
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analyzing the concentration-dependent Soret coefficient minimum in salt solutions: an overview†
Binny A.
Rudani
 a,
W. J.
Briels
*ab and
Simone
Wiegand
*ac
a,
W. J.
Briels
*ab and
Simone
Wiegand
*ac
aIBI-4: Biomacromolecular Systems and Processes, Forschungszentrum Jülich GmbH, D-52428 Jülich, Germany. E-mail: s.wiegand@fz-juelich.de
bUniversity of Twente, Computational Chemical Physics, Postbus 217, 7500 AE Enschede, The Netherlands. E-mail: w.j.briels@utwente.nl
cChemistry Department - Physical Chemistry, University Cologne, D-50939 Cologne, Germany
First published on 31st January 2025
Abstract
Temperature gradients often cause the separation of the components in liquid mixtures by a process called thermodiffusion and quantified by the Soret coefficient. In recent years, the existence of minima in the Soret coefficient as a function of concentration has been investigated by experiments and simulations for various aqueous salt solutions. In this paper, we analyze the data of ten 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolytes (lithium, sodium and potassium chloride, lithium, sodium and potassium iodide, potassium acetate, sodium and potassium thiocyanate and guanidinium chloride) in water, together with those of newly measured Soret coefficients for aqueous cesium iodide solutions. The latter were measured in the temperature range between 15 °C and 45 °C and concentrations between 0.5 and 3 moles per kg of the solvent using thermal diffusion forced Rayleigh scattering. We analyze the data by expressing the Soret coefficients as products of two factors, one purely thermodynamic factor and one being the ratio of two Onsager coefficients. It turns out that the ratio of Onsager coefficients is the main factor responsible for the non-monotonic behavior of the Soret coefficients, contrary to recent findings using computer simulations of binary Lennard-Jones mixtures. Moreover, for salts with the same anion, we find that the thermodynamic factors increase with increasing Pauling radii of the cations, while the Onsager ratios increase monotonically with the radii of the hydrated cations.
:1 electrolytes (lithium, sodium and potassium chloride, lithium, sodium and potassium iodide, potassium acetate, sodium and potassium thiocyanate and guanidinium chloride) in water, together with those of newly measured Soret coefficients for aqueous cesium iodide solutions. The latter were measured in the temperature range between 15 °C and 45 °C and concentrations between 0.5 and 3 moles per kg of the solvent using thermal diffusion forced Rayleigh scattering. We analyze the data by expressing the Soret coefficients as products of two factors, one purely thermodynamic factor and one being the ratio of two Onsager coefficients. It turns out that the ratio of Onsager coefficients is the main factor responsible for the non-monotonic behavior of the Soret coefficients, contrary to recent findings using computer simulations of binary Lennard-Jones mixtures. Moreover, for salts with the same anion, we find that the thermodynamic factors increase with increasing Pauling radii of the cations, while the Onsager ratios increase monotonically with the radii of the hydrated cations.
1 Introduction
Temperature gradients occur in a large number of systems that are investigated in a wide variety of research areas, such as biotechnology, chemical process engineering or energy technology.1,2 Due to the coupling of heat and mass fluxes they often give rise to concentration gradients, a phenomenon known as thermodiffusion or thermophoresis and sometimes also referred to as the Ludwig–Soret effect. The resulting concentration gradients created under these conditions are often of crucial importance, both in natural phenomena and in technical processes involving temperature differences. Examples include the formation of rocks (petrology), the behavior of oil reservoirs and various separation techniques.3–6 Due to its high sensitivity to solute–water interactions, it is commercially used to characterize the binding affinity between the ligand and the protein.7–9 In addition, the investigation of temperature gradients plays a role in the development of new technologies for the conversion of waste heat into electricity, e.g. in the development of thermoelectric liquid cells.10–12Thermodiffusion is accounted for by an off-diagonal term in Onsager's linear relations between heat and mass flows and the corresponding generalized thermodynamic forces.1 In the presence of a temperature gradient and a concentration gradient, the mass flow in a binary fluid can be described as follows:
![[j with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006a_20d1.gif) m = −ρD m = −ρD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) gradc − pc(1 − c)DTgradT gradc − pc(1 − c)DTgradT | (1) |
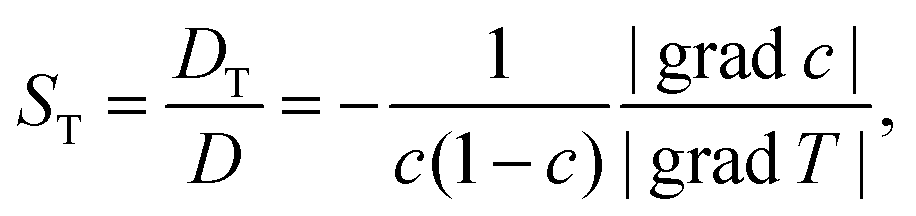 | (2) |
m = ![[0 with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0030_20d1.gif)
The first systematic studies on the Ludwig–Soret effect were carried out for aqueous salt solutions.13–17 An unresolved question in salt solutions is why the Soret coefficient often reaches a minimum value with varying concentrations.16–22 Along the lines of the theory of irreversible thermodynamics23 (see Appendix) Gittus and Bresme suggested writing the Soret coefficient as a product of two factors, one being purely thermodynamic and the other, the ratio of two Onsager coefficients  , being dynamic in principle21
, being dynamic in principle21
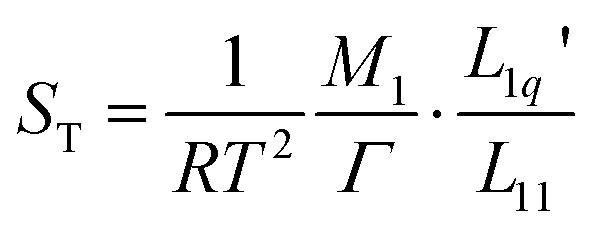 | (3) |
and to investigate their relation with the minimum of the Soret coefficient separately. Here M1 is the molar mass of the salt, m1 is its molality and μ1 is its chemical potential; M2 is the molar mass of the solvent and Ω is its molality. In the following, we refer to M1/(RT2Γ) as the thermodynamic factor. Note that sometimes21Γ itself is called the thermodynamic factor. In this paper, we will calculate the thermodynamic factors using thermodynamic data from the literature. The ratios of Onsager coefficients
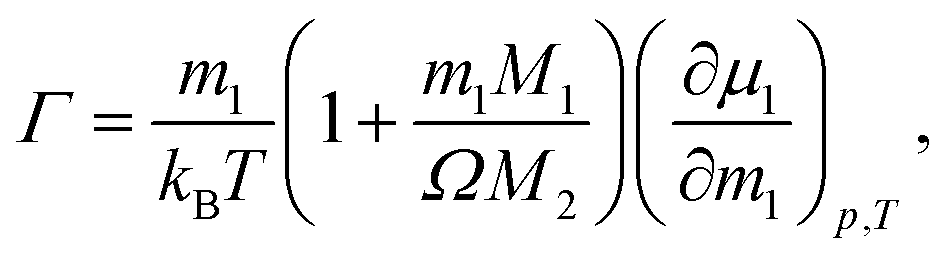
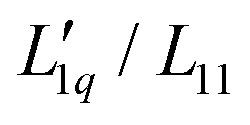 are next obtained by dividing the experimental Soret coefficients by the corresponding thermodynamic factors.
are next obtained by dividing the experimental Soret coefficients by the corresponding thermodynamic factors.
As mentioned above, eqn (3) is derived from the principles of irreversible thermodynamics1,23 and has been widely used by many researchers.2,21,24–26 In the Appendix, it is shown that the Onsager ratio,  , is related to the so called ‘heat of transfer’ Q* according to
, is related to the so called ‘heat of transfer’ Q* according to 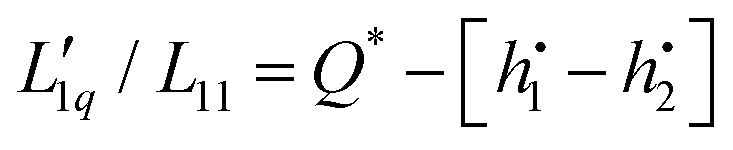 , where
, where  is the specific enthalpy of component i. It is also clear from the discussion in the Appendix that Q* is in principle a kinetic quantity. We therefore would have preferred to separate and discuss Q* from the Onsager ratios, but this is not possible since we do not have information about the specific enthalpies.
is the specific enthalpy of component i. It is also clear from the discussion in the Appendix that Q* is in principle a kinetic quantity. We therefore would have preferred to separate and discuss Q* from the Onsager ratios, but this is not possible since we do not have information about the specific enthalpies.
Simulations of binary (non-ionic) Lennard-Jones mixtures by Gittus and Bresme have shown that in the cases they investigated,  is basically constant as a function of composition, while a minimum occurs in the thermodynamic factor.21 In the case of LiCl, all thermodynamic data are available and we were able to calculate both factors.22 It turned out that
is basically constant as a function of composition, while a minimum occurs in the thermodynamic factor.21 In the case of LiCl, all thermodynamic data are available and we were able to calculate both factors.22 It turned out that 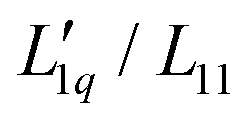 could be expressed as a monotonous exponential function and, in conjunction with the thermodynamic factor, allowed an accurate prediction of the experimental values of the Soret coefficients. It was found that the minimum of the Soret coefficients results from a minimum of the thermodynamic factor, which occurs at concentrations far below the experimental concentrations. Only after multiplication with the (negative) monotonic Onsager ratio did the minimum move into the experimental concentration window.
could be expressed as a monotonous exponential function and, in conjunction with the thermodynamic factor, allowed an accurate prediction of the experimental values of the Soret coefficients. It was found that the minimum of the Soret coefficients results from a minimum of the thermodynamic factor, which occurs at concentrations far below the experimental concentrations. Only after multiplication with the (negative) monotonic Onsager ratio did the minimum move into the experimental concentration window.
