
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of imide side-chain functionality in the doping characteristics of naphthalenediimide derivatives as electron transport materials†
Wai Kin
Yiu
 ,
Lewis
Mackenzie
,
Dylan
Wilkinson
,
Marcin
Giza
,
Benjamin
Vella
,
Michele
Cariello
,
Stephen
Sproules
,
Graeme
Cooke
* and
Pablo
Docampo
*
,
Lewis
Mackenzie
,
Dylan
Wilkinson
,
Marcin
Giza
,
Benjamin
Vella
,
Michele
Cariello
,
Stephen
Sproules
,
Graeme
Cooke
* and
Pablo
Docampo
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Graeme.Cooke@glasgow.ac.uk; Pablo.Docampo@glasgow.ac.uk
First published on 27th May 2025
Abstract
Achieving effective doping in n-type organic molecular charge transport materials is critical for the development of high-performance optoelectronic devices. However, the role of side-chains in doping reactions remains incompletely understood in some systems. This study focuses on naphthalenediimide (NDI) derivatives, which offer simple synthetic protocols and potentially lower costs compared to traditional fullerene-derived materials. In particular, we explore two functionalised NDI derivatives, comparing one with polar ethylene glycol side-chains (NDI-G) to a non-polar variant with branched alkyl side-chains (NDI-EtHx). Our results show that the effectiveness and speed of the doping reaction with (4-(1,3-dimethyl-2,3-dihydro-1H-benzoimidazol-2-yl)phenyl)dimethylamine (N-DMBI) is much higher with the more polar NDI-G derivative. We postulate that this arises partly from the closer interactions between the dopant and the NDI molecule, facilitated by the polar glycol side-chains. As a result, thin films reach conductivities exceeding 10−2 S cm−1. We additionally demonstrate their incorporation into efficient perovskite solar cells, demonstrating the effectiveness of the doping process. We investigate this process with a combination of spectroscopy and density functional theory (DFT) modelling, showing that a complex is likely formed between the resulting N-DMBI cation and the NDI radical anion which then promotes electron transfer to a neutral NDI molecule, thereby generating free charge in the film. These findings underscore the importance of synthetic design on the doping behaviour, with the incorporation of ethylene glycol side-chains emerging as an effective strategy to achieve better electrical conductivity for NDI based systems.
Introduction
Doping is an effective approach to enhance the conductivity of organic semiconductors by generating free charges through interactions between dopant species and the host organic semiconductor.1 This method has been particularly effective in fullerene-based electron transport materials (ETMs) such as C60 and phenyl-C61-butyric acid methyl ester (PCBM), enabling conductivities exceeding 10 S cm−1.2 However, achieving optimal doping is essential to meet the requirements of specific device applications. For instance, excessively high conductivity in transistors can lead to current leakage and reduced low on/off ratios,3 which explains why doped fullerene ETMs in perovskite solar cells typically exhibit conductivity within the range of 10−3–10−4 S cm−1.4,5 Recently, there has been increasing interest in non-fullerene alternatives such as naphthalenediimide (NDI) derivatives due to their simpler synthesis and tuneable energy levels.6–8 These characteristics contribute to reduced chemical usage, lower energy consumption, and decreased production costs. While n-type doping has improved NDI conductivity up to 10−1 S cm−1,9 the doping mechanism in these systems is not yet fully understood.1NDI-based materials face challenges with poor solubility in organic solvents, resulting in poor film morphology and an increased density of defects.10,11 These defects can act as trapping sites for charge carriers, hindering efficient charge transport.12,13 The solubility issue of NDI-based molecules can be addressed by synthetically modifying the imide or naphthalene unit side-chains, which do not require complicated synthesis procedures.14,15 These simple modifications, however, can also impact the material's optoelectronic properties.16,17 For instance, Liu et al. demonstrated that the polarity and the length of glycol side-chain in fullerene derivatives influence performance in n-type organic thermoelectric materials,16 and Vijayakumar et al. investigated how the length of alkyl side-chain affects doping kinetics and charge transport properties in poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophene) (PBTTT) films.18 A systematic investigation of how different side-chains affect the reactivity between host molecules and molecular dopants is generally lacking. Further research is needed to understand how specific synthetic modifications, particularly the choice of side-chains, not only enhance solubility in solvent, but also influence the local doping environment and control the doping behaviour.
In this study, we have developed two NDI-based molecules featuring ethylene glycol (NDI-G) and ethylhexyl (NDI-EtHx) side-chains. Both moieties enhance the solubility in organic solvents but exhibit different reaction kinetics upon addition of (4-(1,3-dimethyl-2,3-dihydro-1H-benzoimidazol-2-yl)phenyl)dimethylamine (N-DMBI) as the n-type dopant. We investigate the origin of the conductivity differences of doped and undoped species as well as the optical and redox properties of these systems. We use UV-vis spectroscopy and cyclic voltammetry (CV), together with density functional theory (DFT) modelling to probe the doping process of NDI derivatives with N-DMBI. Utilising UV-vis and conductivity measurements, we have shown that NDI-G exhibited a more effective doping reaction with N-DMBI, resulting in shorter doping times and superior conductivity. Lastly, we have fabricated perovskite solar cells using NDI-G and NDI-EtHx as ETLs and report that the choice of side-chain critically impacts device performance.
