
Metal pyrazolate frameworks: crystal engineering access to stable functional materials
Xiang-Jing
Kong
 ab,
Guang-Rui
Si
a,
Tao
He
*ab and
Jian-Rong
Li
*a
ab,
Guang-Rui
Si
a,
Tao
He
*ab and
Jian-Rong
Li
*a
aBeijing Key Laboratory for Green Catalysis and Separation and Department of Chemical Engineering, College of Materials Science & Engineering, Beijing University of Technology, 100124, Beijing, China. E-mail: hetao@bjut.edu.cn; jrli@bjut.edu.cn
bDepartment of Chemical Science, Bernal Institute, University of Limerick, Limerick, V94 T9PX, Ireland
First published on 7th March 2025
Abstract
As the focus evolves from structure discovery/characterization (what it is) to property/performance exploration (what it is for), the pursuit of stable functional metal–organic frameworks (MOFs) has been ongoing in terms of both fundamental research and industrial implementation. Under the guidance of crystal engineering principles, a plethora of research has developed pyrazolate MOFs (metal pyrazaolate frameworks, MPFs) featuring strong coordination M–N bonding. This attribution helps them retain their structures and functions under the alkaline conditions required for practical use. Based on poly-topic pyrazolate ligands, various classic MOFs, such as Co(bdp), Fe2(BDP)3, Ni8L6, PCN-601, and BUT-55, to name a few, have revealed fascinating architectures, intriguing properties, and record-breaking performances in applications during the past decade. This review will present the full scope of MPFs to date: (1) the superiority and significance of constructing MPFs through the crystal engineering approach, (2) synthetic strategies adopted in building and/or modifying MPFs, (3) structural features and stability of the MPF community, and (4) potential applications in energy and environmental related fields. The future opportunities of MPFs are also discussed for designing the next-generation of smart materials. Overall, this review attempts to provide insights and guidelines for the customization of pyrazolate-based MOFs for specific purposes, which would also promote the development of stable functional porous materials for addressing societal challenges.
 Xiang-Jing Kong | Xiang-Jing Kong received her PhD from Beijing University of Technology in 2020 under the supervision of Prof. J.-R. Li. From 2021, she was a postdoctoral researcher under the supervision of Prof. M. J. Zaworotko in the crystal engineering research group at the University of Limerick. She then joined Prof. O. K. Farha's group at Northwestern University as a postdoctoral researcher in 2023. Her research interest is focused on the design and synthesis of novel MOFs and their applications in adsorption, catalysis, etc. |
 Guang-Rui Si | Guang-Rui Si received his PhD degree from Beijing University of Technology in 2024 under the supervision of Prof. J.-R. Li. Now he is a postdoctoral researcher under the supervision of Prof. Z.-R. Nie at the same university. His research focuses on the design, synthesis, and application of functionalized MOFs. |
 Tao He | Tao He received his PhD from Beijing University of Technology in 2019 under the supervision of Prof. J.-R. Li. Then he pursued postdoctoral work under the supervision of Prof. Z.-R. Nie at the same university during 2019–2021 before joining Qingdao University in 2021. After completing postdoctoral research at the University of Limerick (2022–2025) under the supervision of Prof. M. J. Zaworotko, he joined Beijing University of Technology as an associate professor. His research interest is focused on stable porous materials for gas storage/separation and heterocatalysis. |
 Jian-Rong Li | Jian-Rong “Jeff” Li obtained his PhD in 2005 from Nankai University under the supervision of Prof. X.-H. Bu. Until 2008, he was an assistant professor at the same University. From 2008 to 2012, he worked as a postdoctoral research associate and then as an assistant research scientist at Texas A&M University in Prof. H.-C. Zhou's group. Since 2011, he has been a full professor at Beijing University of Technology. His research interests are focused on porous materials for chemical engineering, energy, and environmental science. |
1. Introduction
Metal–organic frameworks (MOFs)1,2 have been a booming family of crystalline porous materials since the discovery of HKUST-13 and MOF-54 with permanent porosity. The modular nature of MOFs allows for their design and synthesis amenable to crystal engineering principles across a huge library of organic/inorganic building blocks.5–8 Through decades of rapid development, MOFs have presented extraordinary structure diversity and application versatility spanning gas storage and separation, heterogeneous catalysis, sensing, biomedical diagnosis and therapy, and proton conduction.9–18 For practical use, good stability under real conditions is a prerequisite.19–21 Plenty of MOFs have shown great potential in applications but were eventually demonstrated unsuitable for practical use due to their limited stability. As the emphasis shifts from identifying and characterizing structures to exploring their properties and performances, the pursuit of stable functional MOFs continues to be a priority in both fundamental research and industrial applications.Chemical stability refers to the ability of a MOF to retain crystallinity (intact coordination bonds) under chemical environments/upon chemical treatments, such as exposure to humidity/water, acids, bases, nucleophilic/electrophilic reagents, salt aqueous solutions, and so on.20 The strength of coordination bonds is one of the main reasons among various thermodynamic and kinetic factors defining the stability of frameworks. According to the hard/soft acid/base (HSAB) principle, both combinations of carboxylate ligands with high-valent metals and azolate ligands with low-valent metals can form strong coordination bonds.19 Carboxylate MOFs built with high valent metal ions (i.e. Al(III), Cr(III), Zr(IV), and Ti(IV)) have attracted increasing attention due to their water/acid stability, as well as unique properties.20,22–25 Many recent reviews have discussed the construction and application of carboxylates MOFs.16,26–28 As their base-stable counterparts, MOFs based on azolate ligands are relatively less reported due to the limited ligand scope and immature synthetic protocols.29 Zeolitic imidazolate framework-8 (ZIF-8, also named metal azolate framework-4, MAF-4), as a representative example, is composed of imidazolate linkers and Zn(II) ions. This framework exhibits remarkable stability in aqueous environments, being able to maintain its structure in an 8 M NaOH basic solution.30,31 MAF-X27-Cl as a triazolate-based framework was able to retain its structural integrity in strong alkaline solution (1.0 M KOH) over one week.32 Reactions between pyrazolate ligands and divalent transition metal salts are expected to afford pyrazolate MOFs (metal pyrazaolate frameworks, MPFs) with higher base stability due to the higher pKa value of pyrazoles among azoles.33,34 Nevertheless, the strong metal–pyrazolate (M–N) bonding makes it harder to crystallize in a controlled manner and grow into big crystals before powder or even amorphous products form and precipitate.
In the past two decades, with continuous efforts, advances have been made in the design and application of MPFs. A series of polypyrazolate ligands have been designed, and a series of relevant MOFs, M(bdp) (M = Co, Fe, Zn, Ni, Cu; bdp2− = benzenedipyrazolate), Fe2(BDP)3, Ni8L6, PCN-601, BUT-55, to name a few, have been synthesized, which reveal fascinating architectures, intriguing properties, and record-breaking performance in applications (Fig. 1).35–40 Some of these materials have structures specific for azolate MOFs,41 such as M(bdp) with pts topology and BUT-55 with wuk topology, while others are isoreticular analogs of known carboxylate MOFs, including Fe2(BDP)3 (sct), Ni8L6 (fcu) and PCN-601 (ftw), highlighting the strength of crystal engineering principles in enlarging the MOF library.
 | ||
| Fig. 1 Timeline of representative works on MPFs. | ||
With a better understanding of materials design principles from the perspective of crystal engineering and increasing attention on stable MOFs, research on MPFs has thrived during the last decade (Fig. 2). Although a few reviews have overviewed MPFs or their structures, no systematic review documenting MPFs has been published yet.42–45 We intend to provide a rich palette of MPFs in this review, spanning the significance of their design and synthesis, common and distinct structure features, superior base stability and wide application scope. This review will start by classifying and introducing the structures of MPFs (building blocks and topologies), followed by summarizing synthetic strategies adopted to build and/or modify MPFs. Representative examples will be elaborated to discuss the stability of MPFs. And then, based on physical/chemical properties, potential applications will be explored. The structure–property relationship will also be discussed for insights into the underlying mechanism, which would in turn improve and perfect the design principles. Overall, this review attempts to contribute an up-to-date library for MPFs. It is expected to facilitate the design of next-generation MOFs by taking inspiration from the known structures, which could expedite the discovery of more advanced functional materials for addressing global challenges.
 | ||
| Fig. 2 The number of pyrazolate-MOF publications from 2008 to 2025. | ||
2. Synthesis and structure of pyrazolate-based MOFs
As five-membered aromatic nitrogen-containing heterocycles, azoles are common building blocks of synthetic or natural compounds (Fig. 3a).29 Pyrazole is an azole with two adjacent N atoms. The coordination behaviors of the sp2 N-donors in pyrazoles are basically identical to pyridines, while the other sp3 N in pyrazoles could be deprotonated to form the corresponding pyrazolate anions. Both N atoms share the same environment as those in imidazoles, though their relative positions are different (1,2 for pyrazoles and 1,3 for imidazoles). Pyrazoles also exhibit similar stronger basicity to their imidazole isomers.46,47 In contrast to imidazolate, which has a bridging angle around 140°, pyrazolate exhibits a much smaller bridging angle, approximately 70°, leaving two coordinated metal ions in close proximity (ca. 3.5–4.7 Å depending on the ion radius) (Fig. 3b).48,49 The coordination geometry of exo-bidentate pyrazolate usually resembles the syn,syn-μ-η1,η1-bidentate mode of carboxylate.29 This geometry comparability allows for accessing isoreticular pyrazolate counterparts by employing the crystal engineering approach.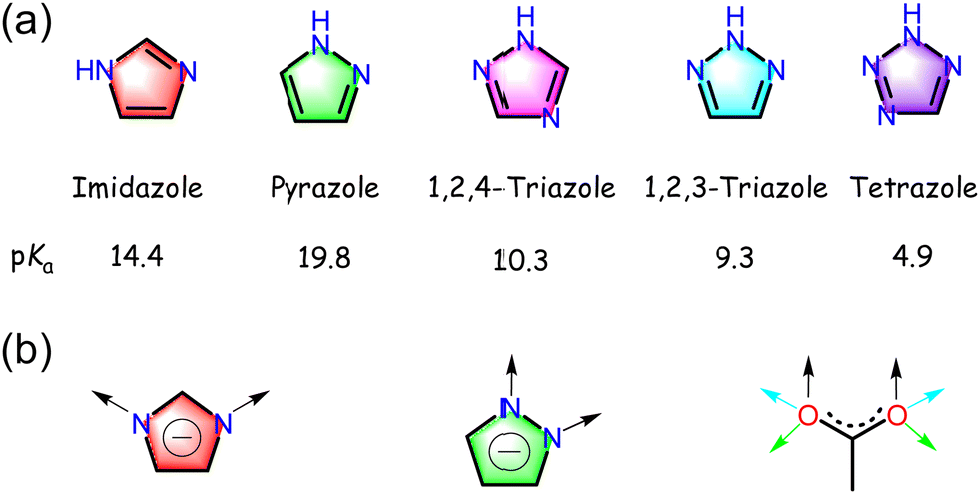 | ||
| Fig. 3 (a) Structure and pKa value of prototypical azoles. (b) Comparison of the coordination modes of imizolate, pyrazolate, and carboxylate. | ||
In addition to the similarity, pyrazolates also present some uniqueness in forming metal-based building blocks specific for pyrazolate MOFs, including zigzag chains (rod building blocks, RBBs) with square-planar/tetrahedral metal ions, helical chains with tetrahedral metal ions, triple zigzag chains with octahedral metal ions, and cubic clusters [M8(OH)6(Pz)12] (Pz = pyrazolate, M = Ni or Co) (Table 1 and Fig. 4).29,43 The structural configurations and internal surface properties of MOFs can be systematically tuned by varying the geometry of spacing ligands and metal ions, as well as building block modification with specific functional groups (Table 1 and Fig. 4, 5).43 Actually, pyrazolate derivatives have been well explored as ligands for the construction of discrete, polynuclear complexes, which is not our focus here.50–55 Similarly, there are numerous reports on the use of pyrazole-containing multifunctional ligands, such as pyrazole carboxylic acid, for the synthesis of MOFs, however, this is also beyond the scope of our review.42,56,57 In this section, we will focus on the reported MPFs constructed from different chains/clusters.
