
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From photocatalysis to photon–phonon co-driven catalysis for methanol reforming to hydrogen and valuable by-products†
Hui Wang
 ab,
Eleana Harkou
c,
Achilleas Constantinou
c,
Sultan M. Al-Salemc
d,
George Manos
b and
Junwang Tang
*bef
ab,
Eleana Harkou
c,
Achilleas Constantinou
c,
Sultan M. Al-Salemc
d,
George Manos
b and
Junwang Tang
*bef
aCollege of Environmental Science and Engineering, Hunan University, Changsha 410082, P. R. China
bDepartment of Chemical Engineering, University College London (UCL), London, WC1E 7JE, UK. E-mail: jwtang@tsinghua.edu.cn
cDepartment of Chemical Engineering Cyprus University of Technology, 57 Corner of Athinon and Anexartisias, Limassol 3036, Cyprus
dEnvironment and Life Sciences Research Centre, Kuwait Institute for Scientific Research, Safat 13109, Kuwait
eIndustrial Catalysis Centre, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China
fOrdos Laboratory, Inner Mongolia, 017000, China
First published on 2nd January 2025
Abstract
Hydrogen energy will play a dominant role in energy transition from fossil fuel to low carbon processes, while economical, efficient, and safe hydrogen storage and transportation technology has become one of the main bottlenecks that currently hinder the application of the hydrogen energy scale. Methanol has widely been regarded as a primary liquid H2 storage medium due to its high hydrogen content, easy storage and transportation and relatively low toxicity. Hydrogen release from methanol using photocatalysis has thus been the focus of intense research and recent years have witnessed its fast progress and drawbacks. This review offers a comprehensive overview of methanol-based hydrogen production via photocatalysis, spotlighting recent developments in photocatalysts referring to thermal catalysts, including efficient semiconductors and cocatalysts, followed by the discussion of mechanistic investigation via advanced techniques and their disadvantages. Beyond this, particular focus has been placed on the discussion of co-driven processes involving coupling of photons (photocatalysis) with phonons (thermal catalysis) – the concept of photon–phonon co-driven catalysis – for methanol reforming and cutting-edge reactor design strategies, in order to enhance the overall process efficiency and applicability. Concluding with forward-looking insights, this review aims to provide valuable guidance for future research on hydrogen release through methanol reforming.
 Hui Wang | Dr Hui Wang is a professor at the College of Environmental Science and Engineering, Hunan University. She received her PhD in Chemical Engineering from University College London under the supervision of Prof. Junwang Tang. Her current research focuses on photo-thermal coupling for hydrogen production from biomass alcohols, as well as the high-value transformation of pollutants. |
 Eleana Harkou | Ms Eleana Harkou is a PhD candidate in the Cyprus University of Technology, with a background in Chemical Engineering. She is skilled in both computational fluid dynamics (CFD) simulations and process modeling, with extensive experience in the modeling, development, and scaling of chemical reactors for diverse applications and in optimising fluid flow, heat/mass transfer, and chemical processes. She specialises in the reaction engineering field with main focus on the reactor design to maximise the efficiency and performance of catalytical reaction processes. |
 Achilleas Constantinou | Dr Achilleas Konstantinou is an assistant Professor at the Cyprus University of Technology (CUT). He specializes in Chemical and Catalytic Reaction Engineering and, in particular, in designing multiphase reactors to intensify and improve their performance for industrial applications. Dr Konstantinou has made extensive use of complex Computational Fluid Dynamics (CFD) tools and successfully validated the prediction of concentrations, temperatures and velocity profiles in micro-units against experimental findings, in order to improve their design and render them suitable for industrial production. Dr Konstantinou also holds the titles of Chartered Chemical Engineer (MIChemE), Chartered Engineer (CEng) and Chartered Scientist (CSci). |
 Sultan M. Al-Salemc | Dr Sultan M. Al-Salem specialises in polymer degradation kinetics, a particular research interest of his. He is also interested in polymer weathering, Plastic Solid Waste (PSW) management, reactor design, downstream intensification processes, Waste Management, thermal engineering, life cycle assessment (LCA), biodegradable polymers, microplastics (MPs) and off-gas engineering. Dr Al-Salem has authored/co-authored a number of book chapters, refereed journal and conference papers. He currently holds the position of a Research Scientist at the Environment & Life Sciences Research Centre (ELSRC) of the Kuwait Institute for Scientific Research (KISR) working on various research projects and pursuing a number of major R&D works. |
 George Manos | Dr George Manos is an Associate Professor in Chemical Engineering at University College London (UCL). His research involves hydrocarbon reaction processes with a focus on plastic waste catalytic pyrolysis and more generally catalysis of renewable energy processes. He is also working on catalyst deactivation by coking and coke characterisation, as well as statistical mechanical modelling of adsorption on microporous materials. Dr Manos is the author of more than one hundred refereed journal papers, as well as numerous book chapters and conference papers. He is the co-editor of the book Adsorption and Phase Behaviour in Nanochannels and Nanotubes. |
 Junwang Tang | Prof. Junwang Tang is a Member of Academia Europaea, a Leverhulme Trust Senior Research Fellow, Fellow of RSC and Fellow of IMMM and formerly a full Professor at Chemical Engineering in University College London. He is currently a Chair Professor of Materials Chemistry and Catalysis in the Department of Chemical Engineering and Director of the Industrial Catalysis Centre at Tsinghua University. He has concentrated on photocatalysis and pioneered in photon–phonon co-driven catalysis for the activation of small molecules (e.g., H2O, CO2, CH4, and N2) as well as microwave catalytic depolymerisation of plastics. He has received many prestigious honors, including the 2022 IChemE Oil and Gas Award, the 2021 IChemE Andrew Medal, the 2021 RSC Corday-Morgan Prize, and the 2021 IChemE Innovative Product Award. |
1. Introduction
H2 has been considered as a primary energy source to reduce our dependence on fossil fuels. It can be obtained from various renewable energy resources and can act as an energy supplier for fuel cells. However, transportation and storage of H2 have been the biggest challenges restricting its instant commercial utilisation. On-site production of H2 has been reported as a promising technology to meet the end-user's requirements and overcome transportation and storage challenges. Unlike organic substrates and some inert raw materials such as ethanol, lignocellulosic biomass, and glycerol, which require higher temperatures for carbon–carbon bond cleavage, methanol's molecular structure lacks carbon–carbon bonds.1 This unique feature allows hydrogen generation from methanol to occur at relatively low temperatures, together with the high hydrogen content in methanol, making it a very practical and energy-efficient option. Kinetically, methanol's small molecular size allows it to easily adsorb and to be activated on catalyst surfaces, thereby increasing the reaction rate. Furthermore, ongoing research in the field of green methanol synthesis has demonstrated the potential of this compound as a sustainable source of hydrogen.Traditional methanol reforming processes have predominantly relied on thermocatalysis, driving chemical reactions at relatively high temperatures. However, it often demands significant energy inputs and produces a large amount of CO2, which can undermine its sustainability. In contrast, photocatalysis offers a more energy-efficient alternative by utilising light energy, typically solar, to activate catalysts for hydrogen production. This approach not only harmonises with renewable energy strategies but also operates under considerably milder conditions compared to thermocatalysis, enhances valuable by-product selectivity and reduces energy consumption. Current progress increasingly highlights the vast potential of photocatalysis, particularly when compared to thermocatalysis, as depicted in Fig. 1. Significant advancements have been made in enhancing hydrogen generation activity and valuable by-product selectivity through photocatalytic processes. The scientific community is vigorously pushing technological boundaries, engineering innovative materials, refining catalytic conditions, and unravelling complex reaction mechanisms. However, despite these advances, a comprehensive summary involving photocatalytic methanol dehydrogenation and practical reactor design referring to thermal catalysis remains notably lacking. This review aims to bridge this gap by starting with the advantages and drawbacks of thermal catalysis as a reference and then detailing photocatalytic hydrogen production from methanol. It presents a thorough examination of the latest developments, highlighting diverse photocatalysts and focusing on by-product selectivity – an aspect to some extent overlooked in prior reviews. Following that, special attention is devoted to strategically coupling photons with phonons for a catalytic process (the concept of photon–phonon co-driven catalysis), which was firstly underlined by our group recently, a combination to address individual limitations, complementary by the reactor design. Therefore, this review concludes with forward-looking insights, aiming to enhance understanding and to spur further innovation in the field of efficient photocatalytic hydrogen production and valuable by-product synthesis.
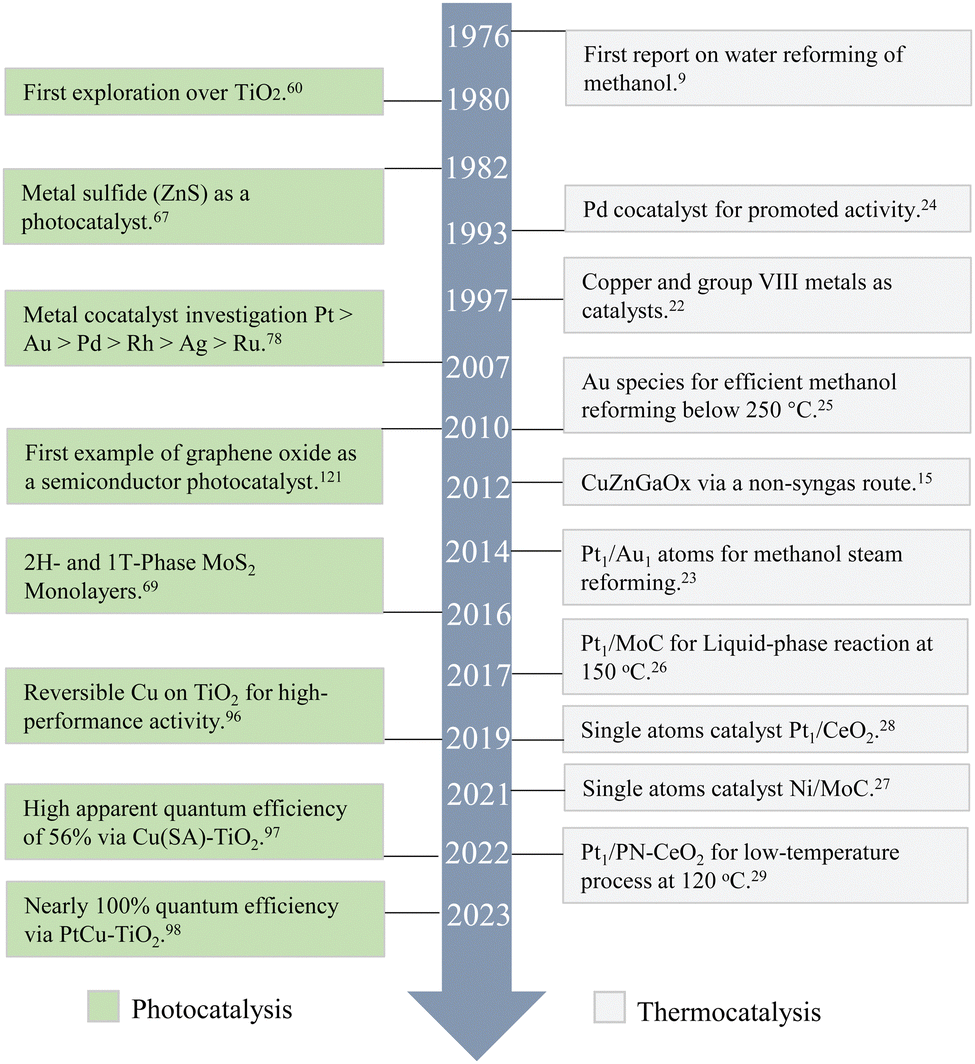 | ||
| Fig. 1 The development of methanol-based hydrogen production via photocatalysis and thermocatalysis. | ||
2. Thermocatalytic methanol-based hydrogen production
The generation of hydrogen from methanol–water reforming under thermal conditions is a conventional method where catalysts with high activities were reported in the literature.2 Thermocatalysis is a widely known development through the years for chemical conversion of various hydrocarbons such as plastic solid waste.3–8 Since the first report regarding methanol-involved hydrogen production in 1976, many efforts have been made to further improve activity.9 Cu catalysts are commonly used at an industrial level for methanol steam reforming due to the low cost and the very high activity at relatively low temperatures (around 250 °C).10 However, it is difficult to control the size and shape of these catalysts because of their high sensitivity to oxidation.11 Moreover, they suffer from loss of specific surface area by sintering of their nanoparticles. To overcome these limitations, PdZn-based catalysts have been proposed. Even though they showed better selectivity towards hydrogen and better stability, their catalytic stability was lower than that of Cu catalysts. Although further improvements for PdZn-based catalysts enhanced their thermal stability, they still suffer from significant CO and CH4 formation or coke formation.12 Al2O3 and ZnO are often used as supports. The addition of ZrO2 to Cu-based catalysts greatly enhances their activity and eliminates the production of CO.13 The main reasons for the deactivation of catalysts during methanol steam reforming are catalyst poisoning and coking.14 CuZnGaOx was applied for thermocatalytic methanol-involved hydrogen production via a non-syngas route and the methanol conversion reached nearly 100%.15 Table S1 (ESI†) shows catalysts used for the methanol steam reforming reaction with reaction conditions and obtained results for H2 yield, selectivity, and activity.Cocatalysts play a crucial role in thermocatalytic methanol reforming, enhancing reaction efficiency and product safety by converting undesirable by-products. Notably, acidic or alkaline cocatalysts help transform unwanted compounds into more valuable or less hazardous substances. Despite the high activity and reduced CO formation reported with finely dispersed Cu-based catalysts, there is no consensus on the promotion mechanism.16,17 Factors like the state of Cu, including its dispersion, valence, and stability, significantly influence catalytic performance and by-product selectivity. For instance, commercial Cu/ZnO/Al2O3 has achieved nearly 100% methanol conversion by manipulating the catalyst to specific nano-sizes.18 Recent studies19,20 also shed light on the in situ reduction process of Cu, from Cu2+ to Cu0, highlighting both Cu+ and Cu0 as active sites that enhance catalytic activity. A similar phenomenon was observed in CeO2 with changeable valence states.21 Noble metals, known for their excellent thermal stability, prevent catalyst deactivation at high temperatures (>300 °C) but tend to drive thermocatalytic methanol conversion towards CO rather than CO2,22 which has been mitigated by introducing second promoters such as Zn, In, Cd, Au and Ga.22,23 Moreover, the choice of support can greatly affect activity and selectivity, as seen with Pd/ZrO2 and Pd/ZnO catalysts, which respectively excel in hydrogen generation and CO2 selectivity.24 In another report, Au serves as an efficient cocatalyst in CeO2 catalysed methanol-involved hydrogen production at low temperatures (<250 °C), where strongly bonded Au–O–Ce species were the main active species.25 Recent developments in single-atom cocatalysts, like Pt and Ni atomically dispersed on α-molybdenum carbide (α-MoC),26,27 demonstrate exceptional relatively low-temperature hydrogen production (⩽150 °C), due to the synergistic action of single atoms and α-MoC. Similarly, single atom Pt1 was deposited on CeO2 to offer high hydrogen activity, which was 40 times higher than 2.5 nm Pt/CeO2.28 Recently, a lower temperature methanol-involved hydrogen production was reported,29 where the synergy of Pt single atoms and Lewis pairs allowed porous CeO2 to realise efficient H2 generation at 120 °C with very low CO levels (0.027%). The optimal Pt1/PN-CeO2 catalysts exhibited a H2 generation rate of 199 molH2 molPt−1 h−1 at 135 °C.
The mechanism investigation for methanol steam reforming has been accomplished by many scientists not only for the most used Cu catalysts but also for various catalysts such as In2O3-, Cu/ZnO-, Ni–Cu-based and M-βMo2C.14,30–33 Surface species were found in methanol steam reforming systems.34 Methanol preferred to adsorb at the top site with an O bond on the clean Cu(111) surface, with the possible pathways of methanol being CH3O or CH2OH.35 The former was generated through direct dissociation due to lower activation energy and higher stability. The reaction mechanism of CeO2 and Ni/CeO2 showed that the preferable reaction process is described as CH3OH → CH3O → CH2O → CH2OOH → CHOOH → CHOO → CO2.35,36 DFT calculations to understand better the catalytic cycles releasing H2 and CO216,37 and steady-state isotopic transient kinetic analysis (SSITKA) to study the detailed process involving methoxyl and CO species adsorbed on the catalyst were also performed.38
Thermocatalysis known for its classical roots and high-temperature efficiency demands substantial energy input. Nevertheless, it provides a well-established and versatile method for hydrogen generation that can operate under various conditions without relying on external energy sources. It boasts high activity, making it suitable for industrial applications. However, the further decrease of energy demands and substantially reducing operating temperature/CO2 emission are considered as key challenges. Additionally, the formation of by-products, such as CO, CH4 and coke, is an inherent issue in thermocatalytic processes, leading to a loss of catalytic selectivity and a shortened catalyst lifespan. Maintaining active sites at elevated temperatures, especially for oxidation reactions involving methanol and reduction reactions involving protons over metal catalysts and metal oxide catalysts, proves to be exceptionally challenging.
