
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of branching in the ultrafast dynamics and two-photon absorption of two pyrimidine push–pull molecules†
Alexandros Katsidasa,
Michaela Feckovábcd,
Filip Bureš cd,
Sylvain Achelleb and
Mihalis Fakis*a
cd,
Sylvain Achelleb and
Mihalis Fakis*a
aDepartment of Physics, University of Patras, Patras, 26504, Greece. E-mail: fakis@upatras.gr; Tel: +30 2610 996794
bUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes)-UMR 6226, F-35000, Rennes, France
cInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czechia
dInstitute of Technology and Business in České Budějovice, Okružní 517/10, České Budějovice, 37001, Czechia
First published on 7th May 2025
Abstract
The dynamics and two-photon absorption (2PA) properties of two pyrimidine chromophores are studied using femtosecond time-resolved fluorescence and two-photon excited fluorescence techniques. The pyrimidine is used as an electron withdrawing group and is substituted at the C2 position with a phenylacridan fragment, while diphenylaministyryl donor moieties are appended at positions C4/6 to afford the pseudo-dipolar and pseudo-quadrupolar molecules 1 and 2, respectively. Chromophore 2 shows more efficient fluorescence emission, while 1 exhibits larger Stokes shifts. Their decay pathways are discussed through an emission from a Franck–Condon charge transfer (FC-CT) and a relaxed charge transfer (R-CT) state. Ultrafast dynamics in tetrahydrofuran show population of the R-CT state for 1 that is faster than solvation, while for 2, due to its pseudo-quadrupolar nature, R-CT population is slower and occurs from the solvated FC-CT state. Finally, molecule 2 shows better 2PA properties with cross sections reaching 560 GM at 820 nm.
Introduction
Intramolecular charge transfer (ICT) in push–pull π-conjugated organic chromophores is a fundamental process, controlling the nature and dynamics of the excited states, the photophysical properties and the potential for future applications of these chromophores in diverse and emerging photonic applications.1–6 It also plays a crucial role in various biophysical phenomena.7–10 The population of ICT states occurs in chromophores of dipolar, quadrupolar and octupolar topology and depends not only on the electron donating/withdrawing groups and the π-conjugated core but also on the molecular environment. In solution, the polarity of the solvent affects the energy level of the ICT state, rendering its population energetically favorable.11–16 Emission by an ICT state is characterized by a low-energy, broad and structureless emission spectrum with a reduced fluorescence quantum yield (Φ), compared to the narrow and vibronically structured emission stemming from a Franck–Condon (FC) state.17–20 Often, the ICT process leads to the population of a number of states with variable degrees of charge-transfer (CT) or to an excited state conformation with a twisted morphology (TICT), resulting in a very broad spectrum with dual peaks.21–27The ICT takes place within the excited state lifetime and especially in the timescale of 100 fs to a few ps. It often occurs simultaneously with solvent relaxation, structural reorganization and isomerization, leading to rich and complicated photodynamics. Separating the above kinetic phenomena is typically a non-trivial task.28–30 In dipolar (D–π–A) molecules, ICT is typically expected and is easily manifested by significant positive emission solvatochromism. In quadrupolar and octupolar molecules, ICT is related to the excited state symmetry breaking (SB), a phenomenon that has attracted significant scientific interest.31–37 SB is decisively dependent on solute–solvent interactions since the solvent's strong reaction field enhances SB and an excited state of quadrupolar nature relaxes to a dipolar state.
Molecules with ICT properties have been the subject of intense scientific interest since a deep understanding of the control and modulation of ICT is imperative for advances in photonic devices. They have demonstrated enhanced electro-optic coefficients,38,39 second harmonic generation efficiency40,41 and two-photon absorption (2PA) properties.17,42–50 Especially, due to their non-linear optical (NLO) properties, they are attractive for a variety of applications in imaging,51 photodynamic therapy,52 optical limiting,53,54 direct laser microfabrication and 3D optical data storage.55–57 Moreover, they have also exhibited reverse intersystem crossing (RISC) and excellent thermally activated delayed fluorescence (TADF) properties due to a small energy gap between singlet and triplet excited states.58–61 Finally, aggregation induced emission (AIE) has also been reported for push–pull structures,62,63 rendering these ICT molecules highly suitable for next-generation organic light emitting diodes (OLEDs), while they are also finding use in organic solar cells as electron acceptors.64,65
As it is easily understood, push–pull molecules with ICT properties are appealing not only from a fundamental point of view but also due to a plethora of applications. Alkyl-amino, phenyl-amino, 9H-carbazol-9-yl and acridan fragments are some of the most well-known groups to be used as electron donors.66–71 On the other hand, various heterocyclic rings have been investigated as electron withdrawing groups in ICT molecules such as benzothiazole and triazine.72–75 The pyrimidine group with two nitrogen atoms has been used as a suitable fragment in NLO chromophores, in TADF emitters and in stimuli responsive fluorescence switches.76–79 In our recent work, we have reported on the synthesis of six 4-styryl or 4,6-distyryl substituted pyrimidines linked to acridan at the C2 position via a phenylene linker. We have focused on their dual emission properties and long-lived ICT states.80
As a continuation of our previous work, we focus herein on the ultrafast dynamics and NLO properties of two previously reported pyrimidines, having diphenylamino donors connected to the styryl fragments. Ultrafast dynamics reveal emission by an initially excited Franck–Condon CT (FC-CT) state and a relaxed CT (R-CT) state due to solvent or structural reorganization, while good 2PA values are found. Based on ultrafast dynamics and 2PA measurements, it is concluded that the acridan group does not play a role in the emission process as it lies almost perpendicular to the adjacent phenyl ring.
Experimental
Steady state spectroscopy
The two molecules were examined for their steady state spectra in hexane (HEX), decane (DEC), toluene (TOL), tetrahydrofuran (THF), dichloromethane (DCM), acetone (ACT) and acetonitrile (MeCN). The absorption spectra were recorded with a Shimadzu UV-3000 spectrometer, while a Horiba S2 Jobin Yvon Fluoromax 4 was used for recording the steady-state fluorescence spectra. The Φ values were determined relative to that of 9,10-bis(phenylethynyl)anthracene in cyclohexane (Φ = 1.00).81Time resolved spectroscopy
The time resolved fluorescence dynamics were studied in the ps–ns and fs–ps timescales by means of the time correlated single photon counting (TCSPC) and the femtosecond upconversion methods.32,82 In the former, a ps diode laser at 400 nm, with a 60 ps pulse duration, was used as the excitation source. The detection was made under magic angle conditions for the determination of the lifetimes, while anisotropy measurements were also conducted to determine the rotational times. The overall temporal resolution of the technique was 80 ps. For the upconversion measurements, a fs mode-locked Ti:sapphire laser with 80 fs pulses was used. The second harmonic beam at 400 nm served as the excitation beam. The temporal resolution of the system was 250 fs. Again, both magic angle and anisotropy measurements were made. For the latter, the polarization plane of the excitation laser was changed using a Berek compensator. In both techniques, the time resolved anisotropy was calculated usingwhere Ipar(t) and Iper(t) are the fluorescence intensities with parallel and perpendicular polarizations to the excitation source's polarization plane. For the TCSPC, a correction factor, G, was determined to compensate for the different responses of the monochromator at the two perpendicular polarizations. For the upconversion technique, this factor is unity.