Recent detailed studies have shown that the hydrophilicity of a salt affects its thermodiffusion properties.27–29 Given the fact that salt molecules in solution are surrounded by a shell of more or less tightly bound water molecules, Mohanakumar et al.20 suggested that the minimum occurs when these hydration shells begin to overlap, i.e. when the hydrated spherical salt molecules are randomly closely packed.30 In order to obtain quantitative results the system was mapped on a Lennard-Jones binary mixture and data from the literature were used.31 Surely this mapping can only be very approximate and the results agreed only modestly with the experimental data.
In the present paper, we will continue our analysis based on the representation given in eqn (3) and discuss the two factors in relation to the hydration of the salts. We will find that for all aqueous salt systems that show a minimum of the Soret coefficient with concentration, the Onsager ratio 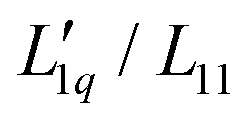 shows a minimum as well, while the thermodynamic factor does not or barely so. This makes it clear that for aqueous salt solutions the minimum in the Soret coefficient with concentration cannot be explained in thermodynamic terms only, but kinetic contributions are mainly responsible. A further indication that kinetic processes play an important role is the observation that the Soret coefficient for sodium and potassium increases monotonously with the hydrodynamic radii of the salt ions and not with their ionic radius.
shows a minimum as well, while the thermodynamic factor does not or barely so. This makes it clear that for aqueous salt solutions the minimum in the Soret coefficient with concentration cannot be explained in thermodynamic terms only, but kinetic contributions are mainly responsible. A further indication that kinetic processes play an important role is the observation that the Soret coefficient for sodium and potassium increases monotonously with the hydrodynamic radii of the salt ions and not with their ionic radius.
This paper is structured as follows. In the experimental part, section 2, we present the thermodiffusion data of cesium iodide (CsI) in water between 15 °C and 45 °C and in the concentration range between 0.5 and 3 mol kg−1 using thermal diffusion forced Rayleigh scattering (TDFRS). In section 3, we collect the thermodiffusion data of ten other salts and for these and for the CsI salt, and we calculate their thermodynamic factor and their Onsager ratio 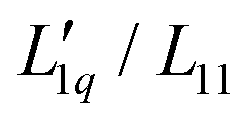 for a range of concentrations at a temperature of 25 °C. We start with simple aqueous alkali halide solutions consisting of spherical anions and cations. First, we discuss sodium chloride (NaCl) and potassium chloride (KCl) in comparison to the recently published lithium chloride (LiCl).16,18,22,32–37 We continue with iodide salts: lithium (Li), sodium (Na) and potassium (K) iodide (I). We next discuss potassium acetate with a compact spherical cation and a larger spherical anion with a relatively low surface charge density, and finally sodium thiocyanate and potassium thiocyanate (SCN). All systems exhibit a minimum of the Soret coefficient with concentration. The anion SCN− looks like a short rod. Finally, we examine the guanidinium cation, a disk-shaped cation with amphiphilic character, to see how it differs from the simple ions. In Section 4, we discuss the salient characteristics of the data, in particular their dependence on the size of the cations. We will find that the thermodynamic factors depend differently on size compared to the Onsager ratios.
for a range of concentrations at a temperature of 25 °C. We start with simple aqueous alkali halide solutions consisting of spherical anions and cations. First, we discuss sodium chloride (NaCl) and potassium chloride (KCl) in comparison to the recently published lithium chloride (LiCl).16,18,22,32–37 We continue with iodide salts: lithium (Li), sodium (Na) and potassium (K) iodide (I). We next discuss potassium acetate with a compact spherical cation and a larger spherical anion with a relatively low surface charge density, and finally sodium thiocyanate and potassium thiocyanate (SCN). All systems exhibit a minimum of the Soret coefficient with concentration. The anion SCN− looks like a short rod. Finally, we examine the guanidinium cation, a disk-shaped cation with amphiphilic character, to see how it differs from the simple ions. In Section 4, we discuss the salient characteristics of the data, in particular their dependence on the size of the cations. We will find that the thermodynamic factors depend differently on size compared to the Onsager ratios.
2 Experiments on cesium iodide
2.1 Methods and materials
CsI was purchased from Sigma-Aldrich, and had a purity of ≥99.999%. Precise amounts of CsI were dissolved in deionized water (Milli-Q water) to prepare transparent solutions of concentrations between 0.5 and 3 mol kg−1. The solutions were then filtered through a 0.2 μm membrane filter (Whatman Anatop 10) transferred to optical quartz cells (Hellma) with an optical path length of 0.2 mm to perform measurements of (∂n/∂c)p,T, (∂n/∂T)p,c, and TDFRS.The auxiliary parameters, concentration and temperature dependence of the refractive index, were measured independently. The refractive index as a function of concentration was measured with an Abbe refractometer (Anton Paar Abbemat MW) at a wavelength of 632.8 nm. We measured the refractive index for five concentrations to determine the (∂n/∂c)p,T. The refractive index change with temperature, (∂n/∂T)p,c was measured interferometrically.38 All data are shown in the ESI.† The TDFRS set-up is described in Section S1 in the ESI.†
We conducted a series of two TDFRS experiments with each concentration in the temperature range between 15 °C and 45 °C, using different cells and freshly prepared samples for each experiment. In each experiment, we collected at least 3000 individual signals, and their average was calculated. We then examined both the on and off phases of each signal, resulting in two different sets of values for ST and D.39,40 For each specific temperature and concentration, the mean values were derived from four measurement curves analyzed according to eqn (S1) in the ESI.† The standard deviation of the mean value is shown as error bars in the figures. The mean pH value for the aqueous CsI solution was approximately 8.1.
2.2 Results
In previous research, we examined the concentration dependence of the Soret coefficient for lithium, sodium, and potassium iodide as a function of temperature and concentration. For all three systems, we identified a minimum in ST as a function of concentration and used a concept of overlapping hydration shells to explain this occurrence.20 According to dielectric measurements and terahertz spectroscopy,41,42 tight hydration shells do not form around larger cations such as cesium. To better understand the relationship between the minimum of the Soret coefficients and the hydration of the cations, we extend the range of data by providing results for cesium iodide.
Fig. 1(a) shows ST of aqueous CsI solutions as a function of concentration for temperatures between 15 °C and 45 °C. At the lowest measured temperature of 15 °C, ST increases monotonically with concentration. At higher temperatures 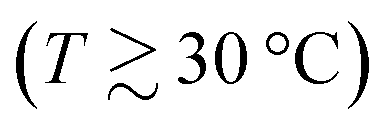 , however, ST develops a non-monotonic dependence on the concentration. This is in agreement with the other iodide salts but in sharp contrast with LiCl solutions where non-monotonic behavior of ST could only be observed at very low temperatures in experiments as well as in a simulation study while it disappeared with increasing temperatures.19,22 The difference between these two systems is most probably related to differences in their hydration shells and related to entropic effects, which typically dominate at higher temperatures. It should be noted that the minimum occurs at 1 m, the same concentration as for the other iodide salts.
, however, ST develops a non-monotonic dependence on the concentration. This is in agreement with the other iodide salts but in sharp contrast with LiCl solutions where non-monotonic behavior of ST could only be observed at very low temperatures in experiments as well as in a simulation study while it disappeared with increasing temperatures.19,22 The difference between these two systems is most probably related to differences in their hydration shells and related to entropic effects, which typically dominate at higher temperatures. It should be noted that the minimum occurs at 1 m, the same concentration as for the other iodide salts.
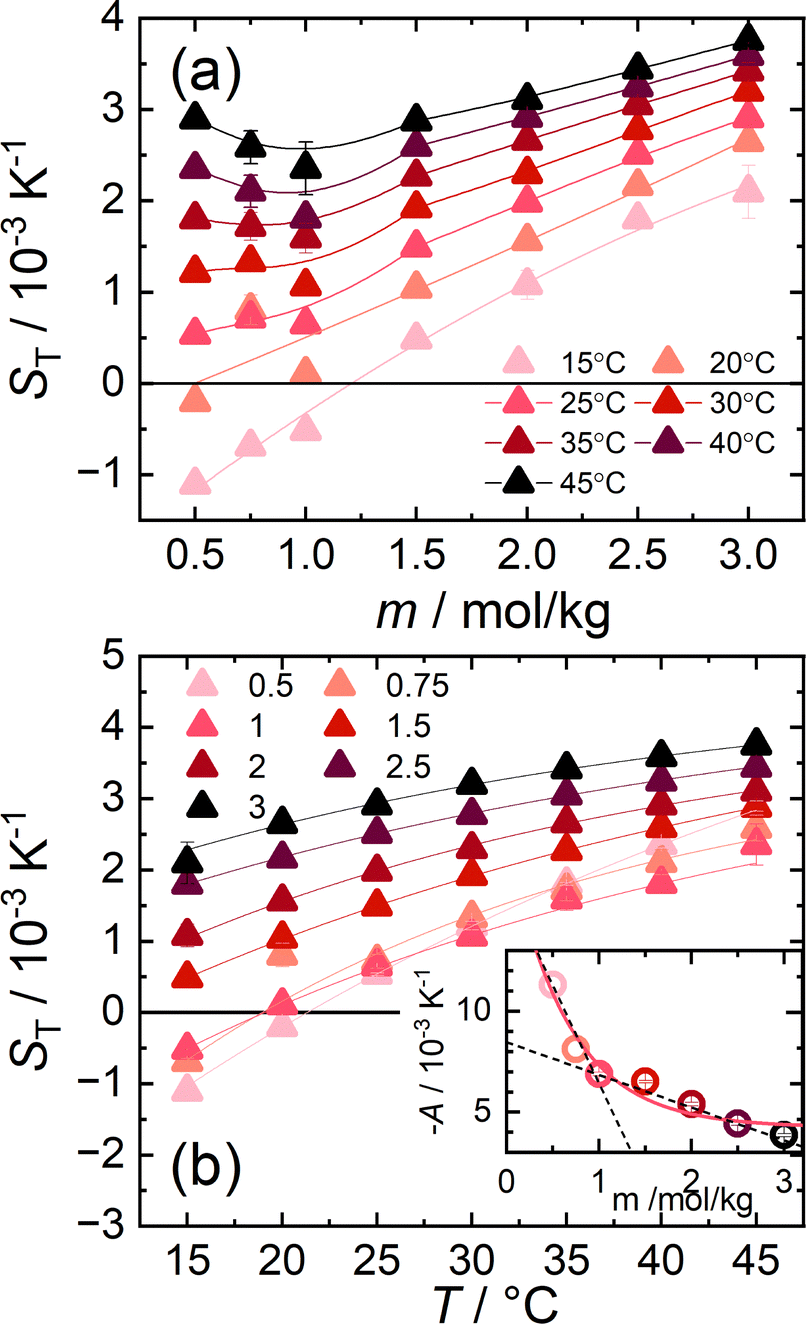 | ||
| Fig. 1 (a) Soret coefficients of aqueous cesium iodide solutions as a function of concentration for temperatures between 15 °C and 45 °C. The Bezier lines connecting the data points are a guide for the eye. (b) ST of aqueous CsI solutions as a function of temperature for concentrations between 0.5 m and 3 m. The lines are fitted according to eqn (4) and their respective fitting parameters are tabulated in ESI,† Table S1. The inset shows the magnitude of A. The amplitude as a function of molality can be described by an exponential fit. The trend of A at low and high concentrations is highlighted by two dashed lines intersecting around 1.0 m. | ||
Fig. 1(b) shows the temperature dependence of ST between 0.5 m and 3 m. For each concentration, the temperature dependence was fitted using an empirical expression43
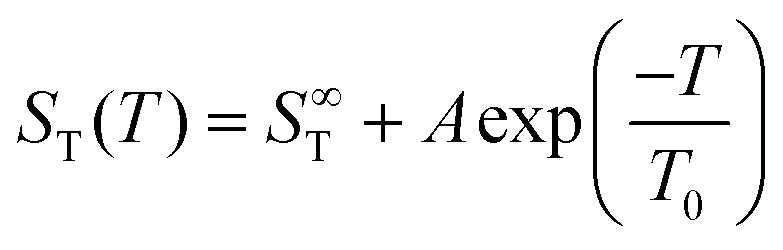 | (4) |
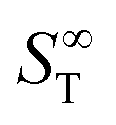 represents the thermophoretic high temperature limit. The amplitude A is a measure of the temperature sensitivity and is equal to
represents the thermophoretic high temperature limit. The amplitude A is a measure of the temperature sensitivity and is equal to 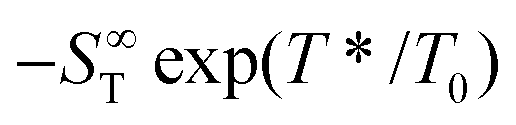 in the form of the equation originally proposed by Iacopini and Piazza.43 In their equation, T* denotes the temperature at which ST changes sign. Here, T0 stands for the exponential growth rate, which quantifies the curvature with temperature. The adjusted fitting parameters are listed in Table S1 in the ESI.† The inset in Fig. 1(b) shows the amplitude A as a function of molality. As in the case of LiCl,22 the amplitude decreases exponentially with increasing molality, and a low concentration region with a steeper slope and a high concentration region with a flatter slope can be recognized. The intersection point is approximately at 1 m.