Results and discussion
In this work, we focus on the impact of the side-chain environment around NDI derivatives has on the doping efficiency with N-DMBI. We postulate that manipulating the polarity of side-chains attached to the molecules will allow the control of the effectiveness and rate of the doping reaction. Here, we have attached an ethylene glycol side-chain to create a more polar environment in the NDI-G derivative, while an ethylhexyl side-chain was incorporated in NDI-EtHx to create a less polar one.The full synthesis of NDI-G and NDI-EtHx is reported in the ESI† and chemical structures of NDI-G, NDI-EtHx, and N-DMBI are shown in Fig. 1a. The solution optical properties of NDI-G and NDI-EtHx were evaluated using UV-vis spectroscopy to characterise the effect of the side-chain functionalisation has on the light absorption properties. Our results show similar spectra for both molecules with no significant influence of side-chains, see Fig. 1b, indicating that this type of functionalization does not significantly influence the electronic properties of their ‘as-synthesised’ form.
 | ||
| Fig. 1 (a) Structures of NDI-G, NDI-EtHx and N-DMBI; (b) solution UV-vis absorption spectra of pristine NDI-G and NDI-EtHx in chlorobenzene, 1 × 10−5 M; (c) cyclic voltammetry measurements of NDI-G and NDI-EtHx with ferrocene as reference (1 × 10−4 M in CH2Cl2; scan rate 0.1 mV s−1); and (d) conductivity measurements of pristine NDI-G, NDI-EtHx and PCBM self-doped in air. | ||
CV was used to determine the solution redox properties of NDI-G and NDI-EtHx (Fig. 1c), as well as the dopant, N-DMBI (Fig. S6, ESI†). Both NDI derivatives exhibited two pseudoreversible reduction waves by comparing with ferrocene as internal reference. The redox waves correspond to two one-electron reductions forming radical anion and dianion states, respectively. The estimated electron affinities (EAs) of NDI-G and NDI-EtHx are −3.67 eV and −3.70 eV, respectively, which are shallower than PCBM (−3.92 eV).19
DFT calculations were performed on an analogue of NDI-G and NDI-EtHx, in which the side chain groups were truncated to methyl units (methyl-NDI) to simplify the systems, facilitate convergence of the geometry optimisations, and focus on the interaction between the dopants and the core NDI moiety. This adjustment is based on the minimal effect that side-chains generally have on the electronic properties of the NDI core.20 The gas phase highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of methyl-NDI were calculated to be −7.22 eV and −3.56 eV, respectively. In combination with the CV results, this indicates that NDIs have similar energy levels to fullerenes derivatives, and hence that NDI-G and NDI-EtHx have the potential to serve in a similar range of optoelectronic device applications from the point of view of energetic alignment. Conductivity was extracted from the resistance values of standard current–voltage curve employing pristine NDI derivatives on patterned-ITO substrates with an inter-digitated contact configuration. NDI derivatives exhibit lower conductivity (∼× 10−7 S cm−1) compared to PCBM (1.57 × 10−6 S cm−1), as shown in Fig. 1d.
To enhance the conductivity of the NDI derivatives, N-DMBI has been employed as an n-type dopant. N-DMBI has well-documented effectiveness in catalysing the reduction of organic compounds21 and is able to facilitate hydrogen and/or electron transfer reactions by forming radicals.22,23 The HOMO of N-DMBI is −4.67 eV, which is deeper than the LUMO of PCBM, NDI-G and NDI-EtHx. As a result, direct electron transfer between N-DMBI and NDI derivatives is not possible due to energy level misalignment. Furthermore, C–H bond dissociation at room temperature is energetically unfavourable due to its high enthalpy change. Hence, photo24 or thermal activation24,25 is required for effective doping.
To confirm the NDI radical anion formation in the doping process, electron spin resonance (ESR) and UV-vis absorption measurements were recorded upon addition of 10 mol% and 30 mol% N-DMBI to NDI-G and NDI-EtHx, respectively. Upon addition of N-DMBI, ESR shows a strong paramagnetic signal with a g value of 2.004 and hyperfine features were observed for NDI-EtHx. These features were less distinct in the NDI-G moiety, possibly due to its larger molecular structure, which slows molecular tumbling and obscures the hyperfine details. The ESR signal further confirms the formation of an NDI radical anion, while there is no appreciable radical signal obtained in the pristine NDI-G and NDI-EtHx samples, as shown in Fig. 2a and b. Experimental solid-state UV-vis absorption spectra for the pristine and N-DMBI doped NDI derivatives are shown in Fig. 2c. Interestingly, the derivatives display different absorption features upon doping (ca. 450 nm versus 475 nm), suggesting that either different reduced species are being formed or that the local environment provided by the differing side-chains shifts the absorption for the NDI radical anion.26,27
 | ||
| Fig. 2 X-band EPR spectrum of pristine and after addition of N-DMBI for (a) NDI-G (3.57 × 10−2 M) and (b) NDI-EtHx (4.08 × 10−2 M) recorded in chlorobenzene; (c) UV-vis absorption spectra of pristine and after addition of N-DMBI for both NDI derivatives (1 × 10−5 M). | ||
Generally, the doping process in molecular electron transporters is described as a two-step process: (i) ionisation of the dopant by transferring a charge to the semi-conducting host material to form the complex and (ii) dissociation of the complex generating free charge carriers by transferring the electron to a neutral nearby host.28 However, step (ii) is not at all obvious in an organic semiconductor due to the strong Coulombic binding energy between host material and additive.28 Here, unlike n-dopants that undergo direct dopant to acceptor electron transfer, N-DMBI doping operates by a more complicated process that may be specific to the acceptor used and the conditions employed. Therefore, a single mechanism will probably not be able to account for all N-DMBI/acceptor combinations.29
The initial step for N-DMBI doping reactions will likely result from C–H bond cleavage and the general consensus appears to be that initial hydride [H−] transfer rather than other mechanisms such as hydrogen radical [H˙] transfer to the acceptor plays the most significant role for most N-DMBI/acceptor combinations.30,31 However, the initial step is not straightforward as other studies have indicated that electron transfer may occur prior to C–H bond homolysis for powerful electron acceptors that have poor affinity for hydride or hydrogen transfer.32,33 Furthermore, when NDI is used as an acceptor, previous studies have shown that N-DMBI gives rise to very sluggish hydride transfer and that the hydrogen transfer may not be energetically favourable.30,34,35 Nevertheless, regardless of which species is initially formed, it is clear from ESR that the radical anion states of NDI-EtHx and NDI-G are formed upon doping with N-DMBI. Presumably this occurs by subsequent reactions of the NDI hydrides (NDI-H−) (or other species) formed initially.
