAC for CO2R to CO. Vijay et al. also investigated the same CO2R to CO reaction mechanisms via *CO2 and *COOH intermediates on both Fe–N–C and Ni–N–C and determined that the rate-limiting step could be either CO2 adsorption or the subsequent proton-coupled electron transfer step of forming *COOH.45 Beyond CO, several computational efforts have been conducted to study CO2R reaction pathways to hydrocarbons. Chen et al. used DFT to explain the experimentally observed high activity of Cu–N–C for CO2R to acetone and proposed that the pyrrolic coordination environment results in an energetically favorable pathway with the rate-limiting step being the first CO2 reduction step of forming *COOH, consistent with spectroscopic characterization of SAC structures using extended x-ray absorption fine structure.46 Zhou et al. confirmed the favorability of pyrrolic coordination environment over the pyridinic structure for Ni–N–C SACs towards CO generation.47 Yang et al. investigated reaction mechanisms of CO2R to methane and methanol also on Cu–N–C and proposed that the selectivity towards different products originates from *COH reduction to *CHOH for methanol vs. to *C for methane, explaining the experimentally observed high activity of methanol production.34 While still focusing on Cu–N–C, Guan et al. studied a different reaction pathway towards forming methane, i.e., through *CO to *CHO to *CH2O, along with C–C coupling mechanism via formation of OC–*COH intermediate towards generating ethylene, and concluded that the selectivity of C1vs. C2 products is dependent on Cu loading density.36 In addition to mechanistic analysis, DFT simulations provide the opportunities of assessing hypothetical catalysts prior to trial-and-error experimental synthesis and verification. For example, Wang et al. investigated 23 N-doped M–N–C SACs with M ranges from 3d to 5d transition metals and concluded that their CO2R reaction mechanisms and reactivities toward methane generation are highly dependent on the outermost d-shell electrons of the catalytically active metal centers with no ubiquitous rate-limiting step.48 Hou et al. investigated 28 N and S co-doped SACs, M–NxS4−x–C SACs (M = Fe, Co, Ni, Cu), toward generating methanol and methane. They considered multiple different reaction pathways and identified Cu–N1S3–C as an active catalyst for CO, CH3OH, and CH4 production, as well as Fe–N3S1–C for formic acid generation.49 Wei et al. assessed stability and catalytic performance of 10 dual metal-site SACs, M1/M2–N–C (M1, M2 = Fe, Co, Ni, Cu), for CO2R to CO, formic acid, methanol, and methane. Considering CO2 reduction to *COOH and *CHOH reduction to *CH as the rate-limiting-steps, they predicted that Fe/Co–N–C and Co–N–C exhibit the best activity and selectivity towards methane formation.50
Despite previous success of using DFT to study and design graphene-based SACs for electrochemical CO2 conversion, there is a fundamental gap in understanding the roles of metals, dopants, and coordinating environment in tuning reactivity of SACs. In addition, the reaction mechanisms of CO2R to hydrocarbons beyond CO product on SACs remain unclear, limiting the opportunities of developing effective design principles for SACs and leaving the question whether descriptors established on metal catalysts are transferrable to SACs a puzzle. In this work, we focus on rationalizing catalytic activities of a large SAC chemical space with various metal centers (i.e., all 3d transition metals) and dopants (2p dopants: B, N, O; 3p dopants: phosphorus or P, S) as electrocatalysts for CO2R towards generating methane and methanol. We first narrow down the selection of potential SAC candidates using CO adsorption energy, which has been previously identified as an effective descriptor for CO2R activity on metal catalysts,51 and then elucidate full reaction mechanisms of CO2 conversion to methane and methanol on identified SAC candidates with an CO adsorption energy close to the optimal value of −0.67 eV.52 Consequently, we propose promising SACs as CO2R electrocatalysts for experimental synthesis and verification. To the best of our knowledge, no theoretical investigation exists heretofore of performing such systematic mechanistic analyses on B-, O-, P-, and S-doped SACs for electrochemical CO2R to hydrocarbons.
II. Computational methods
We performed all spin-polarized DFT calculations within Vienna Ab initio Simulation Package (VASP)53,54 version 6.3.1. We self-consistently simulated valence electrons of 2s and 2p electrons for B, C, N, and O, 3s and 3p electrons for P and S, and 4s and 3d electrons for first-row transition metals (Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn) using the all electron, frozen-core, projector augmented-wave (PAW) method.55 We employed the Perdew–Burke–Ernzerhof (PBE)56 exchange–correlation functional in tandem with Grimme's D357,58 dispersion correction and the Becke–Johnson damping function.59 We applied a kinetic-energy cutoff value of 800 eV for the plane-wave basis set. We obtained the initial SAC geometry, which consists of one metal atom, four dopants, and 26 C atoms in the unit cell from a previous work (Fig. 1).60 Each clean SAC structure was fully optimized (both atomic positions and lattice parameters) until the absolute total force on each atom was less than 0.03 eV Å−1. After adding adsorbates on SACs, we re-optimized atomic positions of all atoms in the supercell at fixed lattice parameters using the same convergence threshold. We confirmed that all periodic cells contain at least 15 Å of vacuum along the c-axis before and after geometry relaxation to avoid artificial interactions between the periodic images in the direction normal to the SAC surface. Locating the most stable geometries of both clean and adsorbed SACs is challenging due to the vacancies created and substitutes doped in graphene. We developed a systematic strategy to ensure that the most stable geometries were discovered in our simulations (Supplementary Method and Table S1, ESI†). In addition to full geometry optimizations, we also performed benchmark calculations of surface-constrained optimizations, in which the position of each atom in SAC along the c-axis (i.e., the direction normal to graphene plane) was fixed to maintain a planar geometry. We employed an automatic, Γ-point-centered Monkhorst–Pack61k-point grid of 3 × 3 × 1 to sample the Brillouin zone. The chosen kinetic energy cutoff and k-point grid converge the total energies to within 1.5 meV per atom (Fig. S1 and S2, ESI†). We used a default initial magnetic moment guess of 1 on each atom. Benchmark calculations indicated that using different initial magnetic moment guesses for the metal atoms results in same final energies and magnetic moments (Tables S2 and S3, ESI†). We applied a Fermi surface smearing with first order Methfessel–Paxton method62 and a smearing width of 0.1 eV to integrate the Brillouin zone and aid self-consistent field convergence. Adding dipole field energy and potential corrections63 yields similar CO adsorption energies with differences less than 0.01 eV on representative N-doped SACs (Table S4, ESI†). Benchmark DFT+U calculations of applying a Hubbard U value of 3.29 eV on Fe, as suggested in previous studies,64,65 showed similar reaction energies for CO2R to methane and methanol pathways on Fe–N–C (Fig. S3, ESI†).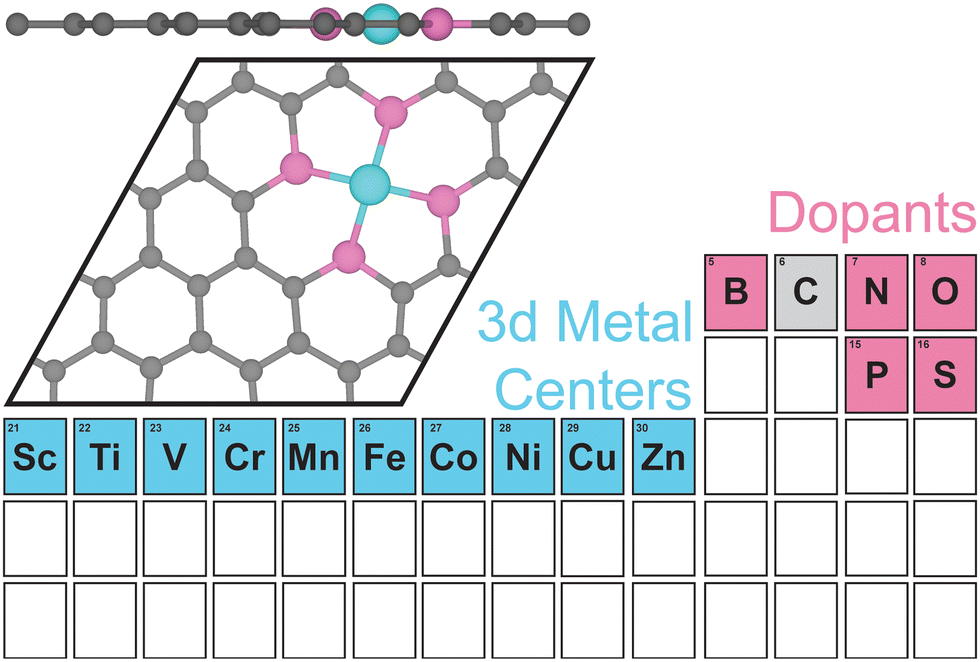 | ||
