
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStriking the balance between the capping ligands for CsPbBr3 nanocrystal synthesis via an emulsion LARP approach†
Govind B. Nair *a,
Sumedha Tambolia,
Sanjay J. Dhobleb and
Hendrik C. Swart*a
*a,
Sumedha Tambolia,
Sanjay J. Dhobleb and
Hendrik C. Swart*a
aDepartment of Physics, University of the Free State, P. O. Box 339, Bloemfontein 9300, South Africa. E-mail: govind1291@yahoo.com; Nair.GB@ufs.ac.za
bDepartment of Physics, R.T.M. Nagpur University, Nagpur-440033, India. E-mail: swarthc@ufs.ac.za
First published on 22nd May 2025
Abstract
The presence of surface ligands plays a vital role in driving the growth kinetics of metal halide perovskite nanocrystals (NCs). Irrespective of the synthesis method, their influence is essential for achieving the desirable optical and morphological properties of such nanocrystals. However, there is still a lot of debate over the selection of the ligands and their functioning during the growth process of halide perovskites. Yet, most of the studies agree that the combination of oleic acid (OA) and oleylamine (OLA) plays a pivotal role in the synthesis of CsPbBr3 NCs. In this study, the impact of OA and OLA ligands on the synthesis and properties of CsPbBr3 NCs has been investigated, and their growth mechanism has been established. An emulsion-based ligand-assisted reprecipitation (emulsion LARP) approach was adopted for the synthesis of these NCs. The findings of this study revealed that a balanced proportion of OA and OLA is crucial for ensuring optimum passivation of surface defects and obtain sharp excitonic emission bands in the green region.
Introduction
CsPbBr3 nanocrystals (NCs) have achieved tremendous progress in the field of optoelectronics with their tunable optical properties and exceptionally high luminescence efficiency.1 Although the synthesis protocols showed a notable effect on the quality and properties of the NCs, it is more noteworthy to underscore the crucial role of ligands in tuning their morphology and optical properties.2–6 Ligand engineering has a crucial role in determining the size, shape and optical properties of the NCs by controlling their growth kinetics and ensuring better surface passivation as well as colloidal stability of the NCs.7,8 To date, several ligands have been employed during the synthesis of CsPbBr3 NCs and they have been extended to the synthesis of other members of the halide perovskite family as well. Some of the noteworthy ligands comprise primary alkyl amines,9 amphiphilic polymers,10 inorganic phosphate and bromide anions,11 oleylammonium bromide (OAmBr),12 short-chain acids,13 phthalimide,14 and zwitterionic ligands.15,16 Even though these ligands offer better surface passivation and colloidal stability to the NCs, each of them is accompanied by certain challenges that affected the overall quality of the NCs. For example, NCs capped with amphiphilic polymers such as octylamine-modified polyacrylic acid (OPA) suffered accelerated halide exchange reactions as compared to hydrophobic ligand-capped NCs when dispersed in polar solvents.17 Similarly, zwitterionic ligands induce complexity in controlling the size distribution and morphology of the NCs, leading to less consistent crystals.16,18,19Upon retrospection of the synthesis of CsPbBr3 NCs, a majority of the syntheses adopted a binary ligand system. Nevertheless, the most frequently used combination consisted of oleic acid (a long-chain carboxylic acid) and oleylamine (an L-type primary alkyl amine ligand).20,21 However, there is less clarity about the ideal quantity of capping ligands that must be used to obtain the best quality NCs. In this context, two primary queries must be defined and addressed: (1) Is it necessary to involve more than one capping ligand for the synthesis of CsPbBr3 NCs? and (2) What quantity of these ligands must be ideally introduced during the synthesis? The objective of this study is to address these questions, and this shall result in refining the synthesis process for gaining optimal quality NCs.
In this study, a detailed investigation was performed to understand the impact of oleylamine (OLA) and oleic acid (OA) on the growth kinetics of CsPbBr3 NCs in an emulsion LARP method and to identify their optimal concentrations for achieving desirable properties of the NCs. An interplay between the OLA and OA ligands, and their influence on the size, shape, and size-distribution of CsPbBr3 NCs has already been established in the hot-injection method.22 However, due to the diverse growth conditions involved in each of the synthesis methods, it is difficult to extend the conclusions from that study to every other method possibly used for the synthesis of CsPbBr3 NCs. The optimized quantity of the ligands cannot be generalized for every synthesis process by just using the investigations and observations derived from a single method. To conclude, such studies must be exclusively conducted for every method. Herein, an emulsion-based ligand-assisted reprecipitation (emulsion LARP) approach has been employed for the synthesis, which follows a solution-based process at room temperature. An emulsion LARP process creates an emulsion by mixing two immiscible solvents comprised of the precursor salts. The demulsification of the mixture provides a controlled environment that finally leads to the formation of perovskite nanocrystals. Since ligands are added to control and stabilize the growth of NCs, this method simultaneously exhibits the peculiarities that are usually observed in the LARP method. Unlike the conventional LARP method, emulsion LARP avoids compromising on the growth control of the NCs by minimizing undesirable side reactions among the precursors.
Here, the distinct role of OLA and OA in the phase formation and recrystallization of CsPbBr3 NCs was established. To reach this understanding, the synthesis process was conducted in two different phases: in the first phase, the amount of OA was fixed, and the quantity of OLA was adjusted. Subsequently, in the second phase, the amount of OLA was kept constant at its optimum (which was determined from the first phase of synthesis) and the quantity of OA was altered. In addition, a sample was prepared without adding both OA and OLA to confirm their role in curtailing the Ostwald ripening of the NCs. Before proceeding with the final combinations of the capping ligands, preliminary experiments were conducted by exploring the different combinations of OA and OLA, wherein one of the capping ligands was kept fixed along with the other parameters and the second capping ligand was varied. The OA and OLA concentrations in these preliminary experiments were decided based on the preceding literature that reported the usage of these ligands for the CsPbX3 NCs synthesized by the LARP method. The exploratory trials involved testing the luminescence intensity of the NCs prepared under different combinations of OA and OLA. The luminescence intensity acted as the deciding factor for optimizing the capping ligands. After narrowing down the optimal range of the capping ligands, the final set of syntheses were performed.
