
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCan we talk about ionic bonds in molecules? Yes, just as we do for covalent bonds†
Ángel
Martín Pendás
 *,
Diogo J. L.
Rodriges
and
Evelio
Francisco
*,
Diogo J. L.
Rodriges
and
Evelio
Francisco
Dpto. Química Física y Analítica, Univ. Oviedo, C/Julián Clavería 8, 33006 Oviedo, Spain. E-mail: ampendas@uniovi.es
First published on 2nd January 2025
Abstract
A claim that ionic bonds exist only in ionic solids is critically analyzed by focusing on the controversial LiH molecule, classified as covalent by non-orthogonal valence bond supporters, polar-covalent by molecular orbital advocates, and ionic by real-space proponents. Using orbital invariant techniques we show that LiH can be regarded ionic in the same manner that dihydrogen is considered covalent.
The chemical bond is the central concept of chemistry, and much of what our society owes to this central science emerges from the ability of its practitioners to tame and manipulate bonds. However, as all theoretical and most experimental chemists know, chemical bonds are emergent objects, not contained in the preceding ontological level, quantum mechanics. In a more pedantic theoretical phrasing, there is no Dirac observable for a chemical bond. Thus, it is no surprise that much work has been devoted to clarifying what a bond is: chemists have devoted their lives to it. Although the taxonomic breadth of bonds has grown over the years with the advent of many types of multicenter and non-covalent interactions,1,2 the simplest covalent and ionic categories, albeit as suitable limiting models, seem to have stood the test of time well. Several recent works coming from the molecular orbital (MO) community,3,4 however, have stressed the impossibility of ionic bonding in isolated molecules, leaving this category for extended solids only. According to this view, ionic bonding stems from the valence bond (VB) ansatz, which has no explicit polar bonds, and the LiF molecule, for instance, is bound by a polar covalent link. Similarly, for these authors the bond in H2 is fully covalent even though some 10% of its binding energy comes from VB ionic terms. Surprisingly, the VB community that favors an ionic view of LiF, denies it for LiH, which is found to be mainly covalent.5 Notice that the existence of the zwitterionic-like symmetric resonance structures H+H−, H−H+ in the VB does not lead to polarity in dihydrogen, an obvious indication of the different meaning that the term ‘ionic’ has in the MO and VB community.6 This also raises the question whether all non-polar homodiatomic bonds should be understood as fully covalent or not, an issue that is directly related to the strength of electron correlation that is not typically considered in MO theory descriptions of chemical bonds.
Given the importance of these issues, some independent clarifications are due. We have already written on the role of electrostatics and covalency in bonding,7 on the origin of the large VB covalency of LiH,6 and on the choice of references in the LiH and LiF cases,8,9 but we think it necessary to reconsider this problem. After all, the 1/r long-range tail of electrostatic interactions makes them major forces that chemists should never overlook. To avoid paradigm-dependent interpretations we stick to orbital invariant descriptions (except when highlighting why one view gives rise to this or that interpretation). Although we firmly think10 that some kind of atoms (or fragments) in-the-molecule are needed to make sense of bonding beyond diatomics, the quantum theory of atoms in molecules (QTAIM)11 being among the less biased options available, we will only use AIMs when necessary to avoid the common criticism regarding the too large charges of Bader atoms (see ref. 12 for an extensive discussion on this issue). We also note that, given the relationship of the claims we are considering to how chemical bonding transitions from molecules to solids, several techniques that allow bonds in condensed systems to be studied from a chemical perspective,13–15 might provide interesting insights into this issue, although we have not explored them here.