DFT modelling was also used to investigate the interactions of the N-DMBI cation and methyl-NDI radical anion that likely result from the doping reaction. The electrostatic potential (ESPs) and spin density maps of the complex indicated that the negative charge is localized on more electronegative oxygen and nitrogen atoms of the NDI units (Fig. S7a, ESI†), and the unpaired electron is highly delocalized on the NDI unit (Fig. S7b, ESI†). The calculated binding energies of [methyl-NDI−/N-DMBI+]˙ and π–π dimer of methyl-NDI are 56.3 kcal mol−1 (2.44 eV) and 0.4 kcal mol−1 (0.02 eV), respectively, showing a stronger binding force of the complex than most organic materials (typically 0.5 eV).36 The [methyl-NDI−/N-DMBI+]˙ is stable and the high coulombic binding energy suggests it is unlikely to dissociate into free charge carriers. We have also modelled [NDI-G−/N-DMBI+]˙ and [NDI-EtHx−/N-DMBI+]˙ to reveal whether the gas phase calculations can reveal any information regarding the role the side-chain moieties play in the complexation process (Fig. S8, ESI†). The calculated binding energies were slightly lower than that observed for [methyl-NDI−/N-DMBI+]˙ ([NDI-G−/N-DMBI+]˙ = 38.52 kcal mol−1 (1.67 eV) and [NDI-EtHx−/N-DMBI+]˙ = 42.57 kcal mol−1 (1.85 eV)).
The computed energy levels of [methyl-NDI−/N-DMBI+]˙, [NDI-G−/N-DMBI+]˙ and [NDI-EtHx−/N-DMBI+]˙ were compared. In all cases the calculated LUMO level of the neutral NDI derivative is deeper than the singly occupied molecular orbital (SOMO) of its complex with N-DMBI, indicating a potential for electron transfer from complex to neutral NDI unit, as shown in Fig. 3 and Fig. S8 (ESI†). Furthermore, the SOMO/LUMO gaps for [NDI-G−/N-DMBI+]˙ (0.4 eV) and [NDI-EtHx−/N-DMBI+]˙ (0.4 eV) and their respective neutral NDI species were slightly smaller than that obtained for [methyl-NDI−/N-DMBI+]˙ (0.9 eV). Therefore, the DFT modelling suggests that the N-DMBI cation and radical anion species of the NDI derivatives form stable complexes due to their strong binding energies.37 Importantly, the energy alignment between the complexes and their neural NDI derivatives enables an electron hopping process, and thus provides a route for effective n-type doping.
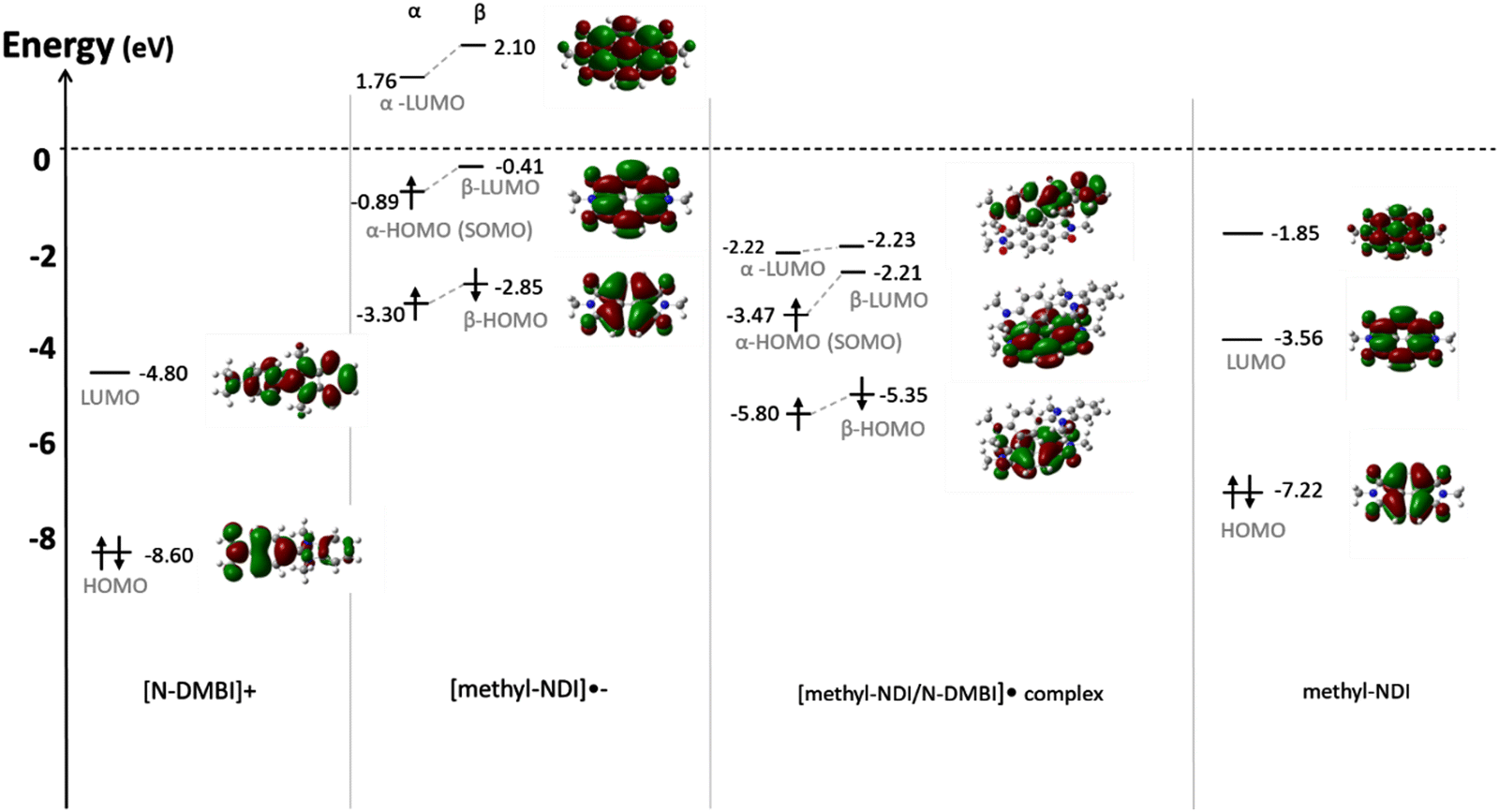 | ||
| Fig. 3 DFT predicted HOMO/LUMO maps and energies of [N-DMBI]+, [methyl-NDI]˙−, [methyl-NDI−/N-DMBI+]˙ complex and neutral methyl-NDI. | ||
We next conducted a series of solid-state UV-vis absorption (Fig. 4) and conductivity (Fig. 5 and Tables S1, S2, ESI†) measurements of NDI-G and NDI-EtHx in terms of additive concentration and reaction time. The UV-vis absorption spectra in Fig. 4a and b illustrate the differing reaction times between the two NDI derivatives upon the addition of 50 mol% N-DMBI. Similar to the solution studies, new absorption peaks at 450 nm for NDI-G, and at 475 nm for NDI-EtHx are observed. NDI-G exhibited faster reaction kinetics than NDI-EtHx, with NDI-G displaying an absorption peak at 2 hours whereas it took approximately 24 hours NDI-EtHx to display a new absorption band. This slower doping process for NDI-EtHx is likely due to sluggish hydride transfer. In contrast, the glycol side-chain in NDI-G enhances its polarity and improves miscibility within the host/dopant blend.16 This enhanced compatibility between NDI-G and N-DMBI molecules contributed to a more effective doping process compared to NDI-EtHx.16,38–40 This observation aligns with previous reports, which have shown that an increase in reduction peak intensity in UV-vis absorption spectra correlates with the progression of the doping reaction time.4,41