in the form of the equation originally proposed by Iacopini and Piazza.43 In their equation, T* denotes the temperature at which ST changes sign. Here, T0 stands for the exponential growth rate, which quantifies the curvature with temperature. The adjusted fitting parameters are listed in Table S1 in the ESI.† The inset in Fig. 1(b) shows the amplitude A as a function of molality. As in the case of LiCl,22 the amplitude decreases exponentially with increasing molality, and a low concentration region with a steeper slope and a high concentration region with a flatter slope can be recognized. The intersection point is approximately at 1 m.
3 Comparison of different salt systems
In this section, we compare the dependence of Soret coefficients on concentration for a large variety of ionic systems, and draw some conclusions about generic aspects which may be of help in a further theoretical understanding of thermodiffusion. We will concentrate on a sequence of cations with increasing ionic radius, specifically, lithium, sodium, potassium and cesium mono-valent ions. In the following (some of) these ions will be combined to give chlorides, iodides, acetate and thiocynates; a final system we treat is guanidinium chloride. In this section all systems will be taken at 25 °C.In Table 1, we have collected some properties of the various cations discussed. The ions are ordered according to their ionic radius rPi (Pauling-type crystal ionic radius).44
| Li+ | Na+ | K+ | Cs+ | |
|---|---|---|---|---|
| M/u | 6.9 | 23 | 39.1 | 132.9 |
| r Pi/Å | 0.74 | 1.02 | 1.38 | 1.7 |
| d i⋯w/Å | 2.1 | 2.4 | 2.8 | 3.1 |
| r hyd/Å | 2.3 | 2.8 | 2.7 | 3.0 |
| S ClT/10−3 K−1 | −2 | 1 | 0.4 | — |
| S IT/10−3 K−1 | −3.32 | 0.15 | −0.25 | 0.65 |
3.1 Chloride salts
In this section, we analyze the thermodiffusion data of LiCl, NaCl and KCl in water. Fig. 2(a) shows the Soret coefficient at 25 °C as a function of molality. We use the literature data for sodium and potassium chloride16,32–37 and omit the sharp minimum observed by Gaeta,16 which was not reproduced by the other groups.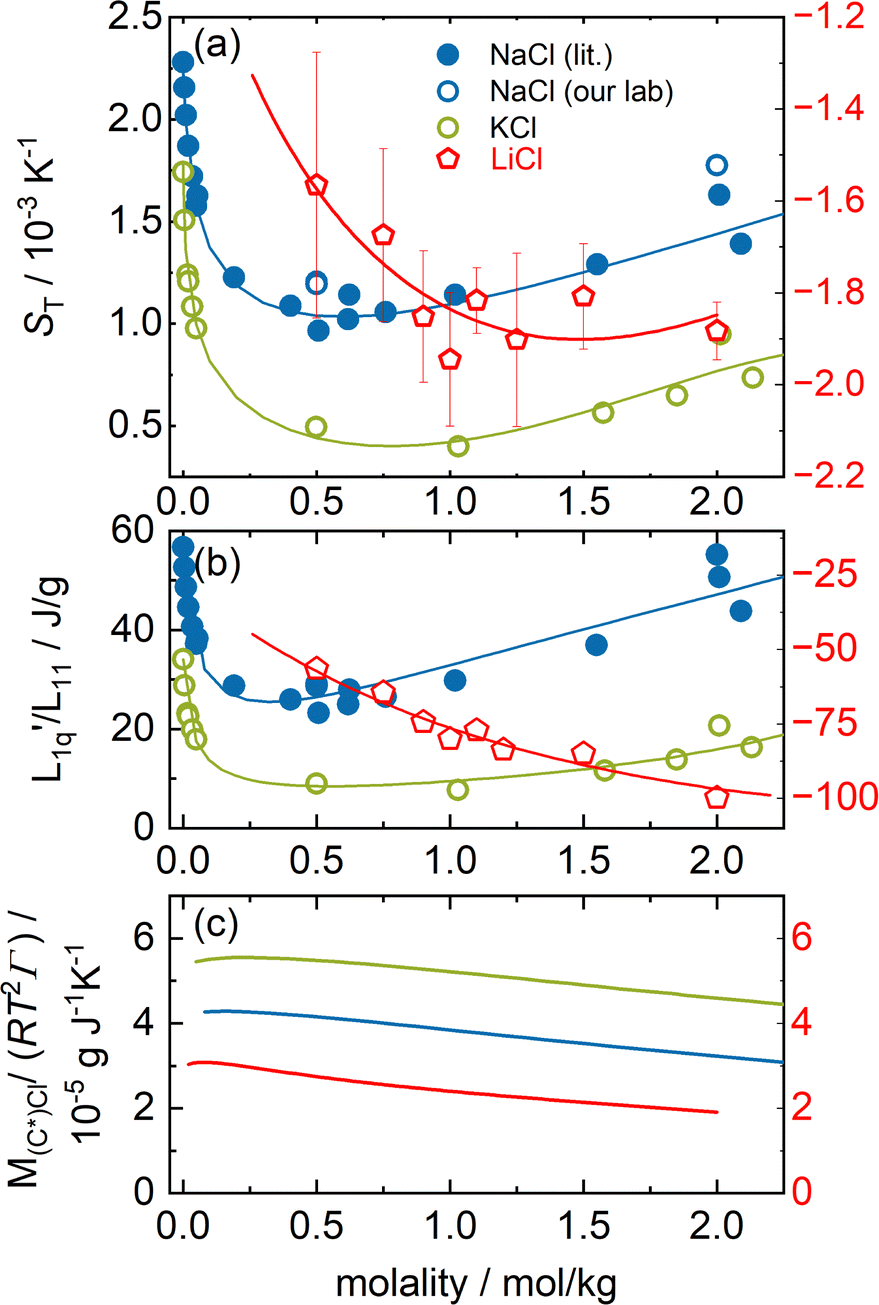 | ||
Fig. 2 (a) Soret coefficients of sodium and potassium chloride in water as a function of molality at T = 25 °C. We used the Soret coefficients of NaCl (filled blue symbols) and KCl (open green symbols) from the literature16,32–37 and we added our TDFRS measurements (open blue symbols). For comparison, we show the recently published data for lithium chloride at T = 25 °C.22 Note that in all three parts of the figure, the data for LiI are plotted against the right red y-axis. The red line in the case of LiCl corresponds to the green line in Fig. 5 in ref. 22, where the Onsager ratio was approximated by an exponential function. The solid lines for NaCl and KCl are a guide for the eye. (b) The ratio of the phenomenological Onsager coefficients 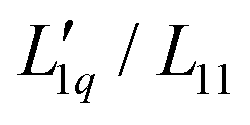 for NaCl and KCl at 25 °C calculated from the experimental data according to eqn (3). The solid lines are a guide to the eye. (c) M(C*)Cl/(RT2Γ) at 25 °C for sodium and potassium chloride. C* stands for the corresponding cation. Further details can be found in the text. for NaCl and KCl at 25 °C calculated from the experimental data according to eqn (3). The solid lines are a guide to the eye. (c) M(C*)Cl/(RT2Γ) at 25 °C for sodium and potassium chloride. C* stands for the corresponding cation. Further details can be found in the text. | ||
We have added two data points from our laboratory that are consistent with the published data. Note that one point is outside the concentration range in Fig. 2. The experimental data of all three systems show a minimum of the Soret coefficient with concentration. While NaCl and KCl show thermophobic behavior (ST > 0), LiCl exhibits thermophilic behavior over the entire concentration range (ST < 0). The concentration, mmin, where the Soret coefficient for LiCl is minimal is almost twice as high as that of the other two salts. Note that mmin for NaCl (mmin ≈ 0.6 mol kg−1) and KCl (mmin ≈ 0.7 mol kg−1) could only be observed using a conductometric method.36,45 With our optical method we cannot examine solutions below 0.5 mol kg−1, so we are not able to resolve the minimum.