We will focus on the controversial LiH case. All our MO and VB computations are performed with the GAMESS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 and XMVB17 codes, respectively, using standard Dunning basis sets from the (aug)-cc-pVnZ series. Quantum chemical topology (QCT) analyses have been performed both with AIMAll18 and our PROMOLDEN19 packages. Note that the inner contracted Gaussian functions of the Dunning basis sets closely correspond to the several atomic orbitals of the neutral reference atoms. At the aug-cc-pVTZ level, the Hartree–Fock (HF) energies of H, Li, and Li+ are −0.49983, −7.43269, and −7.23638 a.u., respectively, providing an ionization potential for Li that deviates by just 1.4 kcal mol−1 from the experiment.20 Deletion of the second 1s contracted Gaussian function from the Li basis set does not alter the energy of Li+ to the sixth decimal place, demonstrating that it builds the 2s orbital of neutral Li. At R(LiH) = 1.5957 Å, the HF energy of LiH is −7.9868 a.u., a value that decreases to −8.0031 a.u. upon performing a complete active space CAS(2,2) calculation. This is the simplest theoretical level that allows for a neutral dissociation to open-shell Li and H radicals. Since we pursue orbital invariant descriptions, we will report natural orbitals, which can be obtained both for MO and VB descriptions, and natural occupation numbers. Besides the basic 1s Li core, the CAS calculation has two active orbitals harbouring two electrons, σg, and σu, that evolve from close to two/close to zero occupation at equilibrium, to equal populations at dissociation. In our case, the natural occupations of the active orbitals equal 1.964, and 0.04e, respectively, at the selected distance. The CAS Mulliken/Löwdin/QTAIM charges of H are −0.23/−0.36/−0.90e.
16 and XMVB17 codes, respectively, using standard Dunning basis sets from the (aug)-cc-pVnZ series. Quantum chemical topology (QCT) analyses have been performed both with AIMAll18 and our PROMOLDEN19 packages. Note that the inner contracted Gaussian functions of the Dunning basis sets closely correspond to the several atomic orbitals of the neutral reference atoms. At the aug-cc-pVTZ level, the Hartree–Fock (HF) energies of H, Li, and Li+ are −0.49983, −7.43269, and −7.23638 a.u., respectively, providing an ionization potential for Li that deviates by just 1.4 kcal mol−1 from the experiment.20 Deletion of the second 1s contracted Gaussian function from the Li basis set does not alter the energy of Li+ to the sixth decimal place, demonstrating that it builds the 2s orbital of neutral Li. At R(LiH) = 1.5957 Å, the HF energy of LiH is −7.9868 a.u., a value that decreases to −8.0031 a.u. upon performing a complete active space CAS(2,2) calculation. This is the simplest theoretical level that allows for a neutral dissociation to open-shell Li and H radicals. Since we pursue orbital invariant descriptions, we will report natural orbitals, which can be obtained both for MO and VB descriptions, and natural occupation numbers. Besides the basic 1s Li core, the CAS calculation has two active orbitals harbouring two electrons, σg, and σu, that evolve from close to two/close to zero occupation at equilibrium, to equal populations at dissociation. In our case, the natural occupations of the active orbitals equal 1.964, and 0.04e, respectively, at the selected distance. The CAS Mulliken/Löwdin/QTAIM charges of H are −0.23/−0.36/−0.90e.
