
Regulation of protein translocation through A Si3N4-CNT stacked nanopore using an embedded gold nanoparticle†
Wei
Si‡
 *a,
Haonan
Chen‡
a and
Gensheng
Wu
b
*a,
Haonan
Chen‡
a and
Gensheng
Wu
b
aJiangsu Key Laboratory for Design and Manufacture of Micro-Nano Biomedical Instruments, School of Mechanical Engineering, Southeast University, Nanjing 211100, China. E-mail: wei.si@seu.edu.cn
bSchool of Mechanical and Electronic Engineering, Nanjing Forestry University, Nanjing 210037, China
First published on 11th June 2025
Abstract
Proteins play a crucial role in the growth, movement, and reproduction of life, and the determination of accurate protein sequences is of great significance in understanding the specific functions of proteins. Nanopore sequencing is currently the most prevalent method due to its speed, cost-effectiveness, and ability to sequence long proteins. However, its accuracy is often compromised by the rapid translocation of the sample, making it challenging to generate effective blocking signals. To address this, a protein translocation regulation device is proposed in this paper. The primary component of this device is a carbon nanotube on a silicon nitride membrane, with two holes in its sidewall to facilitate the lateral passage of proteins. Further, we have incorporated a gold nanoparticle into the carbon nanotube, which can be controlled to move within it. By manipulating the position of the gold nanoparticle, we can alter the conformation of the proteins inside the nanotube, thereby achieving varying degrees of speed reduction. In addition, we made the gold nanoparticle positively charged in our simulation environment. Under the effect of electroosmotic flow, we realized better speed reduction. This proposed device holds significant potential for large-scale protein down-rate sequencing in the future.
Introduction
Proteins are important components of all living things and play an integral role in the human body as it carries out its life activities.1–3 As an efficient molecular machine, proteins perform most of the molecular functions of the cell, including transporting substances,4 providing energy5 and catalyzing chemical reactions.6 The realization of these complex functions is related to the unique three-dimensional structure of proteins.7 When the three-dimensional structure of a protein is disrupted, these specific functions cannot be performed, which in turn produces a range of diseases. For example, Alzheimer's disease and age spots are caused by structural changes in Tau and Aβ proteins, respectively.8 In turn, the formation and alteration of a protein's specific structure depends on the sequence of amino acids that make up the protein.9,10 These different kinds of amino acids polymerize together to form a wide variety of three-dimensional structures. Although it has now been shown that some polypeptide fragments do not need to fold into an ordered three-dimensional structure to fulfill their functions, these disordered polypeptide fragments are still controlled by the amino acid sequence.11,12 Therefore, understanding the amino acid sequences of proteins is of great significance in advancing the fields of proteomics and medicine. However, direct determination of amino acid sequences is a very complex task. Proteins are composed of more than 20 amino acids, and the length of a single protein can reach tens of thousands of amino acids,13 so the endless possibilities make the measurement of amino acid sequence extremely difficult. Therefore, we need some efficient methods to accomplish the measurement of amino acid sequences.Currently, the predominant techniques for protein sequencing are Edman degradation and mass spectrometry. Edman degradation is a protein sequencing method pioneered by Pehr Edman in the 1950s, marking the first successful measurement of a protein sequence.14,15 However, this method is limited to short peptides with free amino acids at the n-terminus and requires an extended measurement period. Mass spectrometry, also referred to as the birdshot method due to its process of fragmenting proteins into multiple short peptides prior to analysis, is the most widely employed sequencing technique.16–18 Although the emergence of mass spectrometry and the subsequent improvement of tandem mass spectrometry have improved the efficiency of protein sequencing, the mass spectrometer can only work properly at a specific range of protein concentration, which greatly increases the cost required for sequencing using mass spectrometry. Given these limitations, there is a pressing need for an innovative method to address the gaps in protein sequencing technology.
The advent of nanopore sequencing technology has, to a significant extent, addressed the challenges inherent in molecular diagnostics. Initially utilized for ion transport and DNA sequencing, nanopore technology has since expanded to other biomolecular detection domains.19 The fundamental principle behind nanopore sequencing involves applying a bias voltage across the nanopore. Analytes are drawn into the nanopore due to this voltage, leading to alterations in the ionic currents within. By analyzing variations in the timing, amplitude, and frequency of these ionic currents, sequence data about the analyte can be obtained.20,21 A notable advantage of this technology is its enhanced sequencing speed compared to previous methods, coupled with its capability to identify a broad spectrum of molecular structures, including enantiomers. Currently, nanopores are categorized into two primary types, biological nanopores and solid-state nanopores. Bio-nanopores are formed by self-assembly of biomolecules within a phospholipid bilayer,18 while solid-state nanopores are fabricated from materials such as silicon nitride and graphene.22 Both of them have a wide range of applications in different scenarios and needs. However, while nanopore sequencing offers distinct benefits, its application to protein sequencing remains challenging. A primary limitation is its suboptimal sensing accuracy and temporal resolution. Given the complex three-dimensional architecture of proteins, a sufficiently high bias voltage is essential for their passage through a nanopore. This often results in an exceedingly rapid translocation time, making it difficult to distinguish individual amino acid types.23 Many researchers have endeavored to prolong the translocation duration of proteins through nanopores. For example, Chan et al.,24 designed a mutant aerosol nanopore. This mutant has a narrower pore size than normal aerosol at some positions, which can effectively extend the transit time of protein molecules through the pore and improve sequencing accuracy. Erik et al.,25 on the other hand, utilized self-assembly to coat the walls of the nanopore with a lipid bilayer. This mobile lipid bilayer is sticky and can interact with the moving protein molecules, thereby reducing their translocation speed and extending their residence time within the nanopore. While all these methods can extend the translocation time of protein molecules, they primarily focus on biomodification, somewhat overlooking the role that the nanopore structure itself plays in speed control.