| Type | Name | Ligand | Metal | Stability | Application |
|---|---|---|---|---|---|
| Single-walled chain | Co(bdp) | 1,4-Benzenedi(4′-pyrazolyl) (H2BDP) | Co | Thermally stable up to 260 °C | H2 adsorption,35,58 catalytic oxidation of cyclohexene,59 CO2/CH4 separation,60 methane storage38,61 |
| Ni(bdp), Ni(bpb), PCF-9 | Ni | pH 3–12 (room temperature (RT), 24 h); thermally stable up to 400 °C | Adsorption of harmful organic vapors,62 oxidation of 5-hydroxymethylfurfural,63 heterogeneous catalysis,64 C2H6/C2H4 separation,65 photo-reduction66 | ||
| Zn(bdp), Zn(bpb), AIST-1 | Zn | pH = 10 (RT, 24 h); thermally stable up to 400 °C | Adsorption of harmful organic vapors,62 amination of 4-chloropyridine,67 hydrogen storage,68 oxygen evolution reaction (OER),69 drug delivery,70 zinc–air batteries71 | ||
| Fe(bdp) | Fe | — | Methane storage,38 switchable thermal conductivity (simulation)72 | ||
| Co(F-bdp) | H2(F-bdp) | Co | — | Gas storage and separation61 | |
| Co(p-F2-bdp) | H2(p-F2-bdp) | ||||
| Co(o-F2-bdp) | H2(o-F2-bdp) | ||||
| Co(D4-bdp) | H2(D4-bdp) | ||||
| Co(p-Me2-bdp) | H2(p-Me2-bdp) | ||||
| Zn-BDP-x | H2BDP-X, X = –NO2, –NH2, –OH | Zn | Thermally stable up to 400–450 °C | Gas separation and purification,73 drug delivery,74 and catalytic cycloaddition of CO275 | |
| Ni-BDP-x | Ni | ||||
| BUT-31 | H2BDP-CHO | Zn | Boiling water; 0.1 M HCl, 4 M NaOH solution (RT, 24 h); thermally stable up to 400 °C | Gas adsorption76 | |
| MFU-2 | 1,4-Bis(3,5-dimethyl-1H-pyrazol-4-yl)benzene (H2DMBDP) | Co | Ethanol, ethanol/water 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (RT, 96 h); thermally stable up to 340 °C :3 (RT, 96 h); thermally stable up to 340 °C |
Catalytic oxidation77 | |
| Zn(BPE) | 1,2-Bis(1H-pyrazol-4-yl)ethyne (H2BPE) | Zn | Water (15 days) | Removal of pollutants in wastewater78 | |
| Ni(BPEB) | 1,4-Bis(1H-pyrazol-4-ylethynyl)benzene (H2BPEB) | Ni | Water vapors, acetone (RT, 5 days); boiling water (2 days); pH 5–9 (RT, 8 h); thermally stable up to 422 °C | Synthesis and structure,79 catalytic oxidation63 | |
| Zn(BPEB) | Zn | Thermally stable up to 410 °C | Synthesis and structure79 | ||
| M-pyNDI | Naphthalene diimide bipyrazole (H2NDI) | M = Zn, Co, Ni | Thermally stable up to 300 °C | Gas sensing80 | |
| ZnNDI | H2NDI | Zn | — | Electrical conductivity enhancement,81 semiconductor/insulator switching,82 electrochromism,83 redox conductivity,84 electrical conductivity (computation)85 | |
| ZnPMDI | Pyromellitic diimide bis-pyrazolate (PMDI) | — | Redox conductivity84 | ||
| Zn(NDI-X) | H2NDI-X (X = H, SC2H5, NH-C2H5) | Water stable | Electrochromism,86 water adsorption87 | ||
| Zn(dmpz)2NDI | (dmpz)2NDI | — | Electrode88 | ||
| Zn-PDI-MOF | perylene diimide (PDI) pyrazolate | — | Electrochromic properties89 | ||
| Fe-pyNDI | 2,7-Bis(1H-pyrazol-4-yl)-benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone (H2pyNDI) | Fe | — | Electrosynthesis of ammonia90 | |
| Zn(BPZ) | 4,4′-Bipyrazole (H2BPZ) | Zn | Air-stable; thermally stable up to 300–450 °C | Synthesis and structure,91 gas separation92,93 | |
| Co(BPZ) | Co | ||||
| Cd(BPZ) | Cd | ||||
| Hg(BPZ) | Hg | ||||
| Cu(BPZ) | Cu | ||||
| Ni(BPZ) | Ni | ||||
| Pd(BPZ) | Pd | ||||
| Cu(H2BPZ)2(NO3)2 | Cu | ||||
| Cd(H2BPZ)(CH3COO)2 | Cd | ||||
| M-CFA-6 | Fe | Thermally stable up to 250 and 300 °C | Synthesis and structure94 | ||
| Ga | |||||
| Zn(BPZX) | H2BPZX, X = H, NO2,NH2 | Zn | Thermally stable up to 435–453 °C | Adsorption of CO295 | |
| M(BPZNO2) | H2BPZNO2 | M = Co, Cu, Zn | Thermally stable up to 390 °C | CO2 capture96 | |
| Co(BPZX) | H2BPZX, X = H, NO2, NH2 | Co | Thermally stable up to 400 °C | Catalytic oxidation97 | |
| M(BPZNH2) | H2BPZNH2 | M = Zn, Ni, Cu | Water vapor stable; thermally stable up to 290–430 °C | CO2 capture and conversion98 | |
| MIXMOFs | 3,5-Diamino-4,4′-bis(1H-pyrazole) (3,5-H2L) | Cu, Ni | Thermally stable up to 377 °C | CO2 electrochemical reduction (CO2RR)99 | |
| FICN-8 | (5,10,15,20-Tetra(1H-pyrazol-4-yl) porphyrin) (H4TPP) | Cu | — | CO2RR100 | |
| PCN-1004 | 1,1,2,2-Tetrakis(4-(1H-pyrazol-4-yl)phenyl) ethene (H4TPPE) | Cu | pH 1–14; thermally stable up to 300 °C | Heterocatalysis101 | |
| Zn(Hazbpz)NO3 | 4,4′-Azobis(3,5-dimethyl-1H-pyrazole) (H2AZBPZ) | Zn | Thermally stable up to 340 °C | Synthesis and structure102,103 | |
| [Cu(NO3)2(H2O)2(H2azbpz)2]·2H2O | Cu | — | Synthesis and structure102,103 | ||
| [Ni(H2O)4(H2azbpz)2](NO3)2·2H2O | Ni | ||||
| Zn(azbpz) | Zn | Water; thermally stable up to 400 °C | C2H2/CO2 separation104 | ||
| Double walled Chain | Zn(1,3-BDP) | 1,3-Benzenedi(4′-pyrazolyl) (1,3-H2BDP) | Zn | pH = 3 (90 °C, 30 min); thermally stable up to 400–500 °C | Hydrogen storage68 |
| Co(1,3-BDP) | Co | ||||
| BUT-53 | pH = 5–14 (RT, 24 h); thermally stable up to 363–432 °C | Trace removal of benzene vapor,40 SF6/N2 separation105 | |||
| BUT-54 | 2,7-Di(1H-pyrazol-4-yl)naphthalene) (H2DPN) | Trace removal of benzene vapor40 | |||
| BUT-55 | H2BDP |
||||
| BUT-56 | 2,5-Di(1H-pyrazol-4-yl)pyridine) (H2DPP) | ||||
| BUT-57 | 2,7-Di(1H-pyrazol-4-yl)pyrene) (H2PDP) | ||||
| BUT-58 | H2BDP | Zn | |||
| JNU-204, HIAM-3004 | 4,7-Di(1H-pyrazol-4-yl)benzo[c][1,2,5]thiadiazole (H2PBT, H2BT-Pz) | pH = 3–14 (RT, 24 h); thermally stable up to 500 °C | Photocatalytic aerobic oxidation,106 luminescence107 | ||
| HIAM-3005 | 4,7-Di(1H-pyrazol-4-yl)benzo[c][1,2,5]-selenadiazole (H2DPBS, H2BS-Pz) | pH = 2–12 (RT, 24 h); thermally stable up to 500 °C | Luminescence107 | ||
| HIAM-3006 | 5,6-Dimethyl-4,7-di(1H-pyrazol-4-yl)benzo[c][1,2,5]thiadiazole (DDPBT) | ||||
| HIAM-213 | 3,5-Di(1H-pyrazol-4-yl)aniline (H2DPA) | pH = 3–14 (RT, 2 days); thermally stable up to 350 °C | Gas separation108 | ||
| HIAM-214 | H3BTP | Thermally stable up to 180 °C | |||
| Fe(II)(bppdi)(DMF)0.5 | (2,6-Bis(1H-pyrazolyl)-pyromellitic diimide) (H2bppdi, H2TET) | Fe | Thermally stable up to 360 °C | NO reduction109 | |
| Fe2(BDP)3 type | Fe2(BDP)3 | H2BDP | Fe | pH = 2 to 10 (100 °C 2 weeks); thermally stable up to 360 °C, | Separation of hexane isomers,37 C2H6/C2H4 adsorption separation,110 CO2 adsorption111,112 |
| A2Fe2(BDP)3 (A = Li +, Na +, K+ | Thermally stable up to 360 °C | H2 adsorption113 | |||
| AxFe2(BDP)3 (A = Na +, K +; 0 < x ≤ 2) | — | O2 adsorption114 | |||
| Fe2(BPEB)3 | 1,4-Bis(1H-pyrazol-4-ylethynyl)benzene (H2BPEB, H2EBDP) | Water vapor (RT, 1 h); thermally stable up to 410 °C | Synthesis and structure79 | ||
| Fe-PF1 | H2EBDP-F, H2EBDP-2F, H2EBDP-4F | Water (RT, 24 h); thermally stable up to 300–350 °C | Adsorption of CO2115 | ||
| Fe-PF2 | |||||
| Fe-PF4 | |||||
| Cu3 | CFA-2 | 3,3′,5,5′-Tetraphenyl-1H,1′H-4,4′-bipyrazole (H2PhBPZ) | Cu | Thermally stable up to 300, 500 °C | Synthesis and structure116 |
| CFA-3 | Ag | ||||
| Cu-BTPP | 1,3,5-Tris((4-pyrozole)phenyl)-benzene) (H3BTPP) | Cu | Thermally stable up to 350 °C | Synthesis and structure117 | |
| FJU-66 | H2NDI | Cu | pH = 3–14, 10 M NaOH (RT, 12 h); thermally stable up to 530 °C | Conductivity118 | |
| M4 (M = Ni, Cu, Co, Cd, Pd) | Ni3(BTP)2 | 1,3,5-Tris(1H-pyrazol-4-yl)-Benzene (H3BTP) | Ni | pH = 2–14 (100 °C, 14 days); thermally stable up to 430–510 °C | Synthesis and structure,33 adsorption and degradation of nerve agent simulant and pesticide,119 electroreduction of CO2120 |
| Cu3(BTP)2 | Cu | ||||
| BUT-124(Cd) | Cd | Unstable | OER121 | ||
| BUT-124(Co) | Co | H2O, pH = 14 (KOH) (RT, 24 h) | |||
| BUT-32 | 2,4,6-Tris(4-(pyrazole-4-yl)phenyl)-1,3,5-triazine (H3TPTA) | Ni | pH = 3, 4 M NaOH, (RT, 1 day); thermally stable up to 398 and 380 °C | C–C coupling122 | |
| BUT-33 | C–C,122 Gold extraction123 | ||||
| BUT-33(Pd) | Pd | pH = 3, 8 M NaOH (RT, 1 day) | Suzuki and Heck coupling124 | ||
| BUT-78 | 3,3′,5,5′-Tetra(1H-pyrazol-4-yl)-1,1′-biphenyl (H4BPTP) | Ni | pH = 2, 5 M NaOH, (RT, 1 day); thermally stable up to 430 °C | Capture of trace benzene and SO2125 | |
| MFU-1 | H2DMBDP | Co | Ethanol, ethanol/water 7:3 (RT, 96 h); thermally stable up to 340 °C |
Catalytic oxidation126 | |
| Ni8/Co8/Zn9 | [Ni8(OH)4(OH2)2(pbp)6] | 4,4′-Bis(1H-pyrazol-4-yl)biphenyl (H2PBP) | Ni | Thermally stable up to 410 °C | Synthesis and structure127 |
| [Ni8(OH)4(OH2)2(tet)6] | 2,6-Bis(1H-pyrazol-4-yl)pyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetrone (H2TET) | ||||
| [Ni8(OH)4(H2O)2(L)6] | H2BDP; H2BYDP; H2EBDP | Stable in base (24 h); thermally stable up to 300–350 °C | The capture of harmful volatile organic compounds,36 SO2 adsorption,128 synthesis and structure129 | ||
| K[Ni8(OH)5(EtO)(H2O)2(BDP_X)5.5] | H2BDP_X, X = H, OH, NH2 | Thermally stable up to 300 °C | CO2 capture from flue gas,130 CO2131 and SO2132 adsorption | ||
| K3[Ni8(OH)3(EtO)(H2O)6(BDP_O)5] | |||||
| Pd@NiBDP | H2BDP | Hydroamination of alkynes133 | |||
| Ni8(BDP)6@Cu | Heterocatalysis134 | ||||
| Ni8(BDP)6@Fe | Oxidase-mimicking H2O2 activation135 | ||||
| Ni8(BDP-X)6 | H2BDP-X, X = CHO, CN, COOH | pH = 3, 4 M NaOH (RT, 1 day); thermally stable up to 300 °C | CO2 adsorption and proton conduction136 | ||