3. Photocatalytic methanol-based hydrogen production
Photocatalysis, a promising alternative to thermocatalysis, uses photons instead of heat to drive chemical reactions under ambient conditions. As shown in Fig. 2(a), thermocatalytic methanol reforming follows an “uphill” thermodynamic pathway, requiring external heat to overcome the activation energy barrier. In contrast, photocatalysis excites the catalyst with photons, generating the energetic charge carriers that lower the barrier, enabling a “downhill” reaction pathway without external heating. This allows photocatalytic methanol reforming to occur at lower temperatures, offering a more energy-efficient and sustainable approach.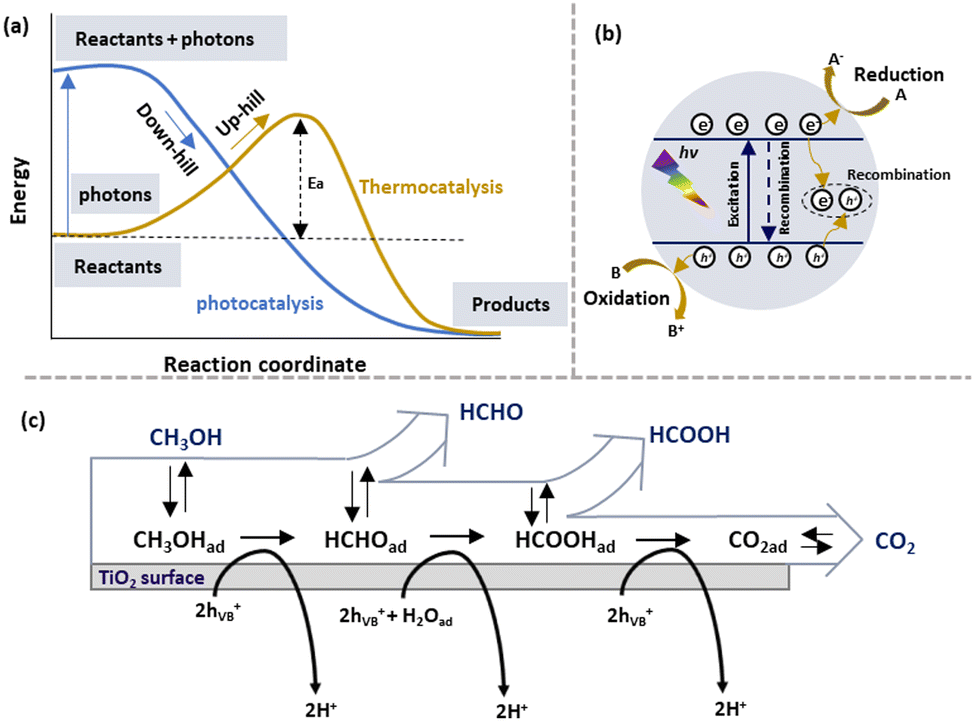 | ||
| Fig. 2 (a) Difference in thermodynamics of photocatalysis and thermal catalysis. (b) Scheme for semiconductor photocatalysis. (c) The pathway for methanol oxidation on the TiO2 surface.39 (c) Reproduced from ref. 39 with permission from Elsevier, Copyright 2011. | ||
3.1 Photocatalytic principles
Semiconductors have served as photocatalysts for water splitting since the 1970s, as they potentially presented favourable electronic properties,40 UV and visible light absorption ability and advantageous charge transport kinetics. Unlike conductors, the semiconductors have a band structure.41,42 The detailed fundamentals of photocatalysis have been widely discussed, as shown in Fig. 2(b).42,43 Under illumination, photons with energy higher than or equal to the semiconductor band gap excite the electrons of the valence band (VB) into the conduction band (CB) and leave the corresponding holes in the VB. Then, these photogenerated charges reach the surface of the semiconductor due to the inbuilt potential followed by entering into the adsorbed molecules to drive reduction or oxidation reactions. Using highly sustainable solar energy, photocatalysis has been used in several research fields, including water splitting,44–46 N2 fixation,47,48 degradation of organic pollutants,49,50 CO2 reduction,51 etc. It is worth mentioning that the recombination of photogenerated electrons and holes is inevitable, resulting in reaction efficiency reduction.52 Hence, it is essential to prevent the charge carrier recombination.Since Fujishima and Honda reported water splitting to produce H2 using the TiO2 photoelectrode, significant attention has been paid to photocatalytic H2 generation.40 Methanol-based hydrogen production is less thermodynamically challenging for H2 generation. The methanol conversion pathways can be illustrated using the following equations (eqn (1)–(4)).
| CH3OH → HCHO + H2 ΔG0 = 64.1 kJ mol−1 | (1) |
| HCHO + H2O → HCOOH + H2 ΔG0 = 47.8 kJ mol−1 | (2) |
| HCOOH → CO2 + H2 ΔG0 = −95.8 kJ mol−1 | (3) |
Overall reaction:
| CH3OH + H2O → CO2 + 3H2 ΔG0 = 16.1 kJ mol−1 | (4) |
3.2 Photocatalysts
Photocatalysts possessing a suitable band gap relative to the redox reactions are of intrinsic importance in photocatalysis, determining their light absorption range. In addition, the desired morphology engineering, long-term stability, and high surface area are favourable factors for activity enhancement. Many semiconductor materials have been employed as photocatalysts, including Cu2O, NiO, MgO, SrTiO3 etc.53–59 Among them, TiO2 is undoubtedly a widely studied semiconductor photocatalyst as it possesses favourable photochemical stability and is low cost. The first report of photocatalytic H2 generation from methanol dates back to the 1980s.60 Since then, several reports have shown H2 production from methanol. For instance, reduced TiO2 (black TiO2) was used to enhance the methanol photo-reforming activity, where Ti(III) and defects were introduced in the anatase surface, exhibiting 10 mmol g−1 h−1 H2 reaction rates with the assistance of Pt as a cocatalyst under simulated solar light irradiation.61 Interestingly, a controlled study revealed that the H2 production activity of Pt/black TiO2 was limited at room temperature for methanol photo-conversion, which could be overcome by increasing reaction temperatures.62 By controlling the crystalline phases of TiO2, the H2 evolution activity could be increased. For example, a mixed-phase of TiO2 was achieved by controlling the annealing conditions and showed high photocatalytic H2 production.63,64 In another report, Pt/TiO2 was optimised by tuning the anatase–rutile phase to obtain an extremely low CO concentration (<5 ppm) as a by-product. Due to the effective charge separation at the phase junction of two TiO2 crystals, enhanced photocatalytic performance was observed, while CO production could be suppressed by adjusting the surface base.65Representative photocatalysts without cocatalysts operated at room temperature shown in Table S2 (ESI†), including sulphides and nitrides, have also been used for methanol photo-reforming. Especially, CdS has received massive interest due to its strong visible-light response, though the poor photostability limits its application. This issue could be in part solved by the assistance of a supported cocatalyst. Also, annealing CdS under air could promote surface hydroxylation for alcohol dehydrogenation under visible light irradiation.66 In 1982, Yanagida et al. observed H2 evolution from a methanol–water mixture using ZnS.67 Interestingly, HOCH2–CH2OH was the main oxidation by-product. Furthermore, they promoted the HOCH2–CH2OH selectivity to 52% in 1984, which was regarded as the first case for photocatalytic C–C coupling via free radical intermediates.68 Moreover, the high selectivity of 95% towards HOCH2–CH2OH was achieved using colloidal ZnS and the primary intermediate was found to be a ˙CH2OH radical.68 MoS2 has been a relatively popular two-dimensional material in recent years for methanol-based hydrogen production.69 The lamellar structure was conducive to the adsorption and desorption of reactants and by-products in catalytic reactions.70 However, ordinary MoS2 showed less marginal activity while defective MoS2 was endowed with efficient H2 evolution activity. Beyond inorganic metal oxide semiconductors, metal–organic frameworks (MOFs) have recently attracted much attention due to their semiconducting character.71 Theoretically, the structure of MOFs allows them to be more versatile in photocatalyst design. A typical MOF UiO-66 (NH2) was reported for H2 generation from methanol photo-reforming thanks to efficient charge separation and prolonged charge lifetime.72 Very recently, Ti-based MOFs were investigated to study the role of various ligands in methanol dehydrogenation. For instance, MIL-125 exhibited 38 times higher activity than NH2-MIL-125 as holes in MIL-125 reacted with methanol, whereas holes in NH2-MIL-125 were likely to be located on its N sites, which restricted methanol oxidation.73 Followed by the catalyst exploration, covalent organic frameworks (COFs) were also used for photocatalytic water reforming of methanol, but they exhibited very limited H2 yield.74
Overall, each type of catalyst presents unique strengths and limitations, requiring further improvement. Metal oxides like TiO2 and ZnO are valued for their durability and cost-effectiveness, ideal for long-term applications. However, modifications such as doping and phase engineering are often necessary to improve visible light absorption and charge separation. Sulphides, including CdS and ZnS, offer excellent visible-light absorption but typically need strategies to ensure long-term stability due to susceptibility to photocorrosion. Polymers, especially C3N4, MOFs and COFs, provide a highly customisable platform with flexible structure that allows for readily tunable light absorption and active site configurations, though they face challenges of short lifetime of excitons and limited charge mobility.
In addition to the required band gap, it is more significant to reduce the reaction overpotential, which can be achieved by loading appropriate cocatalysts on the surface of a semiconductor. In detail, photogenerated electrons from the CB of the semiconductor flow to the Fermi level of cocatalysts until reaching an equilibrium.75 This results in space charge accumulation in the semiconductor, leading to band bending to form the Schottky barrier at the interface if the cocatalyst work function is more positive than the semiconductor work function.76 The Schottky barrier extracts the photogenerated electrons efficiently, thereby reducing the charge recombination. The following thus discusses a population of representative cocatalysts.
3.3 Cocatalysts
Similar to thermocatalysis, high-performing noble metals have been the most common cocatalysts used for photocatalytic conversion due to their positive Fermi levels, high redox potentials for electron extraction and small Schottky barrier.77 The early discovery of photocatalytic H2 generation from methanol-conversion dates back to the 1980s, in which Pd, Pt, and RuO2 on TiO2 were studied.60 Among them, Pt/TiO2 achieved a high quantum efficiency of up to 44%. Various metal cocatalysts have also been studied,78 and the activity followed Pt > Au > Pd > Rh > Ag > Ru, due to the work function of noble metals for Schottky barrier construction. The cocatalyst's particle size has also been considered as one of the ways to increase the photocatalytic H2 generation.79 In a report, the Au/TiO2 system was studied for H2 production from methanol dehydrogenation, in which the H2 production efficiency was inversely proportional to the particle size of the catalyst, suggesting that the smaller particle size could offer more active surface sites and high dispersion that were key to obtaining higher photocatalytic activity.80 A detailed study on various noble metals on TiO2 revealed that Pt was the most effective cocatalyst, followed by Au and Ag for photocatalytic H2 evolution.81 The obtained ESR results indicated that the Ti3+ concentration on Pt/TiO2 was lower than Au/TiO2, suggesting that the photogenerated electrons easily transferred from Ti3+ to Pt. These electrons reacted with the protons on Pt to produce H2. The oxidation state of the metal cocatalysts was also reported to influence photocatalysis. For instance, a novel mesoporous CNT/TiO2 hybrid photocatalyst, enhanced with Pt nanoparticles, achieved a high hydrogen generation rate of 40.6 mmol g−1 h−1.82 This efficiency arose from its unique structure: a network of interconnected TiO2 nanocrystals provided abundant active sites and facilitated continuous charge transfer, while embedded CNTs created intimate interfaces that promoted charge separation, improved electron mobility, and reduced recombination. Additionally, oxygen vacancies formed during annealing introduced inter-bandgap states, lowering the flat band potential and enhancing charge transport. Oros-Ruiz et al. compared the performance of the metallic and oxidised Au as cocatalysts on TiO2.83 The metallic Au enhanced the activity significantly, whereas the oxidised Au showed an adverse effect as it acted as an electron sink. Similarly, in another report, metallic Pd-loaded ZnO was found to show higher activity at 200 °C compared to PdO loaded ZnO.84 Interestingly, the reaction pathway of Pd-mediated methanol conversion was likely similar to Cu. First, CH3OH was decomposed into HCHO followed by the formation of HCOOH, while HCHO reacted with H2O and finally oxidised to CO2 together with H2 production. During this process, a small amount of CO production was inevitable. In addition to ZnO, Pd was also loaded on various substrates such as Al2O3, La2O3 and Nd2O3 and the expected activity was obtained, which again proved the role of Pd. Bowker et al. studied the contribution of the Pd loading amount on the photocatalyst surface and CH3OH concentration for methanol-based hydrogen production.85 Increasing the cocatalyst loading amount to 1% enhanced H2 generation due to the increased interface between the metal and the semiconductor. In addition, atomic Pt assembled on TiO2 (Pt1/def-TiO2) was reported to promote H2 evolution, in which the Pt–O–Ti3+ surface formation was identified as an effective strategy.86 This interface enhances charge transfer, achieving H2 evolution rates of up to 52.7 mmol g−1 h−1. The Pt–O–Ti3+ atomic interface facilitates the transfer of photo-generated electrons from Ti3+ defect sites to individual Pt atoms, thereby improving electron–hole pair separation. A summary of representative cocatalysts for methanol based H2 production is also provided in Table S3 (ESI†).Due to their low cost, earth-abundant transition metals have been applied in photocatalytic methanol conversion.87 Among the various non-noble metals, Cu is an efficient cocatalyst due to its outer electron arrangement, being similar to Au and Ag. The work function of Au (5.1 eV) is greater than that of TiO2 (4.4 eV), and electrons in the CB of TiO2 overcome the energy barrier at the interface and migrate to the Au surface.88,89 Similarly, the high work function (4.6 eV) of Cu compared to TiO2 enabled it to extract the photogenerated electrons from the CB. In addition, its high electrical conductivity and plasmonic properties offered visible-driven methanol-based hydrogen production for H2 generation.90 Apart from the above basic advantages of Cu, altering the morphology of Cu also influenced the catalytic activity. For instance, Cu nanowires fabricated on TiO2 nanorods were reported for H2 production from methanol-based hydrogen production, in which Cu nanowires were obtained by a microwave-assisted thermal strategy. TiO2 harvested the incident light and photogenerated the charge carriers that were successfully extracted by Cu before recombination.91 In another report, Cu was incorporated into ultrafine TiO2, resulting in extraordinary H2 generation activity (2.88 mmol h−1 g−1).92 This enhancement was due to the efficient charge separation by Cu, active site engineering, and high surface area. However, the photocatalytic mechanism for the enhanced activity is still unclear due to the mixed states of Cu (Cu1+ and Cu2+), especially the reversible process (Cu1+/Cu2+) during photocatalysis. The valency of Cu in the sample was identified to be between 0 and +1.93 The high activity was attributed to the optimum valence caused by the oxidation and self-regulation of Cu. Furthermore, Cu0 was highlighted as a potential alternative cocatalyst in H2 generation from methanol based hydrogen generation, resulting in a remarkable increase of H2 evolution (23 mmol h−1 g−1) under UV irradiation.94 An interfacial charge transfer mechanism in the Cu–TiO2 system was proposed, according to which the Cu with multivalent states acted as active species for the photoreaction. In detail, Cu2+ acted as an electron trapping site for efficient charge transfer and was converted to Cu+, in addition to working as the active site.95 In a similar manner, a reversible copper activation was observed in a TiO2 photocatalyst with single copper atoms (Cu/TiO2), leading to a high hydrogen generation rate of 16.6 mmol g−1 h−1 and a significant quantum efficiency of 45% at 340 nm, thanks to its reversible and cooperative photoactivation process.96 Our early work dramatically enhanced single atom Cu loading and dispersion on anatase by using MIL-125 as a precusor, leading to a hydrogen evolution rate of 101.7 mmol g−1 h−1 with the quantum efficiency of up to 56% at 365 nm.97 In situ analysis identified Cu2+ as the active site for electron trap, which was then converted to Cu+ to facilitate proton reduction to H2. While very recent we found that the activity and stability of the photocatalyst could be remarkably improved by cooperating Pt nanodots with single atoms Cu, resulting in the hydrogen evolution rate of 476.8 mmol g−1 h−1 and the quantum efficiency of 99.2%.98 This further enhancement is attributed to the synergy between the Cu single atoms and Pt nanodots, where the reversible Cu acts as an electron bridge between TiO2 and Pt, thus accelerating proton reduction by Pt. Ni-based cocatalysts have received more attention after NiO was used as a cocatalyst with SrTiO3 for H2 generation by the Domen group.99 NiOx could act as a dual-functional cocatalyst, where Ni extracted the photogenerated electrons and NiO captured the holes.100 Ni(OH)2 served a similar role to Ni in facilitating H2 generation by the conversion of N2+/Ni0.101–103 For example, Ni(OH)2 quantum dots loaded on TiO2 nanotubes exhibited a good H2 evolution rate of 4.7 mmol g−1 h−1.103 The strong electronegativity enables Ni(OH)2 to improve the shuttling of photogenerated charges, thus promoting H2 evolution. The nanotubes, with a high surface area and unidirectional electron flow, mitigated charge recombination, while the Ni(OH)2 cocatalyst promoted charge separation and electron transfer for efficient H2 generation. In parallel, non-oxide cocatalysts such as Ni2P have also attracted tremendous interest due to their graphite-like structure. For instance, Ni2P-loaded TiO2 exhibited an outstanding H2 production rate of 9.38 mmol g−1 h−1 under a 300 W Xe lamp, which was 85 times higher than bare TiO2.104 The promoted charge separation was attributed to the upshifted Fermi level caused by the electron injection into Ni2P. Another widely studied non-oxide cocatalyst is MoS2. A high H2 evolution was obtained by MoS2 loaded TiO2, which outperformed analogous Pt/TiO2, Pd/TiO2, and Ru/TiO2. The decisive factor was the intimate contact between MoS2 and TiO2, thus promoting charge separation.105 Additionally, loading MoS2 onto the metal sulphide CdS also improved methanol photoconversion activity, resulting in a hydrogen production rate of 33.2 mmol g−1 h−1—500 times higher than that of pristine MoS2.106 This improvement can be attributed to the interaction between MoS2 as a cocatalyst and CdS as a photoharvester, which facilitates the efficient transport of photo-excited electrons.