Unlike the slow development of protein sequencing technology, DNA sequencing technology has become more mature in recent years. Since DNA consists of only four bases, A, T, G and C, it is relatively easy to sequence.26 So far, the mainstream sequencing methods include Sanger sequencing, illumina sequencing and nanopore sequencing. Sanger sequencing was invented in the 1970s. It randomly terminates the extension of DNA strands by ddNTPs to generate fragments of different lengths, and then determines the base sequences by electrophoretic separation and fluorescence detection.27 It has the advantage of high sequencing accuracy, but the sequencing speed has always been a major constraint to its large-scale application. Illumina sequencing is the most widely used high-throughput sequencing technology, which has realized sequencing while synthesizing for the first time with the help of PCR amplification and fluorescent dye labeling.28 The principle of DNA nanopore sequencing technology is similar to that of protein nanopore sequencing technology, but due to the advantage of fewer types of constituent bases, the identification process is relatively simple. Currently, DNA nanopore sequencing technology can recognize DNA of any base length, and the recognition rate is 80% to 90% correct.29 Therefore, the difficulties in the field of protein sequencing may also be solved if the time of protein crossing the pore can be extended, and the numerous amino acids of proteins can be more appropriately classified and processed by algorithms to recognize the numerous amino acids.
Carbon nanotubes are an emerging nanomaterial with excellent mechanical, optical and electrochemical properties while possessing a very high diameter-to-length ratio.30 In recent years, the advent of functionalized carbon nanotubes has facilitated their application in the medical field.31,32 Owing to their unique shape and cellular uptake capabilities, they are frequently employed as carriers for anticancer drugs.33 For instance, Cao et al.34 successfully loaded doxorubicin onto modified carbon nanotubes and triggered its intracellular release through a pH-responsive mechanism. Beyond drug delivery, carbon nanotubes have been utilized in the development of medical sensors fatoni et al.35 combined nanotubes with chitosan–bovine serum albumin to create a blood glucose detector. The detector was able to accurately measure blood glucose levels in human blood by eliminating interference from other substances in human blood. More recently, Wei et al.36 constructed a nanopore sensor by inserting carbon nanotubes into a phospholipid bilayer. This sensor merges the principles of nanopore sequencing and effectively discerns various amino acids. Although this sequencing method relies on biological structures, it still pioneer a new avenue in nanopore sequencing.
As a rare metal discovered by mankind in ancient times, gold has been highly utilized in the fields of electricity, heat, optics, art and economy.37–39 With the advancement of micro-nano field exploration, numerous properties of gold within this domain have been progressively uncovered. For instance, gold, once perceived as an inert metal, can catalyze the oxidation of carbon monoxide on specific metal oxides when dispersed into sufficiently small gold nanoparticles.40 Furthermore, when gold particles enter the human bloodstream, they interact with proteins to form protein crowns, a phenomenon some scholars attribute to the competitive binding of proteins with gold.41 Consequently, if we can manipulate the gold nanoparticle to the vicinity of the nanopore using existing optical or magnetic tweezers technology,42–44 we may be able to achieve the down-rate translocation of protein molecules due to the interaction between the gold nanoparticle and proteins. Nevertheless, gold nanoparticles suffer from the disadvantages of low stability, difficult dispersion, and high cost, which pose a challenge for the application of gold nanoparticles in experiments. Molecular dynamics simulation methods originated in the 1960s. Since molecular dynamics simulation does not need to rely on harsh experimental environments, molecular dynamics simulation has been applied on a large scale in the biological and materials fields since the 1980s.45 At present, a number of scholars have utilized molecular dynamics simulation methods to study the properties of gold nanoparticles and achieved better results.46,47 Therefore, with the help of molecular dynamics simulation, we propose a protein translocation regulation device based on carbon nanotube and gold nanoparticle. Carbon nanotube was positioned horizontally on a pre-processed silicon nitride platform, with through holes drilled in the center to allow proteins to pass through. Gold nanoparticle was immobilized near the through-hole within the carbon nanotube. As the protein passed through the through-hole, it interacted with the gold nanoparticle, resulting in a reduction in speed. Additionally, we can regulate the position and charge amount of the gold nanoparticle to control the speed reduction effect. Compared with the existing protein down-rate translocation by biological methods, this method has designed a new down-rate device. The device is characterized by high stability, significant rate reduction effect, and relatively low cost, which provides the possibility of large-scale application of protein rate reduction sequencing. Finally, We sincerely hope that this device can promote the development of proteomics in the near future.
Results and discussions
In order to investigate the translocation dynamics of proteins during their lateral passage through carbon nanotube and to develop a method for protein translocation at a reduced rate, the setup of a feasible simulation system was shown in Fig. 1(a), where the upper and lower panels represent the main and top views, respectively. A semi-cylindrical groove, comparable in size to an armchair-type carbon nanotube, was horizontally etched into the silicon nitride substrate. The carbon nanotube was positioned above this recess. Two circular holes with the size of 1.6 nm were created in the sidewall of the carbon nanotube for allowing the peptide to pass through. In addition, a 3 nm circular hole was drilled into the silicon nitride substrate. We ensured that the influence of the silicon nitride on polypeptide translocation was minimized while still accommodating the passage of the polypeptide. The polypeptide, composed of a random sequence of 12 amino acids, was situated directly above the carbon nanotube. When a downward voltage was applied, the overall positively charged polypeptide descended through the carbon nanotube's through-hole, generating a blocking signal. To further validate the device's feasibility, we applied an external bias voltage of 0.04 V Å−1 to a composite system devoid of polypeptides and subsequently analyzed its potential distribution. The statistical results were illustrated in Fig. 1(b) and (c). Fig. 1(b) depicted the potential distribution at the system's center within the yoz plane. It is evident that the central potential was markedly lower than the surrounding potentials at the levels of z = 32 Å and z = −8 Å, with this disparity being more pronounced at z = −8 Å. This can be attributed to the resistance differential between the ionic solution and the solid material at the orifice, which intensifies as the orifice size increases.48 The high similarity between the size of the region where the difference in potential appears and the size of the aperture was well demonstrated. Moreover, the sharp decline in potential along the z-axis in both Fig. 1(b) and (c) can also be ascribed to the high resistance of the solid material. Intriguingly, Fig. 1(c) revealed a certain potential increase in the regions between −20 Å to −10 Å and 10 Å to 30 Å. We hypothesize this was due to the robust ion adsorption by the composite material formed by carbon nanotube and silicon nitride. Nevertheless, given the overall decreasing potential trend along the z-axis, we believe that the composite system can accomplish the transport of peptides in the presence of an external field. | ||
| Fig. 1 Schematic diagram of peptide translocation through carbon nanotube in a simulation system. (a) The front view (top) and top view (bottom) of the simulation system. The system uses armchair shaped carbon nanotube of size (24,24). Carbon nanotube are shown in gray in the figure, silicon nitride is shown in yellow and blue, and peptides are displayed in multiple colors based on different amino acid residues. Among them, lysine, arginine, serine, glutamine, asparagine, cysteine, threonine, and leucine are displayed as ching, pink, orange, red, purple, lime, mauve, and oche, respectively. The ion solution is shown as a cyan transparent surface. Potassium and chloride ions are shown as magenta and green spheres, respectively. (b) Two-dimensional potential distribution of the simulated system in the yoz plane when the electric field is 0.04 V Å−1. (c) Potential distribution of the simulated system along the z-axis under the same conditions as (b). | ||
