
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decoding disorder: unravelling entropy effects in deep eutectic systems with neutron spectroscopy†
Rafael A. F.
Serrano
 a,
Catarina F.
Araújo
a,
Paulo
Ribeiro-Claro
a,
Pedro D.
Vaz
b,
Svemir
Rudić
c,
João A. P.
Coutinho
a and
Mariela M.
Nolasco
*a
a,
Catarina F.
Araújo
a,
Paulo
Ribeiro-Claro
a,
Pedro D.
Vaz
b,
Svemir
Rudić
c,
João A. P.
Coutinho
a and
Mariela M.
Nolasco
*a
aCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Aveiro, 3810-193, Portugal. E-mail: mnolasco@ua.pt
bChampalimaud Foundation, Champalimaud Centre for the Unknown, 1400-038 Lisboa, Portugal
cISIS Neutron & Muon Source, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0QX, UK
First published on 15th May 2025
Abstract
The vibrational dynamics and phase behavior of deep eutectic systems (DES) comprising ammonium salts and urea have been investigated using a combination of computational chemistry with inelastic neutron scattering (INS) and isotopic substitution. The study explores the impact of cation (a)symmetry on the vibrational modes of ammonium salts and urea in eutectic mixtures. By employing deuterated urea in tetraalkylammonium-based DES mixtures, selective observation of the INS spectra of individual components is achieved. Subtracting INS spectra for isotopically substituted urea mixtures provided separate “views” of the salt and urea contributions, highlighting changes in intermolecular interactions. Two types of organization of urea molecules are derived from the INS bands of urea in the mixture with symmetric and asymmetric cations. This is consistent with the view that the cation's asymmetry affects mostly the urea side of the solid–liquid equilibrium diagram and deep eutectic behavior stems from the deviation of the urea solid–liquid equilibrium line from ideality.
1. Introduction
Recent studies have highlighted the significant role of entropy in influencing the behavior of eutectic systems, shifting attention away from the traditionally emphasized enthalpic factors.1–4 While most research on deep eutectic solvents (DES) has focused on how novel intermolecular interactions among DES constituents lower the melting point, recent findings suggest that asymmetric components may lead to deviations from ideal behavior through entropic effects. Specifically, in a urea-tetraalkylammonium DES3 the asymmetry of the cation has been found to be the main factor leading to a large negative deviation from ideality. The melting entropy of urea, acting as solvent, increases significantly due to the presence of asymmetric solute cations and this affects the urea side of the solid–liquid equilibrium (SLE) diagram.Despite growing evidence that asymmetry in cations can increase melting entropy and drive deviations from ideal eutectic melting points, there remains a critical need for further supporting evidence into how these factors relate to the structure and dynamics of the liquid phase in DES. Specifically, how these entropic effects influence molecular interactions, such as hydrogen bonding networks and disorder, within the liquid phase is not yet fully understood. Addressing this gap requires experimental techniques capable of probing molecular-level interactions, and inelastic neutron scattering (INS) emerges as a powerful tool to achieve this.
Previous works demonstrated the usefulness of inelastic neutron scattering (INS) spectroscopy for probing the molecular dynamics of DES.3,5,6 Additionally, discrete and periodic density functional theory (DFT) calculations complement these studies by helping assign vibrational modes and further elucidating the DES structure and dynamics. INS is particularly powerful for exploring vibrational modes sensitive to molecular disorder, providing insights into how molecular asymmetry contributes to observed behavior. The ability of INS to detect methyl torsional modes in tetraalkylammonium salts, for example, allows to measure the degree of free space around methyl groups, shedding light on the hydrogen bonding network in choline chloride:urea DES.5 For the same system, the intensity changes in the 1400 cm−1 region upon the increase of water content reveal the strengthening of hydrogen bonds with the NH donors.6
A critical but underutilized approach in this field is the use of hydrogen/deuterium (H/D) isotopic substitution. As stated by B. S. Hudson7,8 and underlined more recently by W. Langel,9 H/D substitution permits nearly effective masking of components in multi-component systems, and this is the aspect of INS that most clearly distinguishes it from other methods. This technique is commonly applied in quasi-elastic neutron scattering (QENS) to study protein dynamics (see e.g. ref. 10) and there are a few reports on the use of isotopic H/D substitution in the study of polymers, either natural (cellulose)11,12 or synthetic.13–17 However, to the best of our knowledge, it has been less explored in INS studies of systems like DES.18,19 Its application to DES can provide detailed insights into molecular interactions and dynamics by shifting the scattering cross-section without significantly altering the molecular structure.
In this work, the enthalpic and entropic factors affecting the behavior of urea–tetralkylammonium chloride DES are addressed through INS, taking advantage of the H/D isotopic substitution. The urea 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures with the asymmetric cholinium chloride salt, [Ch]Cl, already discussed in the previous work, is compared with mixtures with two new salts (Scheme 1): (i) chlorocholinium chloride, [ChloroCh]Cl, in which the cation keeps the asymmetry of [Ch]Cl, but replaces the strong hydrogen bond donor and acceptor OH group by a chlorine atom, which is a weak acceptor; and (ii) N-ethylcholinium chloride, [EthylCh]Cl, which adds asymmetry to the cation by replacing a methyl group by a longer ethyl fragment. Isotopic H/D substitution of urea allows the (almost) separated analysis of both DES components, as the INS spectra of DES mixtures with urea-d4 are dominated by the cation contribution. On the other hand, the urea contribution to INS spectra of each mixture can be obtained by subtracting the spectrum of (urea-d4)-based mixture from the spectrum (urea-h4)-based mixture.
:1 mixtures with the asymmetric cholinium chloride salt, [Ch]Cl, already discussed in the previous work, is compared with mixtures with two new salts (Scheme 1): (i) chlorocholinium chloride, [ChloroCh]Cl, in which the cation keeps the asymmetry of [Ch]Cl, but replaces the strong hydrogen bond donor and acceptor OH group by a chlorine atom, which is a weak acceptor; and (ii) N-ethylcholinium chloride, [EthylCh]Cl, which adds asymmetry to the cation by replacing a methyl group by a longer ethyl fragment. Isotopic H/D substitution of urea allows the (almost) separated analysis of both DES components, as the INS spectra of DES mixtures with urea-d4 are dominated by the cation contribution. On the other hand, the urea contribution to INS spectra of each mixture can be obtained by subtracting the spectrum of (urea-d4)-based mixture from the spectrum (urea-h4)-based mixture.
 | ||
| Scheme 1 Schematic structures of urea and the cations of the chloride-based salts investigated in this work. From left to right, urea, choline, chlorocholine, N-ethylcholine. Cations are labelled as [Ch], [ChloroCh], and [EthylCh], respectively. | ||
To this end, a step-by-step strategy was developed for using INS spectroscopy to investigate the structure and dynamics of urea:tetraalkylammonium chloride DES. Initially, molecular models that best represent the crystalline forms of each pure component were chosen. Key vibrational modes in the INS spectra of the pure salts and urea were then identified. Next, isotopic substitution was utilized to isolate the spectral contributions of the salts and urea within the DES mixtures. Finally, the changes in the vibrational profiles of both DES components upon mixing were analyzed to assess the impact of asymmetry-driven entropy.
