
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Click and shift: the effect of triazole on solvatochromic dyes†
Jean Rouillon*a,
Carlos Benitez-Martin ab,
Morten Grøtlib and
Joakim Andréasson*a
ab,
Morten Grøtlib and
Joakim Andréasson*a
aDepartment of Chemistry and Chemical Engineering, Chemistry and Biochemistry, Chalmers University of Technology, SE-41296, Göteborg, Sweden. E-mail: a-son@chalmers.se
bDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-40530, Göteborg, Sweden
First published on 4th February 2025
Abstract
Solvatochromic dyes are prime candidates for exploring complex polarity-dependent biological processes. The design of novel dyes for these applications typically presents long synthetic routines. Herein, we report a concise synthesis of azido-functionalised push–pull fluorenes, belonging to the most potent groups of fluorosolvatochromic compounds. These fluorenes can be attached to multiple alkynes via the highly versatile copper-catalysed azide–alkyne cycloaddition (CuAAC). Our study focuses on the beneficial effect of the triazole, formed by CuAAC, comparing the simple push–pull fluorene FR1 with the novel model compound FR1TP. While the triazole in FR1TP is not conjugated to the π-system of the dye, the heterocycle surprisingly produces a bathochromic emission shift of around 50 nm. This effect, caused by triazole-induced LUMO stabilisation, was also observed for two other model compounds: FR2TP and FR1TM. All compounds display photophysical properties that are highly desired for in vivo imaging dyes, including remarkable two-photon absorption properties. The experimental results are supported by theoretical calculations. We anticipate that our findings will enable synthesis of new high-performance fluorosolvatochromic dyes for diverse applications.
Introduction
Modulating the optical properties of fluorescent materials is key to the design of new sensing or imaging technologies.1–5 Prime examples of such materials are solvatochromic dyes, which are characterized by displaying pronounced changes in their photophysical properties with solvent polarity. This distinctive feature has been pivotal to their use in bioimaging to study protein aggregation,6 proteome stress,7 lipid organisation of cell membranes,8 and detection of pathogenic bacteria, among others.9,10 These advanced applications are based on environment-dependent biophysical parameters and require solvatochromic fluorophores suitable for imaging. Desired properties include (i) excitation in the visible region, (ii) high fluorescence quantum yields, (iii) good photostability, and (iv) high two-photon cross-sections (for multiphoton microscopy). Since the development of PRODAN by Weber and Farris in 1979,11 several derivatives with all these properties have been developed.2Fluorene-based dyes, such as FR0, developed by Klymchenko and coworkers are push–pull systems with notable solvatochromic properties, often comparable to or even exceeding those of PRODAN-like dyes (Fig. 1).12,13 In this nomenclature, “FR” denotes “fluorene,” while the following number indicates the number of carbon atoms attached to the carbonyl group; a “0” specifies an aldehyde substituent. The behaviour of these push–pull fluorenes has been investigated by varying the nature of the acceptor groups and donor amines.14 The trends observed in their photophysical properties (solvatochromism, twisted internal charge-transfer, fluorescence in aqueous environments etc.) align with those observed for other classical push–pull systems.15–17
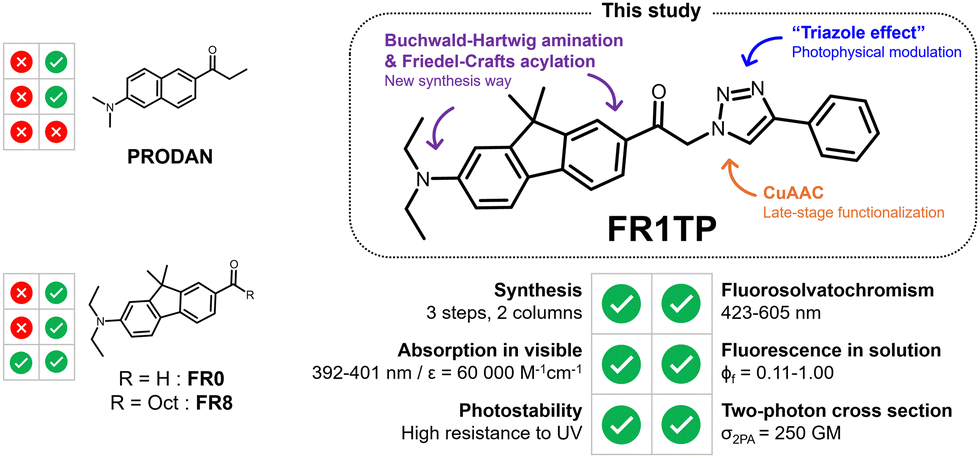 | ||
| Fig. 1 Main features of FR1TP and parent compounds (PRODAN, FR0 and FR8). | ||
However, unlike PRODAN and similar commercially marketed products (e.g., solvatochromic pyrene derivatives18), these compounds remain underexplored, not merely due to the complexity of their syntheses (i.e. five steps for the aldehyde version of FR0 and seven steps for the keto version of FR8; see below for structures),12 but also that the synthetic routes lack flexibility. This limitation implies that ad hoc syntheses must be developed for each new application,19–21 which hinders broader studies. Click chemistry appears to be the most viable approach for overcoming these barriers of synthetic versatility.22,23 In this context, one of the most applied reactions is the Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC), forming a 1,2,3-triazole derivative.
