
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of electrochemical sensors based on quinone derivatives adsorbed on graphene for the detection of [Cd(CN)4]2−†
Golfer
Muedas-Taipe
 *a,
Michael
Badawi
b,
Angélica María
Baena-Moncada
a and
Miguel
Ponce-Vargas
*bc
*a,
Michael
Badawi
b,
Angélica María
Baena-Moncada
a and
Miguel
Ponce-Vargas
*bc
aLaboratorio de Investigación de Electroquímica Aplicada, Facultad de Ciencias de la Universidad Nacional de Ingeniería, Av. Túpac Amaru 210, Rímac, Lima, Peru. E-mail: gmuedast@uni.edu.pe
bLaboratoire Lorrain de Chimie Moléculaire (L2CM) UMR CNRS 7053, Université de Lorraine. Faculté des Sciences et Techniques, 54500 Vandœuvre-lès-Nancy, France. E-mail: miguel-armando.ponce-vargas@univ-lorraine.fr
cUniversité de Reims Champagne-Ardenne, Moulin de la Housse 51687, Reims Cedex 02 BP39, France
First published on 15th April 2025
Abstract
Although cyanide is essential in mining operations, its high toxicity to both human health and the environment makes it an extraction agent that requires continuous in situ monitoring. This can be achieved through electrochemical sensors, which enable optimal detection of cyanide and related species without the need for time-consuming sample preparation steps. Graphene-based electrochemical sensors can be enhanced through non-covalent functionalization, involving the adsorption of a modifier onto the substrate surface via π–π interactions. In this study, we explored the effect of incorporating quinone derivatives onto a graphene substrate using a density functional theory (DFT) approach, coupled with a methodology based on the variation of the electronic density gradient (igmh). This approach aims to identify novel materials for the electrochemical detection of the tetracyanocadmate ion, [Cd(CN)4]2−, from WAD-CN (weak acid dissociable cyanide). First, we quantify the noncovalent contacts between the quinone and the graphene support through a fragment-based calculation. Subsequently, we focus on the coordination bond strength involving Cd2+ and the quinones attached to graphene. Then, we evaluate the effect of incorporating electron-donating substituents, which would directly lead to stronger coordination bonds with the metal center. The results reveal that an optimal balance between the modifier's anchoring on the substrate and its coordination strength toward Cd2+ can be achieved by functionalizing the graphene surface with 3-hydroxy-o-benzoquinones substituted at the 4-position with electron-donating groups. This suggests that experimental efforts conducted in this direction could lead to the development of electrochemical sensors with lower detection limits.
Introduction
The use of cyanide in mining operations, coupled with the limitations of current analysis and monitoring techniques, poses significant risks to both public safety and environmental sustainability.1–5 In this context, electrochemical sensors offer a powerful tool for the rapid, selective, and non-invasive detection of cyanide-related species.6–8 They could be used in real-time monitoring without time-consuming steps in sample preparation.Among the materials used for the manufacture of sensing devices, graphene has been widely employed due to its high electrical conductivity, great stability and extensive surface area.9–15 The performance of graphene sensors can be enhanced through covalent and non-covalent modifications.16 Covalent modification entails the formation of chemical bonds providing a strong link between graphene and organic molecules. However, most covalent functionalization methods result in a reduction in electrical conductivity. Additionally, some of these methods require complex synthesis protocols, which also results in increased costs.17 In contrast, non-covalent modification relies on the adsorption of the modifier onto the graphene surface via π–π interactions, which preserves the graphene π-conjugated system, and thus its electrical conductivity.18–21 Among the vastness of reagents proposed for noncovalent functionalization of graphene, quinones stands out due to their high redox reactivity, excellent electrochemical reversibility and the facile tuning of electrochemical properties achieved by modifying their molecular scaffolds and functional groups.22–26
To understand the effects of the non-covalent quinone attachment to graphene in sensing applications, it is mandatory to obtain an in-depth comprehension of the nature and strength of the interactions involved. Theoretical interpretations based on high-level ab initio calculations have been instrumental in this respect, enabling a better understanding of the interplay between attractive (electrostatic, dispersive, and inductive interactions) and repulsive (exchange repulsion) forces governing the formation of graphene-quinone assemblies at the electrode/solution interphase.27 The proposed models are often supported by computational methods based on quantum mechanics, which explicitly account for the electronic structure of the molecular systems. Recent work conducted by our group concerning the development of electrochemical sensors based on carbonaceous materials decorated with quinone derivatives for the detection of cyanide complexes from WAD-CN (weak acid dissociable cyanide), includes the rationalization of the adsorption process occurring at the electrode/solution interphase by calculations based on density functional theory (DFT).28–30
Herein, the effect of incorporating a panel of quinone derivatives to graphene is explored through a DFT approach complemented by a methodology based on the electronic density gradient variation,31 looking for the rational design of voltammetric sensors oriented to the detection of the tetracyanocadmate ion, [Cd(CN)4]2−, from WAD-CN. It should be noted that [Cd(CN)4]2− is the complex most commonly found in wastewater from mineral extraction activities, and these wastewaters can have pH values as high as 13. However, the optimal performance of some of the sensing platforms developed by our team is achieved at pH 5. Therefore, our current focus is on enhancing the sensing capacity of quinone-functionalized sensors in basic media. To this end, in the first stage, we evaluate the adsorption energies of the quinone-graphene assembly to assess the anchoring of the quinone over the support. We propose a model consisting of a Cd(II) center coordinated to two cyanide ions and two quinone ligands interacting with the graphene sheet, all immersed in an aqueous solvent medium, aiming to replicate the electrode/electrolyte interface under operando conditions. The noncovalent contacts between the complex and the graphene support were quantified, as well as the strength of the coordinative bonds involving the metal center and the surrounding quinone ligands. Next, the effect of incorporating electron-donating substituents into a benzoquinone scaffold, which would directly enhance the coordination bonds with the metal center, was evaluated. Finally, variations in the electronic structure of the functionalized graphene resulting from Cd2+ incorporation were evaluated through a total density of states (TDOS) analysis. This study is part of an ongoing research program focused on developing enhanced electrochemical sensing platforms for detecting cyanide complexes in water. It is anticipated that the computational protocol proposed herein could be extended to related systems aimed at the development of novel electrochemical sensors for detecting water pollutants.