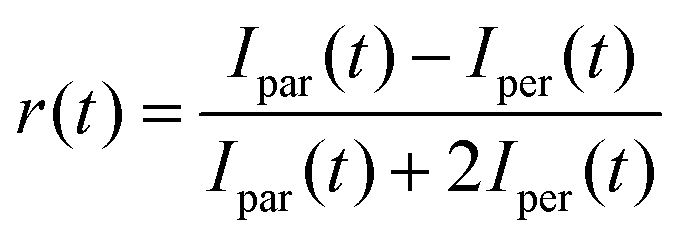
2PA spectroscopy
All the samples were fluorescent chromophores, so their 2PA properties were examined by a two-photon excited fluorescence (2PEF) technique from 730 to 870 nm using the same mode-locked Ti:sapphire fs laser mentioned above.83–85 The 2PA fluorescence intensity was detected as a function of the excitation power, which was changed using a half-wave plate and a polarizer, always validating the square-law dependence. From these measurements, the 2PA cross sections (δ2PA) were calculated using rhodamine 6G in MeOH (2 × 10−5 M) as the reference sample. The measured scattered light from the solvent has been subtracted.Results and discussion
Materials
Fig. 1 shows the chemical structures of the two push–pull molecules 1 and 2 studied herein, both bearing a pyrimidine-based electron-acceptor central core. The pyrimidine is substituted at position C2 with a phenylacridan group. Also, diphenylamino substituted styryl moieties are appended in positions C4/6. The synthesis, using cross-coupling and Knoevenagel reactions, has been described previously, together with thermal properties, X-ray analysis and DFT-calculated parameters.80 It was revealed that the acridan moiety has a perpendicular arrangement with respect to the residual π-system. Besides, non-zero ground state dipole moment was found while the compounds possess no symmetry. | ||
| Fig. 1 The chemical structures of the pyrimidine-based molecules 1 and 2. | ||
Steady state spectroscopy
The fundamental optical properties of chromophores 1 and 2 were recorded in solutions with solvents of different polarity. Fig. 2 shows the absorption and emission spectra of the two molecules. Table 1 summarizes the photophysical results. The absorption spectra show a main peak at 400–430 nm attributed to the HOMO−1 to LUMO transition and account for an ICT transition.80 In the apolar solvents HEX and DEC, both molecules feature slightly narrower and structured absorption spectra, while the structure is lost as the polarity increases. The shift of the spectra is however minimal pointing to an insignificant stabilization of the ground state.86 The broadening, however, can be attributed to a ground state with a small degree of ICT nature.18 The observed absorption solvatochromism is small and does not correlate with the orientational polarisability of the solvent.87 It rather correlates with the refractive index, pointing to dispersion as the main solute–solvent interaction in the ground state. For 2, the absorption spectra are red-shifted compared to 1 (by about 0.216 eV), which is typical for molecules with longer conjugation lengths, indicating an electronic coupling between the branches at positions C4 and C6. Such an electronic coupling is typical for branched compounds with a nitrogen atom as a central core, while it is hindered in the case of a benzene core due to a suppression of coherent interactions among the branches.88–90 | ||
| Fig. 2 Absorption (a) and (c) and emission (b) and (d) spectra of molecules 1 and 2 in various solvents. | ||
| Molecule | Solvent | λαbs (nm)/ε (mM−1 cm−1) | λem (nm) | Φ | Stokes shift (cm−1) | τ (ns)a | δ2PA (GM) |
|---|---|---|---|---|---|---|---|
| a Taken as the average of the lifetimes measured by the TCSPC technique. | |||||||
| 1 | HEX | 396/24.4 | 432, 459 | 0.23 | 2100 | 0.87 | 150 |
| DEC | 399 | 436, 463 | — | 2130 | 1.07 | ||
| TOL | 404/26.1 | 471 | 0.35 | 3520 | 1.44 | 110 | |
| THF | 400/34.0 | 508 | 0.47 | 5310 | 2.42 | 130 | |
| DCM | 406/31.3 | 532 | 0.75 | 5830 | 2.80 | ||
| ACT | 397 | 540 | — | 6670 | 2.96 | ||
| MeCN | 398 | 566 | — | 7460 | 2.91 | ||
| 2 | HEX | 415, 433/65.1 | 451, 479 | 0.52 | 1920 | 1.25 | 560 |
| DEC | 417, 436 | 456, 482 | — | 2050 | 1.36 | ||
| TOL | 426/62.1 | 480 | 0.65 | 2640 | 1.53 | 460 | |
| THF | 424/65.6 | 523 | 0.61 | 4460 | 2.38 | 460 | |
| DCM | 430/58.0 | 553 | 0.94 | 5170 | 2.89 | ||
| ACT | 422 | 563 | — | 5930 | 2.49 | ||
| MeCN | 419 | 592 | — | 6970 | 1.84 | ||
As a quantitative investigation, the absorption transition dipole moments for 1 and 2 were calculated using the equation:91
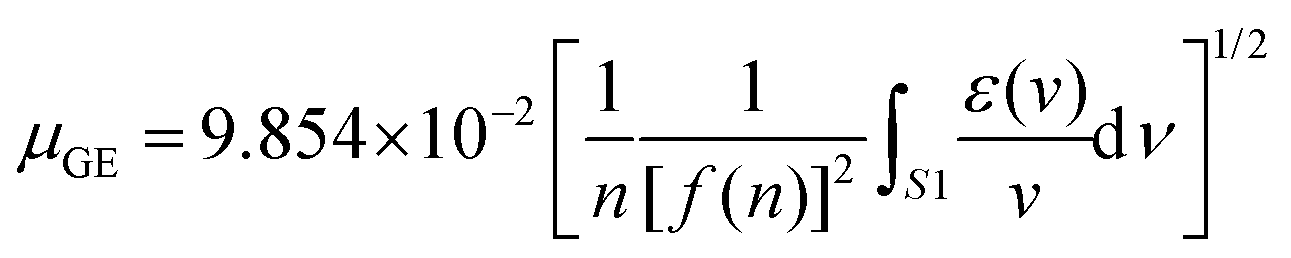
The emission spectra are more sensitive to solvent polarity as shown in Fig. 2. In apolar solvents, both molecules feature structured and narrow spectra with clear vibronic peaks attributed to a FC-CT state. Although only a qualitative comparison of the absorption and emission spectra can be made (a quantitative comparison involves plotting the spectra in cm−1),92 it is obvious that the mirror image rule is far from being obeyed in apolar solvents and this is due to an emission originating from a planar rigid structure due to an increase in the torsional barrier in the excited state.93,94 On the other hand, in the ground state, the barrier is decreased, leading to an absorption from a variety of torsional arrangements. As the polarity increases, the spectra become broader, structureless and the peak is shifted to longer wavelengths, revealing the population of a R-CT state, which becomes the emitting state in polar environments. Finally, the fluorescence Φ values were measured for four representative solvents and were found to be significantly higher for 2 vs. 1, reaching 0.94 in DCM.
To obtain a deeper insight into the effect of polarity on the steady state spectra, the Lippert–Mataga plots were constructed, i.e. the Stokes shift was plotted as a function of solvent polarizability (Fig. S1, ESI†). As can be seen, molecule 2 has a slightly smaller Stokes shift for all the solvents and also a slightly smaller slope of the Lippert–Mataga plot. From the slope of these plots, the change of the permanent dipole moments Δμsolv upon excitation can be calculated using the following equation:18,95,96

where ε is the dielectric constant and n is the refractive index of the solvent. Table S2 (ESI†) summarizes these values for all solvents. For the pseudo-dipolar chromophore 1, the Onsager radius was estimated from its DFT-optimized structure as half of the distance between the Ph2N donor occupied by the HOMO–1 and the LUMO spread over the central pyrimidine acceptor. For the pseudo-quadrupolar molecule 2, the longest distance for the charge separation corresponds to the Ph2N-centered HOMO–1 and the opposite olefinic linker bearing the LUMO (Fig. S2, ESI†).97 Finally, Δμsolv was found to be 9.85 D for 1 and 17.47 D for 2, respectively (Table S3, ESI†).