This synthetic strategy has already demonstrated its relevance in bioimaging, including a wide range of targeted applications such as the synthesis of fluorogenic probes,24–26 the fabrication of complex bioconjugates,27,28 and the visualisation of biologically active small molecules within living cells.29
In this study, we introduce a new group of solvatochromic dyes synthesized from an azide compound (FR1N3) as a key intermediate. This approach facilitates the synthesis of FRT (“T” for triazole) derivatives via CuAAC (Fig. 1). Surprisingly, the triazole moiety, although not a constituent of the donor and acceptor π-system (commonly abbreviated as D–π–A), has a positive effect on the overall optical properties of these new dyes. This “triazole effect” was investigated in a comprehensive spectroscopic study, accompanied by a time-dependent density functional theory (TD-DFT) approach. These results demonstrate the considerable potential of these molecules in bioimaging, due to their excellent two-photon absorption (2PA) properties.
Results and discussion
Synthesis
Building on the seminal methodology developed by Klymchenko and coworkers to synthetise fluorene-based dyes,12 Cho and coworkers notably simplified the synthetic process to obtain FR0 (Fig. 2).19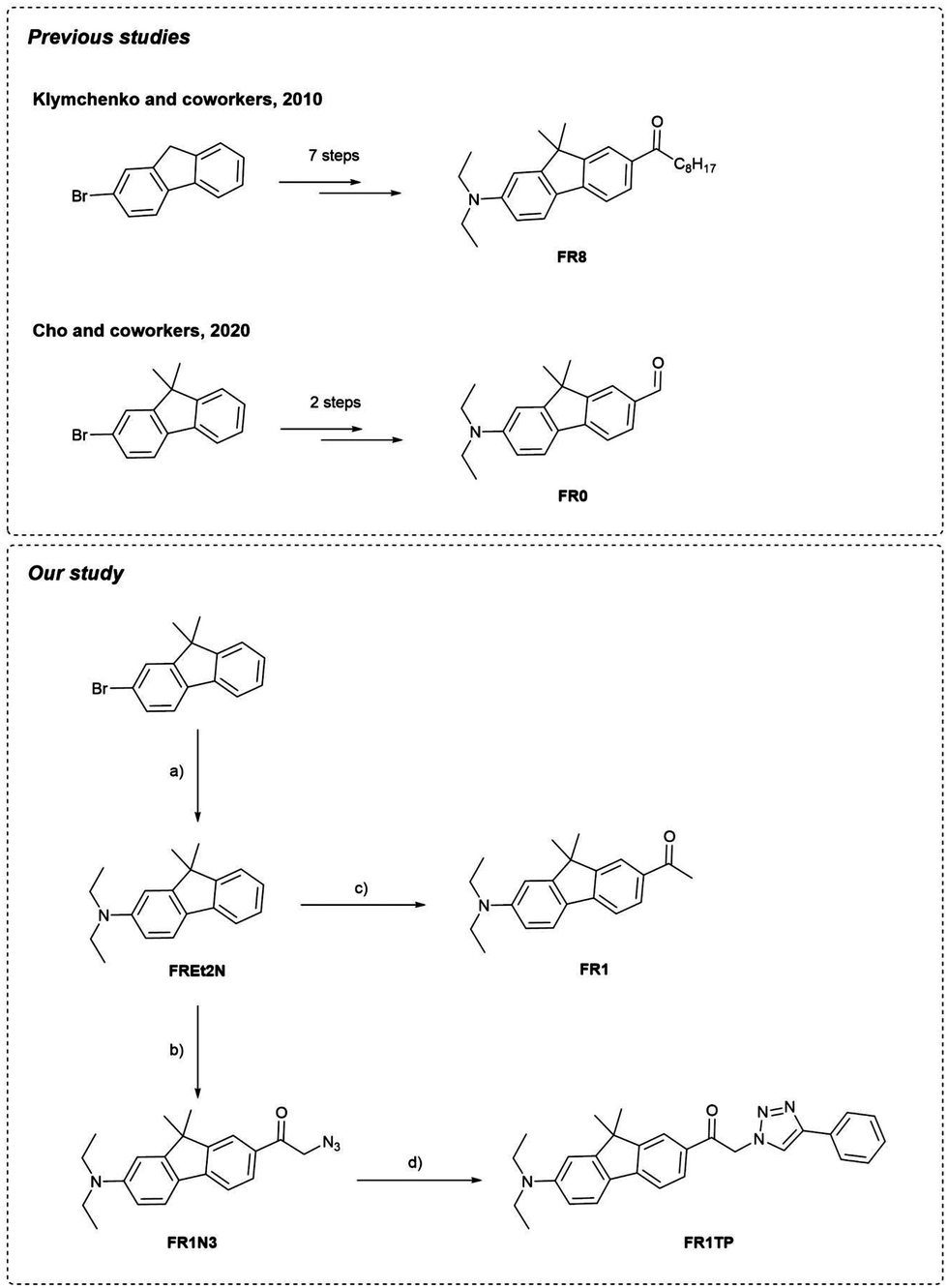 | ||
| Fig. 2 Synthesis of previously described fluorophores FR0, FR8 and new synthesis of FR1 and FR1TP. Reagents and conditions: (a) Et2NH, Pd(OAc)2, BINAP, NaOtBu, toluene, 110 °C, overnight, 76%; (b) 1. Bromoacetyl bromide, TfOH, 0 °C, 1 h; 2. NaN3, DMSO, rt, 10 min., 66%; (c) acetyl chloride, AlCl3, DCM, 0 °C to rt, overnight, 59%; (d) phenylacetylene, [Cu(CH3CN)4]PF6, TBTA, CHCl3, rt, overnight, 86%. | ||
Inspired by this approach, we applied a similar strategy to the commercially available 2-bromo-9,9-dimethyl-9H-fluorene (Fig. 2). Briefly, the Buchwald–Hartwig amination, followed by Friedel–Crafts acylation, provided a shortcut for the synthesis of the keto-terminated FR1, with just two synthetic steps and one chromatography column vs. seven steps and three chromatography columns as required for FR8. Additional details and characterization data are available in the ESI† (Sections S2 and S3, and Fig. S1–S15).
