
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlating reductive vanadium oxide transformations with electrochemical N2 activation and ammonia formation†
Kabirat
Balogun
 a,
Qasim
Adesope
a,
Stella
Amagbor
a,
Agbara
Tochi
a,
Adam
Vass
b,
Guido
Mul
b,
Christoph
Baeumer
b,
Georgios
Katsoukis
*b and
Jeffry A.
Kelber
*a
a,
Qasim
Adesope
a,
Stella
Amagbor
a,
Agbara
Tochi
a,
Adam
Vass
b,
Guido
Mul
b,
Christoph
Baeumer
b,
Georgios
Katsoukis
*b and
Jeffry A.
Kelber
*a
aDept. of Chemistry, University of North Texas, Denton, TX 76203, USA. E-mail: g.katsoukis@utwente.nl
bMESA+ Institute for Nanotechnology, Faculty of Science and Technology, University of Twente, Drienerlolaan 5, 7522 NB, Enschede, The Netherlands
First published on 2nd June 2025
Abstract
The electrochemical reduction of nitrogen to ammonia (E-NRR) could become an environmentally friendly approach, yet its molecular-scale reaction mechanisms remain difficult to elucidate. Here, we use in situ electrochemical infrared reflection–absorption spectroscopy (EC-IRRAS) to examine vanadium oxide electrodes in neutral aqueous electrolyte (pH 7). Ex situ XPS reveals that the vanadium oxide electrode initially consists predominantly of V5+ species in the form of V2O5. However, in neutral aqueous electrolyte (pH 7), the surface evolves into anionic ortho-, meta-, and polyvanadate species at potentials above +0.6 V vs. RHE. Upon cathodic sweeping these anionic vanadates undergo progressive reduction toward V2O4. In an N2 saturated electrolyte, subsequent reduction and redeposition of these anionic vanadates remove a distinct vanadyl (V4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) feature – likely associated with an undercoordinated site, i.e. oxygen vacancies or grain boundaries – while the appearance of a broad, red-shifted band suggests the formation of vanadyl intermediates that interact with N2. Crucially, we find that ammonia (or ammonium) formation initiates at −0.28 V versus RHE, coinciding with a phase transition from V2O4 to V2O3 and continues until this transition completes. This onset is accompanied by the appearance of adsorbed N2 at −0.28 to −0.38 V versus RHE, indicating an associative mechanism. Overall, these findings emphasize the pivotal role of transient redox transitions (V5+ → V4+ → V3+) in enabling N2 activation – beyond the static presence of V2O3 alone – and highlight the promise of vanadium oxides as dynamic platforms for E-NRR.
O) feature – likely associated with an undercoordinated site, i.e. oxygen vacancies or grain boundaries – while the appearance of a broad, red-shifted band suggests the formation of vanadyl intermediates that interact with N2. Crucially, we find that ammonia (or ammonium) formation initiates at −0.28 V versus RHE, coinciding with a phase transition from V2O4 to V2O3 and continues until this transition completes. This onset is accompanied by the appearance of adsorbed N2 at −0.28 to −0.38 V versus RHE, indicating an associative mechanism. Overall, these findings emphasize the pivotal role of transient redox transitions (V5+ → V4+ → V3+) in enabling N2 activation – beyond the static presence of V2O3 alone – and highlight the promise of vanadium oxides as dynamic platforms for E-NRR.
Introduction
Given the essential role of ammonia in industry and society,1–4 the development of sustainable production methods is imperative. Electrochemical nitrogen reduction (E-NRR) presents a sustainable alternative for ammonia production by utilizing renewable energy sources.1,5,6 However, the development of highly selective and efficient (i.e. low overpotential) electrocatalysts for E-NRR remains a significant challenge. This is primarily due to the competitive hydrogen evolution reaction (HER), which occurs at less negative potentials than E-NRR, typically reducing the selectivity and efficiency of ammonia synthesis.3,5,7,8To address the challenges of HER competition, various transition metal-based materials have been explored for NRR, including oxides, oxynitrides, and carbides of Fe, Cu, V, and Nb.1,5,8,9 Both experimental and computational studies on vanadium compounds, including VN,7,8 VON,9,10 and VOx,11,12 as well as vanadium single-atom catalysts,13 have identified the active site as the vanadium center coordinated to lattice oxygen. Additionally, VOx has been identified as highly selective for NRR over HER under acidic or neutral reaction conditions.14 While numerous computational studies8,14,15 propose an associative pathway for nitrogen reduction on metal oxide surfaces – i.e., N2 associative adsorption, followed by protonation – experimental validation of this mechanism remains elusive. In situ electrochemical Fourier-transform (FT)-infrared spectroscopy has been utilized to investigate the adsorption, dissociation, and hydrogenation of N2 on various cathodes.16–18 Despite advancements, peak assignments remain ambiguous, and the oxidation state of vanadium cations, along with the roles of surface vanadyl and lattice oxygen during key steps – particularly N2 chemisorption and surface nitridation – remains unclear.19,20 In this study, we investigated catalyst surface changes and reaction intermediates formed during the E-NRR on nitrogen-free vanadium oxide thin film catalysts using electrochemical FT-infrared reflection–absorption spectroscopy (EC-IRRAS). By identifying reaction intermediates, tracking time-resolved changes in catalyst oxidation states, and detecting new bond formations, we integrated our findings with previous studies to elucidate the NRR mechanism on the vanadium oxide surface. This enabled us to determine the onset potential for vanadium N2 interactions, chemisorbed N2, and ammonia, correlating these processes to the oxidation state of vanadium.
