
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the surface of mesoporous nickel oxide and its interaction with oxygen and water†
Sina Wrede‡
 a,
Qianhui Liu‡a,
Libo Chenb,
Luca D’Amarioa,
Bin Caia,
Mattia Scardamagliac,
Zhi-Bin Zhangb,
Maria Hahlin*ad and
Haining Tian*a
a,
Qianhui Liu‡a,
Libo Chenb,
Luca D’Amarioa,
Bin Caia,
Mattia Scardamagliac,
Zhi-Bin Zhangb,
Maria Hahlin*ad and
Haining Tian*a
aDepartment of Chemistry-Ångström Laboratory, Uppsala University, SE-75120 Uppsala, Sweden. E-mail: maria.hahlin@kemi.uu.se; haining.tian@kemi.uu.se
bDepartment of Electrical Engineering, Uppsala University, SE-75120 Uppsala, Sweden
cMAX IV Laboratory, Lund University, SE-22100 Lund, Sweden
dDepartment of Physics and Astronomy, Uppsala University, SE-75120 Uppsala, Sweden
First published on 26th May 2025
Abstract
The surface chemistry of mesoporous nickel oxide (NiOx) plays a pivotal role in its functionality across various technological applications. Herein, we present a detailed study of NiOx surface states using multiple spectroscopic techniques, including ambient pressure X-ray photoelectron spectroscopy (XPS), ultraviolet-visible (UV-vis) spectroscopy, and Fourier-transform infrared (FTIR) spectroscopy, to observe the removal and reformation of surface species during gas dosing. Our findings reveal the presence of both surface oxygen- and hydroxide-species on the NiOx surface. Furthermore, the results suggest that NiOx surface states consist of approximately 50% hydroxides and the remainder comprising adsorbed oxygen species, likely predominantly diatomic oxygen ions (O2−), that are associated with higher valence Ni states (Ni3+). In situ experiments demonstrate that the formation and stability of hydroxides and diatomic oxygen ions depend on temperature and are significantly influenced by interactions with atmospheric oxygen and water. Our insights into the NiOx surface state composition and reactivity offer a nuanced understanding of its surface chemistry, with implications for enhancing its performance in catalysis, sensing, energy harvesting and energy storage devices where the surface states of nickel oxide are known to dictate electronic and chemical properties of the material.
Introduction
Nickel oxide is as a versatile p-type transition metal oxide, finding diverse applications in gas sensors, catalysis, electronic devices, and energy conversion and storage.1–4 However, the inherent insulating nature of stoichiometric mono-nickel oxide (NiO) renders it unsuitable for these applications.5,6 As such, non-stoichiometric nickel oxide is the primary focus when discussing nickel oxide in such contexts, making it more accurate to refer to it as NiOx. Like many other binary metal oxides, nickel oxide commonly exists in non-stoichiometric forms and exhibits p-type conductivity that is attributed to defects, stemming from nickel vacancies and oxygen interstitials.6 As such, nanostructured nickel oxide, where surface effects dominate, exhibits notably higher conductivity compared to its bulk counterpart.7 Consequently, the electronic and chemical properties of NiOx are intricately linked to its surface characteristics, posing challenges to comprehensive understanding.8 Surface states in metal oxides, typically stemming from defects, vacancies, and adsorbates, thus play a decisive role in shaping the interaction between the material and its environment.While surface studies on sputtered or oriented nickel oxide thin films under controlled conditions exist,8–11 the understanding of mesoporous NiOx films (typically NiOx>1), fabricated under less controlled conditions, remains limited. However, especially for applications where a high surface area is desired, (meso)porous films are attractive – such as in sensors, catalysis or energy conversion and storage.2,6 Typically, for these films, nanoparticles are sintered together to form porous structures. Due to the large surface area, the surface of NiOx fully dictates the electronic and chemical properties of mesoporous films and as such, device properties. For example, in NiOx photocathodes for solar energy harvesting, these surface states have been attributed to the large spreads in device performance in the literature.11–13 In dye-sensitized systems, these surface states play a dual role in photocathode efficiency: they not only act as charge recombination centres but have also been found to be involved in electrolyte and dye regeneration and aid in hole transfer.12,14,15
Even though prior studies have contributed to the understanding of the surface of NiOx films, the scientific discourse still persists regarding the precise chemical nature and role of these states. The sensor community has focused on the reactions of gases with chemisorbed oxygen species at the NiOx surface,3,16,17 largely not considering hydroxides or oxyhydroxides at the surface which the (photo)catalysis field has predominantly focused on due to their possible involvement in the water splitting reaction mechanism.10–12
In contrast, this study embarks on an exploration of the intricate chemical nature of the nickel oxide surface, attempting to gain a holistic understanding of the surface of mesoporous nickel oxide films utilizing various in situ spectroscopic methods. It focuses on the removal and re-formation of surface states, as well as their interactions with water and oxygen which are present under atmospheric conditions in which mesoporous NiOx films are typically prepared and used. Through examination of the mesoporous NiOx films with electrochemistry, UV-vis and IR spectroscopy, and near ambient pressure X-ray photoelectron spectroscopy (AP-XPS) as support, this article contributes valuable insights to the ongoing discourse surrounding the surface properties of this multifaceted material, particularly in the context of its applications as a mesoporous material in many different devices.