2. Materials and methods
2.1 Chemicals
All pure compounds were obtained commercially: chlorocholine chloride ((2-chloroethyl)trimethylammonium chloride, ≥98% purity, Sigma-Aldrich), ethylcholine chloride (N-ethyl-2-hydroxy-N,N-dimethylethanaminium chloride, ≥97% purity, AKOS GmbH custom synthesis), tetramethylammonium chloride (≥98% purity, Sigma-Aldrich), tetraethylammonium chloride (≥99% purity, Sigma-Aldrich) and urea-d4 (98% atom D, Sigma-Aldrich).2.2 Sample preparation for INS and melting temperature measurements
The eutectic compositions of the systems under study were found to have a mole fraction of urea [x(Urea)] corresponding approximately to a urea:salt ratio of 1:2, in agreement with reported values.20,21 The binary mixtures were all prepared by the heating method. Prior to mixture preparation, the individual components were dried during 5 days under vacuum at 60 °C and up to a constant weight by using phosphorus pentoxide and additionally combining vacuum and heating in a Schlenck apparatus. After drying, the components were weighed inside a glovebox into sealed glass vials using a previously reported procedure6 and placed under constant stirring and room temperature until a homogenous transparent liquid was obtained. The mixtures are above their respective melting points at room temperature.
Melting temperatures for the binary mixtures were obtained following the melting point capillary method, as described in the previous work.3,5,6 Heating was performed in an oil bath for [ChloroCh]Urea and in a cryoscopic bath for [EthylCh]Urea mixtures. Temperatures reported correspond to the point at which the last solid phase disappeared, and the associated uncertainty reflects ±2σ based on multiple replicates.
2.3 Spectra acquisition
The INS spectra were collected using the TOSCA22–25 time-of-flight spectrometer at the ISIS Neutron and Muon Source of the STFC Rutherford Appleton Laboratory (Chilton, UK). Each sample, weighing ca. 0.6–1.0 g, was transferred to a flat thin-walled aluminum can, inside a controlled atmosphere glove box, to avoid moisture uptake. The sealed can was then mounted perpendicular to the incident beam using a regular TOSCA centered stick. Before placement in the neutron beam path, the mixtures were “shock-frozen” by quenching samples in liquid nitrogen to avoid phase separation and preserve their room-temperature morphology. This procedure has been previously validated by comparison with a “slow cooling” process.6 Spectra were collected below 15 K and measured in the 0 to 8000 cm−1 region. Data was converted to the conventional scattering law, S(Q, ν) vs. energy transfer (in cm−1), using the MANTID program (version 6.9.1).26 All INS spectra of DES samples and of pure alkylammonium chloride salts were obtained within project RB1810054.27Periodic DFT calculations: calculations were performed using the plane wave pseudopotential method as implemented in CASTEP 8.0 code.28,29 All calculations were done using the Perdew−Burke−Ernzerhof (PBE) functional based on the generalized gradient approximation (GGA).30 The plane-wave cutoff energy was set at 830 eV. Brillouin zone sampling of electronic states was performed on 2 × 4 × 5 Monkhorst–Pack grid. The initial structure of the tetraethylammonium chloride urea layer-type inclusion compound was retrieved from the Cambridge Structural database (CSD code PAMLAQ).31 The initial structures for [ChloroCh]Cl and [EthylCh]Cl were built from the X-ray structure of [Ch]Cl (CSD code CHOCHL0132) by replacing the OH group by the Cl atom or the H atom by the CH3 group, and their optimized structures are made available by the authors in Theoretical Crystallographic Open Database, TCOD entries 30000154 and 30000155, respectively. Geometry optimizations were carried out with fixed cell parameters and accuracy of the optimization requested residual forces to fall below 0.005 eV A−1. Phonon frequencies were obtained by diagonalization of dynamical matrices calculated using density-functional perturbation theory.33 The calculated atomic displacements in each mode that are part of the CASTEP output enable visualization of the atomic motions and support the assignment of vibrational modes. The inelastic neutron scattering simulated intensities were estimated from the calculated eigenvectors using the AbINS software34 a part of the Mantid package.26
2.4 Discrete DFT calculations
Geometry optimizations and vibrational frequency calculations of chloride:urea and urea:urea clusters were computed with the Gaussian 09 software,35 using the B3LYP density functional and the 6-311++G(d,p) basis set as implemented in Gaussian package. The clusters were built following the results of ref. 36. Optimized structures were found to be real minima, with no imaginary frequencies. The inelastic neutron scattering simulated intensities were estimated from the calculated eigenvectors using the AbINS software,34 as above.3. Results
3.1 The pure compounds: calculated vs. experimental spectra
The INS spectra of pure [Ch]Cl and tetraethylammonium chloride, [N2,2,2,2,2]Cl, have been presented and discussed previously.3,37 Both systems are important in this work as reference systems, either for analyzing the effect of symmetry loss or the effects of functional group substitution.The work of Pawlukojć et al.37 shows that the computational model accurately describes the experimental spectrum of [Ch]Cl, allowing for a clear assignment of bands, such as the OH torsion mode (around 623 cm−1), CN stretching and bending modes, and intense CH3 torsion modes. In what concerns [N2,2,2,2]Cl, the computational results enable the comparison between conformational alternatives in the crystal, as described earlier.3
The experimental INS spectrum of [ChloroCh]Cl is compared with the calculated spectrum in Fig. 1. The computational model was built from the [Ch]Cl crystalline structure, replacing the OH group by a chlorine atom. This model yields a calculated INS spectrum matching reasonably with the experimental spectrum, capturing most of the observed vibrational bands. The absence of the OH torsion (observed around 625 cm−1 for [Ch]Cl) is notable, and several bands can be accurately assigned.