Excited state dynamics
 | ||
| Fig. 3 Fluorescence decays in the nanosecond timescale for 1 and 2 in various solvents. Exc.: 400 nm, detection at the peaks of the emission spectra. | ||
Next, the emission transition dipole moment is calculated using the lifetimes obtained by the TCSPC method and the following equation:91

μEG can also provide useful information on the nature of the excited state. The values are summarized in Table S1 (ESI†) and are higher for 2 (e.g. μEG for 2 in DCM is 9.53 vs. 8.26 D for 1) following the changes in the quantum yields.
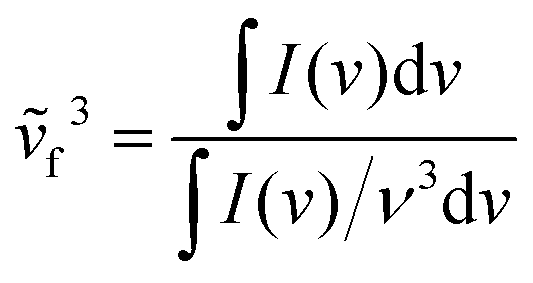
The dynamics along with the time-resolved emission spectra (TRES) and time-resolved area normalized emission spectra (TRANES) for TOL and THF are shown in Fig. 4 and 5 for 1 and 2, respectively.98–102 The 2D plots of the TRES are shown in Fig. S3 (ESI†). In all samples, a fast decay is observed at shorter wavelengths and a slow rise at longer ones. This effect is due to a relaxation of the excited state towards lower energies caused by solvent/structural relaxation and R-CT population and leads to a red-shift of the emission spectrum. The curves were analyzed using a global fit method and the fitting results for the two faster mechanisms are shown in Tables S6 and S7 (ESI†). In all cases, a three-exponential fitting was needed to fit the results. The third (slowest) component was fixed to be equal to the lifetime found by the TCSPC technique. Within the ps timescale, two decay lifetimes of the order of ∼1.3 ps and 5.9–6.6 ps in TOL and 0.6–0.8 and 2.5–2.8 ps in THF were found. The lifetimes correspond to decay mechanisms at short wavelengths (positive amplitudes), while they evolve to rise mechanisms (negative amplitudes) at longer ones. The relaxation times are smaller in THF for both molecules because it is a more polar solvent than TOL and the solute–solvent interactions are stronger. In both solvents, the relaxation times are similar to the solvation times for TOL and THF.103,104
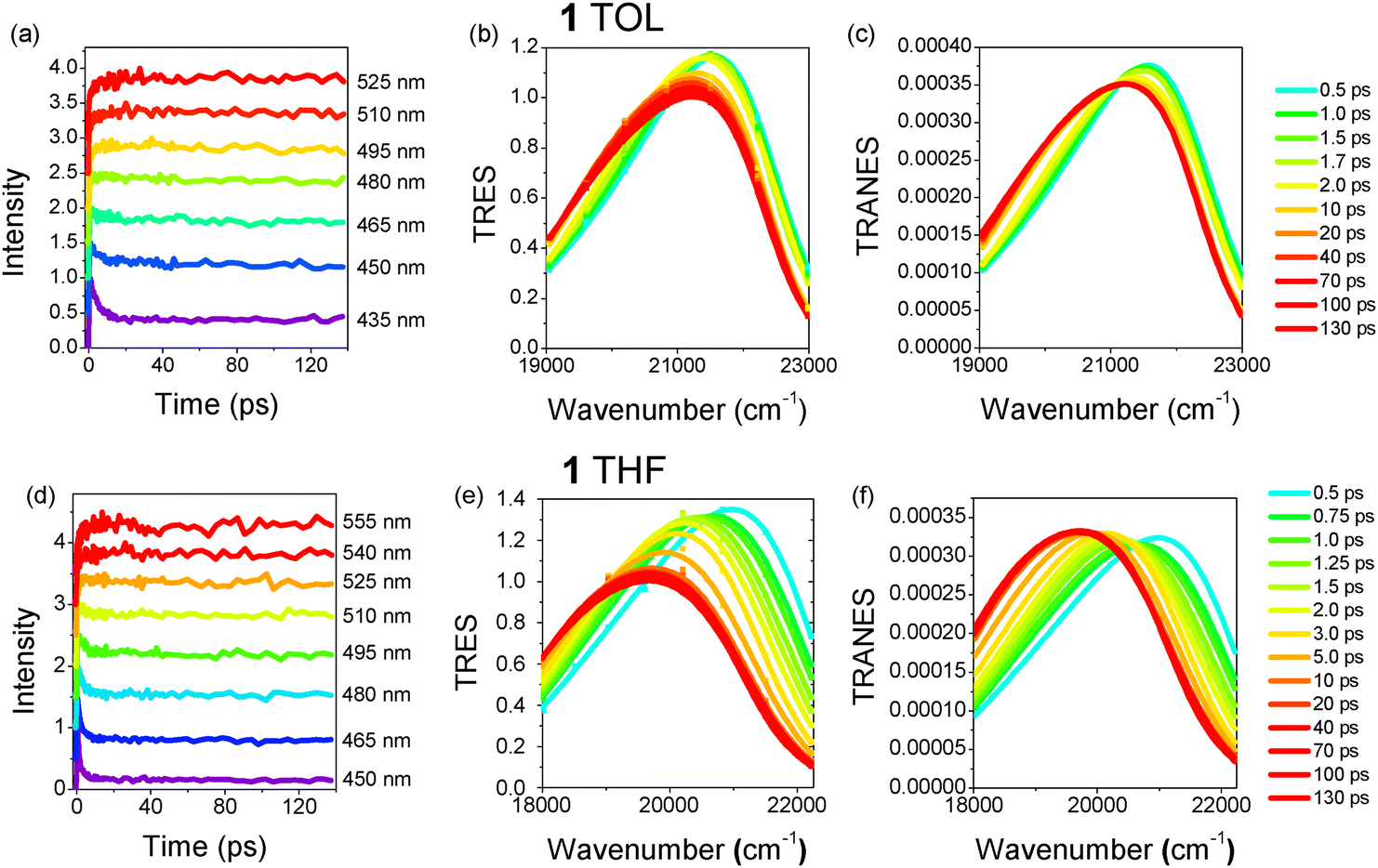 | ||
| Fig. 4 (a) and (d) Fluorescence dynamics at selected wavelengths, (b) and (e) TRES and (c) and (f) TRANES for 1 in TOL and THF. | ||
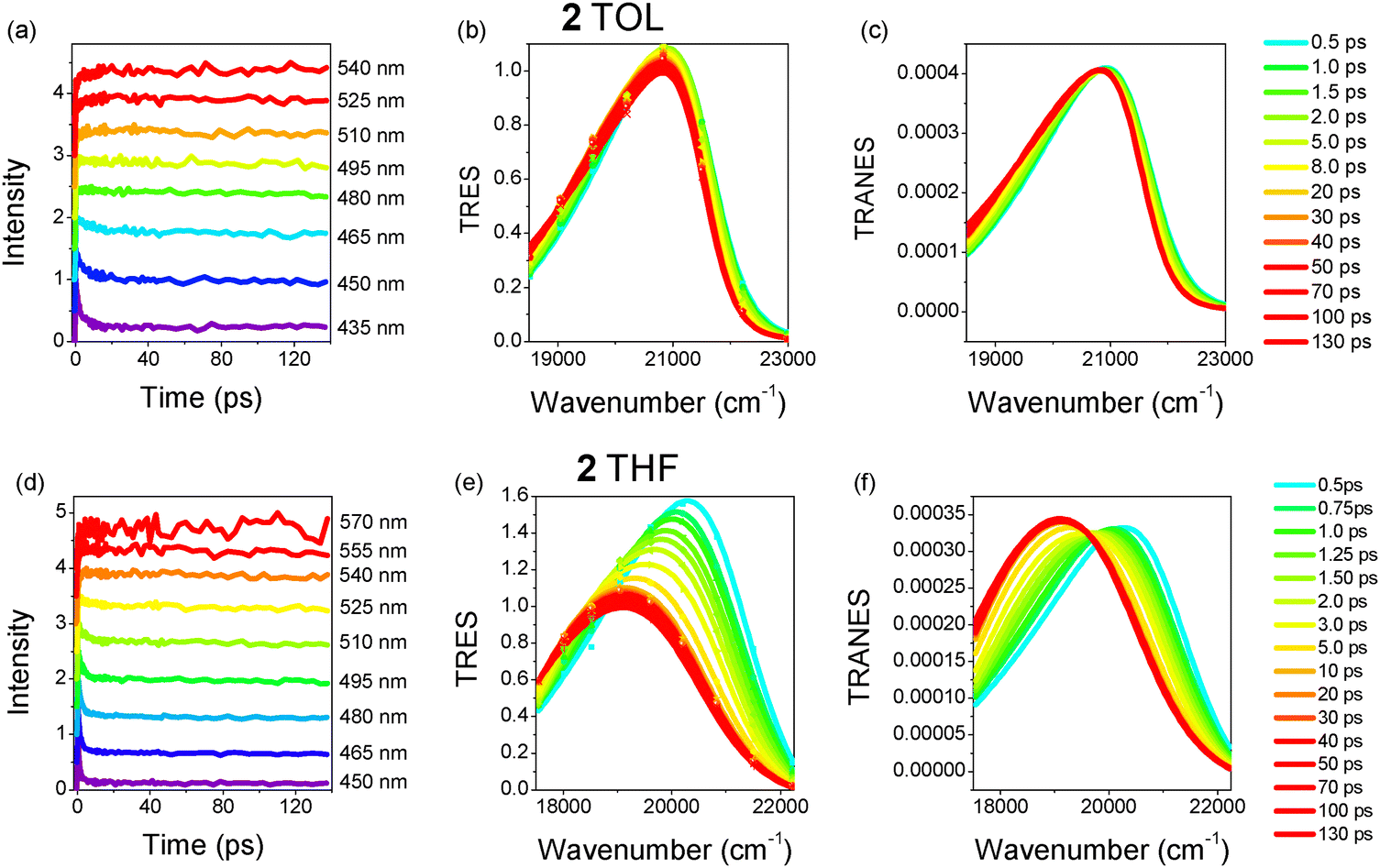 | ||
| Fig. 5 (a) and (d) Fluorescence dynamics at selected wavelengths, (b) and (e) TRES and (c) and (f) TRANES for 2 in TOL and THF. | ||
The TRES of 1 in TOL exhibit a small transient red-shift (374 cm−1), which is completed within ∼10 ps, while the TRES of 2 remain almost unchanged with only a slight broadening. On the other hand, in THF the spectral changes are more obvious with the spectra of both molecules having a significant red-shift (1313 cm−1 and 1171 cm−1 for 1 and 2, respectively) within 10 ps. The solvent response function C(t) for both molecules and solvents is plotted in Fig. S4 (ESI†). C(t) decays faster in THF pointing to a more efficient spectral relaxation. A bi-exponential function was used to fit C(t) with lifetimes similar to those found after global analysis of the individual dynamics pointing to inertial and diffusive solvation mechanisms (Table S8, ESI†). The total area-normalized intensity obtained by the TRES is also plotted in Fig. S4 (ESI†) and its dynamics is visually compared to the C(t). In toluene, the total intensity exhibits a rise up to 2 ps, possibly due to vibronic relaxation. It is obvious that spectral relaxation i.e C(t) is faster than quenching (intensity decrease) in toluene, while in the more polar THF, the spectral relaxation is highly related to the quenching of the excited state. Apart from the transient red-shift, the spectra are also accompanied by a gradual broadening, indicating a decay process involving more than one emitting state.17,29 Fig. S5 (ESI†) shows the FWHM of the TRES as a function of time. Especially in the case of THF, for both compounds, an initial decrease of the FWHM is observed. This is due to a fast decay of the high energy rotamers to a rigid excited state.