In analogy to this procedure, azide functionalisation of these fluorenes was envisaged by first introducing a bromoacetyl unit. Optimised Friedel–Crafts acylation of FREt2N with bromoacetyl bromide, followed by direct bromo-azido substitution, led to the clickable fluorene FR1N3. Finally, submitting FR1N3 to the CuAAc click reaction with phenylacetylene provided an excellent yield of the model compound FR1TP. Importantly, this strategy permits the conjugation of diverse alkynes with our fluorosolvatochromic unit without a significant increase in synthetic effort.
Spectroscopic properties in solution
FR1 and FR1TP photophysics were characterised in a broad range of solvents using UV-vis absorption and steady-state as well as time-resolved fluorescence spectroscopy. The spectra are shown in Fig. 3, and the data are summarised in Table S1 (ESI†). Additional details and spectra are included in the ESI† (Section S1 and Fig. S16–S21). Like FR0, the absorption bands of FR1 and FR1TP do not shift significantly with solvent polarity (Table S1, and Fig. S16–S19, ESI†). Therefore, in Fig. 3, the absorption spectra in toluene only are shown as an example. Based on the spectral position of the emission bands, a pronounced fluorosolvatochromism was observed for FR1 and FR1TP, similar to that of the parent compounds FR0 and FR8.12 The structured emission bands in hexane, ascribed to fluorescence originating from a local excited state, further prove the ICT character of these compounds. Note that by varying the solvent polarity, FR1 and FR1TP display emission spectra across the entire visible region.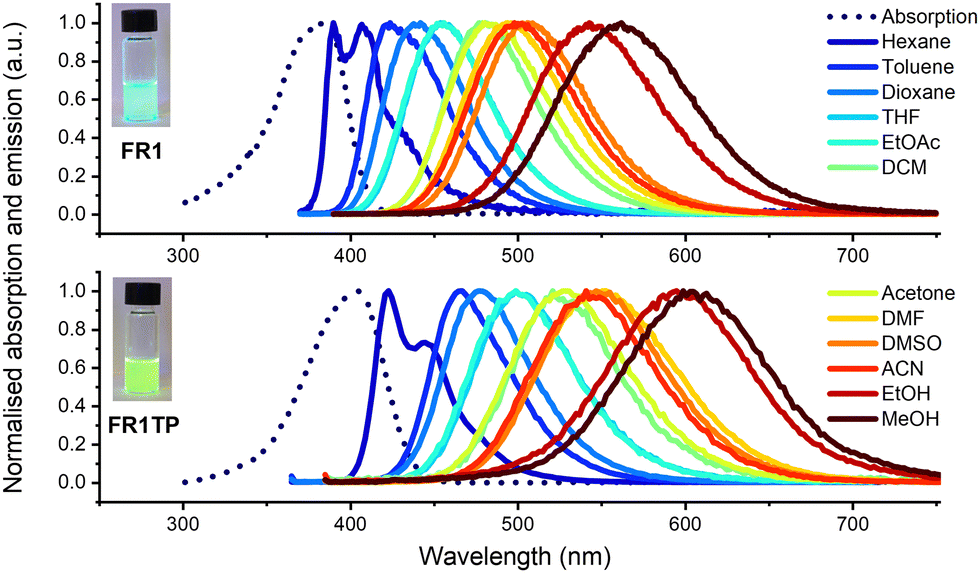 | ||
| Fig. 3 Absorption (dotted line) in toluene and emission (solid lines) of FR1 and FR1TP in a series of solvents. Concentration around 10−5 M. Excitation for emission readout at 360 nm in n-hexane, toluene, and dioxane, and at 380 nm for all other solvents pictures of acetone solutions of FR1 and FR1TP under irradiation at 365 nm are displayed in the left insets. | ||
Surprisingly, the emission bands of FR1TP are systematically redshifted by 47 nm on average, compared to those of FR1 for a given solvent. This desired property was not anticipated since the inherent properties of push–pull dyes are mainly dictated by the composition of the π-system, i.e. (i) the nature of the π-bridge and (ii) the D–A system.30 However, FR1 and FR1TP share a common electronic system: only the additional 4-phenyl-1,2,3-triazole differentiates FR1TP from FR1. Since this unit is isolated from the main π-system by an sp3 carbon, no significant electronic communication is expected between the triazole and the fluorophore moieties. Therefore, the triazole group must influence the π-system without participating in the overall π-conjugation. To our knowledge, such an effect attributed to non-conjugated triazole has not been described previously. To verify this hypothesis, two other derivatives with the same D–π–A structure were synthesised (Fig. 4a): (i) FR1TM, a mesityl derivative, to characterise the impact of the steric hindrance in position 4 of the triazole; and (ii) FR2TP, a derivative containing an ethyl linker, to explore the impact of the triazole–fluorophore distance. The synthetic routes of FR1TM and FR2TP together with the absorption and emission