Results and discussion
Colloidal CsPbBr3 NCs were prepared by the emulsion LARP approach following the procedures published in our previously reported article.23 A detailed description of the synthesis protocols is given in the ESI† and a graphical representation is depicted in Scheme 1. In the initial phase of the synthesis, the ligands (OLA and OA) were added to a non-polar solvent (hexane), whereas the Cs- and Pb-precursors were dissolved in polar solvents (water and dimethyl sulfoxide (DMSO), respectively). The mixing of polar solvents in the non-polar phase resulted in the formation of an emulsion, which confined the precursors inside the micelle-like structures formed by the ligands. These micelle-like structures acted as microenvironments for confining the nucleation and growth of NCs within their limits. The introduction of acetone for demulsification arrested the nanocrystal growth by decreasing the solubility of both precursors and surface ligands. If the reaction was allowed to continue without the addition of acetone, the NCs would have formed with uncontrolled size and shapes.23 To accurately assess the impact of ligands on the growth kinetics, all synthesis conditions were kept consistent. A gradual injection mode was adopted to administer the precursors comparably and the mixing was achieved at a consistent stirring rate. This approach was contrary to the LaMer theory of nucleation and growth, which suggests that burst injection of precursors triggers homogeneous nucleation leading to the simultaneous formation of numerous nuclei, followed by their growth without instigating any additional nucleation.24 The LaMer approach was supposed to help in regulating the size distribution of the nanocrystals during their growth process. However, this concept was found to fail frequently during the synthesis of CsPbBr3 NCs owing to the substantial dispersion of Cs4PbBr6 NCs formed alongside CsPbBr3 NCs due to the inadequate mixing rate provided by magnetic stirring.25 Furthermore, it was observed that the formation of Cs4PbBr6 NCs was favoured in a Cs-rich environment.26 So, Pb-precursors were preferably added first to the non-polar solution before injecting the Cs-precursors. Ng et al. demonstrated that the formation of Cs4PbBr6 can occur as a by-product in the LARP method driven by the Cs+-rich, DMF-rich and amine-rich environments.25 Therefore, DMF was avoided and the amount of DMSO used was kept to a minimum to protect the NCs from any adverse effects. | ||
| Scheme 1 Emulsion LARP synthesis of CsPbBr3 NCs. | ||
In the preliminary stage of experiments, a sample was prepared without the addition of either OA or OLA. This sample produced large crystals exhibiting an orange hue. Subsequently, in the first phase of the synthesis, the amount of OA was kept fixed at 0.5 mL and the amount of OLA was varied from 0 to 1 mL. After optimizing the amount of OLA, a second series of samples were prepared by fixing the optimized quantity of OLA and varying the amount of OA from 0 to 2 mL. The quantities of OLA and OA were optimized based on the optical performance of the resultant NCs. Fig. 2 shows the influence of ligand ratios on the photoluminescence (PL) properties of the CsPbBr3 NCs excited by a 365 nm near-ultraviolet (NUV) beam. This case study can be discussed in two parts: the first one is to identify the impact of OLA (Fig. 2(a)), and the second one is to identify the impact of OA (Fig. 2(b)) on the nucleation and growth of the NCs. When the NCs were prepared without adding either OA or OLA, the PL spectra showed a broader band with dual peaks at 401 and 425 nm, along with a shoulder hump at 453 nm. These emission peaks are indicative of multiple defects that act as emissive states in the NCs. Due to the lack of stabilizing and surface-capping ligands, the surfaces of the NCs were poorly passivated, which led to the formation of defective and unstable NCs with uncontrolled size expansion. As a result, there was an increased probability of non-radiative self-trapped exciton (STE) recombination at defect sites, which caused broad emissions at shorter wavelengths. When only OA (0.5 mL) was introduced into the synthesis (without any OLA), the PL spectrum resembled the one observed for the sample prepared without any OA and OLA. This suggests that the introduction of OA alone is inadequate to passivate the surface and preserve the emissive properties of the NCs. Moreover, it is assumed that the absence of OLA might have decreased the interaction of OA with the NCs and led them to a defect-rich state. However, things turned around with the addition of OLA and the PL spectra started showing the peculiar single emission peak at 515 nm. It was expected that the green emission line would gradually occur with the gradual increase in the amount of OLA, and it might take a certain quantity of OLA to suppress the blue emission lines. Surprisingly, the PL spectrum for one of the lowest amount of OLA (0.02 mL) showed a drastic transformation leading to a sharp emission in the green region and seemingly suppressing the blue emission lines mostly. As the amount of OLA was increased, the intensity of the green emission line became stronger and reached its maximum at 0.1 mL of OLA. This pronounced occurrence of the peak at 515 nm reflected the extent of surface passivation of the NCs facilitated by the presence of OLA. In the presence of OLA, the atoms encountered an orderly arrangement to form highly crystalline NCs with minimal defects, which allowed the excitons to radiatively recombine through a well-defined emissive state. Also, the reduced number of defects ensured to decrease the probability of non-radiative recombination. The addition of OLA also offered better control over the shape and size of the NCs (this will be discussed later in this section) providing highly crystalline NCs with well-passivated surfaces. Interestingly, this behavioural pattern held only up to 0.1 mL of OLA, and when the amount of OLA exceeded this limit, there was a decrease in the PL intensity. This decreasing trend continued with more addition of OLA (until 0.5 mL). But the peak at 515 nm again vanished and the blue emission lines reappeared when 1 mL of OLA was added. This indicates that the NCs returned to a defect-rich state, which might have occurred due to the disrupted crystal growth process in the presence of excessive OLA. It is also possible that the excess amount of OLA might have prevented further nucleation due to over-capping of the NCs or caused the NCs to dissolve in the solution.
The second series of samples were prepared to study the influence of OA on the growth of NCs. When only OLA alone was added during the synthesis, the PL spectra showed two emission peaks centered at 461 nm and 511 nm. However, this was a significant transformation from the case wherein no OA or OLA was added. From earlier studies, it was inferred that excessive OLA would lead to the transformation of CsPbBr3 into Cs4PbBr6.24 This naturally leads to the idea that the 461 nm emission might be corresponding to the Cs4PbBr6 NCs. Nevertheless, there are arguments about the origin of blue emissions from Cs4PbBr6 NCs, which have a bandgap of 3.95 eV (≈313 nm).26 While certain groups of researchers have advocated the blue and green emission lines of Cs4PbBr6 NCs,27–29 some others have voted against this possibility.30,31 Theoretically, Cs4PbBr6 exhibits a zero-dimensional perovskite structure, which is non-luminescent in the visible region due to the large bandgap. Tailoring the insulator bandgap of these zero-dimensional perovskites to the blue spectral region is fundamentally impossible unless the local coordination environment of individual [PbBr6]4− octahedra is intentionally modified by introducing additional defect states by doping ions such as Sn2+ or Sn4+.30 Hence, it is more likely that the 461 nm emission peak corresponds to the defect states generated on the NCs during the synthesis or to the relatively smaller NCs of CsPbBr3.32,33 Proceeding ahead, the PL spectra showed a remarkable transformation with the addition of OA and subsequently, increasing its amount, while keeping the amount of OLA at its optimized level. As the amount of OA was gradually increased, the peak emission in the green spectral region intensified and the blue emission was suppressed progressively. When 0.4 mL of OA was added, the blue emission completely disappeared and only the green emission line remained with a remarkable increase in its intensity. The green emission peak reached its maximum intensity at 0.5 mL of OA, but the intensity dropped at 1 mL of OA. A further increase in OA (2 mL) showed a very weak green emission and witnessed the reappearance of the blue emissions, which were observed in the case of the sample prepared without adding any OA or OLA. From these results, it can be inferred that OA plays a huge role in enhancing the surface passivation of the CsPbBr3 NCs for improving their stability and reducing the non-radiative recombination. The optimal balance between the amounts of OA and OLA was crucial for promoting the uniformity in the size and shape of the NCs and superior passivation of their surface defects. However, excessive OA might have over-passivated the NCs and disrupted the growth dynamics to bring out a significant change in their surface chemistry. This ultimately led to an imbalance between the ligands, and the NCs formed with a lot more defects. Moreover, this affected the growth of the NCs and produced them with a broader size distribution.