| Fig. 1 SAC structure side view (top) and top view (bottom) with different metal centers (Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) shown in blue, dopants (B, N, O, P, and S) shown in magenta, and C shown in gray. | ||
To quantify metal- and dopant-induced distortion within SAC, we computed displacements of metal and dopant atoms along the direction normal to the graphene surface (i.e., z-direction) using the equation,
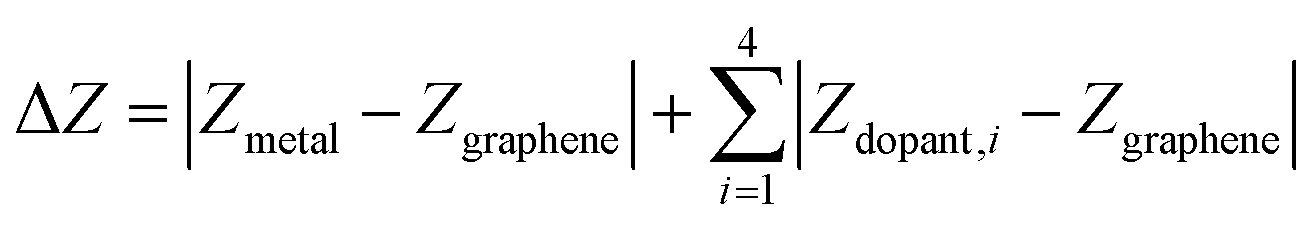
| Ecomplexation = ESAC − Egraphene − Emetal |
| ΔECO = ECO+SAC − ESAC − ECO |
III. Results and discussion
Dopant effects on structural stabilities of SACs
We considered a pyridinic-type SAC moiety, i.e., the central metal atom is coordinated with four pyridinic dopant atoms (Fig. 1). Other structures, such as a pyrrolic-type SAC moiety, might exist, yet how metal centers and dopants fill C vacancies in graphene is not the focus of our work. Starting from planar monolayer of SAC geometries, we fully optimized the structures and observed system-dependent out-of-plane distortion, particularly in the region of metal center and dopants (Table S1, ESI†). Local distortion among active sites is due to the capabilities of dopants to fill in C vacancies on graphene as well as symmetry and rigidity enforced by periodic boundary conditions. Overall, 2p-coordinating SACs with N, B, or O dopants exhibit distortion only among the metal site if any, while 3p-coordinating SACs with P or S dopants experience distortion among both the metal and dopant sites, regardless of the metal center.To quantify the local out-of-plane distortion observed in SACs, we computed displacements of metal and dopant atoms along the direction normal to the graphene surface as described in the Computational methods section (Fig. 2). When performing full geometry optimizations with no constraint, we observed that SACs with 2p dopants (i.e., B, N, or O) either remain planar with a 0.0 Å distortion or exhibit a moderate distortion of less than 2.1 Å concentrated on the metal site. Though the overall structural properties remain similar across all 2p dopants, different dopants exhibit divergent local distortion sensitivities to metal sites. Within N-doped SACs, only early transition metals, Sc–N–C, Ti–N–C, and V–N–C show local distortion around the metal center with displacements of 1.0 to 1.3 Å, while the other M–N–C SACs preserve the planar structure. Interestingly, boron-doped SACs exhibit consistently local distortions with displacements in the range of 1.0 to 1.6 Å for all transition metals, indicating the crucial role of dopants in determining local metal-coordination environment. Oxygen-doped SACs show more variances in out-of-plane distortions that are sensitive to metal center valence electrons. Early and late transition metals exhibit moderate displacements, while mid-row transition metals remain planar in the same plane with graphene substrate. Switching from 2p dopants to 3p dopants (i.e., P or S), we observed out-of-plane distortion concentrated not only on metal sites, but also on dopants, increasing the degree of distortion from less than 2.1 Å for 2p dopants to 3.0 to 6.0 Å for 3p dopants. Although all 3p-coordinating SACs show severe out-of-plane distortion, P-coordinating SACs are more distorted than corresponding S-coordinating SACs for early and mid-row transition metals and vice versa for late transition metals. In addition, we noted that Zn–O–C, Zn–P–C, and Zn–S–C are not structurally stable materials since the metal centers cannot form any chemical bonds with dopants (Table S1, ESI†). Most SACs exhibit structures with all four dopants in the same side of the graphene, while two exceptions (Cu–P–C and Fe–S–C) display structures with dopants on the opposite side of graphene (Table S1, ESI†).
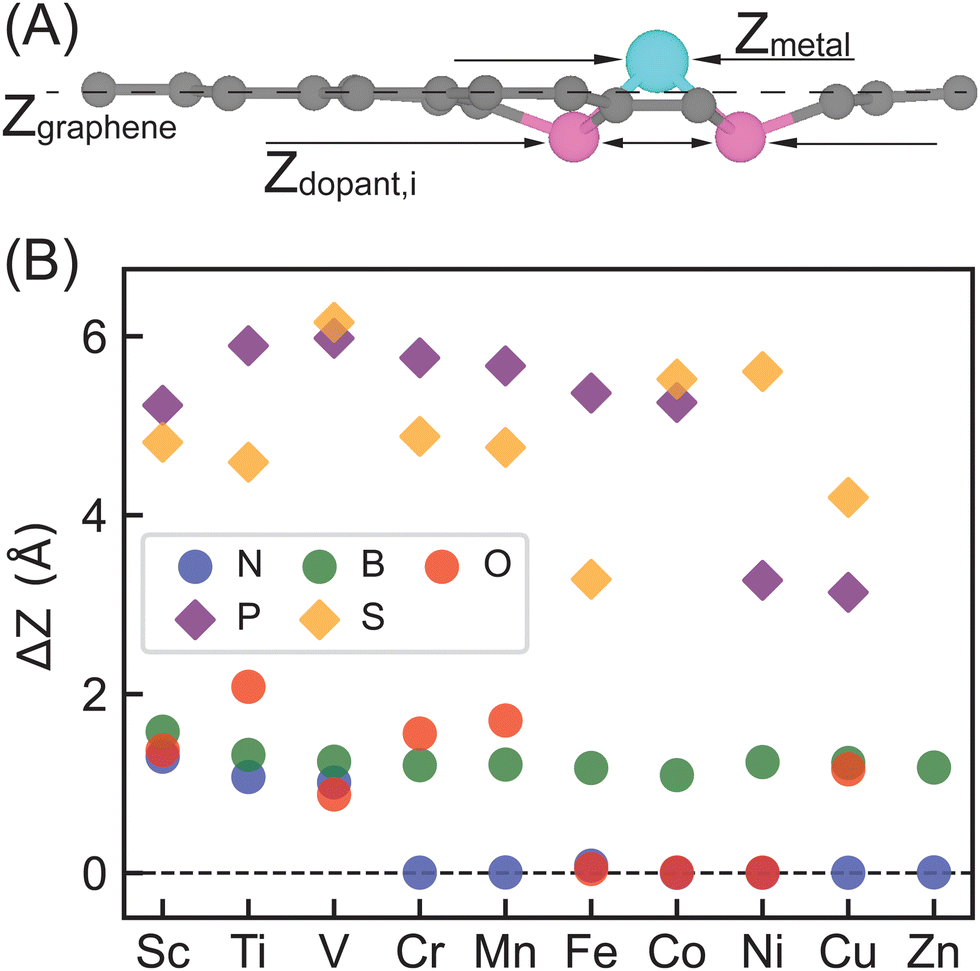 | ||
| Fig. 2 (A) A representative model showing different coordinate values in the z-direction used to quantify out-of-plane distortion of SACs. (B) Out-of-plane distortion (ΔZ) of SACs with different 3d metals and dopants (B: green circles; N: blue circles; O: red circles; P: purple diamonds; S: orange diamonds). | ||
These observations are consistent with previous studies performed on other graphene-based SACs that graphene's ability to accept a dopant is highly dependent on atomic radius of dopant elements,68 and thus dopants with similar atomic radius as carbon are more suited for retaining the 2-dimensional monolayer structure of graphene.69 Our results also indicated that 2p-coordinating systems with dopants possessing similar atomic radius to carbon favor planar configuration compared with 3p-coordinating systems with dopants possessing larger atomic radius than carbon. In addition, we generally expect longer bond distances between 3p elements with C atoms than C–C bond distances in graphene.70,71 To enable enough space for forming 3p-C bonds and 3p-metal bonds within the rigid framework of graphene, 3p dopants and metal sites move along the direction normal to the graphene monolayer to facilitate those longer bond distances. In addition, we computed complexation energy (vide supra) to assess stabilization that the doped graphene provides to the metal center on five representative SACs, including Fe–N–C, Sc–B–C, Ni–O–C, Cu–P–C and Co–S–C (Table S5, ESI†). All SACs exhibit negative complexation energies, showing strong stabilization effects that the support provides to the active metal sites. In general, SACs with 2p pyridinic dopants are likely to be more structurally stable than SACs with 3p pyridinic dopants in experimental synthesis.