The first indication that a covalent interpretation of LiH is defective comes from deleting the 2s orbital from the Li basis set in the molecule. This raises the HF/CAS energy of LiH by only 1.8/1.9 kcal mol−1, meaning that the bonding interaction in LiH (in MO parlance, this is driven by the 2σg orbital) does not require the Li 2s orbital. An exam of the 2σg orbital when the Li 2s contracted function is absent, shows that the former is built from a polarized 1s H function (c2 ≈ 0.4) and a very diffuse 2pz Li function (c2 ≈ 0.5) that extends well into the H nuclear region. Let us now turn to the valence bond framework. A localized VB calculation with the standard two ionic plus one covalent structure – Li+H−, Li−H+, and Li·H· – gives an energy 0.3 kcal mol−1 above the CAS(2,2) calculation, providing renormalized (inverse) weights for the covalent, Li+–H− and Li−–H+ structures of 0.42, 0.59, and 0.00, respectively, and H Mulliken/Löwdin charges of +0.55/−0.354e. Interestingly, restricting the calculation to the covalent VB structure raises the energy by just 0.3 kcal mol−1 (leading to strictly zero Mulliken charges and to a fully VB covalent description). In both VB cases, the natural orbitals (NOs) are very close to each other and to the CAS ones reported above. This very nicely shows the well-known hazard of using extended basis sets to obtain chemical interpretations in localized VB calculations.21
Several revealing computational experiments can be performed thanks to the flexibility of strictly localized VB. For instance, we can force a description of LiH using a single VB structure in which the first and second orbitals (the 1σg and 2σg functions in MO) are built from exclusively Li and H primitives, respectively. This leads to a single determinant, fully ionic, LiH molecule, with −1.0 Mulliken charge for the H atom, and an energy lying 16 kcal mol−1 above the CAS solution (E = −7.9772 a.u.). Reducing the size of the basis set has a profound influence on the VB results. At the cc-pVDZ level, the scenario is rather different. Now, renormalized weights of the covalent and ionic structures are 0.99, 0.01, and 0.00, respectively, leading to a VB-covalent description of LiH and to −0.03/−0.06 Mulliken/Löwdin H charges. The single-determinant ionic VB wavefunction commented above now lies 27.5 kcal mol−1 above the full 3-structures VB energy. This example shows that smaller basis sets thus use Li-centered functions to variationally improve the energy of the molecule, leading to an unphysical exaggerated covalent weight. A more precise breathing orbital VB calculation (BOVB), in which the orbitals of the different resonance structures are different, lowers the VB energy by 0.1 kcal mol−1, changing the renormalized weights to 0.96, 0.03, and 0.01. This decreases only very slightly the anomalous covalent weight. We also notice that in this case, the traditional Chirgwin–Coulson weight of the covalent structure is much smaller, 0.80, falling to 0.74 when using a smaller 6-31++G** basis set, a value very close to that already reported at R(LiH) = 1.6328 Å.5
A second strong argument in favor of an ionic description of LiH lies in the orbitals themselves (see Fig. 1). The CAS 2σg NO (n ≈ 1.96) is a polarized H 1s function with a delocalization tail at the Li core (left panel). However, when we force a VB description, the first of the two localized valence orbitals is an H function (middle panel, blue), while the second, which is built from Li functions by construction, is described as a Li sp hybrid (middle panel, red). This is mostly semantics since it is located mainly in the H region. As shown in our first point, beyond the introduction of delocalization tails that improve the energy, the valence orbital of Li is not needed to describe LiH. Moreover, as commented, diagonalization of the first order density matrix at the VB level leads to almost the CAS NOs. Provided that ∇2ρ faithfully reproduces atomic shells in first- and second-period elements, the right panel of the figure reinforces all of the above. There is no valence shell around Li, and the K shell around the H atom closely reproduces the polarization pattern seen in the NO and VB orbitals.