After confirming the viability of polypeptide translocation through the composite system in the presence of an external field, we performed MD simulations of the system for 12 ns, and the results of the simulations were shown in Fig. 2(a)–(d). Fig. 2(a) depicts a series of microperiodic conformations of a polypeptide located on the top of the device as it traverses the composite system. As can be seen from the figure, when the time is 0.72 ns, the peptide started to enter the upper pore of the carbon nanotube. When the time is 7.2 ns, the polypeptide has completely passed through the carbon nanotube. This phenomenon was further elucidated by the plots of the peptide center of mass over time in Fig. 2(b) and the number of amino acids passing through the nanopore during peptide translocation in Fig. 2(c). As shown in Fig. 2(c), the peptides were swiftly captured by the carbon nanotube and propelled through the carbon nanotube due to the electrophoretic force. From the electric field application to the complete entry of the peptides into the carbon nanotube, only 1.7 ns elapsed. The short translocation time and fast translocation speed of the peptides made it challenging to capture distinct blocking signals for amino acid identification. As depicted in Fig. 2(d), we analyzed the ionic currents during translocation. The statistical results suggested that while there were variations in the opening and blocking currents before and after peptide passage, these differences were not pronounced enough to identify specific amino acid species. And even more unfortunately, this was the case for the vast majority of our simulations. Coincidentally, in our observations of peptide translocation events through carbon nanotube, we found that due to the vertical downward orientation of the electric field, the peptides always passed through the carbon nanotube in a “one” conformation, as seen in Fig. 2(a) and ESI,† Movie S1. This resulted in minimal contact between the peptides and the walls of the carbon nanotube, thereby preventing slow passage through the nanopore due to adsorption with the carbon nanotube. This outcome was contrary to our original intention when designing this device. Therefore, we aim to enhance the device to fully leverage the superior performance of carbon nanotube in micro and nano areas and slow down the translocation speed of peptides through carbon nanotube, which will be explained in detail later.
 | ||
| Fig. 2 MD simulations of peptide capture and translocation through carbon nanotube in a composite system. (a) A series of microperiodic sequences of manipulating polypeptides translocation through the composite system. (b) Changes in the center of mass of the peptide over time during peptide translocation, where the data are counted at 2.4 ps intervals. (c) Change in the number of amino acids passing through the nanopore over time during peptide translocation where the black curve indicates the number of amino acids passing through the upper pore and the red curve indicates the number of amino acids passing through the lower pore. Similarly, the data were counted every 2.4 ps. (d) Variation of ionic current through the nanopore with time during the passage of peptides through the carbon nanotube. In this case, the red solid lines represent the average open pore current and the average blocking current, respectively. The currents were sampled at 2.4 ps intervals and averaged in 0.24 ns blocks. | ||
Carbon nanotube, due to their unique properties and superior diameter-to-length ratio, are frequently employed as carriers for molecular scale substances such as anticancer drugs, proteins, and metal particles.49,50 If we can use carbon nanotube to carry a substance that can interact with proteins at a specific distance, we may be able to slow down the rate of translocation of peptides through carbon nanotube to some extent. A carbon nanotube composite system bearing gold nanoparticle was shown in Fig. 3(a). The gold nanoparticle was located in the left part of the carbon nanotube as a whole, and the distance between its right side and the center of the nanopore was l. The presence of van der Waals forces positioned the gold nanoparticle at a certain distance from the upper and lower walls of the carbon nanotube. As the peptide traversed the carbon nanotube, the gold nanoparticle remain fixed at a specific position, achievable through the use of optical or magnetic tweezers in practical applications. In addition, due to the limitation of computational resources, the peptide was partially placed in the carbon nanotube from the beginning to reduce the translocation time through the carbon nanotube. Fig. 3(b) depicted a series of microscopic sequences of peptide translocation through carbon nanotube at a distance of 1 Å. The diagram revealed that even when the peptide was initially positioned partially within the nanopore, it required 6.72 ns for complete passage, significantly extending the translocation duration. This observation was further supported by comparing the translocation times of gold nanoparticle at varying positions, as shown in Fig. 3(c) and (d). In Fig. 3(d), data from four successful peptide crossings were collected and averaged. These measurements were taken without the inclusion of gold nanoparticle and with distances between the gold nanoparticle and the nanopore centers set at 7 Å, 4 Å, and 1 Å. The detailed data were presented in ESI,† Fig. S1–S4. The statistical translocation times were 3.8 ns, 4 ns, and 6.1 ns respectively, indicating that the average translocation time increased in correlation with the proximity of the gold nanoparticle. Notably, all these times exceeded the 2.2 ns recorded in the absence of gold nanoparticle. This trend was attributed to the van der Waals forces acting between the gold nanoparticle and the peptides. Based on the measurement results, we found that the van der Waals potential energy was incrementally increasing when the distance between the center of the nanopore and the gold nanoparticle decreased from 7 Å to 1 Å. This suggested that the van der Waals force was increasing as the distance between the polypeptide and the gold nanoparticle approached, leading to a decrease in the polypeptide translocation rate. This was also illustrated by the measurements of van der Waals forces for several other simulations in ESI,† Fig. S5–S7. In addition, from the microperiodic sequence diagram of the peptide crossing the composite system at a distance of 1 Å between the nanopore and the gold nanoparticle in Fig. 3(b), we found that the conformation of the gold nanoparticle changed, which did not occur when the gold nanoparticle were farther away from the peptide, see ESI,† Movie S2. This was because at a closer distance between the gold nanoparticle and the peptide, the van der Waals force changed from gravitational to repulsive, driving the protein away from the gold nanoparticle and shifting its conformation towards the shape of the gold nanoparticle. The distance at which the transition occured also fit well with the results of previous scholars’ measurements of the radius of the bottom of the potential energy trap of gold atoms.51 The change in the conformation of the polypeptide not only led to an increase in its cross-sectional area in the horizontal direction, making it difficult to pass through the smaller-sized nanopore, but also increased the contact surface between the polypeptide and the carbon nanotube walls, which enhanced the adsorption effect of the tube wall on the polypeptide. Unlike the previous slowdown of the adsorption of peptides by gold nanoparticle at a longer distance, this slowdown caused by the change in conformation of the peptides was more significant, as illustrated by the significant increase in the average time required for peptide translocation when the distance between the center of the nanopore and the gold nanoparticle was decreased from 4 Å to 1 Å in Fig. 3(d). However, in the current condition, the change in the conformation of the polypeptide was still not prominent enough due to the remaining distance between the gold nanoparticle and the center of the nanopore. Therefore, the challenge lies in how to further leverage the effect of peptide conformational changes to slow down the rate of peptide translocation (Fig. 4).