Materials and methods
The vanadium oxide thin films used for this study were synthesized at the University of North Texas (UNT) using a method described elsewhere.12,21 Briefly, films were deposited using a DC magnetron sputter deposition method in an ultrahigh vacuum chamber with a base pressure of 10−8 Torr. The chamber is also equipped with capacity for inductively coupled plasma treatment, and Auger electron spectroscopy (AES) for in situ sample analysis.22 Films were produced by generating plasma in 4 mTorr Ar gas at 20 W for 20 min at room temperature using a commercially available vanadium metal target (purity 99.7%, Plasma Materials Inc.) mounted on a Meivac/Ferrotac commercially available DC magnetron sputter gun. Samples were analyzed in situ using AES. The AES instrument is a commercial single pass cylindrical mirror analyzer with co-axial electron gun operated at 3 keV (Staib, Inc.).Samples were deposited on glassy carbon substrates for EC-IRRAS experiments. Ex situ XPS was done at University of Twente before electrochemical tests (Omicron XM 1000 Al-Kα, monochromatic X-ray source, 1486.6 eV, Omicron EA 125 energy analyzer). The photoemission analyzer angle with respect to the sample normal was 0°. XPS spectra were analyzed using CasaXPS and a Shirley background was used for peak fitting.23 XPS spectra were referenced to the O 1s binding energy of lattice oxygen of vanadium oxide at 530.0 eV as popularly reported in literature.24–26 In analysis of the vanadium 2p spectra, multiplet splitting due to the final states effects of V photoelectrons was taken into consideration.25,27 The complexity associated with the multiplet splitting was resolved by analysing the V 2p peak as previously reported by Coulston et al., 1996.25,27 The average oxidation state distribution of V sites can be estimated by applying the equation
| Average VOx = 13.82 − 0.68 [O 1s–V 2p3/2] |
For in situ electrochemical studies, a Bruker V80v FTIR instrument was used to acquire spectral data. The instrument is made up of an IR source, an MIR polarizer (KRS-5) in an automatic rotational unit capable of p-polarization and s-polarization, a ZnSe IR transparent hemispherical crystal, an A530/V reflection unit for the electrochemical cells, and a medium band MCT detector. The detector is of medium-bandwidth (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–600 cm−1) cooled with liquid nitrogen during use. The polarizer was set to p-polarization, while the reflective gold mirrors were set at an incident angle of 30°, resulting in a 70–80° refraction angle at the electrolyte/ZnSe interface. These angle settings increased the surface electric field through a “grazing angle” geometry,28 thus ensuring amplification of vibrational modes of constituents in the electrolyte near the metal electrode surface. An estimation of the percentage contributions of the vibrational mode intensity vs. distance to the electrode can be found in our previous work.29 Please note that according to the Fresnel equations ca. 96% of the IR beam is reflected at the VOx interface,30 meaning that up to 4% of the IR signal may also contain VOx bulk information because of an IR beam penetration depth in the 5 to 6 micrometer range (that may reflect at another VOx interface and, hence, reach the detector). The instrument aperture and resolution were set at 1.5 mm and 8 cm−1 respectively. Electrochemical conditions were applied using a VersaSTAT3 potentiostat connected by BNC through a Bruker E525/Z connector box to allow automatic trigger of the potentiostat from the OPUS 3D software to synchronize IR spectra and I–V data collection. Prior to measurement, the ZnSe crystal was polished, rinsed in an ultrasonic bath, and dried. The working electrode was mounted on a holder with electrical back contact to a copper rod placed in otto configuration (upside-down) in close contact with the crystal in the electrochemical cell compartment filled with 4 ml electrolyte solution. The reference (Ag/AgCl) and counter (Pt wire) electrodes were also inserted to form a 3-electrode system. The working electrode was adjusted until its alignment with the ZnSe crytal produced an amplitude of around 2500. This set-up produced a thin layer of ca. 1 to 2 micrometer electrolyte between the working electrode and the ZnSe crystal. Gas was continuously delivered and bubbled through a plastic delivery tube directly into the electrolyte. Scheme S1 (ESI†) shows the schematic representation of the electrochemical FTIR set-up.
000–600 cm−1) cooled with liquid nitrogen during use. The polarizer was set to p-polarization, while the reflective gold mirrors were set at an incident angle of 30°, resulting in a 70–80° refraction angle at the electrolyte/ZnSe interface. These angle settings increased the surface electric field through a “grazing angle” geometry,28 thus ensuring amplification of vibrational modes of constituents in the electrolyte near the metal electrode surface. An estimation of the percentage contributions of the vibrational mode intensity vs. distance to the electrode can be found in our previous work.29 Please note that according to the Fresnel equations ca. 96% of the IR beam is reflected at the VOx interface,30 meaning that up to 4% of the IR signal may also contain VOx bulk information because of an IR beam penetration depth in the 5 to 6 micrometer range (that may reflect at another VOx interface and, hence, reach the detector). The instrument aperture and resolution were set at 1.5 mm and 8 cm−1 respectively. Electrochemical conditions were applied using a VersaSTAT3 potentiostat connected by BNC through a Bruker E525/Z connector box to allow automatic trigger of the potentiostat from the OPUS 3D software to synchronize IR spectra and I–V data collection. Prior to measurement, the ZnSe crystal was polished, rinsed in an ultrasonic bath, and dried. The working electrode was mounted on a holder with electrical back contact to a copper rod placed in otto configuration (upside-down) in close contact with the crystal in the electrochemical cell compartment filled with 4 ml electrolyte solution. The reference (Ag/AgCl) and counter (Pt wire) electrodes were also inserted to form a 3-electrode system. The working electrode was adjusted until its alignment with the ZnSe crytal produced an amplitude of around 2500. This set-up produced a thin layer of ca. 1 to 2 micrometer electrolyte between the working electrode and the ZnSe crystal. Gas was continuously delivered and bubbled through a plastic delivery tube directly into the electrolyte. Scheme S1 (ESI†) shows the schematic representation of the electrochemical FTIR set-up.