Rare earth elements such as Gd3+ and Er3+ were also reported to enhance the H2 evolution activity of SrTiO3, primarily attributed to their up-conversion properties.107,108 The inclusion of Er3+ in SrTiO3 results in a notable increase in the H2 formation rate, achieving up to 3.3 mmol g−1 h−1, due to the up-conversion luminescence properties of Er3+.108 The incorporation of Er3+ leads to a redshift in the absorption edge and a modest improvement in visible light absorption.
Bimetallic cocatalysts have also been reported for H2 evolution from methanol-based hydrogen production. Tunable NiPd bimetallic cocatalysts loaded on TiO2 were reported for methanol based hydrogen generation.109 Interestingly, Ni1Pd10 with large aggregates (30 nm) loaded on TiO2 indicated a higher H2 formation rate (4.4 mmol g−1 h−1) than that of Ni10Pd1/TiO2 with a small cocatalyst size (3 nm). The bimetallic metals provided a synergistic effect contributing to charge transfer and acted as active sites for H2 generation. Loading dual cocatalysts of Ni and Au onto TiO2 resulted in an enhanced activity, yielding an impressive H2 generation rate of 6.36 mmol g−1 h−1 in 50% methanol–water solution.110 Similarly, PdAu bimetallic cocatalysts loaded on TiO2 were reported to enhance the activity and the electronic structure. DFT was used to understand the role of dual cocatalysts111,112 and it was found that the presence of Au suppressed in situ oxidation of Pd, allowing the oxidation ability of holes to be low enough to prevent methanol mineralisation. This way, PdAu promoted the formation of methyl formate, as opposed to CO2, which was typically produced by PdO/TiO2. In another report, the utilisation of Pt and Sn cocatalysts to modify TiO2 was investigated, resulting in an H2 evolution rate of 2 mmol g−1 h−1.113 The introduction of Sn was found not only to reduce the unfavourable OH groups and vacancies but also to assist higher Pt dispersion. Oxygen vacancies contributed to visible light absorption, while Sn introduction created electron trapping sites that improved charge separation and hydrogen production at lower tin contents. However, increased tin loading altered the Sn assemblies, affecting surface properties, acidity, and interfacial charge transfer, leading to diminished efficiency.
Apart from metal-based cocatalysts, nanostructured carbon materials including graphene oxide (GO), graphitic carbon nitride (g-C3N4) and carbon nanotubes (CNT) have also been reported as cocatalysts for methanol-based hydrogen production.46,114,115 Graphene was reported to successfully extract the electrons from TiO2, leading to a 41 times higher H2 evolution rate than bare TiO2.116 Such a role was also observed in CNTs while depositing them on TiO2.53,114,117 The various synthesis methods of CNTs could influence the H2 evolution. The catalysts obtained using the hydrothermal strategy were more efficient than those synthesised by photo-deposition or chemical reduction.118 Depositing GO on the surface of the semiconductors could promote methanol based H2 evolution by acting as an electron acceptor.119,120 In addition, GO with an interlayer spacing of 0.42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm served as an individual photocatalyst for H2 evolution, and the activity in 20% methanol solution reached 2.8 mmol g−1 h−1.121 The removal of oxygen groups during photocatalysis reduces the bandgap and improves conductivity, while maintaining stable H2 production.
nm served as an individual photocatalyst for H2 evolution, and the activity in 20% methanol solution reached 2.8 mmol g−1 h−1.121 The removal of oxygen groups during photocatalysis reduces the bandgap and improves conductivity, while maintaining stable H2 production.
Overall representative photocatalysts loaded with diverse cocatalysts for methanol based H2 production are shown in Fig. 3. Noble metals like Pt and Rh remain the gold standard for methanol reforming due to their high stability, catalytic efficiency, and resistance to overoxidation. However, their high cost and the risk of CO byproduct formation, which can poison the catalyst, remain significant drawbacks. For instance, the photocatalyst Rh/CaNb6 presented a quantum efficiency of up to 65% at 300 nm,122 indicating its ability to efficiently facilitate charge separation and suppress recombination. In contrast, non-noble metals like Cu and Ni offer more economical alternatives with favourable electronic and plasmonic properties for visible-light absorption and charge transfer, though their quantum efficiency was generally below 20%.102,105,106,123–125 In Cu systems, we found that the precise oxidation state control was crucial to mitigate deactivation and photocorrosion, with a single atom Cu system achieving a quantum efficiency of 56% at 365 nm.97 A recent innovation combining Cu single atoms with Pt nanodots has reached a breakthrough quantum efficiency of 99.2% at 365 nm,98 illustrating a promising strategy to enhance methanol reforming efficiency and selectivity by integrating the advantages of both single atoms and nanoparticles.
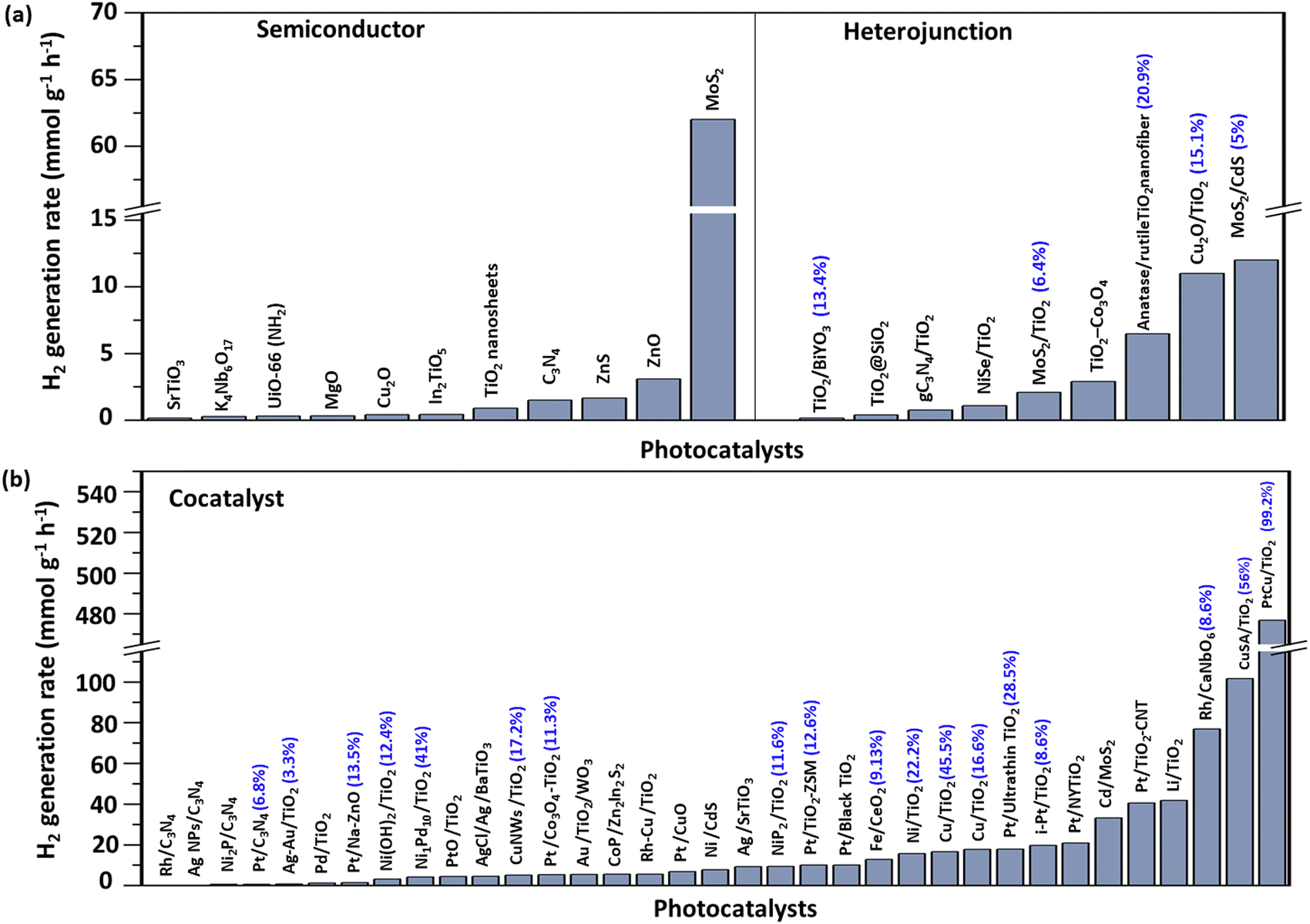 | ||
| Fig. 3 Representative photocatalysts for methanol based H2 production: (a) without cocatalysts or heterojunction structure and (b) loaded with different cocatalysts. The percentages highlighted in parentheses represent the quantum efficiency. | ||
3.4 By-product selectivity to C1/C2
Compared with traditional thermal catalytic technology, the methanol conversion, apart from valuable H2, into C1 or C2 by-products driven by photocatalysis is cleaner and more sustainable, making the catalytic reaction conditions milder. Most of the reports mainly deal with improving H2 production and focus less on the valuable liquid by-products formed during the methanol oxidation reaction, while complete oxidation to CO2 to maximise hydrogen production also results in carbon emissions, undermining the process sustainability. Furthermore, these oxygenates (e.g. formaldehyde, formic acid, and C2 compounds) are much more expensive/valuable than either H2 or CO2 and the current industrial processes to produce these oxygenates are energy- and CO2-intensive. Thus production of high-value organic by-products offers a more sustainable and economically viable alternative to the present industrial processes.Formaldehyde (HCHO) and formic acid (HCOOH) are the two primary liquid intermediates produced from methanol photo-reforming and are more valuable than COx. For example, 37% HCHO and 98% HCOOH are three times more expensive than pure methanol. Typically, methanol reforming produces three main products: HCHO, HCOOH, and CO2. Under irradiation, methoxy species derived from methanol are adsorbed onto the surface of metal oxides, leading to the subsequent formation of HCHO and HCOOH. In 1996, HCHO was detected from methanol photo-reformation using anatase TiO2, demonstrating that the produced hydroxyl radicals oxidise the intermediate. Inspired by the initial finding, various types of TiO2 were developed to explore the photo-formation pathways of HCHO from methanol. The process proved faster under inert conditions than in an O2 atmosphere, with HCHO emerging as the sole oxidized by-product.126 We found that PtCu–TiO2 was highly efficient in catalysing the oxidation of methanol to HCHO, achieving a high selectivity of approximately 98.6% for HCHO. Subsequently, other non-noble metals such as Cu, Ni, and Co were loaded on anatase type TiO2 to study the role of metals in the efficiency of methanol photo-reforming.127 The primary products observed were HCHO and H2, with a minor presence of HOOH. Excess photoexcited electrons migrated to metal sites, enhancing H+ reduction and charge separation, while traces of Cu2+ suggested partial oxidation of Cu by photo-generated holes. Felipe et al. further investigated the influence of Au on TiO2, focusing on by-product selectivity towards HCHO and HCOOH.128 They found that increasing CH3OH concentration boosted catalytic activity by 50%. This enhancement was attributed to the increased availability of reactants at the catalyst surface, facilitating the formation of key intermediates and promoting effective separation of photogenerated electron–hole pairs. Furthermore, higher light intensity improved the selectivity towards HCHO by 38% and HCOOH by 62%. At moderate light intensities, back reactions could overshadow hydrogen production. In contrast, higher intensities generated an excess of electrons that competed with back electron transfer and reduced side reactions, ultimately enhancing the production of hydrogen and intermediates.