| NiBDP-AgS | H2BDP-NH2 | pH 2–13, 1 M DBU solution; thermally stable up to 400 °C | Chemical fixation of CO2137 | ||
| Pd@Ni-MOF | 4,4′-(Benzene-1,4-diyldiethyne-2,1-diyl)bis(1-H-pyrazole) (H2EBDP) | — | Catalytic Heck arylation of functionalized olefins138 | ||
| BUT-2 | H2BDP | — | Synthesis and structure139 | ||
| BUT-3 | H2BDP-CN | ||||
| BUT-4 | H2BDP-CHO | ||||
| BUT-48 | 1,3-H2BDP-CN | pH = 5, 1 M NaOH (RT, 24 h); boiling water (24 h); thermally stable up to 350 °C | |||
| BUT-49 | 1,3-H2BDP-CF3 | ||||
| BUT-123 | H3BTPP | ||||
| Ni8(OH)4(H2O)2(L4)6 | (4,4′-Buta-1,3-diyne-1,4-diyl)bis-pyrazole (H2BYDP) | — | C2H2/CO2 separation140 | ||
| Ni8(OH)4(H2O)2(L5)6 | 4,4′-(Benzene-1,4-diyldiethyne-2,1-diyl)bis-pyrazole (H2EBDP) | ||||
| NiL1 | 4,4′-((2,5-Bis((4-(methylthio)phenyl)ethynyl)-1,4-phenylene)bis(ethyne-2,1-diyl))bis(1H-pyrazole) (H2EBDP-SMe) | pH = 3, 15 M NaOH (24 h); thermally stable up to 300 °C | Catalytic hydrogen evolution reaction (HER),141 proton conduction142 | ||
| NiL2 | 4,4′-((2,5-Di(hex-1-yn-1-yl)-1,4-phenylene)bis(ethyne-2,1-diyl))bis(1H-pyrazole) (H2EBDP-Prop) | pH = 1, 15 M NaOH (RT, 24 h), thermally stable up to 320 °C | Proton conduction142 | ||
| NiL1 | 2,2′-((2,5-Bis((1H-pyrazol-4-yl)ethynyl)-1,4-phenylene)bis(ethyne-2,1-diyl))dianiline (H2EBDP-NH2) | pH = 3–13 (RT, 24 h) | Electrocatalytic hydrogen evolution143 | ||
| NiL1-F | 4,4′-((2,5-Bis((1H-pyrazol-4-yl)ethynyl)-1,4-phenylene)bis(ethyne-2,1-diyl))bis(1-(perfluorophenyl)-1H-1,2,3-triazole) | Thermally stable up to 300 °C | Remove perfluoro pollutants144 | ||
| JNU-212 | H2BDP | 7 M NaOH (24 h, 100 °C); pH = 2 (RT, 24 h) | Catalytic oxidation145 | ||
| JNU-213 | H2BT-Pz | ||||
| JNU-214 | H2BS-Pz | ||||
| JNU-215 | 4,9-Di(1H-pyrazol-4-yl)naphtho[2,3-c][1,2,5]selenadiazole (H2NS-Pz) | ||||
| CFA-25 | 1,4-Bis(1H-pyrazol-4-yl)benzoxadiazole (H2BPBO) | — | Ionic transport146 | ||
| PCN-601 | H4TPP | 0.01 mM HCl (RT, 24 h), 10 M NaOH (100 °C, 24 h); thermally stable up to 300 °C | Synthesis and structure,39 photoreduction,147 electrocatalytic148 | ||
| PCN-602 | 5,10,15,20-Tetrakis(4-(pyrazolate-4-yl)phenyl)porphyrin (H4TPPP) | pH 4-14, 1 M KF, Na2CO3 and K3PO4 (1 day); thermally stable up to 300 °C | C−H bond halogenation149 | ||
| PCN-624 | 5,10,15,20-Tetrakis(2,3,5,6-tetrafluoro-4-(1H-pyrazol4-yl)phenyl)-porphyrin (H4TTFPPP) | Water, ethanol, HCl (1 M), Na2CO3 (3 M) and K3PO4 (3 M) (RT, 24 h); thermally stable up to 400 °C | Heterocatalysis150 | ||
| TPE4Pz-Ni | H4TPPE | pH = 3, saturated NaOH solution (RT, 24 h); thermally stable up to 350 °C | C–S cross-coupling151 | ||
| FICN-18 | 2,2′-((1E,1′E)-(ethane-1,2-diylbis(azaneylylidene))bis(methaneylylidene))bis(4-(1H-pyrazol-4-yl)phenol) (H2EBE) | Boiling water; pH = 1–14 (RT, 21 days); thermally stable up to 200 °C | Heterocatalysis152 | ||
| FICN-19 | Co | ||||
| fcu-L-Co | H2DPP, 3,6-di(1H-pyrazol-4-yl)pyridazine (H2L2), 2,5-di(1H-pyrazol-4-yl)pyrazine (H2L3), and 2,5-di(1H-pyrazol-4-yl)pyrimidine (H2L4) | pH = 5, 4 M NaOH solution (RT, 24 h); thermally stable up to 350–400 °C | Structural transformation and water sorption153 | ||
| Zn9O2(OH)2(L)6 | H2EBE | Zn | pH = 3, 2 M NaOH (RT, 210 days); pH = 3 and H2O (21 days, 100 °C); thermally stable up to 400 °C | Catalytic CO2 cycloaddition154 | |
| Others | BUT-41 | 1,3,6,8-Tetra(1H-pyrazol-4-yl)-9H-carbazole (H4CTP) | Ni | Thermally stable up to 400 °C | Adsorption of CO2, N2, and CH4155 |
| MROF-12 | H2NDI | Cu | pH = 2–13 (RT, 12 h); thermally stable up to 481 °C | Separation of C2H2/CH4 mixture156 | |
| Cu-TPP | H4TPP | Cu | pH = 1–14 (RT, 24 h); thermally stable up to 300 °C | Fluorescence and detection157 | |
| MnTPzP-Mn | Mn | Thermally stable up to 300 °C | Li–CO2 battery cathode material158 | ||
| TMBP·CuI | (3,3′,5,5′-Tetramethyl-4,4′-bipyrazol) (H2BPZ-Me) | Cu | Water-stable; thermally stable up to 300 °C | Immobilization of I2 and ICl159 | |
| [Mn(DMPMB)] (n = 1, 2) | H2DMPMB | M = Co, Zn, Cd, Cu, Ag | Thermally stable up to 300 °C | Synthesis and structure160 | |
| [Cu2(bpz)2(CN)X], (X = Cl; I) | H2BPZ-Me | Cu | — | Catalytic azide–alkyne “Click” reactions161 | |
| [Cu6(bpz)6(CH3CN)3(CN)3Br]·2OH | |||||
| M[CFA-4] | 1,4-Bis(3,5-bis(trifluoromethyl)-1H-pyrazole-4-yl)benzene (H2TFPB) | M = Cu(I), K, Cs, Ca(0.5) | Thermally stable up to 300 °C | Gas sorption and fluorescence162 | |
| PCN-300 | H4TPP | Cu | pH = 1–14 (RT, 24 h); thermally stable up to 300 °C | C–O cross-coupling reaction163 | |
| PCN-1003 | H4TPPE | Ni | pH = 1–12 (RT, 24 h); thermally stable up to 450 °C | Perfluorooctanoic acid concentration and degradation164 | |
| SIFSIX-18-Cd | H2BPZ-Me | Cd | Boiling water (12 h); air (2 months); thermally stable up to 250 °C | Synthesis and structure,165 hydrogen isotope separation166 | |
| SIFSIX-18-Ni | Ni | 75% relative humidity (40 °C, 14 days); thermally stable up to 300 °C | Trace CO2 capture167 |
 | ||
| Fig. 4 Typical chain/cluster structures observed in MPFs. | ||
 | ||
| Fig. 5 Typical pyrazolate ligands used in constructing MOFs. | ||
2.1. Chains/RBBs (Zn/Co/Ni/Fe)
Divalent late transition metal ions with 4-coordinate geometries, including Fe(II), Co(II), Ni(II), Cu(II), and Zn(II), can link the simple pyrazolate (Pz−) ligand to construct infinite one-dimensional (1D) chains or RBBs, represented by the formula [M(Pz)2], which could also be considered as extended vertex-sharing M2(Pz)2 rings.50–53 Therein tetrahedral metal ions form planar M2(Pz)2 rings with two bridging pyrazolate moieties, while square-planar metal ions form bent ones.50–53 Cu(II) is able to adopt both the square-planar and flattened tetrahedral geometries, enabling the formation of two distinct isomeric [Cu(Pz)2] chains.53 These chains/RBBs can be connected to form higher dimensional networks with polypyrazolate ligands by replacing the monopyrazolates.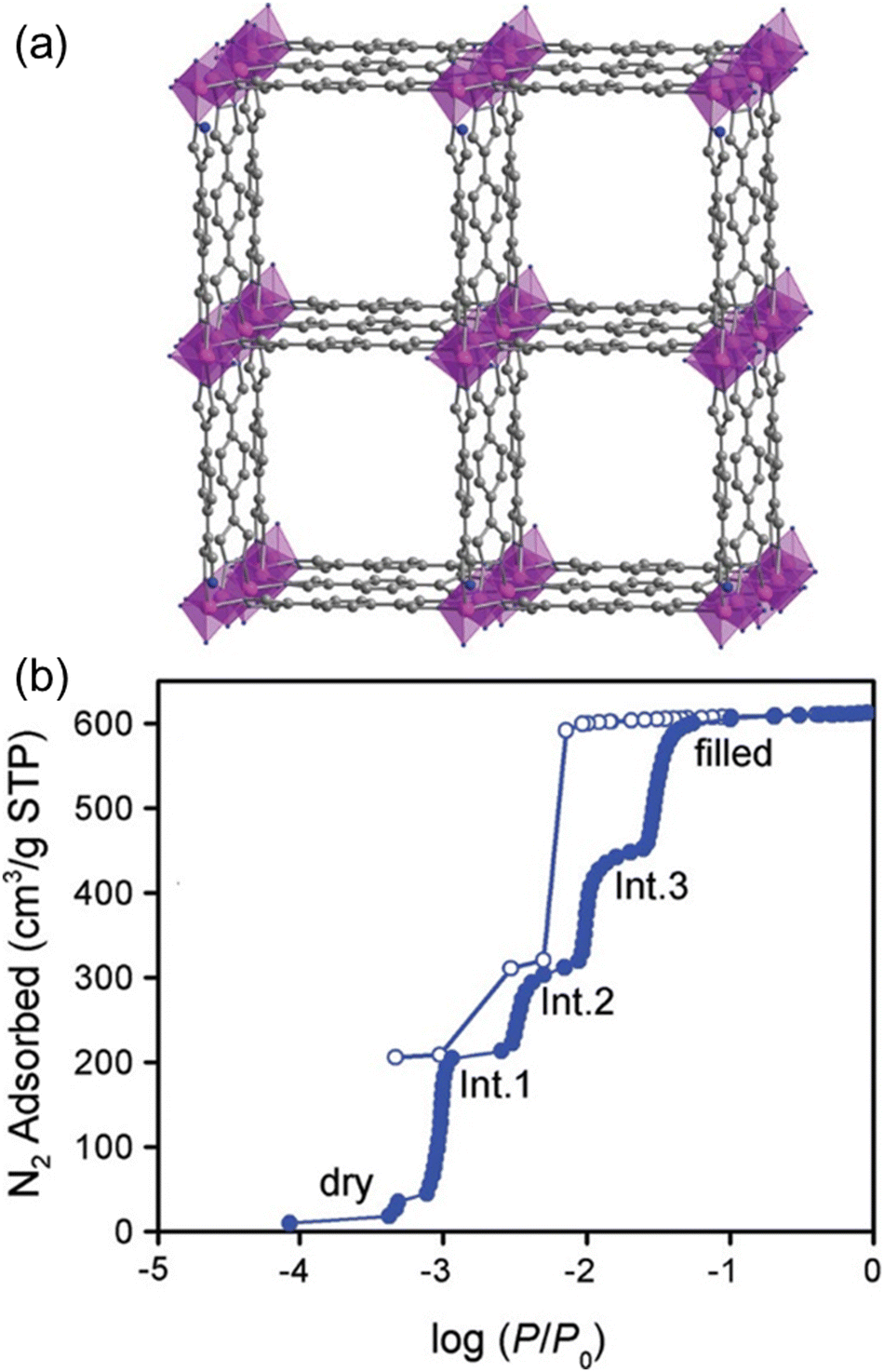 | ||
| Fig. 6 (a) Structure of Co(bdp). (b) N2 adsorption isotherm measured at 77 K, indicating the five pressure-dependent phases.58 Reprinted with permission from American Chemical Society 2010. | ||
After the discovery of prototype Co(bdp), a great number of variants/analogs were synthesized under the guidance of crystal engineering principles in the following decades. The metal ions are amenable to substitution by various species, including Fe(II), Ni(II), Zn(II), and Cu(II), to yield isostructural compounds. In 2010, Long's group reported the synthesis of Zn(bdp) following similar procedures to that for Co(bdp).68 Navarro's group also synthesized powder samples of Zn(bdp) and Ni(bdp) in the same year.62 The overall structures of as-synthesized Zn(bdp) and Ni(bdp) are isoreticular with Co(bdp), however, the coordination modes show slight differences.35,62 The Zn(II) in Zn(bdp) exhibits a similar coordination geometry with Co(bdp), while Ni(II) in Ni(bdp) adopts a square-planner geometry in an orthorhombic structure. In addition, they show different degrees of flexibility upon gas adsorption/desorption, where Zn(bdp) is more rigid compared the other two analogs. In 2015, an iron analog Fe(bdp) was accessed in Long's group.38 Fe(bdp) is also isoreticular with Co(bdp) with Fe(II) irons in tetrahedral coordination geometry as well. Another Cu-based variant Cu(bdp), also named Cu-dpb, was synthesized by Cao and co-workers in 2020.64 There are no structural details about Cu(bdp) available, except that it matched the PXRD pattern of Ni(bdp).38,64
With the M(bdp) platforms in hand, researchers have attempted to functionalize the bdp2− ligand for new isostructural materials with diverse functionality. In 2011, Volkmer and co-workers used the four-methyl functionalized 1,4-bis(3,5-dimethyl-1H-pyrazol-4-yl)benzene (H2dmbdp) ligand to synthesize the Co(bdp) analog, MFU-2, also denoted Co(dmbdp).77 As opposed to the flexible nature of Co(bdp), MFU-2 was demonstrated to be a rigid network exhibiting type I gas sorption isotherms due to the steric interactions between adjacent methyl groups. Viewed along the Co(II) chains/RBBs, these methyl groups also lead to a torsional distortion of planes running through opposite pyrazole rings, while the planes through adjacent pyrazole ring systems are strictly orthogonal to each other in Co(bdp). In 2012, Navarro and co-workers synthesized two series, Mbdp-X, adopting the Ni(bdp) and Zn(bdp) structure, with the tagged organic linkers H2bdp-X (X = –NO2, –NH2, –OH), respectively, as well as a structural isomer of Ni(bdp), Nibdp-SO3H with a sulfonate tagged linker, H2bdp-SO3H.73 In 2016, Long and co-workers introduced various functional groups, including fluorine, deuterium, and methyl to the H2BDP ligand, affording five Co(bdp) analogs, Co(F-bdp), Co(p-F2-bdp), Co(o-F2-bdp), Co(D4-bdp), and Co(p-Me2-bdp).61 After functionalization, all the materials have a high and similar degree of flexibility but different pore size and pore chemistry. The introduction of functional groups has altered the edge-to-face π–π interactions in the collapsed phase, leading to divergent behaviours in methane storage.