Computational details
Calculations in the framework of the density functional theory were conducted by using the Gaussian16 software package,32 applying the M06-2X exchange correlation functional33 and the effective core potential LANL2DZ34 for Cd, and the 6-311G(d)35 basis set for the rest of atoms. The M06-2X functional has been chosen given that it is a functional parametrized taking into account noncovalent assemblies.36 The LANL2DZ pseudopotential, in turn, enables the assessment of cadmium at a low computational cost. For the other atoms, the 6-311G(d) basis set is the largest we can apply with our computational resources. The geometry optimizations were followed by frequency calculations to verify that each final structure represents the energy minimum of the corresponding potential energy surface. SCF convergence was set to 10−6 Hartrees for energy, density, and root convergence. Geometry optimizations were considered converged when the maximum gradient was <0.00045 a.u., RMS gradient <0.00030 a.u., maximum displacement <0.0018 a.u., and RMS displacement <0.0011 a.u. The geometry optimizations were followed by frequency calculations to verify that each final structure represents the energy minimum of the corresponding potential energy surface. To model the graphene surface, a polycyclic aromatic hydrocarbon (C112H26) was considered, given than this cluster is large enough to minimize the edge effects on the details of the electronic structure, and small enough to track the Kohn–Sham equations involved.The interaction energy of the ligand–graphene and complex–graphene assemblies has been calculated according to the following scheme:
| Eadsorption = Eligand(complex)–graphene − Eligand(complex) − Egraphene |
Additionally, the total density of states (TDOS) according to the Hirshfeld partition has been calculated with the Multiwfn code,41 to evaluate variations in the electronic structure of the functionalized graphene as a result of the Cd2+ capture.
Results and discussion
Interaction between quinone derivatives and graphene
Two panels of quinone molecules with scaffolds of different size were considered (Scheme 1). The first set (1–7) enables us to explore the interplay between graphene and the modifier. It includes: 1,8-dihydroxyanthraquinone (1), 1,4-dihydroxyanthraquinone (2), 9,10-phenanthroquinone (3), 1-hydroxy-9,10-phenanthroquinone (4), 2-hydroxy-1,4-naphthoquinone (5), 2-hydroxy-p-benzoquinone (6), and 4-hydroxy-o-benzoquinone (7). The selection of these molecules is based on previous research conducted by our team, focused on the development of electrochemical sensors based on quinone derivatives supported on carbonaceous materials for the detection of cyanide pollutants from mining process.28–30 The second set, oriented to evaluate inductive effects, consists of 4-substituted 3-hydroxy-o-benzoquinone derivatives (8–14). | ||
| Scheme 1 2D representation of the quinone derivatives under consideration in the present work. | ||
The structures of the quinones were fully optimized considering solvent effects, then placed over graphene and relaxed. The 2D representation of all the considered structures is depicted in Scheme 1. To evaluate the extent of the ligand attachment to the support, the interaction between the moieties is quantified through a fragment calculation, and by an igmh analysis. The igmh approach provides a binding score associated with the attenuation of the electronic density gradient when two molecular fragments approach each other.28 It also provides a graphical representation of the noncovalent surface associated with the graphene–ligand interplay. The optimized geometries of the graphene–ligand systems (1a–7a) are depicted in Fig. 1.
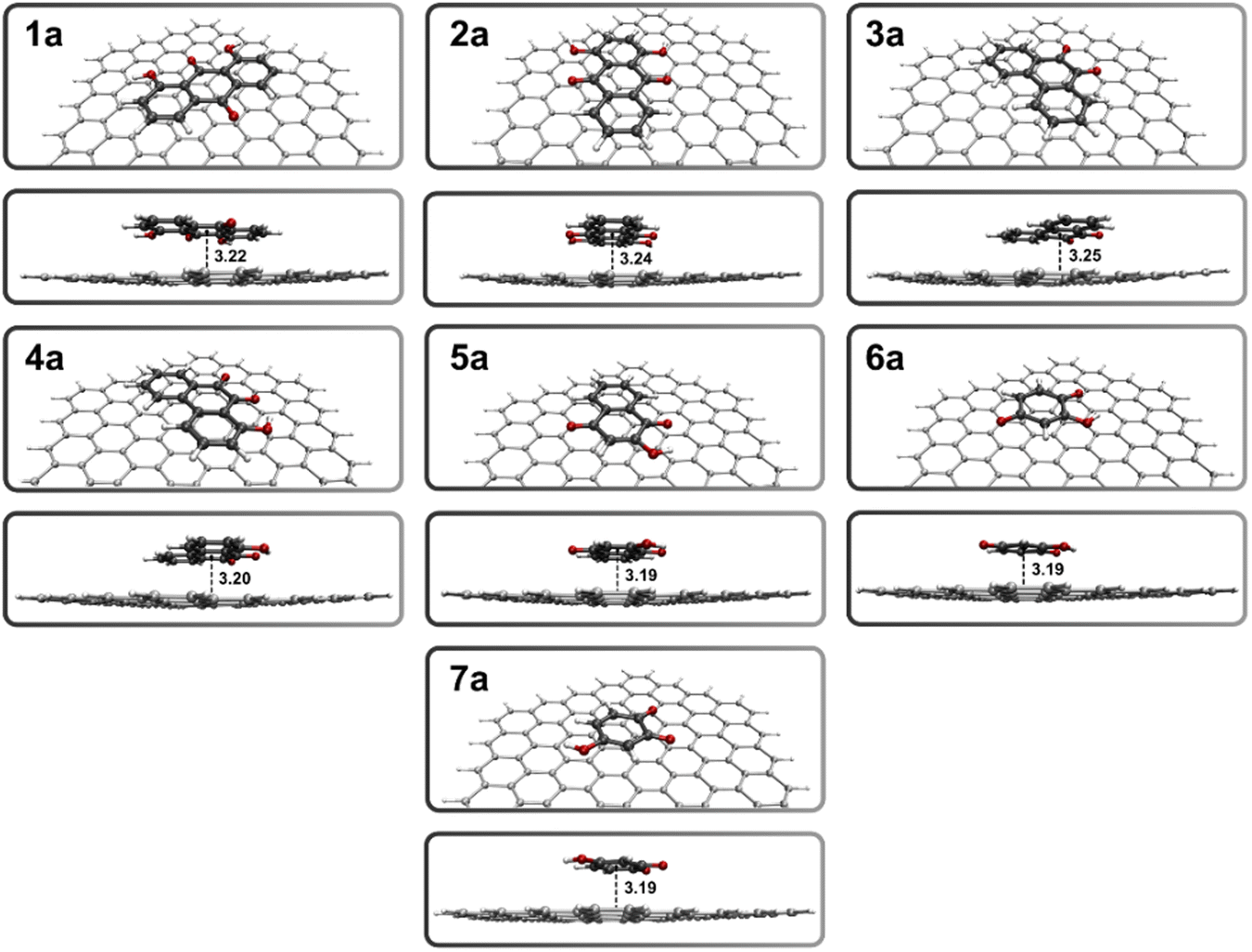 | ||
| Fig. 1 Optimized structures of the graphene–ligand systems involving quinones 1–7 (1a–7a) obtained at the M06-2X/6-311G(d) level of theory. The graphene–ligand distances are expressed in angstroms (Å). | ||