 | ||
| Fig. 3 Manipulation of peptide translocation through MD simulation using a deceleration device composed of carbon nanotube and gold nanoparticle. (a) The front view (top) and top view (bottom) of the compound deceleration system settings. Among them, the diameter of the gold nanoparticle is 20 Å, which is shown in tan in the model diagram. The distance from the rightmost side of the gold nanoparticle to the center of the nanopore on the carbon nanotube is denoted as l. All other settings are the same as those in Fig. 1(a). (b) Under the action of gold nanoparticle, peptides pass through a series of microscopic periodic sequences of carbon nanotube. (l = 1 Å) (c) The changes in the number of amino acids passing through the nanopores during the process of peptide translocation through carbon nanotube under the action of gold nanoparticle over time. (d) The variation of average peptide translocation time with the distance between gold nanoparticle and the center of nanopores. The error bar represents the standard error. (e) The variation of van der Waals potential energy between peptides and gold nanoparticle over time under different distance conditions. | ||
 | ||
| Fig. 4 MD simulation of manipulation of peptide translocation through a rate-lowering system formed by gold nanoparticle deep into carbon nanotube. (a) Main view of the rate-lowering system. The rightmost end of the gold nanoparticle is located to the right of the center of the nanopore, and the distance between the two is still noted as l. (b) A series of microscopic periodic sequences of a peptide translocating through the rate-reducing system formed by a gold nanoparticle deep into a carbon nanotube. (l = −8 Å) (c) Variation of the center of mass position with time as the polypeptide translocates through the rate-reducing system when the gold nanoparticle are in different positions. (d) Variation of the number of amino acids crossing the nanopore during peptide translocation with time when the gold nanoparticle are in different positions. (e) Mean time of peptide translocation through the rate-reducing system when gold nanoparticle are in different positions. Error bars represent standard errors. | ||
When the peptide was in close proximity to the gold nanoparticle, since the conformation of the peptide changed according to the shape of the gold nanoparticle in the contact portion, there was a high possibility to realize a dramatic change in the conformation of the peptide to achieve a better speed reduction by simply increasing the contact area between the gold nanoparticle and the peptide. Fig. 4(a) illustrated a model diagram of the rate reduction system formed by gold nanoparticle deep into carbon nanotube. In this model, the distance between the rightmost end of the gold nanoparticle and the center of the nanopore on the carbon nanotube was represented as l. To distinguish from the previous model, we denoted l as positive when the gold nanoparticle was on the left side, and l as negative when it was on the right side. Subsequently, with the same remaining conditions, we performed MD simulations of the new deceleration system. In order to move the gold nanoparticle to the specified positions, we used the steered molecular dynamics (SMD) method in the simulation, see ESI,† Movie S3. A series of microperiodic sequences derived from the simulation were presented in Fig. 4(b). As observed in Fig. 4(b), the time required for the polypeptide to traverse the composite system significantly increases to 12 ns when the gold nanoparticle were moved inward. The phenomenon was further elucidated by the plots depicting the center of mass over time during peptide translocation, the count of amino acids traversing the pore over time during peptide translocation, and the average duration of peptide crossing the nanopore, as shown in Fig. 4(c)–(e). Consistent with previous methods, the duration of peptide crossing the nanopore in Fig. 4(e) was determined by averaging four data sets, the details of which were provided in Fig. S8–S11 of the ESI.† From the results, it can be seen that the peptide translocation time was prolonging when the distance between the gold nanoparticle and the center of the nanopore increases from 2 Å to 8 Å, which proved our previous inference that the peptide conformation undergoes continuous change as the gold nanoparticle moved deeper into the nanopore. Additionally, our MD simulations revealed that the peptide did not invariably traverse the nanopore as the gold nanoparticle shifted position. When the distance between the gold nanoparticle and the center of the nanopore reached 10 Å, the polypeptide consistently remained within the nanopore, as illustrated in Fig. S12 in the ESI.† We made an analysis of the reasons for this phenomenon. On the one hand, at this point, the rightmost end of the carbon nanotube extended beyond the nanopore boundary, effectively blocking the entire nanopore region by the gold nanoparticle. Consequently, it became challenging for proteins to pass through the nanopore due to the vertical downward electrophoretic force. On the other hand, due to the close contact of the peptide with the gold nanoparticle and carbon nanotube, the peptide was subjected to significant van der Waals forces. Therefore, in practical applications, it is advisable to avoid situations where the position of the gold nanoparticle is excessively deep.