CO adsorption energy as a descriptor for CO2R reactivity
CO remains the dominant product of electrochemical CO2R on most catalysts at lower applied potentials, leading to assumptions that *CO is a key intermediate toward further PCET steps of acquiring hydrocarbons and *CO reduction is likely to be the rate-limiting step.72–75 *CO binding strength has recently been proposed by Liu et al. as a descriptor for CO2R activity due to its linear correlation with reaction barriers of the *CO reduction step on multiple metallic surfaces.51 Zhong et al. further identified the optimal CO adsorption energy being −0.67 eV for CO2R to hydrocarbons on metal catalysts.52 Using this descriptor, we first narrowed down potential SACs for electrochemical CO2R within our focused chemical space, i.e., all possible combinations between 3d metals and dopants (B, N, O, P, S), by calculating CO adsorption energies on them (Fig. 3). Given the DFT errors in predicting adsorption energies,76–78 we used a range of ±0.2 eV to the optimal value of −0.67 eV (i.e., in the range of −0.87 to −0.47 eV) in determining promising candidates for electrochemical CO2R to hydrocarbons. | ||
| Fig. 3 (A) Top view and side view of CO-adsorbed SAC. CO adsorption energies of all 3d-transition metals embedded on (B) nitrogen-, (C) boron-, (D) oxygen-, (E) phosphorus-, and (F) sulfur-doped graphene. The red line indicates the optimal CO adsorption energy identified on metallic surfaces. A range of ±0.2 eV from the optimal line is shown in the shaded green region to indicate the promising candidates. Empty symbols indicate structurally unstable SACs. | ||
Nitrogen-doped SACs, particularly with mid-row or late 3d transition metals, are the most widely studied SACs for electrochemical CO2 reduction both experimentally and computationally.25,35,43 However, unfortunately, none of those materials exhibits optimal CO binding strength within ±0.2 eV from −0.67 eV (Fig. 3B), indicating that none of them are likely to reduce CO2 to hydrocarbons with reasonable efficiencies. Given the extensive investigations of such catalysts, we included Co–N–C with a CO adsorption energy of −0.92 eV, which is close to the optimal region, as well as the most widely studied Fe–N–C in our mechanistic analysis (vide infra). Overall, our DFT simulations predict strong CO binding strength for early and mid-row transition metals and very weak CO binding strength for late transition metals (Ni–N–C, Cu–N–C, and Zn–N–C), indicating that CO is readily desorbed from the surface once it is generated. This is consistent with multiple experimental observations that nitrogen-doped SACs with late 3d transition metals are efficient electrocatalysts for CO2R to CO,26,79 such as Ni–N–C being a superior electrocatalyst for CO generation with nearly 100% faradaic efficiency.31,32
While nitrogen dopants are among the most experimentally synthesized graphene-based catalysts at atomic scale, SACs coordinated with other dopants studied in this work (i.e., B, O, P, S) are mostly hypothetical materials. Screening and narrowing down those materials in silico prior to trial-and-error experimental synthesis and verification can efficiently accelerate the catalyst design process. For boron-doped SACs, we observed a clear sensitivity of CO adsorption energy on metal center d-fillings that CO binding strength becomes stronger from Sc–B–C (−0.72 eV) to Co–B–C (−1.90 eV) and then weaker from Ni–B–C (−1.87 eV) to Zn–B–C (−0.79 eV) with increasing outer shell d electrons, resulting in two potential active and selective electrocatalysts, Sc–B–C and Zn–B–C, for converting CO2 to hydrocarbons (Fig. 3C). Switching to another 2p dopant (i.e., O), unfortunately, we predicted that all of them bind CO too strongly and thus are unlikely effective electrocatalysts to reduce CO2 to hydrocarbons (Fig. 3D). *CO binding strength on 3p-coordinating (i.e., with P or S) SACs exhibits more systematic dependence on d-filling, regardless of dopant type (Fig. 3E and F). We observed more negative CO adsorption energies with increasing outer shell d electrons from Sc (−1.03 eV for Sc–P–C, −1.44 eV for Sc–S–C) to Fe (−2.21 eV for Fe–P–C, −1.95 eV for Fe–S–C), and then less negative CO adsorption energies with increasing outer shell d electrons from Fe to Ni (−0.46 eV for Ni–P–C, −0.62 eV for Ni–S–C). The only exception is Cu SACs (−0.67 eV for Cu–P–C, −1.63 eV for Cu–S–C) showing stronger CO binding strength compared with Ni SACs. Unfortunately, we cannot find stable structures for Zn–P–C, Zn–S–C, and Zn–O–C. Overall, we further identified four 3p-coordinating SACs with optimal CO binding strength, namely Ni–P–C, Cu–P–C, Co–S–C, and Ni–S–C, in our following mechanistic studies.
Comparing the effect of tuning dopants on CO binding strength, we observed that the sensitivity is dependent on d-filling of metal centers (Fig. S4, ESI†). In general, early and mid-row transition metals (Sc, Ti, V, Cr, Mn, and Fe) exhibit a moderate range of CO adsorption energies when changing dopants, while late transition metals (Co, Ni, and Cu) show a larger range of CO adsorption energies. However, there is no systematic trend which dopant results in consistently strong or weak CO binding strength across all 3d transition metals. Motivated by the effects that Ni and Cu SACs have weak CO binding strength with N dopants and moderate or strong CO binding strength with other dopants, we propose a strategy of combining different dopants via heteroatom doping to tune binding energy of key adsorbates for SAC design.
For the widely studied nitrogen-doped SACs which mostly preserve planar geometries (Fig. 2), the natural approach to simulate the geometry is allowing full relaxation of atomic positions in geometry optimizations.47,60 However, for SACs with other dopants which exhibit out-of-plane distortions, a constrained geometry optimization of restricting the movement of atoms in the direction normal to the graphene plane has been used.66 Here, we benchmarked the accuracy of using this approach for SAC study. We constrained the movement of each atom within the graphene plane by fixing the coordinates in z-direction (i.e., direction normal to the graphene plane) in geometry optimizations for both clean and CO-adsorbed surfaces when predicting CO adsorption energies (Fig. S5, ESI†). Overall, constrained optimizations predict less negative CO adsorption energies for most SACs with several exceptions including early transition metals embedded in boron- and phosphorus-doped systems. There are larger variations in CO adsorption energy predictions between performing full and constrained geometry optimizations, ranging from −2.73 to 0.81 eV. Given the possible large discrepancies in energetics predictions, we highlight the importance of performing full relaxation when simulating SACs to incorporate the structural effects.