 | ||
| Fig. 1 Experimental (top line, green) and calculated (bottom line, blue) INS spectra for chlorocholine chloride, [ChloroCh]Cl, pure salt. Some band assignments (approximate description) are shown for illustration purposes. | ||
Fig. 2 compares the experimental spectrum of [EthylCh]Cl with the calculated spectra from two computational models, considering either the trans–trans or gauche–gauche chain orientations. Previous studies3 concluded that the trans–trans orientation is preferred in [N2,2,2,2]Cl crystals, whereas gauche orientations are favored in triethylmethylammonium chloride([N2,2,2,1]Cl). For [EthylCh]Cl, while neither alternative perfectly fits the experimental spectrum, the trans–trans model provides a better match, namely in the description of δNC4 deformation modes in the 400–600 cm−1 region and for the νNC stretching modes near 900 cm−1.
 | ||
| Fig. 2 Experimental (top line, red) and calculated INS spectra for two polymorphs of Ethylcholine chloride, [EthylCh]Cl pure salt: gauche–gauche polymorph (bottom line, grey) and trans–trans polymorph (middle line, blue). Some band assignments (approximate description) are shown for illustration purposes. | ||
3.2 The mixtures: ideal vs. real
As discussed previously38 for a binary mixture with complete immiscibility in the solid phase and full miscibility in the liquid phase, the solid–liquid equilibrium (SLE) phase diagram of each component can be described by the following equation: | (1) |
 | (2) |
Analysing Fig. 3, which presents the ideal lines predicted by eqn (2), it can be concluded that the melting temperature of the ideal eutectic point is primarily determined by the ideal curve of urea. Given that the melting temperatures of the ammonium salts are typically above 500 K and their melting enthalpies are below 10 kJ mol−1, the ideal lines intersect within the 0.4 < xUrea < 0.8 interval. Consequently, the eutectic point temperature for ideal mixtures is predicted to be within the 360 ± 30 K range. For the typical 1:1 and 1:2 salt:urea ratios highlighted in Fig. 3, this interval narrows to approximately 360 ± 10 K (i.e., ca. 90 ± 10 °C).
 | ||
| Fig. 3 Ideal liquidus temperature calculated for urea (black dashed line) and for two hypothetical salts with different melting enthalpy and melting temperature: ΔmH = 10 kJ mol−1, Tm = 500 K (blue line) and: ΔmH = 9 kJ mol−1, Tm = 600 K (magenta line). The shadowed areas represent the expected interval for the cations’ ideal curve, assuming the ΔmH uncertainty of ±4 kJ mol−1. Vertical dashed lines for x = 0.5 and x = 0.67. | ||
Table 1 shows that eutectic systems based on [Ch]Cl and [ChloroCh]Cl exhibit similar melting temperatures at the eutectic point, in contrast to [EthylCh]Cl, which shows a deeper deviation. This suggests that substituting the hydroxyl group in the [Ch] cation with a Cl atom has minimal impact on the eutectic point. Since the hydroxyl group is a strong hydrogen bond donor and acceptor, whereas the covalently bonded Cl atom is at best a weak acceptor, it can be concluded that hydrogen bonds between the cation and urea do not significantly influence the melting enthalpy of the [Ch]Cl–urea eutectic mixture.
:2 salt:urea
On the other hand, replacing a methyl group attached to the nitrogen atom with an ethyl group in the [Ch] cation has a larger impact on the melting temperature of the eutectic point. Relative to the average value obtained from Fig. 3, the difference between ideal and real eutectic temperatures, ΔTm, can be estimated to be ca. −60 °C for [Ch]Cl:urea and [ChoroCh]Cl:urea, and ca. −100 °C for [EthylCh]Cl:urea. The greater decrease in melting point for the latter is then attributed to the entropic effect arising from the increased asymmetry of the N-substituents in the [EthylCh] cation.
3.3 The mixtures: H/D substitution
Fig. 4 illustrates the INS spectra and their analysis for the mixture of [Ch]Cl:urea 1:2, comparing the samples containing urea-h4 and urea-d4. The INS spectrum of the [Ch]Cl:urea 1:2 sample containing urea-d4 is dominated by the bands of [Ch]Cl and provides a view of “[Ch]Cl in the mixture”, to be compared with pure [Ch]Cl. On the other hand, by subtracting this spectrum from the one containing urea-h4, the difference spectrum is mainly due to the urea signals and can be considered the view of “urea in the mixture”. This is the approach used in the following sections, while acknowledging its limitations. The spectrum of the “cation in the mixture” includes signals from deuterated urea, which, as demonstrated from direct comparison between urea-h4 and urea-d4 INS spectra (Fig. S1, ESI†), introduces weak interference. More critical are the constraints affecting the “urea in the mixture” spectrum, obtained from mathematical subtraction. The most significant limitation is the strong dependence of the difference spectrum on the signal-to-noise ratio of the original spectra. Additionally, small variations in the position or width of the subtracted bands can produce derivative-like oscillations in the resulting difference spectrum.
 | ||
| Fig. 4 Experimental INS spectra of [Ch]Cl:urea systems. From top to bottom, [Ch]Cl:urea, [Ch]Cl:urea-d4 and pure [Ch]Cl; Difference spectrum obtained from the subtraction ([Ch]Cl:urea) minus ([Ch]Cl:urea-d4), to be compared with pure urea. | ||
3.4 The ammonium salt view of DES (with urea-d4)
In previous work,3 the effect of cation asymmetry upon the deviation from ideality in a series of tetraalkylammonium chloride–urea mixtures has been related with their respective INS spectra in the region of methyl torsions. It has been shown that the mixture with the asymmetrically substituted cation presents stronger deviation from ideality and significant broadening of INS bands ascribed to torsional motions of the methyl group. Fig. 5 presents the INS spectra in the region of methyl torsions for the three mixtures with urea-d4. | ||
| Fig. 5 Experimental INS spectra, in the region of the methyl torsional modes, of the mixtures with urea-d4 and the three asymmetric alkylammonium chloride salts. Labels identify the cation only: from top to bottom, ethylcholine chloride:urea-d4, chlorocholine chloride:urea-d4, and choline chloride:urea-d4. | ||
The inclusion of [Ch]Cl:urea-d4 system in this figure confirms the origin of the bands in this region: the reported profile of the [Ch]Cl:urea system (Fig. 8, ref. 3) is identical to the one observed herein for [Ch]Cl:urea-d4, ruling out possible contributions from urea in the previous analysis of this region.3,5,6
Fig. 5 confirms the previously suggested hypothesis that broader methyl torsional bands reflect a greater degree of disorder in the mixture which, in turn, leads to a deeper negative deviation from ideality.3 It should be mentioned that a previous study with the [Ch]Cl:urea 1:2 DES (also known as “Reline”) concluded that urea molecules displace the chloride anions away from the cations’ solvation shell, thus increasing methyl group dynamics.5 The three methyl-torsion components identified in [Ch]Cl:urea are also evident in the INS profile of the [ChloroCh]Cl:urea DES. In the case of the [EthylCh]Cl:urea mixture, there is loss of band resolution, ascribed to the additional mobility and molecular disorder brought by the ethyl group.