 | ||
| Fig. 4 UV-vis absorption spectra of (a) NDI-G and (b) NDI-EtHx in film on glass substrates with 50 mol% of N-DMBI in the presence of reaction times ranging from 0 to 24 hours; (c) NDI-G and (d) NDI-EtHx in film on glass substrates in the presence of 0 mol% to 50 mol%. | ||
 | ||
| Fig. 5 Logarithmic plot of conductivity measurements for N-DMBI doped in NDI-G and NDI-EtHx at (a) reaction time ranging from 0 to 24 hours (50 mol% N-DMBI doping) and (b) ranging from 0 mol% to 50 mol% (reaction time: 2 hours for NDI-G and 16 hours for NDI-EtHx) (each data point represents the average of three samples with error bars). | ||
However, the intensity of the UV-vis absorption spectrum of doped NDI-G declined after peaking at 2 hours, indicating a lack of stability of the resulting species. This decline is likely due to the highly reactive nature of organic radicals, which often leads to instability under ambient condition.42 In contrast, NDI-EtHx exhibited a continuous increase in radical formation without a similar decline in intensity. Additional absorption measurements (Fig. S9, ESI†) confirm this radical instability, showing a reduction in the radical peak upon exposure to air.
The investigation was extended to demonstrate how increasing the mole percentages from 0 mol% to 50 mol% of N-DMBI influenced the doping process for NDI-G and NDI-EtHx. The new absorption peaks appeared from 20 mol% of N-DMBI doping for NDI-G and NDI-EtHx. As the concentration of N-DMBI was increased, the equilibrium position shifted towards the products, demonstrating a more pronounced doping reaction at higher dopant concentrations up to 50 mol%.
Conductivity measurements of NDI derivatives after the addition of 50 mol% N-DMBI added were performed to compare the doping efficiency with different side-chains, as shown in Fig. 5a. For NDI-G, the initial conductivity was low at 2.52 × 10−7 S cm−1 and significantly improved to 4.89 × 10−4 S cm−1 after 1 h. This enhancement reached its maximum value of 1.09 × 10−3 S cm−1 at the 2 h mark. After this point, the conductivity declined to 5.8 × 10−5 S cm−1 after 24 hours. In contrast, for NDI-EtHx, a 1 h reaction with N-DMBI led to an improvement in conductivity from an initial value of 2.35 × 10−7 S cm−1 to 3.25 × 10−6 S cm−1. The conductivity continued to increase to around 10−5 S cm−1 and remained constant up to 24 hours. The results align with UV-vis absorption findings and suggest that the glycol side-chain provides a better environment for the n-type doping, allowing NDI-G to achieve maximum conductivity more quickly than NDI-EtHx. However, it is noteworthy that the conductivity in NDI-G dropped if the reaction was allowed to proceed for longer times.