Reaction mechanisms of CO2R to methane and methanol on 2p-coordinating SACs
To further assess the reactivity of identified SAC candidates using CO adsorption energy as a descriptor (vide supra), we elucidated full reaction mechanisms of CO2R to hydrocarbons. We considered eight SACs with varying metal centers and dopants in our mechanistic analysis, including Sc–B–C, Zn–B–C, Ni–P–C, Cu–P–C, Co–S–C, and Ni–S–C with optimal CO adsorption energy, as well as Co–N–C with CO adsorption energy close to optimal value and the most widely studied Fe–N–C. Given the large interatomic distances between active metal centers within SACs, C–C coupling steps toward forming C2+ products are extremely challenging,36,80 and thus here we considered C1 hydrocarbon products, i.e., methane and methanol formation. Typically, electrochemical CO2R proceeds via *CO reduction to either *COH or *CHO, resulting in divergent reaction pathways following these two intermediates.14,19 We focused on five possible reaction pathways towards methane formation, i.e., CO2 → *COOH → *CO → *COH or *CHO → *C or *CHOH → *CH or *CH2OH → *CH2 → *CH3 → CH4, and two reaction pathways toward methanol formation, i.e., CO2 → *COOH → *CO → *COH or *CHO → *CHOH → *CH2OH → CH3OH (Fig. 4). Unlike previous studies that only considered atop adsorption site on metal centers for intermediates,37,48,81 we examined three possible adsorption sites: atop, bridge, and hollow around metal centers for key intermediates (Table S6, ESI†). Surprisingly, the most favorable adsorption site highly depends on both type of intermediates and metal-coordinating environments of SACs (Table S7, ESI†). Atop site is not consistently predicted to be the most favorable adsorption site for many adsorbates, indicating a complete screening of all possible adsorption sites is crucial in mechanistic study of graphene-based single atom catalysts.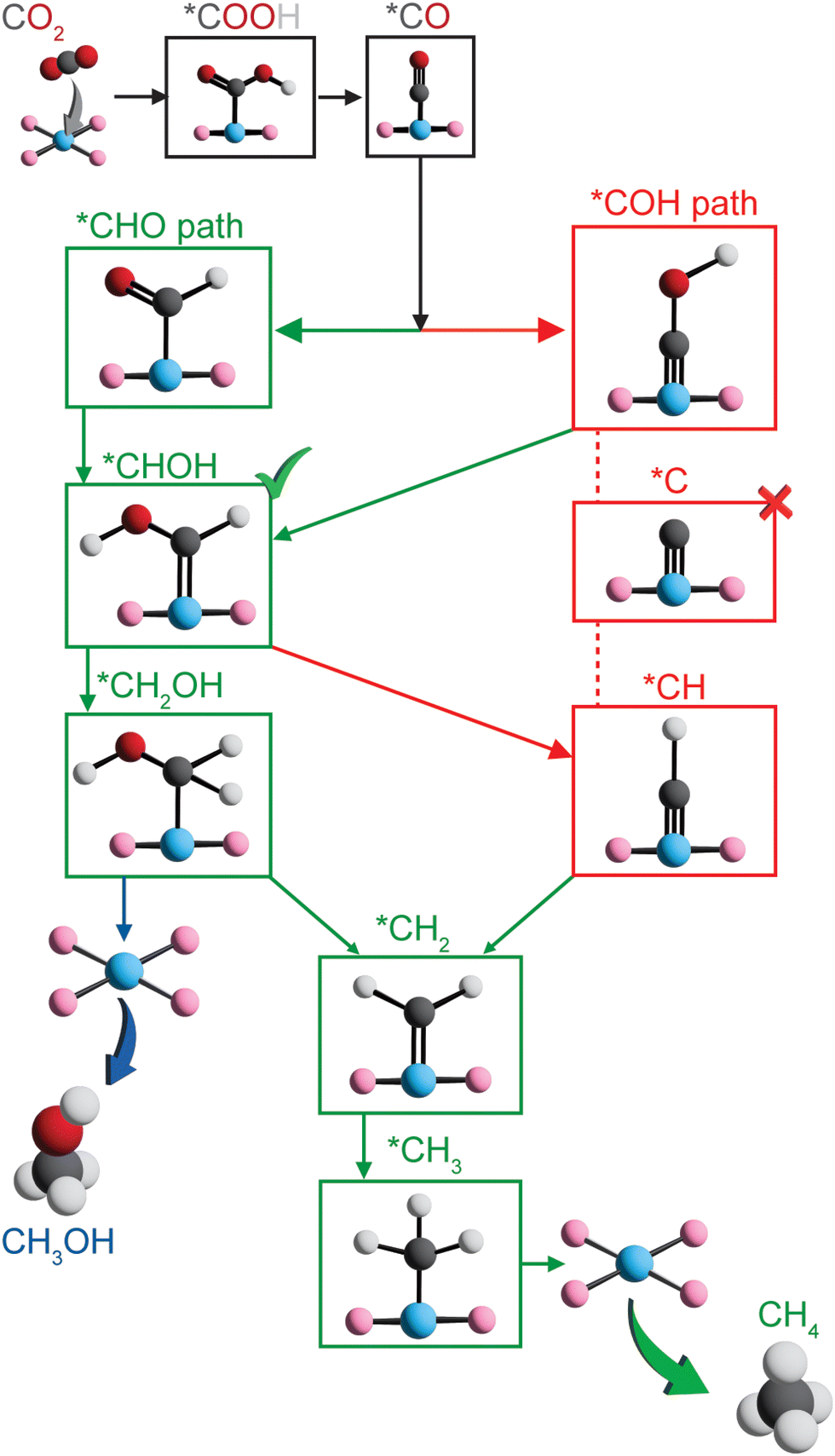 | ||
| Fig. 4 Possible reaction pathways considered for electrochemical CO2R toward methane and methanol formation on graphene-based SACs. The green path denotes the most favorable reaction pathway for most SACs except for Zn–B–C. | ||
We first considered reaction mechanisms of CO2R to methane and methanol on Zn–B–C (Fig. 5A). The initial PECT step of CO2 to *COOH is energetically favorable with a reaction energy of −0.28 eV (Table S8, ESI†). Subsequent PCET step from *COOH to *CO and one water molecule is predicted to have a slightly positive reaction energy of 0.24 eV. Interestingly, we observed thermodynamically favored *CO reduction to form both *CHO and *COH with reaction energies of −0.04 eV and −0.15 eV, respectively. The small quantitative energy difference, i.e., ∼0.1 eV, indicates competitive formation and thus co-existence of *CHO and *COH as key CO2R intermediates on Zn–B–C. Considering the next PCET step, we obtained a high reaction energy of 2.86 eV for *COH reduction to form *C and one water molecule (Fig. S6 and Table S8, ESI†), indicating the infeasibility of this pathway, whereas subsequent PCET steps of both *CHO and *COH to form *CHOH are more favorable with reaction energies of 0.02 eV and 0.13 eV, respectively. Selectivity toward methane and methanol is dependent on the step of *CHOH reduction. Methanol formation proceeds through *CH2OH intermediate with a reaction energy of −1.12 eV and the final reduction step with a reaction energy of zero. Methane is more thermodynamically favorable with a lower reaction energy for *CHOH reduction to *CH (−1.32 eV), but a higher reaction energy for subsequent reduction to *CH2 (0.45 eV), following by favorable further reduction to form *CH3 (−1.47 eV) and CH4 (−0.05 eV).
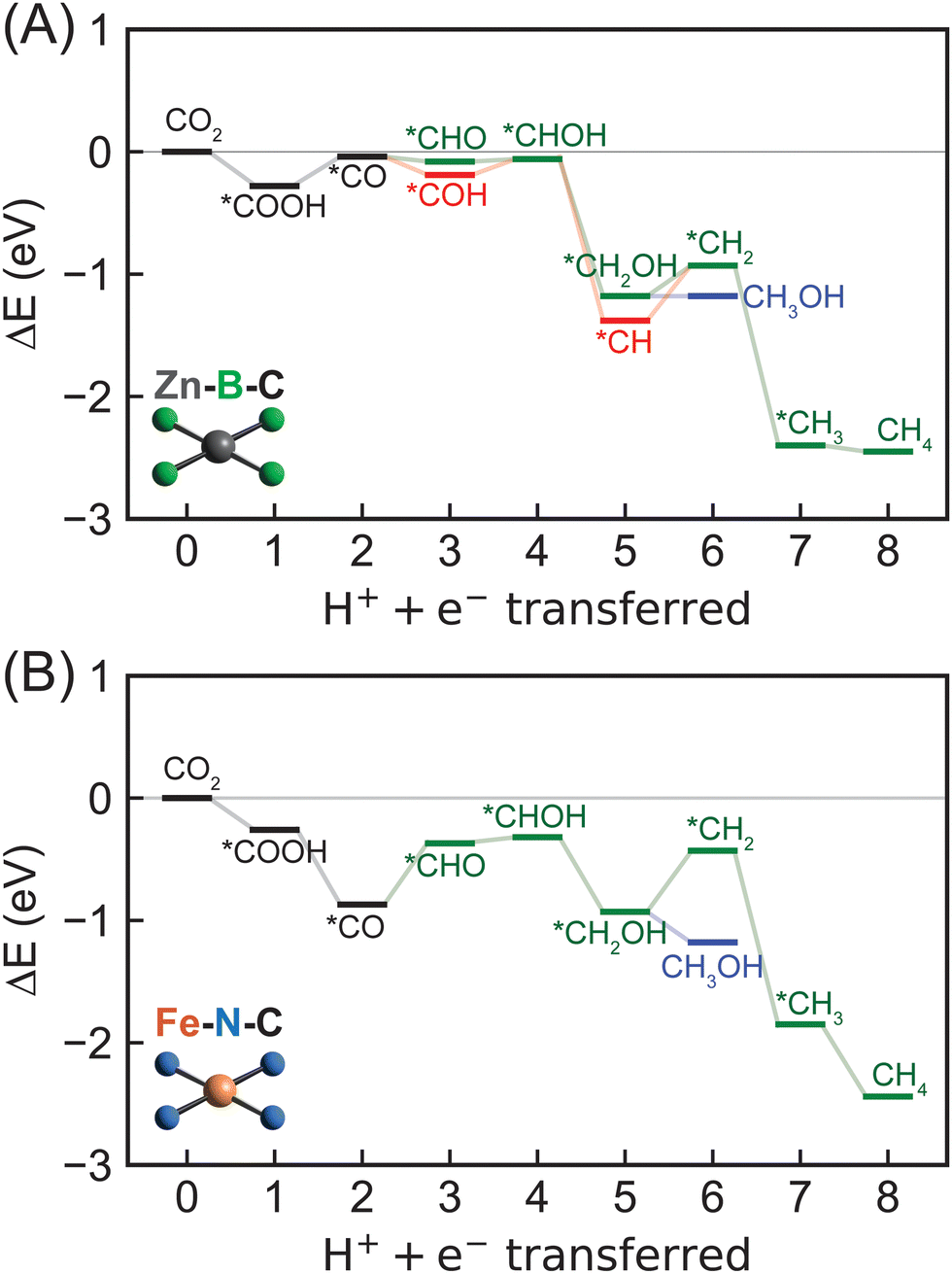 | ||
| Fig. 5 Energetics of CO2R to methane via eight PCET steps and methanol via six PCET steps on (A) Zn–B–C and (B) Fe–N–C SACs predicted by DFT-PBE-D3. The green lines show the most favorable pathway toward methane or methanol formation through a *CHO intermediate identified on most SACs except for Zn–B–C. The red lines denote other favorable pathways on Zn–B–C. Blue lines indicate formation of methanol. | ||
Overall, we found Zn–B–C SAC to be a promising electrocatalyst for CO2R toward generating both methane and methanol. The most favorable reaction pathway proceeds via CO2 → *COOH → *CO → *COH → *CHOH → *CH → *CH2 → *CH3 → CH4 with *CH → *CH2 (0.45 eV) as the potential rate-limiting step for methane generation. Methanol formation is also possible through CO2 → *COOH → *CO → *COH → *CHOH → *CH2OH → CH3OH with *COOH → *CO (0.24 eV) as the potential rate-limiting step. Therefore, Zn–B–C SAC requires very low applied potentials, i.e., −0.45 V vs. the RHE and −0.24 eV vs. the RHE, for electrochemical CO2R to generate methane and methanol, respectively. In addition, the identified rate-limiting steps on Zn–B–C SAC differ from conventional understanding of *CO reduction step reported previously on metallic surface,51 casting doubt whether *CO adsorption energy is an effective descriptor to screen SACs as electrocatalysts for CO2R. To provide a more complete picture of catalytic performance of Zn–B–C SAC, we also investigated its activity for HER. We predicted that the potential rate-limiting step of HER on Zn–B–C is the Heyrovsky step with a reaction energy of 0.33 eV, which is only slightly lower than the reaction energy of 0.45 eV for the rate-limiting step of CO2R to methane (Fig. S7, ESI†). Both reaction energies are easily surmountable with applied potentials, indicating competing CO2R and HER on Zn–B–C.