Finally, following the method described by Vauthey et al.105 and used by Xia et al.,106 the instantaneous emission dipole moment, μEG(t), is calculated at different times for 1 and 2 in THF. The time dependence of μEG(t) is shown in Fig. S6 (ESI†) in non-normalized and normalized units. For both molecules, μEG(t) decreases within the first 10 ps reaching a constant value at longer times, which is equal to the emission dipole moment of the relaxed excited state as shown in Table S1 (ESI†). The decrease of μEG(t) is associated with a change in the nature of the excited state. The initial μEG(t) values of 9.5 and 7.5 D found for 1 and 2, respectively, are considered as the emission dipole moments of the FC-CT state. The more intense decay of μEG(t) for 2 points to a change of its excited state from FC-CT with a pseudo-quadrupolar nature to an excited state with a stronger CT character.
Although the TRES provide a quantitative view of the spectral changes, further insight into the photodynamic mechanisms is obtained by observing the TRANES (Fig. 4 and 5). In TOL, both molecules have an iso-emissive point, indicating that there are two emissive species/states. The transient spectral changes for 1, as mentioned above, are stronger than those of 2, which could be ascribed to its dipolar nature, also revealed by the time resolved anisotropy and 2PA measurements (vide infra). The two emissive species could be considered as the FC-CT and R-CT states, while the contribution of the latter is less significant in the case of 2. On the other hand, the two molecules reveal different and more pronounced behaviors in THF. 1 exhibits an iso-emissive point within the first 2 ps, while a gradual red-shift of the spectrum occurs at longer times and the iso-emissive point is lost (Fig. S7, ESI†). The iso-emissive point at short timescales is due to an emission from FC-CT and R-CT states with peaks at 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 and 20220 cm−1, the latter being populated on a ∼100 fs timescale, i.e. within the inertial solvation time for THF. Then, the emission from the R-CT state dominates and it relaxes at longer times (2–10 ps), leading to the observed transient red-shift to 19700 cm−1. Molecule 2 in THF shows exactly the opposite behavior. Initially, the spectrum experiences a transient shift in <3 ps (without an isosbestic point) due to an inertial and diffusion relaxation of the FC-CT state by the solvent molecules from 20300 cm−1 to 19500 cm−1 (Fig. S8, ESI†). After 3 ps, an iso-emissive point is revealed, which is an indication of the population of a R-CT state (at 19100 cm−1) by energy transfer from the FC-CT state and the simultaneous emission of both states. Fig. S9 (ESI†) shows an energy diagram that is used as a model for the explanation of the dynamics for 1 and 2 in THF. The population of the R-CT state for 2 occurs after solvation and is slower than for 1, while the R-CT population for 1 is within the inertial solvent relaxation. This is attributed to the quadrupolar nature of 2 compared to the dipolar character of 1.
000 cm−1 and 20220 cm−1, the latter being populated on a ∼100 fs timescale, i.e. within the inertial solvation time for THF. Then, the emission from the R-CT state dominates and it relaxes at longer times (2–10 ps), leading to the observed transient red-shift to 19700 cm−1. Molecule 2 in THF shows exactly the opposite behavior. Initially, the spectrum experiences a transient shift in <3 ps (without an isosbestic point) due to an inertial and diffusion relaxation of the FC-CT state by the solvent molecules from 20300 cm−1 to 19500 cm−1 (Fig. S8, ESI†). After 3 ps, an iso-emissive point is revealed, which is an indication of the population of a R-CT state (at 19100 cm−1) by energy transfer from the FC-CT state and the simultaneous emission of both states. Fig. S9 (ESI†) shows an energy diagram that is used as a model for the explanation of the dynamics for 1 and 2 in THF. The population of the R-CT state for 2 occurs after solvation and is slower than for 1, while the R-CT population for 1 is within the inertial solvent relaxation. This is attributed to the quadrupolar nature of 2 compared to the dipolar character of 1.