The behavior of the ammonium cation in the different mixtures is also evident in the 400–1800 cm−1 region of the INS spectra. Fig. 6 compares the spectra of pure ammonium salts with those of their mixture with urea-d4, assumed to reveal the behavior of the “salt in the mixture”. In the case of [Ch]Cl, several noteworthy changes are observed. The disappearance of the OH torsion mode, at ca. 623 cm−1, results from the expected H/D exchange between ND2 and OH groups (O–D modes have very low intensity, as urea-d4 modes). Significant broadening is evident in the skeletal deformation modes, particularly the δNCC mode at approximately 530 cm−1. This broadening can be straightforwardly ascribed to the increased dynamics of the methyl and methylene groups in the mixture.
 | ||
| Fig. 6 Comparison between INS spectra of pure salts (lower black lines) and their eutectic mixtures with urea-d4 (upper lines). From top to bottom, the salts are choline chloride, chlorocholine chloride and ethylcholine chloride. The mixtures with urea-d4 provide the INS view of ammonium chloride salt “in the mixture”. | ||
The INS spectra for [ChloroCh]Cl and [EthylCh]Cl systems further reinforce these observations. In general, the cation in the mixtures exhibits increased dynamics, which is reflected in the broadening of spectral bands and a noticeable loss of resolution. As in [Ch]Cl case above, the most relevant change is the noticeable broadening of the δNC4 and δNCC deformation bands, although differences in the CH3 rocking modes are also observed. In the case of [EthylCh]Cl, a significant change of the band profile is observed for the ca. 1200–1300 cm−1 region. Since the bands in this region are related to bending modes of the CH2 groups (e.g., CH2 twisting modes), the changes can be ascribed to the conformational freedom of the additional ethyl group.
3.5 The urea view of DES (from difference spectra)
The INS spectrum of urea was described by Hudson et al.,39 based on periodic DFT calculations. Fig. 7, bottom line, shows the simulated INS spectrum obtained using periodic DFT calculations with a large crystalline cell containing 16 urea molecules, which provides a slightly improved representation of the experimental spectrum. Both previous39 and present calculations yield assignments of the INS bands consistent with the work of Keuleers et al., who analyzed the infrared and Raman spectra of urea in its crystalline form.40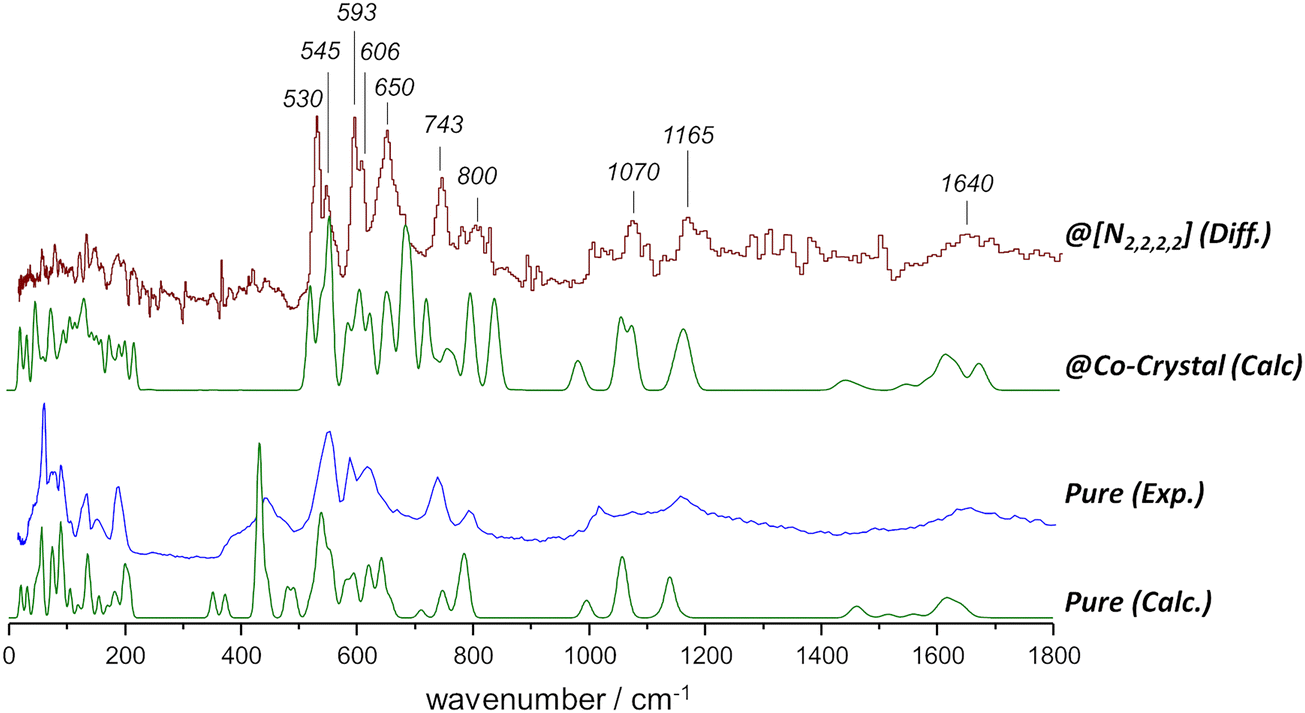 | ||
| Fig. 7 INS spectra of urea, up to 1800 cm−1. From bottom to top: calculated and experimental INS spectra of pure urea; calculated spectrum of urea in its co-crystal with tetraethylammonium chloride; experimental spectrum of urea in its eutectic mixture with tetraethylammonium chloride (difference spectra, see text). | ||
As mentioned above, subtracting the spectrum of urea-d4 mixture from the spectrum of urea-h4 mixture primarily removes the ammonium salt bands, revealing the spectrum of “urea in the mixture”. Fig. 7, top line, also presents the INS spectrum of urea in the mixture with the symmetrical [N2,2,2,2]Cl salt, a system chosen to represent the symmetrical cation because its urea co-crystal structure is well-characterized (CSD code PAMLAQ). This known structure enables periodic-DFT calculations of its INS spectrum, allowing the urea contributions to be isolated by excluding non-urea atomic contributions. The resulting calculated spectrum of “urea in its co-crystal with [N2,2,2,2]Cl” is then compared to the experimental spectrum of “urea in the mixture with [N2,2,2,2]Cl”, obtained via spectral subtraction. Fig. 7 presents this comparison alongside the observed and calculated spectra of pure crystalline urea.