Some relevant geometrical parameters, along with the associated interaction energies and igmh scores, are presented in Table 1. The graphene–ligand distance has been measured from the center of the aromatic ring of the ligand to the closest carbon of the graphene sheet. The graphene–complex distance, in turn, has been calculated as the average of the distances between the aromatic rings of the ligand coordinating the Cd(II) center and the closest carbon of the graphene sheet. The igmh isosurfaces representing the region where noncovalent interactions arise, and the corresponding binding scores for 1a–7a are depicted in Fig. 2.
| Graphene–ligand assembly | 1a | 2a | 3a | 4a | 5a | 6a | 7a | 8a | 9a | 10a | 11a | 12a | 13a | 14a |
| Graphene–ligand distance (Å) | 3.22 | 3.24 | 3.25 | 3.20 | 3.19 | 3.19 | 3.19 | 3.13 | 3.17 | 3.21 | 3.16 | 3.17 | 3.19 | 3.17 |
| Interaction energy (kcal mol−1) | −25.7 | −25.3 | −22.7 | −24.0 | −18.4 | −11.8 | −13.4 | −15.7 | −15.2 | −15.3 | −13.8 | −15.9 | −16.3 | −17.4 |
| igmh binding score (a.u.) | 0.94 | 0.91 | 0.81 | 0.89 | 0.70 | 0.20 | 0.16 | 0.60 | 0.57 | 0.58 | 0.52 | 0.59 | 0.61 | 0.62 |
| Graphene–complex assembly | 1b | 2b | 3b | 4b | 5b | 6b | 7b | 8b | 9b | 10b | 11b | 12b | 13b | 14b |
| Graphene–complex average distance (Å) | 3.26 | 3.29 | 3.23 | 3.29 | 3.25 | 3.17 | 3.12 | 3.21 | 3.22 | 3.23 | 3.22 | 3.20 | 3.21 | 3.18 |
| Interaction energy (kcal mol−1) | −50.6 | −47.1 | −43.4 | −41.3 | −31.4 | −21.8 | −30.2 | −25.6 | −27.4 | −27.2 | −25.2 | −28.3 | −28.4 | −30.9 |
| Dihedral angle (°) | 115 | 109 | 107 | 104 | 123 | 123 | 130 | 133 | 124 | 128 | 127 | 130 | 129 | 127 |
| igmh binding score (a.u.) | 1.79 | 1.73 | 1.57 | 1.54 | 1.31 | 0.96 | 1.02 | 1.15 | 1.15 | 1.14 | 1.02 | 1.36 | 1.13 | 1.14 |
| igmh bonding score (a.u.) | 0.24 | 0.25 | 0.16 | 0.24 | 0.21 | 0.20 | 0.16 | 0.15 | 0.17 | 0.17 | 0.18 | 0.19 | 0.19 | 0.20 |
 | ||
Fig. 2
igmh analysis of the graphene–ligand systems involving quinones 1–7 (1a–7a), with an isosurface of 0.01 a.u., and a BGR color code in the range −0.04 a.u. < ρ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sign(λ2) < 0.04 a.u. sign(λ2) < 0.04 a.u. | ||
The first graphene–ligand assembly herein evaluated, involves the 1,8-dihydroxyanthraquinone (1a, Fig. 1). It exhibits (quasi-)complete planarity of the quinone over the graphene surface, with only the hydroxyl hydrogen atoms lying slightly outside of the anthraquinone plane. An equilibrium distance of 3.22 Å is found between the moieties, which remains almost unaltered along the series. Remarkably, similar distances are obtained by Young-Kyu et al., for a panel of quinone molecules interacting with pristine graphene, through periodic DFT calculations.43 In turn, the interaction energy obtained from the fragment calculations is −25.7 kcal mol−1. In the 1,4-dihydroxyanthraquinone system (2a), two coordinative sites are placed on opposite sides of the anthraquinone scaffold, which in principle could lead to a broader distribution of captured Cd2+ ions, as the steric effects associated with the close binding sites in 1a are ruled out. A graphene–ligand distance of 3.23 Å and an interaction energy of −25.3 kcal mol−1 are found, very similar to results obtained in 1a.
While the interfragment distance (3.23 Å) remains unchanged, a slightly lower interaction energy (−22.7 kcal mol−1) is found in the graphene–ligand assembly involving 9,10-phenanthroquinone (3a). This can be attributed to the absence of –H⋯π contacts due to the lack of hydroxyl groups. To clarify this point, an –OH was introduced at the 1 position of 3a, leading to the 1-hydroxy-9,10-phenanthroquinone structure (4a). As expected, an increase in the magnitude of the graphene–ligand interaction energy occurs (−24.0 kcal mol−1) relative to the parent 3a.