Switching to the other boron-doped SAC, Sc–B–C, we observed consistent negative and positive reaction energies for the first two CO2R PCET steps as on Zn–B–C, but with much larger values (−1.00 eV for CO2 reduction to *COOH and 1.03 eV for *COOH reduction to *CO; Fig. S6, S8 and Table S8, ESI†). However, CO reduction to *CHO is favored over *COH formation by 0.89 eV (−0.52 eV for *CHO formation, 0.37 eV for *COH formation), divergent from the competing steps of *CHO and *COH formation predicted on Zn–B–C. Following *CHO formation, subsequent PCET steps involve *CHOH formation (−0.03 eV), *CH2OH formation (−0.86 eV), leading to methanol (0.20 eV), or *CH2 formation (0.08 eV), *CH3 formation (−0.88 eV), resulting in methane (−0.26 eV), and thus methane is slightly more favorable than methanol on Sc–B–C. To summarize, methane proceeds via CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → *CH2 → *CH3 → CH4 and methanol proceeds via CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → CH3OH. The rate-limiting step is *COOH reduction to *CO with a high reaction energy of 1.03 eV, indicating Sc–B–C might not be an active electrocatalyst for CO2R.
For nitrogen-doped SACs, unfortunately, none of them possesses a CO adsorption energy close to the optimal value of −0.67 eV. However, to understand how CO2 can be electrochemically reduced to hydrocarbons catalyzed by those most widely studied graphene-based SACs, we included Fe–N–C and Co–N–C in our mechanistic analysis. Both methane and methanol production on Fe–N–C follow the same reaction pathways as on Sc–B–C, but with several different trends in energetics, including reaction energy of *COOH reduction to *CO is negative (−0.61 eV) and reaction energy of *CO reduction to *CHO reduction (0.50 eV) is positive (Fig. 5B and Fig. S6, Table S8, ESI†). In addition, the reduction of *CH2OH to CH3OH (−0.25 eV) is more thermodynamically favorable than to *CH2 (0.50 eV), indicating preferred methanol formation on Fe–N–C with a rate-limiting step of *CO reduction to *CHO. The required applied potential for methanol generation on Fe–N–C is predicted to be −0.50 V vs. the RHE, suggesting that it might be an active electrocatalyst for CO2R to hydrocarbons. However, previous experiments82–84 suggested that Fe–N–C can only electrochemically convert CO2 to CO with reasonable efficiencies. This is because *CO readily desorbs from the surface upon generation with a very weak *CO adsorption energy, though the required applied potentials are predicted to be low enough for methanol generation. Therefore, tuning *CO adsorption energy represents an effective strategy to strengthen the activity of Fe–N–C for CO2R to hydrocarbons. For Co–N–C, CO2R to methanol follows the same reaction pathway as Sc–B–C and Fe–N–C, but with a different rate-limiting step of *CHO reduction to *CHOH with a predicted high reaction energy of 0.98 eV (Fig. S6, S8 and Table S8, ESI†). Unfortunately, we were not able to locate stable structures of *CH2 adsorbed on Co–N–C, indicating that methane formation might not be possible on Co–N–C. Our DFT predictions are consistent with experiments33,85 that Co–N–C can only electrochemically reduce CO2 to CO. In addition, comparing reactivity of Fe–N–C and Co–N–C for CO2 to CO conversion, we predicted a much lower *COOH to *CO reaction energy for Fe–N–C (−0.61 eV) than for Co–N–C (0.22 eV) (Fig. S6, S8 and Table S8, ESI†), consistent with previous experiment86 showing that Fe–N–C is more active than Co–N–C for this chemistry.
To summarize, we predicted that Zn–B–C and Fe–N–C might be active electrocatalysts for CO2R to methane and methanol, respectively, given their surmountable energies at moderate applied potentials. However, we observed both metal- and dopant-dependent CO2R mechanistic behaviors on 2p-coordinating SACs (Table 1). Zn–B–C favors *CO reduction to *COH, whereas the other three SACs (i.e., Sc–B–C, Fe–N–C, and Co–N–C) proceeds via *CO reduction to *CHO toward methane or methanol formation. In general, methane formation is more favorable on boron-doped SACs, while methanol generation is more preferred on nitrogen-doped SACs. Unfortunately, rate-limiting steps are fully system-dependent on 2p-coordinating SACs, indicating *CO binding strength might not be an effective descriptor for CO2R reactivity since this proposed descriptor assumes that *CO reduction is the rate-limiting step.