When producing H2, simultaneous photocatalytic C–C coupling of methanol to C2 by-products like ethylene glycol (HOCH2CH2OH) is promising yet challenging, with limited reports citing success using metal sulphides such as ZnS and CdS. Initially, in 1982, H2 was generated from methanol under UV irradiation with low activity (0.17 mmol g−1 h−1).67 Subsequent studies using ZnS achieved a high selectivity for producing HOCH2CH2OH from aqueous methanol, with the selectivity later enhanced to 52%, marking a significant advancement in photo-assisted C–C coupling through free radical intermediates.32 Further investigations into colloidal ZnS increased the selectivity to 95% after 6 hours of irradiation, with characterisation studies indicating that the ˙CH2OH radical was the primary intermediate.129,130 In another report, the porous MoS2/CdS photocatalyst was explored for the conversion of CH3OH to HOCH2CH2OH through efficient activation of the C–H bond.106 This process generated a hydroxymethyl radical (˙CH2OH), which readily desorbed from the catalyst surface to undergo coupling. The system achieved a quantum efficiency of 5% at 450 nm. Advanced characterization and computational analyses confirmed the reaction mechanism, which involved C–H bond scission followed by C–C coupling, competing with the activation of O–H bonds that led to aldehyde formation. Additionally, classic cocatalysts like Pd, Pt, MoS2, and CoP were employed on Zn2In2S5 for methanol photo-conversion, with CoP showing the highest activity, producing HOCH2CH2OH at a rate of 5.5 mmol g−1 h−1.131
Methyl formate (HCOOCH3) is another common C2 product forming via the C–C coupling of methanol. The early photo-assisted HCOOCH3 formation was reported in 1985 using MoO3/TiO2 photocatalysts.132 The process involved primary oxidation of adsorbed CH3O(a) species, converting them into HCOO(a) on plain TiO2, with HCOOCH3 as the main by-product. Incorporating a surface molybdate monolayer significantly enhanced the selectivity, while suppressing secondary oxidation reactions. At lower molybdate loadings, HCOOCH3 still dominated as the primary oxidation product. However, once the molybdate monolayer was fully developed, dimethoxymethane became the predominant product, exhibiting nearly 100% selectivity at low conversions. This change in selectivity underscores the kinetic differences between the TiO2 surface and the molybdate monolayer, with the latter providing a more controlled reaction pathway. By 2010, a peak selectivity of 90% for HCOOCH3 was achieved in gaseous methanol conversion at room temperature, though the conversion rate was only about 10%.133 Fundamental studies using Fourier Transform Infrared Spectroscopy revealed that methanol adsorbed on TiO2 as molecular and dissociated species, with subsequent oxidation to form HCHO intermediates that underwent dimerisation to HCOOCH3. Elevated temperatures also affected the adsorption equilibrium of intermediates on the catalyst surface, decreasing their availability for selective coupling reactions. The photo-oxidation of methanol on preoxidised TiO2(110) yields HCOOCH3 through a two-step photochemical process.134 Initially, methanol thermally dissociates into methoxy groups (CH3O) and water. Upon UV light irradiation, methoxy undergoes photo-oxidation to produce HCHO, which further reacts with transient formyl species to form HCOOCH3. Mass spectrometry and scanning tunnelling microscopy confirmed this mechanism, showing methyl formate formation only when both methoxy and HCHO were present on the surface. In parallel, oxygen adatoms healed surface defects and reduced charge recombination but were not directly involved in the reaction. Silver nanoparticles (Ag NPs) on TiO2 (P25) and SiO2 significantly enhanced photocatalytic methanol oxidation under UV light.135 On TiO2, Schottky barriers at the Ag–TiO2 interface prolonged charge carrier lifetimes, promoting methoxy oxidation to intermediates that coupled to form HCOOCH3. On SiO2, the plasmonic resonance of Ag NPs induced localised electric fields, driving methanol oxidation through a distinct mechanism. Both systems outperformed their bare counterparts, with Ag/SiO2 achieving a peak MF production rate of 23.46 mmol g−1 h−1, highlighting the role of Ag NPs in improving selectivity and activity. Additionally, TiO2-supported Cu catalysts facilitated methanol oxidation to HCOOCH3 in the gas phase, achieving a HCOOCH3 production rate of 56.4 mmol g−1 h−1.136 The ultra-small CuO improved the charge carrier transfer to promote activity. A similar mechanism of CuO was reported on CuZnAl and ZnO photocatalysts, showing a HCOOCH3 selectivity of 50%.137 Besides, ethanol production from methanol conversion was also reported using GaN, in which ˙CH2OH reacted with methanol to form ethanol.138 Very recently, ethene production from two CH3OH molecules was reported using surface engineered TiO2,139 with the high Ti3+ concentration facilitating methanol oxidation to HCHO and subsequent coupling to ethene, with HCHO as the main by-product and the remaining methanol reacting to produce ethene.
Table S4 (ESI†) provides a summary of the oxidized by-products and selectivity for various photocatalysts. Methanol conversion typically includes proton reduction to H2, methanol oxidation, and C–C coupling. Enhancing photoexcited charge separation and the oxidation capacity of holes is crucial for achieving high activity. Reaction conditions such as pH, temperature, and reactant concentration significantly influence the adsorption of organic intermediates, thereby affecting by-product selectivity. Cocatalysts are pivotal in improving charge transfer, serving as active sites for activity enhancement and controlling oxidation processes. Additionally, the inclusion of suitable cocatalysts can alter surface affinity towards reactants or products, influencing the coupling pathway and the selectivity of the byproducts. Thus, the design of photocatalysts with a cocatalyst and the precise control of reaction conditions are vital for optimizing by-product selectivity. Various characterization techniques are essential to elucidate the mechanisms of C–H activation and C–C coupling. While some studies have shown successful conversion of methanol into valuable chemicals, they often come at the expense of reduced H2 generation activity or selectivity. Therefore, developing efficient photocatalysts that maintain high selectivity while also ensuring favourable H2 yields remains a significant challenge.
3.5 Process control
Several factors influence H2 production from methanol reforming, including morphology, crystallinity, surface area, synthesis conditions and reaction atmosphere. Morphology and crystallinity of photocatalysts can be affected by the synthesis technology employed. Smaller photocatalysts typically facilitate rapid transfer of photo-induced charge carriers to the material surface, decreasing recombination rates and increasing surface area, thus offering more reactive sites and boosting H2 evolution. For instance, protonated g-C3N4, treated with HNO3, showed improved hydrogen production due to structural exfoliation that created ultra-small pores enhancing charge transfer and surface area. Additionally, the calcination temperature of the catalyst significantly affects hydrogen production. An example is the Ni–TiO2 catalyst, which exhibited a hydrogen production rate of 1.0 mmol−1 g−1 h−1 when calcined at 550 °C, due to improved crystallinity and surface activation.140 However, higher calcination temperatures (above 650 °C) were found to negatively impact the hydrogen production rate, dropping to 0.150 mmol g−1 h−1, possibly due to detrimental changes in catalyst shape and particle size. This observation was further supported by a study, where an N-doped TiO2 catalyst calcined at 450 °C displayed a higher hydrogen generation rate (4.4 mmol−1 g−1 h−1) compared to the same catalyst calcined at 550 °C (3.8 mmol−1 g−1 h−1), with performance deteriorating further at 650 °C (3.4 mmol−1 g−1 h−1).141 These findings suggest that elevated calcination temperatures increase the particle size, reducing the catalyst's dispersion and electron availability, which in turn affect its efficiency in hydrogen production.While mild reaction temperatures mildly influence the thermodynamics of photocatalytic reactions, they significantly enhance the desorption of by-products from the catalyst surface, thus boosting photocatalytic activity. Huaxu et al. observed that increasing the reaction temperature from 45 °C to 55 °C led to a significant enhancement in the Pt/TiO2 photocatalyst's H2 generation rate, rising from 4.71 mmol−1 g−1 to 15.18 mmol−1 g−1 within 4 hours.142 Similarly, Maggard noted optimal activity for a TiO2 photocatalyst within the temperature range of 60–80 °C.143 Conversely, lower temperatures tended to reduce H2 generation activity, largely due to slower by-product desorption rates compared to reactant adsorption rates on the catalyst surface. Higher temperatures facilitated charge carrier transfer from the valence band to higher energy states, helping to prevent charge recombination. Velázquez et al.144 used 2 wt% Pt on TiO2 with bio-renewable oxygenated methanol to achieve 13 mmol−1 g−1 h−1 at 20 °C, with increases to 19.5 µmol−1 g−1 h−1 and 38 mmol−1 g−1 h−1 at 40 °C and 60 °C, respectively, due to synergistic effects of light and thermal energy aiding electron excitation. Moreover, the pH of the methanol/water medium impacted photocatalytic H2 production. Lin et al.145 found a 2.25-fold activity increase for Pt–TiO2−xNx as pH rose from 3 to 6.3, correlating to peak methanol adsorption at a pH matching the point of zero charge.146 This condition maximised surface –OH groups essential for H2 formation. Additionally, incident photon absorption crucially influences photocatalytic activity. Tambago et al. reported doubled hydrogen evolution activity with increased irradiation intensity from 33 mW cm−2 to 70 mW cm−2.147 This effect was confirmed by Baniasadi,148 who saw a 20% increase in hydrogen generation activity by boosting light intensity from 900 W cm−2 to 1000 W cm−2.
3.6 Mechanistic understanding
According to early mechanistic research on intermediates from photocatalytic methanol-involved hydrogen generation proposed by Kawai et al.,60 methanol was progressively degraded on the TiO2 surface to form HCHO, HCOOH, and finally CO2, accompanied by H2 production as the reduction product. Fig. 2(c) illustrates the pathway for methanol photooxidation on the TiO2 surface,39 highlighting stages of adsorption/desorption and chemical transformation. The multistep oxidation process introduces complexity due to the formation of liquid intermediates. Rapid production of HCHO with no detection of other liquid products suggested it as the initial step in methanol photo-reforming. Further studies on Pt/TiO2 and Au/TiO2 catalysts revealed a mechanism of CH3OH → HCHO → HCOOH → H2 and CO2, with Au's surface plasmon resonance enabling Au–Pt/TiO2 to operate under visible light. However, CO and CH4 were observed when switching from UV light to visible light irradiation, with CO adsorption deactivating active species on the semiconductor. Thus investigating surface species and photocatalytic charge mobility plays a pivotal role in advancing the understanding of photocatalytic processes. It helps unravel the complex surface chemistry involved, including adsorption, activation, and desorption processes, leading to improved catalyst performance and reaction kinetics. Transient absorption spectroscopy (TAS) is a reliable technique to investigate photophysics during methanol-based hydrogen production.149 Recently, a unique electron-accepting photocatalyst, Cu single atom loaded TiO2 (CuSA–TiO2), was reported.97 The transient absorption spectroscopy (TAS) spectra indicated the electron features, where modified TiO2 exhibited a reduced electron signal after loading CuSA, suggesting that electrons were effectively trapped from TiO2 to CuSA. When adding Ag+ as an electron scavenger, a decreased signal of CuSA–TiO2 was observed due to the consumption of electrons by Ag+. As expected, CuSA–TiO2 showed a similar profile of photoelectron decay to pure TiO2, indicating the electron extraction by CuSA. Femtosecond TAS was also used to investigate the transportation dynamics of the twin Z-scheme catalyst CN/H–TiO2.150 Compared with CN and H–TiO2, the TSP sample (Fig. 4(a)) exhibited the highest τ value, revealing the longest life of the carrier. Such a phenomenon was attributed to fast electron capture and slow optical carrier restructuring. In addition, in situ extended X-ray absorption fine structure (EXAFS) was used to investigate the density of orbital states involved in electron transitions, revealing the charge transfer pathway over PtCu–TiO2 for methanol-based hydrogen production, as shown in Fig. 4(b).98 By analysing Ti L-edge and O K-edge Extended X-ray Absorption Fine Structure (EXAFS) absorption features in the dark and during irradiation, the excitation of electrons from O 2p to Ti 3d under irradiation was observed. Cu was the most effective in abstracting electrons from TiO2, followed by Pt as an electron-trapping site. The combination of Cu and Pt in PtCu–TiO2 exhibited irreversible electron transfer characteristics. Furthermore, EXAFS can be used to reveal the electronic features of catalytic materials. For example, the k2-weighted EXAFS spectra of MoS2/CdS indicated an intensity reduction in Mo–Mo coordination, which was ascribed to the sheet-edges serving as H2 production sites.106 Jacquelin et al.151 applied in situ electron paramagnetic resonance (EPR) spectroscopy to monitor electron transfer over Au/TiO2, revealing that different wavelengths of light stimulated the transfer through distinct electron excitation pathways within the Au particle. In another study of applying in situ EPR for charge mobility investigation, it was shown that pristine g-C3N4 favoured negative reduction potential, while pristine TiO2 favoured positive oxidation potential.150 In the case of the g-C3N4/H–TiO2 heterojunction catalyst both oxidation and reduction abilities were significantly enhanced compared to pristine catalysts, confirming the spatial distribution of oxidation and reduction sites and the effective separation and transfer of photogenerated carriers through a twin Z-scheme charge transfer path. Time-resolved Raman and IR spectroscopy techniques have emerged as valuable tools in capturing ultrafast molecular transformations and charge transfer dynamics, essential for advancing the understanding of photocatalytic mechanisms. Time-resolved Raman spectroscopy, for instance, was effectively employed to explore photochemical processes on plasmonic metal nanoparticle surfaces. In a study conducted by Baumberg et al.,152 metal nanoparticles generated energetic charges through nonradiative plasmon relaxation, enabling hot-electron-induced photoreduction reactions. By monitoring temporal changes in surface-enhanced Raman scattering signals from molecules adsorbed on Au nanoparticles, researchers observed real-time hot charge production, transport, and single-molecule redox events at plasmonic hotspots. Time-resolved IR absorption spectroscopy was used to monitor electron- and hole-capture reactions on TiO2 and Pt/TiO2 photocatalysts in the presence of dioxygen, water vapour, and methanol vapour. After a 355 nm UV pulse, a transient IR absorption band from 3000 to 1000 cm−1 appeared, attributed to photogenerated electrons in shallow mid-gap states.153,154 Under vacuum, these electrons recombined with holes at a multi-exponential rate, while the presence of reactants altered electron decay rates, indicating reactant-driven capture reactions. This observation highlighted the influence of methanol on electron decay kinetics, advancing the understanding of reaction pathways in photocatalytic water-splitting. Expanding on this dynamic view, step-scan time-resolved FTIR spectroscopy provided a time-resolved approach to studying methanol oxidation on TiO2 surfaces, indicating that long-lived electron decays correlated with photocatalytic activity.155 This decay was linked to electron consumption for hydrogen production, facilitated by methanol or water adsorption on TiO2. By examining proton transfer mechanisms, this approach offered mechanistic insights that enhanced those obtained from steady-state observations.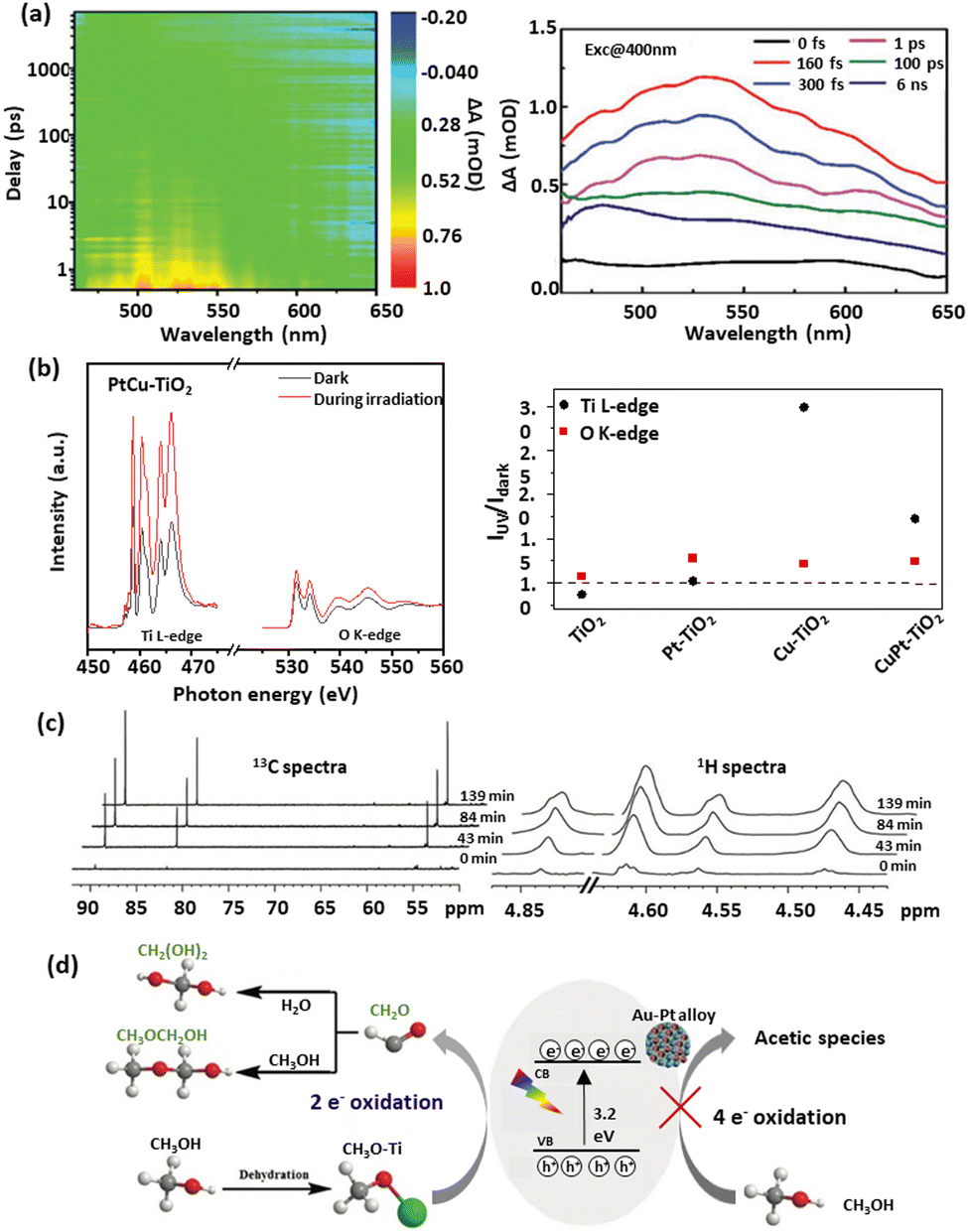 | ||
| Fig. 4 Representative mechanism investigation via advanced techniques: (a) Transient absorption spectra of TSP.150 (b) In situ Ti L-edge and O K-edge EXAFS spectra of PtCu–TiO2 in the dark and during irradiation and the ratio of intensity under light irradiation to that under dark conditions.98 (c) In situ NMR scheme of 13C and 1H over Au–Pt/TiO2 for H2 generation from selective methanol oxidation, and (d) the proposed mechanism156 (a) Reproduced from ref. 150 with permission from John Wiley and Sons, Copyright 2023. (b) Reproduced from ref. 98 with permission from Springer Nature Limited, Copyright 2023. (c) and (d) Reproduced from ref. 156 with permission from Elsevier, Copyright 2023. | ||
While time-resolved IR captures rapid transformations, in situ FTIR enables the identification of surface-bound intermediates throughout the reaction. Studies on Pt/TiO2 during methanol photooxidation, for example, identified intermediates such as CH2O(a), CH2OO(a), and HCOO(a).155 Furthermore, Haselmann et al.157 developed a liquid-cell in situ attenuated total reflection (ATR)-FTIR setup, utilising top-irradiated ultraviolet light to investigate surface species of methanol-based hydrogen production over Pt/TiO2. The FTIR spectra acquired during the photocatalytic reaction unveiled five noteworthy spectral components, including the following vibrations: (i) ν(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) bands, which originate from H2O, methanol, and Ti–OH in TiO2; (ii) ν(C–H) vibrations, derived from organic species; (iii) ν(CO) signals, arising from gaseous and dissolved CO2; (iv) bands of CO coordinated to Pt0; and (v) ν(CO) signals, stemming from carbonyl functionalities. To further explore the roles of free radicals and reaction intermediates, nuclear magnetic resonance (NMR) spectroscopy can be employed. For instance, the in situ NMR (13C, 1H) on an Au–Pt/TiO2 system identified key methanol oxidation intermediates, with the 13C NMR spectrum showing peaks at 55, 83, and 90 ppm corresponding to chemisorbed methoxy species, methanediol, and methoxymethanol, respectively.156 As the reaction progressed (Fig. 4(c)), the increasing intensities of these peaks indicated the accumulation of methanediol and methoxymethanol, a trend corroborated by 1H NMR spectra. This evidence supported a primary two-electron dissociation pathway in methanol oxidation in this work, leading to HCHO rather than a more complex four-electron route (Fig. 4(d)). In other words, methanol was initially adsorbed onto titanium hydroxyl (Ti–OH) sites, forming surface methoxy species (CH3–O–Ti), which were then oxidised by photogenerated holes or hydroxyl radicals (˙OH) to produce HCHO. Due to its high reactivity, HCHO quickly reacted with water to form methanediol or with methanol to produce methoxymethanol on the Au–Pt/TiO2 catalyst.