Although the variation of OA and OLA showed a noticeable trend in the peak intensity of the PL emissions, there was no trend observed in the shifting of the peak position. The peak position of the NCs fluctuated between 510 and 515 nm with the variation in the quantities of OA or OLA. The peak shift was more pronounced when the amount of OA was varied. The complexity of the interactions between the ligands and the NCs can be attributed to the absence of a clear trend in the peak-shifting. These complex interactions influenced multiple aspects such as surface passivation by ligands, variation in growth dynamics and size fluctuations of NCs, strain and reconstruction of the NC surface, modification of the local electronic structure of the NCs over their surface, and ligand exchange events. All these aspects manifested as factors responsible for influencing the peak shifts, but they operated simultaneously often counteracting one another. As a result, the peak-shifting showed a non-systematic fluctuation. Moreover, these factors influenced the free exciton (FE) and self-trapped exciton (STE) recombination, which affected the Stokes shift and the full width at half maximum (FWHM) of the emission bands. When the radiative excitonic recombination occurs without being localized by the lattice, FE emissions are observed. Therefore, FE emissions were dominant when the ligands successfully achieved the surface passivation of the NCs. On the other hand, STE recombination was dominant when the exciton recombined under the influence of lattice distortions or defect states. A dominant FE recombination led to narrow FWHM and smaller Stokes shift, whereas a dominant STE led to broader FWHM and larger Stokes shift in their respective PL emission bands. STE recombination was dominant in all those cases where the ligand capping was insufficient or excessive, which led to poor surface-passivated NCs and higher defect states on the NCs. The optimized concentration of both the ligands was crucial for suppressing the STEs and promoting the FEs in these NCs. The variation in the color emissions of the CsPbBr3 NCs prepared under the two series can be seen from the photographs captured under a fluorescent lamp and a 365 nm NUV lamp, as shown in Fig. 1. The CIE chromaticity diagram depicting the overall color variation of the NCs is shown in Fig. S3 and S4 (ESI†). Their corresponding chromaticity coordinates are listed in Tables S1 and S2 (ESI†), respectively. From the observations made from the PL studies, it is concluded that the NCs prepared with 0.5 mL OA and 0.1 mL OLA were the optimum sample. The internal photoluminescence quantum yield (PLQY) for the optimum sample was found to be 72%, as shown in Fig. S5 (ESI†).
 | ||
| Fig. 1 Photographs of CsPbBr3 NCs captured under a fluorescent lamp and 360 nm UV lamp: (a) prepared with different amounts of OA and (b) prepared with different amounts of OLA. | ||
 | ||
| Fig. 2 PL spectra of CsPbBr3 NCs excited at 365 nm, showing the influence of varying ligand concentrations: (a) with fixed OA and varying OLA concentrations and (b) with fixed OLA and varying OA concentrations. | ||
Fig. 3 shows the absorbance spectra of the CsPbBr3 NCs at varying concentrations of the two ligands. The impact of OA and OLA on the absorption properties of CsPbBr3 NCs was individually examined as shown in Fig. S2 and S3 (ESI†), respectively. For a systematic comparison, samples prepared without adding OA and OLA are shown in Fig. S2(a) and S3(a) (ESI†). This sample showed a very weak absorbance in the UV region, but no absorbance across the visible region. This is indicative of the poorly stabilized NCs that must have aggregated due to the absence of these ligands. While observing the absorbance spectra for the first set of samples (wherein the amount of OA was kept fixed and the amount of OLA was varied), it could be seen that the absorbance went on improving with increasing OLA concentration and showed an absorption edge at 500 nm. But this trend was found to be valid only up to OLA = 0.1 mL. Beyond this critical limit, the absorbance began to weaken, and at OLA = 1 mL, the absorbance spectrum showed an abrupt change with the shift in the absorption edge from 500 to 321 nm. Except for the variation in the intensity, there was no shift in the absorption edge for the NCs prepared with OLA = 0.02 to 0.5 mL. This indicates that the variation of OLA might not have substantially altered the dimensions or the electronic structure of the NCs.
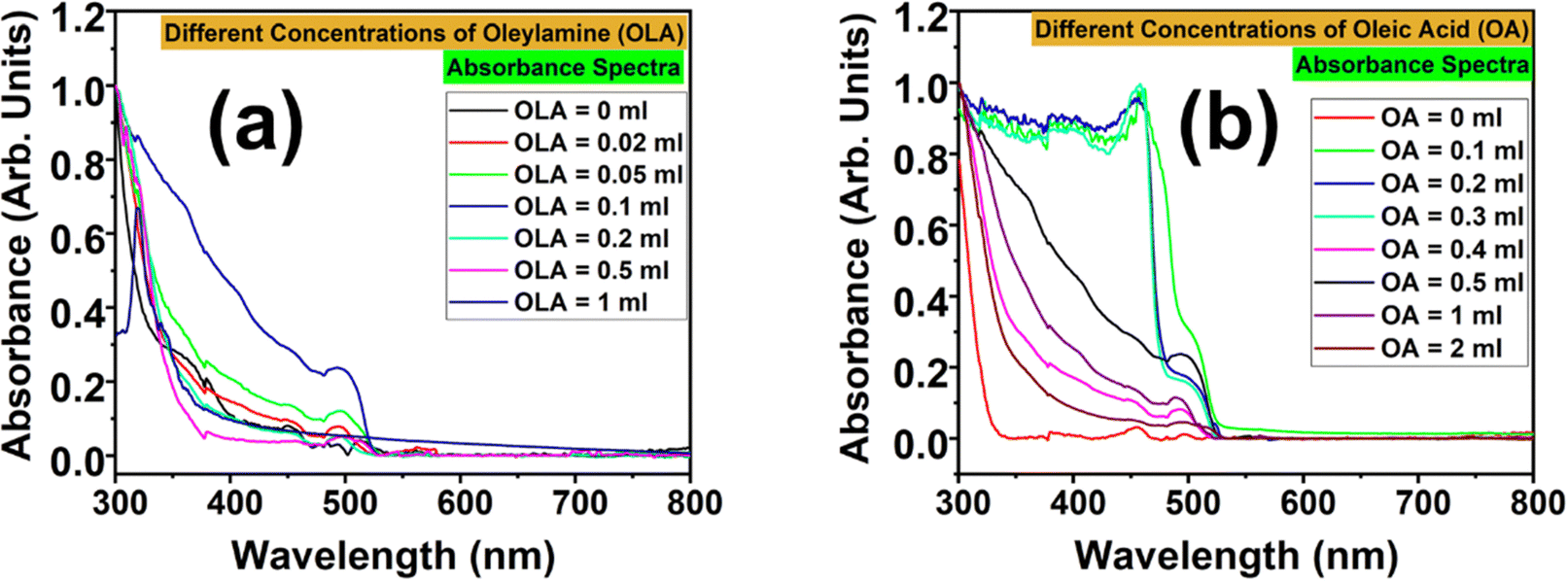 | ||
| Fig. 3 UV-visible absorbance spectra of CsPbBr3 NCs, showing the influence of varying ligand concentrations: (a) with fixed OA and varying OLA concentrations and (b) with fixed OLA and varying OA concentrations. | ||
In the second set of samples (wherein the amount of OLA was kept fixed, and the amount of OA was varied), the absorbance spectrum for OA = 0 mL showed a resemblance with that of OA = OLA = 0 mL. In fact, the absorption edge started appearing only when the NCs were prepared with the addition of OA. However, the absorbance spectra showed dual absorption edges for lower amounts of OA: the first edge was at 456 nm and the second one was at 500 nm. For NCs prepared with OA = 0.1 to 0.3 mL, the absorption edge at 456 nm was more prominent than that at 500 nm. But as the OA concentration was increased, the absorption edge at 456 nm got suppressed and that at 500 nm progressively increased. The NCs showed optimal absorbance for OA = 0.5 mL. With a further increase in OA concentration, the absorbance became weaker but without any shift in the absorption edge. From these observations, the optimum amounts for OLA and OA were found to be 0.1 mL and 0.5 mL, respectively. From the optical analysis of the NCs, it must be inferred that OA and OLA play a crucial role in achieving highly stable CsPbBr3 NCs with optimum luminescence and optical properties. The absence of either one or both could seriously damage the quality of the NCs and lead to poor colloidal stability. This is evident from the PL and absorbance spectra of the NCs prepared without using OA or OLA (OA = OLA = 0 mL, OA = 0 mL and OLA = 0 mL). This can be attributed to the increase in surface defects, lack of stabilization and increased aggregation of NCs in the absence of ligands. The presence of both ligands is essential for the stabilization of the NCs and passivation of their surface defects. They together provide steric hindrance and passivation that prevents the occurrence of aggregation of NCs. In the absence of these ligands, NCs suffer from poor dispersion in the solution, and they are prone to cluster together into larger lumps. This results in the weaker absorption of the NCs in the visible region and they suffer due to altered electronic properties. Moreover, the surface defects (that remain unpassivated) act as non-radiative recombination centres, which decrease the probability of radiative recombination, and thus lead to weaker optical absorption and PL intensity. However, based on the observations from the PL and optical absorption properties, it can be hypothesized that OLA mainly acts in the passivation of surface defects, whereas OA contributes to the stabilization of NCs in colloidal solution. Nevertheless, the quantity of OLA and OA needs to be precisely optimum. In case their amount exceeds the prescribed levels, then excess OLA will lead to agglomeration or dissolution of the NCs, while excess OA will result in the crowding of ligands that effectively reduce the defect passivation. Nevertheless, the optimal concentration of both these ligands is vital for the surface defect passivation and stabilization of the NCs for the enhancement of their PL intensity, whereas excessive ligand concentrations would negatively affect the PL intensity.