Fig. 5b shows the conductivity for NDI-G and NDI-EtHx with varying mole percentages of N-DMBI, ranging from 0 mol% to 50 mol%, with reaction times determined from the maxima in Fig. 4a and b (NDI-G for 2 hours and NDI-EtHx for 16 hours). The initial conductivity of pristine NDI-G and NDI-EtHx ranged from 2 to 3 × 10−7 S cm−1. In the case of NDI-EtHx, the conductivity increased to 6.21 × 10−6 S cm−1 with 5 mol% N-DMBI and reached the maximum of 2.38 × 10−5 S cm−1 at 30 mol%. However, the conductivity started to decline beyond this optimal additive concentration level, from 1.82 × 10−5 S cm−1 at 40 mol% and further decreasing to 1.40 × 10−5 S cm−1 at 50 mol%. In the case of NDI-G, the conductivity dramatically increased to 1.84 × 10−3 S cm−1 with 2 mol% N-DMBI and peaked at 1.00 × 10−2 S cm−1 with 10 mol% N-DMBI doping. However, a further increase in additive concentration led to a decline in conductivity, which dropped to 4.50 × 10−4 S cm−1 at 50 mol%.
These results indicate that the successful doping of the NDI derivatives, with optimal conditions for NDI-G yielding conductivity values up to 10−2 S cm−1. However, excessive additive concentration had a detrimental effect. High additive concentration levels likely introduce trap states within the material,43,44 which can capture electrons and lead to a reduction in the electron conductivity45,46 and mobility,43,45 as well as increased recombination.47 Consequently, this behaviour reduces the number of free charges within the material. Based on our results, the optimised doping conditions for these materials are: 10 mol% N-DMBI for a 2 h reaction for NDI-G, and 30 mol% N-DMBI for a 16 h reaction for NDI-EtHx. Notably, the doping kinetics of NDI-G are competitive with other materials, achieving peak conductivity within 3 hours.41,48
To evaluate the effectiveness of n-doping of NDI derivatives and their suitability in hybrid optoelectronic devices, we fabricated inverted PSCs with a configuration of glass/ITO/MeO-2PACz/Al2O3 NPs/perovskite/ETL/BCP/Ag. The performance of PSCs incorporating the pristine and doped NDI derivatives was compared to PCBM-based PSCs. Fig. 6 illustrates the J–V curves for these solar cells, while Table 1 provides a summary of their photovoltaic performance. Doping NDI-G with N-DMBI successfully enhances both conductivity and photovoltaic performance, with power conversion efficiency (PCE) increasing from 10.44% to 14.12%. After doping, the short circuit current density (Jsc) of NDI-G PSCs rises from 21.25 mA cm−2 to 22.15 mA cm−2, approaching the performance of PCBM-based PSCs. The improved conductivity in doped NDI-G facilitates better charge extraction towards the electrode, resulting in higher Jsc. Additionally, the fill factor (FF) of doped NDI-G PSCs increased from 0.53 to 0.68 compared to undoped NDI-G PSCs. This enhancement is associated with a reduction of the series resistance, as evidenced by the slopes of the J–V curves, see Fig. 6. After doping, the values extracted for the series resistance are similar to PCBM-based PSCs, demonstrating the success of the approach. In contrast, doped NDI-EtHx does not exhibit any improvement in device performance. This lack of improvement is likely a result of the less effective doping process which does not yield a high enough conductivity value to overcome the detrimental effects of the addition of the dopant molecules.
 | ||
| Fig. 6 J–V characteristics curves of PCBM, pristine and doped of NDI-G and NDI-EtHx based PSCs (lines represent to fitting curves of each J–V curves). | ||
| ETL | N-DMBI (mol%) | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|
| PCBM | — | 21.86 ± 0.38 | 1.07 ± 0.01 | 0.78 ± 0.02 | 18.26 ± 0.69 |
| NDI-G | 0 | 21.25 ± 0.58 | 0.93 ± 0.06 | 0.53 ± 0.02 | 10.44 ± 0.80 |
| 10 | 22.15 ± 1.14 | 0.94 ± 0.04 | 0.68 ± 0.07 | 14.12 ± 1.05 | |
| NDI-EtHx | 0 | 18.56 ± 0.63 | 1.00 ± 0.07 | 0.50 ± 0.02 | 9.18 ± 0.83 |
| 30 | 18.47 ± 1.43 | 0.95 ± 0.04 | 0.48 ± 0.02 | 8.45 ± 0.77 |
However, despite the higher conductivity in doped NDI-G compared to PBCM, its overall performance is still lower than this reference system. Clearly, there are more elements at play at the interface than simple energy-level related arguments. In particular, there is a growing body of knowledge highlighting that fullerenes derivatives play additional roles to facilitate charge extraction, for instance in the form of surface passivation.49 This effect is attributed to the interaction at the perovskite/PCBM interface, which reduces interfacial recombination and lowers the interfacial energy barrier.50 Similar benefits have been observed in regular PSCs where C60 self-assembled monolayers (SAMs) were used to passivate SnO2, reducing interfacial defects.51 This defect mitigation is further supported by minimized hysteresis in PCBM-based devices compared to NDI-based devices, as shown in Fig. S10 (ESI†). Moreover, PCBM enhances performance by increasing Jsc, raising the shunt resistance (RSH), and decreasing the series resistance (Rs), all of which contribute to an improved FF.