H) bands, which originate from H2O, methanol, and Ti–OH in TiO2; (ii) ν(C–H) vibrations, derived from organic species; (iii) ν(CO) signals, arising from gaseous and dissolved CO2; (iv) bands of CO coordinated to Pt0; and (v) ν(CO) signals, stemming from carbonyl functionalities. To further explore the roles of free radicals and reaction intermediates, nuclear magnetic resonance (NMR) spectroscopy can be employed. For instance, the in situ NMR (13C, 1H) on an Au–Pt/TiO2 system identified key methanol oxidation intermediates, with the 13C NMR spectrum showing peaks at 55, 83, and 90 ppm corresponding to chemisorbed methoxy species, methanediol, and methoxymethanol, respectively.156 As the reaction progressed (Fig. 4(c)), the increasing intensities of these peaks indicated the accumulation of methanediol and methoxymethanol, a trend corroborated by 1H NMR spectra. This evidence supported a primary two-electron dissociation pathway in methanol oxidation in this work, leading to HCHO rather than a more complex four-electron route (Fig. 4(d)). In other words, methanol was initially adsorbed onto titanium hydroxyl (Ti–OH) sites, forming surface methoxy species (CH3–O–Ti), which were then oxidised by photogenerated holes or hydroxyl radicals (˙OH) to produce HCHO. Due to its high reactivity, HCHO quickly reacted with water to form methanediol or with methanol to produce methoxymethanol on the Au–Pt/TiO2 catalyst.
4. Integrating photocatalysis and thermocatalysis
With the above success, traditional photocatalysis still presents a low H2 production rate and moderate selectivity for valuable by-products due to fast charge recombination and overoxidation. From a thermodynamic standpoint, methanol reforming is an endothermic reaction where heat is necessary. In parallel photochemical processes lower the activation energy barrier by facilitating the formation of key intermediates. Kinetically, according to the Arrhenius equation, the reaction activity increases with the temperature. Thus enhancements in photocatalytic performance via thermal effects should be favourable. Photo-thermo catalysis is divided into three categories: one involves photon energy to heating that is directly used to activate the reactants, another uses photon-induced surface plasma to drive the chemical reactions, and the last is to couple heating with photocatalysis (the concept of photon–phonon co-driven catalysis). The first two are widely reported and the last one is the most attractive fundamentally as it combines the advantages of the photocatalysis (high selectivity) and thermocatalysis (high activity).158,159 Thus, the following focuses on the process of photon–phonon co-driven catalysis, which was firstly underlined by us about two years ago and we believe that it is the emerging and future research field to replace only photocatalysis, while distinguishing it from related concepts such as localized surface plasmon resonance (LSPR).The photon–phonon co-driven catalytic process likely operates through a synergistic mechanism that enhances the efficiency and selectivity of chemical reactions. Photons generate electron–hole pairs, activating surface catalytic sites to drive redox reactions, while phonons may provide localised thermal energy to promote bond dissociation and improve intermediate hopping and product desorption dynamics. This interplay could create pathways that might be challenging to achieve with photon-driven or phonon-driven systems alone. The heating in this co-driven process can originate from two alternative sources: (i) heat induced by infrared light or (ii) externally supplied heating, which is dependent on the application environment. If the process takes place indoors, external heating is needed. If it takes place outdoors, IR from sunlight can provide heating energy.
A pre-heating is necessary for the vaporisation of methanol and water by the conventional gas phase reforming reaction.160 The photo-thermo catalytic methanol reforming can potentially reduce the reaction temperature to as low as 100 °C. Nevertheless, lower conversion rates at lower temperatures and poor stability are some of the limitations of the early photothermal methanol reforming.161 A solar-to-hydrogen production from methanol photothermal conversion achieved an efficiency of almost 33%, indicating the potential implementation of the synergistic effect of photocatalysis and thermocatalysis on the industrial methanol dehydrogenation.162 It was later reported that the photo-thermocatalysis could dramatically enhance the H2 production rates.163,164 Photo-thermocatalytic materials are a complex of two different types of materials that ideally have to possess full-spectrum light harvesting ability, effective photo-to-thermal conversion, and abundant active sites. Some of the materials that can be utilised are the inorganic semiconductors, plasmonic metals, metal–organic framework catalysts and even polymers.
Domen's group has shown that raising reaction temperature from 25 °C to 58 °C increased the solar-to-hydrogen (STH) efficiency of the SrTiO3:Al photocatalyst from 0.4% to 0.6%, as shown in Fig. 5(a).165 Additionally, higher temperatures facilitated the generation of hydrogen through photocatalysis, whereas in most cases the temperature was within a lower range (from room temperature to up to 100 °C). This enhancement was attributed to the lower apparent activation energy (7.6 kJ mol−1), reflecting the light-driven nature of the reaction, where electron–hole pairs drove redox processes. The observed reaction rates demonstrated stable hydrogen and oxygen evolution, with small gas bubbles released efficiently due to the thin water layer and hydrophilic panel design. This temperature dependence and sustained activity highlight the importance of optimising both catalyst performance and reactor design to maximise solar hydrogen production efficiency. CoO NPs were reported to show a significant increase in hydrogen production efficiency, ranging from 0.34 to 1.96 mmolh−1g−1, as the temperature was raised from 25 °C to 100 °C. Importantly, halting light irradiation ceased hydrogen generation, indicating the neglect of thermally induced hydrogen evolution.166 A cost-effective NiOx-enhanced TiO2 catalyst was developed for hydrogen production from methanol dehydrogenation. Optimized with 5 wt% Ni, the catalyst produced hydrogen at 53.7 mmol h−1 g−1 under simulated AM1.5G sunlight at 260 °C, more than doubling the output without the light irradiation. Quantum efficiency measurements showed 66.24% at 380 nm, decreasing to 15.35% at 500 nm. In addition under visible light (>420 nm) at 260 °C, the yield increased dramatically to 26.9 mmolh−1g−1 from 1.1 µmol h−1 g−1 at room temperature.167 CuInS2 offered a remarkable activity in photon–phonon co-driven conversion of methanol, with the hydrogen generation rate of 36 mmol g−1 h−1. Its exceptional low-temperature H2O molecule dissociation ability facilitated the formation of abundant interfacial OH radicals, thereby enhancing the C–H single bond breakage in methanol, reducing the apparent activation energy by 26%. Encouragingly, CuInS2@MIL-101(Cr) demonstrated an excellent total turnover number (TON), reaching up to 16775 within 65 hours of operation without any deactivation of the catalyst.168 Recently, a nickel–iron bimetal catalyst supported by gallium nitride nanowires on a silicon substrate, NiFe/GaN, achieved a notable hydrogen evolution rate of 61.2 mmol h−1 g−1 from methanol–water under light illumination.169 As the reaction temperature decreased from 90 °C to 10 °C, the hydrogen production rate dropped by a factor of four, and no hydrogen was detected when heating in the absence of light, highlighting the combined effects of photo- and thermal catalyses, as shown in Fig. 5(b). This result underscores the catalyst's ability to synchronise ultraviolet-driven charge carrier excitation with photothermal effects from visible and infrared light, maximising sunlight utilisation. The synergistic combination of Ni and Fe dramatically lowered the energy barrier of the potential-limiting step (*CHO → *CO), as confirmed by operando spectroscopy and density functional theory (DFT) calculations. The GaN NWs/Si platform enhanced light absorption, charge separation, and catalytic site dispersion while leveraging photothermal effects to further improve efficiency. The reaction pathway (*CH3O → *CH2O/*CHO → *CO → *CO2) proceeded alongside water dissociation into reactive ˙OH species, enabling sustained hydrogen production. In a study that combined photocatalytic and thermocatalytic effects across the full solar spectrum to efficiently convert methanol into hydrogen, a CuZnAl-LDH precursor was used to fabricate a CuO/ZnO/Al2O3 nanocatalyst.170 This catalyst demonstrated an impressive hydrogen production rate of 144.6 mmol g−1 h−1 at 130 °C, outperforming systems based solely on either photocatalysis or thermocatalysis. The most attractive was that the dual reaction sites of PtCu–TiO2 exhibited extremely high hydrogen generation from a methanol/water mixture.98 The catalyst's activity soared to 476.8 mmol g−1 h−1 when increasing the temperature from 25 °C to 70 °C, with no hydrogen observed in the absence of illumination, as shown in Fig. 5(c).98 The synergistic approach leveraging both photons and phonons significantly enhanced the catalytic performance and efficiency in methanol reforming, which outperforms all photocatalysts and is comparable to the best thermal catalysts. Another example is a recent study that revealed photocatalytic hydrogen evolution boosted by the solar-heat, where SAAg-g-C3N4 demonstrated a good activity and stability.171 The observed catalytic enhancement of SAAg-g-C3N4 was attributed to the favourable Gibbs free energy of the adsorbed hydrogen atom and the formed N–Ag bonds. When the temperature was elevated from 25 °C to 55 °C, the hydrogen generation rate significantly increased, underscoring the positive impact of solar heat on the photocatalytic process.