spectra of these compounds are provided in the ESI† (Scheme S1 and Fig. S20 and S21). The absorption bands of these fluorene derivatives show only small variations with solvent polarity (Fig. S22 and Table S2, ESI†). Importantly, the fluorosolvatochromism trends outlined above for FR1 and FR1TP are observed also for these new analogues (see Fig. S20 and S21, ESI†). The fluorescence properties of all the newly synthesised FR-analogues were further investigated, and their fluorescence quantum yields were measured across all the solvents studied. From the data presented in Fig. 4b, the following observations can be made:
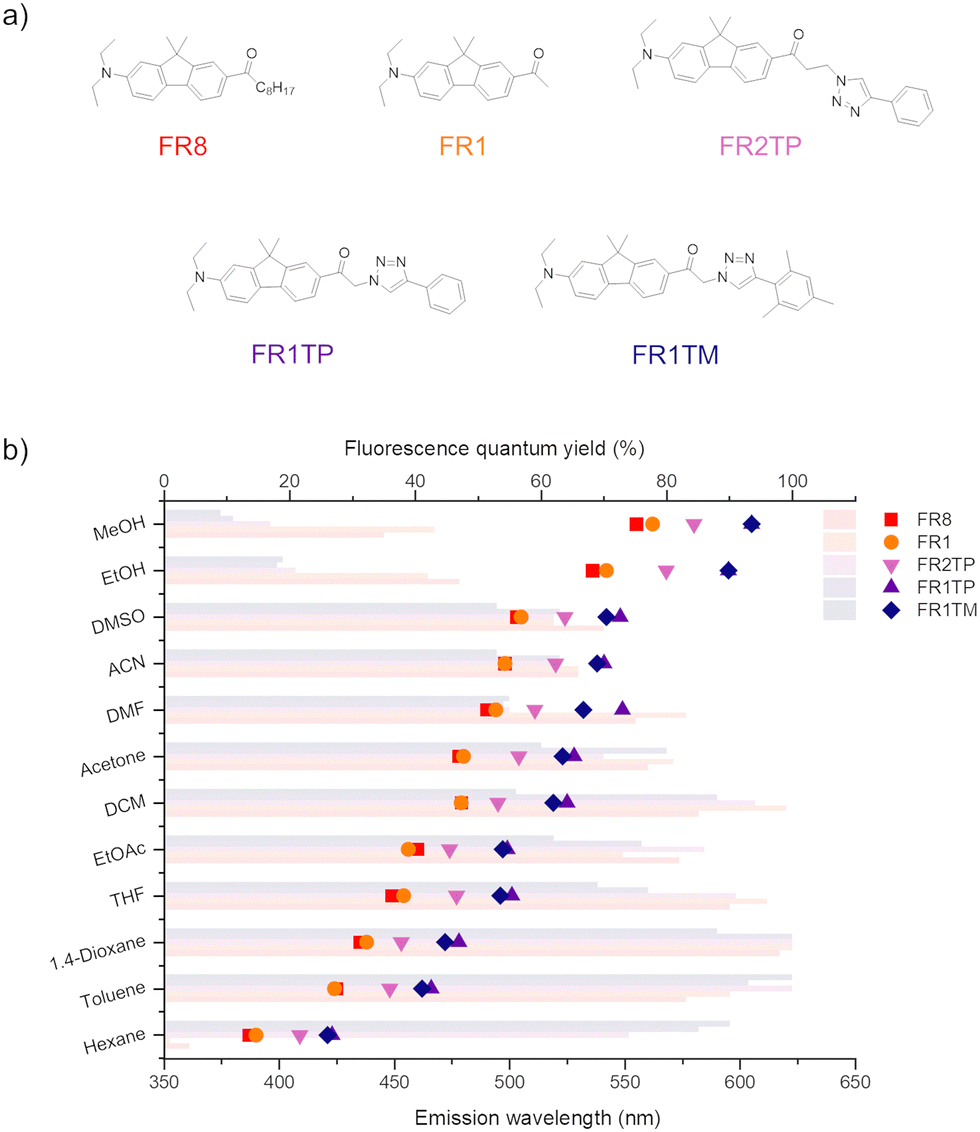 | ||
| Fig. 4 (a) Structures of the fluorene derivatives studied in this work. (b) Fluorescence quantum yields (faded horizontal bars) and emission maxima (symbols) of FR8 (red), FR1 (orange), FR1TP (pink), FR2TP (purple) and FR1TM (dark blue) in different solvents. | ||
(i) The influence of the alkyl chain in the α-position on the ketone is negligible: both FR1 and FR8 display similar emission bands and fluorescence quantum yields.
(ii) The effect of substituting the triazole at position 4 is marginal: FR1TM has a slightly blue-shifted emission compared to FR1TP (by 3 nm on average for the same solvent), indicating that steric hindrance in this position is not relevant to the fluorescence properties.
(iii) The “triazole effect” depends strongly on the fluorene–triazole distance: the emission maxima of FR2TP and FR1TP are redshifted by 20 nm and 47 nm on average, respectively, for a given solvent compared to FR1 (lacking the triazole). This observation clearly demonstrates that an increase in the fluorene–triazole distance significantly reduces the redshift, and strengthens the hypothesis that the triazole unit induces the bathochromic effect.
(iv) In hexane, FR1 (as FR8) display weak fluorescence only (ϕf < 0.05), whereas the triazole derivatives show a substantial increase in the fluorescence quantum yield (ϕf > 0.70). FR0 and PRODAN were previously found to be prone to aggregation in hexane.31 The induced excitonic couplings also observed for FR1, which strongly favour non-radiative decay, are significantly inhibited by the triazole for FR1TP (see the fluorescence lifetime study below).
(v) All analogues are characterised by exceptional fluorescence quantum yields in nonpolar or slightly polar solvents, which gradually decrease with increasing solvent polarity. This behaviour, classically observed in push–pull architectures,12,32 is observed also for the triazole-decorated derivatives.
Additionally, fluorescence lifetimes were determined in a few representative solvents and are shown in Fig. S23 and Tables S3 and S4 (ESI†). In 1,4-dioxane, the radiative and non-radiative rate constants (kr and knr, respectively) of these compounds, with or without triazole, are of the same order globally (〈kr〉 = 0.43 ns−1 and 〈knr〉 = 0.55 ns−1). In hexane, the triazole compounds showed on average a slightly more efficient non-radiative deactivation compared to 1,4-dioxane (〈kr〉 = 0.52 ns−1 and 〈knr〉 = 0.94 ns−1), likely due to their lower solubility in hexane. The negative influence of hexane on FR1 resulted in a very short lifetime (<0.05 ns), with a non-radiative rate at least 6 times higher than the triazole derivatives.