The next step was to understand the carrier dynamics in the NCs, which is crucial to create a window into the fundamental photophysics of the NCs for optimizing their optical and electronic properties. Clarifying the NCs’ optical characteristics and energy transfer mechanisms, describing their recombination dynamics and defect chemistry, evaluating their photostability and quantum efficiency, optimizing their surface passivation, and customizing their material properties that are desired for particular photonic and optoelectronic applications are all made possible by the PL decay analysis. The PL decay curves for the green and blue emission lines of the NCs under 365 nm excitation were examined in the current context. Fig. 4 shows the PL decay curves for the green emission lines of the CsPbBr3 NCs that were prepared with fixed OA concentration and different OLA concentrations, whereas Fig. 5 shows the PL decay curves for the NCs prepared with fixed OLA and different OA concentrations. All the decay curves were fit with a tri-exponential decay model as given below:


 | ||
| Fig. 4 PL decay curves for the green emission lines of CsPbBr3 NCs excited at 365 nm showing the variation of the decay kinetics for fixed OA concentration and different OLA concentrations: (a) OLA = 0.02 mL, (b) OLA = 0.05 mL, (c) OLA = 0.1 mL, (d) OLA = 0.2 mL, (e) OLA = 0.5 mL, and (f) OLA = 1 mL. | ||
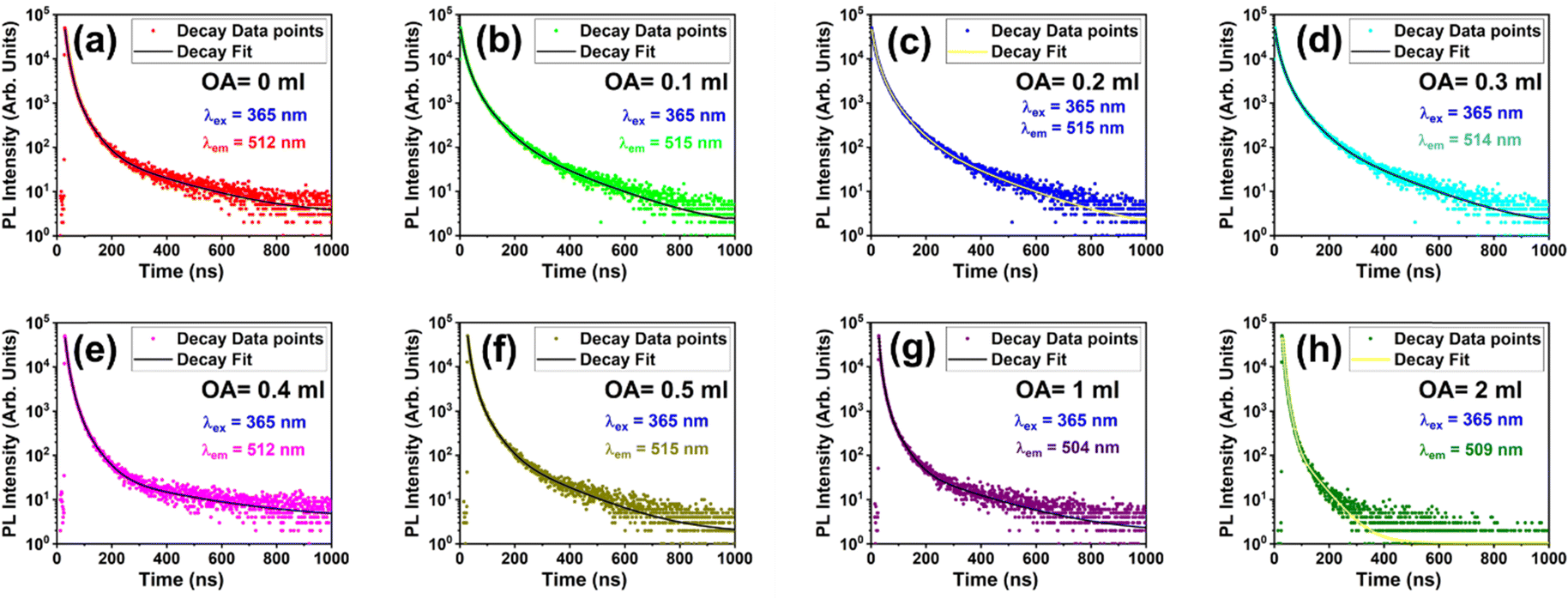 | ||
| Fig. 5 PL decay curves for the green emission lines of CsPbBr3 NCs excited at 365 nm showing the variation of the decay kinetics for fixed OLA concentration and different OA concentrations: (a) OA = 0 mL, (b) OA = 0.1 mL, (c) OA = 0.2 mL, (d) OA = 0.3 mL, (e) OA = 0.4 mL, (f) OA = 0.5 mL, (g) OA = 1 mL, and (h) OLA = 2 mL. | ||
The best-fit parameters for the green emission lines of the NCs are shown in Tables S3 and S4 (ESI†). As per the fitting, the decay lifetime consisted of three decay components that correspond to different carrier dynamics in the NCs. The first component showed the shortest lifetime that ranged between 5 and 7 ns, which can be attributed to the non-radiative recombination due to the unpassivated surface defects or trap states. The trap states quench the excitonic emission and promote the non-radiative recombination of the charge carriers. As a result, the overall PL efficiency is adversely affected, and the average lifetime suffers a dip in its value due to the shortest component of the lifetime. The second component showed an intermediate lifetime that ranged between 10 and 20 ns, which may be attributed to the radiative recombination within the NCs. However, there is a probability that this component is partially influenced by the moderately passivated surfaces or some shallow traps present in the NCs. The third components showed the longest lifetime that ranges between 30 and 60 ns, which corresponds to the radiative recombination of the charge carriers in well-passivated NCs. Such efficient radiative recombination can be associated with the high-quality NCs that are isolated from their environments and are less influenced by the surface defects. As a result, the excitons get sufficient time to recombine slowly and emit photons more efficiently. The three lifetime components give a measure of the non-radiative and radiative recombination pathways occurring in the NCs, which indicates the level of surface passivation and control over the defect states in the NCs. Those NCs with higher average lifetime are highly suited for optoelectronic applications. Fig. S6 and S7 (ESI†) show the decay curves for the blue emission lines of the NCs prepared under the two series. Their corresponding decay lifetime values are listed in Tables S5 and S6 (ESI†), respectively. The blue emission lines decay rapidly and show two components: one with the shorter lifetime and one component with the longer lifetime. However, the shorter lifetime components had a dominant presence with their contribution around 90%, while the longer lifetime component had not more than 10% contribution. This indicates that the defect states are primarily responsible for the blue emission lines.