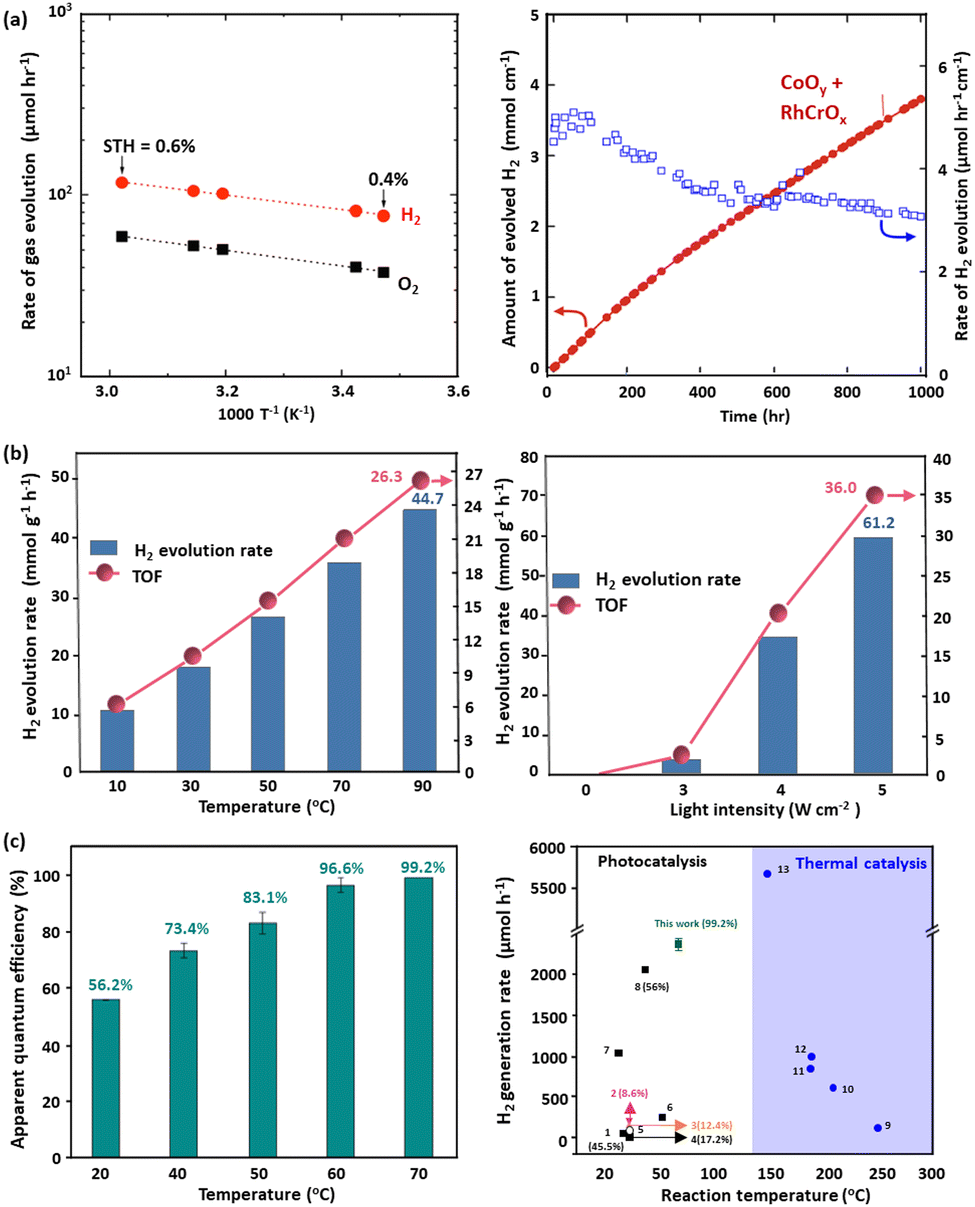 | ||
| Fig. 5 (a) Temperature dependence of the water splitting activity and reaction time courses of RhCrOx/SrTiO3:Al under AM 1.5G simulated sunlight,165 (b) photocatalytic effects of NiFe/GaN on chamber temperature and light intensity,169 (c) The quantum efficiency of PtCu–TiO2 at various temperatures and the activity comparison of diverse photocatalysts and thermal catalysts.98 (a) Reproduced from ref. 165 with permission from Elsevier, copyright 2017. (b) Reproduced from ref. 169 with permission from the American Chemical Society, Copyright 2019. (c) Reproduced from ref. 98 with permission from Springer Nature Limited, Copyright 2023. | ||
5. Reactor design
A meticulously engineered reactor can significantly enhance light absorption and mass transfer, thereby substantially increasing the yield and specificity of desired by-products. The design parameters of reactors, such as shape, size, thickness, and materials (e.g., quartz and borosilicate), critically influence photocatalytic methanol-based hydrogen production.172,173 The conventional packed bed reactor, often used in thermocatalysis for hydrogen production from methanol, is favoured industrially for its simpler design and construction, despite a high pressure drop across the reactor.174,175 Recent studies, including extensive experimental and theoretical simulations like computational fluid dynamics (CFD), have been conducted.27,30,34,35,176–178 For instance, Karim et al.179 investigated how deviations from isothermality in a packed bed reactor affected methanol steam reforming rates using a commercial CuO/ZnO/Al2O3 catalyst. They identified heat transfer limitations within the reactor bed and implemented a reduction in reactor diameter to enhance heat transfer, achieving near-isothermal operation and higher apparent catalyst activity. Chougule and Sonde explored the effects of temperature, steam-to-carbon (S/C) ratio, and operating conditions on methanol conversion in a tubular packed bed reactor (Fig. 6(a)).180 Their findings, based on both simulation and experimental data, indicated optimal hydrogen generation and methanol conversion at a temperature of 300 °C with an S/C ratio of 1.4. Comparatively, Karim et al. assessed a coated-wall reactor where the catalyst bed is affixed to the reactor wall, noting advantages such as lower pressure drop, improved mass and heat transfer, and minimal catalyst usage.181,182 In contrast, packed bed reactors with internal diameters between 1 and 4.1 mm exhibited limited heat transfer and temperature gradients up to 40 K. Hafeez et al. conducted a simulation study comparing the performance of packed bed and coated wall microreactors using a CuO/ZnO/Al2O3-based catalyst (BASF F3-01), observing comparable performance at equivalent temperatures.183 Their case studies included variables such as temperature, residence time, steam to methanol ratio, and catalyst coating thickness, revealing that larger catalyst pellet sizes led to internal mass transfer resistance and decreased methanol conversion. Additionally, thicker catalyst wall-coatings resulted in higher volumetric productivity for the same reactor diameter.181 Another reactor type utilised for methanol-based hydrogen production is the multi-tubular packed-bed reformer, which also serves as a heat exchanger, shown in Fig. 6(b).184 This reformer's structure includes baffles, tubes, and a shell, typically insulated to minimize heat loss. The tubes, filled with the CuO/ZnO/Al2O3 catalyst, were arranged in equilateral triangle bundles, supported by baffle plates that enhanced flow distribution and heat transfer efficiency between the tube and shell sides.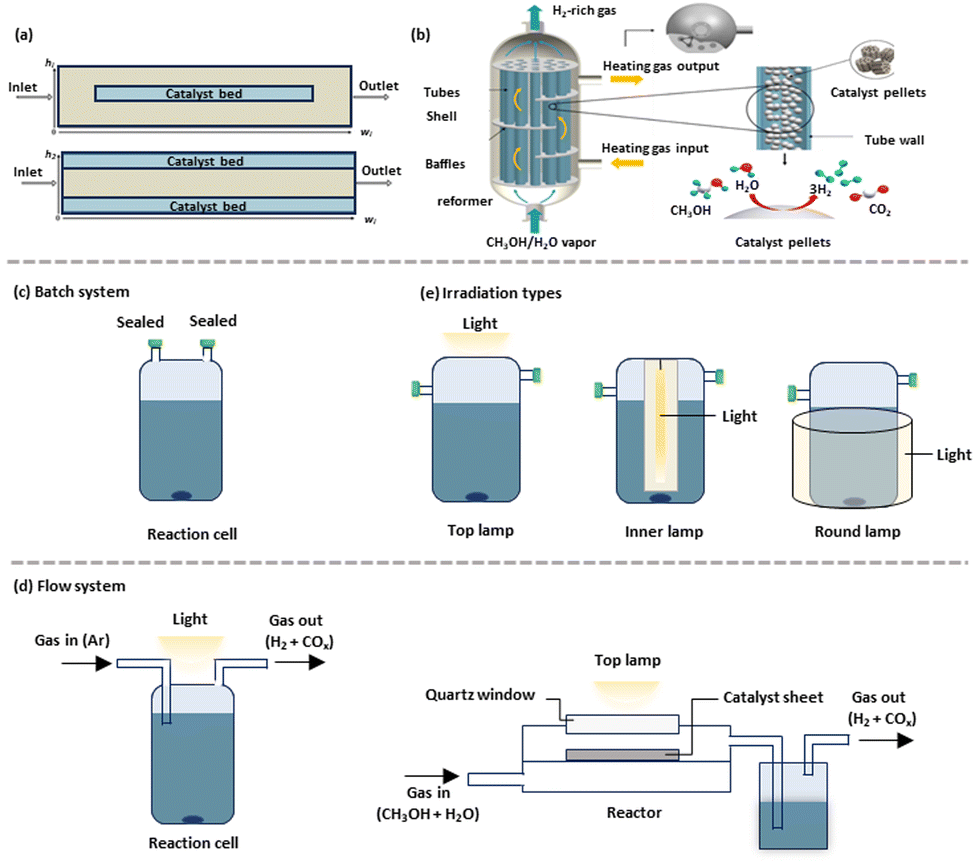 | ||
| Fig. 6 Schematic representation of the reforming reactor: (a) packed bed and coated-wall packed bed microreactors, (b) a multi-tubular packed-bed reactor.184 (c) Batch system and (d) flow systems. (e) The irradiation types. (b) Reproduced from ref. 184, no permission required. | ||
Inspired from the design of thermal catalytic reactors, photocatalytic reactors have evolved to include diverse configurations that enhance light utilization and catalytic efficiency. Batch reactors (Fig. 6(c)), a primary type, are often employed in the suspension systems. In the suspended systems, small catalyst particles are uniformly dispersed in solution. This configuration, widely used in batch reactors, enhances the external surface area exposed to irradiation and maintains a consistent temperature through uniform mixing, which improves the selectivity of desired products. However, suspended systems face challenges with mass transfer limitations between reactants and the catalyst surface, often restricting overall photocatalytic efficiency. Additionally, separating catalyst particles from the solution for recycling can be both costly and time-consuming. Flow reactors (Fig. 6(d)) present another configuration, where photocatalysts can either be suspended in an aqueous methanol solution or immobilized on substrates. These reactors often include a liquefaction section to capture intermediates produced during hydrogen generation from methanol. While the flow systems offer enhanced catalyst exposure, they may suffer from issues related to reactant accumulation and by-product formation, potentially impacting the selectivity and efficiency. In contrast, gas-phase reactors eliminate the need for catalyst separation, as the photocatalyst is immobilized on substrates via methods like suction or drop casting, simplifying recovery and enhancing sustainability. For example, Chiarello et al. reported that a flow reactor achieved 30% higher activity compared to a batch reactor due to enhanced mass transfer and photon utilization.185 In these systems, a single oxidized product was typically produced in the gas phase, whereas more intermediates were found in the liquid phase. A similar gas phase system was reported over Cu–TiO2 under ambient conditions,186 where the H2 generation activity from methanol-based hydrogen production was enhanced by a factor of 1.63 compared to that of the liquid phase counterpart. Such accessible activity was ascribed to promoted mass transfer and regulated reaction time when using a gas phase flow system.
Irradiation pathways are crucial for achieving uniform light distribution across the reactor and the catalytic surface, particularly when scaling up. As shown in Fig. 6(e), top irradiation is suitable for small systems, allowing light to penetrate directly through the catalyst bed. Inner irradiation, with the light source positioned inside the reactor, provides radial light distribution along the reactor, ensuring the relatively uniform activation of all catalyst particles. Round irradiation utilises light sources encircling the reactor, providing multidirectional exposure and is particularly advantageous in a heated reactor. In scaled-up systems, where light penetration is often limited to less than 1 mm in thicker catalyst beds, light guides or optical fibres are essential for directing light to specific photocatalytic sites, enhancing light harvesting.187 Additionally, fixed-bed reactors can be scaled up by increasing the diameter while optimising light distribution, thus improving efficiency. Further improvements in flow reactors have been demonstrated by Goto et al., who developed a panel-type reactor using Al-doped SrTiO3, achieving a 10% solar hydrogen efficiency and scalable to at least one square metre.188 The panels were angled at 10–20 degrees for optimal light capture, and a thin water layer facilitated consistent gas release while preventing pressure buildup. To further ensure efficient gas escape, the reactor's interior was hydrophilic, allowing stable gas bubble release. In both batch and flow reactors, advanced designs, such as fixed-bed setups with parabolic mirrors or solar concentrators, can further concentrate light onto the catalytic surface, boosting light intensity without extra energy input. Flow reactors also enable better control over methanol residence time, product desorption, and mass transfer. However, the limited reaction time on active sites in flow systems can sometimes reduce methanol conversion, requiring a balance between the residence time and the conversion efficiency.
Reactors for integrating thermocatalysis with photocatalysis have also been reported, thereby harnessing the synergistic effects of both methods to enhance reaction efficiency and output. One approach is to use a heating jacket if external heating is needed, allowing for uniform irradiation of the catalyst bed by an external heat source. Another approach involves direct photo-thermo systems, which minimize heat transfer distances compared to traditional methods, resulting in faster start-up times and enhanced load flexibility.189 Very recently, Erwin et al. developed a novel system integrating an aluminium-doped strontium titanate (Al:SrTiO3) photocatalyst with a solar vapour generator.190 This novel setup achieved highly efficient hydrogen generation through a dual mechanism that involved photocatalysis driven by UV-visible light and thermal energy from infrared (IR) light. This innovative design featured a floating photocatalyst layer above the solar vapor generator, ensuring that it only interacts with water vapor and not with liquid water. This strategic separation greatly enhanced the photocatalyst's longevity and effectiveness by protecting it from potential contaminants in natural water sources, thereby enhancing the hydrogen production process. The advancement of tandem reactors featuring high-performance catalysts is crucial for enhancing catalytic activity. Sun and Kim et al. developed a sophisticated dual-chamber microreactor incorporating metal-doped MOFs@COFs,191 designed specifically for complex liquid–gas tandem reactions. This innovation highlighted the potential of advanced reactor designs in effectively handling intricate chemical processes. Moreover, progress in photo- and thermocatalysis notably enhances the catalyst efficiency and the specificity for activating chemical bonds. A prime example of this is the use of g-C3N4 nanosheets modified with AuCu alloy nanoparticles for the photothermal reduction of CO2 to ethanol in a CEL-HPR reactor, which achieved a remarkable 93% selectivity. This high level of selectivity was achieved under photo-phonon synergistic conditions, with optimal ethanol production occurring at a reaction temperature of 120 °C. This underscored the benefits of incorporating precise thermal control in photocatalytic strategies.188
6. Challenges and perspectives
6.1 Developing catalysts with enhanced activity and selectivity
Exploring efficient photocatalysts is pivotal for advancing methanol-based hydrogen generation. Since the pioneering use of TiO2 for photocatalytic hydrogen production in 1980, significant advances have been achieved, particularly at wavelengths ⩽380 nm, nearing 100% quantum efficiency. Nonetheless, photocatalysts modified to absorb visible photons, e.g. up to 500 nm, despite enhancements like co-catalysts or vacancies, struggle to exceed 20% quantum efficiency. The main hurdles are rapid charge recombination and undesirable electron transfer pathways that compromise efficiency and selectivity.Single-atom catalysts, especially those involving noble metals such as Pt and Pd, as well as non-noble metals like Cu, have demonstrated potential to boost methanol-based hydrogen production. Dual active site catalysts present a viable alternative to traditional noble metal catalysts by operating at lower temperatures and delivering higher hydrogen generation rates and stability. Future research should prioritize creating highly active photocatalysts that respond to both visible and infrared light. Modulating the selectivity of desired valuable products remains a challenging yet attractive goal. By adjusting the wavelength of incident light and carefully controlling the reaction pathways, the selectivity towards by-products can be significantly improved. Furthermore, the use of tandem catalysts can enhance by-product selectivity by enabling intermediates to be further converted into the desired products. Ensuring the structural integrity and long-term durability of these catalyst systems is essential for practical applications. Durability tests for methanol-based hydrogen generation typically span only tens of hours, and the in-depth deactivation analyses are scarce. Persistent challenges include the hundred- or thousand-hour stability and recyclability of catalysts.
6.2 Reactor design optimisation
Achieving high hydrogen production rates while maintaining operational stability necessitates precise reactor design. Batch reactors, although preferred for small-scale trials due to their precise control over experimental conditions, lack scalability and continuous operation capabilities. Conversely, flow reactors address these limitations by ensuring continuous reactant flow and enhanced mass transfer. Traditional reactors face significant challenges such as suboptimal light absorption due to the limited surface area or poor distribution of light in photocatalysis, high pressure drops and poor heat management. Recent advancements, including advanced microreactors and coated-wall reactors, even continuous stirred tank reactors (CSTR) begun to overcome these issues by improving mass and heat transfer, reducing pressure drops, and facilitating better temperature control for more efficient and rapid reactions. Additionally, advanced heat exchangers that recuperate and reuse thermal energy contribute to increased system efficiency. Innovative reactor configurations, such as spiral or honeycomb structures, can also optimize light distribution and thermal management.6.3 Unravelling the underlying mechanisms
Identifying catalytic active sites and understanding reaction mechanisms are critical for future advancements in photocatalysis. Exploring the activity of C–H and O–H bond scission, as well as C–C coupling in various metals like Cu, Ni, Pd, Pt, and Ru, remains a significant area of interest. It is important to note that the deactivation mechanisms of catalysts under light exposure have been seldom explored; yet understanding these mechanisms could provide valuable insights for developing more effective and durable catalysts. Microkinetic modelling and quantitative structure–activity relationships will be crucial for designing more efficient catalysts.Developing in situ detection methods is crucial for further understanding mechanisms. Techniques such as in situ infrared (IR) and Raman spectroscopy are effective for identifying and characterizing surface intermediates during catalytic reactions, aiding in the tracking of methanol's conversion to hydrogen. Techniques like time resolved transient absorption spectroscopy, in situ X-ray photoelectron spectroscopy (XPS) and extended X-ray absorption fine structure (EXAFS) offer more valuable insights into charge carrier dynamics and recombination processes, which should be extensively used in the emerging area.
6.4 Photon–phonon co-driven catalysis
Integrating photocatalysis with thermocatalysis utilizes the unique benefits of both systems. The energy from photons can be used to generate active charges, while thermal energy can aid in overcoming the small activation barriers of intermediate transformation and product desorption that limit the product selectivity. The photon–phonon co-driven process also intends to utilize the full solar spectrum for catalysis, i.e. IR photons to generate phonons for thermal catalysis and UV-visible photons for photocatalysis. As such the photon–phonon co-driven process would overcome the drawbacks of individual processes, enhancing the overall reaction kinetics. Thus the photon–phonon co-driven catalytic methanol reforming would dramatically decrease the temperature needed for the conventional thermal catalytic conversion, simultaneously maintaining a high selectivity for valuable products.Integrating photons with other energies such as phonons, ultrasonic energy, microwave energy through a tandem system can also improve the selectivity of high-value liquid by-products. For example, an intermediate product is first generated through photocatalysis, and then phonon/ultrasonic energy is used to convert this intermediate into the desired liquid by-product. This approach requires a thorough understanding of the reaction mechanisms and the ability to control the reaction pathways to achieve high selectivity for the final product. Moreover, the integration of advanced characterization techniques to investigate the photothermal effect, elucidate detailed photon–phonon co-driven catalytic mechanisms, and precisely determine the distribution of various forms of energy conversion in the multi-energy-coupled catalysis will represent a critical area of research in the future.
6.5 AI promoted photon–phonon co-driven catalysis
Designing catalytic materials for photocatalytic methanol reforming presents numerous challenges due to the complex interactions that occur. This would be more complex in a photon–phonon co-driven process, such as surface chemical reactions, material restructuring, diverse energy coupling and so on. Integrating artificial intelligence (AI) into the design and optimisation process of these catalysts significantly enhances the accuracy and efficiency of screening potential catalyst materials. Advanced AI models, such as the sure independence screening and sparsifying operator, can identify key active sites that dictate catalyst performance in methanol reforming by analysing features such as the reducibility of active species and the adsorption strength of reaction intermediates and products. These models can predict catalyst selectivity for liquid products and emphasize the importance of the chemical properties of additive metals used as co-catalysts.In addition to catalyst design, reactor optimisation is crucial for improving the efficiency of methanol reforming. Applying real-time monitoring technologies and computational modelling, including AI-based tools like COMSOL, facilitates dynamic adjustments to reactor conditions tailored to specific process requirements. This AI-driven approach, combined with experimental data, establishes a robust framework for optimising the photon–phonon co-driven catalytic process. It makes methanol reforming a more viable and economically attractive method for sustainable hydrogen production and the generation of valuable liquid byproducts. By leveraging the capabilities of AI, researchers can uncover patterns and relationships that might be overlooked using traditional methods, accelerating the discovery of high-performance catalysts, the exploration of the understanding of catalysis mechanisms and the selection of scalable reactors.