 | ||
| Fig. 1 Left, CAS(2,2)/aug-cc-pVTZ 2σg NO. Middle, VB/cc-pVDZ localized orbitals. In both cases, |ϕ| = 0.08 a.u. Right, CAS(2,2)/aug-cc-pVTZ ∇2ρ isolines (negative/positive values indicated by red/blue colors) in a plane containing the Li (left) and H (right) nuclei. | ||
Our third line of reasoning contradicts the claim of no-ionic-bonds-except-in-solids by analyzing bond formation in LiH. Although we believe that we should be able to unveil the nature of bonding without resorting to references, for many researchers chemical bonds are inseparable from the path that leads to them.22 This route is very well known in spectroscopy: the following paragraphs would have been rather obvious to people like R. S. Mulliken. Fig. 2 shows an aug-cc-pVTZ full configuration interaction (FCI) calculation of the evolution of the first four 1Σ states of LiH with internuclear distance. As already reported,23 the low electron affinity of H impedes the first singlet excited state, 21Σ, to dissociate to the ionic Li+–H− reference and enforces a series of avoided crossings. For future reference, all these states dissociate to a ground state 1s1–2S H atom and a Li atom of varying character: 1s22s1–2S in 11Σ, 1s22p1–2P0 in 21Σ, 1s23s1–2S in 31Σ, and 1s23p1–2P0 in 41Σ, after other crossings at larger distances that are not shown. Notice that other real-space accounts paying attention to the electron transfer mechanism in LiH, have also been presented.24
 | ||
| Fig. 2 FCI/aug-cc-pVTZ potential energy curves of the first four 1Σ states of LiH. The −1/R Coulombic tail, with zero at the Li+–H− reference energy, is also shown as a dashed line. The approximate position of the three avoided crossings is indicated with circles, and the distances at which a detailed analysis is provided in the text, are shown with dotted vertical lines. | ||
A simple visual inspection is revealing. The Coulombic tail demonstrates (in the distance range covered by the figure) three sequential avoided crossings (ACi) of the ionic state when R decreases as we go down to the ground state. Thus, the 41Σ state is ionic until AC1, then this character is inherited by 31Σ until AC2, by 21Σ until AC3 at about 3.4 Å, and finally by the ground state. At equilibrium, and despite some obvious mixing, the ground state should be labelled as ionic. This is nothing but basic quantum mechanics. In the CAS(2,2)/aug-cc-pVTZ calculation, for instance, the binding energy of the ground state measured from the ionic reference is −167.5 kcal mol−1. If QTAIM charges are used to estimate the purely Coulombic stabilization (−Q2/R, Q = 0.898 a.u.) we get −167.8 kcal mol−1, a rather remarkable coincidence that agrees with the rule of thumb that, in ionic solids, the Madelung contribution typically represents about 90% of the total lattice energy.
We now follow the nature of these states. Trying to avoid paradigm-dependent interpretations as much as possible, we use very basic tools. First, we show that NOs can be used to follow the nature of the states. In all of them, at all distances, the first NO ϕ1 corresponds to a Li core, with occupation very close to n1 = 2.0, so we mainly report the n2, and n3 occupations in the following. At R = 20 Å, all 11Σ, 21Σ, and 31Σ display n2 ≈ n3 ≈ 1e, with ϕ2 corresponding closely to the 1s H orbital and ϕ3 to a 2s, 2pz, and 3s Li function, respectively. All are in agreement with the final dissociation fragments of each state. On the contrary, the occupations in the 41Σ state are n2,n3 ≈ 2.0,0.0e, so its wavefunction is close to a single determinant in which the Li 2l functions are absent, i.e., the ionic state with an energy evolving as −1/R. At R = 8.0 Å, after AC1, 11Σ displays n2 = 1.005e, and n3 = 0.995e, 21Σ has n2,n3 = 1.039,0.961. In both cases, the ϕ's maintain their nature, an H 1s, and a Li 2s/2pz orbital. However, 31Σ has changed, and now n2,n3 ≈ 1.967,0.026e. Its ϕ2 is an H 1s orbital, and the low-occupation ϕ3 function still has a large Li 2pz character. The contrary occurs in the 41Σ state. This example demonstrates how, save some unavoidable mixing, the third and fourth Σ singlets have exchanged their nature after AC1 takes place. This is revealed neatly by the NOs, and no atomic partition is needed to come to this conclusion. Now we show that the nature of the states is also faithfully encoded by the Laplacian of the electron density.