We use the published mean ionic activity coefficients γ±46 to calculate the thermodynamic factor RT2Γ/Msalt of all three salts, and next obtain from these and the experimental values of the Soret coefficients the ratios of their Onsager coefficients 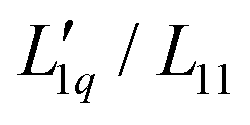 (see eqn (3)). The mean ionic activity coefficients γ± and also the calculated rational activity coefficient γW of all salts are shown in the ESI,† Fig. S1. The procedure for calculating the rational activity coefficients assuming complete dissociation or complete association is described in ESI,† Section S3. Further information including values of Γ, diffusion coefficient D and degrees of dissociation α for all chloride salts can be found in ESI,† Fig. S2–S4.
(see eqn (3)). The mean ionic activity coefficients γ± and also the calculated rational activity coefficient γW of all salts are shown in the ESI,† Fig. S1. The procedure for calculating the rational activity coefficients assuming complete dissociation or complete association is described in ESI,† Section S3. Further information including values of Γ, diffusion coefficient D and degrees of dissociation α for all chloride salts can be found in ESI,† Fig. S2–S4.
First we observe that the thermodynamic factors in all three cases exhibit a very small maximum at very low concentrations, while beyond these they monotonously decay (see Fig. 2(c)). Furthermore we notice that the thermodynamic factors increase with increasing Pauling radii.
Secondly, the ratio of Onsager factors 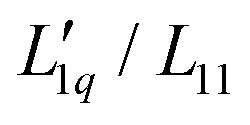 , called L-ratio from now on, for NaCl and KCl exhibit a minimum as a function of concentration (see Fig. 2(b)). The concentrations at which
, called L-ratio from now on, for NaCl and KCl exhibit a minimum as a function of concentration (see Fig. 2(b)). The concentrations at which 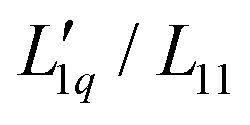 reaches a minimum are mLmin ≈ 0.25 mol kg−1 and mLmin ≈ 0.4 mol kg−1 for NaCl and KCl respectively. These are slightly lower than those for ST. The L-ratio of LiCl shows only an exponential decay with concentration and no minimum. Even so, in combination with the thermodynamic factor, it produces a very shallow minimum.22 The behavior of Li salts often deviates from that of the other alkali earth metals due to its small size and high ionization energy.47 In contrast to the thermodynamic factor, L-ratio does not increase monotonously with the size of the Pauling radius of the cations. Actually, the order of L-ratio curves for NaCl and KCl is reversed. This gives rise to a similar swap of the ST curves. Such a swapping was already observed by Balos et al.48 when they analyzed the concentration dependence of the amplitude of the THz Kerr effect. This effect might be because the exchange of water molecules becomes easier with K compared to the other ions.42
reaches a minimum are mLmin ≈ 0.25 mol kg−1 and mLmin ≈ 0.4 mol kg−1 for NaCl and KCl respectively. These are slightly lower than those for ST. The L-ratio of LiCl shows only an exponential decay with concentration and no minimum. Even so, in combination with the thermodynamic factor, it produces a very shallow minimum.22 The behavior of Li salts often deviates from that of the other alkali earth metals due to its small size and high ionization energy.47 In contrast to the thermodynamic factor, L-ratio does not increase monotonously with the size of the Pauling radius of the cations. Actually, the order of L-ratio curves for NaCl and KCl is reversed. This gives rise to a similar swap of the ST curves. Such a swapping was already observed by Balos et al.48 when they analyzed the concentration dependence of the amplitude of the THz Kerr effect. This effect might be because the exchange of water molecules becomes easier with K compared to the other ions.42
3.2 Iodide salts
In a recent publication20 we discussed the iodide salts (LiI, NaI and KI) emphasizing the role of overlapping hydration shells. Here, we will concentrate on the two factors that emerge naturally in the irreversible thermodynamics description of the Soret coefficient. In the next section we will address the importance of hydration shells again in relation to the thermodynamic factors and L-ratios described here. We extend the systems discussed in our previous paper by including the experimental results that we described in Section 2.As before, we use the published mean ionic activity coefficients γ±46 of all four salts at T = 25 °C to calculate the thermodynamic factor RT2Γ/Msalt and next the ratio of Onsager coefficients L-ratio. The mean ionic activity coefficients γ± and also the calculated rational activity coefficient of all salts are shown in ESI,† Fig. S5. Further information including values of Γ and α for all iodide salts can be found in the ESI,† Fig. S6 and S7.
In Fig. 3(a) the Soret coefficient of NaI and KI shows a clear minimum as a function of concentration, also LiI has a shallow minimum. On the other hand, the Soret coefficient of CsI is seen to increase monotonously, almost linearly with concentration at this temperature. In Section 2 we have seen that with increasing temperatures also CsI develops a minimum in its Soret coefficient at particular concentrations.
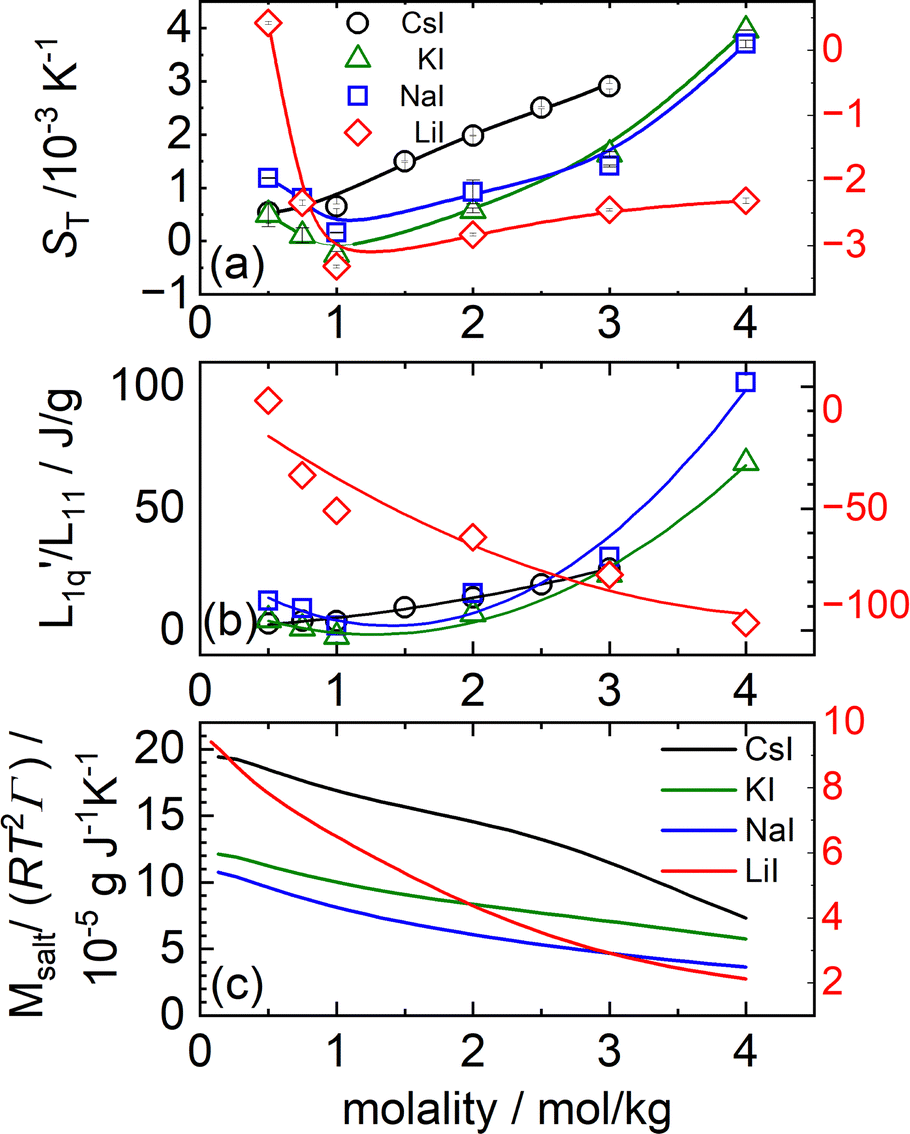 | ||
Fig. 3 (a) Soret coefficients of LiI, NaI, KI and CsI in water as a function of molality at T = 25 °C. The solid lines are B-splines and serve as a guide for the eye. Note that in all three parts of the figure, the data for LiI are plotted against the right red y-axis. (b) The ratio of the phenomenological Onsager coefficients  for NaI, KI and CsI at 25 °C calculated from the experimental data according to eqn (3). The solid lines are second-order polynomial fits. (c) Msalt/(RT2Γ) at 25 °C for different iodide salts. for NaI, KI and CsI at 25 °C calculated from the experimental data according to eqn (3). The solid lines are second-order polynomial fits. (c) Msalt/(RT2Γ) at 25 °C for different iodide salts. | ||
As with the chlorides all thermodynamic factors are smoothly decaying with concentration (see Fig. 3(c)). Even the very weak maximum at low concentrations is absent in this case.
In Fig. 3(b), we see that L-ratio, 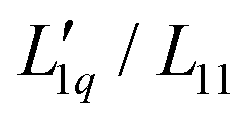 , as a function of concentration exhibits a minimum for NaI and KI. The concentrations at which the L-ratio reaches its minimum in these cases are mLmin ≈ 1.4 mol kg−1 and mLmin ≈ 1.2 mol kg−1 for NaI and KI, respectively. In contrast to the various chloride systems discussed above, these are slightly larger than the concentrations at which ST reaches its minima. As with LiCl, LiI also has an exponentially decaying L-ratio, while still showing a shallow minimum in the Soret curve. Apparently Li salts are intrinsically different from those with the other cations, as corroborated by numerical values of L-ratio, which are very different from those in the other cases. The L-ratio of CsI increases monotonically, in fact linearly with concentration.