In the case of samples prepared with fixed OA and varying OLA concentrations, the average lifetime increased from 22.52 to 30.57 ns with increasing OLA concentration (OLA = 0.02 to 0.1 mL). However, a further increase in OLA resulted in the decrease of lifetime from 30.57 to 18.66 ns (OLA = 0.1 to 1 mL). Similarly, the average lifetime increased from 16.99 to 29.21 ns with increasing OA concentration (OA = 0 to 0.5 mL), and a further decrease in OA resulted in a shortening of the lifetime. In both the series of samples, it was noted that the third lifetime component had a major contribution in deciding the average lifetime. The first component barely varied with the variation in OA or OLA concentration, while the second component showed slightly more variation than the first component. On the other hand, the third component showed significant variation with the change in the ligand concentration. Furthermore, the relative percentages of the second and the third components were well above 30% and comparable to each other, whereas the first component had a marginal contribution of less than 10%. However, this was not the case observed for the decay of the blue emission lines. The blue emission lines were observed only for a few samples that had either highly scarce or excessive quantities of ligands. Consequently, only a few samples were able to show the decay lifetime for the blue emission lines, and those samples were labelled as poorly passivated and abnormally formed NCs. For the blue emission lines, the first two lifetime components comprised shorter lifetimes, and each of them contributed around 40–50% to the average lifetime. In contrast, the third component showed a moderately long lifetime, and it contributed around 5% of the average lifetime.
From the observed decay lifetime values for the green emission line, it can be concluded that the lifetime improved when optimum quantities of OA and OLA were used. This resulted from the suppression of the non-radiative recombination centres by passivating the surface defects. In order to suppress the blue emission lines and increase the lifespan of the green emission lines, the OA and OLA capping ligands have to be properly balanced. The blue emission lines mostly occurred due to the presence of defect states in the NCs, which is reflected by the dominance of the shorter lifetime components in their average lifetime. Hence, they were not observed for all the samples in the series. The defect states leading to blue emission lines can be associated with the surface traps and halide vacancies in the NCs that can capture the excited charge carriers, viz. holes and electrons. Optimum levels of both OA and OLA ensured that the density of these traps was reduced by passivating the NC surface, and thereby suppressing the defect-mediated blue lines. The carboxylic acid group (–COOH) and the primary amine group (–NH2) from OA and OLA, respectively, played the key role in stabilizing the NCs and passivating their surface. The carboxylic acid group neutralized the positive surface charges by forming a strong ionic bond with the undercoordinated metal ions on the NC surface. On the other hand, the primary amine group neutralized the negative charges by coordinating with the anionic sites on the surface such as uncoordinated lattice anions and the oxygen adsorbed from the environment. These mechanisms suppressed the defect-mediated emission lines in the blue region and channelled the energy to promote radiative emissions in the green region.
After investigating the influence of the capping ligands on the optical properties of the NCs, the next step was studying their influence on the dimensional and morphological aspects of the NCs. The four primary cases were considered for this purpose: (1) samples prepared without adding either OA or OLA, (2) samples prepared using only OA (0.5 mL), (3) samples prepared using only OLA (0.1 mL), and (4) samples prepared using the optimized quantities of both OA (0.5 mL) and OLA (0.1 mL). The HR-TEM images of the samples falling under these four cases are shown in Fig. 6(a)–(d), respectively. These images provided a better understanding and insights into the influence of the capping ligands on the crystallinity, morphology and structure of the NCs. Fig. 6(a) shows that large lumps of CsPbBr3 crystals were formed when no capping ligands were introduced during the synthesis. The dimensions of these crystals were around 300 nm in diameter. From Fig. 6(b), it can be seen that the introduction of only OA resulted in the formation of relatively smaller-sized truncated tetrahedron structures with a broader particle-size distribution. Despite the dimensions being larger than the desired levels, these crystals showed isolation from one another without any traces of agglomeration. Fig. 6(c) shows that the introduction of OLA alone resulted in clusters of crystals with variable size and shape. A closer look into the image revealed that these clusters were formed by the agglomeration of smaller-sized crystals with indefinite shapes. However, Fig. 6(d) shows that the optimum concentrations of OA and OLA together can steer the growth kinetics and lead to the formation of high-quality CsPbBr3 NCs with desirable dimensions. It can be seen that the NCs formed in this case showed well-defined structures with rectangular shapes and relatively narrower particle-size distribution ranging between 4 and 18 nm. The average size of the NCs was approximately 10 nm, as seen in Fig. S8 (ESI†). The elemental map obtained using EDS (Fig. S9, ESI†) showed that Cs, Pb and Br were evenly distributed in the micrograph. From the uniformity of the shape and size of the NCs, it is clear that both OA and OLA in their optimized quantities must be present during the synthesis of the NCs to maintain a controlled growth condition. Fig. 6(e) shows the HR-TEM image with atomic lattice fringes within a randomly selected nanocrystal from Fig. 6(d). The interplanar spacing (d) between the lattice fringes was found to be 2.9 Å, which corresponds to the (200) plane of the monoclinic CsPbBr3. The atomic lattice fringes confirmed the high crystallinity of the NCs and thereby proved that optimum quantities of OA and OLA are crucial for yielding well-defined crystal structures at the atomic level. The crystal structural phase and the crystallographic orientation were further confirmed from the selected area electron diffraction (SAED) pattern, as shown in Fig. 6(f). The SAED pattern showed concentric rings, which is indicative of the polycrystalline nature of the NCs. There were four prominent diffraction rings observed in the pattern that can be specifically associated with the (001), (110), (200), and (202) planes of the monoclinic CsPbBr3 (JCPDS Card No. 18-0364). These planes confirmed the monoclinic crystal structure of CsPbBr3, which seemingly appears as a near-cubic structure with only a slight distortion in the lattice parameters and lattice angles (a = b = 5.827 Å, c = 5.891 Å, α = β = 90°, γ = 89.65°). The SAED pattern suggests that the NCs exhibited a consistent crystallographic phase with a well-defined orientation.