In addition to the metal substitution and ligand functionalization, there are plenty of publications on reticular contraction/expansion by shortening or lengthening the ligand. The simple rigid spacer 4,4′-bipyrazole (H2BPZ) has been employed to synthesize MOFs with late transition metal ions by Galli, Civalleri and co-workers in 2012.91,171 A series of M(BPZ) (M = Zn, Co, Cu, Ni) were obtained under conventional routes (room-temperature stirring for Zn and Cu with base), solvothermal (for Co) or reflux (for Ni with base) conditions. The four materials are isoreticular 3D porous networks containing square and rhombic 1D channels, respectively. In 2019, Galli, Rossin, and co-workers used the amino-functionalized organic linker 3-amino-4,4′-bipyrazole (H2BPZNH2) to synthesize three MOFs M(BPZNH2) (M = Zn, Ni, Cu).95 The Zn(II) compound was formed as a 3D structure featuring tetrahedral Zn(II) nodes and bridging BPZNH22−, which define the vertices and edges of square-shaped channels. The Ni(II) and Cu(II) analogs, which are isostructural, feature square-planar metal centers and BPZNH22− linkers acting as connectors at the vertices and edges of rhombic channels within a 3D framework. These findings are in agreement with their parent M(bdp) materials.
In 2013, Dincă and co-workers extended the dmbdp2− linker with redox-active 1,4,5,8-naphthalene diimide (NDI) containing cores to get three new ligands with different functionalities, H2NDI-X (X = H, S-C2H5, NH-C2H5).87 Solvothermal reaction of these ligands with zinc salt yielded an extending isostructural series of MOFs, designated as Zn(NDI-X). These MPFs feature RBBs of tetrahedral Zn(II) and NDI linker functionalized channels (width of ∼16 Å). Post-synthetic oxidation of Zn(NDI–SEt) using dimethyldioxirane was deployed to introduce ethyl sulfoxide and ethyl sulfone groups, thereby increasing the hydrophilicity of the channels. In 2022, Diring and co-workers substituted the central phenyl group of dmbdp2− linker with highly electro-deficient perylene diimide (PDI) cores, resulting in two new PDI ligands with their four-bay positions functionalized by either a chlorine (1,6,7,12-tetrachloro-perylene-3,4,9,10-tetracarboxylic dianhydride, PDI-Cl) or bridged ethyl-1,2-disulfone (PDI-SO2eth) group.172 In 2023, based on these two ligands, they reported the in situ preparation of two Zn-PDI-MOF thin films with interesting electrochromic properties.89 These two MOFs, Zn-PDI-Cl and Zn-PDI-SO2 are expected to be isostructural to Zn(NDI-X) as reported by Dincă and co-workers, featuring the infinite chains/RBBs, in which Zn(II) ions are in a tetrahedral geometry and spaced by the long ligand.
In terms of alkynyl functionalization, Galli and co-workers employed a long ligand 1,4-bis(1H-pyrazol-4-ylethynyl)benzene (H2BPEB) to react with a number of transition metal ions (M = Zn(II), Ni(II), Fe(II)).79 Comprising the same structural motif (square planar metal ions), Ni(BPEB) and Ni(BDP-X) constitute an isoreticular family. While an attempt at the isoreticular expansion of Zn(BDP-X) failed with the insurgence of interpenetration in α-Zn(BPEB), which might be promoted by more space spared from the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds due to the tetrahedral geometry adopted by Zn(II). The two interpenetrated networks finally cross between them. However, the reaction with Fe(II) in N,N′-dimethylformamide (DMF) yields a different phase Fe2(BPEB)3, which will be discussed later (the section on Fe2(BDP)3 and derivatives). In addition, two MOFs, Zn(BPE) and Zn(BPE)·nDMF (interpenetrated i-Zn and non-interpenetrated ni-Zn·S) were synthesized based on a 1,2-bis(1H-pyrazol-4-yl)ethyne (H2BPE) ligand by Galli and co-workers in 2023.78 Interestingly, a HgCl2-triggered interpenetrated to non-interpenetrated phase transformation (i-Zn-to-HgCl2@ni-Zn) was observed at the molecular level, representing the first example of pristine phase decomposition and concomitant assembly of the other phase upon the inclusion of a specific guest.
C triple bonds due to the tetrahedral geometry adopted by Zn(II). The two interpenetrated networks finally cross between them. However, the reaction with Fe(II) in N,N′-dimethylformamide (DMF) yields a different phase Fe2(BPEB)3, which will be discussed later (the section on Fe2(BDP)3 and derivatives). In addition, two MOFs, Zn(BPE) and Zn(BPE)·nDMF (interpenetrated i-Zn and non-interpenetrated ni-Zn·S) were synthesized based on a 1,2-bis(1H-pyrazol-4-yl)ethyne (H2BPE) ligand by Galli and co-workers in 2023.78 Interestingly, a HgCl2-triggered interpenetrated to non-interpenetrated phase transformation (i-Zn-to-HgCl2@ni-Zn) was observed at the molecular level, representing the first example of pristine phase decomposition and concomitant assembly of the other phase upon the inclusion of a specific guest.
In 2015, Volkmer and co-workers synthesized a new type of chain/RBB based MOFs M-CFA-6 (M = Ga, Fe, CFA = termed Coordination Framework Augsburg University).94 The structure motif of the M-CFA-6 framework is similar to that of MIL-53, featuring trivalent metal centers in octahedral coordination connected by the bifunctional BPZ2− ligands and hydroxyl groups.170 MIL-53 and CFA-6 show similar thermal stabilities, surface areas and pore sizes. The key difference lies in the breathing behavior of MIL-53, which is not observed in the more rigid network of CFA-6. In 2018, the authors reported the Mn(III)-based analog of M-CFA-6 (M = Ga, Fe), Mn-CFA-6.173 The Jahn–Teller distortion of Mn(III) centers causes asymmetric distortion of the hydroxyl bonds and results in a distortion of the unit cell. As opposite to Fe-CFA-6 and Ga-CFA-6, Mn-CFA-6 exhibits similar structural flexibility with MIL-53.
In 2024, Cao and co-workers synthesized FICN-8 by combining a tetra-pyrazole ligand, (5,10,15,20-tetra(1H-pyrazol-4-yl) porphyrin) (H4(H2TPP)), with Cu RBBs and elucidated its structure using continuous rotation electron diffraction (cRED).100 The analysis revealed that FICN-8 crystallizes in the orthorhombic Cmmm space group. The structure features fully deprotonated pyrazolate groups from CuTPP4−, which was formed by chelating the H2TPP4− ligand to a Cu(II) ion in the center. The CuTPP4− ligand connects to eight different Cu(II) centers via eight pyrazolate N atoms. Each Cu(II) center, in turn, coordinates to four pyrazolate N atoms, forming square planar units. These units link into 1D [Cu(Pz)2]n chains, which are interconnected by CuTPP4− ligands to create a 3D framework (Fig. 7a and b). This structure is notably different from the 2D MOF, PCN-300,163 despite using the same porphyrinic ligand, marking the first instance of forming 1D pyrazolate chains with a tetratopic pyrazole ligand. In the same year, Zhou et al. reported the synthesis of PCN-1004 via a solvothermal reaction between Cu(OAc)2 and 1,1,2,2-tetrakis(4-(1H-pyrazol-4-yl)phenyl) ethene (TPPE).101 TPPE is also a tetrapyrazole ligand, but its structure is entirely different from FICN-8. Each TPPE ligand connects to eight copper ions through four bidentate μ2-pyrazolate. The copper ions exhibit square planar coordination geometry, with each copper ion coordinating to four nitrogen atoms from four TPPE ligands, resulting in Cu(Pz)2 chains. The interconnection of TPPE with the Cu(Pz)2 chains creates a 3D network, featuring two types of 1D rhombic channels with different sizes (Fig. 7c and d).
 | ||
| Fig. 7 Structures of FICN-8 (a) and (b) and PCN-1004 (c) and (d).100,101 Reprinted with permission from Wiley-VCH 2024. | ||
In 2022, Li and co-workers also reported a series of MPFs, BUT-53 to BUT-58 (BUT = Beijing University of Technology; BUT-53 = CoDPB, H2DPB = 1,3-H2BDP; BUT-54 = CoDPN, H2DPN = 2,7-di(1H-pyrazol-4-yl)naphthalene; BUT-55 = CoBDP; BUT-56 = CoDPP, H2DPP = 2,5-di(1H-pyrazol-4-yl)pyridine; BUT-57 = CoPDP, H2PDP = 2,7-di(1H-pyrazol-4-yl)pyrene; BUT-58 = ZnBDP), constructed from Co(II) or Zn(II) ions and dipyrazolate ligands under solvothermal reactions (Fig. 8).40 They are all double-walled structures as pairs of pyrazolate moieties link adjacent tetrahedral metal cations to form helical chains/RBBs that are cross-linked by pairs of BDP2− ligands. BUT-55/BUT-58 and Co(bdp)/Zn(bdp) share the same formula and are supramolecular isomers, yet they differ in pore structures and pore chemistry. The rigid BUT-55 exhibits smaller 1D square channels (width ∼8.0 Å) than those of the flexible Co(bdp) (width ∼13.2 Å). Replacing the BDP2− ligand with longer ones DPP2− and PDP2− afforded isostructural BUT-56 and BUT-57 with larger pores, while using angular variants of BDP2− and DPN2− yielded BUT-53 and BUT-54 with smaller pores. Unlike the cross-wise arrangement of ligands in BUT-55 to BUT-58, the ligands in BUT-53 and BUT-54 adopt a nose-to-nose configuration with their central aromatic rings aligned in an eclipsed orientation. Li and co-workers developed another double-walled MOF, JNU-204 (JNU = Jinan University), with a 4,7-di(1H-pyrazol-4-yl)benzo[c][1,2,5]thiadiazole (H2PBT) ligand, where a photosensitive pyrrolo[2,1-a]isoquinoline unit was integrated with coordinating pyrazolate moieties.106 JNU-204 is a robust microporous Zn-MPF of the wuk topology, isostructural with the aforementioned BUT-55 to BUT-58.