Anisotropy dynamics
In our previous work, DFT analysis in molecules 1 and 2 revealed that the acridan moiety is rotated and lies off plane with respect to the pyrimidine core; therefore, it is not expected to play a crucial role in the optical properties and ICT process. To confirm this assumption, anisotropy measurements in the fs–ps timescale were conducted and the plots are shown in Fig. 6. | ||
| Fig. 6 Anisotropy dynamics within the first 40 ps measured by the up-conversion technique for 1 (a) and 2 (b) in TOL and THF. | ||
The initial anisotropy r0 provides an aspect of the molecular symmetry after excitation. For dipolar molecules, r0 is expected to be close to 0.4, while for quadrupolar and octupolar ones it is expected to be close to 0.2 and 0.1, respectively. For 1 and 2, r0 is 0.35 and 0.24–0.27, i.e. close to the values expected for dipolar (D–π–A) and quadrupolar (D–π–A–π–D) symmetry.107–109 Also, no decay of the anisotropy in the 100 fs to ps timescale was observed for 2, as expected for octupolar molecules due to coherent and incoherent energy delocalization mechanisms among the branches. Anisotropy decay measurements in the ns timescale were also performed to determine the rotational correlation time τcor as a function of solvent viscosity. τcor ranges from 0.15 ns to 0.38 ns for 1 and from 0.25 to 0.67 for 2 and is linearly dependent on the solvent viscosity (Fig. S10 and S11 and Table S9, ESI†).
Two-photon absorption properties
The 2PA spectra are presented in Fig. 7 for both molecules in HEX, TOL and THF. The two-photon induced fluorescence as a function of excitation intensity is shown in Fig. S12 (ESI†) exhibiting the quadratic dependence as expected for a 2PA process. Due to its longer conjugation length, 2 has larger 2PA cross sections than its dipolar counterpart reaching 560 GM in HEX and 450 GM in TOL and THF. Apart from the difference in the 2PA values, the 2PA peak of 2 falls within the same spectral region as that of 1, meaning that the spectral shift observed in their 1PA spectra is not maintained. 2PA spectroscopy is, in general, useful in identifying the nature and molecular geometry of the excited states.110 A comparison of 2PA spectra with the rescaled 1PA ones is shown in Fig. S13 (ESI†). For 2, the 2PA peak is blue-shifted compared to twice the wavelength of the rescaled 1PA peak, following the expected behavior of a quadrupolar molecule based on the Frenkel exciton model.33,87,111 Besides, the 2PA and rescaled 1PA spectra of 1 coincide, providing further evidence for its dipolar behavior as also concluded by the fs-anisotropy measurements. However, even in non-centrosymmetric chromophores, a blue-shift of the 2PA band relative to the 1PA one can be observed due to an involvement of enhanced vibronic transitions in the 2PA spectrum.112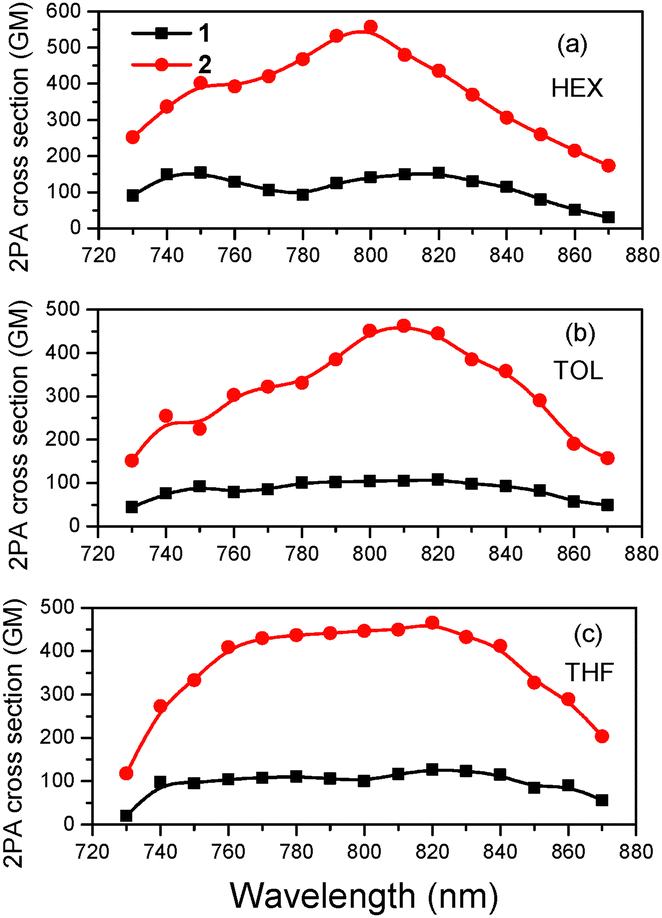 | ||
| Fig. 7 2PA spectra of 1 and 2 in (a) HEX, (b) TOL and (c) THF. | ||
Mono- and di(diphenylamino)styrylpyrimidines have been extensively studied in the past for their 2PA properties.76 In this frame, the effects of the electron donating group, branching and π-conjugated bridge have been addressed.113–115 Surprisingly, very large 2PA cross section values reaching 3000 GM have been found. However, great care must be taken when discussing and comparing different results since in certain cases very high excitation powers and sample concentrations have been used. On the other hand, a comparison of the 2PA values of 1 and 2 can be made with those of the molecules presented in our previous work,50 where the acridan group is replaced by another diphenylamino substituent, addressing the influence of the C2 substitution. Interestingly, the three-branched A–(π–D)3 compound with diphenylamino groups at the periphery (2c in ref. 50) has very similar 2PA cross section values and peak wavelengths compared to 2, showing that the replacement of the diphenylamino with the out-of-plane oriented acridan does not play a decisive role in the 2PA properties.
Finally, the 2PA results are also useful in calculating again the difference between the excited and ground state dipole moments using the equation:19,116,117
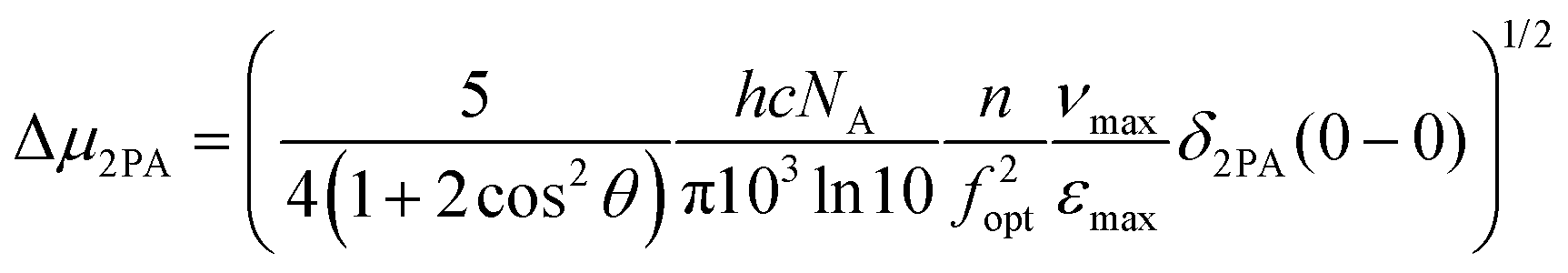
Conclusions
In summary, we have investigated the role of branching in the steady-state, excited state dynamics and 2PA properties of two pyrimidine molecules with a phenylacridan group at the C2 position. The pyrimidine was also substituted at the C4/6 positions with styryl groups attached to diphenylamino donors to obtain molecules 1 and 2. Steady state spectroscopy shows a larger Stokes shift for molecule 1, while higher fluorescence quantum yields were obtained for 2. The time-resolved fluorescence measurements revealed emission pathways attributed to a FC-CT and a R-CT state. For 1 in THF, the R-CT population occurs as fast as inertial solvation, followed by diffusive solvation of the R-CT state. On the other hand, for 2, the FC-CT to R-CT energy transfer is slower than that for 1, occurring by the solvated FC-CT state. Time resolved anisotropy measurements in the fs–ps timescale showed an excited state of dipolar and pseudo-quadrupolar nature for 1 and 2, respectively. Finally, 2PA characterization showed a more than 4 times increase of the 2PA cross sections for 2 vs. 1, reaching 560 GM at 820 nm in HEX.Author contributions
Investigation: AK, MFec, MF; methodology: MF; writing – original draft: MF; writing – review and editing: MF, SA and FB.Data availability
The data supporting this article are available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this work was supported by the ERDF “Innovative materials suitable for high added value applications (INMA)” (No. CZ.02.01.01/00/23_021/0008593). M. F. acknowledges the Région Bretagne, France, for her PhD funding (HOLED project).References
- P. K. Samanta and R. Misra, J. Appl. Phys., 2023, 133, 020901 CrossRef CAS.