Removal and re-formation of surface states
Mesoporous NiOx films were fabricated from a nanoparticular paste that has been sintered under ambient atmosphere at 450 °C. It is a common way to prepare nickel oxide mesoporous films.13,18 A cathodic potential scan from the open-circuit potential (OCP) to the flat region in cyclic voltammetry (CV in ESI,† Fig. S4) results in bleach that covers the whole visible region with a slight feature around 500 nm, as shown in Fig. 1A.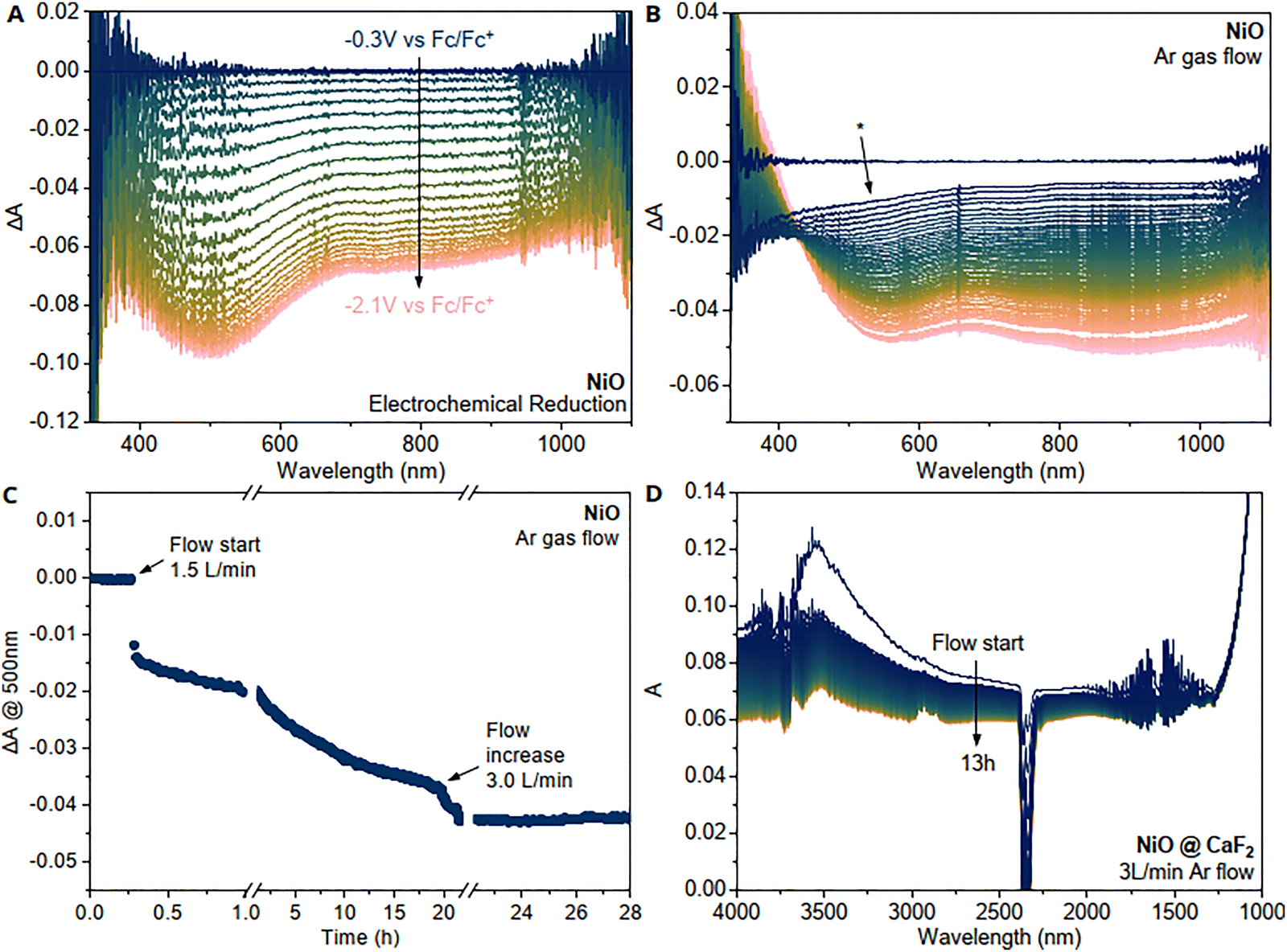 | ||
| Fig. 1 Absorption change of as-prepared mesoporous NiOx on conductive (FTO) glass substrates during (A) electrochemical reduction from the OCP and (B) Ar gas flow. Initial featureless loss in intensity marked by asterisk * is due to the loss of scattering signal of water at the surface. (C) Kinetic trace of NiOx absorption at 500 nm during Ar gas flow. (D) IR absorption change with Ar gas flow of mesoporous NiOx on CaF2. | ||
In the literature, it is generally accepted that the visible bleach to a lighter colour of mesoporous NiOx films during electrochemical reduction is related to the reduction of nickel oxide surface states that lie energetically within the bandgap.19 We and others have also found that this bleach is visible upon chemical reduction (by NaBH4) of mesoporous NiOx20 or by passivating the surface through targeted atomic deposition of boron14 or atomic layer deposition (ALD) of Al2O3.15 It is worth noting that during ALD, the applied vacuum and heating may also contribute to the partial removal of surface states in addition to deposition process.
In contrast to these more aggressive methods, we have found that even a flow of inert gas, such as argon or nitrogen, can also remove some of these surface states and cause the characteristic bleach of nickel oxide that is associated to the removal of nickel oxide surface states (Fig. 1B). Furthermore, we also observed that a prolonged exposure to vacuum (ca. 100 mbar) can also remove some of these surface states (ESI,† Fig. S5). A steady stream of argon through a lab-made gas cell (ESI,† Fig. S1) shows an initial featureless bleach of NiOx (marked by asterisk *) and then a continuous bleach that spans the whole visible spectrum together with the characteristic feature at 500 nm. The isosbestic point at 450 nm that shows up after a time delay, indicating that two processes likely happen during the flow of inert gas. The initial featureless bleach (marked with *) can be assigned to water since it matches the scattering signal of water on glass (see ESI,† Fig. S6) and a drastic lowering of water bands in the infrared spectrum upon Ar flow (Fig. 1D) can also be observed. Due to the large surface area of the mesoporous film, the total amount of adsorbed water can be quite large which leads to the significant scattering signal. The kinetic trace of the bleach during Ar flow (Fig. 1C) also shows that this initial desorption of water is much faster (within minutes) than the consequent bleach that exhibits the typical NiOx surface state character. An increase in flow helped speed up the removal of surface species until no significant change in absorption was observed, and the fully “bleached” film was obtained. By comparing the amplitude of difference absorption spectra obtained from electrochemical reduction and Ar gas flow, we can also estimate that roughly half of the surface states can be removed with Ar gas flow if we assume all surface states are electrochemically responsive and have similar extinction coefficient.
It is generally worth noting that hydroxide species are more stable than absorbed oxygen species, starting to decompose over 250 °C,21 and should therefore not be removed during Ar gas flow. After the initial loss of adsorbed water, the IR spectra also show a remaining signal around 3600 cm−1 even after 13 h Ar flow which can likely be assigned to hydroxides on the surface (Fig. 1D). Contrary, oxygen related species are less stable and adsorbed oxygen (O2(ads)) readily desorbs at room temperature and as temperatures rise, superoxide O2−(ads) (adsorbed and charged) leaves the surface or is transformed to O−(ads).16 We therefore propose that surface oxygen species are mainly responsible for the observed change in absorption during Ar gas flow after the removal of water and make up approximately half of the surface states estimated the absorption change between Fig. 1A and B by assuming the same extinction coefficient of all surface states. In comparison, electrochemical reduction can also access species that are not oxygen related, such as hydroxides and oxihydroxides. Based on these considerations, we propose that hydroxides and related species make up the remaining half of surface states.
A similar magnitude of the bleach as with Ar gas flow could be reached by heating the film to 200 °C in air for 2 min (ESI,† Fig. S7) which further supports this hypothesis, as hydroxides should be more stable at this temperature, while many surface oxygen ion species should not. It is worth noting that there is a spectral difference between Fig. 1A and B in the region below 450 nm. The absorption feature of NiOx that has been treated with gas flow shows an increase in absorption in this region, whereas the electrochemically reduced one does not. Looking at the kinetic traces of individual wavelengths (see ESI,† Fig. S12), it also becomes apparent that this increased absorption feature rises at the same time as the bleach at 500 nm, which is after the removal of water. This increased absorption (also visible upon heating to 300 °C in ESI,† Fig. S7) is similar to what has been previously reported by our group20,22 and is indicative of changes in the nickel oxide bandgap.
It is known that the bandgap slope in this energy range becomes less steep when going from stochiometric nickel oxide to non-stoichiometric nickel oxide (NiOx>1), resulting in a higher absorption of more stochiometric nickel oxide between approximately 350 to 500 nm.23 It is therefore likely that the flow of Ar leads to a more stoichiometric character of nickel oxide, which would explain the increase in absorption at lower wavelengths at the absorption onset of the bandgap.