On the other hand, using protic solvents such as methanol or ethanol implies solvent-specific interactions for the triazole derivatives.33 In ethanol, FR1TP and FR1TM display shorter fluorescence lifetimes than FR1 (1.89 and 1.85 ns for FR1TP and FR1TP vs. 2.99 ns for FR1). More interestingly, FR2TP has a similar pattern to FR1: consequently, the proximity of the triazole to the fluorene moiety is detrimental to proticity-induced non-radiative relaxation. In aprotic polar solvents however, such as dimethylsulfoxide, the differences in the lifetimes are negligible, proving the isolated effect of the proticity. To further investigate the impact of solvent proticity on the fluorescence properties, deuterated methanol was employed to minimize proton interaction with the dyes.34 The shape and the spectral position of the emission bands of FR1 and FR1TP were unchanged in deuterated methanol compared to regular methanol, indicating no influence on the electronic properties (Fig. S24a, ESI†). However, both FR1 and FR1TP experienced a significant increases in the fluorescence quantum yields when changing from methanol to deuterated methanol: from 0.43 to 0.62 for FR1 and from 0.11 to 0.26 for FR1TP (Fig. S24b, ESI†). This suggests that solvent proton interaction mainly contributes to fluorescence quenching in polar protic solvents, aligning with other dye systems.34 Additionally, this effect was more pronounced for FR1TP than for FR1, with an increased quantum yield by factors of 2.4 for FR1TP and 1.4 for FR1. Consequently, the more efficient fluorescence quenching of FR1TP compared to FR1 implies a distinctive interaction between the carbonyl/triazole system of FR1TP and the solvent proton.
Characterisation of the triazole effect
The experimental results described above show that the introduction of the triazole at different positions significantly influences the photophysical properties. To support the experimental observations, theoretical calculations were performed using the mPW1PW91/6-311+G(2d,p) level of theory. Calculations were conducted in vacuum and with various solvents (toluene, dichloromethane, and acetonitrile). The results are summarised in Table S9 (ESI†), and more details are provided in the ESI.† The experimental trends are satisfactorily reproduced, and it is also encouraging to note the good agreement between the calculated and the experimental values.The lowest transition, S0 → S1, is dominated in all cases by HOMO and LUMO frontier orbitals, as represented in Fig. 5. For all derivatives, the HOMO is mainly localised around the diethylamino moiety, whereas the electron density is strongly shifted towards the carbonyl moiety in the LUMO. This observation confirms the D–π–A configuration of these compounds. However, the LUMO distribution varies between the analogues, and a minor triazole contribution is identified from these calculations, especially for derivatives FR1TP and FR1TM. Importantly, the electronic distribution of these orbitals along the scaffolds remains consistent, irrespective of the solvent considered in the calculations (Fig. S32–S34, ESI†). A better representation of the electron-density changes and the intramolecular charge transfer (ICT) phenomena is given by the electron density difference (EDD) plots (Fig. S30, ESI†).
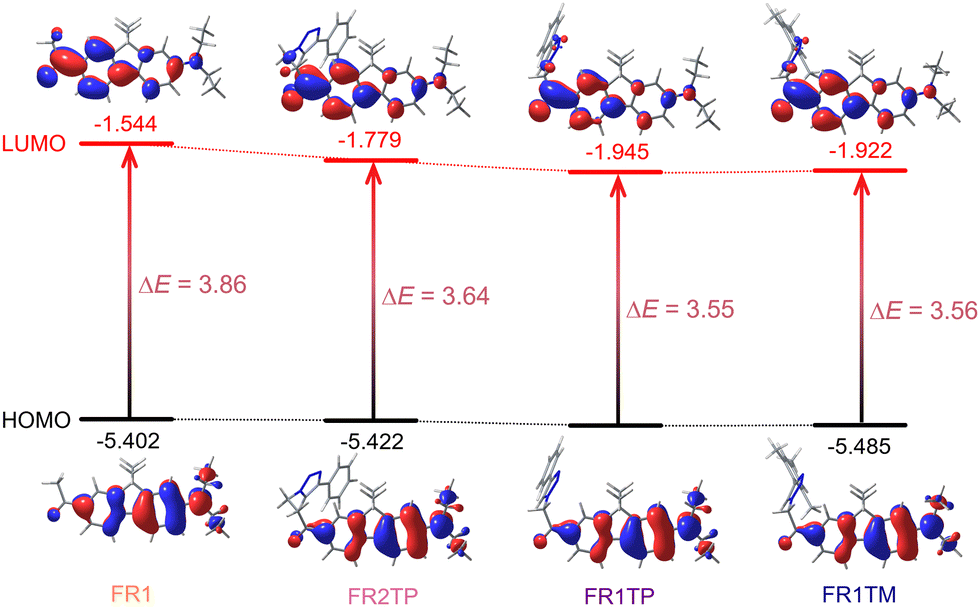 | ||
| Fig. 5 Frontier molecular orbitals contour plots and energies for FR1, FR2TP, FR1TP and FR1TM at the mPW1PW91/6-311+G(2d,p)//mPW1PW91/6-31+G(d) level (isosurface value: 0.03 a.u.). Orbital energies indicated are in eV. | ||
Analysis of the orbital energies further supports the role of triazole in the bathochromic shift. Notably, the relative energies of the HOMO levels are almost identical for all studied compounds. However, the LUMO levels differ substantially, clearly showing stabilisation by the triazole unit. More specifically, in vacuum, the energy associated with the LUMO of FR1 was –1.544 eV, with a decrease of 14% for FR2TP and 24% for FR1TP and FR1TM. Thus, the proximity of the triazole motif to the fluorene part induces a decrease in the excitation energy. This behaviour is also well reproduced by TDDFT calculations performed in vacuum: both absorption and emission wavelengths are redshifted upon the incorporation of the triazole unit onto the scaffolds (Table S9, ESI†). For instance, the excitation energy is reduced from 3.86 eV for FR1 to 3.56 eV for FR1TP. Furthermore, similar trends are observed in the relative energies of frontier molecular orbitals when solvent effects are included in the DFT calculations (Fig. S32–S34, ESI†).