Moving to ligands featuring a naphthoquinone scaffold, the assembly involving 2-hydroxy-1,4-naphthoquinone (5a) yields a fragment energy of −18.4 kcal mol−1, consistent with a smaller extent of π-stacking. This trend becomes more pronounced when moving to the benzoquinone derivatives: 2-hydroxy-p-benzoquinone (6a) and 4-hydroxy-o-benzoquinone (7a). The interaction energies for these systems are lower than those of the previously tested assemblies, specifically −11.8 kcal mol−1 for 6a and −13.4 kcal mol−1 for 7a, as a consequence of their smaller scaffold. The igmh analysis is able to capture the extent of the π-stacking along the series, with larger igmh scores for the anthraquinone-based systems (0.94 for 1a, and 0.91 for 2a), followed by the phenanthroquinones (0.81 for 3a, and 0.89 for 4a), naphthoquinone (0.70 for 5a), and benzoquinones (0.20 for 6a, and 0.16 for 7a) graphene–ligand assemblies.
The results obtained in this first part suggest that the recovery of the graphene support by the quinone modifiers is plausible in terms of the interaction energy involved, which is approximately −10 kcal mol−1 per molecule ring. Furthermore, the graphene–ligand interfragment distance remains almost constant throughout the series (∼3.2 Å), irrespective of the number of cycles featured by the modifier. It is also noteworthy that the igmh approach is able to quantify the π–π interactions along the considered series.
Toward the detection of [Cd(CN)4]2−
In the second part of this study, the detection of the tetracyanocadmate ion, [Cd(CN)4]2−, is modeled by considering a complex in which one Cd2+ is chelated by two quinone ligands and coordinated by two cyanide ions, all positioned over the graphene sheet. In this model, the quinone derivative is assumed to be already fixed to the graphene electrode surface prior to the formation of the graphene–complex assembly, which occurs when the electrode is immersed in a WAD-CN solution. In mineral extraction processes, the wastewater contains [Cd(CN)4]2− in basic media. For safety reasons, these solutions should be kept at high pH values to avoid the formation of the highly toxic HCN. For this reason, we are working to improve the performance of our quinone-based electrodes in basic media. Currently, the experimentally obtained electrodes can detect at pH values as high as 9, but the maximum response is obtained at pH 5. This is an ongoing research effort. In our model, we consider a distorted octahedral local geometry for the Cd(II) center, as it is the most likely local environment for a Cd(II) center coordinated by two quinones interacting through π–π stacking with the graphene surface and two cyanide ligands. Without considering the two cyanide ligands, a square-planar local geometry for Cd(II) would need to be considered, which is a less likely scenario. Given the coordinative strength characteristic of chelating ligands, we consider a low-spin for the Cd(II) complex.The noncovalent interaction energy between the complex and graphene is quantified using a fragment calculation (considering graphene-quinone, and [Cd(CN)2] as interacting moieties) and the igmh binding score. The metal–ligand coordination strength, in turn, is quantified by the sum of the four igmh scores associated with the bonds involving the quinone oxygen atoms and the metal center, which is hereafter referred to as the igmh coordinative bonding score.
The optimized geometries for the graphene–complex assemblies are shown in Fig. 3, and the igmh isosurfaces, along with the binding scores, are depicted in Fig. 4.
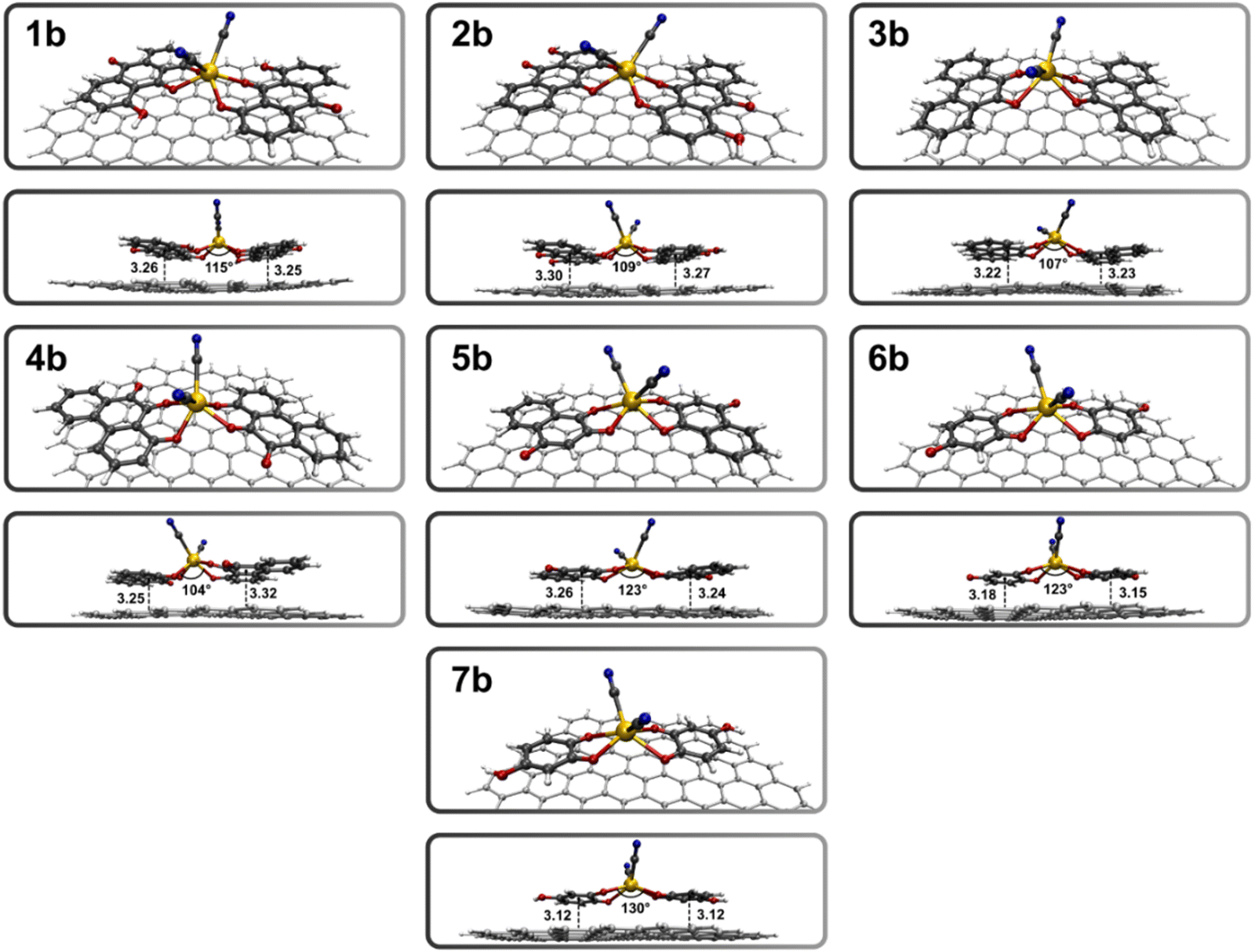 | ||
| Fig. 3 Optimized structures of the graphene–complex systems involving quinones 1–7 (1b–7b), obtained at the M06-2X/LANL2DZ 6-311G(d) level of theory. The graphene–complex distances are expressed in Å, and the dihedral angles in degrees. | ||