Cyclic voltammograms between +1.0 V and −0.3 V vs. RHE were run for 50 cycles as a surface cleaning step to remove adsorbed contaminants and generate a clean and stable catalyst surface before electrolyte saturation with gas. Gas was delivered for 30 minutes to ensure saturation before running CV from +0.68 V to −0.83 V. Experiments were carried out in N2 saturated, Ar saturated and deuterated electrolyte solution. Obtained spectra were processed using OriginPro. Electrolyte used was 0.1 M NaCl aqueous and deuterated solution. We previously reported the E-NRR activity of vanadium oxide thin films in a 0.1 M Na2SO4 solution at pH 7.12 In the present study, however, we employed a 0.1 M NaCl solution instead. This change was made due to the strongly absorbing asymmetric stretch of the sulfate anion in the region of interest between 1300 and 1000 cm−1.
Results
Thin film deposition and characterization.
In situ AES spectra of a vanadium film on glassy carbon substrate before and after deposition are shown in Fig. 1a. The spectrum before deposition, in green, shows a C KLL peak at 265 eV and an O KLL feature at 503 eV that can be assigned to C and adsorbed oxygen from the glassy carbon substrate prior to V deposition.23 The spectrum in red was acquired after vanadium deposition, and shows V LMM and O KLL peaks only, without a C KLL peak. The presence of a O KLL peak with such low intensity can be explained to be due to VOx formed due to residual oxygen in the deposition chamber at a base pressure of ∼10−8 Torr. The thickness of the film is estimated to be 50 nm.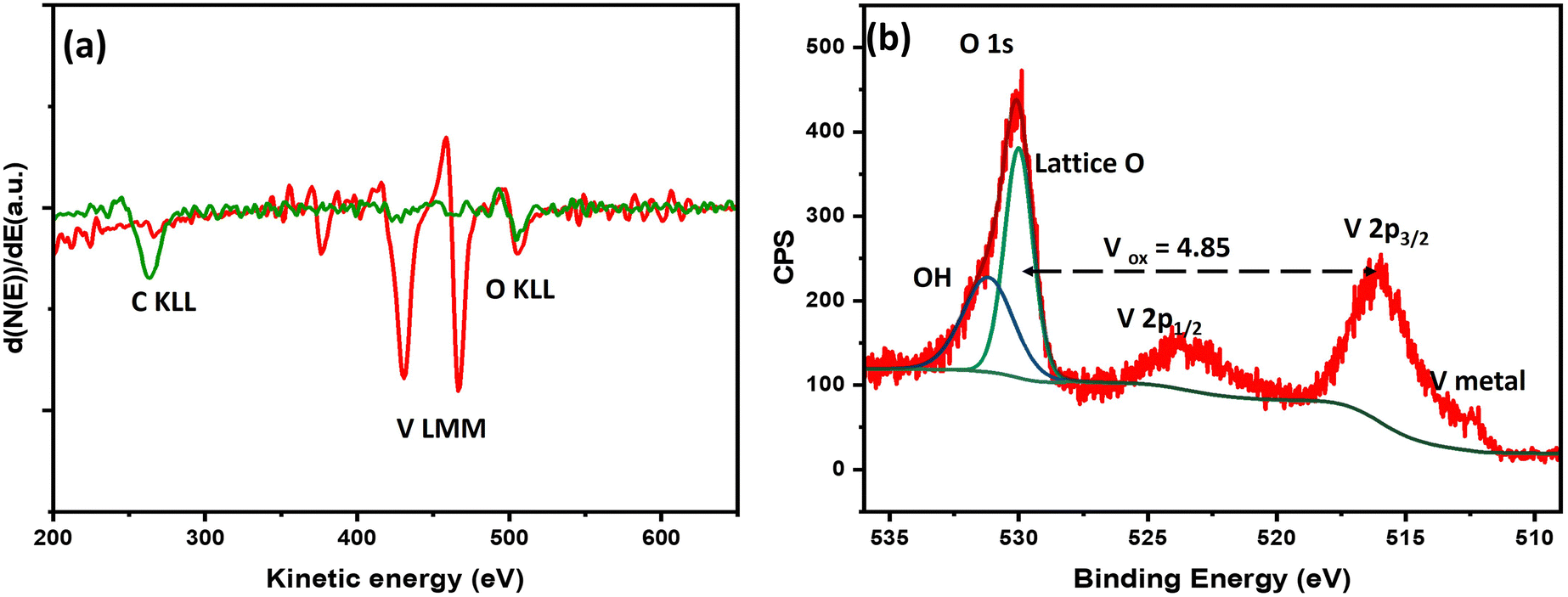 | ||
| Fig. 1 (a) In situ AES spectra of a vanadium thin film on glassy carbon electrode (green trace before, red trace after deposition). (b) Ex situ XPS spectra of a vanadium thin film on glassy carbon prior to EC-IRRAS measurement, exhibiting metallic and oxidized vanadium species. | ||
Ex situ XPS analysis prior to electrochemical testing and IRRAS measurements is shown in Fig. 1b. The average V oxidation state was estimated to be 4.85, suggesting highly oxidized vanadium oxide at the surface with the presence of some oxygen vacancies. From the spectral features, the V 2p3/2 peak maximum is centered above 516 eV which is a well-reported binding energy for V in +5 oxidation state.24,26,27 The peak at 512.3 eV corresponds to V(0), i.e. V metal, thus suggesting a thin layer of vanadium oxide on pure V metal. Thus, the sample exhibits a layered VOx/V structure.26 The long ambient exposure of the sample during shipping led to such oxidation at the surface.