 | ||
| Fig. 6 HR-TEM images of the CsPbBr3 NCs prepared by the emulsion-LARP method: (a) without adding either OA or OLA, (b) with only OA (0.5 mL), (c) with only OLA (0.1 mL), and (d) with optimized quantities of both OA (0.5 mL) and OLA (0.1 mL), (e) determination of the interplanar spacing from the HR-TEM image of CsPbBr3 nanocrystals prepared using optimized quantities of OA and OLA. (f) Corresponding SAED pattern and the indexation of the diffraction rings. | ||
The crystal structures were further validated from the XRD patterns shown in Fig. S10 and S11 (ESI†), which conveyed the structural evolution of the NCs with varying concentrations of OLA and OA. In the absence of both OA and OLA, the XRD pattern showed a series of reflections corresponding to CsPbBr3 and other residual unreacted precursors. This suggests that a phase-pure CsPbBr3 was not achieved in the absence of the capping ligands. The monoclinic phase was more apparent, and the reflections were brighter and crisper when OA was introduced alone during the synthesis. This suggested that OA alone could stabilize the monoclinic phase to a certain extent. The three reflections corresponding to the (001), (110), and (200) planes appeared prominently in the pattern. The XRD patterns improved when 0.02 mL of OLA was added during the synthesis. The three reflections started broadening with the increasing OLA concentration, which is indicative of the role of OLA in confining the growth of the NCs and inhibiting the Ostwald ripening process. These samples showed phase-pure monoclinic CsPbBr3, thus indicating the synergistic effect of OA and OLA in producing phase-pure NCs with high crystallinity. At OLA = 0.1 mL, the reflections were consistent and free from any impurities, which suggested the formation of well-stabilized NCs with optimal passivation. This consistency was observed even for OLA = 0.2 mL. However, there was a drastic change in the pattern when the OLA concentration exceeded 0.2 mL. The XRD patterns for OLA = 0.5 mL showed decreased crystallinity and a series of reflections differing from the monoclinic CsPbBr3 phase. This pattern showed more resemblance with the trigonal Cs4PbBr6 phase (JCPDS Card No. 75-2251). At OLA = 1 mL, the pattern completely matched with the trigonal Cs4PbBr6 phase, and there were no more traces of monoclinic CsPbBr3. This observation is in line with the previous reports claiming the transformation of the CsPbBr3 phase into the Cs4PbBr6 phase in the presence of excess OLA.24 In the case of samples prepared by varying the OA concentration, the XRD pattern continued to show a mixed phase until OA = 0.2 mL. However, with the increasing OA concentration, the phase purity and crystallinity of the NCs were found to improve. From OA = 0.3 mL, the reflections became more well-defined and showed better match with the monoclinic CsPbBr3 phase. The XRD patterns continued to show consistency for higher OA concentrations too. Unlike OLA, the addition of excess OA did not induce phase transformation of the monoclinic CsPbBr3 NCs.
The chemical states and the bonding between the Cs 3d, Pb 4f, and Br 3d core levels were studied using X-ray photoelectron spectroscopy (XPS). Fig. S12 (ESI†) shows the full-survey scan for the CsPbBr3 samples prepared without adding any ligands, prepared by adding either OA or OLA, and prepared using the optimized quantities of both OA and OLA. The high-resolution XPS scans for the core levels of Cs 3d, Pb 4f, and Br 3d for these four samples are shown in Fig. S13 (ESI†). The XPS peak fitting was performed for each of these core levels and their corresponding binding energies are shown in Table S7 (ESI†). There were slight differences observed in the binding energies of the core levels due to the surface passivation caused by the ligands and the presence of certain defects in the NCs. The XPS profiles for the Cs 3d and Br 3d core levels appeared to be more consistent when at least one ligand was introduced during the synthesis. However, the Pb 4f core levels showed drastic variations across the four samples. The Cs 3d levels showed spin–orbit splitting into Cs 3d5/2 and Cs 3d3/2 levels with binding energies around 724.2 and 738.3 eV. Br 3d also showed spin–orbit splitting into Br 3d5/2 and Br 3d3/2 levels with binding energies around 68.7 and 69.5 eV, respectively. Pb 4f core levels showed spin–orbit splitting into Pb 4f7/2 and Pb 4f5/2 components. However, Pb 4f showed binding energies corresponding to both Pb2+ and Pb0 when at least one of the capping ligands was absent during the synthesis.34 This indicates the tendency of Pb2+ to transform into metallic Pb0 if both the capping ligands are not present during the synthesis. Due to similar binding energies of Cs 3d in CsBr and CsPbBr3, it is difficult to identify the formation of additional phases through their XPS scan. A similar reason applies to Br 3d core levels too. However, for the samples prepared without any capping ligands, both Cs 3d and Br 3d core levels showed additional peaks that indicated the presence of additional species. Nevertheless, the additional peaks observed in the Pb 4f core level scans were sufficient to deduce the presence of multiple phases when both the ligands were not optimally present during the synthesis. The binding mode of the ligands and the presence of different functional groups in the four samples were elucidated using their Fourier Transform Infrared (FTIR) spectra in the mid-IR region (4000 to 400 cm−1), as shown in Fig. S14 (ESI†). Table S8 (ESI†) shows the vibrational modes responsible for the absorption peaks in the FTIR spectra recorded for the CsPbBr3 NCs prepared without OA or OLA, with only OA, with only OLA, and with both optimized quantities of OA and OLA.35–38 The sample prepared without any ligands showed no absorption in this region. However, the sample prepared using only OA showed absorption peaks corresponding to the carboxylic group that confirmed the presence of OA. The sample prepared using only OLA showed absorption peaks due to C–N stretching vibrations, in addition to the peaks corresponding to the vibrations in the carboxylic group. Nevertheless, the Pb–Br stretching vibrations of the lattice were less prominently seen in the samples prepared in the first three cases. On the other hand, the sample prepared with optimized quantities of OA and OLA showed strong absorption due to the lattice vibrations of Pb–Br stretching. In this case, the presence of the vibrational modes of amide and the carboxylic groups indicated the strong interaction of the ligands with the CsPbBr3 NCs.
To further elucidate the effect of capping ligands on the growth mechanism of CsPbBr3 NCs in the solution-based emulsion LARP method, it was essential to understand the core dynamics of the reaction solution. In the preliminary stage, the capping ligands are supposed to mix well with the non-polar solvent, whereas the Cs- and Pb-precursors must dissolve well in the polar solvent. When OA and OLA were dissolved in a non-polar organic solvent such as hexane, an acid–base reaction occurred that resulted in the deprotonation of OA and protonation of OLA, as shown in Fig. S15(a) (ESI†). Consequently, OA transformed into oleate (OA−) ions while OLA transformed into oleylammonium (OLA+) ions.39 The next stage was preparing the solutions of CsBr and PbBr2 in distilled water and DMSO, respectively. CsBr was readily soluble in distilled water; however, PbBr2 was not easily soluble in DMSO, and it required a few minutes of sonication to completely dissolve them. As shown in Fig. S15(b) (ESI†), CsBr completely dissociated into Cs+ and Br− ions, whereas PbBr2 dissociated partially into Pb2+ and Br− ions along with PbBr+ species. The addition of these precursor solutions to the non-polar solvent resulted in the formation of an emulsion. Fig. 7 shows the schematic representation of the nucleation and growth kinetics of CsPbBr3 NCs synthesized by an emulsion-LARP method. For the nucleation to take place, it was necessary to break the barrier between the liquid solvents in the emulsion and facilitate the reaction between all the precursors in the solution. This was achieved by introducing an adequate amount of acetone that demulsified the emulsion. After vigorously stirring the solution, the oleate and oleylammonium ions became available to react and bond with the metal cations and the bromide ions. Due to the strong electrostatic interactions, OA− ions reacted with Pb2+ and Cs+ ions to form Cs-oleate and Pb-oleate complexes. In addition, PbBr+ species also reacted with OA− ions to form their corresponding oleate complex. Simultaneously, OLA+ ions reacted with Br− ions to form oleylammonium bromide through hydrogen bonding.40 Under vigorous stirring, these complexes interacted with each other and spontaneously self-assembled themselves in such a way that their head consisting of metal ions or bromide ions interacted with each other, and their hydrophobic tails pointed outwards.