| Product | Rate-limiting step | Reaction energy | |
|---|---|---|---|
| Zn–B–C | Methane | *CH + H+ + e− → *CH2 | 0.45 |
| Sc–B–C | Methane | *COOH + H+ + e− → *CO + H2O | 1.03 |
| Fe–N–C | Methanol | *CO + H+ + e− → *CHO | 0.50 |
| Co–N–C | Methanol | *CHO + H+ + e− → *CHOH | 0.98 |
| Cu–P–C | Methanol | *CO + H+ + e− → *CHO | 0.56 |
| Ni–P–C | Methanol | CO2 + H+ + e− → *COOH | 0.74 |
| Ni–S–C | Methanol | CO2 + H+ + e− → *COOH | 1.02 |
| Co–S–C | Methanol | CO2 + H+ + e− → *COOH | 1.37 |
Reaction mechanisms of CO2R to methane and methanol on 3p-coordinating SACs
Moving from 2p-coordinating SACs to 3p-coordinating SACs, we next considered Cu–P–C (Fig. 6A), which has a CO adsorption energy (−0.67 eV) close to the optimal value (Fig. 3). Consistent with most 2p-coordinating SACs, Cu–P–C favors reaction pathways of CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → *CH2 → *CH3 → CH4 and CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → CH3OH toward methane and methanol formation, respectively (Fig. S5, ESI†). Due to the large reaction energy difference (0.65 eV) between *CH2OH reduction to form *CH2 (−0.49 eV) and methanol (−1.14 eV; Table S8, ESI†), Cu–P–C is predicted to exhibit a strong preference towards methanol formation with an early rate-limiting step of *CO reduction to *CHO (Table 1). To overcome the reaction energy (0.56 eV) of such step, an applied potential of −0.56 V vs. the RHE is needed for methanol generation on Cu–P–C, indicating an active electrocatalyst for CO2R to hydrocarbons. Moving to the other phosphorous-doped SAC (Ni–P–C) with promising CO binding strength, we observed same reaction pathways for CO2R to methane and methanol with a same preference for methanol formation (Fig. S6 and S8, ESI†). However, the rate-limiting step changes from *CO reduction to the very first PCET step of CO2 reduction to *COOH with a reaction energy of 0.74 eV (Table 1 and Table S8, ESI†). We have identified two sulfur-doped SACs with promising CO binding strength, namely Ni–S–C and Co–S–C (Fig. 3). Their mechanistic behaviors as electrocatalysts for CO2R are similar to Ni–P–C (Fig. 6B and Fig. S6, S8, ESI†). Unfortunately, the reaction energies of the rate-limiting step, CO2 reduction to *COOH, are starkly high on both Ni–S–C (1.02 eV) and Co–S–C (1.37 eV; Table 1), indicating their low activities to activate CO2.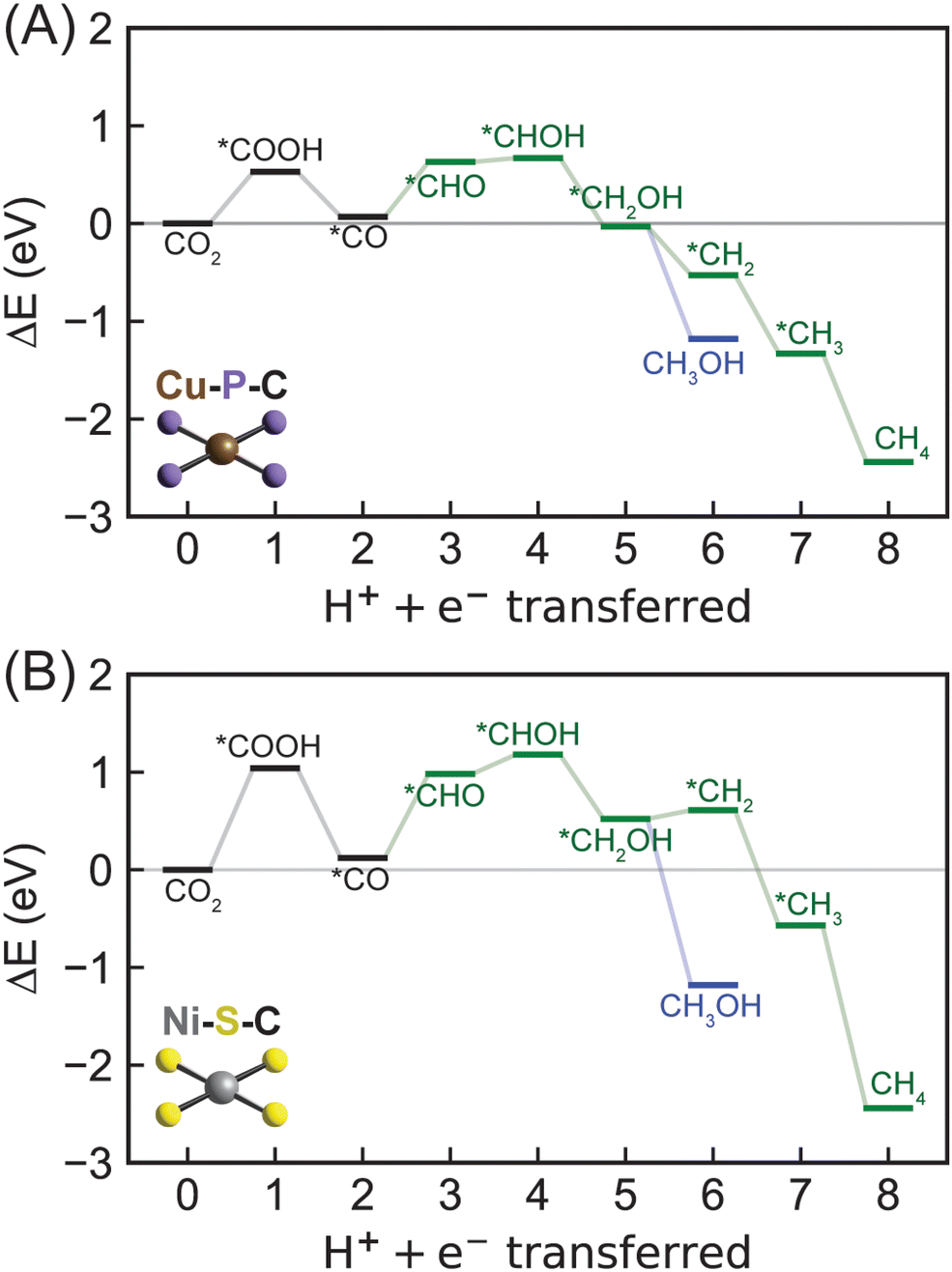 | ||
| Fig. 6 Energetics of CO2R to methane via eight PCET steps and methanol via six PCET steps on (A) Cu–P–C and (B) Ni–S–C SACs predicted by DFT-PBE-D3. The green lines show the most favorable pathway toward methane or methanol formation through a *CHO intermediate identified on most SACs except for Zn–B–C. Blue lines indicate formation of methanol. | ||
Overall, all 3p-coordinating SACs favor methanol formation over methane, yet Cu–P–C is the only one with surmountable energies at moderate applied potentials. 3p-coordinating SACs exhibit early rate-limiting steps, i.e., *CO reduction for Cu–P–C and CO2 reduction for Ni–P–C, Ni–S–C, and Co–S–C. This prediction is consistent with previous computational work showing that CO2 adsorption and reduction is critical in electrochemical CO2R.37,45 In addition, we observed relatively high reaction energies for the very first PCET steps, especially on sulfur-doped SACs, indicating challenges in activating CO2 even towards generating CO, which is different from 2p-coordinating SACs and conventional metal surfaces. This discrepancy in mechanistic behavior between SACs and metal catalysts again suggests the inefficiency of using CO binding strength as a descriptor for CO2R reactivity.
IV. Summary and conclusions
Motivated by recent experimental success in using nitrogen-doped SACs for electrochemical CO2R to generate CO and enhancing subsequent proton-coupled electron transfer steps using other dopants, we applied computational modeling with DFT to enable a systematic understanding of the roles of metal-coordination environment in tuning SAC activity for CO2 conversion. We screened a large chemical space containing all possible combinations of 10 active metals (i.e., 3d transition metals) and five dopants (i.e., B, N, O, P, and S). We first investigated their structural stabilities and noted dopant-dependent rigidities that 2p-coordinating SACs preserve planar geometries or at most display a minor degree of out-of-plane distortion in the metal center, while 3p-coordinating SACs exhibit severe out-of-plane distortion among the metal-coordination region. We then used *CO binding strength, which was previously identified on metal catalysts as a descriptor for CO2R activity, to narrow down the potential SAC candidates for converting CO2 to hydrocarbons. We identified six hypothetical SACs that might be efficient electrocatalysts for CO2R, namely Sc–B–C, Zn–B–C, Ni–P–C, Cu–P–C, Co–S–C, and Ni–S–C.To further assess their activity, we elucidated reaction mechanisms of CO2R to methane and methanol on the identified SACs. Two nitrogen-doped SACs (Co–N–C and Fe–N–C) were also included in the mechanistic analysis to understand dopant effects. We observed consistent preferred reaction pathways for methane and methanol formation proceeding via CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → *CH2 → *CH3 → CH4 and CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → CH3OH on most SACs except for Zn–B–C. Surprisingly, unlike metallic surfaces, the predicted rate-limiting steps on SACs are system-dependent and vary with both dopants and metals, challenging the development of efficient design principles for SACs. In addition, it indicates infeasibility of using CO adsorption energy as a descriptor for CO2R reactivity since *CO reduction is not a consistent rate-limiting step on SACs. Regarding CO2R selectivity, boron-doped SACs favor methane formation, while SACs with other dopants (i.e., N, P, and S) prefer methanol formation. Overall, we identified Zn–B–C as a promising electrocatalyst for CO2R to methane, as well as Fe–N–C and Cu–P–C for CO2R to methanol due to their surmountable energies at moderate applied potentials. We believe our systematic studies reveal fundamental insights into the role of dopants and metals in tuning SAC reactivity and inspire new design principles for CO2R electrocatalysts. Moving forward, we are extending full mechanistic analyses of CO2R to valuable hydrocarbons on graphene-based SACs from selected 3d transition metals to all 3d–5d transition metals, with the ultimate goal of developing new and effective descriptors to guide activity and selectivity.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge support by the National Science Foundation under Award No. 2349619, by the Advanced Research Projects Agency-Energy (ARPA-E), U.S. Department of Energy, under Award Number DE-AR0001786, and by Northeastern University, Chemical Engineering Department under start-up funding. The views and options of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. This work was carried out using computational resources from Discovery, Research Computing at Northeastern University.References
- T. R. Karl and K. E. Trenberth, Modern Global Climate Change, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- A. Terando, D. Reidmiller, S. W. Hostetler, J. S. Littell, T. D. Beard Jr., S. R. Weiskopf, J. Belnap and G. S. Plumlee, Using Information from Global Climate Models to Inform Policymaking—The Role of the U.S. Geological Survey, Open-File Rep., 2020, 1–32 Search PubMed.