 | ||
| Fig. 4
igmh analysis of the graphene–complex systems involving quinones 1–7 (1b–7b), with an isosurface of 0.01 a.u., and a BGR color code in the range −0.04 < ρsign(λ2) < 0.04 a.u. | ||
In all optimized structures a distorted octahedral sphere for the metal center is observed, caused by the π–π interactions between the ligand aromatic rings and graphene. In the graphene–complex assembly featuring two 1,8-dihydroxyanthraquinone ligands (1b), an interaction energy of −50.6 kcal mol−1 is found, which is twice the value obtained with the single ligand over graphene (−25.7 kcal mol−1 in 1a). This suggests that the central cation does not significantly contribute to stabilizing the graphene–complex array. Remarkably, the graphene–complex distances (∼3.2 Å) do not vary with respect to the graphene–ligand series, confirming that π–π interactions rule the formation of the assemblies. Lower interaction energies are obtained for 2b (−47.1 kcal mol−1), 3b (−43.4 kcal mol−1), and 4b (−41.3 kcal mol−1), suggesting less stable structures compared to 1b, despite the same number of rings in the ligands.
Moving to 5b, generated with two 2-hydroxy-1,4-naphthoquinones, the binding energy (−31.4 kcal mol−1) is significantly lower compared to the previous systems, consistent with the presence of two rings in the involved ligands, which result in a less pronounced π-stacking. Following this trend, 6b and 7b, involving benzoquinone ligands, exhibit the lowest interaction energies, i.e., −21.8 kcal mol−1 and −30.2 kcal mol−1, respectively. The difference between their binding energies can be attributed to the absence of –H⋯π interactions in 6b, as the deprotonated hydroxyl groups of the ligands are already involved in Cd2+ coordination.
The igmh approach is applied to evaluate the extent of π-stacking along the graphene–complex series. This approach proves capable of capturing the two-fold increase in the interacting ligand surface, as well as the decrease in π-stacking, ranging from 1.79 for 1b, to 0.96/1.02 for 6b/7b.
The detection of [Cd(CN)4]2− implies the oxidation of the complex at high potentials, resulting in the production of cyanate and the release of Cd2+ ions.28–30 According to the proposed model, the Cd2+ ions are chelated by the adsorbed quinones, forming four coordinative bonds. The igmh score bonding associated with the sum of these bonds (see Table 1) reveals a stronger coordination in the anthraquinone complexes (0.25 for 2b and 0.24 for 1b), and 1-hydroxy-9,10-phenanthroquinone (4b, 0.24), followed by those generated with 2-hydroxy-1,4-naphthoquinone (5b, 0.21) and 2-hydroxy-p-benzoquinone (6b, 0.20). The lower values correspond to 9,10-phenanthroquine (3b, 0.16) and 4-hydroxy-o-benzoquinone (7b, 0.16) complexes.
An appealing result from the igmh bonding analysis concerns the deprotonated hydroxyl group in 1-hydroxy-9,10-phenanthroquinone (4b), which is involved in the chelation of the metal center along with the keto group. This group, in tandem with a carbonyl, leads to a stronger coordination (bonding score of 0.24) relative to 9,10-phenanthroquinone (3b, 0.16), where two carbonyl groups coordinate the metal center. This can be attributed to the more localized electronic density of –O− compared to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen. Similarly, the presence of a coordinated deprotonated hydroxyl group in 2-hydroxy-p-benzoquinone (6b, 0.20) enhances the bonding score in comparison to 4-hydroxy-o-benzoquinone (7b, 0.16).
O oxygen. Similarly, the presence of a coordinated deprotonated hydroxyl group in 2-hydroxy-p-benzoquinone (6b, 0.20) enhances the bonding score in comparison to 4-hydroxy-o-benzoquinone (7b, 0.16).