As expected, the “urea in co-crystal” calculated spectrum does not entirely match the spectrum of urea in the mixture. The “shock freezing” procedure used to avoid the formation of pure solids (see Fig. S2, ESI†) is also incompatible with the slow evaporation process required for the formation of a co-crystal.31 Nonetheless, there are notable similarities between the spectra, and several vibrational modes of urea in the mixture with [N2,2,2,2]Cl can be confidently assigned from this comparison.
Table 2 presents the proposed assignments, based on periodic DFT calculations, for urea in the mixture with tetraethylammonium chloride, compared with pure urea. The disruption of the head-to-tail hydrogen bond network of pure urea is quite evident in the NH2 wagging modes, namely, in the disappearance of the intense symmetric NH2 wag band below ca. 500 cm−1. In addition, the symmetric and asymmetric NH2 wagging modes significantly mix with NH2 torsional modes and CO bending modes up to ca. 750 cm−1. These changes can be ascribed to the formation of new urea–chloride and urea–urea associations in the mixture.
| INS maxima/cm−1 | Approximate descriptiona | |
|---|---|---|
| Pure39 | @[N2,2,2,2]Cl | |
| a Same notation as Keuleers et al.:40ν – stretching; δ – deformation; w – wagging; ρ – rocking; π – out-of-plane deformation; τ – torsion. | ||
| 445 | w sNH2 | |
| 530 | 530 | δNCN |
| 553 | 545 | w sNH2 |
| 590 | 593 | δCO + wsNH2 |
| 606 | w sNH2 | |
| 621 | w asNH2 + τNH2 | |
| 650 | w asNH2 + τNH2 | |
| 743 | 743 | w sNH2 + πCO |
| 798 | 800 (broad) | τNH2 + πCO |
| 1020 | 1002 | ν sCN |
| 1079 | 1070 | ρ asNH2 |
| 1162 | 1165 | ρ sNH2 |
| 1662 | 1640 (broad) | δ asNH2; δsNH2 |
Fig. 8 illustrates the changes in the spectrum of pure urea when observed in eutectic mixtures with asymmetric salts. Unlike the behavior seen in the eutectic mixture with the symmetric salt shown in Fig. 7, mixtures with asymmetric salts produce INS spectra with broader bands, giving their increased dynamics. For instance, from Fig. 7 and 8, doublets observed in the 500–600 cm−1 range merge into single bands at ca. 535 cm−1 and 590 cm−1, respectively. And the intense band at 650 cm−1 is blurred to a broad intensity shoulder in the mixture with [Ch]Cl, and further depreciated in the mixture with [EthylCh]Cl.
 | ||
| Fig. 8 INS spectrum, up to 1800 cm−1, of pure urea compared with its INS spectra in different eutectic mixtures, obtained from difference spectra (see text). From bottom to top: pure urea, urea in the eutectic mixture with choline chloride [Ch]Cl, and with ethylcholine chloride [EthylCh]Cl. | ||
Other significant effects are seen in the 1000–1260 cm−1 range. The NH2 rocking modes, observed at approximately 1075 cm−1 and 1177 cm−1 in pure urea,39,40 and at around 1070 cm−1 and 1165 cm−1 in the urea:[N2,2,2,2]Cl eutectic mixture (Fig. 7) seem to split into several components in the mixtures with the asymmetric salts [Ch]Cl and [EthylCh]Cl. The lowest-wavenumber band in this range, ca. 1002 cm−1, coincides with the expected position of the νCN stretching mode. However, the increased intensity of this mode without any wavenumber shift appears somewhat improbable. An alternative explanation is the splitting of the asymmetric NH2 rocking mode at 1075 cm−1 into two components at 1002 cm−1 and 1060 cm−1. The splitting of the symmetric NH2 rocking mode is also an easy explanation for the two bands at ca. 1135 cm−1 and 1200 cm−1 (mixture with [Ch]Cl) and 1135 cm−1 and 1180 cm−1 (mixture with [EthylCh]Cl). The origin of the high-wavenumber band observed at ca. 1260 cm−1 is less straightforward. Excluding the possibility of a combination band, the assignment to a NH2 rocking mode remains the most reasonable, despite the large blue shift involved.
Regarding the NH2 deformation (“scissoring”) modes around 1660 cm−1, a general shift to higher wavenumbers is observed, but conclusions are hampered by the low signal-to-noise ratio in the difference spectra. Nonetheless, this shift aligns with the changes reported above for the NH2 rocking modes.
To assist the assignment of INS spectra of urea in these mixtures, it is important to consider the most relevant intermolecular contacts available in the liquid mixture. This has been done with discrete DFT calculations for several possible associations between urea molecules and chloride anions, following the comprehensive procedure by Hunt et al.34 The most relevant urea–chloride clusters found in this context, as well as the corresponding simulated INS spectra, are shown in Fig. S3 and S4 (ESI†), respectively. The limitations of these simple cluster models do not allow definite conclusions, as the calculated spectra are largely dependent on the cluster size (each urea molecule allows the formation of six intermolecular contacts). Nevertheless, some general tendencies are observed. For instance, and confirming the assignments in Table 2, NH2 wagging modes tend to mix with NH2 torsions below ca. 700 cm−1 and with out-of-plane bend of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group above this limit. These modes are highly dependent on intermolecular contacts and thus spread for a wide wavenumber range. On the other hand, the behavior of the NH2 rocking modes is noteworthy. The formation of NH⋯Cl and NH⋯N hydrogen bonds leads to the splitting of the modes and, in some cases, to the arising of a component above 1200 cm−1. It is interesting to note that NH2 rocking modes above 1200 cm−1 are observed for the less organized cluster models, e.g., distorted urea dimer, single urea–chloride pair and distorted urea–urea–chloride associations.