In situ EC-IRRAS: staircase cyclic voltammetry (SCCV)
Staircase cyclic voltammetry (SCCV) was performed in N2- and Ar-saturated 0.1 M NaCl aqueous solutions (Fig. S1, ESI†) with a 37.25 s hold time per 100 mV step, corresponding to an effective scan rate of 1.34 mV s−1. FTIR scans were collected in situ. An initial cathodic current, detected in both N2 and Ar at +0.5 V versus RHE, corresponds to the reversible reduction of vanadium oxide. A second irreversible reduction current is observed predominantly in N2-saturated electrolyte from −0.1 V versus RHE. Due to the open configuration of our setup, rapid exchange between ambient air and Ar occurs, preventing the complete removal of N2 from the electrolyte (see Note S1 in the ESI†). Hence, the experiments denoted with Ar and N2 therefore reflect experiments with ‘low’ and ‘high’ concentrations of N2.Prior to further interpretation, we performed a detailed analysis of the infrared spectra to rule out contributions from potential contaminants (e.g., NO, N2O, and NO2). Further details are provided in Note S2 in the ESI.†
O stretching vibrations characteristic of vanadate (V5+) and vanadyl (V4+) species.31,32 The spectra were recorded in deuterated water to minimize interference from the H2O libration modes typically observed below 1000 cm−1. Terminal VO bonds, located at the edges of the vanadium polyhedra, serve as reliable markers of the oxidation state.33,34 Our working electrode was conditioned at +0.72 V versus RHE (+0.31 V versus SHE), which served as the experimental baseline. At this potential, the vanadium oxide electrode predominantly undergoes corrosion, resulting in the formation of anionic species such as HVO4− (orthovanadate) and HV10O285− (polyvanadate), in agreement with the Pourbaix diagram for the V–H2O system at pH 7.35
 | ||
| Fig. 2 EC-IRRAS data illustrating the reduction of vanadium oxide under cathodic polarization in D2O (0.1 M NaCl). (a) Spectra recorded in Ar-purged (top) and N2-purged (bottom) solutions using +0.72 V vs. RHE as the baseline. (b) Spectra referenced to −0.18 V vs. RHE, highlighting the evolution of key vibrational features. (c) Gaussian fitting parameters for the spectra in the bottom panel of (b). Insets summarize the main conclusion. “m-vanadate” denotes the V5+O stretching vibrations of anionic dissolved vanadium oxide species, while “a-V4+O” refers to the vanadyl vibrations that emerge as adjacent V4+ centers are reduced to V3+, leading to subsequent amorphization. | ||
Upon cathodic sweeping, in Fig. 2a a broad negative peak appears at 988 cm−1 at +0.62 V versus RHE, gradually shifting to 952 cm−1 as the potential is swept to −0.23 V versus RHE. We attribute these features to the redeposition of dissolved vanadate species that exhibit V5+O vibrations significantly red-shifted compared to solid V2O5:36 the initial peak is associated with polyvanadate, and the shift to 952 cm−1 corresponds to the redeposition and reduction of meta- and orthovanadate. Given the initial IR spectra baseline at +0.72 V already includes features from anionic vanadate species at the interface, their reduction would result in reduced IR peak intensity resulting in a negative peak in the IR spectra as observed in Fig. 2a. Importantly, no negative peaks are observed at 1016 cm−1 (TO mode) or 1035 cm−1 (LO mode), which would indicate the reduction of solid V2O5.37 Concurrent with these changes, new broad, kinetically co-evolving bands emerge with maxima at 872 cm−1 and 1001 cm−1. We assign these bands to the formation of V4+–O and V4+O stretching vibrations of V2O4, respectively.34,38
At −0.18 V versus RHE, a marked change in spectral evolution is observed: the reduction of vanadate species reflected by the growth of the V4+O feature at 1001 cm−1 stops. In Fig. 2b, spectra are referenced to a baseline at −0.18 V versus RHE, which clearly reveals the emergence of a dominant band at 966 cm−1. We assign this feature to the formation of oxovanadium species (V3+–O), consistent with the phase transitions expected from the Pourbaix diagram.33 Since V2O3 is not known to exhibit VO double bonds, we cannot exclude that the 966 cm−1 band instead represents a red-shifted V4+O vibration influenced by neighboring V3+ sites, where the shift reflects bond weakening.39 The continued presence of the 1001 cm−1 band suggests that the V4+O species remain intact, while the new oxovanadium species form at distinct surface or subsurface sites.