- L. E. Doman, V. Arora, L. E. Singer, V. Zaretskaya, A. Jones, T. Huetteman, M. Bowman, N. Slater-Thompson, B. Hojjati, D. Peterson, et al., International Energy Outlook 2016, U.S. Energy Inf. Adm., 2016, pp. 7–17.
- BP, BP Energy Outlook, 2017, pp. 12–21.
- D. Panepinto, V. A. Riggio and M. Zanetti, Analysis of the Emergent Climate Change Mitigation Technologies, Int. J. Environ. Res. Public Health, 2021, 18, 6767 CrossRef CAS PubMed.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Sustainable Hydrocarbon Fuels by Recycling CO2 and H2O with Renewable or Nuclear Energy, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu and Y. Yan, et al., Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS.
- C. E. Creissen and M. Fontecave, Solar-Driven Electrochemical CO2 Reduction with Heterogeneous Catalysts, Adv. Energy Mater., 2020, 11, 2002652 CrossRef.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, Selective Formation of C2 Compounds from Electrochemical Reduction of CO2 at a Series of Copper Single Crystal Electrodes, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- C. H. Lee and M. W. Kanan, Controlling H+vs. CO2 Reduction Selectivity on Pb Electrodes, ACS Catal., 2014, 5, 465–469 CrossRef.
- H. Mistry, Y. W. Choi, A. Bagger, F. Scholten, C. S. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon and K. Kisslinger, et al., Enhanced Carbon Dioxide Electroreduction to Carbon Monoxide over Defect-Rich Plasma-Activated Silver Catalysts, Angew. Chem., Int. Ed., 2017, 56, 11394–11398 CrossRef CAS PubMed.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, Ag-Sn Bimetallic Catalyst with a Core-Shell Structure for CO2 Reduction, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan and C. Hahn, et al., Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Q. Zhao and E. A. Carter, Revisiting Competing Paths in Electrochemical CO2 Reduction on Copper via Embedded Correlated Wavefunction Theory, J. Chem. Theory Comput., 2020, 16, 6528–6538 CrossRef CAS.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, Charting C–C Coupling Pathways in Electrochemical CO2 Reduction on Cu(111) using Embedded Correlated Wavefunction Theory, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202931119 CrossRef CAS.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, Electrochemical Hydrogenation of CO on Cu(100): Insights from Accurate Multiconfigurational Wavefunction Methods, J. Phys. Chem. Lett., 2022, 13, 10282–10290 CrossRef CAS PubMed.
- J. Cai, Q. Zhao, W.-Y. Hsu, C. Choi, Y. Liu, J. M. P. Martirez, C. Chen, J. Huang, E. A. Carter and Y. Huang, Highly Selective Electrochemical Reduction of CO2 into Methane on Nanotwinned Cu, J. Am. Chem. Soc., 2023, 145, 9136–9143 CrossRef CAS.
- S. Xu and E. A. Carter, Theoretical Insights into Heterogeneous (Photo)electrochemical CO2 Reduction, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, Revisiting Understanding of Electrochemical CO2 Reduction on Cu(111): Competing Proton-Coupled Electron Transfer Reaction Mechanisms Revealed by Embedded Correlated Wavefunction Theory, J. Am. Chem. Soc., 2021, 143, 6152–6164 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Heterogeneous Single-Atom Catalysts for Electrochemical CO2 Reduction Reaction, Adv. Mater., 2020, 32, 2001848 CrossRef CAS.
- C. Gallagher, W. Siddiqui, T. Arnold, C. Cheng, E. Su and Q. Zhao, Benchmarking a Molecular Flake Model on the Road to Programmable Graphene-Based Single-Atom Catalysts, J. Phys. Chem. C, 2024, 128, 2876–2883 CrossRef CAS.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Single Site Porphyrine-Like Structures Advantages over Metals for Selective Electrochemical CO2 Reduction, Catal. Today, 2017, 288, 74–78 CrossRef CAS.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Identification of Champion Transition Metals Centers in Metal and Nitrogen-Codoped Carbon Catalysts for CO2 Reduction, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- H. Cheng, X. Wu, X. Li, X. Nie, S. Fan, M. Feng, Z. Fan, M. Tan, Y. Chen and G. He, Construction of Atomically Dispersed Cu-N4 Sites via Engineered Coordination Environment for High-Efficient CO2 Electroreduction, Chem. Eng. J., 2021, 407, 126842 CrossRef CAS.
- P. Wang, Z. Jin, P. Li and G. Yu, Design Principles of Hydrogen-Evolution-Suppressing Single-Atom Catalysts for Aqueous Electrosynthesis, Chem. Catal., 2022, 2, 1277–1287 CrossRef CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Modulating the Local Coordination Environment of Single-Atom Catalysts for Enhanced Catalytic Performance, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- L. T. Menisa, P. Cheng, X. Qiu, Y. Zheng, X. Huang, Y. Gao and Z. Tang, Single Atomic Fe–N4 Active Sites and Neighboring Graphitic Nitrogen for Efficient and Stable Electrochemical CO2 Reduction, Nanoscale Horiz., 2022, 7, 916–923 RSC.
- G. Qu, K. Wei, K. Pan, J. Qin, J. Lv, J. Li and P. Ning, Emerging Materials for Electrochemical CO2 Reduction: Progress and Optimization Strategies of Carbon-Based Single-Atom Catalysts, Nanoscale, 2023, 15, 3666–3692 RSC.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Large-Scale and Highly Selective CO2 Electrocatalytic Reduction on Nickel Single-Atom Catalyst, Joule, 2019, 3, 265–278 CrossRef CAS.
- W. Zheng, F. Chen, Q. Zeng, Z. Li, B. Yang, L. Lei, Q. Zhang, F. He, X. Wu and Y. Hou, A Universal Principle to Accurately Synthesize Atomically Dispersed Metal-N4 Sites for CO2 Electroreduction, Nano-Micro Lett., 2020, 12, 108 CrossRef CAS PubMed.
- C. Wang, Y. Liu, H. Ren, Q. Guan, S. Chou and W. Li, Diminishing the Uncoordinated N Species in Co–N–C Catalysts toward Highly Efficient Electrochemical CO2 Reduction, ACS Catal., 2022, 12, 2513–2521 CrossRef CAS.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, Scalable Production of Efficient Single-Atom Copper Decorated Carbon Membranes for CO2 Electroreduction to Methanol, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin and G. Qi, et al., Stable and Efficient Single-Atom Zn Catalyst for CO2 Reduction to CH4, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian and X. Xu, et al., Boosting CO2 Electroreduction to CH4 via Tuning Neighboring Single-Copper Sites, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- Y. Cai, J. Fu, Y. Zhou, Y. C. Chang, Q. Min, J. J. Zhu, Y. Lin and W. Zhu, Insights on Forming N,O-Coordinated Cu Single-Atom Catalysts for Electrochemical Reduction CO2 to Methane, Nat. Commun., 2021, 12, 586 CrossRef CAS PubMed.
- Y. Dai, H. Li, C. Wang, W. Xue, M. Zhang, D. Zhao, J. Xue, J. Li, L. Luo and C. Liu, et al., Manipulating Local Coordination of Copper Single Atom Catalyst Enables Efficient CO2-to-CH4 Conversion, Nat. Commun., 2023, 14, 3382 CrossRef CAS PubMed.
- Z. Wei, Y. Liu, J. Ding, Q. He, Q. Zhang and Y. Zhai, Promoting Electrocatalytic CO2 Reduction to CO via Sulfur-Doped Co–N–C Single-Atom Catalyst, Chin. J. Chem., 2023, 41, 3553–3559 CrossRef CAS.
- K. S. Adarsh, N. Chandrasekaran and V. Chakrapani, In situ Spectroscopic Techniques as Critical Evaluation Tools for Electrochemical Carbon dioxide Reduction: A Mini Review, Front. Chem., 2020, 8, 137 CrossRef CAS PubMed.