To further explore the reasons behind these performance differences, the film morphology was examined using scanning electron microscopy (SEM) (Fig. 7). No significant morphological changes were observed between PCBM, pristine NDI-G, and NDI-EtHx. After doping, the morphology of NDI-G remained unchanged, suggesting that the enhancement in solar cell performance can be attributed to increased charge transport as a result of the doping process. In contrast, doped NDI-EtHx exhibited the formation of needle-shaped features, suggesting that the dopant molecule may influence molecular packing or induce aggregation. The polarity difference between NDI-EtHx and N-DMBI likely leads to dopant residue aligning with the alkyl side-chain in crystalline regions. This disrupts the π–π stacking of NDI molecules or dopant self-aggregates, as observed in SEM images.16 This behaviour is similar to that observed in reported PCBM systems,52,53 where large aggregate formation negatively impacts solar cell performance. It is also possible that the steric hindrance of the alkyl side-chains in NDI-EtHx contributes to the aggregation. This explains why the open circuit voltage (Voc) of doped NDI-EtHx decreased from 1.00 V to 0.95 V, likely due to surface recombination caused by the needle-shaped features, whereas NDI-G maintained a consistent Voc value.
 | ||
| Fig. 7 SEM images of (a) PCBM; (b) pristine NDI-G; (c) pristine NDI-EtHx, (d) doped NDI-G and (e) doped NDI-EtHx. | ||
Conclusions
In this study, we have investigated two non-fullerene ETMs based on NDI analogues with solubility-enhancing side-chains: glycol (NDI-G) and alkyl (NDI-EtHx). The two NDI derivatives exhibit similar optical and electrochemical properties. Their initial pristine conductivities were low, and to address this, we employed n-type doping using N-DMBI as the dopant. However, the HOMO of N-DMBI is deeper than the LUMO of the NDI derivatives and thermal activation was required to generate the NDI radical anion species which was confirmed by EPR. DFT calculations suggest that complex formation occurs between the resulting N-DMBI cation and the NDI radical anion driven by strong binding energies. Importantly, energy alignment between the complex and neutral NDI molecules enables effective electron transfer to the latter, ultimately generating free charges. Through a series of UV-vis absorption and conductivity measurements, we observed that the polar glycol groups in NDI-G facilitate a faster doping reaction compared to the non-polar alkyl chains in NDI-EtHx. This difference is largely attributed to the polarity compatibility between the glycol side-chains and dopant within the film matrix, which promotes intermolecular interactions and enhances the doping reaction. Our optimised NDI-G doped material achieved a conductivity exceeding 10−2 S cm−1. Integrating these derivatives in PSCs increased the PCE from 10.44% to 14.12% after N-DMBI doping, primarily by reducing the Rs. Although NDI-G films outperform PCBM in terms of conductivity, the overall devices are less efficient than those with PCBM due to higher interfacial recombination which also leads to increased hysteresis. This indicates that interface passivation is more critical than reducing Rs in the charge extraction layer. Overall, these findings further underscore the importance of side-chain engineering in the design of new dopant/semiconductor combinations.Author contributions
WKY and BV performed the analytical and device studies. LM, DW and MC synthesised and purified NDI-G and NDI-EtHx. LM and DW performed DFT calculations. MG performed SEM measurements. SS recorded the ESI spectra. GC and PD conceived and supervised the research project. All authors contributed to the writing and reviewing of this manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interests.Acknowledgements
WKY and MG thank the University of Glasgow for a College of Science and Engineering PhD scholarship. GC thanks Leverhulme Trust for a Research Fellowship and the EPSRC (EP/E036244/1). MC, DW and GC thank EPSRC (EP/V027425/1). PD acknowledges EP/T010568/1.References
- T. H. Kim, J. H. Kim and K. Kang, Jpn J. Appl. Phys., 2023, 62, SE0803 CrossRef CAS
.