To elucidate the structural changes occurring in the vanadium oxide electrode from −0.18 V versus RHE onward, we fitted the 966 cm−1 peak using a Gaussian function (see Fig. 2c). First, the peak maximum shifts from 976 to 966 cm−1, corresponding to a stark shift of approximately 50 cm−1 V−1, confirming the bond weakening of the V4+O species when the electron density is increasing. Second, the peak height increases to 17 mOD, indicating the formation of VO bonds not only at the surface but also within the subsurface region. This suggests that structural changes, such as subsurface exposure or increased surface roughness, generate additional VO modes. For comparison, previous studies on ultrathin alumina electrocatalyst overlayers reported a linear LO mode intensity–thickness dependence of 3 mOD nm−1,40 implying that a monolayer alone would not account for the observed intensity. Third, the full width at half maximum (FWHM) steadily increases from about 40 to 65 cm−1, suggesting progressive amorphization or increase in grain boundaries.34 Finally, the integrated peak area stabilizes at −0.7 V, indicating that the overall process reaches completion at that potential. Consequently, we attribute the observed spectral features to the formation of VOx polyhedra, denoted as a-V4+O, that are defect- and grain-boundary-rich.34,41
To ensure that subtle differences between the Argon-purged and N2-purged experiments were captured, we subtracted the spectra from the Argon-purged experiment from those obtained under N2 purging as shown in Fig. 3a. The spectral evolution over time and potential is highly dependent on the electrode pretreatment and initial conditions. These factors were carefully controlled and are taken into account in our data processing to ensure that the subtraction accurately reflects the impact of dissolved N2.
 | ||
| Fig. 3 (a) EC-IRRAS difference spectra generated by subtracting the spectra recorded under Ar purging (top panels in Fig. 2) from those obtained under N2 purging (bottom panels of Fig. 2). The Gaussian fits of the 994 cm−1, 962 cm−1, and 911 cm−1 bands as a function of applied potential are shown next to it. Peak assignments and FWHM are shown above. (b) Peak maxima versus applied potential: the red line shows the linear anti-correlation of the bands at 994 cm−1 and 962 cm−1. The black vertical line indicates the V4+/V3+ redox potential under the experimental conditions. Experiments have been done in D2O (0.1 M NaCl). | ||
In the resulting difference spectra, a subtle, sharp minimum with a full width at half maximum (FWHM) of 20 cm−1 becomes apparent at 994 cm−1, emerging with an onset at +0.52 V versus RHE. Concurrently, a broad feature appears at 962 cm−1 with a FWHM of 52 cm−1. These observations suggest that N2 interacts with the vanadium oxide electrode during the redeposition of corroded vanadate species. We tentatively interpret the sharp minimum as a removal of a specific undercoordinated []-V4+O vanadyl moiety associated with a nearby oxygen vacancy or a reactive grain boundary. The broad positive band is consistent with a red shift of this vanadyl feature. This shift could reflect the filling of the vacancy with a nitrogen atom, leading to the formation of [N]–V4+O or interaction of the vacancy with N2 ([N2]–V4+O). To provide a more direct evidence for these processes, further experiments conducted at higher N2 concentrations (e.g., elevated pressures), with enhanced time resolution and varied time-potential profiles, will be essential.
Another broad feature emerges at 911 cm−1 with a FWHM of 89 cm−1, which we attribute to the NH2 bending mode of neutral (unprotonated) ammonia. We discuss this feature in greater detail in the next section.
Experiments performed in normal water (see Fig. S2, ESI†) are significantly distorted by dynamic changes in the water vibration modes below 1000 cm−1. Although these experiments are not directly comparable to those in deuterated water – since the corrosion and redeposition processes between +0.6 V and −0.2 V vs. RHE are not well captured – we nonetheless observe a weak band emerging at 980 cm−1. We attribute this band to the interaction between a vanadyl moiety and N2 as previously mentioned.
To obtain more robust kinetic information, we deconvoluted the observed features using three Gaussian functions – keeping the frequency positions and FWHM fixed – and extracted the peak maxima, as shown in Fig. 3b. The negative growth of the band at 994 cm−1 proceeds until −0.28 V versus RHE, which coincides with the V4+/V3+ redox potential and marks the onset of ammonia formation. Interestingly, the band at 962 cm−1 initially follows similar growth kinetics (see the red line in Fig. 3b); however, at −0.28 V vs. RHE there is a transient decrease in its intensity as ammonia production starts, followed by a subsequent increase. This behavior suggests that the feature at 962 cm−1 is correlated with ammonia formation and reflects the interaction of chemisorbed N2 or nitride species. At −0.7 V, ammonia formation comes to a halt, which matches the a-V4+O growth stop.
These observations indicate that the formation of [N2]– or [N]–VO is not driven solely by the applied potential; instead, the redeposition and reduction of vanadate (V5+) to vanadyl (V4+) and V3+ appear to be predominant factors. The onset of ammonia formation occurs at a potential nearly identical to that for the V2O4-to-V2O3 phase transition, and ceases once this transition is complete. This close correlation implies that the reductive transformation may play a critical role in facilitating N2 reduction. Consequently, future studies should focus on (1) investigating the energetics and oxygen vacancy generation associated with the corrosion and redeposition of vanadate species, and (2) elucidating the influence of the V2O4-to-V2O3 phase transition on ammonia formation. These efforts will be essential for understanding the mechanistic pathways of N2-to-NH3 conversion in our system.