Rational design of novel sensing platforms
In principle, using an electrochemical sensor based on a benzoquinone derivative could lead to the formation of a greater number of coordinative sites for Cd2+ ions from WAD-CN. This is because a larger amount of this small ligand may facilitate the recovery of the graphene surface compared to quinones with larger scaffolds. Furthermore, smaller molecules typically allow for more densely packed adsorption due to reduced steric hindrance. This feature is particularly appealing when designing electrochemical devices with lower detection limits. This was experimentally verified using the electrochemical technique of chronocoulometry to determine the charge capacity of quinones on the surface of a carbonaceous material (glassy carbon). Table 2 and Fig. S1 (see ESI†) presents the results of this electrochemical technique applied to electrodes based on quinones with two (5) and three fused rings (1, 2, 3) supported on glassy carbon (the experimental procedure to obtain these quinone-functionalized electrodes supported on glassy carbon can be obtained in ref. 30). The highest charge of 35.3 μC was obtained for the sensor functionalized with 5 due to the smaller size of the molecule, while lower capacities were observed for 3 (20.7 μC), 1 (9.28 μC), and 2 (2.59 μC). Additionally, the limits of detection (LOD) and limit of quantification (LOQ) of these sensors were obtained by square wave voltammetry and the calibration curve method (see results in Table 2 and Fig. S2 in ESI†). The sensor prepared using 5 appears to be more sensitive for detecting the Cd(II) complex, with the lowest LOD = 0.16 mg L−1 and LOQ = 0.47 mg L−1 values. This again can be attributed to the smaller size of the benzoquinone, compared to the three-fused-ring ligands, which exhibit higher LOD (LOQ) values. These values are 0.30 mg L−1 (0.89 mg L−1) for 3 (almost twice the value of the sensor prepared with 5), 0.41 mg L−1 (1.24 mg L−1) for 2, and 0.70 mg L−1 (2.13 mg L−1) for 1.| Charge capacity/μC | LOD/mg L−1 | LOQ/mg L−1 | ||
|---|---|---|---|---|
| Glassy carbon (GC) | Functionalized electrode | |||
| 1 | 5.31 | 9.28 | 0.70 | 2.13 |
| 2 | 1.37 | 2.59 | 0.41 | 1.24 |
| 3 | 14.1 | 20.7 | 0.30 | 0.89 |
| 5 | 23.1 | 35.3 | 0.16 | 0.47 |
In light of these considerations, a series of 3-hydroxy-o-benzoquinone derivatives (8–14) is proposed, with an average graphene–ligand interaction energy of approximately −16 kcal mol−1 (see graphene–ligand structures and the corresponding igmh analysis in Fig. S3 and S4 of ESI†). In the graphene–complex panel of benzoquinones 8b–14b, we take advantage of the stronger coordination toward Cd2+ provided by a deprotonated –OH group compared to a keto group, as previously demonstrated in the 1-hydroxy-9,10-phenanthroquinone (4b) and 2-hydroxy-p-benzoquinone (6b) graphene–complex assemblies. In addition, the 3-hydroxy-o-benzoquinone moiety permits the incorporation of electron donor substituents at the 4-position, able to electron density donation by π-conjugation toward the adjacent deprotonated hydroxyl, thereby enhancing the coordinative strength of the ligands. The optimized structures for the graphene–complex systems involving the benzoquinone derivatives 8–14 (8b–14b) are presented in Fig. 5, along with the corresponding igmh analysis. The substituents attached to the 4-position of 3-hydroxy-o-benzoquinone (11) range from the electron-releasing –CN (8), –Cl (9) and –Br (10) to the electron-donating –CH3 (12), –O–CH3 (13), and –NH–CH3 (14). The bonding score associated with the metal–ligand coordination varies according to the electron releasing/donating behavior, as illustrated in the following order: –CN (8b, 0.15) < –Cl (9b, 0.17) ∼ –Br (10b, 0.17) < –H (11b, 0.18) < –CH3 (12b, 0.19) ∼ –O–CH3 (13b, 0.19) < –NH–CH3 (14b, 0.20). This demonstrates the impact caused in the Cd2+ coordination by the appending groups, that is well-captured by the igmh bonding score. This is verified by comparing the igmh bonding scores and the interaction energies with the Hammett parameters of the considered substituents (see Table S1 and Fig. S5 in ESI†). It is observed that as the Hammett values become more negative (indicating a stronger electron-donating behavior of the substituent), the igmh bonding score (which reflects the coordinative strength of the quinone ligands) increases. Additionally, the interaction energy becomes more negative (indicating stronger binding) as the electron-donating behavior of the substituents intensifies. For those assemblies where a good balance is found between the metal–ligand interaction and binding energy (12a–14a), a total density of states (TDOS) analysis was performed to evaluate the effect of cadmium complex capture on their electronic properties. We compared the TDOS of the graphene–ligand systems (12a–14a) with those resulting from the incorporation of the cadmium complex (graphene–complex systems 12b–14b). This graphical comparison is presented in Fig. 6, where it can be observed in all cases the Fermi level at −4.3 eV, and a significant variation in the TDOS profile upon cadmium complex capture, particularly at −17 eV, −13 eV, −10 eV (HOMO), and 2.7 eV (LUMO).