 | ||
| Fig. 8 Double-walled MPFs featuring dipyrazolate ligands and Co(II)/Zn(II)-based RBBs.40 Reprinted with permission from Springer Nature 2022. | ||
In 2021, Wade and co-workers synthesized a novel Fe-MOF 1 with helical 1D Fe(II) chains/RBBs bridged by dipyrazolate linkers and terminal DMF ligands linking every other two adjacent Fe(II) centers (Fig. 9).109 Solvothermal reaction of Fe(OTf)2(THF)2 (OTf = triflate) and 2,6-bis(1H-pyrazolyl)-pyromellitic diimide (H2bppdi/H2TET), with triethylamine as the modulator, results in the formation of 1. Similar synthesis in the presence of trace air/O2 affords the oxidized analog 2-OH of 1. 2-OH is composed of helical Fe(III) chains/RBBs linked by linear bppdi2− linkers as well as μ2-OH ligands to form triangular channels (∼10 Å in diameter). This structure is reminiscent of Fe2(BDP)3, but presents a double-walled network where the μ2-OH groups linking neighbouring Fe(III) ions are cis to each other. This arrangement leads to a disphenoidal arrangement of pyrazolate N atoms and the formation of helical chains. Fe2(BDP)3 contains octahedral Fe(III) centers coordinated by six pyrazolate linkers, forming a single-walled structure; however, each Fe(III) site in 2-OH adopts a pseudo-octahedral geometry with 4 coordination to pyrazolates and two to μ2-OH ligands. 1 adopts a similar framework structure with 2-OH. For 1, the presence of Fe(II) centers necessitates the coordination of neutral DMF molecules as bridges along the chains, instead of the μ2-OH groups present in 2-OH. The distinctive chains/RBBs have accessible coordination sites between adjacent metal centers, creating motifs that resemble the active sites in non-hemediiron enzymes.
 | ||
| Fig. 9 (a) Synthesis of 1 and 2-OH. (b) Single crystal structure of 2-OH.109 Reprinted with permission from Wiley-VCH 2021. | ||
These results indicate that the linear chains/RBBs are prone to forming single-walled structures with linear ligands, such as M(bdp), while helical chains/RBBs prefer to generate double-walled compounds, such as BUT-55 and Fe-MOF 1. Based on these findings, it is promising that the coordination number and geometry of metal nodes could be finely tuned by employing different ligands and synthetic conditions to regulate the composition, structure and thereby properties of the resulting MPFs.
 | ||
| Fig. 10 (a) Structure of Fe2(BDP)3 as viewed down the (001) direction. (b) Chains of pyrazolate-bridged iron(III) ions in Fe2(BDP)3. (c) Schematic of the reduction of Fe2(BDP)3 with potassium naphthalenide to obtain KxFe2(BDP)3, 0 < x ≤ 2. (d) and (e) Scanning electron microscopy (SEM) micrograph of Fe2(BDP)3 microcrystallites and a single crystal.175 Reprinted with permission from Springer Nature 2018. | ||
In 2018, Long's group established a novel strategy for partial reduction of Fe(III) centers in Fe2(BDP)3 to Fe(II) species, yielding a conductive mix-valence MPF, wherein chains of pyrazolate-bridged Fe(II)/Fe(III) ions would provide a pathway for electron transfer, while the large pores would accommodate stoichiometric intercalation of charge-balancing cations (Fig. 10).175 The KxFe2(BDP)3 (0 < x ≤ 2.0) materials were prepared by reducing the pre-synthesized Fe2(BDP)3 with potassium naphthalenide in tetrahydrofuran, exhibiting full charge delocalization within the framework and great charge mobility. The authors further demonstrated the growth of sizable single-crystals of the robust Fe2(BDP)3 and accessed the solvated A2Fe2(BDP)3·yTHF (A = Li+, Na+, K+) series through chemical reductions.113,114 The cation positions were observed to differ in the solvated and activated Na-containing structures: cations were located near the center of the triangular framework channels and were stabilized by weak cation–π interactions with the framework ligands in the former, while cation migration occurred upon solvent removal in the latter to maximize stabilizing cation–π interactions. They also partially reduced the expanded Fe2(BPED)3 to obtain KxFe2(BPED)3, to support the generality of their proposed chemical reduction strategy to access conductive, guest-selective, and mixed-valence Fe-pyrazolate MOFs.114
As mentioned above, when Galli and co-workers tried to expand the M(bdp) (M = Zn(II), Ni(II), Fe(III)) compounds with an elongated H2BPEB ligand, Fe2(BPEB)3 was synthesized as a different phase in the reaction with Fe(III), which could be regarded as the reticular expansion of Fe2(BDP)3.79 The structure reported by Long's group is identical to this compound.114
2.2. Polynuclear metal cluster
In addition to the 1D chains/RBBs, a number of discrete high-symmetry polynuclear clusters formed, such as [Co4(μ4-O)(μ2-dmpz)6], (3,5-dmpz = 3,5-dimethylpyrazolate), structurally similar to those constructed by carboxylate ligands, Zn/Co(μ4-O)(COOH)6, as found in MOF-5.4,126,176 Meanwhile, there are many clusters structurally specific to pyrazolate MOFs, including the cubic octanuclear [Ni8(μ4-OH)6(pz)12]2− and [Co8(μ4-OH)6(pz)12]2− clusters, square tetranuclear M4(pz)8 cluster, and triangular trinuclear Cu3(μ3-X)(μ2-pz)3]n+ (X = OH−, O2−, halide, etc.) cluster.33,127,152,177With bulky substituents, the reaction between univalent coinage metal ions and pyrazolates tends to form larger, distorted rings such as tetranuclear saddle-shaped cycles.180 Alternatively, infinite zigzag chains/RBBs could be accessed without steric substituent groups. Reactions between Cu(II) salts and pyrazoles, in the presence of secondary ligands, typically result in trinuclear [Cu3(μ3-X)(μ2-pz)3]n+ complexes. These complexes feature a central μ3-X entity that connects three 4-coordinate Cu centers, with X representing OH−, O2−, halides, and others.177 Thompson and co-workers reported the heat-promoted preparation of a mixed-valence complex Cu(I)Cu(II)2(F6dmpz)5 (F6dmpz = 3,5-bis-(trifluoromethyl)-pyrazolate).181 In this compound, three Cu ions form a trigonal-planar Cu3 cluster, where a F6dmpz ligand bridges two adjacent Cu ions. The Cu(I) ion is linear 2-coordinate, while two Cu(II) ions additionally coordinate with another two F6dmpz ligands above and below the trigonal planar array, respectively.
Starting from an extended ligand of 1,3,5-tris(1H-pyrazol-4-yl)benzene (H3BTP), 1,3,5-tris (1H-pyrazol-4-yl)phenyl)benzene (H3BTPP), a mixed-valence 2D MPF, Cu-BTPP, Cu(I)2Cu(II)(OH)(BTPP) was obtained after temperature optimization.117 Three Cu ions form a trigonal-planar Cu3 cluster with a central μ3-OH entity, reminiscent of the Cu3(μ3-X)(μ2-pz)3]n+ (X = OH−, O2−, halide, etc.) complexes.177
In 2011, Long and co-workers constructed M3(BTP)2 (M = Ni and Cu) composed of the M4 cluster and tritopic pyrazole ligand H3BTP.33 M3(BTP)2 presents an expanded sodalite-like structure, analogous to the tetrazolate-based MOFs Mn3[(Mn4Cl)3(BTT)8]2 (Mn-BTT, H3BTT = 1,3,5-tris(2H-tetrazol-5-yl)benzene) and triazolate-based H3[(Cu4Cl)3(BTTri)8] (Cu-BTTri, H3BTTri = 1,3,5-tris(1H-1,2,3-triazol-5-yl)benzene).182,183 Mn-BTT was formed with chloride-centered 8-coordinate [Mn4(μ4-Cl)]7+ squares (in a cuboid coordination geometry) linked via triangular BTT3− ligands, and the anionic framework is balanced by [Mn(DMF)6]2+ cations included in the tetrahedral pores. Cu-BTTri is isostructural with Mn-BTT, whereas the anionic framework is balanced by protons. In contrast, M3(BTP)2 comprises of M4 squares, representing a neutral framework. These compounds share a the-a topology with accessible metal centers.
In 2020, Li and co-workers developed a mesoporous Ni(II)-based MPF (BUT-33) as the reticular expansion of Ni3(BTP)2 (Fig. 11).122 BUT-33 is composed of an elongated planar ligand, 2,4,6-tris(4-(pyrazolate-4-yl)phenyl)-1,3,5-triazine (TPTA3−) and square Ni4 cluster. The planar configuration of TPTA3− is critical for the reticular expansion of Ni3(BTP)2, which was not available with the non-planar extending 1,3,5-tris((1H-pyrazol-4-yl)phenyl)benzene (H3BTPP) ligand. BUT-32 has the same composition as BUT-33, but presents a two-fold network interpenetration, which was confirmed to be a thermodynamically stable product. In contrast, the kinetically favored BUT-33 is non-interpenetrated, which features preferred mesopores for catalysis. In the following year, the authors synthesized the first Pd(II)-azolate MOF, BUT-33(Pd), using a deuterated solvent-assisted metal metathesis approach.124 BUT-33(Pd) preserves the sodalite network and mesoporosity of the original BUT-33(Ni) template while exhibiting improved chemical stability and excellent catalytic activity due to the robust Pd(II)–pyrazolate bond and accessible Pd(II) centers. In 2024, Li and co-workers reported the access to BUT-124(Co), as the Co-based variant of M3(BTP)2. Comprising the Co4 cluster and BTP3− ligand, BUT-124(Co) was obtained in a two-step synthesis approach,121 by performing a post-synthetic metal metathesis in a template framework BUT-124(Cd).
 | ||
| Fig. 11 Images of (a) the cubic crystals of BUT-32 and (c) the microcrystalline powder of BUT-33. (b) Ni4pz8 cluster and TPTA3− ligand. Structure of (d) BUT-32 and (f) BUT-33. (e) The sodalite network (the-a) of BUT-32 and BUT-33.122 Reprinted with permission from American Chemical Society 2020. | ||
In addition to the 8-connected Co4 clusters, there were also 6-connected Co4O clusters discovered in MPFs. In 2009, Volkmer and co-workers constructed MFU-1 with the dmbdp2− ligand and Co4O unit.126 Isostructural with MOF-5 with a pcu topology, the MFU-1 network contains octahedral [Co4O(dmpz)6] clusters that are reminiscent of the [Zn4O(CO2)6] clusters in MOF-5.4 This is a rare sample based on [Co4O] units and pyrazolate linkers to date, against the big M(bdp) family amenable to elaborate reticular/crystal engineering contraction/expansion/functionalization/modification.
In 2010, Bordiga and co-workers integrated the cubic Ni(II) node with pyrazolate linkers to build MPFs.127 This marked the beginning of research prevalence in this family, as the pyrzolate counterparts of the famous carboxylate UiO series.185 Two pyrazolyl ligands, 2,6-di(1H-pyrazol-4-yl)pyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetraone (H2TET) and 4,4′-di(1H-pyrazol-4-yl)-1,1′-biphenyl (H2PBP) were designed to react with Ni(OAc)2, affording [Ni8(OH)4(OH2)2(pbp)6] [shortened as Ni8(pbp)6] and [Ni8(OH)4(OH2)2(tet)6] [shortened as Ni8(tet)6]. Although the authors claimed that the octanuclear node resembles that of the [Ni8(μ4-OH)6(μ2-pz)12]2− anion reported by Journaux and co-workers,184 this cluster had been defined as a neutral one [Ni8(OH)4(OH2)2(pz)12] (shortened as Ni8). Two new isostructural MPFs exhibit the fcu topology of the UiO series, but have different pore sizes and chemistry.
In 2013, Navarro and co-workers reported a series of isoreticular materials based on different pyrazole ligands: including two dual-functional pyrazole/carboxylic ligands, H2L1 = 1H-pyrazole-4-carboxylic acid and H2L2 = 4-(1H-pyrazole-4-yl)benzoic acid, and five di-pyrazole ligands, H2L3 = H2BDP, H2L4 = 4,4′-buta-1,3-diyne-1,4-diylbis(1H-pyrazole), H2L5= H2BPEB, and H2L5-R (R = methyl, trifluoromethyl).36 The combination of pyrazolate linkers and 12-connected Ni8 nodes yielded the series of Ni8(L)6 MPFs, which present highly porous 3D networks without interpenetration, therefore, allow for the accessibility of cavities (Fig. 12).
 | ||
| Fig. 12 (a) The crystal structure of Ni8(L5-CF3)6 is viewed as a combination of octahedral and tetrahedral cavities. (b) View of the tetrahedral (top) and octahedral (bottom) cages found in the crystal structures of Ni8(L3)6, Ni8(L4)6, and Ni8(L5)6, and the corresponding metric descriptors.36 Reprinted with permission from Wiley-VCH 2013. | ||
Following this work, they developed a sequential post-synthetic strategy to introduce missing-linker defects and extra-framework cations into Ni8(L)6 to tune their properties in gas adsorption and conductivity.45,130,132,134,140 Li and co-workers also introduced three different functional groups (CHO, CN, and COOH) onto the backbone of H2BDP to build the functionalized Ni8(BDP-X)6 (X = CHO, CN, and COOH) MOFs in 2017.136 These MPFs share the fcu network, but exhibit different pore chemistry with various functional groups. In 2018, Fei and co-workers grafted an amino group on the bdp2− ligand (2-amino-[1,4-bis(1H-pyrazol-4-yl)benzene], H2BDP-NH2).137 Ni8(BDP-NH2)6 was then synthesized, followed by post-synthetic mercaptoacetylation and Ag(I) anchoring, giving rise to the final product Ni8(BDP-NH2)6-AgS with high alkaline resistance and CO2 affinity.