- X. Zhao, J. Shi, Y. Ji and Y. Liu, The Electronic Structure Underlying Electrocatalysts of Two-Dimensional Materials, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1418 CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Norskov, Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Understanding Activity and Selectivity of Metal-Nitrogen-Doped Carbon Catalysts for Electrochemical Reduction of CO2, Nat. Commun., 2017, 8, 944 CrossRef.
- X. Zhao and Y. Liu, Unveiling the Active Structure of Single Nickel Atom Catalysis: Critical Roles of Charge Capacity and Hydrogen Bonding, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS.
- S. Vijay, W. Ju, S. Brückner, S.-C. Tsang, P. Strasser and K. Chan, Unified Mechanistic Understanding of CO2 Reduction to CO on Transition Metal and Single Atom Catalysts, Nat. Catal., 2021, 4, 1024–1031 CrossRef CAS.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Selective Electroreduction of CO2 to Acetone by Single Copper Atoms Anchored on N-Doped Porous Carbon, Nat. Commun., 2020, 11, 2455 CrossRef CAS.
- Y. Wang, L. You and K. Zhou, Origin of the N-coordinated single-atom Ni sites in heterogeneous electrocatalysts for CO2 reduction reaction, Chem. Sci., 2021, 12, 14065–14073 RSC.
- J. Wang, M. Zheng, X. Zhao and W. Fan, Structure-Performance Descriptors and the Role of the Axial Oxygen Atom on M–N4–C Single-Atom Catalysts for Electrochemical CO2 Reduction, ACS Catal., 2022, 12, 5441–5454 CrossRef CAS.
- P. Hou, Y. Huang, F. Ma, X. Wei, R. Du, G. Zhu, J. Zhang and M. Wang, S and N Coordinated Single-Atom Catalysts for Electrochemical CO2 Reduction with Superior Activity and Selectivity, Appl. Surf. Sci., 2023, 619, 156747 CrossRef CAS.
- X. Wei, S. Cao, S. Wei, S. Liu, Z. Wang, F. Dai and X. Lu, Theoretical Investigation on Electrocatalytic Reduction of CO2 to Methanol and Methane by Bimetallic Atoms TM1/TM2-N@Gra (TM = Fe, Co, Ni, Cu), Appl. Surf. Sci., 2022, 593, 153377 CrossRef CAS.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Understanding Trends in Electrochemical Carbon Dioxide Reduction Rates, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, et al., Accelerated Discovery of CO2 Electrocatalysts Using Active Machine Learning, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. D. Becke and E. R. Johnson, A Density-Functional Model of the Dispersion Interaction, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- S. Vijay, J. A. Gauthier, H. H. Heenen, V. J. Bukas, H. H. Kristoffersen and K. Chan, Dipole-Field Interactions Determine the CO2 Reduction Activity of 2D Fe–N–C Single-Atom Catalysts, ACS Catal., 2020, 10, 7826–7835 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- M. Methfessel and A. T. Paxton, High-Precision Sampling for Brillouin-Zone Integration in Metals, Phys. Rev. B:Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS.
- G. Makov and M. C. Payne, Periodic Boundary Conditions in ab initio Calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Revisiting the Universal Principle for the Rational Design of Single-Atom Electrocatalysts, Nat. Catal., 2024, 7, 207–218 CrossRef CAS.
- C. Y. Lin, L. Zhang, Z. Zhao and Z. Xia, Design Principles for Covalent Organic Frameworks as Efficient Electrocatalysts in Clean Energy Conversion and Green Oxidizer Production, Adv. Mater., 2017, 29, 1606635 CrossRef PubMed.
- H. Jia, A. Nandy, M. Liu and H. J. Kulik, Modeling the Roles of Rigidity and Dopants in Single-Atom Methane-to-Methanol Catalysts, J. Mater. Chem. A, 2022, 10, 6193–6203 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- I. H. Kim, J. Lim and S. O. Kim, Discovery of Single-Atom Catalyst: Customized Heteroelement Dopants on Graphene, Acc. Mater. Res., 2021, 2, 394–406 CrossRef CAS.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, Sulfur-Doped Graphene as an Efficient Metal-free Cathode Catalyst for Oxygen Reduction, ACS Nano, 2012, 6, 205–211 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of Bond Lengths Determined by X-ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- D. M. Chen, P. M. Shenai and Y. Zhao, Tight Binding Description on the Band Gap Opening of Pyrene-Dispersed Graphene, Phys. Chem. Chem. Phys., 2011, 13, 1515–1520 RSC.
- A. A. Peterson and J. K. Nørskov, Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, A New Mechanism for the Selectivity to C1 and C2 Species in the Electrochemical Reduction of Carbon Dioxide on Copper Electrodes, Chem. Sci., 2011, 2, 1902 RSC.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, Reaction Mechanisms of CO2 Electrochemical Reduction on Cu(111) Determined with Density Functional Theory, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- Y. Hori, I. Takahashi, Y. Yoshinami and A. Murata, Electrochemical Reduction of CO at a Copper Electrode, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- J. P. Janet, Q. Zhao, E. I. Ioannidis and H. J. Kulik, Density Functional Theory for Modelling Large Molecular Adsorbate–Surface Interactions: A Mini-Review and Worked Example, Mol. Simul., 2016, 43, 327–345 CrossRef.
- Q. Zhao and H. J. Kulik, Where Does the Density Localize in the Solid State? Divergent Behavior for Hybrids and DFT+U, J. Chem. Theory Comput., 2018, 14, 670–683 CrossRef CAS PubMed.
- Q. Zhao and H. J. Kulik, Stable Surfaces That Bind Too Tightly: Can Range-Separated Hybrids or DFT+U Improve Paradoxical Descriptions of Surface Chemistry?, J. Phys. Chem. Lett., 2019, 10, 5090–5098 CrossRef CAS PubMed.
- J. Chen, Z. Li, X. Wang, X. Sang, S. Zheng, S. Liu, B. Yang, Q. Zhang, L. Lei and L. Dai, et al., Promoting CO2 Electroreduction Kinetics on Atomically Dispersed Monovalent ZnI Sites by Rationally Engineering Proton-Feeding Centers, Angew. Chem., Int. Ed., 2022, 61, e202111683 CrossRef CAS PubMed.
- J. Zhang, Y. Wang and Y. Li, Not One, Not Two, But at Least Three: Activity Origin of Copper Single-Atom Catalysts toward CO2/CO Electroreduction to C2+ Products, J. Am. Chem. Soc., 2024, 146, 14954–14958 CrossRef CAS PubMed.
- L. Wang, Z. Liu, R. Li, H. Gao, P. Yang, W. Wang, X. Xue, S. Feng, L. Yu and G. Wang, The Intrinsic Activity Descriptor of TM–N3–C Single-Atom Catalysts for Electrochemical CO2 Reduction: A DFT Study, J. Mater. Chem. A, 2023, 11, 24836–24853 RSC.
- X. Li, Y. Zeng, C.-W. Tung, Y.-R. Lu, S. Baskaran, S.-F. Hung, S. Wang, C.-Q. Xu, J. Wang and T.-S. Chan, et al., Unveiling the In Situ Generation of a Monovalent Fe(I) Site in the Single-Fe-Atom Catalyst for Electrochemical CO2 Reduction, ACS Catal., 2021, 11, 7292–7301 CrossRef CAS.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, Electrochemical Reduction of CO2 Catalyzed by Fe–N–C Materials: A Structure–Selectivity Study, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, Electrochemical Reduction of CO2 on Metal-Nitrogen-Doped Carbon Catalysts, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- C. Wang, H. Ren, Z. Wang, Q. Guan, Y. Liu and W. Li, A Promising Single-Atom Co–N–C Catalyst for Efficient CO2 Electroreduction and High-Current Solar Conversion of CO2 to CO, Appl. Catal., B, 2022, 304, 120958 CrossRef CAS.
- X.-M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M.-M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Baasbjerg, Selective CO2 Reduction to CO in Water using Earth-Abundant Metal and Nitrogen-Doped Carbon Electrocatalysts, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods to locate the most stable SAC geometries; benchmark calculations of kinetic energy cutoff and k-point grid selections; initial magnetic moment guess, dipole correction, and DFT+U benchmarks; geometries of all optimized clean SACs and representative adsorption sites; comparison of dopant effects in tuning CO adsorption energies; constrained geometry optimizations for CO adsorption energy predictions; complexation energies of SACs; most favorable adsorption sites of key intermediates; energetics of HER; energetics of CO2R to methane and methanol reaction pathways on SACs (PDF). Compressed VASP structure files (ZIP). See DOI: https://doi.org/10.1039/d4cp04212c |
| This journal is © the Owner Societies 2025 |