Fig. 3 shows isocontours of ∇2ρ for the first three Σ singlets at R = 8.0 Å. The K and L shells of Li (the number of regions with negative Laplacians) are visible in the first two states, with clear s and p characters, respectively. 31Σ lacks any L shell around Li, while a bigger K hydrogen shell is discerned. Notice that the Laplacian is sensitive to the p-like contamination of Li in this ionic state. Without any need for an atomic partition, at R = 8 Å the 11Σ and 31Σ states can be safely classified as covalent and ionic, respectively. If a QTAIM partition is introduced, Q(H) = 0.000, −0.892e, in the same order.
 | ||
| Fig. 3 Isocontours (blue, positive; red, negative) of ∇2ρ for the aug-cc-pVDZ/FCI 11Σ (top), 21Σ (middle), and 31Σ (bottom) states of LiH at R = 8.0 Å. | ||
AC2 and AC3 occur in a relatively narrow distance range, so we expect mixing and exchange of roles for the first three Σ singlets. At R = 4 Å (Fig. 4), AC2 has already taken place, and AC3 is beginning to occur. 21Σ inherits much of the ionic character of 31Σ at larger distances. The values of the n2,n3 pair in 21Σ are 1.726,0.271, respectively, ϕ2,ϕ3 is a polarized H 1s orbital and an orbital that keeps much of the Li 2pz character that was prevalent before the crossing. In the case of 31Σ, n2,n3 become 1.814,0.179, with the ϕ2,ϕ3 roles mostly reversed. 11Σ is already impacted by the crossings, with n2,n3 being 1.379,0.621, and ϕ2,ϕ3 corresponding to the somewhat delocalized H 1s and Li 2s orbitals.
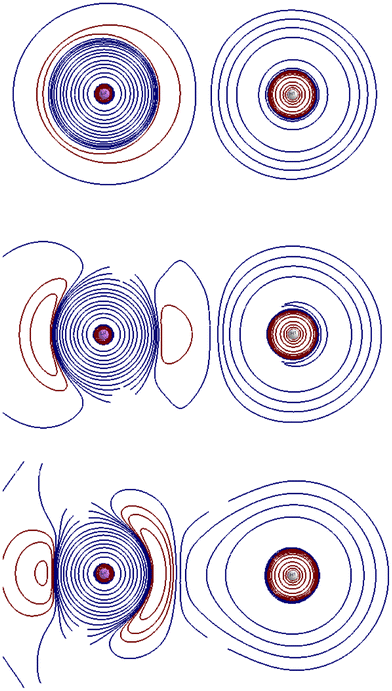 | ||
| Fig. 4 Isocontours (blue, positive; red, negative) of ∇2ρ for the aug-cc-pVDZ/FCI 11Σ (top), 21Σ (middle), and 31Σ (bottom) states of LiH at R = 4.0 Å. | ||
Fig. 4 reveals that, at R = 4.0 Å, the L shell of Li in 11Σ is polarized towards the H atom, and a heavy mixing of the pre-AC2 21Σ and 31Σ features, leaving a backward/forward pz polarized Li atom in the post-AC2 21Σ and 31Σ states. It should be noted that the QTAIM Q(H) charges are −0.083, −0.512, −0.503e in the R = 4 Å 11Σ, 21Σ, and 31Σ states, respectively. At R = 2.0 Å, Fig. 5, the low-lying Σ singlets already show the scenario found at the equilibrium geometry. Since in the high energy region another AC has occurred between 31Σ and 41Σ, we will not consider 31Σ. Now the n2,n3 occupations in 11Σ are 1.933,0.050e, respectively, and the ϕ2,ϕ3 NOs resemble those of 21Σ at R = 4.0 Å. Q(H) = −0.895. The occupations change to 1.373,0.623 in 21Σ, with Q(H) = −0.084. The collapse of the mixed pre-AC3 21Σ and 31Σ partially ionic states to a post-AC3 ionic 11Σ state and a neutral 21Σ, is clear. Fig. 5 shows no Li L shell in the post-AC3 11Σ.