- J. Liu, B. van der Zee, R. Alessandri, S. Sami, J. Dong, M. I. Nugraha, A. J. Barker, S. Rousseva, L. Qiu, X. Qiu, N. Klasen, R. C. Chiechi, D. Baran, M. Caironi, T. D. Anthopoulos, G. Portale, R. W. A. Havenith, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Nat. Commun., 2020, 11, 5694 CrossRef CAS PubMed
- J. Song, F. J. Xu, X. D. Yan, F. Lin, C. C. Huang, L. P. You, T. J. Yu, X. Q. Wang, B. Shen, K. Wei and X. Y. Liu, Appl. Phys. Lett., 2010, 97, 232106 CrossRef
- L. Hu, T. Liu, J. Duan, X. Ma, C. Ge, Y. Jiang, F. Qin, S. Xiong, F. Jiang, B. Hu, X. Gao, Y. Yi, Y. Zhou, L. Hu, T. F. Liu, J. S. Duan, Y. Y. Jiang, F. Qin, S. X. Xiong, F. Y. Jiang, B. Hu, Y. H. Zhou, X. Y. Ma, Y. P. Yi, C. W. Ge and X. K. Gao, Adv. Funct. Mater., 2017, 27, 1703254 CrossRef
- Z. Wang, D. P. McMeekin, N. Sakai, S. van Reenen, K. Wojciechowski, J. B. Patel, M. B. Johnston, H. J. Snaith, Z. Wang, D. P. McMeekin, N. Sakai, S. van Reenen, K. Wojciechowski, J. B. Patel, M. B. Johnston and H. J. Snaith, Adv. Mater., 2017, 29, 1604186 Search PubMed
- S. K. Jung, J. H. Heo, D. W. Lee, S. C. Lee, S. H. Lee, W. Yoon, H. Yun, S. H. Im, J. H. Kim and O. P. Kwon, Adv. Funct. Mater., 2018, 28, 1800346 Search PubMed
- M. Korzec, S. Kotowicz, K. Łaba, M. Łapkowski, J. G. Małecki, K. Smolarek, S. Maćkowski and E. Schab-Balcerzak, Eur. J. Org. Chem., 2018, 1756–1760 CrossRef CAS
- D. Zhao, Q. Jiang, Y. Jia, J. Zhou, N. Zheng, D. Hu and Y. Ma, Mater. Today Energy, 2021, 21, 100710 CrossRef CAS
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed
- D. Wang, T. Ye and Y. Zhang, J. Mater. Chem. A, 2020, 8, 20819–20848 RSC
- S. K. Jung, J. H. Heo, D. W. Lee, S. H. Lee, S. C. Lee, W. Yoon, H. Yun, D. Kim, J. H. Kim, S. H. Im and O. P. Kwon, ChemSusChem, 2019, 12, 224–230 CrossRef CAS PubMed
- J. L. Wu, W. K. Huang, Y. C. Chang, B. C. Tsai, Y. C. Hsiao, C. Y. Chang, C. T. Chen and C. T. Chen, J. Mater. Chem. A, 2017, 5, 12811–12821 RSC
- P. Karuppuswamy, C. Hanmandlu, K. M. Boopathi, P. Perumal, C. C. Liu, Y. F. Chen, Y. C. Chang, P. C. Wang, C. S. Lai and C. W. Chu, Solar Energy Mater. Solar Cells, 2017, 169, 78–85 CrossRef CAS
- J. Hyuck Heo, S.-C. Lee, S.-K. Jung, O.-P. Kwon and S. H. Im, J. Mater. Chem. A, 2017, 13, 20615–20622 Search PubMed
- L. M. Svirskaite, S. Mandati, N. Spalatu, V. Malinauskiene, S. Karazhanov, V. Getautis and T. Malinauskas, Synth. Met., 2022, 291, 117214 CrossRef CAS
- J. Liu, L. Qiu, G. Portale, S. Torabi, M. C. A. Stuart, X. Qiu, M. Koopmans, R. C. Chiechi, J. C. Hummelen, L. Jan and A. Koster, Nano Energy, 2018, 52, 183–191 CrossRef CAS
- M. Comin, V. Lemaur, A. Giunchi, D. Beljonne, X. Blase and G. D’Avino, J. Mater. Chem. C, 2022, 10, 13815–13825 RSC
- V. Vijayakumar, E. Zaborova, L. Biniek, H. Zeng, L. Herrmann, A. Carvalho, O. Boyron, N. Leclerc and M. Brinkmann, ACS Appl. Mater. Interfaces, 2019, 11, 4942–4953 Search PubMed
- M. Lenes, G.-J. A. H. Wetzelaer, F. B. Kooistra, S. C. Veenstra, J. C. Hummelen and P. W. M. Blom, Adv. Mater., 2008, 20, 2116–2119 CrossRef CAS
- M. Khalid, M. Khan, K. Mahmood, M. Arshad, M. Imran, A. A. C. Braga and R. Hussain, Sci. Rep., 2022, 12, 20220 CrossRef CAS PubMed
- P. Wei, J. H. Oh, G. Dong and Z. Bao, J. Am. Chem. Soc., 2010, 132, 8852–8853 CrossRef CAS PubMed
- D. D. Tanner and J. J. Chen, J. Org. Chem., 1989, 54, 3842–3846 Search PubMed
- D. D. Tanner, J. J. Chen, L. Chen and C. Luelo, J. Am. Chem. Soc., 1991, 113, 8074–8081 CrossRef CAS
- O. Bardagot, C. Aumaître, A. Monmagnon, J. Pécaut, P.-A. Bayle and R. Demadrille, Appl. Phys. Lett., 2021, 118, 203904 CrossRef CAS
- S. Riera-Galindo, A. Orbelli Biroli, A. Forni, Y. Puttisong, F. Tessore, M. Pizzotti, E. Pavlopoulou, E. Solano, S. Wang, G. Wang, T. P. Ruoko, W. M. Chen, M. Kemerink, M. Berggren, G. Di Carlo and S. Fabiano, ACS Appl. Mater. Interfaces, 2019, 11, 37981–37990 CrossRef CAS PubMed
- Y. S. Shi, D. D. Yang, T. Xiao, Z. G. Xia, Y. H. Fang and X. J. Zheng, Chem. Eng. J., 2023, 472, 145152 CrossRef CAS
- S. Caby, L. M. Bouchet, J. E. Argüello, R. A. Rossi and J. I. Bardagi, ChemCatChem, 2021, 13, 3001–3009 CrossRef CAS