The “triazole effect” on the fluorosolvatochromism of FRT derivatives can also be quantified at the experimental level. To evaluate the amplitude of the solvatochromic character of a given chromophore, two models are commonly used:12 (i) the correlation between the solvent's ET(30) parameter and the corresponding emission maxima, or (ii) the correlation between the Stokes shift and the orientation polarizability function for each solvent (known as the Lippert–Mataga plot35,36). Using both approaches, the linear correlations obtained for all compounds suggest a noticeable effect of solvent polarity on their respective spectroscopic properties (Fig. S25 and S26/Tables S5–S7, ESI†). Furthermore, the difference in dipole moments between the ground- and excited states extracted from the Lippert–Mataga model reveals that the triazole derivatives display a slightly increased charge-transfer (CT) character: 16 D for FR1, 17 D for FR2TP vs. 20 D for FR1TP and FR1TM. Therefore, the triazole substitution in this chromophore family significantly affects the fluorosolvatochromism.
In addition to the bathochromic shift caused by the “triazole effect”, the molar absorption coefficients in ethanol are also substantially increased for the triazole-substituted analogues: 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 60000, and 57000 M−1 cm−1 for FR1, FR1TP, and FR1TM, respectively. The photostability of these dyes was investigated by monitoring the decrease in fluorescence intensity over time under constant irradiation at 365 nm. This data shows an overall equal or superior photostability of the fluorene dyes compared to PRODAN (see Fig. S32 and Table S8, ESI†). In ethanol, FR1TP shows minimal photodecomposition compared to PRODAN or FR1. FR1 and FR1TP are more stable than PRODAN in toluene. Only the photostability of FR1TP is comparable to PRODAN in THF.
000, 60000, and 57000 M−1 cm−1 for FR1, FR1TP, and FR1TM, respectively. The photostability of these dyes was investigated by monitoring the decrease in fluorescence intensity over time under constant irradiation at 365 nm. This data shows an overall equal or superior photostability of the fluorene dyes compared to PRODAN (see Fig. S32 and Table S8, ESI†). In ethanol, FR1TP shows minimal photodecomposition compared to PRODAN or FR1. FR1 and FR1TP are more stable than PRODAN in toluene. Only the photostability of FR1TP is comparable to PRODAN in THF.
Altogether, these compounds display photophysical properties highly desired for applications in bioimaging. This led us to study the 2PA properties (Fig. 6). FR0 has a high cross section (σ2PA up to 350 GM in ethanol), indicating that FR1TP and its derivatives could be suitable for applications such as two-photon fluorescence microscopy (2PFM). The two-photon excited fluorescence spectra of FR1, FR1TP, FR1TM and FR2TP were recorded in ethanol solution in the 700–950 nm range using a femtosecond pulsed laser source, and the two-photon absorption cross section was calculated according to the two-photon induced fluorescence (2PIF) method (more details are provided in the ESI†). The 2PA spectra of all analogues feature a maximum between 780 and 800 nm, essentially coinciding with the respective S0 → S1 absorption bands. These 2PA bands show high intensity, manifested in the high cross section values: 400 GM for FR1 (in agreement with that observed for FR8),12 250 GM for FR1TP and FR2TP, and 170 GM for FR1TM. These values indicate that the presence of triazole slightly reduces the 2PA capability. The emission observed was generated in all cases by 2PA, as demonstrated by the slopes of ca. 2 determined from the double logarithmic plot representing emission intensity vs. laser excitation power (Fig. S28 and Table S9, ESI†). Furthermore, the fluorescence bands were not affected in spectral position or shape by the excitation mechanism. These observations confirm that the same electronic states are involved in both the absorption and emission processes, regardless of the nature of the excitation (Fig. S29, ESI†).