Data availability
No new data were generated or analysed in this study. The data underpinning the findings of this review are accessible through the original publications cited in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors acknowledge the NSFC project (grant no: U23B20162 and 22250710677) and Beijing Municipal Project (C2022007). The project is also supported by Tsinghua University Initiative Scientific Research Program and UK EPSRC (EP/S018204/2).References
- X. K. Gu and W. X. Li, J. Phys. Chem. C, 2010, 114, 21539–21547 CrossRef CAS.
- L. Li, W. Ouyang, Z. Zheng, K. Ye, Y. Guo, Y. Qin, Z. Wu, Z. Lin, T. Wang and S. Zhang, Chin. J. Catal., 2022, 43, 1258–1266 CrossRef CAS.
- S. M. Al-Salem, A. Antelava, A. Constantinou, G. Manos and A. Dutta, J. Environ. Manage., 2017, 197, 177–198 CrossRef CAS PubMed.
- S. M. Al-Salem, M. Van Haute, H. J. Karam, A. Hakeem, W. Meuldermans, J. Patel, S. Hafeez, G. Manos and A. Constantinou, Ind. Eng. Chem. Res., 2022, 61, 16383–16392 CAS.
- A. Antelava, S. Damilos, S. Hafeez, G. Manos, S. M. Al-Salem, B. K. Sharma, K. Kohli and A. Constantinou, Environ. Manage., 2019, 64, 230–244 CrossRef.
- A. Antelava, N. Jablonska, A. Constantinou, G. Manos, S. A. Salaudeen, A. Dutta and S. M. Al-Salem, Energy Fuels, 2021, 35, 3558–3571 CrossRef CAS.
- S. Hafeez, E. Pallari, G. Manos and A. Constantinou, Catalytic conversion and chemical recovery, Elsevier Inc., 2019 Search PubMed.
- A. Antelava, E. Pallari, G. Manos and A. Constantinou, Design and limitations in polymer cracking fluidized beds for energy recovery, Elsevier Inc., 2019 Search PubMed.
- H. N. C. Kobayashi, Chem. Lett., 1976, 1347–1350 CrossRef CAS.
- J. Zhao, R. Shi, Z. Li, C. Zhou and T. Zhang, Nano Select, 2020, 1, 12–29 CrossRef.
- X. He, Y. Wang, X. Zhang, M. Dong, G. Wang, B. Zhang, Y. Niu, S. Yao, X. He and H. Liu, ACS Catal., 2019, 9, 2213–2221 CrossRef CAS.
- J. L. C. Fajín and M. N. D. S. Cordeiro, ACS Catal., 2022, 12, 512–526 CrossRef.
- A. A. Lytkina, N. A. Zhilyaeva, M. M. Ermilova, N. V. Orekhova and A. B. Yaroslavtsev, Int. J. Hydrogen Energy, 2015, 40, 9677–9684 CrossRef CAS.
- M. V. Twigg and M. S. Spencer, Top. Catal., 2003, 22, 191–203 CrossRef CAS.
- K. M. K. Yu, W. Tong, A. West, K. Cheung, T. Li, G. Smith, Y. Guo and S. C. E. Tsang, Nat. Commun., 2012, 3, 1230 CrossRef PubMed.
- N. Murakami, T. Chiyoya, T. Tsubota and T. Ohno, Appl. Catal., A, 2008, 348, 148–152 CrossRef CAS.
- S. Neubert, D. Mitoraj, S. A. Shevlin, P. Pulisova, M. Heimann, Y. Du, G. K. L. Goh, M. Pacia, K. Kruczała, S. Turner, W. MacYk, Z. X. Guo, R. K. Hocking and R. Beranek, J. Mater. Chem. A, 2016, 4, 3127–3138 RSC.
- L. Pan and S. Wang, Chem. Eng. J., 2005, 108, 51–58 CrossRef CAS.
- H. Meng, Y. Yang, T. Shen, Z. Yin, L. Wang, W. Liu, P. Yin, Z. Ren, L. Zheng, J. Zhang, F. S. Xiao and M. Wei, Nat. Commun., 2023, 14, 7980 CrossRef CAS.
- D. Li, F. Xu, X. Tang, S. Dai, T. Pu, X. Liu, P. Tian, F. Xuan, Z. Xu, I. E. Wachs and M. Zhu, Nat. Catal., 2022, 5, 99–108 CrossRef CAS.
- J. Gao, J. Guo, D. Liang, Z. Hou, J. Fei and X. Zheng, Int. J. Hydrogen Energy, 2008, 33, 5493–5500 CrossRef CAS.
- N. Takezawa and N. Iwasa, Catal. Today, 1997, 36, 45–56 CrossRef CAS.
- X. K. Gu, B. Qiao, C. Q. Huang, W. C. Ding, K. Sun, E. Zhan, T. Zhang, J. Liu and W. X. Li, ACS Catal., 2014, 4, 3886–3890 CrossRef CAS.
- N. Iwasa, S. Kudo, H. Takahashi, S. Masuda and N. Takezawa, Catal. Lett., 1993, 19, 211–216 CrossRef CAS.
- B. W. L. Jang, R. Gläser, M. Dong and C. J. Liu, Energy Environ. Sci., 2010, 3, 253 RSC.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- L. Lin, Q. Yu, M. Peng, A. Li, S. Yao, S. Tian, X. Liu, A. Li, Z. Jiang, R. Gao, X. Han, Y. W. Li, X. D. Wen, W. Zhou and D. Ma, J. Am. Chem. Soc., 2021, 143, 309–317 CrossRef CAS.
- L. N. Chen, K. P. Hou, Y. S. Liu, Z. Y. Qi, Q. Zheng, Y. H. Lu, J. Y. Chen, J. L. Chen, C. W. Pao, S. B. Wang, Y. Bin Li, S. H. Xie, F. D. Liu, D. Prendergast, L. E. Klebanoff, V. Stavila, M. D. Allendorf, J. Guo, L. S. Zheng, J. Su and G. A. Somorjai, J. Am. Chem. Soc., 2019, 141, 17995–17999 CrossRef CAS PubMed.
- S. Zhang, Y. Liu, M. Zhang, Y. Ma, J. Hu and Y. Qu, Nat. Commun., 2022, 13, 5527 CrossRef CAS PubMed.
- X. Tang, J. Li, Z. Fang, X. Dong, C. Sun, X. Qiao and X. Li, Appl. Surf. Sci., 2022, 596, 153635 CrossRef CAS.
- P. Tahay, Y. Khani, M. Jabari, F. Bahadoran and N. Safari, Appl. Catal., A, 2018, 554, 44–53 CrossRef CAS.
- C. Mateos-Pedrero, C. Azenha, P. T. Pacheco, J. M. Sousa and A. Mendes, Appl. Catal., B, 2020, 277, 119243 CrossRef CAS.
- V. Shanmugam, S. Neuberg, R. Zapf, H. Pennemann and G. Kolb, Int. J. Hydrogen Energy, 2020, 45, 1658–1670 CrossRef CAS.
- S. T. Yong, C. W. Ooi, S. P. Chai and X. S. Wu, Int. J. Hydrogen Energy, 2013, 38, 9541–9552 CrossRef CAS.
- Z. J. Zuo, L. Wang, P. De Han and W. Huang, Int. J. Hydrogen Energy, 2014, 39, 1664–1679 CrossRef CAS.
- J. Reyna-Alvarado, O. A. López-Galán, M. Ramos, J. Rodríguez and R. Pérez-Hernández, Catal. Today, 2022, 392–393, 146–153 CrossRef CAS.
- X. Yang, ACS Catal., 2014, 4, 1129–1133 CrossRef CAS.
- X. Wang, D. Li, Z. Gao, Y. Guo, H. Zhang and D. Ma, J. Am. Chem. Soc., 2023, 145, 905–918 CrossRef CAS PubMed.
- G. Luca, D. Ferri and E. Selli, J. Catal., 2011, 280, 168–177 CrossRef.
- K. Fujishima and A. Honda, Nature, 1972, 238, 37–38 CrossRef.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. J. Bard, Science, 1980, 207, 139–144 CrossRef CAS PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- M. Thangamuthu, Q. Ruan, P. O. Ohemeng, B. Luo, D. Jing, R. Godin and J. Tang, Chem. Rev., 2022, 122, 11778–11829 CrossRef CAS.
- X. Chen, N. Li, Z. Kong, W. J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- H. Wang, X. Li, Q. Ruan and J. Tang, Nanoscale, 2020, 12, 12329–12335 RSC.
- D. S. Bhatkhande, V. G. Pangarkar and A. A. C. M. Beenackers, J. Chem. Technol. Biotechnol., 2002, 77, 102–116 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- A. Furube, T. Asahi, H. Masuhara, H. Yamashita and M. Anpo, J. Phys. Chem. B, 1999, 103, 3120–3127 CrossRef CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- S. Kakuta and T. Abe, ACS Appl. Mater. Interfaces, 2009, 1, 2707–2710 CrossRef CAS PubMed.
- Z. Liu, Z. Yin, C. Cox, M. Bosman, X. Qian, N. Li, H. Zhao, Y. Du, J. Li and D. G. Nocera, Sci. Adv., 2016, 2, e1501425 CrossRef.
- A. Hameed and M. A. Gondal, J. Mol. Catal. A: Chem., 2005, 233, 35–41 CrossRef CAS.
- C. Jiang, K. Yip, C. M. A. Parlett, M. K. Bayazit, C. Ching, Q. Ruan, S. J. A. Moniz, A. F. Lee and J. Tang, Appl. Catal., A, 2016, 521, 133–139 CrossRef CAS.
- A. Shoneye, J. Sen Chang, M. N. Chong and J. Tang, Int. Mater. Rev., 2022, 67, 47–64 CrossRef CAS.
- H. Wang, X. Yuan, H. Wang, X. Chen, Z. Wu, L. Jiang, W. Xiong and G. Zeng, Appl. Catal., B, 2016, 193, 36–46 CrossRef CAS.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 764, 694–695 RSC.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- B. Han and Y. H. Hu, J. Phys. Chem. C, 2015, 119, 18927–18934 CrossRef CAS.
- C. H. Lin, J. H. Chao, C. H. Liu, J. C. Chang and F. C. Wang, Langmuir, 2008, 24, 9907–9915 CrossRef CAS.
- J. Jitputti, Y. Suzuki and S. Yoshikawa, Catal. Commun., 2008, 9, 1265–1271 CrossRef CAS.
- Q. Xu, Y. Ma, J. Zhang, X. Wang, Z. Feng and C. Li, J. Catal., 2011, 278, 329–335 CrossRef CAS.
- Z. Jin, Q. Li, X. Zheng, C. Xi, C. Wang, H. Zhang, L. Feng, H. Wang, Z. Chen and Z. Jiang, J. Photochem. Photobiol., A, 1993, 71, 85–96 CrossRef CAS.
- S. Yanagida, T. Azuma and H. Sakurai, Chem. Lett., 1982, 1069–1070 CrossRef CAS.
- J. Wang, P. Yang, B. Cao, J. Zhao and Z. Zhu, Appl. Surf. Sci., 2015, 325, 86–90 CrossRef CAS.
- K. Chang, X. Hai, H. Pang, H. Zhang, L. Shi, G. Liu, H. Liu, G. Zhao, M. Li and J. Ye, Adv. Mater., 2016, 28, 10033–10041 CrossRef CAS PubMed.
- X. Huang, Z. Zeng and H. Zhang, Chem. Soc. Rev., 2013, 42, 1934–1946 RSC.
- A. Corma and H. Garc, J. Mater. Chem., 2010, 20, 3141–3156 RSC.
- C. G. Silva, I. Luz, F. X. Llabrøs and A. Corma, Chem. – Eur. J., 2010, 16, 11133–11138 CrossRef CAS.
- J. Wang, A. S. Cherevan, C. Hannecart, S. Naghdi, S. P. Nandan, T. Gupta and D. Eder, Appl. Catal., B, 2021, 283, 119626 CrossRef CAS.
- Z. L. Wu, C. H. Wang, B. Zhao, J. Dong, F. Lu, W. H. Wang, W. C. Wang, G. J. Wu, J. Z. Cui and P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 4938–4942 CrossRef CAS PubMed.
- M. R. Khan, T. W. Chuan, A. Yousuf, M. N. K. Chowdhury and C. K. Cheng, Catal. Sci. Technol., 2015, 5, 2522–2531 RSC.
- X. Wang, G. Zhang, L. Yang, E. Sharman and J. Jiang, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–22 RSC.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- L. S. Al-Mazroai, M. Bowker, P. Davies, A. Dickinson, J. Greaves, D. James and L. Millard, Catal. Today, 2007, 122, 46–50 CrossRef CAS.
- G. Wu, T. Chen, W. Su, G. Zhou, X. Zong, Z. Lei and C. Li, Int. J. Hydrogen Energy, 2008, 33, 1243–1251 CrossRef CAS.
- G. L. Chiarello, M. H. Aguirre and E. Selli, J. Catal., 2010, 273, 182–190 CrossRef CAS.
- A. Naldoni, M. D’Arienzo, M. Altomare, M. Marelli, R. Scotti, F. Morazzoni, E. Selli and V. Dal Santo, Appl. Catal., B, 2013, 130–131, 239–248 CrossRef CAS.
- A. Moya, A. Cherevan, S. Marchesan, P. Gebhardt, M. Prato, D. Eder and J. J. Vilatela, Appl. Catal., B, 2015, 179, 574–582 CrossRef CAS.
- S. Oros-Ruiz, R. Zanella, R. López, A. Hernández-Gordillo and R. Gómez, J. Hazard. Mater., 2013, 263, 2–10 CrossRef CAS PubMed.
- N. Aas, T. J. Pringle and M. Bowker, J. Chem. Soc., Faraday Trans., 1994, 90, 1015 RSC.
- A. Dickinson, D. James, N. Perkins, T. Cassidy and M. Bowker, J. Mol. Catal. A: Chem., 1999, 146, 211–221 CrossRef CAS.
- Y. Chen, S. Ji, W. Sun, Y. Lei, Q. Wang, A. Li, W. Chen, G. Zhou, Z. Zhang, Y. Wang, L. Zheng, Q. Zhang, L. Gu, X. Han and D. Wang, Angew. Chem., Int. Ed., 2020, 59, 1295–1301 CrossRef CAS PubMed.
- S. S. Yi, X. B. Zhang, B. R. Wulan, J. M. Yan and Q. Jiang, Energy Environ. Sci., 2018, 11, 3128–3156 RSC.
- S. Kashiwaya, J. Morasch, V. Streibel, T. Toupance, W. Jaegermann and A. Klein, Surfaces, 2018, 1, 73–89 CrossRef.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 CrossRef CAS.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- S. Xiao, P. Liu, W. Zhu, G. Li, D. Zhang and H. Li, Nano Lett., 2015, 15, 4853–4858 CrossRef CAS.
- Q. Liu, Q. Zhang, B. Liu and S. Li, Chin. J. Catal., 2018, 39, 542–548 CrossRef CAS.
- N. L. Wu and M. S. Lee, Int. J. Hydrogen Energy, 2004, 29, 1601–1605 CrossRef CAS.
- W. T. Chen, Y. Dong, P. Yadav, R. D. Aughterson, D. Sun-Waterhouse and G. I. N. Waterhouse, Appl. Catal., A, 2020, 602, 117703 CrossRef CAS.
- Y. Nosaka, S. Takahashi, H. Sakamoto and A. Y. Nosaka, J. Phys. Chem. C, 2011, 115, 21283–21290 CrossRef CAS.
- B. H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K. S. Lee, J. H. Kim, S. P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- Y. Zhang, J. Zhao, H. Wang, B. Xiao, W. Zhang, X. Zhao, T. Lv, M. Thangamuthu, J. Zhang, Y. Guo, J. Ma, L. Lin, J. Tang, R. Huang and Q. Liu, Nat. Commun., 2022, 13, 58 CrossRef CAS PubMed.