 | ||
| Fig. 5 Isocontours (blue, positive; red, negative) of ∇2ρ for the aug-cc-pVDZ/FCI 11Σ (top) and 21Σ (bottom) states of LiH at R = 2.0 Å. | ||
A dissection of the bond formation process clarifies the ionic nature of 11Σ–LiH at equilibrium, as well as the neutral or covalent one in 21Σ. Moreover, since the last AC occurs between a state that dissociates to 1s22pz1 Li and another whose limit is 1s22s1 Li, the ionic ground state of LiH inherits some 2pz1, not 2s1, character, after the crossing. This explains why the 2s function of Li is not needed in conventional calculations. Needless to say, these conclusions are even clearer in other systems like LiF, where a single AC changes the character of the ionic and neutral states abruptly.8 No atomic partition is needed to reach these findings, although using AIMs helps their quantification and allows reaching similar insights from a single shot analysis in the equilibrium ground state. Using a valence CCSD/aug-cc-pVTZ IQA analysis of LiH and LiF, for instance,8 we have quantified that the energy of the Li-in-the-molecule atom lies only 2.2 and 8.9 kcal mol−1 away from that of an isolated Li+ cation, respectively, and that the in situ binding energy in both systems is 159 and 179 kcal mol−1, with similar exchange–correlation contributions (covalent energies in the interacting quantum atoms terminology) of 24 and 29 kcal mol−1, respectively. These are ionic molecular systems, whether understood through the whole bond formation process or via atomic partitioning techniques at the equilibrium geometry.
A final strong, independent argumentation that is also invariant under orbital transformations comes from the analysis of the spatial maxima of the square of the wavefunction |Ψ|2.25–28 These maxima offer vivid representations of the most likely positions of all the electrons in a system, providing a picture very close to that of Lewis’ theory. In H2, for instance, two spin-degenerate maxima, with the α electron located at one of the H nuclei and the β electron at the other, are found: a covalent maximum. At any level of theory, from HF to FCI, the global maximum in LiH has a pair of spin-coupled electrons at the Li nucleus and another pair at the H nucleus: an ionic situation. Visually impactful examples can be found in Fig. 6, which shows the Born maximum in LiF and NaCl as obtained from Jastrow-optimized variational quantum Monte Carlo calculations performed with the AMOLQC suite,29 starting with an HF cc-pVTZ guess.
 | ||
| Fig. 6 Born maxima in the LiF (top) and NaCl (bottom) molecules. Different spin electrons are pictured as white/yellow spheres. A pair of spin-coupled electrons lying at each nucleus is not shown. | ||
It is known that the Born maximum of an isolated s2p6 closed shell is a cube formed by two interpenetrated tetrahedra of α and β electrons, much as in Lewis’ cubical atom model. At the Born maxima of both LiF and NaCl, the valence shell electron of the alkali atom has been transferred to the halide one. All LiH, LiF, and NaCl, are thus ionic.
There is no need to invoke an atomic partition to show that ionic bonds (i.e. extremely polar covalent bonds) exist in molecules. Since it is a universal rule that the sum of the ionization potential and the electron affinities for any pair of atoms in the periodic table is an endothermic process, all bonds break homolytically in the gas phase. However, as is well known, state crossings, or avoided crossings in diatomics, can change the nature of a forming bond. We have shown through a battery of orbital invariant techniques that LiH, one of the most controversial systems, should be classified as ionic, as much as dihydrogen is covalent. Our arguments are even more persuasive in other molecules, like LiF. There is no reason to deny ionic bonding in molecules. Real space analyses, which provide consistent answers across a wide range of bonding situations, may help us to critically examine controversial claims such as the one considered here.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. van Schilfgaarde and W. A. Harrison, Phys. Rev. B, 1986, 33, 2653–2659 CrossRef.
- T. Clark, J. Mol. Model., 2023, 29, 66 CrossRef PubMed.