, as a function of concentration exhibits a minimum for NaI and KI. The concentrations at which the L-ratio reaches its minimum in these cases are mLmin ≈ 1.4 mol kg−1 and mLmin ≈ 1.2 mol kg−1 for NaI and KI, respectively. In contrast to the various chloride systems discussed above, these are slightly larger than the concentrations at which ST reaches its minima. As with LiCl, LiI also has an exponentially decaying L-ratio, while still showing a shallow minimum in the Soret curve. Apparently Li salts are intrinsically different from those with the other cations, as corroborated by numerical values of L-ratio, which are very different from those in the other cases. The L-ratio of CsI increases monotonically, in fact linearly with concentration.
Finally, we notice a similar behavior with increasing Pauling radii as found with the chlorides. All thermodynamic factors increase with increasing Pauling radius, including one of the additional CsI. On the other hand, L-ratios increase with Pauling radius from LiI to NaI, after which those of KI decrease, falling below those of NaI. After that the increasing trend of the L-ratio with the Pauling radius seems to resume, with CsI, having the largest Pauling radius, also having the largest L-ratio, at least in the intermediate range of concentrations. At the very lowest and the very largest concentrations, the L-ratio of CsI becomes smaller than those of KI and NaI. In this respect it is interesting to notice that CsI seems to behave more in line with the other salts at higher temperatures (see Section 2.2).
3.3 Potassium acetate, potassium thiocyanate and sodium thiocyanate
In this section we investigate two slightly non-spherical anions, the acetate anion, which is only approximately spherical, and the thiocyanate anion, which is more rodlike.As can be seen in Fig. 4(a), contrary27,28 to sodium acetate, the Soret coefficient of potassium acetate does exhibit a minimum as a function of concentration for the given temperature T = 25 °C, although barely so. For the thiocyanates, both sodium thiocyanate and potassium thioacyanates exhibit such a minimum.
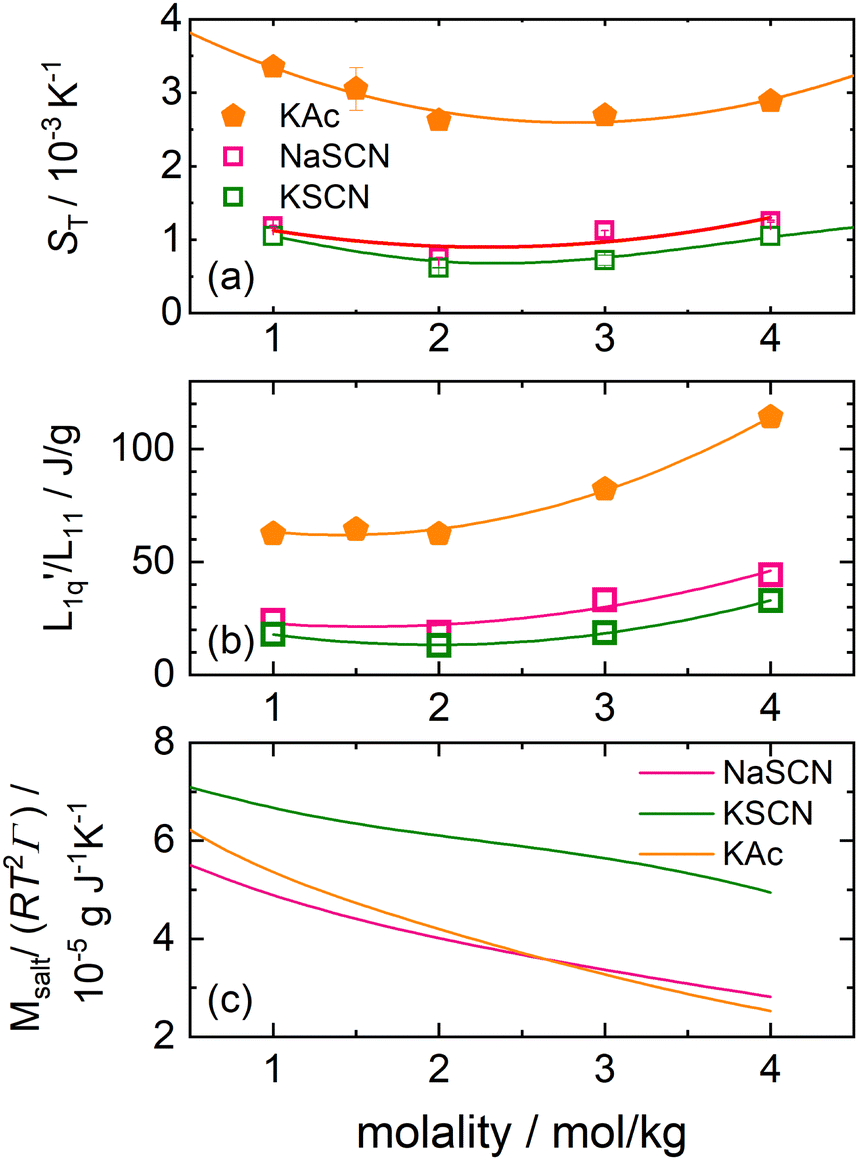 | ||
Fig. 4 (a) Soret coefficient of potassium acetate, sodium thiocyanate and potassium thiocyanate in water as a function of concentration at T = 25 °C.28 The solid lines are second order polynomial fits. (b) The ratio of the phenomenological Onsager coefficients  for KAc, NaSCN and KSCN at 25 °C is calculated from the experimental data according to eqn (3). The solid lines are second-order polynomial fits. (c) Msalt/(RT2Γ) at 25 °C for KAc, NaSCN and KSCN in water. for KAc, NaSCN and KSCN at 25 °C is calculated from the experimental data according to eqn (3). The solid lines are second-order polynomial fits. (c) Msalt/(RT2Γ) at 25 °C for KAc, NaSCN and KSCN in water. | ||
As with the other salts, we have calculated for all three cases the thermodynamic factors, using data from Hamer and Wu,46 and next the L-ratios (see the ESI,† Section S2 for further information). The values for γ± and γW of all salts are shown in ESI,† Fig. S8 and S9.
The thermodynamic factors are monotonously decreasing in all three cases, while the L-ratios exhibit a minimum (see Fig. 4(b) and (c)). For the cyanates, the thermodynamic factors are the largest for the largest Pauling ratio, while for the corresponding Onsager ratios  the opposite is true. This is in agreement with what we have found for the other salts.
the opposite is true. This is in agreement with what we have found for the other salts.
3.4 Guanidinium chloride
In the following, we will discuss the concentration dependence of the Soret coefficients of aqueous GdmCl solutions in more detail. The guanidinium cation is not spherical but disk-shaped. It is characterized by flat hydrophobic surfaces and three amine groups that enable directional hydrogen bonding along the edges. Fig. 5(a) shows the concentration dependence of the Soret coefficient for aqueous solutions of GdmCl at different temperatures between 15 °C and 35 °C. The lines fit with a second order polynomial. Among the monovalent guanidinium salts studied so far, GdmCl is the only salt that shows a non-monotonic behavior of ST in the investigated concentration range.29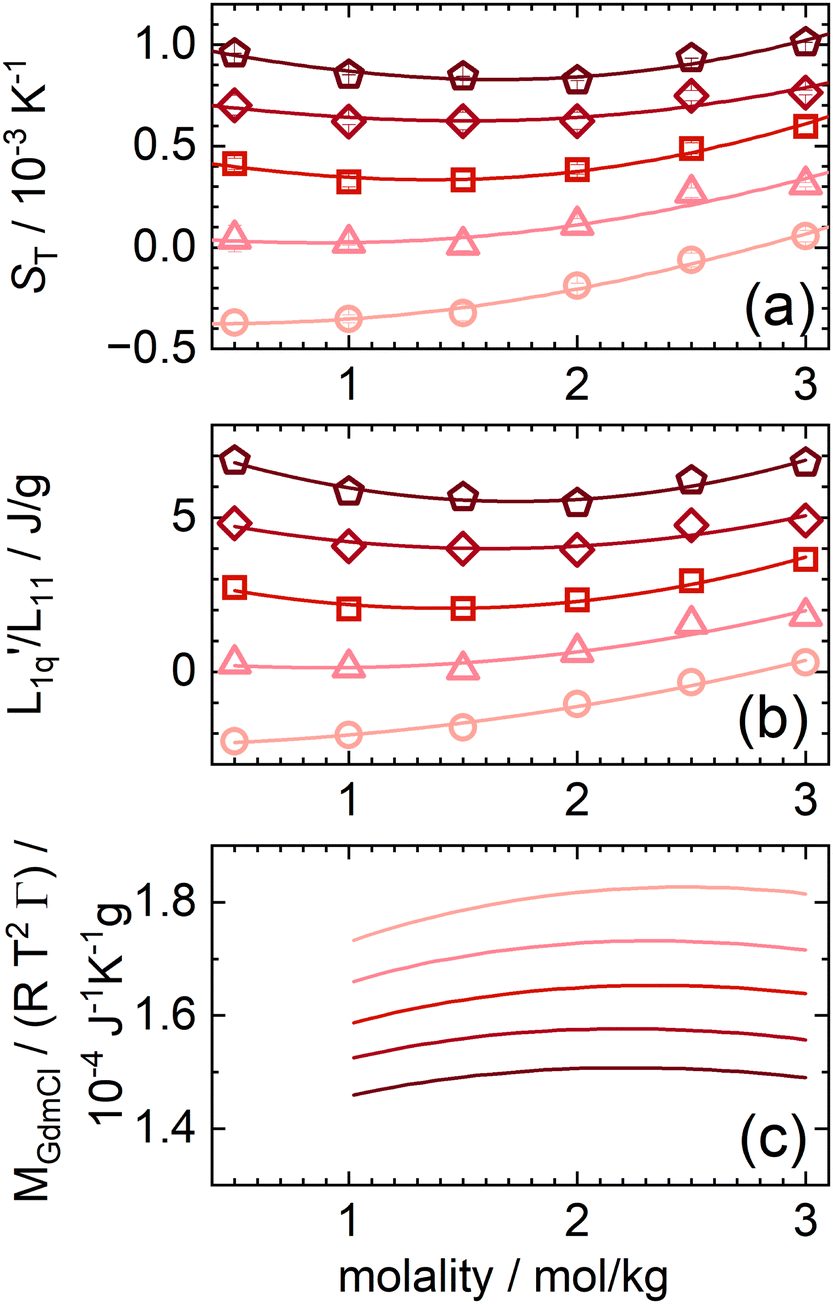 | ||
Fig. 5 (a) Soret coefficients of guanidinium chloride solutions as a function of concentration for temperatures between 15 °C and 35 °C. The lines are quadratic fits. (b) The ratio of the phenomenological Onsager coefficients 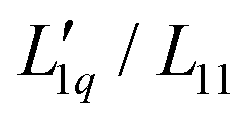 , calculated from the experimental data according to eqn (3). The solid lines are quadratic fits. The temperature dependent red-like symbols correspond to the mean activity coefficients of Makhatadze et al.49 (c) MGdmCl/(RT2Γ) for different temperatures between 15 °C (light color) and 35 °C (dark color).49 , calculated from the experimental data according to eqn (3). The solid lines are quadratic fits. The temperature dependent red-like symbols correspond to the mean activity coefficients of Makhatadze et al.49 (c) MGdmCl/(RT2Γ) for different temperatures between 15 °C (light color) and 35 °C (dark color).49 | ||
We used the mean ionic activity coefficient γ± from Makhatadze et al.49 to calculate MGdmCl/(RT2Γ) for all investigated temperatures and concentrations and plotted them in Fig. 5(c). The values for γ± and also γW of all salts are shown in ESI,† Fig. S10. Fig. 5(b) shows the calculated values of L-ratio (symbols) for all investigated temperatures as a function of concentration, which are well described by a polynomial fit. Compared to the 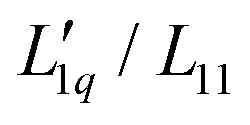 ratio, the factor MGdmCl/(RT2Γ) is smaller by two orders of magnitude, which preserves the minimum when the two factors are multiplied. In this study, we observe an increase in the
ratio, the factor MGdmCl/(RT2Γ) is smaller by two orders of magnitude, which preserves the minimum when the two factors are multiplied. In this study, we observe an increase in the 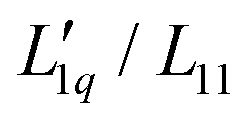 ratio at lower temperatures (T < 25 °C) and a passage through a minimum with increasing concentration at higher temperatures (T ≥ 25 °C). At T = 25 °C the shape of the concentration dependence of L-ratio is consistent with the spherical salts, except for lithium. The factor MGdmCl/(RT2Γ) shows a maximum at 2.3 mol kg−1, which is a much higher concentration compared to the spherical salts. At the same time, the variation of MGdmCl/(RT2Γ) is only ±2% around its mean value, while the factor of the other salts shows a more pronounced concentration dependence between 10–50%. Whether this is related to the non-spherical shape or the amphiphilic character of GdmCl needs further investigation.