O group above this limit. These modes are highly dependent on intermolecular contacts and thus spread for a wide wavenumber range. On the other hand, the behavior of the NH2 rocking modes is noteworthy. The formation of NH⋯Cl and NH⋯N hydrogen bonds leads to the splitting of the modes and, in some cases, to the arising of a component above 1200 cm−1. It is interesting to note that NH2 rocking modes above 1200 cm−1 are observed for the less organized cluster models, e.g., distorted urea dimer, single urea–chloride pair and distorted urea–urea–chloride associations.
4. Discussion
Comparing the experimental melting points of the three eutectic mixtures, [Ch]Cl:urea, [ChloroCh]Cl:urea, and [EthylCh]Cl:urea, with the expected value for ideal behavior, ΔTm values of ca. −60 °C were found for [Ch]Cl and [ChloroCh]Cl systems, while [EthylCh]Cl mixtures presents a ΔTm of ca. −100 °C. The similar behavior of [Ch]Cl and [ChloroCh]Cl strongly suggests that the OH group of choline does not play a significant role in the intermolecular interactions in the eutectic. In other words, potential O–H⋯urea hydrogen bonds do not significantly contribute to the excess enthalpy of mixing. On the other hand, the deeper eutectic behavior of [EthylCh]Cl:urea, with a ΔTm of ca. −100 °C cannot be attributed to enthalpic factors, as the additional methyl group in [EthylCh]Cl introduces no significant interactions or enthalpy differences relative to [Ch]Cl. Being so, it must be ascribed to the positive excess entropy of mixing brought by the increased asymmetry of the cations.In a recent work, Migliorati and D’Angelo41 drew somewhat different conclusions regarding the role of the OH group, based on molecular dynamics simulations of urea-based DES with benzyl-substituted derivatives of choline cation, specifically benzyl(2-hydroxyethyl)dimethylammonium and benzyltrimethylammonium. By comparing the number and type of hydrogen bond interactions with the reported DES melting points,20 Migliorati and D’Angelo concluded that the presence or absence of a hydroxyl group, or a bulky benzyl, on the organic cation, have an impact on the DES properties.
However, the reported melting points for their cation series can also be explained using the same arguments applied above to the [Ch], [ChloroCh] and [EthylCh] series. For instance, replacing the CH2CH2OH group by a benzyl group (as in changing from choline to benzyltrimethylammonium) has little effect on the reported melting temperature of the DES, which shifts from 12 °C to 26 °C, respectively (the melting temperature of the dry [Ch]-based DES is actually 32 °C,21 12 °C corresponding to a hydrated mixture). This behavior is analogous to that observed for [Ch]/[ChloroCh] pair described above. In contrast, a much more significant deviation is observed when the benzyl group replaces a methyl group, thereby introducing greater cation asymmetry. This is evident in the case of benzyl(2-hydroxyethyl)dimethylammonium, which parallels the [EthylCh] case and leads to a much lower DES melting temperature of −33 °C.
By comparing the spectra of the three distinct cations in the mixture—[Ch], [ChloroCh], and [EthylCh]—in the region of the methyl torsions, the observed broadening of the INS bands is consistent with the high molecular disorder in the eutectic mixture, as already noted in a previous work.3 Similar effects are observed in the 400–1800 cm−1 region, where band broadening is the most prominent effect when transitioning from pure salts to eutectic mixtures. These effects are correlated with the increased space around the cation, as urea molecules displace chloride anions from the cations’ solvation shell. The increased space results in a lower frequency of methyl torsions, noticeable broadening of the δNC4 and δNCC deformation bands, and greater conformational freedom for ethyl and hydroxyethyl groups.
A somewhat different situation arises when considering the effects on the INS spectrum of the urea counterpart. The formation of eutectic mixtures leads to more significant changes compared to the pure solid, and these changes are also dependent on the cation's symmetry. In all cases, the disappearance of the band structure at approximately 445 cm−1 can be ascribed to the disruption of the urea's crystalline structure. Simultaneously, the emergence of new bands must be associated with the formation of urea–chloride and distinct urea–urea interactions within the mixture.
For the symmetric [N2,2,2,2] cation, a band appears at 650 cm−1, which broadens into a wide band in the presence of asymmetric cations. This band at 650 cm−1 seems related to the mode at 620 cm−1 in pure urea and calculated at 690 cm−1 for the [N2,2,2,2]:urea inclusion compound structure. The shift is ascribed to the structural transition of urea from a tetragonal crystalline arrangement in pure urea to a centrosymmetric arrangement with urea–chloride contacts in the layer-type host structure.31 Thus, the presence of a well-defined band at 650 cm−1 suggests the existence of nano-domains of urea with important contribution from urea–chloride motifs with a quasi-planar structure. In the presence of asymmetric cations, the composition of these urea nano-domains becomes more diverse, leading to the observed broadening, and apparent vanishing, of this band. At the same time, in the spectra of mixtures with asymmetric cations, the NH2 rocking modes exhibit splitting, which simple model calculations suggest is associated with distorted structures of urea–chloride complexes. This is consistent with the view that the cation's asymmetry affects mostly the urea side of the solid–liquid equilibrium diagram and deep eutectic behavior stems from the deviation of the urea SLE ideal solubility line.3
The spectral changes observed for the asymmetric cation cases seem to be slightly more pronounced for [EthylCh] compared to [Ch], though the difference is not particularly impressive. This may be explained by the fact that the [Ch] cations are already asymmetric, and the additional asymmetry introduced by the ethyl group in [EthylCh] is less disruptive compared to the transition from a completely symmetric to an asymmetric cation.
5. Conclusion
The results presented in this work contribute to elucidating the influence of cation structural modifications on the physicochemical property spectra of DES systems, by correlating enthalpic and entropic effects with the observed changes in vibrational spectra.The analysis of the pure compounds demonstrates the ability of computational models to accurately describe experimental INS spectra, enabling a detailed interpretation of vibrational modes and cation symmetry effects.
Isotopic H/D substitution in urea allows for the isolated observation of the INS spectrum of each component in the eutectic mixture. The independent spectra of the “salt in the eutectic mixture” and “urea in the eutectic mixture” were evaluated to infer the thermodynamic implications of cation asymmetry (entropy effects) and OH-by-Cl group substitution (enthalpy effects).