It was also observed that the removal of surface states through heat or Ar gas flow and subsequent exposure to air lead to the recovery of the bleach. We therefore performed gas flow experiments with different gases after the removal of surface states. For the experiment, NiOx films were first bleached with Ar gas flow before different atmospheres were introduced into the gas flow cell and cycled with Ar. As expected, cycling with an inert gas led to no visible change in the absorption at 500 nm in the performed measured time scale (ESI,† Fig. S9), however exposure to water with Ar as carrier gas or oxygen showed a visible absorption change (absorption traces in Fig. 2, full spectra in ESI,† Fig. S11). Interestingly, water was not able to reform any surface states but instead resulted in large absorption changes due to the water scattering signal that was quickly removed upon switching to an Ar stream and did not leave a recovery of the bleach behind (Fig. 2A).
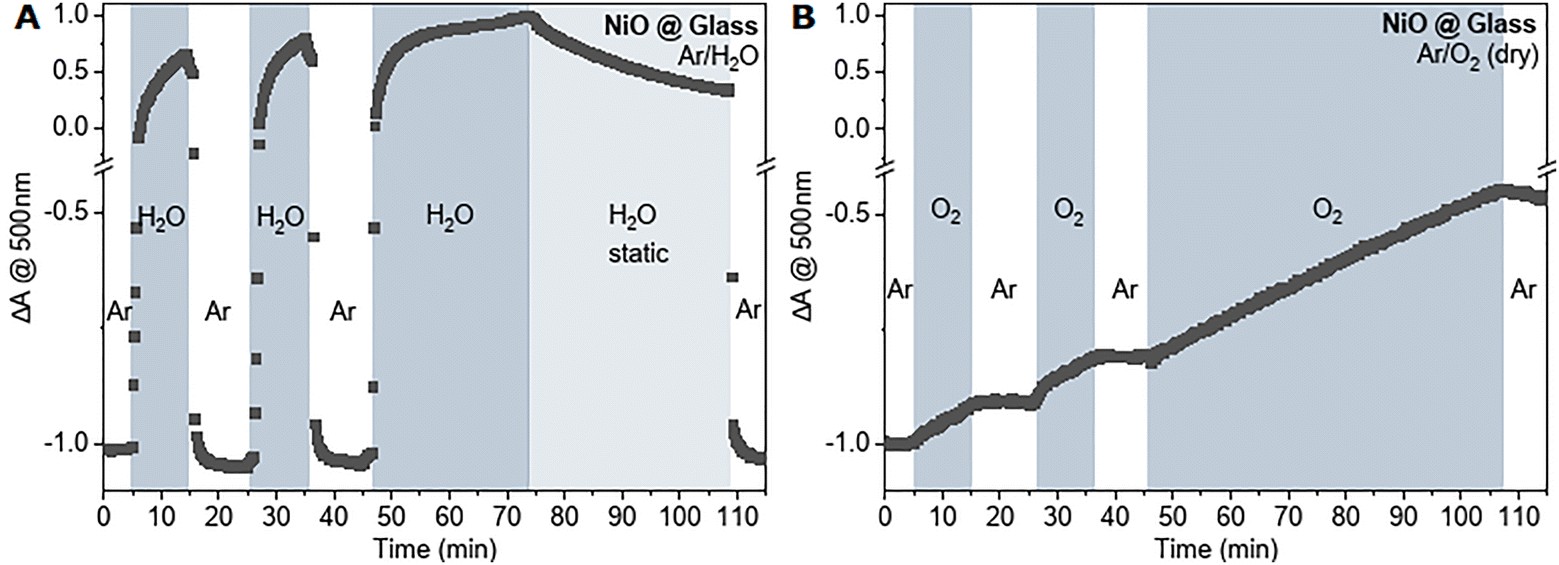 | ||
| Fig. 2 Kinetic traces of NiOx absorption at 500 nm of bleached films via Ar gas flow upon introduction of (A) water with Ar as carrier gas and cycling with pure Ar and (B) oxygen gas flow that is cycled with Ar. Absorption changes are normalized such that the bleached films are at −1. | ||
In contrast, oxygen was able to recover the bleach at 500 nm, visible by the rise of the baseline at 500 nm after switching back to Ar gas flow (Fig. 2B). This suggests that oxygen readily adsorbs to the bleached nickel oxide surface and likely leads to the formation of higher-valence nickel states that causes the rise in absorption (eqn (1)).
| Ni2+ + O2 ↔ [Ni3+⋯O2−] | (1) |
In situ FTIR spectroscopy performed during removal and re-formation of surface states was also employed to gain further insight into interfacial processes during gas flow (Fig. 3A). Upon Ar gas flow, the time-resolved spectra show a bleach in the water region at 3400 cm−1 and a broad baseline drop over the whole spectrum.
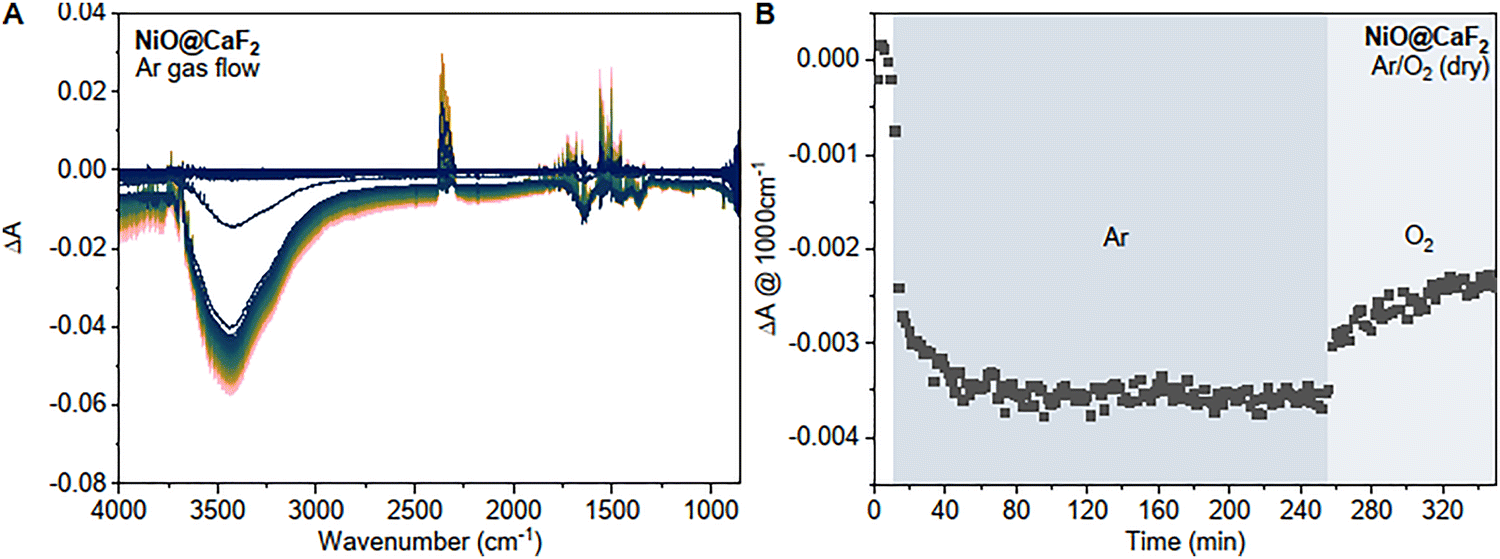 | ||
| Fig. 3 (A) Change in IR absorption of NiOx on CaF2 during Ar gas flow and (B) corresponding kinetic trace at 1000 cm−1 during Ar gas flow and subsequent O2 gas flow. | ||
As previously mentioned, the full IR spectra (ESI,† Fig. S13) show a remaining band in the OH stretch region after prolonged Ar gas exposure which we assign to surface hydroxyl species remaining on the surface. The instantaneous bleach in the water region supports our hypothesis of physiosorbed water that easily desorbs from the surface. A broad absorption over the entire IR region has previously been associated with the presence of free carriers in semiconductors in the literature.25 The broad baseline drop therefore is likely linked to a loss of free carriers and suggests that the removal of surface species is resulting in changes in the electronic properties of nickel oxide. Interestingly, switching for Ar gas flow to O2 flow resulted in the recovery of the baseline (Fig. 3B) which indicates that oxygen can bind and lead to the formation of free carriers (see eqn (2) where m+ symbolizes a free carrier) and diatomic oxygen ions are likely one of the removed species during Ar gas flow.