In our previous MD simulations, we achieved a significant speed reduction effect by altering the position of gold nanoparticle within carbon nanotube. However, as the gold nanoparticle penetrated deeper, they increasingly occupied space near the nanopore, causing its blockage. This blockage hindered the detection of protein sequences through the blockage signal. To address this, we aimed to refine the device so that the gold nanoparticle can induce a more effective rate reduction without excessively penetrating the carbon nanotube. Based on our earlier findings, the primary source of protein speed reduction stemmed from conformational changes due to van der Waals repulsion between the gold nanoparticle and proteins. To prevent nanopore blockage, and considering that the protein resided at the nanopore's center, it was imperative for the gold nanoparticle to induce conformational changes in proteins from a greater distance. This necessitated a stronger repulsion between the gold nanoparticle and the protein. A composite speed reduction system comprisng charged gold nanoparticle and carbon nanotube was shown in Fig. 5(a). Given that the peptide also carries a positive charge, an electrostatic repulsion arises between them. With an appropriate charge magnitude, this electrostatic force will surpass the van der Waals force, potentially meeting our requirements. Fig. 5(b) depicted the potential distribution of the simulated system in the yoz plane. The positive charge of the gold nanoparticle resulted in a significant increase in potential around them. In addition, according to the theory related to electrostatic barriers,52,53 the insertion of the gold nanoparticles into the carbon nanotube medium also induces a further change in the electrostatic potential. These phenomena cause a redistribution of ions. Chloride ions were drawn closer to the nanopore due to attraction, while potassium ions were repelled and move further away. This ionic selectivity subsequently alters the flow field within the system.54 As demonstrated in Fig. 3(c), the chloride ions, which outnumber the potassium ions around the nanopore, induced more pronounced changes in the flow field. Furthermore, since the flow field formed by chloride ions was overall upward and that formed by potassium ions was overall downward due to the electric potential, the trend of the overall flow field in the solution was upward, as shown in ESI,† Fig. S13. This may be another reason for the decrease in the rate of peptide translocation.
 | ||
| Fig. 5 MD simulation of peptide translocation through a new rate-lowering system composed of charged gold nanoparticle and carbon nanotube. (a) Main view of the charged rate-lowering system. In this case, the surface particles of the gold nanoparticle are positively charged with a surface charge density of 0.0023 e Å−2, which is shown in blue color in the figure, and the rest of the setup is the same as that in Fig. 1(a). (b) Two-dimensional potential distribution of the charged simulation system in the yoz plane at an electric field strength of 0.04 V Å−1. (c) Steady-state localized densities of current generation by chloride and potassium ions in the yoz plane of the charged simulated system. Arrows indicate the direction of the local fluxes and colors indicate the magnitude of the local fluxes. The plots were calculated from 24-ns-long MD trajectories sampled at a frequency of 2.4 ps. (d) Variation of the center of mass position with time during peptide translocation through the rate-reducing system when the positions of the charged gold nanoparticle are different. (e) Variation of the number of amino acids passing through the nanopore with time during peptide translocation when the positions of the charged gold nanoparticle are different. (f) Variation of electrostatic energy between peptide and gold nanoparticle with time when the positions of charged gold nanoparticle are different. (g) Average time of peptide translocation through the composite system when the positions of the charged gold nanoparticle and uncharged gold nanoparticle are different. Error bars indicate standard errors. | ||
Subsequently, we performed MD simulations of the composite device, and the results of the simulations were shown in Fig. 5(d) and (e). These figures demonstrated that the total time required for peptide translocation through the composite system continued to increase as the gold nanoparticle drew closer. We believed that this was due to the amplification of electrostatic forces as well as the escalation in the magnitude of peptide conformational changes. In order to verify our conjecture, we statistically analyzed the electrostatic potential energy between the peptide and the gold nanoparticle, as shown in Fig. 5(f). Our findings revealed that the electrostatic potential energy between the peptide and the gold nanoparticle can reach nearly 30 kcal mol−1 at the current charge density, which surpasses the van der Waals energy between the two (8 kcal mol−1). In addition, as the distance between the center of the nanopore and the gold nanoparticle decreased from 7 Å to 3 Å, the electrostatic potential energy between the gold nanoparticle and the polypeptide increased. However, when the distance between the center of the nanopore and the gold nanoparticle decreased from 3 Å to 1 Å, the electrostatic potential energy remained essentially unchanged. This suggested that the distance between the gold nanoparticle and the peptide remained largely stable when the distance of the gold nanoparticle from the nanopore decreased from 3 Å to 1 Å. This finding corroborated that the gold nanoparticle can induce a change in the peptide's conformation when they were 3 Å away from the center of the nanopore, as also demonstrated in the ESI,† Movie S4. To further substantiate these conclusions, we have also calculated the electrostatic potential energy in several other sets of simulations, as detailed in Fig. S14–S17 of the ESI.† Finally, to further validate the significant speed reduction effect of the device following the addition of charge to the gold nanoparticle, we conducted comprehensive data acquisition and averaging, see Fig. S18–S21 of the ESI† for the detailed data. As illustrated in Fig. 5(g), a comparison of the translocation duration between systems with uncharged gold nanoparticle and those with positively charged gold nanoparticle revealed that the latter exhibited a markedly longer translocation time for polypeptides. This aligned with our expectations. Furthermore, there existed a large enhancement in the translocation time when the distance between charged gold nanoparticle and nanopores was reduced from 5 Å to 3 Å, which laterally confirmed the inference that the change in the conformation of the peptide caused a significant decrease in the translocation speed. In conclusion, enhancing the speed reduction can be achieved by introducing additional charges to the gold nanoparticle with the same polarity as that of the polypeptide.