- A. Mityashin, Y. Olivier, T. Van Regemorter, C. Rolin, S. Verlaak, N. G. Martinelli, D. Beljonne, J. Cornil, J. Genoe and P. Heremans, Adv. Mater., 2012, 24, 1535–1539 CrossRef CAS PubMed
- A. D. Scaccabarozzi, A. Basu, F. Aniés, J. Liu, O. Zapata-Arteaga, R. Warren, Y. Firdaus, M. I. Nugraha, Y. Lin, M. Campoy-Quiles, N. Koch, C. Müller, L. Tsetseris, M. Heeney and T. D. Anthopoulos, Chem. Rev., 2022, 122, 4420–4492 CrossRef CAS PubMed
- S. Jhulki, H. I. Un, Y. F. Ding, C. Risko, S. K. Mohapatra, J. Pei, S. Barlow and S. R. Marder, Chemistry, 2021, 7, 1050–1065 CrossRef CAS
- B. D. Naab, S. Guo, S. Olthof, E. G. B. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, J. Am. Chem. Soc., 2013, 135, 15018–15025 CrossRef CAS PubMed
- X. Q. Zhu, M. T. Zhang, A. Yu, C. H. Wang and J. P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed
- S. Yamamoto, T. Sakurai, L. Yingjin and Y. Sueishi, Phys. Chem. Chem. Phys., 1999, 1, 833–837 RSC
- B. D. Naab, S. Zhang, K. Vandewal, A. Salleo, S. Barlow, S. R. Marder, Z. Bao, B. D. Naab, Z. Bao, S. Zhang, S. Barlow, S. R. Marder, K. Vandewal and A. Salleo, Adv. Mater., 2014, 26, 4268–4272 CrossRef CAS PubMed
- K. Al Kurdi, S. A. Gregory, S. Jhulki, M. Conte, S. Barlow, S. K. Yee and S. R. Marder, Mater. Adv., 2020, 1, 1829–1834 RSC
- M. L. Tietze, J. Benduhn, P. Pahner, B. Nell, M. Schwarze, H. Kleemann, M. Krammer, K. Zojer, K. Vandewal and K. Leo, Nat. Commun., 2018, 9, 1182 CrossRef PubMed
- F. Pallini, S. Mattiello, N. Manfredi, S. Mecca, A. Fedorov, M. Sassi, K. Al Kurdi, Y. F. Ding, C. K. Pan, J. Pei, S. Barlow, S. R. Marder, T. Q. Nguyen and L. Beverina, J. Mater. Chem. A, 2023, 11, 8192–8201 RSC
- J. Liu, M. P. Garman, J. Dong, B. Van Der Zee, L. Qiu, G. Portale, J. C. Hummelen and L. J. A. Koster, ACS Appl. Energy Mater., 2019, 2, 6664–6671 CrossRef CAS
- J. Liu, S. Maity, N. Roosloot, X. Qiu, L. Qiu, R. C. Chiechi, J. C. Hummelen, E. von Hauff and L. J. A. Koster, Adv. Electron. Mater., 2019, 5, 1800959 CrossRef CAS
- J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. Akbar Sengrian, P. C. T. Souza, M. Antonietta Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen, L. Jan Anton Koster, J. Liu, L. Qiu, R. Alessandri, X. Qiu, G. Portale, J. Dong, W. Talsma, G. Ye, A. A. Sengrian, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen, L. J. A. Koster and P. C. T. Souza, Adv. Mater., 2018, 30, 1704630 CrossRef PubMed
- S. Wang, H. Sun, U. Ail, M. Vagin, P. O. Å. Persson, J. W. Andreasen, W. Thiel, M. Berggren, X. Crispin, D. Fazzi and S. Fabiano, Adv. Mater., 2016, 28, 10764–10771 CrossRef CAS PubMed
- Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342–3346 RSC
- G. Zuo, Z. Li, O. Andersson, H. Abdalla, E. Wang and M. Kemerink, J. Phys. Chem. C, 2017, 121, 7767–7775 CrossRef CAS
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759–787 RSC
- S. Olthof, S. Mehraeen, S. K. Mohapatra, S. Barlow, V. Coropceanu, J.-L. Brédas, S. R. Marder and A. Kahn, Phys. Rev. Lett., 2012, 109, 176601 CrossRef PubMed
- B. Lü, M. Riede and K. Leo, Phys. Status Solidi A, 2007, 210, 9–43 Search PubMed
- X. Ma, R. A. J. Janssen and G. H. Gelinck, Adv. Mater. Technol., 2023, 8, 2300234 CrossRef CAS
- T. L. Dexter Tam, T. T. Lin, M. I. Omer, X. Wang and J. Xu, J. Mater. Chem. A, 2020, 8, 18916–18924 RSC
- A. F. Akbulatov, O. A. Kraevaya, V. S. Bolshakova, L. A. Frolova and P. A. Troshin, Synth. Met., 2023, 299, 117468 CrossRef CAS
- W. Zhang, Y.-C. Wang, X. Li, C. Song, L. Wan, K. Usman, J. Fang, W. Zhang, Y. Wang, X. Li, C. Song, L. Wan, K. Usman and J. Fang, Adv. Sci., 2018, 5, 1800159 CrossRef PubMed
- C. Wang, D. Zhao, C. R. Grice, W. Liao, Y. Yu, A. Cimaroli, N. Shrestha, P. J. Roland, J. Chen, Z. Yu, P. Liu, N. Cheng, R. J. Ellingson, X. Zhao and Y. Yan, J. Mater. Chem. A, 2016, 4, 12080–12087 RSC
- S. Sung Kim, S. Bae and W. Ho Jo, Chem. Commun., 2015, 51, 17413 RSC
- J. H. Bae, Y. J. Noh, M. Kang, D. Y. Kim, H. Bin Kim, S. H. Oh, J. M. Yun and S. I. Na, RSC Adv., 2016, 6, 64962–64966 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00828j |
| This journal is © the Owner Societies 2025 |