| NiO + O2 ↔ [NiO⋯O2−] + m+ | (2) |
 | ||
| Fig. 4 (A) CV of NiO films on FTO in 0.1 M TBAPF6 in dry acetonitrile with and without purging of Ar or O2 and (B) the corresponding change in absorption at 500 nm during the first CV scan. | ||
In another sample, the NiOx was measured directly from air in pre-purged electrolyte which should allow for oxygen at the surface. In the CV of the non-purged sample, there is a clear peak at −1.6 V vs. Fc/Fc+ and a brief increase in the absorption change at 500 nm of the NiOx film (Fig. 4). The CV peak coincides with the brief increase in absorption. A control purge with oxygen clearly reveals this peak in the CV and brief increase in absorption is due to the presence of oxygen (compare to the Ar purged sample). The peak in the CV should therefore correspond to the reduction of oxygen to the superoxide anion, O2−. Due to the observed increase in absorption at this potential, the previously electrochemically reduced nickel states seem to react with oxygen and may be catalysing oxygen reduction, visible by the recovery of the bleach which suggests the formation of higher valence nickel states. This formation seems to be initially faster than the electrochemical reduction of Ni3+/4+ to Ni2+/3+.
Another interesting observation from the CV and its corresponding absorption change of both purged and non-purged samples is the need of a significantly more positive potential than the OCP to reach the initial absorption of the film. This could be indicative of a small fraction of some higher valence Ni states present in the as-prepared film that cannot be reformed with the potential of the OCP (which is above the first redox peak which likely corresponds to Ni2+/Ni3+) or that the surface reforms.
Considering the previous observation from UV-vis and FTIR experiments, we therefore propose that approximately 50% of the surface states are hydroxides and possibly other states that cannot be removed with room temperature gas flow and cause the remaining hydroxide signal in FTIR spectroscopy, while the other half is mainly adsorbed oxygen species on the NiOx surface (likely forming diatomic oxygen ions such as O2−) which results in a higher valence Ni state (likely mostly Ni3+) upon binding. The oxygen could be end-on chemisorbed or bridge chemisorbed where the oxygen would be bound to two Ni sites. Water is also present at the surface, however, does not seem to cause any change in Ni valence state or change surface properties.
Insights on the influence of temperature and gas injection gained with ambient pressure X-ray photoelectron spectroscopy
 | ||
| Fig. 5 (A) XPS spectra of mesoporous NiOx films at increasing temperature and deconvolution of the O 1s, (B) the relative fitted peak ratio with respect to temperature, and (C) corresponding proposed surface species. | ||
Fig. 5 shows a schematic overview of the fitted XPS peaks with the proposed adsorbates. The most prominent of the fitted surface peaks at 531.4 eV at RT (peak 3) is typically assigned to nickel hydroxides in the literature.11,12,26,27 The same assignment here is supported both by the presence of a significant feature for hydroxyl groups in the IR spectra of NiOx films, and a stable ratio of peak 1 to peak 3 until 200 °C, around which nickel hydroxides are known to decompose.21 The peak at highest binding energies at 533.0 eV (peak 5) is also well documented in the literature and is assigned to adsorbed water.11,12 The same assignment here is supported both by the disappearance of this peak at temperatures above 200 °C, and the fact that this peak become more prominent upon exposure to water (ESI,† Fig. S22).
The two remaining peaks at 530.8 eV (peak 2) and 532.0 eV (peak 4) are more difficult to assign. In WO3, a well-studied gas sensor, the fitted O 1s peak located 1 eV above the lattice oxygen peak (which aligns well with peak 2 in our case) has been assigned to chemisorbed oxygen ions.16 Blume et al.11 have also assigned a peak at this binding energy to surface O2− in sputtered NiO. Furthermore, in CeO2, AP-XPS O 1s peaks at 530.5 and 532.2 eV have been previously assigned to peroxide (O22−) and superoxide (O2−), respectively.28 The C 1s shows carbon impurities on the surface which might be a source of surface oxygen species. However, the totally different temporal response between the carbon species and peak 4 exclude the possible relation between them (Fig. S24, ESI†). On this basis, we assign peak 2 and peak 4 to surface oxygen ions. Their response to O2 gas doing in later section further strengthened the assignment.
A concrete assignment of adsorption coordination of these oxygen ions to the NiO surface is difficult since the spectral contributions of these different adsorption coordination likely overlap and therefore a multitude of species might be hidden within one peak. However, there are some observations that might help in narrowing down the possible components for peak 2 and 4 when comparing the spectra from different temperature. While peak 2 is decreasing immediately when the temperature increases to 75 °C (Fig. 5A and ESI,† Fig. S15b), the corresponding intensity of peak 4 increases with a comparable magnitude, which may suggest a transformation of a species in peak 2 to the species of peak 4. Furthermore, while the intensity of peak 2 is quickly decreasing with temperature, the intensity of peak 4 only starts to decline above 200 °C, suggesting that the transformed species is relatively stable. Out of the considered oxygen species, O−(ads) should be the least stable, followed by O22−(ads), which both can dissociate into O−(ads), which is an even more stable oxygen species.16
Out of the possible surface oxygen ions for peak 2 and peak 4, we therefore cautiously assign peak 4 to O−(ads) due to the observed rise with temperature, while peak 2 is assigned to O2−(ads) and/or O22−(ads). Surface O2− is even more stable,16 however such states are expected at lower binding energies than O−(ads), and would thus be expected in peak 2 if present on the NiOx sample. It should be kept in mind that the nanoparticular nickel oxide surface is highly irregular in reality and different sites, with kinks, steps and corners, which gives rise to a multitude of possible binding modes for surface species. Different facets have a varying amount of “free” bonding sites and can also form different oxidation states and offer binding spots for one or more absorbents, especially for nickel.29 Furthermore, across the surface, this might possibly result in in-between formal oxidation states and that the proposed oxidation state should merely be seen as an indication. The above identification of surface states should therefore best be viewed as interpretations that provide a framework for understanding the overall trends and behaviours rather than definitive descriptions of the surface chemistry.