- F. Bureš, RSC Adv., 2014, 4, 58826 Search PubMed.
- H. Miranda-Salinas, Y.-T. Hung, Y.-S. Chen, D. Luo, H.-C. Kao, C.-H. Chang, K.-T. Wong and A. Monkman, J. Mater. Chem. C, 2021, 9, 8819 RSC.
- C. Zhu, T. Wei, Y. Wei, L. Wang, M. Lu, Y. Yuan, L. Yin and L. Huang, J. Mater. Chem. A, 2021, 9, 1207 Search PubMed.
- Z. Zhao, C. Zeng, X. Peng, Y. Liu, H. Zhao, L. Hua, S.-J. Su, S. Yan and Z. Ren, Angew. Chem., Int. Ed., 2022, 61, e202210864 Search PubMed.
- C. S. Abeywickrama, Chem. Commun., 2022, 58, 9855 RSC.
- N. Yukihira, C. Uragami, K. Horiuchi, D. Kosumi, A. T. Gardiner, R. J. Cogdell and H. Hashimoto, Commun. Chem., 2022, 5, 135 Search PubMed.
- Y.-X. Tan, X. Zhang, Y. Wang and J. Yao, Acc. Mater. Res., 2024, 5, 1377 CrossRef CAS.
- A. M. Gutiérrez-Vílchez, C. V. Ileperuma, V. Navarro-Pérez, P. A. Karr, F. Fernández-Lázaro and F. D’Souza, ChemPlusChem, 2024, 89, e202400348 CrossRef.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933 CrossRef CAS PubMed.
- V. Maffeis, R. Brisse, V. Labet, B. Jousselme and T. Gustavsson, J. Phys. Chem. A, 2018, 122, 5533 CrossRef CAS PubMed.
- D. Fan, Y. Yi, Z. Li, W. Liu, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2015, 119, 5233 CrossRef CAS PubMed.
- Y. Hu, C. Neumann, L. Scholtz, A. Turchanin, U. Resch-Genger and S. Eigler, Nano Res., 2023, 16, 45 CrossRef CAS.
- F. Barati-Darband, M. Izadyar and F. Arkan, J. Phys. Chem. A, 2019, 123, 2831 CrossRef CAS PubMed.
- Y. Li, M. Zhou, Y. Niu, Q. Guo and A. Xia, J. Chem. Phys., 2015, 143, 034309 CrossRef.
- Y. Zhang, M. Jiang, G.-C. Han, K. Zhao, B. Z. Tang and K. S. Wong, J. Phys. Chem. C, 2015, 119, 27630 CrossRef CAS.
- D. J. Stewart, M. J. Dalton, R. N. Swiger, J. L. Fore, M. A. Walker, T. M. Cooper, J. E. Haley and L.-S. Tan, J. Phys. Chem. A, 2014, 118, 5228 CrossRef CAS.
- B. Carlotti, E. Benassi, C. G. Fortuna, V. Barone, A. Spalletti and F. Elisei, Chem. Phys. Chem., 2016, 17, 136 CrossRef CAS PubMed.
- B. Carlotti, E. Benassi, A. Spalletti, C. G. Fortuna, F. Eliseia and V. Barone, Phys. Chem. Chem. Phys., 2014, 16, 13984 RSC.
- F. Xiao, X. Liu, K. Lin, Y. Zhou, W. Gao, Y. Lei, M. Liu, X. Huang and H. Wu, J. Phys. Chem. C, 2021, 125, 16792 CrossRef CAS.
- S. Sasaki, G. P. C. Drummen and G. Konishiac, J. Mater. Chem. C, 2016, 4, 2731 RSC.
- S. Kumar, P. Singh, P. Kumar, R. Srivastava, S. Kalyan Pal and S. Ghosh, J. Phys. Chem. C, 2016, 120, 12723 CrossRef CAS.
- H.-Y. Fu, X.-J. Liu, H. Zha, X.-X. Li, Y. Xu, F. Yanga and M. Xia, Phys. Chem. Chem. Phys., 2019, 21, 1399 RSC.
- K. Hanaoka, S. Iwaki, K. Yagi, T. Myochin, T. Ikeno, H. Ohno, E. Sasaki, T. Komatsu, T. Ueno, M. Uchigashima, T. Mikuni, K. Tainaka, S. Tahara, S. Takeuchi, T. Tahara, M. Uchiyama, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2022, 144, 19778 CrossRef CAS PubMed.
- R. Ghosh, Phys. Chem. Chem. Phys., 2018, 20, 6347 RSC.
- D.-G. Chen, T.-C. Lin, Y.-A. Chen, Y.-H. Chen, T.-C. Lin, Y.-T. Chen and P.-T. Chou, J. Phys. Chem. C, 2018, 122, 12215 CrossRef CAS.
- L. Martinez-Fernandez, T. Gustavsson, U. Diederichsen and R. Improta, Molecules, 2020, 25, 824 CrossRef CAS.
- P. K. Singh, S. Nath, M. Kumbhakar, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. A, 2008, 112, 5598 CrossRef CAS PubMed.
- J. Sung, P. Kim, Y. O. Lee, J. S. Kim and D. Kim, J. Phys. Chem. Lett., 2011, 2, 818 CrossRef CAS.
- B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643 CrossRef CAS PubMed.
- B. Dereka, A. Rosspeintner, R. Stężycki, C. Ruckebusch, D. T. Gryko and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 6029 CrossRef CAS PubMed.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742 CrossRef CAS.
- M. Fakis, V. Petropoulos, P. Hrobárik, J. Nociarová, P. Osuský, M. Maiuri and G. Cerullo, J. Phys. Chem. B, 2022, 126, 8532 CrossRef CAS PubMed.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, J. Phys. Chem. Lett., 2020, 11, 10329 CrossRef CAS.
- R. Roy, S. Chawla, V. Sharma, A. K. Pal, Y. Silori, A. Datta, A. K. De and A. Lal Koner, Chem. Sci., 2024, 15, 6363–6377 RSC.
- A. Cesaretti, A. Spalletti, F. Elisei, P. Foggi, R. Germani, C. G. Fortuna and B. Carlotti, Phys. Chem. Chem. Phys., 2021, 23, 16739 RSC.
- J. Wu, W. Wang, C. Gong, Q. Li, Z. Li, G. Deng, X. Zhang, K. Chen, Y. Gong and K. S. Chiangac, J. Mater. Chem. C, 2017, 5, 7472 RSC.