 | ||
| Fig. 7 Schematic representation of the nucleation and growth kinetics of CsPbBr3 NCs synthesized by an emulsion-LARP method. | ||
The major objective of this study was to understand how the capping ligands changed the reaction environment. Fig. 8(a) shows a graphical illustration of the growth and formation of CsPbBr3 NCs in the presence and absence of the capping ligands OA and OLA. When the Cs- and Pb-precursors were introduced into the reaction solution, they interacted with the Br− ions to undergo rapid nucleation and growth. However, the equilibrium between the OA and OLA ligands was crucial for controlling the reaction and stabilizing the desired phase. In the absence of these ligands, there was no control over the reaction, and the precursors randomly interacted to produce larger-sized crystals with mixed phases. On the other hand, the presence of OA and OLA ensured adequate stabilization of the NCs and a higher degree of coordination to achieve a single-phase formation. OLA is a weak base that acts as a growth regulator for the NCs. OLA readily reacted with Br− ions and modulated the Pb–Br interaction. In the absence of OLA or at low OLA concentrations, the bromide ions remained uncoordinated with ligands and there was high acidity in the reaction solution due to the excessive presence of OA. As a result, the NCs were unable to passivate the surface defects. Moreover, due to the excess amount of OA, the acidity of the solution increased, and this resulted in the dissociation of OA releasing excess H+ ions in the solution. The excess H+ ions bonded with the available Br− ions and thus generated Br-vacancies in the NCs. However, in the presence of excess OLA, they formed strongly coordinated amine-Pb complexes that firmly held the Pb ions without releasing them periodically. This eventually led to a Cs+-rich solution that resulted in the crystallization of the Cs4PbBr6 phase.41,42 An adequate amount of OLA was crucial for consistently releasing the Pb ions and promoting controlled nucleation and growth of CsPbBr3 NCs. On the other hand, OA is a weak base that acts as a stabilizing ligand for the NCs. They bind to the NCs to provide passivation to the crystal facets and control the growth kinetics by slowing down the reaction. When the concentration of OA was far less (0 to 0.2 mL), NCs were inadequately stabilized and they were not able to achieve uniform crystal growth. Yet, the NCs were able to crystallize in a monoclinic CsPbBr3 phase in the presence of OA alone. When OA was introduced at low concentration in conjunction with the optimized amount of OLA, the NCs showed a mixed phase. This can be attributed to the amide-rich reaction environment that created unfavourable conditions for the nucleation and consistent growth of NCs.42 However, the quality of the NCs improved with higher OA concentrations, and surprisingly excess OA also ensured that the NCs crystallized in a single phase. Nonetheless, excess OA affected the optical properties of the NCs, even though there was no significant change in the crystal phase observed in the XRD pattern. However, NCs prepared with OLA alone resulted in their anisotropic growth due to insufficient surface passivation caused by the absence of OA.1 Against all these odds, CsPbBr3 NCs formed with optimum optical and morphological results when optimum quantities of both OA and OLA were utilized. The synergistic functioning of both OA and OLA was essential in achieving the surface passivation and colloidal stability of the NCs. The presence of OA was crucial for the protonation of OLA into OLA+ ions and thereby produced strong electrostatic interaction between the uncoordinated Br− ions and OLA+ ions. This interaction, consequently, led to better coverage of OLA over the NCs and facilitated growth inhibition beyond a certain limit. Moreover, the NCs preserved their colloidal stability and prevented aggregation due to the steric hindrance provided by the long alkyl chains of OA and OLA. In addition, the balance between the acid and amide concentrations in the reaction solution was critical for reducing the defect formation in the NCs (Fig. 8(b)).
 | ||
| Fig. 8 (a) Graphical illustration showing the influence of capping ligands on the morphology of CsPbBr3 NCs. (b) Schematic representation showing the influence of the varying quantities of OA and OLA on the growth kinetics of CsPbBr3 NCs. | ||
Conclusions
This study highlights the significant roles played by oleic acid and oleylamine in controlling the growth kinetics of CsPbBr3 nanocrystals. From the optical and microscopy analysis, it is inferred that the presence of both the stabilizing agents in their optimized quantities is essential for achieving desirable luminescence, optical and morphological properties in the emulsion-LARP-synthesized CsPbBr3 NCs. The NCs had defect-dominated PL emissions, which included many peaks in the blue area, when none of these ligands was present. This feature was also observed when neither of the two ligands were used. Furthermore, the NCs grew and aggregated uncontrollably when both ligands were absent, which negatively impacted their luminescence and optical characteristics. The NCs culminated with definite size and shape only when both the ligands were present in their optimized quantities. However, excess quantities of either of the two ligands disrupted the growth process and resulted in poor luminescence properties. In addition, excess OLA resulted in a phase transition of the three-dimensional CsPbBr3 into a zero-dimensional Cs4PbBr6 perovskite phase. The interaction between the OA and OLA, which followed an acid–base reaction for binding with the halide and metal ions, affected the luminescence intensity and the spectrum purity of the NCs. The variation of OA and OLA led to the tuning of the color emission from the NCs between green and blue. From the PL decay analyses, the green emission was found to originate from the radiative recombination of the excitons, whereas the blue emissions originated from the radiative recombination of the defect states. When the quantities of OA and OLA approached their optimized levels, the blue emissions were suppressed, and the green emissions showed more dominance in the luminescence spectra. This indicated the significance of these capping ligands in suppressing the defect states through surface passivation and improving the radiative recombination as evident from the increase in the PL intensity of the green color emissions. The inferences drawn from this study maintains their exclusivity to only the emulsion-LARP method, and further studies are required to be performed with other synthesis techniques to extend these conclusions to those methods. The findings of this study established the significant roles of both OA and OLA in optimizing the optical and morphological properties of CsPbBr3 NCs. Future studies can be focussed on the emulsion-LARP method's extension for the synthesis of other mixed-halide and hybrid organic–inorganic perovskite NCs using the tunable ligand environment. Moreover, it would be interesting to study the performance of other types of ligands at their optimized level for the synthesis of metal-halide perovskite NCs.Author contributions
GBN conceived and designed the work, conducted the experiments and characterization, wrote the original draft; ST conducted the experiments, wrote the original draft; SJD reviewed and edited the draft; HCS supervised the work, acquired funding, resources for the experiments, and reviewed and edited the draft. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research is supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (Grant 84415). The PL system used is supported by the rental pool programme of the National Laser Centre (NLC) and funded by the National Research Foundation of South Africa (Grant EQP14080486021, 93214). The financial assistance from the University of the Free State is highly recognized.References
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- Y. Li, X. Zhang, H. Huang, S. V. Kershaw and A. L. Rogach, Mater. Today, 2020, 32, 204–221 CrossRef CAS.