 | ||
| Fig. 4 (a) EC-IRRAS spectra showing the formation of ammonium, identified by the NH2 bending mode at 1425 cm−1, during cathodic polarization in H2O (0.1 M NaCl) using +0.72 V vs. RHE as the baseline. The top panel shows measurements under Ar, while the bottom panel shows data collected under N2. (b) Control experiment performed in D2O reveals no NH2 nor ND2 bending modes of ammonium, supporting that ammonia formation occurred earlier as evidenced in Fig. 3. (c) Anodic back sweep following the D2O experiment in (b), showing the emergence of ND4+ attributed to a shift in the interfacial pD. (d) Potential- and time-resolved evolution of the ammonium feature alongside a band at 1556 cm−1, tentatively assigned to hydroxylamine formation. | ||
Fig. 4b presents the same experiment performed in deuterated water. As expected, the ammonium band normally observed at 1425 cm−1 is absent. However, we also do not detect the anticipated ND4+ feature around 1090–1070 cm−1 (which should be shifted by a factor of approximately 1.31).43,44 Instead, as discussed earlier, we observe the formation of ammonia-d3, which typically exhibits a symmetric bending mode in the 870–840 cm−1 range (the more intense asymmetric bending mode is usually superimposed by water bending modes).45 In our case it is blueshifted to 911 cm−1, which commonly occurs when ammonia acts as a ligand and, hence, may be interacting with vanadium cations.46 The NH bending mode of cationic ammonium is much more intense than that of neutral ammonia. Although our experimental setup does not allow precise control of the interfacial pH, an anodic sweep should reacidify the interface, thereby promoting the conversion of ammonia to ammonium. This effect is demonstrated in Fig. 4c, where a new band emerges at 1093 cm−1 upon cycling back to the initial potential at the start of the SCCV, which we attribute to the ND bending mode of ammonium-d4.43
Fig. 4d shows the spectral evolution of the bands at 1425 cm−1 and 1556 cm−1, respectively. The formation of ammonium coinsides with the formation of ammonia in the deuterated water experiment (Fig. 3). The band at 1556 cm−1 has an earlier onset at −0.2 V, does not exhibit any growth and therefore might also originate from a carboxylate impurity.
O stretching vibration appearing at 976 cm−1.47 In addition, two distinct peaks at 2175 cm−1 and 2131 cm−1 are detected. Based on IRRAS studies of N2 adsorption on Li-preadsorbed Ni{110},48 theoretical calculations for N2 chemisorption on V(110),49 and DRIFTS studies on aluminovanadate oxynitride catalysts46 these bands are assigned to the NN stretching mode of chemisorbed N2 on vanadium sites, rendered IR-active by symmetry breaking. Because IRRAS selectively detects vibrations perpendicular to the surface, these results suggest that adsorbed N2 adopts either a head-on or tilted orientation relative to the vanadium oxide surface. This is consistent with previous near ambient pressure XPS studies21 of V oxide surfaces in the presence N2 and H2O vapor, and with previous DFT calculations14 indicating that V3+ surface sites most strongly interact with N2.
 | ||
| Fig. 5 (a) EC-IRRAS spectra recorded during cathodic polarization in H2O (0.1 M NaCl), using +0.72 V vs. RHE as the baseline. The top panel shows measurements under Ar, while the bottom panel shows data collected under N2. The band at 1952 cm−1 is attributed to the first overtone of vanadyl (VO) vibrations. The bands at 2175 and 2131 cm−1 are assigned to end-on adsorption of N2 on the vanadium oxide surface. (b) Potential trace of the intensities at 2175 cm−1 and 1425 cm−1 (from Fig. 4d). | ||
Although we cannot completely exclude the formation of nitrile species – which can display similar doublet-like features in this spectral region50,51 – it would require the prior cleavage of N2 followed by a reaction with carbon impurities.52 We also cannot exclude azides that may evolve due to the reaction of N2 with nitride.46 Nevertheless, all cases involve N2 chemisorption as a key step. The observed spectral features emerge at around −0.38 V versus RHE slightly after the transition from V4+ (V2O4-type) to V3+ (V2O3-type) starts and concurrent with ammonium formation (Fig. 5b). The appearance of adsorbed N2 when V3+ centers are present has been observed previously.10,12
The top panel in Fig. 5 presents a similar experiment conducted in an Ar-saturated electrolyte, which confirms the N2 concentration dependence of the features at 2175 cm−1 and 2131 cm−1. As noted earlier, due to the open configuration of our setup, rapid exchange between ambient air and Ar occurs, preventing the complete removal of N2 from the electrolyte (see Note S1, ESI†).
Discussion
The observed spectral changes reveal a complex evolution of vanadium oxide during electrochemical reduction. Under our experimental conditions at pH 7, the electrode corrodes and gradually dissolves to form anionic species – including m-vanadate, orthovanadate, and polyvanadate – at potentials more positive than +0.6 V versus RHE at the interface. Upon reduction and redeposition of these anionic species, we can observe that N2 interactions are leading to a redshift of a vanadyl feature. Notably, when the phase transition between V2O4 and V2O3 occurs at −0.28 V versus RHE, ammonia/ammonium formation begins and continues until the phase transition is finalized. Concurrently, this phase transition is accompanied by the formation of adsorbed N2.These transformations in the vanadium oxide are critical for N2 reduction. As V5+ is reduced to V4+ and then to V3+, the lattice undergoes structural reorganization, resulting in progressive amorphization of the surface and the creation of undercoordinated vanadium sites with additional oxygen vacancies and grain boundaries. These changes eventually facilitate the adsorption and dissociation of N2 in an associative mechanism. The process is inherently disordered and appears to extend into the subsurface of the electrode, as evidenced by the emergence of new vanadyl features even as reduction advances toward V3+.
The weak spectral features suggest that the current experimental conditions do not adequately capture the reaction kinetics. In particular we cannot observe the protonation and dissociation of N2, which must be on much shorter time-scales. To obtain more robust kinetic data, it is necessary to operate under higher pressure conditions, enhance time resolution, and incorporate chronoamperometric measurements as well as pulsed electrolysis experiments. The correlation that exists between the phase transition and the conversion of N2 to ammonia appears to be transient, requiring both a rapid, abundant supply of N2 at the electrode and potentially continuous phase switching.