In 2019, Li and co-workers obtained single crystals of six Ni8 MPFs under solvothermal conditions for the first time, including three Ni8(BDP-X)6 analogues: Ni8(BDP)6 (BUT-2), Ni8(BDP-CN)6 (BUT-3), Ni8(BDP-CHO)6 (BUT-4); and two disordered pcu MOFs with functional 1,3-BDP linkers, Ni8(1,3-BDP-CN)6 (BUT-48, 1,3-BDP-CN2− = 5-cyan-1,3-benzenedipyrozolate), Ni8(1,3-BDP-CF3)6 (BUT-49, 1,3-BDP-CF32− = 5-trifluoromethyl-1,3-benzenedipyrozolate), and one with a trigonal ligand, Ni8(BTPP)4 (BUT-123, BTPP3− = 1,3,5-tris((4-pyrozolate)phenyl)-benzene).139 BUT-123, featuring a 3-connected trigonal ligand and 12-connected Ni8 cluster, exhibits a new topology, which has not been observed in carboxylate MOFs yet due to unmatched symmetry.
In 2023, Li and co-workers extended the Ni8(BDP)6 family with three new ligands (4,7-di(1H-pyrazol-4-yl)benzo[c][1,2,5]thiadiazole, H2BT-Pz; 4,7-di(1H-pyrazol-4-yl)benzo[c][1,2,5]-selenadiazole, H2BS-Pz; 4,9-di(1H-pyrazol-4-yl)naphtho[2,3-c][1,2,5]selenadiazole, H2NS-Pz), where the central phenyl ring of H2BDP was replaced by benzothiadiazole, benzoselenadiazole, and naphthoselenadiazole, respectively.145 Four fcu MPFs were synthesized accordingly (JNU-212, JNU-213, JNU-214, and JNU-215), where JNU-212 represents Ni8(BDP)6 as the parent compound. The linker engineering by tuning the electron-accepting capacity of the pyrazole linkers was demonstrated to render these Ni8-based MPFs with enhanced charge separation and transfer efficiency under visible-light irradiation. Based on these materials, He and co-workers performed systematic research on employing cruciform pyrazolate ligands featuring pyrazole donors for framework construction, and the presence of bulk arms facilitated the formation of defects (missing linker on the node) in the resulting Ni8-MPFs.141–144
In 2016, Zhou and co-workers brought the Ni8 cluster to a tetra-pyrazolate MOF and constructed the first pyrazolate-porphyrinic MOF, PCN-601, which is composed of the Ni8 cluster and TPP4− ligand (Fig. 13).39 PCN-601 is isostructural with PCN-221, both of which are formed in the ftw-a topology. PCN-601 has small cage windows (∼2.1 × 8.0 Å after deducting van der Waals radii) and strong base-resistance in aqueous media. In the next year, they expanded this MPF by using a longer ligand.149 The 5,10,15,20-tetrakis(4-(1H-pyrazol-4-yl)-phenyl)porphyrin (H4TPPP) ligand was designed by inserting one phenyl between the central porphyrin cycle and peripheral pyrazolate groups in TPP4−. TPPP4−, as one of the shortest elongated versions of TPP4−, maintains the D4h symmetry. The resulting PCN-602 is isostructural with PCN-601 but shows greater pore dimensions (6.0 × 14.0 Å) without compromising the robustness of the framework.
 | ||
| Fig. 13 (a) Ftw-a topological net; (b) structure of PCN-601 and (c) PCN-602.149 Reprinted with permission from American Chemical Society 2017. | ||
Zhou and co-workers further functionalized H4TPPP to obtain a fluorinated ligand 5,10,15,20-tetrakis-(2,3,5,6-tetrafluoro-4-(1H-pyrazol-4-yl)phenyl)-porphyrin (TTFPPP).150 Based on this ligand, a perfluorophenylene functionalized metalloporphyrinic MPF, PCN-624, was synthesized. PCN-624 is isostructural with PCN-601 and PCN-602. Thanks to the pore surface of PCN-624 decorated with pendant perfluorophenylene groups, the framework stability was improved, and selective guest capture was endowed upon this material.
In 2024, Cao reported the synthesis and catalytic applications of two highly chemically stable isoreticular pyrazolate MOFs, FICN-18 and FICN-19.152 These MOFs are composed of 12-connected M8 (M = Ni and Co) clusters and linear metalloligands, forming two-fold interpenetrated structures. The Co8 cluster, Co8(OH)4(OH2)2(pz)12, is rare in MOFs. Li and co-workers subsequently reported four isostructural MOFs, fcu-L-Co, built from anionic the Co8 cluster, [Co8(OH)6(pz)12]2−, and four dipyrazolate ligands, including one recently reported ligand: H2DPP,40 and three new ligands: 3,6-di(1H-pyrazol-4-yl)pyridazine (2), 2,5-di(1H-pyrazol-4-yl)pyrazine (3), and 2,5-di(1H-pyrazol-4-yl)pyrimidine (4).153 These fcu-L-Co frameworks are able to spontaneously transform from Co(II)8 to Co(III)8 in the air, marking the first example of such behavior in MOFs.
In 2022, Cao and co-workers constructed a Zn9 MPF composed of an unprecedented [Zn9O2(OH)2(pyz)12] cluster and a Ni(salen)-based bis(pyrazolate) ligand.154 The Zn9 cluster consists of eight zinc atoms positioned at the vertexes of a cube, with an additional Zn atom sitting at its geometric center. The central zinc atom features a distorted tetrahedral ZnO4 coordination geometry, where the four oxygen atoms link eight peripheral zinc atoms in a μ3-O coordination mode. The geometry and connectivity of the Zn9 cluster are reminiscent of the Ni8/Co8 cluster, as well as the overall framework topology. In contrast, the distance between two adjacent vertex Zn atoms is 3.58 Å, longer than those reported in Ni8 (2.96–3.01 Å) and Co8 (3.02–3.11 Å) clusters, indicating the larger size of the Zn9 cluster. In addition, a two-fold interpenetration occurred in this MOF.
2.3. Unconventional types (Ni/Cu/Cu5/Cu12)
In addition to the typical frameworks assembled with commonly encountered chains/RBBs or clusters, there are some intriguing structures formed by unconventional building blocks.102,103,155,156 In 2016, Li and co-workers designed a tetra-pyrazole carbazole ligand 1,3,6,8-tetra(1H-pyrazol-4-yl)-9H-carbazole (H4CTP).155 Solvothermal reaction of this ligand and NiCl2·6H2O gave a 3D MOF, [Ni(H4CTP)Cl2] (BUT-41), which crystallized in the cubic space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) although H4CTP is in a low Cs symmetry. The framework of BUT-41 shows a rhr topology when both the organic ligand and the single metal center are considered as four-connected nodes. Nanocages are enclosed by interconnecting 12 H4CTP ligands and 20 Ni(II) ions. Each nanocage connects with six adjacent ones through sharing hexagonal windows, resulting in 3D intersecting channels. In 2024, Zhou and co-workers reported a novel MOF named PCN-300, which is constructed from the H4TPPP ligand and [CuPz4Cl2].163 The framework adopts a layered architecture and forms 1D open channels.
although H4CTP is in a low Cs symmetry. The framework of BUT-41 shows a rhr topology when both the organic ligand and the single metal center are considered as four-connected nodes. Nanocages are enclosed by interconnecting 12 H4CTP ligands and 20 Ni(II) ions. Each nanocage connects with six adjacent ones through sharing hexagonal windows, resulting in 3D intersecting channels. In 2024, Zhou and co-workers reported a novel MOF named PCN-300, which is constructed from the H4TPPP ligand and [CuPz4Cl2].163 The framework adopts a layered architecture and forms 1D open channels.
In 2017, Volkmer and co-workers synthesized a fluorinated linker 1,4-bis(3,5-bis(trifluoromethyl)-1H-pyrazole-4-yl)benzene (H2tfpb) and the Cu(I)-based MPF family M[CFA-4], M[Cu5(tfpb)3], (M = Cu(I), K(I), Cs(I), Ca(0.5)).162 Cu(I)[CFA-4] features a 3D framework composed of an unusual pentanuclear [Cu(I)5pz6]− (Cu5) cluster. The Cu5 unit consists of two distorted trigonal-planar coordinated copper(I) ions, and three linear 2-coordinate copper(I) ions. Five Cu(I) ions are connected by twelve nitrogen atoms from six pyrazolate moieties of different tfpb2− ligands. M[CFA-4] represents the first MOF family in which the copper(I)-based Cu5 cluster was observed, showing a new chiral topology thanks to its distinct symmetry. The Cu5 clusters of the M[CFA-4] family are interconnected by the tfpb2− ligand, generating 1D channels along the c-axis direction. In the as-synthesized Cu(I)[CFA-4], the channels are filled with disordered DMF molecules and Cu(I) ions, which balance the framework's negative charge. The latter could be exchanged with various metal ions [K(I), Cs(I), Ca(II)] to get the substituted M[CFA-4] compounds via post-synthetic cation substitution.
In 2018, Xiang and co-workers reported a hierarchical pyrazolate framework MROF-12, which was constructed from an elongated rigid ligand (H2NDI) and a metal–ligand ring cluster [Cu12(μ2-OH)12(dmpz)12] (dmpz = 3,5-dimethyl-pyrazole, shortened as Cu12).156 Adjacent Cu(II) ions are linked by one bidentate bridging dmpz moiety and one μ2-OH group. Twelve Cu ions form a puckered cyclic Cu12 cluster, where the μ2-OH group resides on the inner surface, while dmpz is located on the outer surface. Each cluster is connected to 12 neighboring clusters through NDI2− linkers to produce two types of polyhedral cages: dodecahedron and octahedron cages. The interconnection of two types of polyhedral cages extends to form a 3D framework, generating three different channels.
We discussed above representative examples to show the present scope of high-dimensional MPFs constructed by adopting or beyond the crystal engineering/reticular chemistry principles. It is inevitable to miss some publications out due to having a different focus. The enormous library of MPFs has contributed great structure diversity, which provides opportunities for inducting the general rules of stable compounds synthesis and exploring their properties.
2.4. Synthesis of pyrazolate-based MOFs
Coincident with the inherent high stability of MPFs, researchers have encountered great difficulties in their general synthesis and structure determination.139,186 Unlike the well-developed synthetic protocols for carboxylate MOFs, such as the acid-modulated solvethermal synthesis to harvest large high-quality single crystals, it is still harder to get single crystals of MPFs to date, especially for those involving multinuclear clusters.26,122,139 Possible reasons include the short bridging length and small bending angle of the N⋯NH moiety, the difficult deprotonation of the pyrazolate group, and the easy precipitation of insoluble and amorphous products due to the formation of strong M–N coordination bond. As a result, their structure analysis and prediction are facing great challenges.Nonetheless, some design strategies and synthetic approaches have been established and some single crystals of MPFs have been harvested, offering pivotal references for the discovery of new compounds.35,59,122,139 In this section, we highlight some established synthetic strategies, which are frequently implemented to construct MPFs, including methods for the direct synthesis of powder samples and single crystals, isoreticular expansion, topology-guided design (crystal engineering design), and known structure functionalization/modification.
H2BDP is a simple and popular pyrazole linker, which has been used for the synthesis of a great number of classical MPFs, while most of them have only been obtained as powder samples. The flexible and stable MOFs, Ni(bdp) and Zn(bdp), were harvested as microcrystalline powders.62 When a solution of Ni(CH3COO)2 was refluxed with H2BDP in acetonitrile in the presence of Et3N, Ni(bdp) was separated as a dark orange precipitate. Zn(bdp) is a white product obtained by reacting H2BDP in benzonitrile, also in the presence of Et3N. Similarly, microcrystalline powders of Fe2(BDP)3 and Fe(II)(bppdi)(DMF)0.5 were available from solvothermal reactions between Fe(II)/Fe(III) salts and pyrazole ligands in DMF.37,109
Another classic example is Ni8(BDP)6, which was prepared from the reaction between Ni(OAc)2·4H2O and H2BDP in mixed DMF and H2O.36 Noteworthy, extending/prolonging the H2BDP ligand can improve the porosity of MOF, but also reduce the solubility and affect the crystallinity.36,127 To facilitate the precipitation of powders with high crystallinity, boc-protection of pyrazole ligands was leveraged in the case of extended bipyrazole linkers (Boc2L4, Boc2L5-R; R = H, CH3, CF3; Boc = tert-butoxycarbonyl), which favor the slow release of ligands in the reaction system and enhance sample quality.