- K. Senthilkumar, M. Pizzotti, K. Thirumoorthy, G. Di Carlo, S. Righetto, A. O. Biroli, M. Haukka and N. Palanisami, J. Phys. Chem. C, 2016, 120, 20277 CrossRef CAS.
- J. M. Cole, R. Soc. A, 2003, 361, 2751 CrossRef CAS PubMed.
- Y. Peng, Y. Yan, P. Li, B. Li, H. Jiang, B. Guo, Q. Yuan and W. Gan, Mater. Chem. Front., 2023, 7, 502 RSC.
- L. Sun, W. Zhu, W. Wang, F. Yang, C. Zhang, S. Wang, X. Zhang, R. Li, H. Dong and W. Hu, Angew. Chem., 2017, 56, 7831 CrossRef CAS.
- S. Sarkar, M. Santra, S. Singha, Y. W. Jun, Y. J. Reo, H. R. Kima and K. H. Ahn, J. Mater. Chem. B, 2018, 6, 4446 RSC.
- S. Pascal, Q. Bellier, S. David, P.-A. Bouit, S.-H. Chi, N. S. Makarov, B. Le Guennic, S. Chibani, G. Berginc, P. Feneyrou, D. Jacquemin, J. W. Perry, O. Maury and C. Andraud, J. Phys. Chem. C, 2019, 123, 23661 CrossRef CAS.
- L. Lescos, P. Beaujean, C. Tonnelé, P. Aurel, M. Blanchard-Desce, V. Rodriguez, M. de Wergifosse, B. Champagne, L. Muccioli and F. Castet, Phys. Chem. Chem. Phys., 2021, 23, 23643 RSC.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641 CrossRef CAS.
- M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zaleśny, W. Bartkowiak, H. Ågren, S. Canuto, L. De Boni, E. Ishow and C. R. Mendonca, Sci. Rep., 2014, 4, 4447 CrossRef.
- J. Nociarová, P. Osuský, E. Rakovský, D. Georgiou, I. Polyzos, M. Fakis and P. Hrobárik, Org. Lett., 2021, 23, 3460 CrossRef.
- P. Osusky, J. Nociarova, M. Smolicek, R. Gyepes, D. Georgiou, I. Polyzos, M. Fakis and P. Hrobarik, Org. Lett., 2021, 23, 5512 CrossRef CAS PubMed.
- F. Kournoutas, A. Fihey, J.-P. Malval, A. Spangenberg, M. Fecková, P. le Poul, C. Katan, F. Robin-le Guen, F. Bureš, S. Achelle and M. Fakis, Phys. Chem. Chem. Phys., 2020, 22, 4165 RSC.
- D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781 CrossRef CAS.
- Y. Shen, A. J. Shuhendler, D. Ye, J.-J. Xua and H.-Y. Chen, Chem. Soc. Rev., 2016, 45, 6725 RSC.
- Y. Morel, A. Irimia, P. Najechalski, Y. Kervella, O. Stephan, P. L. Baldeck and C. Andraud, J. Chem. Phys., 2001, 114, 5391 CrossRef CAS.
- Q. Zheng, S. K. Gupta, G. S. He, L.-S. Tan and P. N. Prasad, Adv. Funct. Mater., 2008, 18, 2770 CrossRef CAS.
- G. Williams, M. Hunt, B. Boehm, A. May, M. Taverne, D. Ho, S. Giblin, D. Read, J. Rarity, R. Allenspach and S. Ladak, Nano Res., 2018, 11, 845 CrossRef.
- Q. Geng, D. Wang, P. Chen and S.-C. Chen, Nat. Commun., 2019, 10, 2179 CrossRef PubMed.
- M. Gu, Q. Zhang and S. Lamon, Nat. Rev. Mater., 2016, 1, 16070 CrossRef CAS.
- J. M. Dos Santos, D. Hall, B. Basumatary, M. Bryden, D. Chen, P. Choudhary, T. Comerford, E. Crovini, A. Danos, J. De, S. Diesing, M. Fatahi, M. Griffin, A. K. Gupta, H. Hafeez, L. Hämmerling, E. Hanover, J. Haug, T. Heil, D. Karthik, S. Kumar, O. Lee, H. Li, F. Lucas, C. F. R. Mackenzie, A. Mariko, T. Matulaitis, F. Millward, Y. Olivier, Q. Qi, I. D. W. Samuel, N. Sharma, C. Si, L. Spierling, P. Sudhakar, D. Sun, E. Tankelevičiūtė, M. D. Tonet, J. Wang, T. Wang, S. Wu, Y. Xu, L. Zhang and E. Zysman-Colman, Chem. Rev., 2024, 124, 13736 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Shimomura and G. Konishi, Chem. – Eur. J., 2023, 29, e202301191 CrossRef CAS PubMed.
- M. Mahato, T. Sultana, R. Sahoo, S. Ahamed, N. Tohora, A. Maitia and S. K. Das, Phys. Chem. Chem. Phys., 2025, 27, 1366 RSC.
- Z. Kang, S.-C. Chen, Y. Ma, J. Wang and Q. Zheng, ACS Appl. Mater. Interfaces, 2017, 9, 24771 CrossRef CAS.
- A. A. Raheem, S. Kamaraj, V. Sannasi and C. Praveen, Org. Chem. Front., 2018, 5, 777 RSC.
- N. Altinolcek, A. Battal, C. Nur Vardalli, M. Tavasli, H. A. Yu, W. J. Peveler and P. J. Skabara, J. Mol. Struct., 2021, 1239, 130494 CrossRef CAS.
- G. Tang, W. Yang and J. Zhao, J. Phys. Chem. A, 2022, 126, 3653 CrossRef CAS PubMed.
- A. Karatay, H. Yılmaz, E. A. Yildiz, G. Sevinç, M. Hayvali, B. Boyacioglu, H. Unvere and A. Elmalia, Phys. Chem. Chem. Phys., 2022, 24, 25495 RSC.
- D. Cvejn, E. Michail, K. Seintis, M. Klikar, O. Pytela, T. Mikysek, N. Almonasy, M. Ludwig, V. Giannetas, M. Fakis and F. Bureš, RSC Adv., 2016, 6, 12819 Search PubMed.
- Y. Gong, G.-L. Hou, X. Bi, N. Kuthirummal, A. A. Teklu, J. Koenemann, N. Harris, P. Wei, K. Devera and M. Hu, J. Phys. Chem. A, 2021, 125, 1870 CrossRef CAS PubMed.
- X. Liang, B. Dong, H. Wang, Z. Zhang, S. Wang, J. Li, B. Zhao, Z. Li, Y. Xinga and K. Guo, J. Mater. Chem. C, 2022, 10, 7857 RSC.
- B. K. Kundu, G. Han and Y. Sun, J. Am. Chem. Soc., 2023, 145, 3535 CrossRef CAS PubMed.
- D. J. Stewart, R. Kannan, T. A. Grusenmeyer, J. M. Artz, S. L. Long, Z. Yu, T. M. Cooper, J. E. Haleya and L.-S. Tan, Phys. Chem. Chem. Phys., 2018, 20, 19398 RSC.
- P. Šimon, M. Klikar, Z. Burešová, C. Vourdaki, A. Katsidas, J. Tydlitát, J. Kulhánek, J. Zelenka, M. Fakis and F. Bureš, J. Mater. Chem. C, 2023, 11, 7252 RSC.
- M. Tromayer, P. Gruber, A. Rosspeintner, A. Ajami, W. Husinsky, F. Plasser, L. González, E. Vauthey, A. Ovsianikov and R. Liska, Sci. Rep., 2018, 8, 17273 CrossRef PubMed.