To understand what is happening on the Ni side, we turned to X-ray Absorption Spectroscopy (XAS) in electron yield mode. In our XAS measurements (see ESI,† Fig. S17), we primarily observed an absorption peak at 852.5 eV, with a shoulder at 854.5 eV. According to literature, Ni L edge spectra from NiO exhibits a primary XAS absorption feature around 853 eV, accompanied by a minor shoulder at approximately 855 eV, thus matching the obtained spectra reasonably well.30 In literature, both Ni(OH)2 and NiOOH present significant absorption features at 855 eV. Based on its relative intensity to the main peak, the observed shoulder is likely primarily the NiO shoulder, but with potential contributions from other nickel species such as Ni(OH)2 and NiOOH. Furthermore, Ni4+ species are in literature characterized by absorption features at even higher photon energies (around 856.5 eV), however these are not noticed in the measured spectra.31 The observed variations across different temperatures were marginal, which makes it challenging to draw definitive conclusions regarding the oxidation state or chemical species of nickel. The slight loss of intensity of the 854.5 eV shoulder and the shift towards lower photon energies do, however, suggest a more reduced character of the nickel species with increasing temperature.
To gain further insights, we also performed Ni 2p XPS measurements (see Fig. 6). Assigning the chemical contributions of NiOx from the Ni 2p spectra with multi-component analysis is complex, especially due to the double-peak feature of the main line, which complicates the analysis. The lowest binding energy feature (around 853.5 eV) is assigned to NiOx lattice. The high-energy peak (1.5–2 eV from the NiOx lattice) was previously attributed to surface defects or higher oxidation states of Ni.32,33 However, theoretical calculations using a multi-NiO6 cluster model, combined with experimental results, have also demonstrated that due to strong Coulomb repulsion in the localized d orbital, the creation of a core hole in the 2p level results in an electron transfer from a neighboring NiO6 unit, forming 2p 3dn+1 from the ground state 3dn. This extra hole on the neighboring unit gives rise to the shoulder feature on the main line.34–36 Recent studies have also shown that the shoulder peak has a notable contribution from surface effect as shown by changes in the spectra with varying take-off angles during XPS measurements.37
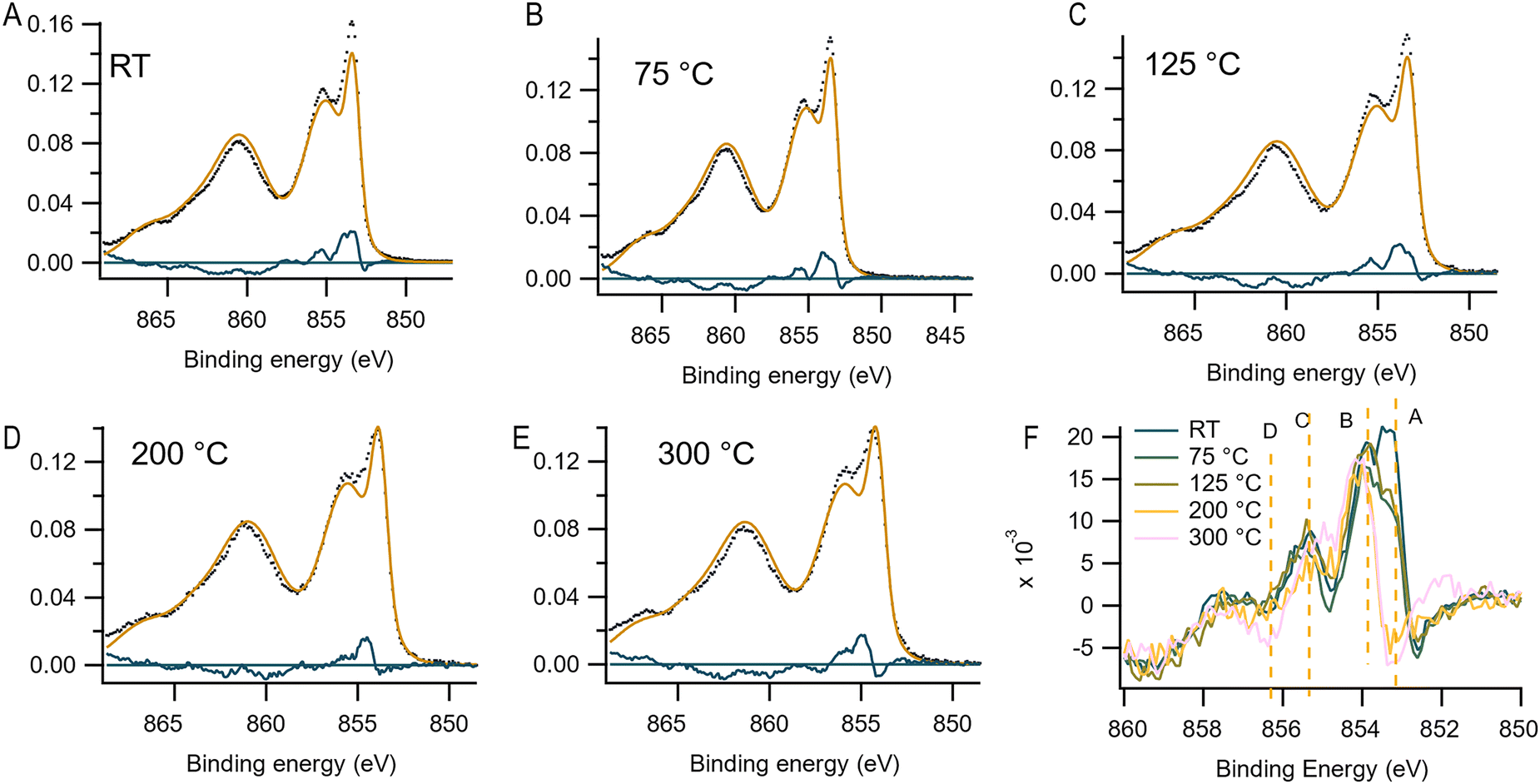 | ||
| Fig. 6 (A)–(E) Area normalized Ni 2p spectra of mesoporous NiOx films (black dots) from synchrotron XPS, the bulk NiO from HAXPES with Ga source (orange dashed) and the residue of the bulk spectra from HAXPES to the data from synchrotron XPS (blue line) with increasing temperatures. (F) Direct comparison of the subtracted spectra to the surface contribution at increasing temperatures. | ||
To extract the surface Ni compounds from the Ni 2p spectra, which are mixed between physical energy splitting and chemical shifts, a reference Ni 2p spectrum is needed to subtract the pure bulk NiO signal. High-energy X-ray photoelectron spectroscopy (HAXPES) with a Ga Kα source (9225 eV) was applied to obtain bulk NiO spectra, using a pass energy of 500 eV under UHV conditions. Fig. S18 (ESI†) shows the O 1s, C 1s, and Ni 2p core level raw spectra in comparison with those obtained from an Al Kα source recorded immediately after the HAXPES at the same instrument. The O 1s spectrum recorded with the Ga source shows a main contribution at 529 eV, attributed to the lattice NiO, whereas the Al source shows a high energy shoulder (Fig. S25, ESI†). The very low C 1s intensity (100 times lower in counts than O 1s) with no spectral features, together with the single feature from O 1s, indicates that the intensity are mainly from the bulk NiO feature using the Ga source HAXPES. Thus, the HAXPES spectra with a Ga source mainly show bulk contributions. The Ni 2p HAXPES spectra reveal a double-peak main line as expected, and the high-energy peak significantly increases in the Ni 2p spectra measured with the Al source, indicating contribution from surface compounds.