Based on the tri-topic H3BTP ligand, Ni3(BTP)2 and Cu3(BTP)2, as yellow and brown microcrystalline powders, were obtained from the reaction of Ni(OAc)2·4H2O or Cu(OAc)2·H2O in DMF at 160 °C.33 They feature sodalite-like frameworks with accessible open metal sites. To access pore-expanding analogs of the M3(BTP)2 type, Long and co-workers synthesized the prolonged ligand H3BTPP by inserting one phenyl group between the central phenyl ring and the peripheral pyrazolate groups.117 Both Ni-BTPP and Cu-BTPP precipitated in high yields as air-stable powders by reacting H3BTPP with metal salts. The diffraction patterns indicate that the two structures are not isostructural nor isomorphous with M3(BTP)2. In 2020, Li and co-workers synthesized another extended ligand, 2,4,6-tris(4-(pyrazol-4-yl)phenyl)-1,3,5-triazine (H3TPTA), with a non-steric triazine core.122 The conventional reaction of Ni(OAc)2·4H2O and H3TPTA under heating at 150 °C and stirring for 2 h yielded a yellow microcrystalline powder of BUT-33. c-RED187 was applied to determine the structure of BUT-33, which is isostructural with Ni3(BTP)2 and the desired mesopores (Fig. 14).
 | ||
| Fig. 14 3D reciprocal lattice presenting the (a) hk0 and (b) hhl planes of the BUT-33 crystal from c-RED analysis, and (c) Pawley refinements of BUT-33.122 Reprinted with permission from American Chemical Society 2020. | ||
Although the authors were unable to obtain single crystals of Fe(II)(bppdi)(DMF)0.5 suitable for X-ray diffraction as aforementioned above, the framework structure has been established from single crystals of an oxidized Fe(III) analogue, Fe(III)(bppdi)(OH).109 In an N2-filled glovebox, a borosilicate glass tube was charged with Fe(OTf)2(THF)2, H2bppdi, DMF, as well as one drop of Et3N. The tube was then sealed with a glass stopcock adapter, degassed, and flame-sealed under a static vacuum. After heating the sealed tube at 170 °C for 2 days, a few small red crystals of Fe(III)(bppdi)(OH) were obtained, offering great opportunities for the structure determination of this MPF. CFA-2 was also synthesized as colorless crystals by heating a DEF–MeOH solution of H2phbpz and Cu(OAc)2·H2O in the presence of Et3N, which is necessary for deprotonating the ligand.116 Mn-CFA-6 and Fe-CFA-6 were also synthesized from solvothermal reactions between trivalent metal salts and the H2BPZ ligand.94,173 For Mn-CFA-6, H2BPZ, N,N-dimethylacetamide (DMA), and 0.1 mL of 2,6-lutidine were mixed in a tube, which was capped and heated at 130 °C for three days. Fe-CFA-6 crystals were obtained by adjusting the molar ratios of the ligand, metal salts, and base in a DMA solution.
The above reactions were performed with the assistance of trace base modulators. In some cases, acid modulators could also help get single crystals of MPFs. Single crystals of Fe2(BDP)3 were obtained by modifying the reported procedure for the synthesis of microcrystalline powder, where an acidic modulator was incorporated to increase the crystallite size.113 Reaction of Fe(acac)3 with H2BDP and acetylacetone in DMF produced dark yellow, needle-like crystals.
The Long group harvested single crystals of Co(bdp) by reacting Co(CF3SO3)2 and H2BDP in DEF at 130 °C for 4 days under air-free conditions along with programmable heating and cooling.35 Volkmer's group obtained a similar crystal, named MFU-3, under different conditions: the ligand reacting with Co(NO3)2·6H2O in DMF at 120 °C for 3 days, where a trace amount of diluted HCl aqueous solution was used as a modulator.59 In 2022, Li's group further modified the synthesis of Co(bdp): reacting the H2bdp ligand with Co(NO3)2·6H2O in DMF at 130 °C, using deionized water as a modulator.40 This method yielded single crystals suitable for SCXRD within 12 hours. Double-walled Co(bdp) analogous crystals of BUT-53 to BUT-58 were also harvested from solvothermal reactions using a DMF and water-mixed solvent in the temperature range of 100–150 °C.40 Among these compounds, crystals of BUT-53, BUT-54 and BUT-57 were accessed with trace acetic acid as the modulator; otherwise, only tiny microcrystals were available. The double-walled isostructural framework, JNU-204, was synthesized as orange prismatic crystals via a solvothermal reaction in DMF and water at 120 °C for 72 h, where HNO3 was added as the modulator.106 In these syntheses, the addition of acidic modulators alters the pH value of the reaction systems, resembling their role in the reaction with carboxylic ligands, including inhibiting the deprotonation of pyrazole ligands and slowing down the nucleation rate. This favors their competitive coordination with the metal centers, contributing to the growth of large crystals.
Preparing single crystals for polynuclear cluster-based MPFs (such as Ni8 MOFs) has consistently been challenging, and the Li group addressed this issue in 2019. Single crystals of Ni8 MPFs (BUT-2 to BUT-4, BUT-48, BUT-49, and BUT-123) were obtained from the solvothermal reaction between Ni(NO3)2·6H2O and respective ligands in a mixed solvent of DMF or DMA and H2O (Fig. 15).139 The formation and size of single crystals were significantly influenced by the metal-to-ligand ratio, solvent composition (particularly water content), and the reaction time, all of which also impacted crystal quality and yield. Similarly, a solvothermal reaction involving H3TPTA and Ni(NO3)2·6H2O in a mixture of DMF and H2O at 150 °C for 72 h, with a small amount of acetic acid as the modulator, yielded yellow cubic crystals of BUT-32.122 They present the first Ni8-/Ni4-based MPFs whose structures have been unambiguously identified by SXRD.
 | ||
| Fig. 15 Optical microscopy images of six Ni8 MOF single crystals. The radius of the crystal centering cycle is 0.1 mm.139 Reprinted with permission from American Chemical Society 2019. | ||
These successful examples are evidence that the synthesis of MPF crystals is highly sensitive to synthetic conditions, particularly for growing single crystals suitable for SCXRD. Subtle variations in synthetic parameters can significantly affect the quality of the resulting MPF crystals and even alter the product species. A wide range of modulators, including weak acids and bases such as acetic acid, Et3N, ammonia solution and 2,6-lutidine, as well as some strong acids such as HCl, HNO3, and trifluoroacetic acid, and even neutral water, would make a difference in yielding good crystals by facilitating the deprotonation of pyrazole ligands and regulating the reaction equilibrium through competitive coordination.
The development of solidate-type MPFs, including M3(BTP)2, BUT-33, and BUT-124(Co), demonstrates the power of these principles in varying the pore size and composition.33,121,122 In 2011, Long and co-workers constructed M3(BTP)2 (M = Ni, Cu) with M4 clusters and tritopic pyrazole ligand H3BTP, creating a sodalite-like structure with accessible metal centers.33 In 2020, Li and co-workers obtained the mesoporous analog BUT-33 using the elongated planar ligand TPTA3−, where the active sites are more accessible than in the microporous M3(BTP)2.122 In 2024, they synthesized BUT-124(Co) from the parent framework, BUT-124(Cd), via post-synthetic metal metathesis.121 The M3(BTP)2 family then has great variety in terms of nodal metal species, including Ni, Cu, Cd and Co.
Based on the first discovery of Ni8 MPFs, Ni8(pbp)6 and Ni8(tet)6,127 Navarro and co-workers expanded this type of structure by using pyrazolate ligands with various functionalities and lengths, to synthesize a series of isoreticular MPFs Ni8(L)6.36 These MPFs exhibit highly porous, non-interpenetrated 3D networks, maintaining accessible pores with different size and chemical environments.
As a continuation of their interest in constructing pyrazolate-porphyrinic MPFs, Zhou and co-workers constructed PCN-601 composed of the Ni8 cluster and H4TPP ligand.39 In 2017, they expanded it to PCN-602 by using an elongated ligand H4TPPP, creating larger pores suitable for catalysis under basic conditions.149 Using a fluorinated ligand, TTFPPP, they further developed PCN-624, which maintains the structure robustness of its parent framework PCN-602 and exhibits an inner pore surface covered with fluorine (Fig. 16).150 All three MOFs share the ftw-a topology, highlighting the strength of crystal engineering/reticular chemistry principles in enriching the diversity of the MOF community, varying chemical composition and pore dimension, and attaching functionalities for specific purposes.
 | ||
| Fig. 16 (a) Ftw-a topological network. (b) Construction of PCN-602. (c) Synthesis and structure of PCN-624.150 Reprinted with permission from American Chemical Society 2018. | ||
A tandem post-synthetic modification approach was employed to build the first Ag(I)-functionalized MPF.137 To introduce thiol functionality, the microcrystalline Ni8(BDP-NH2)6 was incubated with mercaptoacetic acid in a CH2Cl2 solution at room temperature. The covalently modified MOF was then treated with an aqueous solution of 6 mM AgNO3 to anchor Ag(I) sites. This resulting MOF catalyst exhibited high activity and efficiency in the cyclic carboxylation of propargyl amines and CO2 under ambient conditions, due to the incorporation of Ag(I) centers.
An ideal crystal should have good structural ordering/periodicity, but thermodynamics favor deviations from this order, creating “intrinsic defects” that impact various properties of solid-state materials.188,189 Navarro and co-workers investigated the effect of post-synthetic introduction of missing linker defects in Ni8(OH)5(EtO)(H2O)2(BDP-X)6 (X = H, OH, NH2) on their performance in SO2/CO2 sorption.130 Treating MOFs with ethanolic KOH solutions deprotonated coordinated water and hydroxo tags, created defects, and added extra-framework potassium ions, yielding 1@KOH, 2@KOH, and 3@KOH (Fig. 17). The treated materials have higher SO2 adsorption capacities and SO2/CO2 partition coefficients. The countering potassium ions can be further exchangeable with Ba and Cu, and the resulting materials behave differently against their K-based analogs.132,134
 | ||
| Fig. 17 (a) Schematic representation of the successive PSMs, from pristine nickel pyrazolate frameworks to the missing linker defective 1@KOH, 2@KOH 3@KOH and subsequently, the ion-exchanged 1@Ba(OH)2, 2@Ba(OH)2), and 3@Ba(OH)2. (b) Overview of the structures of the studied materials. Reprinted with permission from Springer Nature 2017. | ||
To access the completely reduced A2Fe2(BDP)3 (A = Li+, Na+, K+) compounds from the parent Fe2(BDP)3, single-crystal-to-single-crystal reductions were performed using lithium, sodium, or potassium naphthalenide.113 THF solutions of Na(C10H8) or K(C10H8) were slowly added to Fe2(BDP)3 crystals in THF, with excess naphthalenide to address the possible issues of incomplete solvent exchange and uncertainties in the formula unit. Single crystals of A2Fe2(BDP)3·yTHF were obtained after suspensions were stored for one to two months. For the preparation of Li2Fe2(BDP)3·yTHF, the direct reduction did not afford reduced single crystals suitable for diffraction. Alternatively, Fe2(BDP)3 crystals on a fritted filter were suspended in a vial containing a slurry of microcrystalline Fe2(BDP)3 stirred in a THF solution of lithium naphthalenide. Crystals of Li2Fe2(BDP)3·yTHF could be isolated over half to one month.
Developing MOF-based noble metal catalysts typically involves incorporating metal nanoparticles or anchoring metal-containing molecules onto the framework, which can narrow the pore apertures and limit mass transfer, impairing catalytic performance.16 Uneven dispersion of catalysts may also lower activity. Constructing MOFs with noble metal nodes ensures a high density of uniformly distributed metal sites without compromising porosity. The similar square planar coordination of Pd(II) and Ni(II) allows Pd(II) to replace Ni(II) in BUT-33.124 A six-day reaction in deuterated chloroform at 60 °C achieved this metathesis, resulting in the mesoporous Pd-MPF, BUT-33(Pd), with preserved crystallinity. BUT-33(Pd) retains the structure of BUT-33, demonstrates excellent chemical stability, and performs well as a heterogeneous Pd(II) catalyst.
3. Stability of pyrazolate MOFs
MOFs incorporating pyrazolate ligands with late transition metals generally exhibit high chemical robustness, attributed to the strong donating properties of pyrazolates.47 Various pyrazolate ligands have been used to build highly base-resistant materials, including PCN-601, Ni3(BTP)2, and BUT-33.33,122,149 These MPFs demonstrate significantly better stability in basic environments compared to carboxylate MOFs, which are able to withstand even saturated NaOH solutions at 100 °C. This underscores the power of pyrazolate ligands in the rational construction of base-stable MOFs. Here we introduce some typical examples and analyze the factors that influence their stability.3.1. Base-stable pyrazolate MOFs
The group and the Navarro group reported MOFs featuring the linear bipyrazole ligand H2BDP, such as Fe2(BDP)3, Co(bdp), Ni(bdp), and Zn(bdp).33,35,37,38,62 These MOFs contain linear chains of metal ions with varying coordination geometries, which affect not only the structural flexibility but also framework stability. Fe2(BDP)3, Ni(bdp), and Zn(bdp) are robust in alkaline aqueous solutions, among which Fe2(BDP)3 can notably maintain integrity in saturated NaOH.33,37,62 However, Co(bdp) is not that stable, whose handling needs to be done in a glove box.38 The isomer of Co(bdp), the double-walled BUT-55, as well as the whole BUT-55 series, BUT-53 to BUT-58, has helical RBBs of Co(II) ions linked by BDP2− ligands and shows stability in pH 14 NaOH for 24 hours and maintains integrity in the air after a year.40Featuring cubic M4Pz8 clusters connected to triangular planar ligands, sodalite-type MPFs, M3(BTP)2 (M = Ni2+ and Cu2+) demonstrate exceptional stability in boiling water and alkaline solutions. For instance, Ni3(BTP)2 retains its crystalline structure in saturated NaOH.33 The extended version of Ni3(BTP)2, BUT-33, is a rare example of mesoporous pyrazolate MOFs.122 Both BUT-33 and its interpenetrated microporous analog BUT-32 exhibit excellent resistance to highly basic solutions with a pH of 14.