ratio at lower temperatures (T < 25 °C) and a passage through a minimum with increasing concentration at higher temperatures (T ≥ 25 °C). At T = 25 °C the shape of the concentration dependence of L-ratio is consistent with the spherical salts, except for lithium. The factor MGdmCl/(RT2Γ) shows a maximum at 2.3 mol kg−1, which is a much higher concentration compared to the spherical salts. At the same time, the variation of MGdmCl/(RT2Γ) is only ±2% around its mean value, while the factor of the other salts shows a more pronounced concentration dependence between 10–50%. Whether this is related to the non-spherical shape or the amphiphilic character of GdmCl needs further investigation.
4 Discussion
In the previous section, we analyzed Soret coefficients for a variety of salt solutions by factorising them into a product of two factors, a thermodynamic factor and a kinetic factor expressed as the ratio of two Onsager coefficients. Our interest is in systems that exhibit a minimum for Soret as a function of concentration.In all cases we found that the thermodynamic factors are smoothly decaying or increasing (guanidinium chloride) functions of concentrations. The L-ratio curves on the other hand are convex in all cases, and transmit their minimum to the Soret curves after multiplication with the thermodynamic factors. Only LiCl is exceptional in this respect.20 We conclude that: the non-monotonous behavior of Soret coefficients with concentration is caused by a non-monotonous behavior of the Onsager ratios.
A second interesting feature that we found is related to the dependence of the various curves on the size of the anions. For those cases where the anion was combined with more than one cation – the chlorides, iodides and the thiocyanides – we found that all curves of thermodynamic factors move upwards with increasing Pauling radius of the cations. A similar trend was found for the L-ratio curves, which was interrupted, however, when the L-ratio curves of potassium salts dropped below those of the sodium salts. After this swap, in the case of the iodides, the CsI curve moved up again above that of KI. Although the data are admittedly rather scarce we want to discuss this point in relation to the hydration shells around the various cations.
In Fig. 6 we have plotted the radial distribution functions of oxygen atoms around Li+, Na+, K+ and Cs+-cations in aqueous solutions. The data were scanned from those in a paper by Singh et al.42 With increasing radius the Li+-oxygen radial distribution clearly has a rather strong first peak, which corresponds to a well defined hydration shell. Continuing to larger values of r a very deep extended minimum follows and next a weak second peak. When going to the heavier atoms the whole curve gradually shifts to the right, while at the same time the two peaks become lower and broader and the minimum gradually disappears.
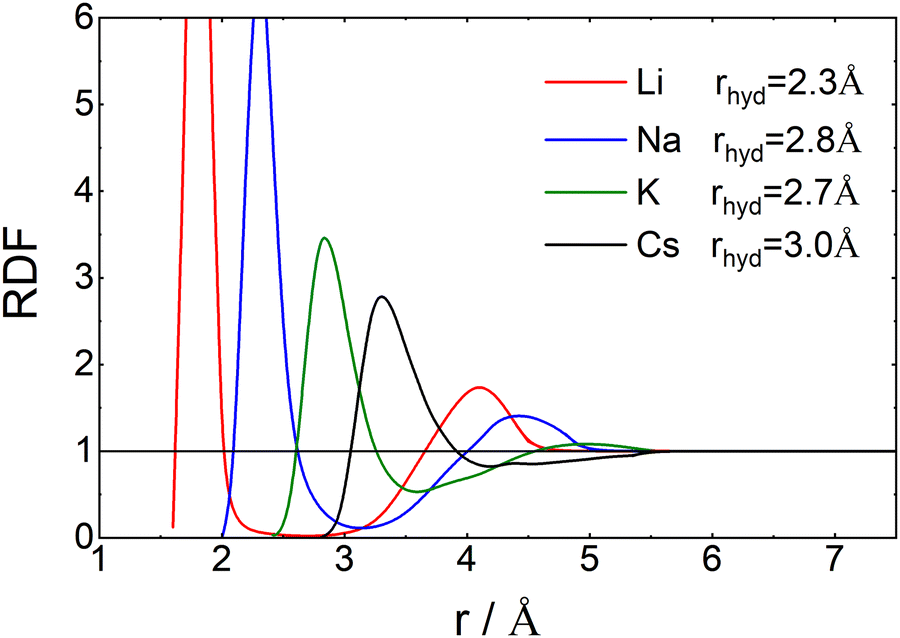 | ||
| Fig. 6 Radial distribution functions, RDF(r), as a function of the distance between the oxygen for Li, Na, K and Cs in water. The data points were scanned by hand from Fig. 4a of Singh et al.42 For large radii, the scanned function was smoothed with an exponential tail to estimate the mean radius of hydration. For further information see ESI,† Section S4. | ||
In order to discuss the thermodynamic factor, we notice that it is proportional to the inverse of the derivative of the chemical potential of salt with respect to its concentration. Although energetic contributions are important, especially at high concentrations, it is to a large extent the size of the salt, represented here by its cation, that determines the chemical potential; this can be inferred from the Widom insertion equation.50 As a consequence, when going to heavier ions it is their excluded volume that matters. By that, we mean the volume that the ion excludes for the water molecules. This volume is characterized by its radius, which is equal to the Pauling radius of the ion plus that of an oxygen atom; values are given in the third row of Table 1. These values correspond roughly to the point where g(r) goes through unity for the first time. This explains why thermodynamic factors increase monotonously with Pauling radii for all concentrations.
Now let's consider the Li+–O radial distribution curve again. The extended and very deep minimum after the first peak implies that the rate of exchange of water molecules between the first and second peak, and next to the bulk will be very small. Already in 1906 Einstein51,52 realized that in order to explain data on the diffusion of sugar molecules in aqueous solutions and viscosity coefficients of such solutions, the radius of a sugar molecule must be taken to be that of the pure sugar molecule augmented with the thickness of shell of attached water. We will take the same point of view and consider how the radius of an ion including its attached water molecules must be estimated when the radial distribution function gradually becomes less pronounced as we found above with increasing mass of the anion involved. What really matters for such an estimate is the surplus of oxygen density around the ion over its bulk value. We therefore suggest to define the ‘mean hydrated radius’ of the ion as
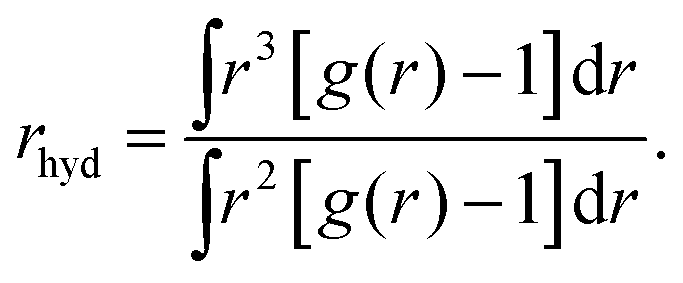 | (5) |
For colloids it is customary to talk about the ‘hydrodynamic radius’ and treat it as an empirical parameter to describe the experimental data.
The results of our calculations are given in the fourth row of Table 1. Clearly rhyd increases when going from Li+ to Na+, next it decreases when going to K+ and increases again when going to Cs+. This behavior is in agreement with the changes that occur in L-ratio and Soret curves when going through the list of ions from Li+ to Cs+. We therefore summarize that the sequence of L-ratio versus concentration curves increases monotonously with rhyd.