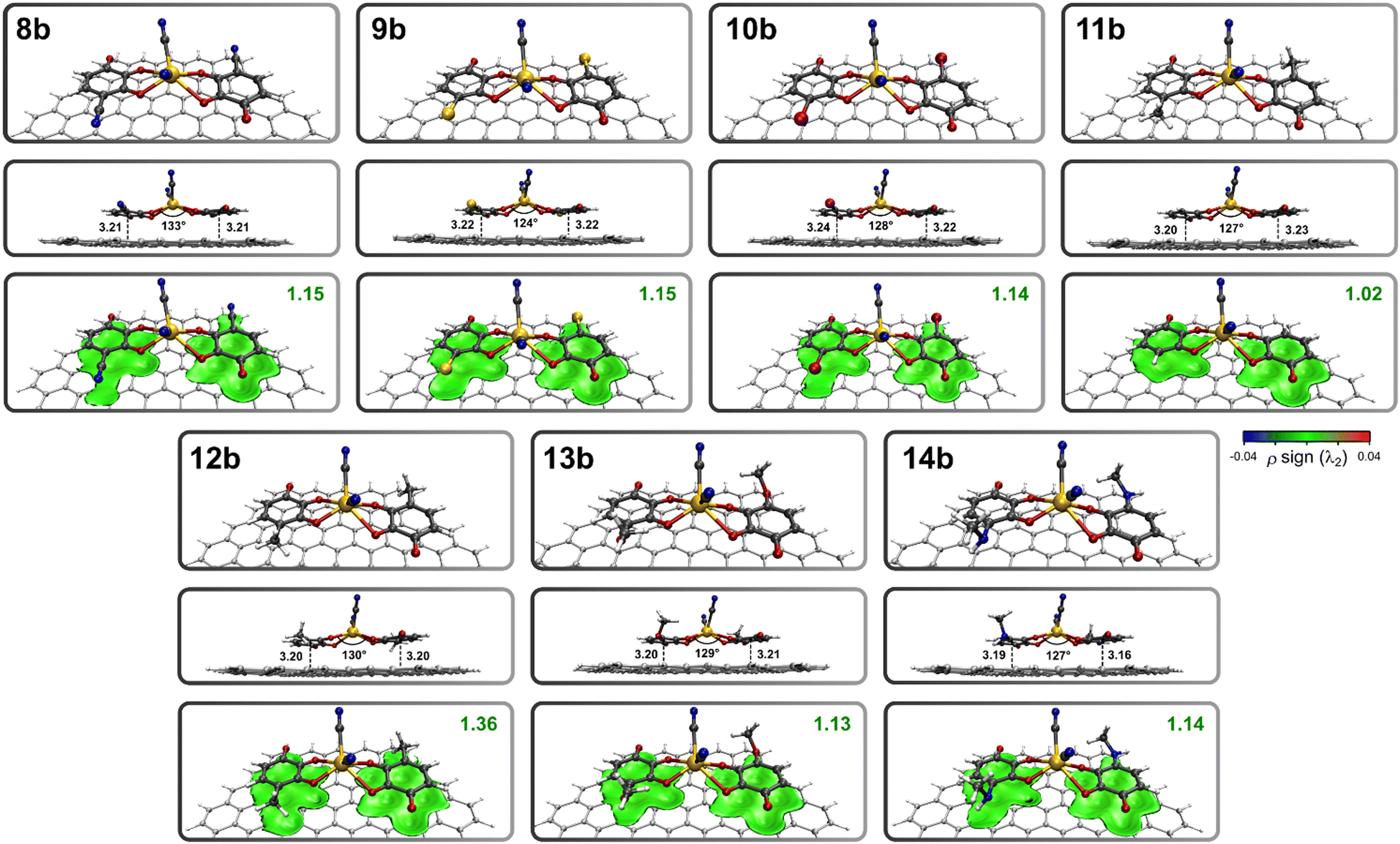 | ||
| Fig. 5 Optimized structures of the graphene–complex systems involving quinones 8b–14b, obtained at the M06-2X/6-311G(d) level of theory, and the igmh analysis with an isosurface of 0.01 a.u., and a BGR color code in the range −0.04 a.u. < ρsign(λ2) < 0.04 a.u. The graphene–ligand distances are expressed in Å. | ||
 | ||
| Fig. 6 Total density of states analysis (TDOS) of graphene–ligand (12a–14a) and graphene–complex (12b–14b) assemblies. | ||
Although the most notable variation occurs in the HOMO region, which can be attributed to the coordination of the metal center by the oxygen atoms, the variation of the LUMO at 2.7 eV can, in turn, be attributed to back-donation from the Cd(II) center to the p orbitals of the carbon atoms bonded to the ketone groups of the quinone ligand upon complex formation. These results demonstrate the sensitivity of the proposed functionalized materials towards WAD-cyanide, making them promising candidates for sensing applications. Moreover, when the TDOS of the graphene support without modifiers is compared to that of quinone-functionalized graphene (see Fig. S6 in ESI†), the profiles are very similar, ensuring that the conductivity of the support is maintained after modification through quinone incorporation.
In summary, the results obtained through the fragment calculations and the igmh approach suggest that a good compromise can be achieved between the number of ligands adsorbed on the graphene support and the coordination strength (which is associated with more effective Cd2+ capture).
This balance can be attained by functionalizing the graphene surface with 4-substituted 3-hydroxy-o-benzoquinones bearing electron-donor groups (12–14), taking advantage of their small size among quinone derivatives and the strong coordination toward the cadmium center, which is enhanced by incorporating electron-donor groups at the 4-position of the benzoquinone ring.
Additionally, the TDOS results reveal that the capture of the cadmium complex generates a variation in the electronic profile, which demonstrates the sensing capability of the proposed functionalized materials.
Conclusions
The DFT protocol employed herein provided valuable guidelines for the design of electrochemical sensors based on quinones adsorbed on graphene. The attachment of the modifier to the support is governed by π–π interactions, which result in an interaction energy of about −10 kcal mol−1 per ligand cycle. Using a model featuring one Cd2+ ion coordinated by two quinone ligands and two cyanide ions, all placed over the graphene sheet, it was found that incorporating a deprotonated hydroxyl group into a benzoquinone scaffold enhances the coordination strength toward Cd2+ due to the more localized electronic density of this substituent compared to a carbonyl group. Additionally, a strategy was employed to increase the number of active sites, which involves the use of benzoquinone derivatives to achieve greater coverage of the graphene surface. In this case, by modifying the substituent at the 4-position of 3-hydroxy-o-benzoquinone, the electronic density of the –O− group is enhanced, translating into more effective Cd2+ capture. Finally, the results of the TDOS analysis demonstrate the sensitivity of the proposed functionalized materials toward [Cd(CN)4]2−. Based on these results, we propose that a good balance between anchoring and Cd2+ capture can be achieved with 4-substituted 3-hydroxy-o-benzoquinone derivatives. We expect that this computational protocol could be extrapolated to other relevant systems toward the development of novel functionalized carbonaceous sensing devices for water pollutants.Author contributions
Golfer Muedas-Taipe: investigation, formal analysis, visualization. Michael Badawi: methodology, formal analysis, validation, resources, writing review and editing. Angélica Baena-Moncada: conceptualization, supervision, project administration. Miguel Ponce-Vargas: conceptualization, methodology, writing original draft, writing review and editing, resources, supervision, project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the ECOS-Nord France-Peru project P22P01 for financial support oriented to international deployments for M. P.-V., G. M. T. and A. M. B. M. G. M. T. and A. M. B. M. also acknowledge CONCYTEC and PROCIENCIA agencies from Peru in the framework of the call for Mobilizations with ECOS NORD-Peru 2021 (contract number 002-2022-PROCIENCIA). The authors also acknowledge the Centre de Calcul Régional ROMEO, France for computing facilities (https://romeo.univ-reims.fr), and particularly Yolain Erre and Fabien Berini for the technical support.Notes and references
- A. Brüger, G. Fafilek, O. J. Restrepo and L. Rojas-Mendoza, Sci. Total Environ., 2018, 627, 1167 CrossRef PubMed.
- K. Dong, F. Xie, W. Wang, Y. Chang, D. Lu, X. Gu and C. Chen, J. Cleaner Prod., 2021, 302, 126946 CrossRef.