Conclusions
In this study, we investigated the electrochemical nitrogen reduction reaction (E-NRR) on vanadium oxide electrodes under ambient conditions using in situ EC-IRRAS. Our results reveal that at pH 7 the electrode corrodes and gradually dissolves to form anionic species – including m-vanadate, orthovanadate, and polyvanadate – at potentials more positive than +0.6 V vs. RHE. We observe that the presence of N2 upon reduction and redeposition of these dissolved species, leads to the removal of a distinct vanadyl mode that may originate from oxygen vacancies and grain boundaries. The appearance of a broad redshifted band suggests that N2 interacts with this ‘defect’. When the phase transition between V2O4 and V2O3 occurs at −0.28 V versus RHE, ammonia/ammonium formation begins and continues until the phase transition is finalized. Concurrently, this phase transition is accompanied by the formation of adsorbed N2.Ammonia/ammonium formation initiates at −0.28 V versus RHE, coinciding with the phase transition from V2O4 to V2O3, and continues until the transition is complete. This observation, along with the formation of adsorbed N2, implies an associative reduction mechanism, albeit we do not observe the intermediate protonation and dissociation steps.
Overall, our findings indicate that dynamic redox transitions from V5+ through V4+ to V3+ are critical for N2 activation. This suggests that the kinetics of ammonia formation are transiently dependent on these phase transitions rather than solely on the presence of a distinct V2O3 phase. In summary, this work underscores the significant influence of redox transitions and applied potential on the mechanistic pathways of E-NRR, highlighting the potential of vanadium oxides as platforms for nitrogen reduction and the importance of in situ techniques for elucidating reaction dynamics and catalyst behavior under operational conditions.
Author contributions
Kabirat Balogun: investigation, methodology, data curation, formal analysis, writing (original draft, review and editing). Qasim Adesope: investigation, methodology. Stella Amagbor: methodology, investigation, writing (review and editing). Agbara Tochi: methodology, investigation. Adam Vass: investigation, methodology, data curation. Guido Mul: conceptualization, and resources. Christoph Baeumer: resources, writing (review and editing). Georgios Katsoukis: investigation, methodology, data curation, formal analysis, writing (original draft, review and editing), conceptualization, validation, resources, supervision. Jeffry A. Kelber: writing (original draft, review and editing), conceptualization, resources, supervision.Data availability
The raw and processed data supporting this study will be publicly available on Zenodo upon acceptance of the manuscript, with a DOI assigned at that time. All data presented in the figures are derived from this dataset.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work at the University of North Texas was partially supported by NSF grant DMR2112864. KB gratefully acknowledges a supplement to the above grant supporting travel to and studies at the University of Twente. This work was also funded by the Dutch Research Council (NWO) (ECCM.TT.ECCM.004). C.B. has received co-funding from the European Union (ERC, 101040669 – Interfaces at Work). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. We thank Prof. Leon Lefferts and Prof. Aayan Banerjee for fruitful discussion about the potential role of vanadium oxide in heterogeneous catalysis.References
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- J. Hou, M. Yang and J. Zhang, Nanoscale, 2020, 12, 69–692 Search PubMed.
- H. Du, T. R. Gengenbach, R. Hodgetts, D. R. MacFarlane and A. N. Simonov, ACS Sustainable Chem. Eng., 2019, 7, 6839–6850 CrossRef CAS.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. J. Xu, Am. Chem. Soc., 2018, 140, 13387 CrossRef CAS PubMed.
- S. D. Young, B. M. Ceballos, A. Banerjee, R. Mukundan, G. Pilania and B. R. Goldsmith, J. Phys. Chem. C, 2022, 126, 12980 CrossRef CAS.
- J. Pan, H. A. Hansen and T. Vegge, J. Mater. Chem. A, 2020, 8, 24098–24107 RSC.
- A. Osonkie, A. Ganesan, P. Chukwunenye, F. Anwar, K. Balogun, M. Gharaee, I. Rashed, T. R. Cundari, F. D’Souza and J. A. Kelber, ACS Appl. Mater. Interfaces, 2022, 14, 531–542 CrossRef CAS PubMed.
- K. K. Dey, S. Jha, A. Kumar, G. Gupta, A. K. Srivastava and P. P. Ingole, Electrochim. Acta, 2019, 312, 89–99 CrossRef CAS.
- A. Ganesan, A. Osonkie, P. Chukwunenye, I. Rashed, T. Cundari, F. D’Souza and J. Kelber, J. Electrochem. Soc., 2021, 168, 026504 CrossRef CAS.
- L. Wang, Y. Liu, H. Wang, T. Yang, Y. Luo, S. Lee, M. G. Kim, T. T. T. Nga, C. Dong and H. Lee, ACS Nano, 2023, 17, 7406 CrossRef CAS PubMed.
- K. Balogun, A. Ganesan, P. Chukwunenye, M. Gharaee, Q. Adesope, S. Nemšák, P. S. Bagus, T. R. Cundari, F. D’Souza and J. A. Kelber, J. Phys.: Condens. Matter, 2023, 35, 333002 CrossRef CAS PubMed.
- T. Hu, C. Wang, M. Wang, C. Li and C. Guo, ACS Catal., 2021, 11, 14417–14427 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496 CrossRef CAS PubMed.
- Y. Yao, H. Wang, X. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- J. Wu, S. Wang, R. Ji, D. Kai, J. Kong, S. Liu, W. Thitsartarn, B. H. Tan, M. H. Chua, J. Xu, X. J. Loh, Q. Yan and Q. Zhu, ACS Nano, 2024, 18, 20934 CrossRef CAS PubMed.