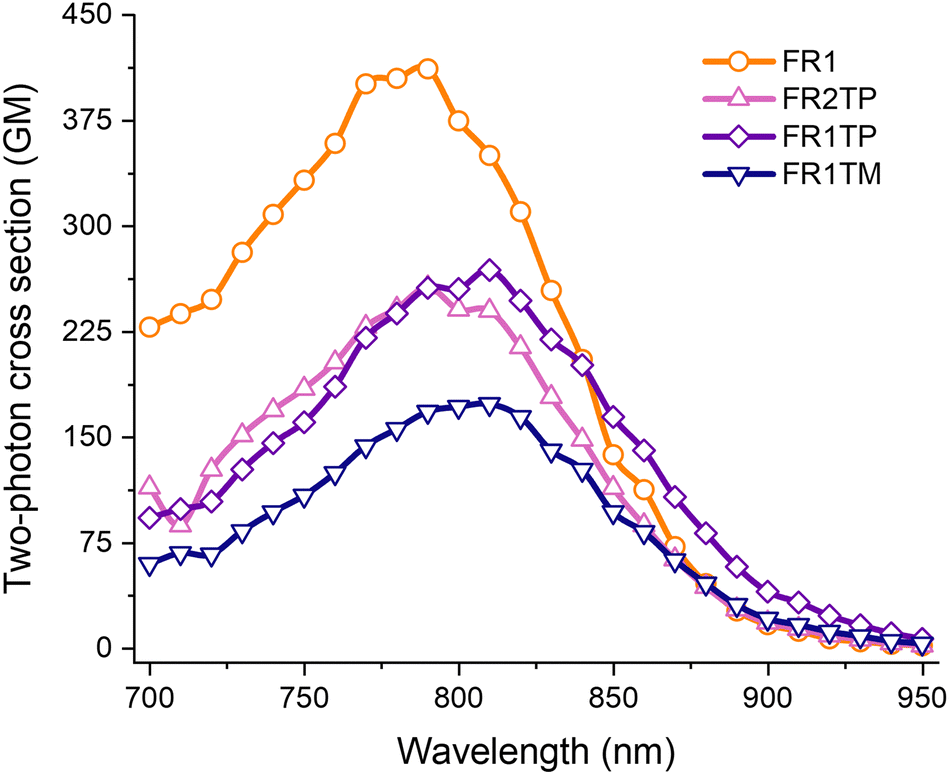 | ||
| Fig. 6 2PA spectra of FR1 (orange), FR2TP (pink), FR1TP (purple) and FR1TM (dark blue) in ethanol (ca.1 μM air-equilibrated solutions). | ||
Conclusions
In summary, we have developed a short synthesis of clickable solvatochromic fluorene derivatives. The new model derivative FR1TP shows a dramatic effect of the triazole on the spectroscopic properties. We demonstrated experimentally and by TD-DFT that the non-conjugated triazole is responsible for a bathochromic shift in the fluorosolvatochromism of FR1TP and similar analogues. Specifically, the triazole stabilizes the LUMO of the fluorene moiety, reducing the overall excitation energy. The pronounced photostability, high molar absorption coefficients and two-photon cross section of these triazole derivatives make them excellent candidates for bioimaging. We anticipate that these results will facilitate the development of new bioconjugates or other types of dyads, to modulate the photophysics of these fluorophores. Studies in this direction are underway in our group.Author contributions
J. R.: conceptualization, formal analysis, investigation, methodology, visualization, writing – original draft. C. B. M.: formal analysis, investigation, methodology, visualization, writing – review & editing. J. A.: formal analysis, funding acquisition, methodology, project administration, supervision, writing – review & editing. M. G.: formal analysis, funding acquisition, methodology, project administration, supervision, writing – review & editing.Data availability
Relevant data selected for open access will be shared via Zenodo.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the European Union's Horizon Europe research and innovation program under grant agreement No 101098934. All the authors want to thank Dr Daniel Tietze for measuring HRMS, Prof. Karl Börjesson for providing access to fluorescence lifetime measurement setup, and Prof. Francisco Najera for fruitful discussions about the theoretical calculations. CBM acknowledges the Carl Trygger Foundation for the Postdoctoral fellowship. JA acknowledges the Swedish Research Council VR for a project grant (#2021-05311).Notes and references
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Förster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC.
- A. S. Klymchenko, Solvatochromic and Fluorogenic Dyes as Environment-Sensitive Probes: Design and Biological Applications, Acc. Chem. Res., 2017, 50, 366–375 CrossRef PubMed.
- M. Hee Lee, J. Seung Kim and J. L. Sessler, Small molecule-based ratiometric fluorescence probes for cations, anions, and biomolecules, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- D. Su, C. Lean Teoh, L. Wang, X. Liu and Y.-T. Chang, Motion-induced change in emission (MICE) for developing fluorescent probes, Chem. Soc. Rev., 2017, 46, 4833–4844 RSC.
- A. Chevalier, P.-Y. Renard and A. Romieu, Azo-Based Fluorogenic Probes for Biosensing and Bioimaging: Recent Advances and Upcoming Challenges, Chem. – Asian J., 2017, 12, 2008–2028 CrossRef PubMed.
- L. Wang, C.-H. Hsiung, X. Liu, S. Wang, A. Loredo, X. Zhang and H. Xiao, Xanthone-based solvatochromic fluorophores for quantifying micropolarity of protein aggregates, Chem. Sci., 2022, 13, 12540–12549 RSC.
- Q. Xia, W. Wan, W. Jin, Y. Huang, R. Sun, M. Wang, B. Jing, C. Peng, X. Dong, R. Zhang, Z. Gao and Y. Liu, Solvatochromic Cellular Stress Sensors Reveal the Compactness Heterogeneity and Dynamics of Aggregated Proteome, ACS Sens., 2022, 7, 1919–1925 CrossRef.
- D. I. Danylchuk, P.-H. Jouard and A. S. Klymchenko, Targeted Solvatochromic Fluorescent Probes for Imaging Lipid Order in Organelles under Oxidative and Mechanical Stress, J. Am. Chem. Soc., 2021, 143, 912–924 CrossRef.
- S. Sahu, V. Parthasarathy and A. K. Mishra, Phenylethynylanthracene based push–pull molecular systems: tuning the photophysics through para-substituents on the phenyl ring, Phys. Chem. Chem. Phys., 2023, 25, 1957–1969 RSC.
- A. K. Pradhan, M. Ray, V. Parthasarathy and A. K. Mishra, Effects of donor and acceptor substituents on the photophysics of 4-ethynyl-2,1,3-benzothiadiazole derivatives, Phys. Chem. Chem. Phys., 2023, 25, 29327–29340 RSC.
- G. Weber and F. J. Farris, Synthesis and spectral properties of a hydrophobic fluorescent probe: 6-propionyl-2-(dimethylamino)naphthalene, Biochemistry, 1979, 18, 3075–3078 CrossRef.