Conclusions
In this article, we designed a protein translocation modulator based on the carbon nanotube and gold nanoparticle. Firstly, we formulated a deceleration model that did not incorporate the gold nanoparticle. When employing this device for protein detection, due to the “one” conformation of the peptide inside the carbon nanotube and the fact that the peptide hardly interacted with the carbon nanotube wall, the translocation rate of the peptide was relatively rapid. Therefore, we introduced the gold nanoparticle into the carbon nanotube. Initially positioned to the left of the nanopore's center, the gold nanoparticle caused a continuous decrease in the peptide translocation rate as they approached the peptide. Interestingly, when the initial distance between the peptide and the gold nanoparticle was 1 Å, there was a significant prolongation in the peptide translocation time. This phenomenon can be attributed to the van der Waals force between the gold nanoparticle and peptides transitioning from attractive to repulsive at this distance, leading to a change in peptide conformation. This further led to close contact between peptides and the carbon nanotube, as well as blockage of peptides in nanopores, resulting in a significant extension of translocation time. Subsequently, we used steered molecular dynamics (SMD) method to position the gold nanoparticle on the right side of the center of the nanopore. This adjustment induced more pronounced changes in protein conformation, enhancing the slowing effect. However, the insertion of gold nanoparticle also led to nanopore blockage, complicating the differentiation of through-hole signals of amino acid sequences. To mitigate this, we added charges of identical polarity to those of the peptides onto the surface of the gold nanoparticle, replacing the previous van der Waals forces with stronger electrostatic forces. This enhancement not only facilitated conformational changes in peptides at extended distances, but also induced an upward electroosmotic flow. Consequently, when the distance between the gold nanoparticle and the nanopore's center remained constant, the charged deceleration device exhibited a superior deceleration effect compared to its uncharged counterpart. It was worth noting that while our simulations utilized the carbon nanotube and gold nanoparticle of a singular size, practical applications may necessitate adjustments in size based on the dimensions of the protein and the desired deceleration effect. This flexibility offers potential for the large-scale implementation of this deceleration device in production settings. At last, we earnestly anticipate that the proposal of this device will provide innovative approaches to nanopore deceleration sequencing and contribute to the advancement of nanopore sequencing technology.Experimental section
General MD methods
In this paper, all simulations with a time step of 2 fs were performed using program NAMD2;55 periodic boundary conditions along the x and y axis were applied. The polypeptides, carbon nanotube, Si3N4, TIP3P water, and ions were described by the CHARMM3656 force field with CUFIX57,58 corrections. As we have done in our previous work, all atoms of the carbon nanotube were treated as aromatic carbons with atom type CA, and were constrained at the center of the system. A Langevin thermostat was applied to all non-hydrogen atoms of the system with a damping coefficient of 1 ps−1 for temperature coupling and to maintain the temperature of the system at 295 K. The particle mesh Ewald (PME) method with a 1-Å-spaced grid was used to describe the long-range electrostatic interactions between atoms in all systems performed in this work, and the calculation of full electrostatics was performed every three time steps. Covalent bonds that involve hydrogen atoms in protein and water molecules were described using the RATTLE and SETTLE algorithms. A smooth 10–12 Å cutoff was applied to evaluate the van der Waals interactions. VMD Topology Toolkit are used to generate and process topology files. NAMD Force expansion package are used to generate grid-force and steered molecular dynamics (SMD) large-scale force fields. VMD59 and Tcl scripts were used to view and analyze the trajectories of all the simulated systems.Force fields in MD simulations
In our MD simulations, we use the CHARMM36 force field to categorize the non-bonding potentials into three categories, van der Waals forces, electrostatic forces and hydrogen bonding. The van der Waals force is used to describe the interaction between uncharged molecules or atoms, and its exact value is derived from the formula for the Lerner-Jones potential. | (1) |
| σij = σ | (2) |
| εij = ε | (3) |
For the calculation of van der Waals forces between two different atoms
| σij = (σi + σj)/2 | (4) |
 | (5) |
 | (6) |
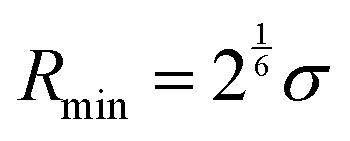 | (7) |
Hydrogen bonding is used to describe the forces between some of the more electronegative atoms. Therefore, hydrogen bonding is not considered when calculating the forces between gold atoms and other atoms. In particular, in MD simulations, its specific value is obtained by superposition of van der Waals forces and Coulomb forces.
MD simulation of peptide translocation through a deceleration system composed of carbon nanotube
The carbon nanotube are armchair shaped (24,24) carbon nanotube with a length of 7.85 nm. The length of the silicon nitride substrate is the same as that of carbon nanotube, with a width of 4.55 nm. There are semi-circular grooves with the same outer diameter as carbon nanotube on the silicon nitride substrate. The size of nanopores on carbon nanotube is 1.6 nm, and the size of nanopores on silicon nitride substrates is 3 nm. Polypeptides are composed of 12 random sequences of amino acids located 0.6 nm directly above carbon nanotube, and the specific amino acid sequence of the polypeptide is Lys–Ser–Arg–Lys–Gln–Asn–Arg–Cys–Lys–Thr–Leu–Arg. A grid-force was added between the peptide and silicon nitride to further eliminate the interference of silicon nitride on peptide translocation. Then the systems were solvated using the VMD's Solvate plugin; water molecules overlapping with the silicon nitride membrane were removed. Potassium and chloride ions were added to neutralize the system and bring the ion concentration to 2 M. The final system contained 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 114 atoms. Subsequently, the conjugate gradient method was used to minimize each system by 9600 steps, and then equilibrated in the number of atoms, pressure, and temperature (NPT) ensemble for approximately 24 ns. In the process of minimizing and equilibrating, constraints are imposed on the deceleration system and peptides to avoid arbitrary movement. The spring constant of carbon nanotube and silicon nitride is 10 kcal mol−1 Å−2, and the spring constant of peptides is 1 kcal mol−1 Å−2. The Nose–Hoover Langevin piston pressure control was used to maintain the pressure of the system at 1 atm by adjusting the system's dimension along the z direction. Finally, Production simulations under applied electric field were performed in the constant number of atoms, volume and temperature (NVT) ensemble for approximately 12 ns. At this point, the fixation of the peptide is released, and the electric field strength applied by the system is 0.04 V Å−1.
114 atoms. Subsequently, the conjugate gradient method was used to minimize each system by 9600 steps, and then equilibrated in the number of atoms, pressure, and temperature (NPT) ensemble for approximately 24 ns. In the process of minimizing and equilibrating, constraints are imposed on the deceleration system and peptides to avoid arbitrary movement. The spring constant of carbon nanotube and silicon nitride is 10 kcal mol−1 Å−2, and the spring constant of peptides is 1 kcal mol−1 Å−2. The Nose–Hoover Langevin piston pressure control was used to maintain the pressure of the system at 1 atm by adjusting the system's dimension along the z direction. Finally, Production simulations under applied electric field were performed in the constant number of atoms, volume and temperature (NVT) ensemble for approximately 12 ns. At this point, the fixation of the peptide is released, and the electric field strength applied by the system is 0.04 V Å−1.