- L.-J. Cui, Y.-Q. Liu, S. Pan, Z.-H. Cui and G. Frenking, Chem. Sci., 2024, 15, 14705–14720 RSC.
- S. Pan and G. Frenking, Molecules, 2021, 26, 4695 CrossRef.
- S. Shaik, D. Danovich and P. C. Hiberty, J. Chem. Phys., 2022, 157, 090901 CrossRef PubMed.
- A. Martín Pendás and E. Francisco, Phys. Chem. Chem. Phys., 2018, 20, 12368–12372 RSC.
- A. Martín Pendás, J. L. Casals-Sainz and E. Francisco, Chem. – Eur. J., 2019, 25, 309–314 CrossRef PubMed.
- D. Menéndez-Crespo, A. Costales, E. Francisco and A. Martín Pendás, Chem. – Eur. J., 2018, 24, 9101–9112 CrossRef PubMed.
- A. Martín Pendás and E. Francisco, Nat. Commun., 2022, 13, 3327 CrossRef PubMed.
- K. Ruedenberg, Rev. Mod. Phys., 1962, 34, 326–376 CrossRef.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon Press, Oxford, 1990 Search PubMed.
- R. F. W. Bader and C. F. Matta, J. Phys. Chem. A, 2004, 108, 8385–8394 CrossRef.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef.
- C. Lian, Z. A. Ali, H. Kwon and B. M. Wong, J. Phys. Chem. Lett., 2019, 10, 3402–3407 CrossRef.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef.
- Z. Chen, F. Ying, X. Chen, J. Song, P. Su, L. Song, Y. Mo, Q. Zhang and W. Wu, Int. J. Quantum Chem., 2015, 115, 731–737 CrossRef.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019. https://aim.tkgristmill.com Search PubMed.
- A. Martn Pendás and E. Francisco, PROMOLDEN. A QTAIM/IQA code (Available from the authors upon request).
- A. Kramida and Y. Ralchenko, NIST Atomic Spectra Database, NIST Standard Reference Database 78, 1999, https://www.nist.gov/pml/data/asd.cfm Search PubMed.
- B. J. Duke and R. W. A. Havenith, Theor. Chem. Acc., 2016, 135, 82 Search PubMed.
- W. H. E. Schwarz and H. Schmidbaur, Chem. – Eur. J., 2012, 18, 4470–4479 CrossRef.
- J. L. Casals-Sainz, J. Jara-Cortés, J. Hernández-Trujillo, J. M. Guevara-Vela, E. Francisco and A. Martín Pendás, Chem. – Eur. J., 2019, 25, 12169–12179 CrossRef PubMed.
- M. Rodríguez-Mayorga, E. Ramos-Cordoba, P. Salvador, M. Solà and E. Matito, Mol. Phys., 2016, 114, 1345–1355 CrossRef.
- A. Lüchow, J. Comput. Chem., 2014, 35, 854–864 CrossRef PubMed.
- L. Reuter and A. Lüchow, Phys. Chem. Chem. Phys., 2020, 22, 25892–25903 RSC.
- L. Reuter and A. Lüchow, Nat. Commun., 2021, 12, 4820–4828 CrossRef PubMed.
- M. Menéndez-Herrero and A. Martín Pendás, IUCrJ, 2024, 11, 210–223 CrossRef PubMed.
- A. Lüchow, S. Manten, C. Diedrich, A. Bande, T. C. Scott, A. Schwarz, R. Berner, R. Petz, A. Sturm, M. Hermsen, K. H. Mood, C. Schulte, L. Reuter, M. A. Heuer and J. Ludovicy, Amolqc (v7.1.0), Zenodo, 2021 DOI:10.5281/zenodo.4562745.
Footnote |
| † Electronic supplementary information (ESI) available: Natural occupations, natural orbitals, and QTAIM charges of the FCI calculations described in the text. See DOI: https://doi.org/10.1039/d4cp04353g |
| This journal is © the Owner Societies 2025 |