The Ni 2p spectra obtained from the synchrotron facility at varying temperatures were then analyzed with the HAXPES bulk NiO as a reference. To curve fit the spectra, a Shirley type background was used encompassing the 2p3/2 spin–orbital to subtract the contribution from inelastic-scattered electrons. To extract the surface species, a self-consistent method is here used by subtracting all the Ni 2p with the bulk NiO from HAXPES after background removal. Subtraction is applied by firstly normalizing all the Ni 2p3/2 spectra after background removal to their peak area. A peak broadening refinement is needed due to the different Gaussian broadening from X-ray source and analyzer between HAXPES and synchrotron facility. Energies are calibrated to the mainline peak position.
Fig. 6(A)–(E) shows the Ni 2p spectra under corresponding temperatures (black dots), and its bulk NiO contribution from Ga-source measurement (orange dashed), and the blue curve gives the residue of the spectra after subtraction to bulk NiO. Fig. 6(F) stacks the subtracted spectra for the surface contribution comparison. As the temperature increases, a clear decrease in the intensity of residue is observed, indicating the general removal of surface species. At room temperature (RT), a surface component at ΔBE = 0 eV vs. NiO (point A) dominates the spectrum and is very sensitive to temperature. Interestingly, the surface contribution of peak A is not observed in the XPS from Al under UHV (10−9 mbar). This suggests that the component at point A is likely Ni linked to surface-adsorbed water (H2O), which does not affect the Ni oxidation state and can be removed under vacuum. Given that the synchrotron data were obtained under a 10−5 mbar environment, the presence of water is plausible. This also explains why water-related species are absent under UHV conditions. The temperature sensitivity of species A is evident, as it significantly diminishes even at 75 °C and disappears fully by 200 °C, consistent with the disappearance of the water signal in the O 1s spectra.
The intensity around point B at ΔBE = 0.5 eV vs. NiO is another region that is changing with temperature. While this has not been discussed much in the literature, based on the above results this might be connected to a nickel species weakly bound to an oxygen species. Considering the temperature stability of this species and observations in the O 1s spectra, and that O2−/O22− are likely less strongly bound, peak B could possibly be nickel bound to these surface oxygen species.
In the literature, Ni(OH)2 has a large contribution around ΔBE = 2.0 eV vs. NiO, with a minor contribution from NiOOH in that range.39 Therefore, peak C is likely dominated by Ni(OH)2 species. The stability of this peak at lower temperatures before decreasing in intensity supports its assignment to nickel hydroxides, which is also in line with observations in the O 1s spectra. In contrast, NiOOH has a large contribution approximately at ΔBE = 2.8 eV vs. NiO.38 This is around the energy of point D, which is very temperature stable and therefore most consistent with O− species bound to nickel. This peak also only decreases at 300 °C, suggesting it represents more stable nickel-bound oxygen species. It should be noted that the appearing shoulder at 852 eV at 300 °C is likely linked to the formation of oxygen vacancies and eventually the formation of metallic nickel that can be seen at 450 °C in vacuum (see ESI,† Fig. S16). However, it is completely absent at lower temperatures which means it is not a species that is present under ambient conditions.
The stable feature around ΔBE = 3.5 eV has previously been linked to the presence of nonlocal charge fluctuations (screening) in nickel oxide.33,35,36
In general, at temperatures above 200 °C, a decrease in the higher binding energies of the residual contribution can be observed in the XPS spectra, indicating that these are the most temperature stable Ni species. The lower binding energy signals are Ni linked to more weakly adsorbed surface oxygen which leave while the more strongly chemical bonded oxide species stays. This trend can be rationalized by considering the behavior of oxygen species bound to the surface. As the temperature increases, these species are proposed to transition from O2− to O− to O2− and ultimately lead to the formation of oxygen vacancies.16
Changes on gas exposure
To study the removal and re-formation of surface states, in situ AP-XPS measurements of mesoporous NiOx films were carried out during a sequence of heating/cooling (RT-200 °C-RT) in vacuum (removal) and subsequent gas dosing (re-formation). Similar to the step-wise heating of the sample, the peak shifts to higher binding energies with increasing temperature (ESI,† Fig. S20) can clearly be seen in the 2D-map of the data in Fig. 7A. It is also evident that the overall intensity and position of the spectra are influenced by the exposure to oxygen, while a mere cooling imposed less changes. | ||
| Fig. 7 O 1s spectra during a sequence of heating to 200 °C and cooling to RT in vacuum followed by (A) oxygen or (C) water gas dosing and subsequent return to vacuum and (B) and (D) the corresponding intensity traces of the fitted peaks during the sequence. | ||
To gain more detailed knowledge, each spectrum in the heatmap (Fig. 7A) is curve fitted (see method) with the same fitting parameter as used above after a linear background subtraction, and the evolution of the fitted peak intensities as a function of applied local temperature and pressure are shown in Fig. 7B, with an identical colour code as previously used. During the heating cycle, the components behave similar to the stepwise heating, with an immediate drop of peak 2 (O2−(ads) and/or O22−(ads)) intensity upon heating, while peak 4 (O−(ads)) steadily rises.
Upon gas dosing with oxygen, the binding energies of all peaks quickly shift towards lower binding energies (Fig. 7A) even at very low oxygen levels. The change in binding energy of the lattice oxygen peak over the whole sequence (ESI,† Fig. S23) also clearly shows this shift to lower binding energies after the initial shift to higher binding energies during heating. This not only shows that nickel oxide surface is very reactive towards oxygen, but particularly the shift of the bulk NiO peak shows that exposure to oxygen and the formation of oxygen states affect the Fermi-level of nickel oxide, and thus, affect electronic properties. The traces of the fitted peaks during the sequence show that peak 2 (O2−(ads) and/or O22−(ads)) is most responsive to the onset of gas dosing even at low oxygen content, as can be expected from the nature of the oxygen gas (Fig. 7B, snapshots of the O 1s spectra during oxygen gas dosing in ESI,† Fig. S20). This further strengthens the proposed assignment of peak 2 to adsorbed diatomic oxygen ions. Theoretical calculations for oxygen adsorption to TiO2 have shown that for a low ratio of oxygen to surface defects, O2 adsorbs as a peroxide (O22−), and for a higher number of oxygen, superoxide species (O2−) are forming.39 Based on fact that the surface should have much fewer adsorbed species after the heating/cooling cycle, it is very likely that O22− is forming initially (hence a quick rise of peak 2) which would also the be fitting for the binding energy that is proposed for O22− in the literature.28 However, under atmospheric conditions, it is likely that O2− is the predominant species.
The same sequence of heating–cooling-gas dosing was repeated for a new sample with water dosing instead of oxygen and the spectra showed a similar heating/cooling behaviour (Fig. 7C and D and snapshots of the O 1s spectra during water dosing in ESI,† Fig. S22). However, in contrast to oxygen, peak 2 and 4 remained unaffected with water dosing while an increase in peak 3 and 5 was visible. This strengthens the previous assignment of peak 2 and 4 to oxygen ions and peak 3 to hydroxides and peak 5 to water. It also suggests that even though water does not seem to affect the electronic properties of NiOx directly, it leads to the formation of hydroxide surface states.