A comparison of the behavior of [Ch] and [ChloroCh] reveals that the OH group plays a limited role in the formation of the [Ch]Cl:urea DES system. The contribution of OH⋯X interactions to both the melting enthalpy—and hence to ΔTm—and to the INS spectra is found to be negligible. This observation is deserving of further inquiry since it runs counter to previous studies which have identified OH⋯chloride contacts as significant players contributing to the melting point depression in tetraalkylammonium chloride:urea systems.41–43
Considering the series [Ch]Cl, [ChloroCh]Cl, and [EthylCh]Cl, the addition of an ethyl group increases molecular asymmetry, amplifies conformational flexibility, and introduces greater deviations from ideal melting behavior. This is evident in the ΔTm values as well as in the changes observed in the INS spectra. The increased flexibility of ammonium cations results in broader spectral bands and reduced resolution, reflecting the entropic contributions to eutectic point depression. However, the cation asymmetry is already reflected in the melting points of pure salts in salt-rich mixtures. Thus, its effects are primarily observed on the urea side of the solid–liquid equilibrium diagram. This becomes evident in the INS spectra of “urea in the eutectic mixture” with symmetric and asymmetric cations. Comparing the “urea view” in the mixture with [N2,2,2,2] and those with [Ch]Cl, [ChloroCh]Cl, and [EthylCh]Cl, it is possible to identify two distinct urea structures, which are assumed to reflect the entropic effects of cation asymmetry in melting urea.
Author contributions
Rafael Serrano: writing – review & editing, validation, investigation, data curation. Catarina F. Araújo: writing – review & editing, validation, investigation, data curation; Paulo Ribeiro-Claro: writing – review & editing, writing – original draft, supervision, funding acquisition, conceptualization. Pedro D. Vaz: writing – review & editing, investigation; Svemir Rudić: writing – review & editing, investigation; João A. P. Coutinho: writing – review & editing, funding acquisition; Mariela Nolasco: writing – review & editing, writing – original draft, supervision, funding acquisition, formal analysis, conceptualization.Data availability
Data for this article, namely the INS spectra, are available at ISIS Neutron and Muon Source Data Journal, at https://doi.org/10.5286/ISIS.E.92921697. Calculated crystal structure for [ChloroCh]Cl and [EthylCh]Cl has been deposited at the TCOD (Theoretical Crystallography Open Data) repository, under TCOD entries 30000154 and 30000155, respectively.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI 10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI 10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI 10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). FCT is also acknowledged for the PhD grant to CFA (SFRH/BD/129040/2017). The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities (TOSCA/RB1810054, https://doi.org/10.5286/ISIS.E.RB1810054). CASTEP calculations were made possible due to the computing resources provided by STFC Scientific Computing Department's SCARF cluster.References
- L. J. B. M. Kollau, M. Vis, A. van den Bruinhorst, R. Tuinier and G. de With, Entropy models for the description of the solid–liquid regime of deep eutectic solutions, J. Mol. Liq., 2020, 302, 112155 CrossRef CAS.
- A. van den Bruinhorst and M. C. Gomes, Is there depth to eutectic solvents?, Curr. Opin. Green Sustainable Chem., 2022, 37, 100659 CrossRef CAS.
- C. F. Araújo, P. Ribeiro-Claro, P. D. Vaz, S. Rudić, R. A. F. Serrano and L. P. Silva, et al., Exploring asymmetry induced entropy in tetraalkylammonium–urea DES systems: what can be learned from inelastic neutron scattering?, Phys. Chem. Chem. Phys., 2024, 26(7), 5969–5977, 10.1039/D3CP04961B.
- A. van den Bruinhorst, C. Corsini, G. Depraetère, N. Cam, A. Pádua and M. Costa Gomes, Deep eutectic solvents on a tightrope: balancing the entropy and enthalpy of mixing, Faraday Discuss, 2024, 253, 273–288, 10.1039/D4FD00048J.
- C. F. Araujo, J. A. P. Coutinho, M. M. Nolasco, S. F. Parker, P. J. A. Ribeiro-Claro and S. Rudić, et al., Inelastic neutron scattering study of reline: Shedding light on the hydrogen bonding network of deep eutectic solvents, Phys. Chem. Chem. Phys., 2017, 19(27), 17998–18009 RSC.
- M. M. Nolasco, S. N. Pedro, C. Vilela, P. D. Vaz, P. Ribeiro-Claro and S. Rudic, et al., Water in Deep Eutectic Solvents: New Insights From Inelastic Neutron Scattering Spectroscopy, Front. Phys., 2022, 10, 834571 CrossRef.
- B. S. Hudson, Vibrational Spectroscopy via Inelastic Neutron Scattering, in Frontiers of Molecular Spectroscopy, ed. J. Laane, Elsevier, Amsterdam, 2009, ch. 17, pp. 597–622 Search PubMed.
- B. S. Hudson, Vibrational spectroscopy using inelastic neutron scattering: Overview and outlook, Vib. Spectrosc., 2006, 42(1, SI), 25–32 CrossRef CAS.
- W. Langel, Introduction to neutron scattering, ChemTexts, 2023, 9(4), 12 CrossRef.
- A. Nidriche, M. Moulin, P. Oger, J. R. Stewart, L. Mangin-Thro and W. Schmidt, et al., Impact of Isotopic Exchange on Hydrated Protein Dynamics Revealed by Polarized Neutron Scattering. PRX, Life, 2024, 2(1), 13005 Search PubMed.
- M. Müller, G. Vogl, H. Schober, H. Chanzy and L. Heux, Inelastic Neutron Scattering study on different types of celulose, in Biological Macromolecular Dynamics, ed. S. Cusack, H. Büttner, M. Ferrand, P. Langan and P. Timmins, Adenine Press, New York, 1997 Search PubMed.
- C. Araujo, C. S. R. Freire, M. M. Nolasco, P. J. A. Ribeiro-Claro, S. Rudić and A. J. D. Silvestre, et al., Hydrogen bond dynamics of cellulose through Inelastic Neutron Scattering Spectroscopy, Biomacromolecules, 2018, 19(4), 1305–1313 CrossRef CAS.
- S. F. F. Parker, J. E. E. Trevelyan and H. Cavaye, Vibrational spectra of neutral and doped oligothiophenes and polythiophene, RSC Adv., 2023, 13(8), 5419–5427 RSC.
- F. Fillaux, S. F. Parker and L. T. Yu, Inelastic neutron scattering studies of polypyrroles and partially deuterated analogues, Solid State Ionics, 2001, 145(1–4), 451–457 CrossRef CAS.