- O. A. Kucherak, P. Didier, Y. Mély and A. S. Klymchenko, Fluorene Analogues of Prodan with Superior Fluorescence Brightness and Solvatochromism, J. Phys. Chem. Lett., 2010, 1, 616–620 CrossRef.
- T. Tanaka, A. Matsumoto, A. S. Klymchenko, E. Tsurumaki, J. Ikenouchi and G. Konishi, Fluorescent Solvatochromic Probes for Long-Term Imaging of Lipid Order in Living Cells, Adv. Sci., 2024, 11, 2309721 CrossRef PubMed.
- J. Shaya, F. Fontaine-Vive, B. Y. Michel and A. Burger, Rational Design of Push–Pull Fluorene Dyes: Synthesis and Structure–Photophysics Relationship, Chem. – Eur. J., 2016, 22, 10627–10637 CrossRef PubMed.
- S. Singha, D. Kim, B. Roy, S. Sambasivan, H. Moon, A. S. Rao, J. Y. Kim, T. Joo, J. W. Park, Y. M. Rhee, T. Wang, K. H. Kim, Y. H. Shin, J. Jung and K. H. Ahn, A structural remedy toward bright dipolar fluorophores in aqueous media, Chem. Sci., 2015, 6, 4335–4342 RSC.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, Aziridinyl Fluorophores Demonstrate Bright Fluorescence and Superior Photostability by Effectively Inhibiting Twisted Intramolecular Charge Transfer, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS.
- J. Zhou, X. Lin, X. Ji, S. Xu, C. Liu, X. Dong and W. Zhao, Azetidine-Containing Heterospirocycles Enhance the Performance of Fluorophores, Org. Lett., 2020, 22, 4413–4417 CrossRef CAS.
- Y. Niko, S. Kawauchi and G. Konishi, Solvatochromic Pyrene Analogues of Prodan Exhibiting Extremely High Fluorescence Quantum Yields in Apolar and Polar Solvents, Chem. – Eur. J., 2013, 19, 9760–9765 CrossRef CAS.
- A. Sharma, J. Sun, I. Singaram, A. Ralko, D. Lee and W. Cho, Photostable and Orthogonal Solvatochromic Fluorophores for Simultaneous In Situ Quantification of Multiple Cellular Signaling Molecules, ACS Chem. Biol., 2020, 15, 1913–1920 CrossRef CAS PubMed.
- C. Benitez-Martin, S. Li, A. Dominguez-Alfaro, F. Najera, E. Pérez-Inestrosa, U. Pischel and J. Andréasson, Toward Two-Photon Absorbing Dyes with Unusually Potentiated Nonlinear Fluorescence Response, J. Am. Chem. Soc., 2020, 142, 14854–14858 CrossRef CAS PubMed.
- L. Y. Chiang, P. A. Padmawar, T. Canteenwala, L.-S. Tan, G. S. He, R. Kannan, R. Vaia, T.-C. Lin, Q. Zheng and P. N. Prasad, Synthesis of C60-diphenylaminofluorene dyad with large 2PA cross-sections and efficient intramolecular two-photon energy transfer, Chem. Commun., 2002, 1854–1855 RSC.
- J. Kaur, M. Saxena and N. Rishi, An Overview of Recent Advances in Biomedical Applications of Click Chemistry, Bioconjugate Chem., 2021, 32, 1455–1471 CrossRef CAS.
- Kenry and B. Liu, Bio-orthogonal Click Chemistry for In Vivo Bioimaging, Trends Chem., 2019, 1, 763–778 CrossRef CAS.
- J. Qi, M.-S. Han, Y.-C. Chang and C.-H. Tung, Developing Visible Fluorogenic ‘Click-On’ Dyes for Cellular Imaging, Bioconjugate Chem., 2011, 22, 1758–1762 CrossRef CAS PubMed.
- P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, CalFluors: A Universal Motif for Fluorogenic Azide Probes across the Visible Spectrum, J. Am. Chem. Soc., 2015, 137, 7145–7151 CrossRef CAS PubMed.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, A Fluorogenic 1,3-Dipolar Cycloaddition Reaction of 3-Azidocoumarins and Acetylenes, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS.
- C. S. McKay and M. G. Finn, Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation, Chem. Biol., 2014, 21, 1075–1101 Search PubMed.
- B. Stump, Click Bioconjugation: Modifying Proteins Using Click-Like Chemistry, ChemBioChem, 2022, 23, e202200016 CrossRef CAS.
- T. Cañeque, S. Müller and R. Rodriguez, Visualizing biologically active small molecules in cells using click chemistry, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- F. Bureš, Fundamental aspects of property tuning in push–pull molecules, RSC Adv., 2014, 4, 58826–58851 RSC.
- A. Marini, A. Muñoz-Losa, A. Biancardi and B. Mennucci, What is Solvatochromism?, J. Phys. Chem. B, 2010, 114, 17128–17135 CrossRef CAS PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures, Chem. Rev., 2003, 103, 3899–4032 CrossRef.
- S. Saha and A. Samanta, Influence of the Structure of the Amino Group and Polarity of the Medium on the Photophysical Behavior of 4-Amino-1,8-naphthalimide Derivatives, J. Phys. Chem. A, 2002, 106, 4763–4771 CrossRef CAS.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Universal quenching of common fluorescent probes by water and alcohols, Chem. Sci., 2021, 12, 1352–1362 RSC.
- E. Lippert, Spektroskopische Bestimmung des Dipolmomentes aromatischer Verbindungen im ersten angeregten Singulettzustand, Z. Elektrochem. Ber. Bunsenges. Phys. Chem., 1957, 61, 962–975 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Solvent Effects upon Fluorescence Spectra and the Dipolemoments of Excited Molecules, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04642k |
| This journal is © the Owner Societies 2025 |