MD simulation of peptide translocation through a deceleration system composed of carbon nanotube and gold nanoparticle
The setup of the simulation system is very similar to the setup of peptide translocation through a deceleration system composed of carbon nanotube. However, spherical gold nanoparticle with a diameter of 2.038 nm were added to carbon nanotube. Its initial coordinates are (0, −17.2, −17) and the crystal orientation is [1,0,0], which carries no charge in this simulation. At this time, the total number of atoms in the system is 43681. Subsequently, 9600 steps of minimization simulation, 24 ns of equilibration simulation, and 24 ns of production simulation were conducted with different positions of gold nanoparticle. The position movement of gold nanoparticle is achieved through steered molecular dynamics (SMD).
MD simulation of peptide translocation through a deceleration system composed of carbon nanotube and charged gold nanoparticle
The initial setup of the simulation system is similar to the setup of peptide translocation through a deceleration system composed of carbon nanotube and gold nanoparticle. However, the surface of gold nanoparticle carries charges with a surface charge density of 0.0023 e Å−2. Subsequently, 9600 steps of minimization simulation, 24 ns of equilibration simulation, and 24 ns of production simulation were conducted with different positions of gold nanoparticle. The position movement of gold nanoparticle is still achieved through steered molecular dynamics (SMD).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the financial support from the National Key R&D Program of China (2023YFF1205801), the National Natural Science Foundation of China (grants no. 52275558 & 52035003) and the Natural Science Foundation of Jiangsu Province-Outstanding Youth Foundation (grant no. BK20230084). Dr Wei Si acknowledges financial support from the Zhishan Young Scholar Award Program of Southeast University and the Fundamental Research Funds for the Central Universities (grant no. 2242024RCB0002).References
- B. P. Ismail, L. Senaratne-Lenagala, A. Stube and A. Brackenridge, Animal Front., 2020, 10, 53–63 CrossRef.
- Z. S. Zhu, A. P. Bassey, Y. Q. Cao, Y. L. Ma, M. Huang and H. S. Yang, Food Res. Int., 2022, 160, 111725 CrossRef CAS PubMed.
- K. J. Han, Y. Zhao, Y. H. Sun and Y. Li, Plant Biotechnol. J., 2023, 21, 2433–2457 CrossRef PubMed.
- M. B. E. Schaaf, T. G. Keulers, M. A. Vooijs and K. M. A. Rouschop, FASEB J., 2016, 30, 3961–3978 CrossRef CAS PubMed.
- M. Kulmanov, M. A. Khan and R. Hoehndorf, Bioinformatics, 2018, 34, 660–668 CrossRef CAS PubMed.
- V. Gligorijevic, P. D. Renfrew, T. Kosciolek, J. K. Leman, D. Berenberg, T. Vatanen, C. Chandler, B. C. Taylor, I. M. Fisk, H. Vlamakis, R. J. Xavier, R. Knight, K. Cho and R. Bonneau, Nat. Commun., 2021, 12, 3168 CrossRef CAS PubMed.
- B. Kuhlman and P. Bradley, Nat. Rev. Mol. Cell Biol., 2019, 20, 681–697 CrossRef CAS PubMed.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N. V. Buchete, S. Coté, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. G. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Y. Wang and P. Derreumaux, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS.
- T. Bepler and B. Berger, Cell Syst., 2021, 12, 654–669 CrossRef CAS.
- D. N. Woolfson, J. Biol. Chem., 2023, 299, 104579 CrossRef CAS.
- M. M. Babu, Biochem. Soc. Trans., 2016, 44, 1185–1200 CrossRef CAS PubMed.
- R. van der Lee, M. Buljan, B. Lang, R. J. Weatheritt, G. W. Daughdrill, A. K. Dunker, M. Fuxreiter, J. Gough, J. Gsponer, D. T. Jones, P. M. Kim, R. W. Kriwacki, C. J. Oldfield, R. V. Pappu, P. Tompa, V. N. Uversky, P. E. Wright and M. M. Babu, Chem. Rev., 2014, 114, 6589–6631 CrossRef CAS PubMed.
- X. J. Pan and T. Kortemme, J. Biol. Chem., 2021, 296, 100558 CrossRef CAS.
- A. W. Brauer, C. L. Oman and M. N. Margolies, Anal. Biochem., 1984, 137, 134–142 CrossRef CAS PubMed.
- K. K. Han, D. Tetaert, B. Debuire, M. Dautrevaux and G. Biserte, Biochimie, 1977, 59, 557–576 Search PubMed.
- J. D. Chapman, D. R. Goodlett and C. D. Masselon, Mass Spectrom. Rev., 2014, 33, 452–470 CrossRef CAS PubMed.
- J. R. Yates, J. Am. Chem. Soc., 2013, 135, 1629–1640 CrossRef CAS PubMed.
- Z. L. Hu, M. Z. Huo, Y. L. Ying and Y. T. Long, Angew. Chem., Int. Ed., 2021, 60, 14738–14749 CrossRef CAS PubMed.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 Search PubMed.
- Y. L. Ying, Z. L. Hu, S. L. Zhang, Y. J. Qing, A. Fragasso, G. Maglia, A. Meller, H. Bayley, C. Dekker and Y. T. Long, Nat. Nanotechnol., 2022, 17, 1136–1146 CrossRef CAS.
- J. A. Alfaro, P. Bohländer, M. J. Dai, M. Filius, C. J. Howard, X. F. van Kooten, S. Ohayon, A. Pomorski, S. Schmid, A. Aksimentiev, E. V. Anslyn, G. Bedran, C. Cao, M. Chinappi, E. Coyaud, C. Dekker, G. Dittmar, N. Drachman, R. Eelkema, D. Goodlett, S. Hentz, U. Kalathiya, N. L. Kelleher, R. T. Kelly, Z. Kelman, S. H. Kim, B. Kuster, D. Rodriguez-Larrea, S. Lindsay, G. Maglia, E. M. Marcotte, J. P. Marino, C. Masselon, M. Mayer, P. Samaras, K. Sarthak, L. Sepiashvili, D. Stein, M. Wanunu, M. Wilhelm, P. Yin, A. Meller and C. Joo, Nat. Methods, 2021, 18, 604–617 Search PubMed.