It is interesting to mention at this point the results of Singh et al.42 who studied various cations using terahertz spectroscopy and observed a rapid exchange between hydrated water and bulk water for larger cations.42 For cesium (Cs) they found that the amplitudes of the two fast modes associated with hydrogen bond breaking increase, while the slow mode related to tightly bound water disappears. These results agree with dielectric measurements by Buchner et al.41 The absence of a strongly attached hydration layer in this case may be related to the absence of a minimum in ST as a function of concentration for CsI for a temperature of 25 °C. Only at higher temperatures, in this case also a minimum develops.
5 Summary and conclusions
In this work, we investigate the concentration dependence of ST for 11 different 1:1 aqueous electrolytes in water, with special attention to the non-monotonic behavior of ST as a function of concentration. With the exception of the thermodiffusive measurements of aqueous CsI solutions, all data have been published either by other researchers or by us. We continued our analysis based on the representation given in eqn (3) and discussed the two factors Msalt/(RT2Γ) and 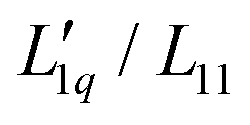 in relation to the hydration of the salts.
in relation to the hydration of the salts.
Our results show that with the exception of GdmCl, the thermodynamic factor Msalt/(RT2Γ) decreases with increasing salt concentration. Interestingly, it varies less for GdmCl compared to the other spherical salts and shows a flat maximum. In addition, the thermodynamic factor increases with increasing Pauling radius of the cation. For salts with the same anion, Msalt/(RT2Γ) follows the sequence: Li < Na < K < Cs. In contrast to previous findings, our analysis has shown that for most of the salts discussed (chlorides, iodides, acetate and thiocyanates) the Onsager ratio (L-ratio) itself has a minimum as a function of salt concentration. Exceptions are lithium salts, which show an exponential decrease, and CsI, which shows a linear increase at 25 °C. These deviations could be related to the unique hydration properties of Li and Cs ions, which are characterized by the densest and loosest hydration shells, respectively. The minimum in L-ratio of GdmCl (disk-shaped) evolves only at high temperatures and the minimum of KAc is not so clear, but overall, we can conclude that the non-monotonic behavior of the Soret coefficients with concentration is caused by a non-monotonic behavior of the Onsager ratio and not solely by the thermodynamic factor.
The hydration of salts plays an important role in their thermodiffusive behavior. Thus, the sequence of the L-ratio increases monotonically with rhyd of the cation versus concentration curves. Here, rhyd is the radius of the ion plus the attached water molecules. For cations with the same anion, rhyd follows the order Li < K < Na < Cs. Note that K has a slightly larger hydration radius compared to Na, which means that the curves for the L-ratio and ST are reversed for chloride and iodide salts.
Finally, we conclude that further systematic simulations and the development of new theoretical approaches are required for a deeper understanding of the non-monotonic behavior of ST in electrolyte solutions.
Author contributions
All authors conceived and planned the experiments. BR carried out the experiments and analyzed the data. All authors contributed to the interpretation of the results and writing the manuscript. All authors provided critical feedback and helped shape the research, analysis and manuscript.Data availability
Data for this article are available on Zenodo at https://doi.org/10.5281/zenodo.14793534.Conflicts of interest
There are no conflicts to declare.Appendix
As is well known,53,54 if we subdivide a homogeneous binary system into two pieces of constant volume and redistribute energy and masses among the two, the change of entropy is given by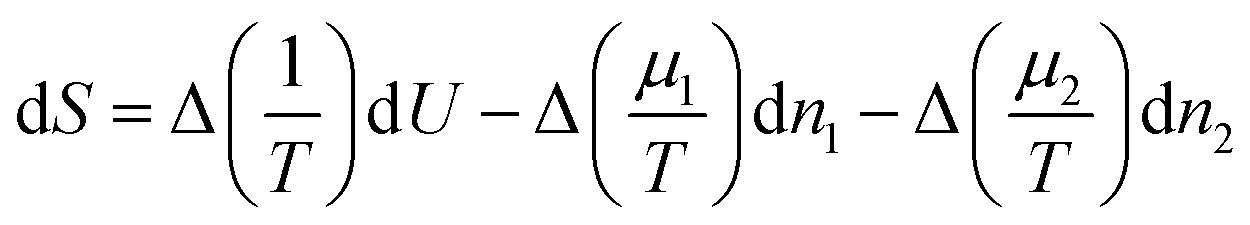 | (6) |
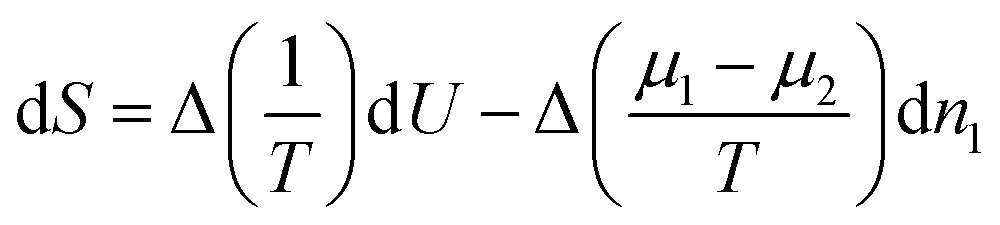 | (7) |
Equilibrium will only occur if Δ(1/T) and Δ((μ1−μ2)/T) are zero, i.e. when the temperature and both chemical potentials are equal in both subsystems; the pressure will then automatically be equal in both subsystems as well. If this is not the case, energy and masses will re-distribute such that the entropy increases. So, the factors in front of dU and dn1 may be considered as driving forces for fluxes of energy and mass.1 In case the system is only slightly out of equilibrium these fluxes may be assumed to be linear in the driving forces.
In the more general case of smoothly varying temperatures and chemical potentials, gradients replace the differences in eqn (7). Moreover, to include the possibility of flowing matter, we consider mass elements as is done in hydrodynamics. So 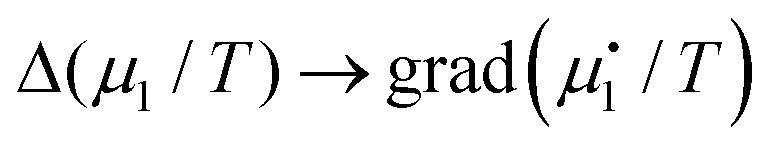 etc., where the dot refers to ‘specific’ etc. The mass flux of component number one then reads
etc., where the dot refers to ‘specific’ etc. The mass flux of component number one then reads
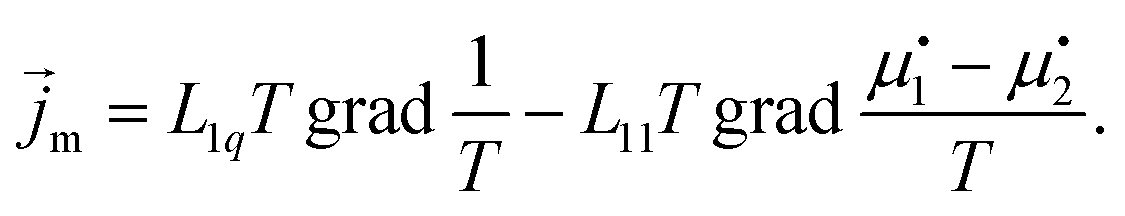 | (8) |
The additional factor of T is conventional. In agreement with the experimental existence of thermodiffusion we also allow the driving force for energy transport to give rise to mass transport. The coefficients Lij are called Onsager coefficients. Changing to weight fractions c and temperature T as independent variables we get
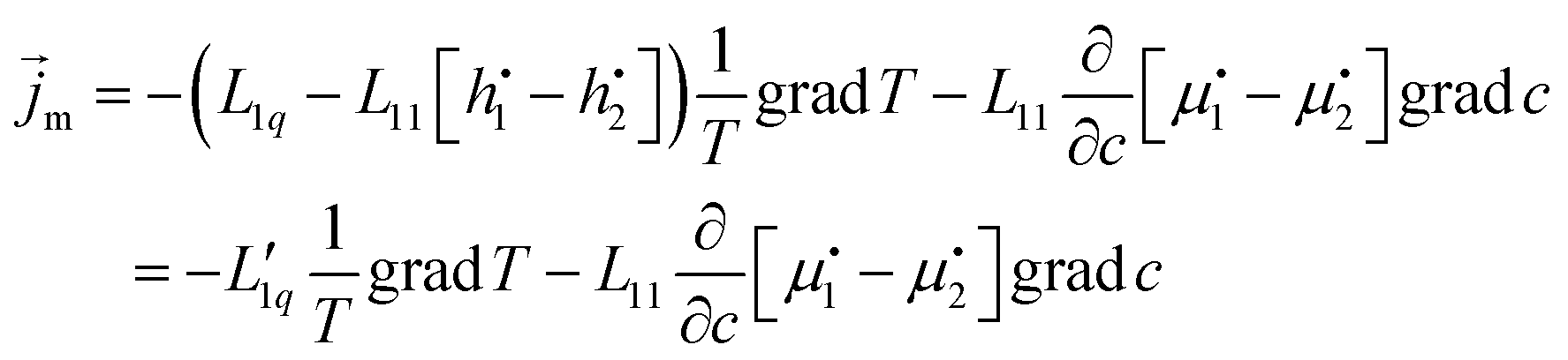 | (9) |
The second equal sign defines 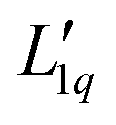 . Setting the flux equal to zero and using the final version in eqn (2) we calculate the Soret coefficient. Eqn (3) is then obtained after using the Gibbs–Duhem equation to transform the derivative of
. Setting the flux equal to zero and using the final version in eqn (2) we calculate the Soret coefficient. Eqn (3) is then obtained after using the Gibbs–Duhem equation to transform the derivative of 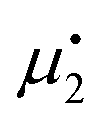 into a derivative of
into a derivative of 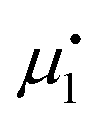 and transforming specific quantities into molar quantities. Note that
and transforming specific quantities into molar quantities. Note that 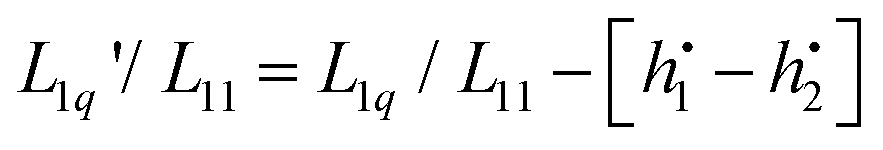 , where L1q/L11 is often called ‘heat of transfer’ and is denoted by Q*.