- H. Wang, H. Qi, X. Sun, S. Jia, X. Li, T. J. Miao, L. Xiong, S. Wang, X. Zhang, X. Liu, A. Wang, T. Zhang, W. Huang and J. Tang, Nat. Mater., 2023, 22, 619–626 CrossRef CAS.
- I. Novotny and C. P. Bianchi, Pflügers Arch. Eur. J. Physiol., 1973, 339, 113–124 CrossRef CAS PubMed.
- M. A. Melo, S. A. Carminati, J. Bettini and A. F. Nogueira, Sustain. Energy Fuels, 2018, 2, 958–967 RSC.
- M. Hojamberdiev, M. Mansoob and Z. Kadirova, Renewable Energy, 2019, 138, 434–444 CrossRef CAS.
- J. Yu, Y. Hai and B. Cheng, J. Phys. Chem. C, 2011, 115, 4953–4958 CrossRef CAS.
- N. Lakshmana Reddy, K. K. Cheralathan, V. Durga Kumari, B. Neppolian and S. Muthukonda Venkatakrishnan, ACS Sustainable Chem. Eng., 2018, 6, 3754–3764 CrossRef CAS.
- Y. Chen and Z. Qin, Catal. Sci. Technol., 2016, 6, 8212–8221 RSC.
- Y. J. Yuan, Z. J. Ye, H. W. Lu, B. Hu, Y. H. Li, D. Q. Chen, J. S. Zhong, Z. T. Yu and Z. G. Zou, ACS Catal., 2016, 6, 532–541 CrossRef CAS.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- Y. Yang, L. Kang and H. Li, Ceram. Int., 2019, 45, 8017–8022 CrossRef CAS.
- D. Zhou, P. Zhai, G. Hu and J. Yang, Chem. Phys. Lett., 2018, 711, 77–80 CrossRef CAS.
- A. L. Luna, D. Dragoe, K. Wang, P. Beaunier, E. Kowalska, B. Ohtani, D. Bahena Uribe, M. A. Valenzuela, H. Remita and C. Colbeau-Justin, J. Phys. Chem. C, 2017, 121, 14302–14311 CrossRef CAS.
- A. L. Luna, E. Novoseltceva, E. Louarn, P. Beaunier, E. Kowalska, B. Ohtani, M. A. Valenzuela, H. Remita and C. Colbeau-Justin, Appl. Catal., B, 2016, 191, 18–28 CrossRef CAS.
- K. Czelej, K. Cwieka, J. C. Colmenares, K. J. Kurzydlowski and Y. J. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 31825–31833 CrossRef CAS.
- J. C. Colmenares, P. Lisowski, D. Łomot, O. Chernyayeva and D. Lisovytskiy, ChemSusChem, 2015, 8, 1676–1685 CrossRef CAS.
- K. Majrik, E. Tálas, Z. Pászti, I. Sajó, J. Mihály, L. Korecz, E. Drotár and A. Tompos, Appl. Catal., A, 2013, 466, 169–178 CrossRef CAS.
- C. G. Silva, M. J. Sampaio, R. R. N. Marques, L. A. Ferreira, P. B. Tavares, A. M. T. Silva and J. L. Faria, Appl. Catal., B, 2015, 178, 82–90 CrossRef CAS.
- H. Wang, M. Thangamuthu, Z. Wu, J. Yang and H. Yuan, Chem. Eng. J., 2022, 445, 136790 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Nanoscale, 2011, 3, 3670–3678 RSC.
- N. Naffati, M. J. Sampaio, E. S. Da Silva, M. F. Nsib, Y. Arfaoui, A. Houas, J. L. Faria and C. G. Silva, Mater. Sci. Semicond. Process., 2020, 115, 105098 CrossRef CAS.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS.
- H. El Marouazi, P. Jiménez-Calvo, E. Breniaux, C. Colbeau-Justin, I. Janowska and V. Keller, ACS Sustainable Chem. Eng., 2021, 9, 3633–3646 CrossRef CAS.
- S. Hu, J. Shi, B. Luo, C. Ai and D. Jing, J. Colloid Interface Sci., 2022, 608, 2058–2065 CrossRef CAS PubMed.
- T. Yeh, J. Syu, C. Cheng, T. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- Y. Okamoto, S. Ida, J. Hyodo, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2011, 133, 18034–18037 CrossRef CAS.
- Z. Ding, H. Hu, J. Xu, P. Lin, C. Cui, D. Qian, P. Wang, L. Xu, J. Pan and C. Li, Int. J. Hydrogen Energy, 2018, 43, 13190–13199 CrossRef CAS.
- J. Liu, G. Liu, M. Li, W. Shen, Z. Liu, J. Wang, J. Zhao, L. Jiang and Y. Song, Energy Environ. Sci., 2010, 3, 1503–1506 RSC.
- S. Mansingh, D. K. Padhi and K. M. Parida, Int. J. Hydrogen Energy, 2016, 41, 14133–14146 CrossRef CAS.
- C. Y. Wang, R. Pagel, D. W. Bahnemann and J. K. Dohrmann, J. Phys. Chem. B, 2004, 108, 14082–14092 CrossRef CAS.
- K. Hirano, H. Asayama, A. Hoshino and H. Wakatsuki, J. Photochem. Photobiol., A, 1997, 110, 307–311 CrossRef CAS.
- F. Varas-Concha, D. Guzmán, M. Isaacs and C. Sáez-Navarrete, Energy Technol., 2018, 6, 1871–1884 CrossRef CAS.
- S. Yanagida, T. Azuma, H. Kawakami, H. Kizumoto and H. Sakurai, J. Chem. Soc., Chem. Commun., 1984, 325, 21–22 RSC.
- L. Chen, W. Gu, X. Zhu, F. Wang, Y. Song and J. Hu, J. Photochem. Photobiol., A, 1993, 74, 85–89 CrossRef CAS.
- H. Zhang, S. Xie, J. Hu, X. Wu, Q. Zhang, J. Cheng and Y. Wang, Chem. Commun., 2020, 56, 1776–1779 RSC.
- Y. C. Liu, G. L. Griffin, S. S. Chan and I. E. Wachs, J. Catal., 1985, 94, 108–119 CrossRef CAS.
- H. Kominami, H. Sugahara and K. Hashimoto, Catal. Commun., 2010, 11, 426–429 CrossRef CAS.
- K. R. Phillips, S. C. Jensen, M. Baron, S. C. Li and C. M. Friend, J. Am. Chem. Soc., 2013, 135, 574–577 CrossRef CAS PubMed.
- X. Yang, A. Zhang, G. Gao, D. Han, C. Han, J. Wang, H. Lu, J. Liu and M. Tong, Catal. Commun., 2014, 43, 192–196 CrossRef CAS.
- J. Liu, C. Han, X. Yang, G. Gao, Q. Shi, M. Tong, X. Liang and C. Li, J. Catal., 2016, 333, 162–170 CrossRef CAS.
- X. Liang, X. Yang, G. Gao, C. Li, Y. Li, W. Zhang, X. Chen, Y. Zhang, B. Zhang, Y. Lei and Q. Shi, J. Catal., 2016, 339, 68–76 CrossRef CAS.
- M. Liu, Y. Wang, M. Liu, Y. Wang, X. Kong, R. T. Rashid, S. Chu and C. Li, Chem, 2019, 5, 858–867 CAS.
- L. Mohrhusen, J. Kra and K. Al-shamery, Phys. Chem. Chem. Phys., 2021, 2, 12148–12157 RSC.
- D. G. Calatayud, O. G. Díaz, A. C. Caballero and J. Ara, Appl. Catal., B, 2014, 153, 192–201 Search PubMed.
- M. Taherinia, M. Nasiri and E. Abedini, Environ. Dev. Sustain., 2019, 21, 1963–1975 CrossRef.
- L. Huaxu, W. Fuqiang, C. Ziming, H. Shengpeng, X. Bing, G. Xiangtao, L. bo, T. Jianyu, L. Xiangzheng, C. Ruiyang, L. Wen and L. Linhua, Int. J. Hydrogen Energy, 2017, 42, 12133–12142 CrossRef.
- Z. Zhang and P. A. Maggard, J. Photochem. Photobiol., A, 2007, 186, 8–13 CrossRef CAS.
- J. J. Velázquez, R. Fernández-González, L. Díaz, E. Pulido Melián, V. D. Rodríguez and P. Núñez, J. Alloys Compd., 2017, 721, 405–410 CrossRef.
- W. Lin, W. Yang, I. Huang, T. Wu and Z. Chung, Energy Fuels, 2009, 23, 2192–2196 CrossRef CAS.
- M. Li, Y. Li, S. Peng, G. Lu and S. Li, Front. Chem., 2009, 4, 32–38 Search PubMed.
- H. M. G. Tambago and R. L. de Leon, Int. J. Chem. Eng. Appl., 2015, 6, 220–227 CAS.
- E. Baniasadi, I. Dincer and G. F. Naterer, Int. J. Hydrogen Energy, 2013, 38, 9158–9168 CrossRef CAS.
- J. Ma, T. J. Miao and J. Tang, Chem. Soc. Rev., 2022, 51, 5777–5794 RSC.
- X. Ruan, C. Huang, H. Cheng, Z. Zhang, Y. Cui, Z. Li, T. Xie, K. Ba, H. Zhang, L. Zhang, X. Zhao, J. Leng, S. Jin, W. Zhang, W. Zheng, S. K. Ravi, Z. Jiang, X. Cui and J. Yu, Adv. Mater., 2023, 35, 1–9 Search PubMed.
- J. B. Priebe, M. Karnahl, H. Junge, M. Beller, D. Hollmann and A. Brückner, Angew. Chem., Int. Ed., 2013, 52, 11420–11424 CrossRef CAS.
- B. De Nijs, F. Benz, S. J. Barrow, D. O. Sigle, R. Chikkaraddy, A. Palma, C. Carnegie, M. Kamp, R. Sundararaman, P. Narang, O. A. Scherman and J. J. Baumberg, Nat. Commun., 2017, 8, 994 CrossRef.
- A. Yamakata, T. A. Ishibashi and H. Onishi, J. Mol. Catal. A: Chem., 2003, 199, 85–94 CrossRef CAS.
- A. Yamakata, T. A. Ishibashi and H. Onishi, Chem. Phys. Lett., 2001, 333, 271–277 CrossRef CAS.
- T. Chen, Z. Feng, G. Wu, J. Shi, G. Ma, P. Ying and C. Li, J. Phys. Chem. C, 2007, 111, 8005–8014 CrossRef CAS.
- F. Wang, Y. Jiang, D. J. Lawes, G. E. Ball, C. Zhou, Z. Liu and R. Amal, ACS Catal., 2015, 5, 3924–3931 CrossRef CAS.
- G. M. Haselmann, B. Baumgartner, J. Wang, K. Wieland, T. Gupta, C. Herzig, A. Limbeck, B. Lendl and D. Eder, ACS Catal., 2020, 10, 2964–2977 CrossRef CAS.
- C. Wang, Y. Xu, L. Xiong, X. Li, E. Chen, T. J. Miao, T. Zhang, Y. Lan and J. Tang, Nat. Commun., 2024, 15, 7535 CrossRef CAS.
- Y. Xu, C. Wang, X. Li, L. Xiong, T. Zhang, L. Zhang, Q. Zhang, L. Gu, Y. Lan and J. Tang, Nat. Sustain., 2024, 7, 1171–1181 CrossRef.
- X. Yu, J. Zeng and Y. Xuan, Energy Technol., 2019, 7, 1–9 CrossRef.
- Y. Pi, Z. Zengcai, W. Lin, B. Zhang and T. Wang, AIChE J., 2023, 69, 1–12 CrossRef.
- X. Bai, D. Yuan, Y. Li, H. Song, Y. Lu, X. San, J. Lu, G. Fu, S. Wang and J. Ye, iScience, 2021, 24, 102056 CrossRef CAS.
- X. Liu, L. Ye, Z. Ma, C. Han, L. Wang, Z. Jia, F. Su and H. Xie, Catal. Commun., 2017, 102, 13–16 CrossRef CAS.
- W. Chen, Y. X. Liu, X. Liang, S. Wang, X. Gao, Z. Zhang and Y. Fang, J. Energy Storage, 2022, 55, 105405 CrossRef.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- S. Guo, X. Li, J. Li and B. Wei, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- S. Fang, Z. Sun and Y. H. Hu, ACS Catal., 2019, 9, 5047–5056 CrossRef CAS.
- W. Lin, J. Li, Z. Zengcai, B. Zhang, X. Wu, Y. Pi and T. Wang, Fuel, 2024, 357, 129990 CrossRef CAS.
- J. Li, B. Sheng, Y. Chen, J. Yang, T. Ma, C. You, Y. Li, T. Yu, J. Song, H. Pan, X. Wang and B. Zhou, ACS Catal., 2023, 13, 10153–10160 CrossRef CAS.
- X. Yu, L. Yang, Y. Xuan, X. L. Liu and K. Zhang, Nano Energy, 2021, 84, 105953 CrossRef CAS.
- X. Li, S. Zhao, X. Duan, H. Zhang, S. Ze Yang, P. Zhang, S. P. Jiang, S. Liu, H. Sun and S. Wang, Appl. Catal., B, 2021, 283, 119660 CrossRef CAS.
- A. G. Variar, M. S. Ramyashree, V. U. Ail, S. P. S, K. Sudhakar and M. Tahir, J. Ind. Eng. Chem., 2021, 99, 19–47 CrossRef CAS.
- C. McCullagh, N. Skillen, M. Adams and P. K. J. Robertson, J. Chem. Technol. Biotechnol., 2011, 86, 1002–1017 CrossRef CAS.
- J. Kang, Y. Song, T. Kim and S. Kim, Int. J. Hydrogen Energy, 2022, 47, 3587–3610 CrossRef CAS.
- D. Iranshahi, A. Golrokh, E. Pourazadi, S. Saeidi and F. Gallucci, Chem. Eng. Process.: Process Intensif., 2018, 132, 16–24 CrossRef CAS.
- H. K. Huang, Y. K. Chih, W. H. Chen, C. Y. Hsu, K. J. Lin, H. P. Lin and C. H. Hsu, Int. J. Hydrogen Energy, 2022, 47, 37542–37551 CrossRef CAS.
- J. Wang, H. Wang and P. Hu, Sci. China: Chem., 2018, 61, 336–343 CrossRef CAS.
- Z. J. Zuo, X. Y. Gao, P. De Han, S. Z. Liu and W. Huang, J. Phys. Chem. C, 2016, 120, 27500–27508 CrossRef CAS.
- A. Karim, J. Bravo and A. Datye, Appl. Catal., A, 2005, 282, 101–109 CrossRef CAS.
- A. Chougule and R. R. Sonde, Int. J. Hydrogen Energy, 2019, 44, 29937–29945 CrossRef CAS.
- A. Karim, J. Bravo, D. Gorm, T. Conant and A. Datye, Catal. Today, 2005, 110, 86–91 CrossRef CAS.
- P. Dokamaingam, S. Assabumrungrat, A. Soottitantawat, I. Sramala and N. Laosiripojana, Int. J. Hydrogen Energy, 2009, 34, 410–421 CrossRef CAS.
- S. Hafeez, E. Aristodemou, G. Manos, S. M. Al-Salem and A. Constantinou, RSC Adv., 2020, 10, 41680–41692 RSC.
- J. Zhu, S. S. Araya, X. Cui, S. L. Sahlin and S. K. Kær, Energies, 2020, 13, 610 CrossRef CAS.
- G. L. Chiarello, L. Forni and E. Selli, Catal. Today, 2009, 144, 69–74 CrossRef CAS.
- H. Jiao, J. Yang, X. Li, C. Wang and J. Tang, Green Chem., 2022, 24, 8345–8354 RSC.
- P. Martínez Molina, K. W. Bossers, J. D. Wienk, J. Rohlfs, N. Meulendijks, M. A. Verheijen, P. Buskens and F. Sastre, Chem. – Asian J., 2023, 18, e202300405 CrossRef PubMed.
- P. Li, L. Liu, W. An, H. Wang, H. Guo, Y. Liang and W. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- C. Pornrungroj, A. Bin Mohamad Annuar, Q. Wang, M. Rahaman, S. Bhattacharjee, V. Andrei and E. Reisner, Nat. Water, 2023, 1, 952–960 CrossRef CAS.
- D. Sun, S. Jang, S. J. Yim, L. Ye and D. P. Kim, Adv. Funct. Mater., 2018, 28, 1–7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cs00551a |
| This journal is © The Royal Society of Chemistry 2025 |