Conclusion
In light of the data acquired from XPS, UV-vis, and FTIR spectroscopy, valuable insights on the surface chemistry of NiOx were achieved. The XPS results affirm our initial observations, indicating that approximately half of the surface states are hydroxides, which persist despite room temperature gas flow, as evidenced by the residual hydroxide signal in FTIR spectroscopy. The remaining surface states are predominantly adsorbed oxygen species, likely diatomic oxygen ions. Both surface species are likely associated with a higher valence Ni states.The distinct reactivity of oxygen with NiOx is evident, as reflected in the response of the oxygen ion peak in the XPS spectra upon oxygen dosing, even at very low oxygen levels. Conversely, water, while present on the surface, does not appear to result in any significant electronic modification to the NiOx. However, its presence aids in the formation of hydroxides, as suggested by the increase of the hydroxide peak in the XPS spectra upon water dosing.
Therefore, we propose that during the fabrication process of nickel oxide, atmospheric oxygen and water lead to the formation of hydroxides and surface oxygen ions (predominantly diatomic at room temperature) in an approximately equal ratio. This insight into the surface state composition provides a more comprehensive understanding of the NiOx surface, demonstrating the importance of both hydroxides and oxygen ions on the nickel oxide properties.
Methods
Materials
All chemicals were purchased from Sigma-Aldrich and used directly unless stated otherwise. Fluorine-doped tin-oxide coated glass (FTO, TEC15) was purchased from Pilkington. Hexafluorophosphate (TCI Europe, ≥98%) was recrystallized and dried at 80 °C in vacuum before use. Acetonitrile was purified by a solvent purification system and dried with 3 Å molecular sieves for 48 h. NiO screen printing paste were lab-made according to a literature procedure40 and available from a previous study. In short, a slurry of NiO nanopowder (<50 nm particle size) and ethanol was mixed with an ethanolic ethyl cellulose solution in terpineol, before ethanol was removed slowly with rotary evaporation.Sample preparation
Except for samples on CaF2 substrates, the mesoporous NiO films were prepared on fluorine-doped tin oxide (FTO)-coated glass substrates or glass that was carefully cleaned in successive ultrasonic baths of detergents, deionized water, acetone and ethanol. For Spectroelectrochemistry and UV-vis measurements, the mesoporous NiO films were either coated directly onto the FTO side or glassy side of the substrates through screen printing of two layers of the lab-made NiO paste based on nanoparticular NiO particles in an ethyl cellulose solution. Between each layer, the films were heated at 150 °C for 5 min on the hotplate. For the conductivity measurements, only one layer was deposited. The films were thermally annealed at 450 °C for 30 min and cooled down to room temperature, resulting in a mesoporous NiO layer of 700 nm for single layer deposition and 900 nm for two layers (measured with Profilometer, Dektak 150 surface profiler). The films on CaF2 substrates for IR spectroscopy were prepared by doctor-blading a single layer of the lab-made NiO paste directly on the CaF2 films and then heated to 150 °C for 5 min on the hotplate before the thermal annealing at 450 °C for 30 min.(Spectro)electrochemistry
Cyclic voltammetry (CV) measurements were performed with a three-electrode electrochemical setup in a cuvette, utilizing an AUTOLAB potentiostat (PGSTAT302N) controlled the software Nova. As reference electrode, Ag/AgNO3 (1.0 mM AgNO3 acetonitrile solution) was utilized and platinum wire (Pt) as counter electrode. FTO-NiO electrodes were used directly as working electrodes, submerged in the electrolyte, 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in dry acetonitrile from a solvent-purification system. A scanning rate of 100 mV s−1 was applied for CV measurements and ferrocene was used as an internal standard. If the measurements were not carried out in the glovebox, measurements were carried out in argon-saturated electrolyte solution. UV-vis spectroelectrochemistry was performed with the same three-electrode electrochemical setup and electrolyte as for electrochemistry measurements, as described previously. Measurements were carried out in a home-made electrochemical setup, consisting of a glass cuvette with 1 cm pass-length and stopper designed to hold the three electrodes, while only having the FTO-NiO films in the optical path.UV-vis and FTIR spectroscopy
UV-vis absorption spectra were recorded with a diode array spectrophotometer (Agilent 8453) that could either be connected to the glovebox for spectroelectrochemistry or used separately. Gas-flow UV-vis experiments were carried out with a lab-made gas cell and that had a film holder in the optical path (see picture of setup in ESI,† Fig. S1). Fourier-transformed infra-red (FTIR) measurements were done in transmission with a TensorII (DLATGS detector, Bruker) and a modified film holder that utilized the CaF2–NiO as one of the optical windows and another CaF2 substrate as the second window, creating a cell through which the gas was flown (see picture of setup in ESI,† Fig. S1).X-ray spectroscopy
To avoid charging as much as possible, a thinner mesoporous NiO film was used for X-ray spectroscopy by diluting the lab-made NiO paste with terpineol (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 w/w) and screen printing a single layer of the diluted paste onto cleaned FTO glass. The films were first heated to 150 °C for 5 min on the hotplate and then thermal annealed at 450 °C for 30 min. The resulting thickness of films was 350 nm (measured with Profilometer, Dektak 150 surface profiler). For the X-ray measurements, the film was mounted onto a metal sled with a thermal couple (see Fig. S3, ESI†).
:3 w/w) and screen printing a single layer of the diluted paste onto cleaned FTO glass. The films were first heated to 150 °C for 5 min on the hotplate and then thermal annealed at 450 °C for 30 min. The resulting thickness of films was 350 nm (measured with Profilometer, Dektak 150 surface profiler). For the X-ray measurements, the film was mounted onto a metal sled with a thermal couple (see Fig. S3, ESI†).
Instrumentation
Ambient pressure XPS measurement was conducted at HIPPIE beamline solid–gas branch in MAX IV synchrotron radiation facility in Lund, Sweden.41 A cell-in-cell design is used for conducting the AP-XPS measurement, of which a small cell (ca. 1L) is provided for ambient application which sits inside of a UHV chamber (base pressure 10−10 mbar). The in situ APXPS measurements with O2, H2O gas dosing were taken at 3 mbar through a gas line with individual mass flow controllers. HAXPES (Kaj Siegbahn Laboratory, Uppsala University) is applied for obtaining the NiO bulk contribution for Ni 2p analysis. The experiment was operated under 10−8 mbar. To obtain a high signal-to-noise ratio of the spectra. 50 scans of each core level (Ni 2p, O 1s, and C 1s) under sweep mode were acquired.More information on the intensity calibration, curve fitting for O1s and Ni 2p analysis can be found in the ESI.†
Author contributions
S. W. conducted the experiments, performed data analysis and interpretation, Q. L. contributed to X-ray measurements, data analysis and interpretation, L. C. aided in conductivity experiments, L. A. contributed to conducting X-ray experiments, B. C. contributed to X-ray experiments, M. S. aided in X-ray experiments and methodology, Z. Z. contributed with providing instrumentation resources, M. H. contributed to X-ray measurements, provided supervision and X-ray methodology and interpretation, H. T. acquired funding, provided resources and supervision and contributed to interpretation of results. Q. L. and S. W. created the original draft. All authors reviewed the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The raw data is available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to gratefully thank the Swedish Energy Agency (49278-1), K&A Wallenberg Foundation (2019.0156) and the Swedish Research Council (2020-04512, 2022-06076) for financial support. We acknowledge MAX IV Laboratory for time on Beamline HIPPIE under Proposal 20221421. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. The authors would also like to thank Pedro Berastegui and Anna Arkhypchuk for their help with the fabrication of our lab-made gas cell.References
- Y. Gong, S. Zhang, H. Gao, Z. Ma, S. Hu and Z. A. Tan, Sustainable Energy Fuels, 2020, 4, 4415–4458 RSC.