- S. Gallagher, J. Kline, F. Jahanbakhshi, J. C. Sadighian, I. Lyons, G. Shen, B. F. Hammel, S. Yazdi, G. Dukovic, A. M. Rappe and D. S. Ginger, ACS Nano, 2024, 18, 19208–19219 CrossRef CAS PubMed.
- F. Zhang, J. Chen, Y. Zhou, R. He and K. Zheng, J. Lumin., 2020, 220, 117023 CrossRef CAS.
- X. Xu, H. He, Z. Fang, H. Lou, C. Lin, L. Chen and Z. Ye, ACS Appl. Nano Mater., 2019, 2, 6874–6879 CrossRef CAS.
- G. B. Nair, R. Krishnan, A. Janse van Vuuren and H. C. Swart, Dalton Trans., 2023, 52, 70–80 RSC.
- N. K. Kumawat, A. Swarnkar, A. Nag and D. Kabra, J. Phys. Chem. C, 2018, 122, 13767–13773 CrossRef CAS.
- H. Wu, Z. Kang, Z. Zhang, H. Si, S. Zhang, Z. Zhang, Q. Liao and Y. Zhang, Small Methods, 2019, 3(7), 1900117, DOI:10.1002/smtd.201900117.
- Q. Zhong, M. Cao, Y. Y. Xu, P. Li, Y. Zhang, H. Hu, D. Yang, Y. Y. Xu, L. Wang, Y. Li, X. Zhang and Q. Zhang, Nano Lett., 2019, 19, 4151–4157 CrossRef CAS PubMed.
- Y. Shu, Y. Wang, J. Guan, Z. Ji, Q. Xu and X. Hu, Anal. Chem., 2022, 94, 5415–5424 CrossRef CAS PubMed.
- S. Chen, Q. Zhong, J. Liu, W. Guan, P. Li, I. Mahmood, M. Cao and Q. Zhang, Nanoscale, 2021, 13, 9634–9640 RSC.
- J. Yin, H. Yang, L. Gutiérrez-Arzaluz, Y. Zhou, J.-L. Brédas, O. M. Bakr and O. F. Mohammed, ACS Nano, 2021, 15, 17998–18005 CrossRef CAS PubMed.
- S. Biswas, S. Akhil, N. Kumar, M. Palabathuni, R. Singh, V. G. V. Dutt and N. Mishra, J. Phys. Chem. Lett., 2023, 14, 1910–1917 CrossRef CAS PubMed.
- V. G. V. Dutt, S. Akhil and N. Mishra, Nanoscale, 2021, 13, 14442–14449 RSC.
- R. Grisorio, F. Fasulo, A. B. Muñoz-García, M. Pavone, D. Conelli, E. Fanizza, M. Striccoli, I. Allegretta, R. Terzano, N. Margiotta, P. Vivo and G. P. Suranna, Nano Lett., 2022, 22, 4437–4444 CrossRef CAS PubMed.
- F. Krieg, P. C. Sercel, M. Burian, H. Andrusiv, M. I. Bodnarchuk, T. Stöferle, R. F. Mahrt, D. Naumenko, H. Amenitsch, G. Rainò and M. V. Kovalenko, ACS Cent. Sci., 2021, 7, 135–144 CrossRef CAS PubMed.
- S. Ghimire, M. F. Khatun, B. M. Sachith, T. Okamoto, J. Sobhanan, C. Subrahmanyam and V. Biju, ACS Appl. Mater. Interfaces, 2023, 15, 41081–41091 CrossRef CAS PubMed.
- S. Guo, H. Liu, H. He, W. Wang, L. Jiang, X. Xiong and L. Wang, Langmuir, 2020, 36, 6775–6781 CrossRef CAS PubMed.
- H. Zhu, M. Kick, M. Ginterseder, C. J. Krajewska, T. Šverko, R. Li, Y. Lu, M. Shih, T. Van Voorhis and M. G. Bawendi, Adv. Mater., 2023, 35(39), 2304069, DOI:10.1002/adma.202304069.
- M. Li, X. Zhang, T. Dong, P. Wang, K. Matras-Postolek and P. Yang, J. Phys. Chem. C, 2018, 122, 28968–28976 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS PubMed.
- G. B. Nair, S. Tamboli, R. E. Kroon, S. J. Dhoble and H. C. Swart, J. Alloys Compd., 2022, 928, 167249 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- C. K. Ng, H. Deng, H. Li, W. Yin, T. Alan and J. J. Jasieniak, J. Mater. Chem. C, 2021, 9, 313–321 RSC.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- Y. Duan, P. Li, Y. Lu, S. Xu and J. Zhang, Ceram. Int., 2021, 47, 13381–13390 CrossRef CAS.
- H. Shi, X. Zhang, X. Sun and X. Zhang, J. Phys. Chem. C, 2020, 124, 1617–1622 CrossRef CAS.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 840–845 CrossRef CAS.
- S. Zou, C. Liu, R. Li, F. Jiang, X. Chen, Y. Liu and M. Hong, Adv. Mater., 2019, 31(24), 1900606, DOI:10.1002/adma.201900606.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- K. Enomoto, R. Oizumi, N. Aizawa, T. Chiba and Y.-J. Pu, J. Phys. Chem. C, 2021, 125, 19368–19373 CrossRef CAS.
- X. Zhang, Y. Zhou, L. Peng, Z. Lin, Y. Chang, Z. Zhou and Y. Li, ACS Appl. Nano Mater., 2022, 5, 17012–17021 CrossRef CAS.
- Y. Xie, B. Peng, I. Bravić, Y. Yu, Y. Dong, R. Liang, Q. Ou, B. Monserrat and S. Zhang, Adv. Sci., 2020, 7(20), 2001698, DOI:10.1002/advs.202001698.
- S. Xiang, W. Chen, F. Wan, L. Du, Z. Zhang, F. Zhou and Z. Jiang, AIP Adv., 2021, 11, 125120, DOI:10.1063/5.0077414.
- C. K. Ng, W. Yin, H. Li and J. J. Jasieniak, Nanoscale, 2020, 12, 4859–4867 RSC.
- J. Mei, F. Wang, Y. Wang, C. Tian, H. Liu and D. Zhao, J. Mater. Chem. C, 2017, 5, 11076–11082 RSC.
- S. Kachhap, S. Fatima, A. Yadav, A. K. Singh and S. K. Singh, ACS Appl. Opt. Mater., 2023, 1, 1974–1986 CrossRef CAS.
- S. Mourdikoudis, M. Menelaou, N. Fiuza-Maneiro, G. Zheng, S. Wei, J. Pérez-Juste, L. Polavarapu and Z. Sofer, Nanoscale Horiz., 2022, 7, 941–1015 RSC.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS PubMed.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S.-T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS PubMed.
- S. L. Sanchez, Y. Tang, B. Hu, J. Yang and M. Ahmadi, Matter, 2023, 6, 2900–2918 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00853k |
| This journal is © the Owner Societies 2025 |