- N. Kuyucak and A. Akcil, Minerals Eng., 2013, 50–51, 13 CrossRef CAS.
- G. Hilson and A. J. Monhemius, J. Cleaner Prod., 2006, 14, 1158 CrossRef.
- B. Guo, Y. Peng and R. Espinosa-Gomez, Miner. Eng., 2014, 66–68, 25 CrossRef CAS.
- A. E. Lindsay and D. O'Hare, Anal. Chim. Acta, 2006, 558, 158 CrossRef CAS.
- M. Shamsipur, Z. Karimi and M. A. Amouzadeh Tabrizi, Microchem. J., 2017, 133, 485 CrossRef CAS.
- A. A. Cárdenas Riojas, A. Wong, G. A. Planes, M. D. P. T. Sotomayor, A. La Rosa-Toro and A. M. Baena-Moncada, Sens. Actuators, B, 2019, 287, 544 CrossRef.
- C. W. Lee, T. H. Eom, S. H. Cho and H. W. Jang, Adv. Sensor. Res., 2023, 2, 2200057 CrossRef.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061 CrossRef CAS.
- F. Yavari and N. Koratkar, J. Phys. Chem. Lett., 2012, 3, 1746 CrossRef CAS PubMed.
- O. He, S. Wu, Z. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764 RSC.
- X. V. Zhen, E. G. Swanson, J. T. Nelson, Y. Zhang, Q. Su, S. J. Koester and P. Bühlmann, ACS Appl. Nano Mater., 2018, 1, 2718 CrossRef CAS.
- A. Aligayev, U. Jabbarli, U. Samadova, F. J. Dominguez-Gutierrez, S. Papanikolaou and Q. Huang, Appl. Surf. Sci., 2025, 686, 162022 CrossRef CAS.
- Y. Li, W. Ju, L. Yang, L. Zhang and Y. Sun, Appl. Surf. Sci., 2020, 529, 147112 CrossRef CAS.
- M. J. Szary, Appl. Surf. Sci., 2025, 679, 161175 CrossRef CAS.
- M. Pykal, P. Jurečka, F. Karlický and M. Otyepka, Phys. Chem. Chem. Phys., 2016, 18, 6351 RSC.
- L. Hou, C. Kong, Z. Hu, Y. Yang, H. Wu, Z. Li, X. Wang, P. Yan and X. Feng, Appl. Surf. Sci., 2020, 508, 145192 CrossRef CAS.
- M. Boota, C. Chen, M. Bécuwe, L. Miao and Y. Gogotsi, Energy Environ. Sci., 2016, 9, 2586 RSC.
- K. Greben, S. Kovalchuk, A. M. Valencia, J. N. Kirchhof, S. Heeg, P. Rietsch, S. Reich, C. Cocchi, S. Eigler and K. I. Bolotin, 2D Mater., 2021, 8, 015022 CrossRef CAS.
- Z. Guo, S. Chakraborty, F. A. Monikh, D.-D. Varsou, A. J. Chetwynd, A. Afantitis, I. Lynch and P. Zhang, Adv. Biol., 2021, 5, 2100637 CrossRef CAS PubMed.
- R. C. Prince, P. L. Dutton and M. R. Gunner, Biochim. Biophys. Acta, Bioenerg., 2022, 1863, 148558 CrossRef CAS PubMed.
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, J. Mater. Chem. A, 2019, 7, 23378 RSC.
- Y. Zhou, H. Lu, J. Wang, J. Zhou, X. Leng and G. Liu, J. Hazard. Mater., 2018, 356, 82 CrossRef CAS PubMed.
- G. C. Sedenho, D. De Porcellinis, Y. Jing, E. Kerr, L. M. Mejia-Mendoza, A. Vasquez-Mayagoitia, A. Aspuru-Guzik, R. G. Gordon, F. N. Crespilho and M. J. Aziz, ACS Appl. Energy Mater., 2020, 3, 1933 CrossRef CAS.
- R. Shi, C. Han, H. Duan, L. Xu, D. Zhou, H. Li, J. Li, F. Kang, B. Li and G. Wang, Adv. Energy Mater., 2018, 8, 1802088 CrossRef.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- A. Cárdenas-Riojas, S. L. Calderón-Zavaleta, U. Quiroz-Aguinaga, G. Muedas-Taipe, A. La Rosa-Toro, M. Ponce-Vargas and A. M. Baena-Moncada, Electroanalysis, 2024, 36, e202300208 CrossRef.
- A. A. Cárdenas-Riojas, G. Muedas-Taipe, A. La Rosa-Toro, M. D. P. Taboada Sotomayor, M. Ponce-Vargas and A. M. Baena-Moncada, J. Appl. Electrochem., 2022, 52, 1053 CrossRef.
- A. A. Cárdenas-Riojas, A. F. Cornejo-Herrera, G. Muedas-Taipe, A. La Rosa-Toro, M. D. P. Taboada Sotomayor, M. Ponce-Vargas and A. M. Baena-Moncada, J. Electroanal. Chem., 2021, 880, 114909 CrossRef.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- J. A. Pople, J. W. McIver and M. J. Frisch, J. Chem. Phys., 1987, 87, 3953 CrossRef.
- D. Josa, J. Rodríguez-Otero, E. M. Cabaleiro-Lago and M. Rellán-Piñeiro, Chem. Phys. Lett., 2013, 557, 170 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102 CrossRef CAS.
- W. Evangelista-Falcón, C. Denhez, A. Baena-Moncada and M. Ponce-Vargas, J. Phys. Chem. B, 2023, 127, 1110 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- H.-J. Kim and Y.-K. Han, Curr. Appl. Phys., 2016, 16, 1437 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental chronocoulometry, LOD and LOQ results for electrodes based on 1, 2, 3 and 5, optimized structures and igmh analysis of graphene–ligand systems involving quinones 8–14, and total density of states analysis (TDOS) of graphene–ligand and graphene–complex assemblies involving quinones 12–14b. See DOI: https://doi.org/10.1039/d5cp00218d |
| This journal is © the Owner Societies 2025 |