- M.-H. Li, J.-H. Yum, S.-J. Moon and P. Chen, Energies, 2016, 9(5), 331 CrossRef.
- T. P. Mokoena, H. C. Swart and D. E. Motaung, J. Alloys Compd., 2019, 805, 267–294 CrossRef.
- J. Zhao, Y. Tian, A. Liu, L. Song and Z. Zhao, Mater. Sci. Semicond. Process., 2019, 96, 78–90 CrossRef.
- T. M. Schuler, D. L. Ederer, S. Itza-Ortiz, G. T. Woods, T. A. Callcott and J. C. Woicik, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115113 CrossRef.
- Y.-J. Lin, T.-H. Su, P.-C. Kuo and H.-C. Chang, Mater. Chem. Phys., 2022, 276, 125345 CrossRef.
- V. Biju and M. Abdul Khadar, Mater. Res. Bull., 2001, 36, 21–33 CrossRef.
- R. Poulain, G. Lumbeeck, J. Hunka, J. Proost, H. Savolainen, H. Idrissi, D. Schryvers, N. Gauquelin and A. Klein, ACS Appl. Electron. Mater., 2022, 4, 2718–2728 CrossRef.
- R. Poulain, A. Klein and J. Proost, J. Phys. Chem. C, 2018, 122, 22252–22263 CrossRef.
- R. Poulain, J. Rohrer, Y. Hermans, C. Dietz, J. Brötz, J. Proost, M. Chatenet and A. Klein, J. Phys. Chem. C, 2022, 126, 1303–1315 CrossRef.
- A. R. Blume, W. Calvet, A. Ghafari, T. Mayer, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2023, 25, 25552–25565 RSC.
- K. Zhu, S. K. Frehan, G. Mul and A. Huijser, J. Am. Chem. Soc., 2022, 144, 11010–11018 CrossRef.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farré, S. Narbey, F. Ostwald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- C. J. Flynn, S. M. McCullough, L. Li, C. L. Donley, Y. Kanai and J. F. Cahoon, J. Phys. Chem. C, 2016, 120, 16568–16576 CrossRef CAS.
- L. Tian, R. Tyburski, C. Wen, R. Sun, M. Abdellah, J. Huang, L. D’Amario, G. Boschloo, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2020, 142, 18668–18678 CrossRef CAS.
- E. Ciftyurek, Z. Li and K. Schierbaum, Sensors, 2023, 23(1), 29 CrossRef CAS.
- C. Li, P. G. Choi and Y. Masuda, Adv. Sci., 2022, 9, 2202442 CrossRef CAS PubMed.
- M. Bonomo, G. Naponiello, I. Venditti, V. Zardetto, A. D. Carlo and D. Dini, J. Electrochem. Soc., 2017, 164, H137 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef.
- L. D’Amario, R. Jiang, U. B. Cappel, E. A. Gibson, G. Boschloo, H. Rensmo, L. Sun, L. Hammarström and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 33470–33477 CrossRef.
- C. S. Carney, R. E. Chinn, Ö. N. Doğan and M. C. Gao, J. Alloys Compd., 2015, 644, 968–974 CrossRef.
- L. D'Amario, J. Föhlinger, G. Boschloo and L. Hammarström, Chem. Sci., 2018, 9, 223–230 RSC.
- M. D. Dong, J. Y. Shen, C. Y. Hong, P. X. Ran, R. H. He, H. W. Chen, Q. Y. Lu and J. Wu, J. Appl. Phys., 2022, 132, 173901 CrossRef.
- C. E. Simion, B. Junker, U. Weimar, A. Stanoiu and N. Bârsan, Sens. Actuators, B, 2023, 390, 134028 CrossRef.
- J. Sá, P. Friedli, R. Geiger, P. Lerch, M. H. Rittmann-Frank, C. J. Milne, J. Szlachetko, F. G. Santomauro, J. A. van Bokhoven, M. Chergui, M. J. Rossi and H. Sigg, Analyst, 2013, 138, 1966–1970 RSC.
- H. Idriss, Surf. Sci., 2021, 712, 121894 CrossRef.
- Y. Koshtyal, D. Nazarov, I. Ezhov, I. Mitrofanov, A. Kim, A. Rymyantsev, O. Lyutakov, A. Popovich and M. Maximov, Coatings, 2019, 9(5), 301 CrossRef.
- L. Soler, A. Casanovas, C. Escudero, V. Pérez-Dieste, E. Aneggi, A. Trovarelli and J. Llorca, ChemCatChem, 2016, 8, 2748–2751 CrossRef.
- A. G. Marrani, V. Novelli, S. Sheehan, D. P. Dowling and D. Dini, ACS Appl. Mater. Interfaces, 2014, 6, 143–152 CrossRef PubMed.
- M. Al Samarai, A. W. Hahn, A. Beheshti Askari, Y.-T. Cui, K. Yamazoe, J. Miyawaki, Y. Harada, O. Rüdiger and S. DeBeer, ACS Appl. Mater. Interfaces, 2019, 11, 38595–38605 CrossRef.
- F. Massel, B. Aktekin, Y.-S. Liu, J. Guo, M. H. Sørby, D. Brandell, R. Younesi, M. Hahlin and L.-C. Duda, Energy Adv., 2023, 2, 375–384 RSC.
- M. W. Roberts and R. S. C. Smart, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2957–2968 RSC.
- I. Preda, R. J. O. Mossanek, M. Abbate, L. Alvarez, J. Méndez, A. Gutiérrez and L. Soriano, Surf. Sci., 2012, 606, 1426–1430 CrossRef.
- M. Oku, H. Tokuda and K. Hirokawa, J. Electron Spectrosc. Relat. Phenom., 1991, 53, 201–211 CrossRef.
- M. Tomellini, J. Electron Spectrosc. Relat. Phenom., 1992, 58, 75–78 CrossRef.
- M. A. van Veenendaal and G. A. Sawatzky, Phys. Rev. Lett., 1993, 70, 2459–2462 CrossRef.
- L. Soriano, I. Preda, A. Gutiérrez, S. Palacín, M. Abbate and A. Vollmer, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 233417 CrossRef.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef.
- U. Aschauer, J. Chen and A. Selloni, Phys. Chem. Chem. Phys., 2010, 12, 12956–12960 RSC.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef PubMed.
- S. Zhu, M. Scardamaglia, J. Kundsen, R. Sankari, H. Tarawneh, R. Temperton, L. Pickworth, F. Cavalca, C. Wang and H. Tissot, J. Synchrotron Radiat., 2021, 28, 624–636 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00137d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2025 |