, where L1q/L11 is often called ‘heat of transfer’ and is denoted by Q*.
Since we do not have access to enthalpies for all of our systems, we cannot use L1q, but must restrict ourselves to using 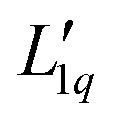 .
.
As a final remark, let us mention that the non-primed Onsager coefficients can be calculated through so called Green–Kubo integrals.21,55,56 These are integrals of time correlation functions of products of the fluxes corresponding to the indexes of the Onsager coefficient being studied. So the correlation functions describe the decay of one flux given the initial value of the other flux, and as such comprise the physics laying behind the Onsager coefficients showing that these are dynamic properties. Nevertheless, in case L1q and L11 have a common dynamic factor while otherwise being described by thermodynamic properties, the heat of transfer becomes a thermodynamic property. In the present paper, it is shown that such is not the case for ionic salt solutions.
Acknowledgements
The authors are thankful to Jan Dhont and Holger Gohlke for fruitful discussions. The authors are grateful to Shilpa Mohanakumar for her help. The authors thank Peter Lang for his generous support of our work. BR acknowledges the support from the International Helmholtz Research School of Biophysics and Soft Matter (BioSoft).Notes and references
- S. R. de Groot, Thermodynamics of irreversible processes, North Holland, Amsterdam, 1966 Search PubMed.
- S. Kjelstrup, D. Bedeaux, E. Johannessen and J. Gross, Non-equilibrium thermodynamics for engineers, World Scientific, Hackensack, NJ, 2nd edn, 2017 Search PubMed.
- F. Montel, J. Bickert, A. Lagisquet and G. Galliero, J. Pet. Sci. Eng., 2007, 58, 391–402 CrossRef CAS.
- G. Galliero and F. Montel, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 041203 CrossRef PubMed.
- W. Köhler and K. I. Morozov, J. Non-Equilib. Thermodyn., 2016, 41, 151–197 Search PubMed.
- H. Pasch, Chromatographia, 2021, 84, 525–530 CrossRef CAS.
- P. Baaske, F. M. Weinert, S. Duhr, K. H. Lemke, M. J. Russell and D. Braun, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9346–9351 CrossRef CAS PubMed.
- S. A. I. Seidel, P. M. Dijkman, W. A. Lea, G. van den Bogaart, M. Jerabek-Willemsen, A. Lazic, J. S. Joseph, P. Srinivasan, P. Baaske, A. Simeonov, I. Katritch, F. A. Melo, J. E. Ladbury, G. Schreiber, A. Watts, D. Braun and S. Duhr, Methods, 2013, 59, 301–315 CrossRef CAS PubMed.
- D. Niether and S. Wiegand, J. Phys.: Condens. Matter, 2019, 31, 503003 CrossRef CAS PubMed.
- T. J. Salez, B. T. Huang, M. Rietjens, M. Bonetti, C. Wiertel-Gasquet, M. Roger, C. L. Filomeno, E. Dubois, R. Perzynski and S. Nakamae, Phys. Chem. Chem. Phys., 2017, 19, 9409–9416 RSC.
- C.-G. Han, X. Qian, Q. Li, B. Deng, Y. Zhu, Z. Han, W. Zhang, W. Wang, S.-P. Feng, G. Chen and W. Liu, Science, 2020, 368, 1091–1098 CrossRef CAS PubMed.
- Y. Jia, Q. Jiang, H. Sun, P. Liu, D. Hu, Y. Pei, W. Liu, X. Crispin, S. Fabiano, Y. Ma and Y. Cao, Adv. Mater., 2021, 33, e2102990 CrossRef PubMed.
- C. Soret, Arch. Geneve, 1879, 3, 48–64 Search PubMed.
- C. C. Tanner, Trans. Faraday Soc., 1927, 23, 75–95 RSC.
- D. R. Caldwell, J. Phys. Chem., 1975, 79, 1882–1884 CrossRef CAS.
- F. S. Gaeta, G. Perna, G. Scala and F. Bellucci, J. Phys. Chem., 1982, 86, 2967–2974 CrossRef CAS.
- J. Colombani, J. Bert and J. Dupuy-Philon, J. Chem. Phys., 1999, 110, 8622–8627 CrossRef CAS.
- M. Jokinen, J. A. Manzanares, K. Kontturi and L. Murtomäki, J. Membr. Sci., 2016, 499, 234–244 CrossRef CAS.
- S. Di Lecce, T. Albrecht and F. Bresme, Phys. Chem. Chem. Phys., 2017, 19, 9575–9583 RSC.
- S. Mohanakumar, H. Kriegs, W. J. Briels and S. Wiegand, Phys. Chem. Chem. Phys., 2022, 24, 27380–27387 RSC.
- O. R. Gittus and F. Bresme, Phys. Chem. Chem. Phys., 2023, 25, 1606–1611 RSC.
- N. Lee, S. Mohanakumar, W. J. Briels and S. Wiegand, Phys. Chem. Chem. Phys., 2024, 26, 7830–7836 RSC.
- S. R. de Groot and P. Mazur, Non-equilibrium Thermodynamics, Dover, New York, 1984 Search PubMed.
- R. Haase, Thermodynamik der Irreversiblen Prozesse, Steinkopff, Heidelberg, 1963, vol. 8 Search PubMed.
- L. J. T. M. Kempers, J. Chem. Phys., 1989, 90, 6541–6548 CrossRef CAS.
- J. Dhont and W. J. Briels, J. Colloid Interface Sci., 2024, 666, 457–471 CrossRef CAS PubMed.
- S. Mohanakumar, J. Luettmer-Strathmann and S. Wiegand, J. Chem. Phys., 2021, 154, 084506 CrossRef CAS PubMed.
- S. Mohanakumar and S. Wiegand, Eur. Phys. J. E: Soft Matter Biol. Phys., 2022, 45, 10 CrossRef CAS PubMed.
- B. A. Rudani, A. Jakubowski, H. Kriegs and S. Wiegand, J. Chem. Phys., 2024, 160, 214502 CrossRef CAS PubMed.
- G. D. Scott and D. M. Kilgour, J. Phys. D: Appl. Phys., 1969, 2, 863–866 CrossRef.
- P. A. Artola and B. Rousseau, Phys. Rev. Lett., 2007, 98, 125901-1 CrossRef PubMed.
- J. Chanu, J. Chim. Phys. Phys.-Chim. Biol., 1958, 55, 743–753 CrossRef.
- P. N. Snowdon and J. C. R. Turner, Trans. Faraday Soc., 1960, 56, 1812–1819 RSC.
- J. N. Agar and J. C. R. Turner, Proc. R. Soc. London, Ser. A, 1960, 255, 307–330 Search PubMed.
- C. D. Price, Tech. Rep. U.S.A.F, 1961, Contract no. AF(052)-99 (AD0276280) Search PubMed.
- D. G. Leaist and L. Hui, J. Phys. Chem., 1990, 94, 447–451 CrossRef CAS.
- F. Römer, Z. Wang, S. Wiegand and F. Bresme, J. Phys. Chem. B, 2013, 117, 8209–8222 CrossRef PubMed.
- A. Becker, W. Köhler and B. Müller, Ber. Bunsen-Ges. Phys. Chem. Chem. Phys., 1995, 99, 600–608 CrossRef CAS.
- P. Rossmanith and W. Köhler, Macromolecules, 1996, 29, 3203–3211 CrossRef CAS.
- S. Wiegand, H. Ning and H. Kriegs, J. Phys. Chem. B, 2007, 111, 14169–14174 CrossRef CAS PubMed.
- R. Buchner, W. Wachter and G. Hefter, J. Phys. Chem. B, 2019, 123, 10868–10876 CrossRef CAS PubMed.
- A. K. Singh, L. C. Doan, D. Lou, C. Wen and N. Q. Vinh, J. Chem. Phys., 2022, 157, 054501 CrossRef CAS PubMed.
- S. Iacopini, R. Rusconi and R. Piazza, Eur. Phys. J. E: Soft Matter Biol. Phys., 2006, 19, 59–67 CrossRef CAS PubMed.
- R. Heyrovská, Chem. Phys. Lett., 1989, 163, 207–211 CrossRef.
- J. N. Agar and J. C. R. Turner, J. Phys. Chem., 1960, 64, 1000–1003 CrossRef CAS.
- W. J. Hamer and Y.-C. Wu, J. Phys. Chem. Ref. Data, 1972, 1, 1047–1100 CrossRef CAS.
- CRC handbook of chemistry and physics: A ready-reference book of chemical and physical data, ed. D. R. Lide, CRC Press, Boca Raton, 84th edn, 2003 Search PubMed.
- V. Balos, N. K. Kaliannan, H. Elgabarty, M. Wolf, T. D. Kühne and M. Sajadi, Nat. Chem., 2022, 14, 1031–1037 CrossRef CAS PubMed.
- G. I. Makhatadze, J. Fernandez, E. Freire, T. H. Lilley and P. L. Privalov, J. Chem. Eng. Data, 1993, 38, 83–87 CrossRef CAS.
- D. Frenkel and B. Smit, Understanding molecular simulation: From algorithms to applications, Acad. Press, San Diego, 2nd edn, 2011, vol. 1 Search PubMed.
- A. Einstein, Ann. Phys. Chem., 1906, 19, 289–306 CrossRef CAS.
- A. Einstein, Investigations on the theory of the Brownian movement, Repr. of ed. Methuen, Dover Publ, New York, 1926 edn, 1946 Search PubMed.
- D. ter Haar and H. Wergeland, Elements of Thermodynamics, Addison-Wesley, 1966 Search PubMed.
- M. L. McGlashan, Chemical thermodynamics, Academic Press, London and New York, 1979 Search PubMed.
- R. Zwanzig, Annu. Rev. Phys. Chem., 1965, 16, 67–102 CrossRef CAS.
- J. M. Ortiz de Zárate and J. Sengers, Hydrodynamic Fluctuations in Fluids and Fluid Mixtures, Elsevier, Amsterdam, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04477k |
| This journal is © the Owner Societies 2025 |