- S. Ioppolo, G. Fedoseev, M. Minissale, E. Congiu, F. Dulieu and H. Linnartz, Phys. Chem. Chem. Phys., 2014, 16, 8270–8282 RSC.
- M. Minissale, G. Fedoseev, E. Congiu, S. Ioppolo, F. Dulieu and H. Linnartz, Phys. Chem. Chem. Phys., 2014, 16, 8257–8269 RSC.
- K. Balogun, P. Chukwunenye, F. Anwar, A. Ganesan, Q. Adesope, D. Willadsen, S. Nemšák, T. R. Cundari, P. S. Bagus, F. D’Souza and J. A. Kelber, J. Chem. Phys., 2022, 157, 104701 CrossRef CAS PubMed.
- P. Chukwunenye, A. Ganesan, M. Gharaee, K. Balogun, F. Anwar, Q. Adesope, T. R. Cundari, F. D’Souza and J. A. Kelber, J. Mater. Chem. A, 2022, 10, 21401 RSC.
- Practical Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, ed. D. Briggs and M. P. Seah, John Wiley & Sons, Chichester, UK, 1990, vol. 1 Search PubMed.
- J. Chastain and J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, ULVAC-PHI, Incorporated, 1995.
- D. Goodacre, M. Blum, C. Buechner, H. Hoek, S. M. Gericke, V. Jovic, J. B. Franklin, S. Kittiwatanakul, T. Söhnel, H. Bluhm and K. E. Smith, J. Chem. Phys., 2020, 152, 044715 CrossRef CAS PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- G. W. Coulston, E. A. Thompson and N. Herron, J. Catal., 1996, 163, 122–129 CrossRef CAS.
- W. Suetaka, Surface infrared and Raman spectroscopy: methods and applications, Springer Science & Business Media, 2013, vol. 3 Search PubMed.
- G. Katsoukis, H. Heida, M. Gutgesell and G. Mul, ACS Catal., 2024, 14, 13867–13876 CrossRef CAS PubMed.
- R. Beaini, B. Baloukas, S. Loquai, J. E. Klemberg-Sapieha and L. Martinu, Sol. Energy Mater. Sol. Cells, 2020, 205, 110260 CrossRef CAS.
- L. Abello, E. Husson, Y. Repelin and G. Lucazeau, Spectrochim. Acta, Part A, 1983, 39, 641–651 CrossRef.
- T. Chirayil, P. Zavalij and M. S. Whittingham, Solid State Ionics, 1996, 84, 163–168 CrossRef CAS.
- I. L. Botto, M. B. Vassallo, E. J. Baran and G. Minelli, Mater. Chem. Phys., 1997, 50(3), 267–270 CrossRef CAS.
- P. Shvets, K. Maksimova and A. Goikhman, Phys. B, 2021, 613, 412995 CrossRef CAS.
- F. Hernández-Pérez, R. Antaño-López and F. Espejel, Rev. Int. Contam. Ambiental, 2021, 37, 501–502 Search PubMed.
- C. J. Vessey, M. P. Schmidt, M. Abdolahnezhad, D. Peak and M. B. J. Lindsay, ACS Earth Space Chem., 2020, 4, 641–649 CrossRef CAS.
- A. Šurca and B. Orel, Electrochim. Acta, 1999, 44, 3051–3057 CrossRef.
- L. Schumacher and C. Hess, J. Phys. Chem. C, 2024, 128, 9317–9330 CrossRef CAS.
- P. Shvets, O. Dikaya, K. Maksimova and A. Goikhman, J. Raman Spectrosc., 2019, 50, 1226–1244 CrossRef CAS.
- D. Leon-Chaparro, M. D. Nguyen, C. Baeumer, G. Mul and G. Katsoukis, Adv. Mater. Interfaces, 2025, 2400846 CrossRef.
- M. Schindler, F. C. Hawthorne and W. H. Baur, Chem. Mater., 2000, 12, 1248–1259 CrossRef CAS.
- M. N. Hughes and K. Shrimanker, Inorg. Chim. Acta, 1976, 18, 69–76 CrossRef CAS.
- M. E. Jacox and W. E. Thompson, Phys. Chem. Chem. Phys., 2005, 7, 768–775 RSC.
- D. de Waal, A. M. Heyns, K. Range and C. Eglmeier, Spectrochim. Acta, Part A, 1990, 46, 1639–1648 CrossRef.
- J. E. Bertie and M. R. Shehata, J. Chem. Phys., 1985, 83, 1449–1456 CrossRef CAS.
- H. Wiame, C. Cellier and P. Grange, J. Catal., 2000, 190, 406–418 CrossRef CAS.
- T. Liu, I. Temprano and S. J. Jenkins, Phys. Chem. Chem. Phys., 2017, 19, 21848–21855 RSC.
- T. Liu, I. Temprano, S. J. Jenkins and D. A. King, J. Chem. Phys., 2013, 139, 184708 CrossRef PubMed.
- P. Rochana, K. Lee and J. Wilcox, J. Phys. Chem. C., 2014, 118(8), 4238–4249 CrossRef CAS.
- A. A. Tsyganenko, A. M. Chizhik and A. I. Chizhik, Phys. Chem. Chem. Phys., 2010, 12, 6387–6395 RSC.
- R. J. G. Nuguid, M. Elsener, D. Ferri and O. Kröcher, Appl. Catal., B, 2021, 298, 120629 CrossRef CAS.
- J. Song, Q. Liao, X. Hong, L. Jin and N. Mézailles, Angew. Chem., Int. Ed., 2021, 60, 12242–12247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00554j |
| This journal is © the Owner Societies 2025 |