- A. A. Y. Guilbert, M. Zbiri, P. A. Finn, M. Jenart, P. Fouquet and V. Cristiglio, et al., Mapping Microstructural Dynamics up to the Nanosecond of the Conjugated Polymer P3HT in the Solid State, Chem. Mater., 2019, 31(23), 9635–9651 CrossRef CAS.
- M. Zbiri, P. A. Finn, C. B. Nielsen and A. A. Y. Guilbert, Quantitative insights into the phase behaviour and miscibility of organic photovoltaic active layers from the perspective of neutron spectroscopy, J. Mater. Chem. C, 2021, 9(35), 11873–11881 RSC.
- C. F. Araújo, S. V. Pandeirada, I. M. Oliveira, G. B. Rosa, B. Agostinho and A. J. D. Silvestre, et al., Crystal structure of poly(trimethylene 2,5-furandicarboxylate) redux – a new model supported by computational spectroscopy, Polym. Chem., 2024, 15(42), 4349–4363 RSC.
- M. M. Nolasco, A. Ribeiro, P. J. A. Ribeiro-Claro, S. Rudic, C. F. Araujo and P. D. Vaz, Exploring asymmetry-induced entropy in deep eutectic solvents, STFC ISIS Neutron Muon Source, 2018, 5969–5977 Search PubMed.
- C. F. Araujo, D. O. Abranches, J. A. P. Coutinho, P. D. Vaz, P. Ribeiro-Claro and M. M. Nolasco, Good vibrations: understanding deep eutectic solvents through the lens of vibrational spectroscopy, Appl. Spectrosc. Rev., 2024, 60, 137–192 CrossRef.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Novel solvent properties of choline chloride/urea mixtures, Chem. Commun., 2003,(1), 70–71, 10.1039/B210714G.
- M. Gilmore, M. Swadzba-Kwasny and J. D. Holbrey, Thermal Properties of Choline Chloride/Urea System Studied under Moisture-Free Atmosphere, J. Chem. Eng. Data, 2019, 64(12), 5248–5255, DOI:10.1021/acs.jced.9b00474.
- ISIS Facility INS/TOSCA. [cited 2022 Sep 15]. Available from: https://www.isis.stfc.ac.uk/Pages/tosca.aspx.
- S. F. Parker, D. Lennon and P. W. Albers, Vibrational Spectroscopy with Neutrons: A Review of New Directions, Appl. Spectrosc., 2011, 65(12), 1325–1341 CrossRef CAS.
- S. F. Parker, F. Fernandez-Alonso, A. J. Ramirez-Cuesta, J. Tomkinson, S. Rudic and R. S. Pinna, et al., Recent and future developments on TOSCA at ISIS, in J. Phys.: Conf. Ser., ed. M. JimenezRuiz and S. Parker, 2014, vol. 554, p. 012003 Search PubMed.
- R. S. Pinna, S. Rudić, S. F. Parker, J. Armstrong, M. Zanetti and G. Škoro, et al., The neutron guide upgrade of the TOSCA spectrometer, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 896, 68–74 CrossRef CAS.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell and L. Chapon, et al., Mantid-Data analysis and visualization package for neutron scattering and mu SR experiments, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- M. M. Nolasco, A. Ribeiro, P. J. A. Ribeiro-Claro, S. Rudic, C. F. Araujo and P. D. Vaz, Exploring asymmetry-induced entropy in deep eutectic solvents, STFC ISIS Neutron Muon Source, 2018 DOI:10.5286/ISIS.E.92921697.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert and K. Refson, et al., First principles methods using CASTEP, Z. Kristallogr., 2005, 220(5–6), 567–570 CAS.
- K. Refson, P. R. Tulip and S. J. Clark, Variational density-functional perturbation theory for dielectrics and lattice dynamics, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(15), 155114 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS.
- Q. I. Li and T. C. W. Mak, Hydrogen-Bonded Urea-Anion Host Lattices. Part 4. Comparative Study of Inclusion Compounds of Urea with Tetraethylammonium and Tetraethylphosphonium Chlorides, J. Inclusion Phenom. Mol. Recognit. Chem., 1997, 28(2), 151–161, DOI:10.1023/A:1007985329632.
- J. Hjortas and H. Sorum, Re-investigation of crystal structure of choline chloride, Acta Crystallogr., Sect. B, 1971, B 27, 1320 CrossRef.
- V. Milman, A. Perlov, K. Refson, S. J. Clark, J. Gavartin and B. Winkler, Structural, electronic and vibrational properties of tetragonal zirconia under pressure: a density functional theory study, J. Phys.: Condens. Matter, 2009, 21(48), 485404 CrossRef.
- K. Dymkowski, S. F. Parker, F. Fernandez-Alonso and S. Mukhopadhyay, AbINS: The modern software for INS interpretation, Phys. B, 2018, 551, 443–448 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009. Available from: https://www.gaussian.com Search PubMed.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Doubly ionic hydrogen bond interactions within the choline chloride-urea deep eutectic solvent, Phys. Chem. Chem. Phys., 2016, 18(27), 18145–18160 RSC.
- A. Pawlukojc and L. Hetmanczyk, INS, DFT and temperature dependent IR investigations of dynamical properties of low temperature phase of choline chloride, Chem. Phys., 2014, 445, 31–37 CrossRef CAS.
- D. O. Abranches and J. A. P. Coutinho, Everything You Wanted to Know about Deep Eutectic Solvents but Were Afraid to Be Told, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 141–163 CrossRef CAS.
- M. R. Johnson, K. Parlinski, I. Natkaniec and B. S. Hudson, Ab initio calculations and INS measurements of phonons and molecular vibrations in a model peptide compound – urea, Chem. Phys., 2003, 291(1), 53–60 CrossRef CAS.
- R. Keuleers, H. O. Desseyn, B. Rousseau and C. Van Alsenoy, Vibrational Analysis of Urea, J. Phys. Chem. A, 1999, 103(24), 4621–4630, DOI:10.1021/jp984180z.
- V. Migliorati and P. D’Angelo, How does the cation nature affect the DES structure?, Chem. Phys. Lett., 2025, 858, 141731 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Liquid structure of the choline chloride-urea deep eutectic solvent (reline) from neutron diffraction and atomistic modelling, Green Chem., 2016, 18(9), 2736–2744 RSC.
- V. Migliorati, F. Sessa and P. D’Angelo, Deep eutectic solvents: a structural point of view on the role of the cation, Chem. Phys. Lett., 2019, 737, 100001 CrossRef . Available from: https://www.sciencedirect.com/science/article/pii/S2590141918300011.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01144b |
| This journal is © the Owner Societies 2025 |