- X. J. Zhang, Y. Dai, J. L. Sun, J. L. Shen, M. H. Lin and F. Xia, Anal. Chem., 2024, 96, 2277–2285 CrossRef CAS PubMed.
- H. Brinkerhoff, A. S. W. Kang, J. Q. Liu, A. Aksimentiev and C. Dekker, Science, 2021, 374, 1509–1513 CrossRef CAS.
- C. Cao, N. Cirauqui, M. J. Marcaida, E. Buglakova, A. Duperrex, A. Radenovic and M. Dal Peraro, Nat. Commun., 2019, 10, 4918 CrossRef PubMed.
- E. C. Yusko, J. M. Johnson, S. Majd, P. Prangkio, R. C. Rollings, J. L. Li, J. Yang and M. Mayer, Nat. Nanotechnol., 2011, 6, 253–260 CrossRef CAS PubMed.
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, Science, 2023, 382, 6673 Search PubMed.
- Y. H. Wang, Y. Zhao, A. Bollas, Y. R. Wang and K. F. Au, Nat. Biotechnol., 2021, 39, 1348–1365 CrossRef CAS PubMed.
- H. Satam, K. Joshi, U. Mangrolia, S. Waghoo, G. Zaidi, S. Rawool, R. P. Thakare, S. Banday, A. K. Mishra, G. Das and S. K. Malonia, Biology, 2023, 12, 997 Search PubMed.
- B. Y. Guo, T. Zeng and H. C. Wu, Sci. Bull., 2015, 60, 287–295 CrossRef CAS.
- A. Eatemadi, H. Daraee, H. Karimkhanloo, M. Kouhi, N. Zarghami, A. Akbarzadeh, M. Abasi, Y. Hanifehpour and S. W. Joo, Nanoscale Res. Lett., 2014, 9, 383 CrossRef PubMed.
- D. C. Ferrier and K. C. Honeychurch, Biosensors, 2021, 11, 486 Search PubMed.
- M. S. Khan, B. H. J. Gowda, N. Hasan, G. Gupta, T. Singh, S. Md and P. Kesharwani, Eur. Polym. J., 2024, 206, 112800 CrossRef.
- H. Zare, S. Ahmadi, A. Ghasemi, M. Ghanbari, N. Rabiee, M. Bagherzadeh, M. Karimi, T. J. Webster, M. R. Hamblin and E. Mostafavi, Int. J. Nanomed., 2021, 16, 1681–1706 CrossRef.
- B. Li, X. X. Zhang, H. Y. Huang, L. Q. Chen, J. H. Cui, Y. L. Liu, H. H. Jin, B. J. Lee and Q. R. Cao, Int. J. Pharm., 2018, 543, 8–20 CrossRef CAS PubMed.
- A. H. Pourasl, M. T. Ahmadi, M. Rahmani, H. C. Chin, C. S. Lim, R. Ismail and M. L. P. Tan, Nanoscale Res. Lett., 2014, 9, 33 CrossRef PubMed.
- W. C. Peng, S. H. Yan, K. Zhou, H. C. Wu, L. Liu and Y. L. Zhao, Nat. Commun., 2023, 14, 2662 Search PubMed.
- N. Li, P. X. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
- S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS PubMed.
- S. J. H. Shahzad, E. Bouri, D. Roubaud and L. Kristoufek, Econ. Model., 2020, 87, 212–224 CrossRef.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2020, 120, 464–525 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- Y. Z. Shi, Q. H. Song, I. Toftul, T. T. Zhu, Y. F. Yu, W. M. Zhu, D. P. Tsai, Y. Kivshar and A. Q. Liu, Appl. Phys. Rev., 2022, 9, 031303 CAS.
- D. L. Gao, W. Q. Ding, M. Nieto-Vesperinas, X. M. Ding, M. Rahman, T. H. Zhang, C. Lim and C. W. Qiu, Light: Sci. Appl., 2017, 6, e17039 Search PubMed.
- I. De Vlaminck and C. Dekker, Annu. Rev. Biophys., 2012, 41, 453–472 CrossRef CAS.
- S. A. Hollingsworth and R. O. Dror, Neuron, 2018, 99, 1129–1143 Search PubMed.
- X. L. Chen, A. Munjiza, K. Zhang and D. S. Wen, J. Phys. Chem. C, 2014, 118, 1285–1293 Search PubMed.
- C. T. Lai, W. Q. Sun, R. U. Palekar, C. S. Thaxton and G. C. Schatz, ACS Appl. Mater. Interfaces, 2017, 9, 1247–1254 Search PubMed.
- C. Hyun, R. Rollings and J. L. Li, Small, 2012, 8, 385–392 Search PubMed.
- C. Y. Yan, C. Q. Chen, L. Hou, H. J. Zhang, Y. Y. Che, Y. D. Qi, X. J. Zhang, J. L. Cheng and Z. Z. Zhang, J. Drug Targeting, 2017, 25, 163–171 CrossRef CAS PubMed.
- Y. H. Li, Z. W. Li, R. P. Misra, C. X. Liang, A. J. Gillen, S. Zhao, J. Abdullah, T. Laurence, J. A. Fagan, N. Aluru, D. Blankschtein and A. Noy, Nat. Mater., 2024, 23, 1123–1130 Search PubMed.
- T. Halicioglu and G. M. Pound, Phys. Status Solidi A, 1975, 30, 619–623 CrossRef CAS.
- A. G. Cherstvy, J. Phys. Chem. B, 2006, 110, 14503–14506 CrossRef CAS.
- A. G. Cherstvy, J. Phys. Chem. B, 2007, 111, 7914–7927 Search PubMed.
- N. Laohakunakorn, V. V. Thacker, M. Muthukumar and U. F. Keyser, Nano Lett., 2015, 15, 695–702 Search PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS.
- Y. Jejoong and A. Aleksei, J. Phys. Chem. Lett., 2012, 3, 45–50 Search PubMed.
- Y. Jejoong and A. Aleksei, Biophys. J., 2016, 110, 646a Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01017a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2025 |