- M. Fecková, P. le Poul, F. Bureš, F. Robin-le Guen and S. Achelle, Dyes Pigm., 2020, 182, 108659 CrossRef.
- S. Achelle, M. Hodée, J. Massue, A. Fihey and C. Katan, Dyes Pigm., 2022, 200, 110157 CrossRef CAS.
- S. Achelle, J. Rodríguez-López, F. Bureš and F. Robin-le Guen, Chem. Rec., 2020, 20, 440 CrossRef CAS.
- S. Achelle, J. Rodríguez-López and F. Robin-le Guen, Org. Biomol. Chem., 2023, 21, 39 Search PubMed.
- M. Fecková, I. K. Kalis, T. Roisnel, P. le Poul, O. Pytela, M. Klikar, F. Robin-le Guen, F. Bureš, M. Fakis and S. Achelle, Chem. Eur. J., 2021, 27, 1145 Search PubMed.
- M. Taniguchi and J. S. Lindsey, Photochem. Photobiol., 2018, 94, 290 CrossRef CAS.
- V. Petropoulos, I. Georgoulis, C. Vourdaki, P. Hrobárik, I. Sigmundová, J. Nociarová, M. Maiuri, G. Cerullo and M. Fakis, Chem. Phys. Chem., 2023, 24, e202300127 CrossRef CAS.
- P. Hrobarik, V. Hrobarikova, I. Sigmundova, P. Zahradnik, M. Fakis, I. Polyzos and P. Persephonis, J. Org. Chem., 2011, 76, 8726 CrossRef CAS PubMed.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express., 2008, 16, 4029 CrossRef CAS.
- S. de Reguardati, J. Pahapill, A. Mikhailov, Y. Stepanenko and A. Rebane, Opt. Express., 2016, 24, 9053 CrossRef CAS PubMed.
- U. Subuddhi, S. Haldar, S. Sankararaman and A. K. Mishra, Photochem. Photobiol. Sci., 2006, 5, 459 CrossRef CAS.
- P. Suppan, J. Photochem. Photobiol., A, 1990, 50, 293 CrossRef CAS.
- C. Katan, F. Terenziani, O. Mongin, M. H. V. Werts, L. T. Porrès, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024 CrossRef CAS PubMed.
- L. Yan, X. Chen, Q. He, Y. Wang, X. Wang, Q. Guo, F. Bai, A. Xia, D. Aumiler, S. Vdović and S. H. Lin, J. Phys. Chem. A, 2012, 116, 8693 CrossRef CAS PubMed.
- F. Terenziani, C. LeDroumaguet, C. Katan, O. Mongin and M. Blanchard-Desce, Chem. Phys. Chem., 2007, 8, 723 CrossRef CAS.
- J. E. Lewis and M. Maroncelli, Chem. Phys. Lett., 1998, 282, 197 CrossRef CAS.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814 CrossRef CAS.
- M. I. Sluch, A. Godt, U. H. F. Bunz and M. A. Berg, J. Am. Chem. Soc., 2001, 123, 6447 CrossRef CAS.
- B. Dereka, A. Rosspeintner, R. Stężycki, C. Ruckebusch, D. T. Gryko and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 6029 CrossRef CAS PubMed.
- J. R. Lacowicz, Principles of Fluorescence Spectroscopy, Springer, 2006 Search PubMed.
- M. Jia, X. Ma, L. Yan, H. Wang, Q. Guo, X. Wang, Y. Wang, X. Zhan and A. Xia, J. Phys. Chem. A, 2010, 114, 7345 CrossRef CAS.
- M. Eugenio Vazquez, J. B. Blanco and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 1300–1306 CrossRef PubMed.
- N. Periasamy and A. S. R. Koti, Proc. Indian Natn. Sci. Acad., 2003, 69, A41 Search PubMed.
- P. K. Singh, M. Kumbhakar, H. Pal and S. Nath, J. Phys. Chem. B, 2010, 114, 5920 CrossRef CAS.
- A. K. Mora and S. Nath, J. Phys. Chem. B, 2019, 123, 8767 CrossRef CAS PubMed.
- S. R. Rather and P. Sen, J. Chem. Phys., 2013, 138, 084308 CrossRef PubMed.
- S. Santra, A. K. Mora and S. Nath, J. Photochem. Photobiol., A, 2023, 437, 114474 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311 CrossRef CAS.
- L. Reynolds, J. A. Gardecki, S. J. V. Frankland, M. L. Horng and M. Maroncelli, J. Phys. Chem., 1996, 100, 10337 CrossRef CAS.
- J. S. Beckwith, A. Rosspeintner, G. Licari, M. Lunzer, B. Holzer, J. Fröhlich and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 5878 CrossRef CAS PubMed.
- X. Niu, Z. Kuang, M. Planells, Y. Guo, N. Robertson and A. Xia, Phys. Chem. Chem. Phys., 2020, 22, 15743 RSC.
- N. A. Montgomery, G. J. Hedley, A. Ruseckas, J. C. Denis, S. Schumacher, A. L. Kanibolotsky, P. J. Skabara, I. Galbraith, G. A. Turnbull and I. D. W. Samuel, Phys. Chem. Chem. Phys., 2012, 14, 9176–9184 RSC.
- O. P. Varnavski, J. C. Ostrowski, L. Sukhomlinova, R. J. Twieg, G. C. Bazan and T. Goodson III, J. Am. Chem. Soc., 2002, 124, 1736–1743 CrossRef CAS.
- K. Seintis, D. Agathangelou, D. Cvejn, N. Almonasy, F. Bureš, V. Giannetas and M. Fakis, Phys. Chem. Chem. Phys., 2017, 19, 16485–16497 RSC.
- N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J.-L. Brédas, J. W. Perry, A. Pron, M. Kivala and K. Müllen, J. Phys. Chem. A, 2012, 116, 3781 CrossRef CAS PubMed.
- C. Sissa, V. Parthasarathy, D. Drouin-Kucma, M. H. V. Werts, M. Blanchard-Desce and F. Terenziani, Phys. Chem. Chem. Phys., 2010, 12, 11715 RSC.
- M. Drobizhev, N. S. Makarov, S. E. Tillo, T. E. Hughes and A. Rebane, Nat. Methods, 2011, 8, 393 CrossRef CAS.
- L. Li, Y.-P. Tian, J.-X. Yang, P.-P. Sun, J.-Y. Wu, H.-P. Zhou, S.-Y. Zhang, B.-K. Jin, X.-J. Xing, C.-K. Wang, M. Li, G.-H. Cheng, H.-H. Tang, W.-H. Huang, X.-T. Tao and M.-H. Jiang, Chem. – Asian J., 2009, 4, 668 CrossRef CAS PubMed.
- B. Liu, X.-L. Hu, J. Liu, Y.-D. Zhao and Z.-L. Huang, Tetrahedron Lett., 2007, 48, 5958 CrossRef CAS.
- Q. Zhang, L. Luo, H. Xu, Z. Hu, C. Brommesson, J. Wu, Z. Sun, Y. Tian and K. Uvdal, New J. Chem., 2016, 40, 3456 RSC.
- A. Rebane, M. Drobizhev, N. S. Makarov, G. Wicks, P. Wnuk, Y. Stepanenko, J. E. Haley, D. M. Krein, J. L. Fore, A. R. Burke, J. E. Slagle, D. G. McLean and T. M. Cooper, J. Phys. Chem. A, 2014, 118, 3749 CrossRef CAS PubMed.
- A. Rebane, M. Drobizhev, N. S. Makarov, E. Beuerman, J. E. Haley, D. M. Krein, A. R. Burke, J. L. Flikkema and T. M. Cooper, J. Phys. Chem. A, 2011, 115, 4255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00589b |
| This journal is © the Owner Societies 2025 |