The 12-connected Ni8/Co8 clusters are prominent inorganic nodes in base-stable pyrazolate MOFs. Integrating Ni8/Co8 nodes with dipyrazolate linkers, the typical M8(L)6 family shares the fcu topology but exhibits different pore sizes and functionalities.36,127,139 Stability tests confirmed that these MOFs maintain their structure in NaOH aqueous solution, demonstrating excellent base resistance. In 2016, Zhou, Li, and co-workers developed PCN-601, the most base-stable pyrazolate-based MOF, using a top-down approach.39 When the Ni8 clusters meet tetratopic ligands, PCN-601 and PCN-602 were synthesized with the ftw-a topology (Fig. 18a and b), isostructural with MOF-525 (or PCN-221).190,191 PCN-601 demonstrated exceptional stability in 20 M NaOH at 100 °C, highlighting its robustness under concentrated alkaline conditions. Although PCN-602 with larger pores is somewhat less resistant to bases compared to PCN-601, it maintains structural integrity in 1 M NaOH and remains stable in 1 M solutions of KF, Na2CO3, and K3PO4, which, as coordinating anions, can easily disintegrate MOFs (Fig. 18c and d).149
 | ||
| Fig. 18 (a) Image of PCN-601 samples being treated with 0.1 mM HCl solution and saturated NaOH for 24 h, respectively. (b) UV-vis spectra of the samples in (a). (c) PXRD patterns and (d) N2 adsorption/desorption isotherms of PCN-602 and of those treated in different aqueous solutions.39,149 Reprinted with permission from American Chemical Society 2016 and 2017. | ||
3.2. Factors contributing to the stability of pyrazolate MOFs
Similar to the whole MOF community, the stability of MPFs can be affected by multiple factors, including the strength of the coordination bond, connectivity of building blocks, length and rigidity of ligands, hydrophobicity of the pore surface, the operating environment, etc.A strong coordination bond forms the thermodynamic foundation for the stability of MOFs, generally providing these materials with high resistance to various competing entities.19 The strength of coordination bonds is influenced by the charge density of the metal ions.20 Higher metal ion charges and smaller ionic radii contribute to greater charge density, thereby increasing metal–ligand bond strength. This concept aligns with the HSAB principle and is supported by extensive research in MOF chemistry.20,192 According to the HSAB principle, stable MPFs can be constructed using a combination of soft azolate ligands (such as imidazolates, pyrazolates, and triazolates) and soft divalent metal ions (like Zn2+, Cu2+, Ni2+, Mn2+, and Ag+). The metal–nitrogen (M–N) bonds typically exhibit greater resistance to bases compared to the M–O bonds formed in carboxylate MOFs.47 Such enhanced base resistance is due to the stronger orbital interactions between nitrogen donors in azolates and the metal ions. Increasing the donor strength of the ligand, which is measured by its basicity, further strengthens the metal–ligand bonds. Among azolaes, pyrazolates exhibit the highest pKa values, i.e. the highest basicity, promising to form strong coordination bonds. This effect is particularly pronounced with late transition metals, leading to improved framework stability.
However, the stability of MPFs is not solely determined by the strength of the M–N bonds. The thermodynamic stability of the metal hydroxides formed in alkaline conditions also plays a significant role.47 The pKsp values of various metal hydroxides, which indicate the metal ions’ affinity for hydroxide ions (OH−), are critical in this context. Higher pKsp values imply a stronger affinity for OH−, making MOFs with such metal ions more susceptible to degradation in alkaline environments. Late transition metals, Co2+, Ni2+, Cu2+, and Zn2+, have a lower affinity toward OH−.47 Therefore, MOFs incorporating pyrazolate ligands and late transition metals often exhibit exceptional base stability. This stability is attributed to both the strong metal–ligand interactions and the lower susceptibility of these bonds in competition with OH− in highly alkaline conditions.
The kinetic inertness of MOFs is also a crucial indicator of their utility in practical applications, which is primarily influenced by the activation energy required to break the coordination bonds.46 High kinetic inertness means that MOFs can resist decomposition under moderate conditions with less energy input, such as lower temperatures or less extreme chemical environments. In kinetics, this is related to the ligand exchange rates of the metal ions (Fig. 19).46 For example, even though Pd and Ni have similar charge–radius ratios and affinities for hydroxide anions, Pd-MPFs exhibit slower ligand exchange rates compared to Ni-MOFs. Experimental data show that Pd-MPFs are more inert than their Ni-based counterparts.124 Similarly, Co-MPFs display higher kinetic inertness compared to Cd-MPFs of similar structures.121 In addition to ligand exchange rates, other structural factors also affect the kinetic inertness of MOFs, which will be explored further in the next section.
 | ||
| Fig. 19 Water exchange rate of metal ions.46 Reprinted with permission from Springer Nature 2019. | ||
3.2.2.1. Connectivity of building units. Higher connectivity in MPFs, whether from organic ligands or metal nodes, enhances structural stability by facilitating the repair of defects and preventing further decomposition.20 This principle helps explain the remarkable chemical stability of PCN-601, which features a ftw-a network.39 Both the Ni8 cluster (12-connected node) and the porphyrinic ligand TPP4− (4-connected linker) exhibit relatively high connectivity compared to other inorganic clusters and organic ligands. From a kinetic standpoint, MPF decomposition in solution can be viewed as a process where coordination moieties are replaced by small molecules or ions, leading to the formation and accumulation of structural defects. Higher connectivity in the framework helps suppress ligand dissociation and enhances the rate of ligand association, which promotes faster repair of these defects. This effect, analogous to the chelating effect observed with multi-dentate ligands, is referred to as the 3D chelating effect.193 The same principles apply to highly connected metal nodes, reinforcing the stability of MPFs. Consequently, MPFs with more interconnected fragments are significantly more stable.
3.2.2.2. Length and rigidity of ligands. In addition to the 3D chelating effect, the length and rigidity of ligands play a significant role in determining the stability of MOF materials. PCN-601 demonstrated that the length and rigidity of the ligand are associated with the activation energy required for ligand dissociation, impacting the overall decomposition of the MPF.39 As illustrated in Fig. 20, two isoreticular frameworks are compared, one constructed with a long ligand and the other with a short one. From a kinetic perspective, the decomposition of MPFs requires activation energy. Whether the substitution reactions proceed through a dissociative or associative pathway, metal ions or clusters and ligands undergo displacement in the transition states due to the addition of molecules or anions to the metal coordination sphere. For MPFs with isostructures, we can reasonably assume that the displacement of ligand coordination terminals remains consistent during these processes (ds = dl). However, shorter ligands will bend more significantly in the transition state (with a greater bending angle θs > θl), resulting in a higher energy barrier and making decomposition kinetically more challenging. Consequently, MPFs with shorter ligands exhibit greater chemical stability and higher kinetic inertness under alkaline conditions, a trend confirmed by experimental data. For instance, PCN-601 and Ni3(BTP)2, which have shorter ligands, retain their structural integrity even in saturated NaOH solutions.33,39 In contrast, their reticular expansions PCN-602 and BUT33, which feature elongated ligands, completely decompose when exposed to the same conditions.122,149 These findings demonstrate that shorter rigid ligands enhance the chemical stability of MOFs, especially in basic environments.
 | ||
| Fig. 20 Illustration of the kinetic stability of MOFs with ligands of different lengths.39 Reprinted with permission from American Chemical Society 2016. | ||
3.2.2.3. Hydrophobic groups. Modifying MOFs’ surfaces to increase their hydrophobicity has proven to be an effective strategy for enhancing their stability in basic environments.194 In 2011, Volkmer and colleagues synthesized the Co(bdp) analog, MFU-2, utilizing the four-methyl functionalized dmbdp ligand.77 Unlike the flexible Co(bdp), MFU-2 exhibits a rigid framework, characterized by a type I gas sorption isotherm and enhanced stability. Introducing hydrophobic groups, such as methyl groups, makes the materials less accessible to water molecules and hydroxide ions (OH−), which are common sources of framework degradation in aqueous solutions.19,20,126 The presence of methyl groups has been shown to increase the relative pressure of water vapor condensation within the frameworks, demonstrating their impact on hydrophobicity. Consequently, these modifications reduce the probability that vulnerable sites within the MOFs are exposed to destructive species, leading to greater kinetic inertness and improved stability under basic conditions.
4. Applications of pyrazolate MOFs
Thanks to their customizable structures, expansive surface areas, adjustable pore sizes, and exceptional stability, MOFs have been extensively researched and have demonstrated remarkable performance across various applications. This section will summarize the application of MPFs in gas adsorption and separation, heterocatalysis, sensing, biomedicine, etc.4.1. Gas adsorption/separation
Gas adsorption and separation have garnered significant interest among chemists and engineers due to their low energy requirements in addressing social, environmental, and industrial challenges.11,13,17,27,195 MOFs, with their adjustable porous structures and large specific surface areas, are ideal for molecular adsorption and separation. MPFs, in particular, have been extensively studied for these applications, demonstrating excellent performance. They have been applied in the adsorption and separation of hydrocarbons, the removal of harmful gases from the air (such as SO2 and VOCs), and the storage of energy-related gases like CH4 and H2.37,38,40,62,65,113,114,132Navarro et al. demonstrated that the Ni8(L)6 series can serve as sorbents for the capture of warfare agents. Their performance is influenced by the pore size and surface polarity, which are determined by the length and functionality of the linkers.36 Analysis of the adsorbate phase after the adsorption of diethylsulfide (DES) in 80% RH humid streams showed that only Ni8(L5-CF3)6 can capture DES in high efficiency. Ni8(L5-CF3)6 also outperforms the hydrophobic activated carbon Blücher-101408, demonstrating the superiority of the Ni8(L)6 family in DES capture. Furthermore, Navarro et al. introduced a defect engineering strategy that deliberately incorporated missing-linker defects and uncoordinated extra-framework Ba2+ into the Ni8-MPF, resulting in 1@Ba(OH)2, 2@Ba(OH)2, and 3@Ba(OH)2 to enhance SO2 adsorption.132 These modified materials exhibited high SO2 adsorption capacities at 303 K and low SO2 partial pressures (0.025 bar). Dynamic adsorption experiments revealed that these treatments enhanced SO2 capture capacity by increasing the basicity of the nickel hydroxide clusters and introducing more amino and hydroxyl functional groups on the linkers, which are binding sites of SO2, as well as by the formation of stable MSO3 (M = Ba, 2K) species near defect sites.
Earlier work by Navarro et al. also reported that Ni(bdp) can be used to capture benzene and cyclohexane.62 This material also demonstrated the ability to capture tetrahydrothiophene (up to 0.34 g per gram of material) from CH4–CO2 mixtures under dynamic conditions, even in the presence of 60% humidity. However, current regulations on the low concentration of aromatic-based VOCs in indoor air and industrial effluent streams have increased the demand for structurally designed MOF-based adsorbents, which need to show enhanced binding strength to capture VOCs at trace levels.196–198 In this regard, the 1D channel apertures in the double-walled MPF family (BUT-53–58) were fine-tuned by modifying the linker length and geometry, resulting in channels ranging from 7.8 to 11 Å. These MOFs demonstrated high benzene uptakes at 298 K ranging from 2.47 to 3.28 mmol g−1 at pressures below 10 Pa (Fig. 21). Notably, with an inlet concentration of 10 ppm and a flow rate of 10 mL min−1, BUT-55 achieves a breakthrough time of approximately 8000 h g−1. The benzene residual in the effluent streams was reduced to 2.82 ng L−1, well below the indoor air limits of 3–100 μg m−3. SCXRD analysis revealed that the adsorption mechanism for benzene binding in BUT-55 involves C–H⋯N/C–H⋯π interactions within the cavities and C–H⋯N, C–H⋯π, and π⋯π interactions within the channels. Among this family, BUT-55 shows the highest adsorption capacity at pressures below 0.01 kPa, significantly surpassing the performance of the best carbon-based materials like carboxen for capturing aromatic-based VOCs. This kind of performance, combined with facile regeneration and good recyclability, makes BUT-55 a promising adsorbent for removing aromatic VOCs from indoor air.
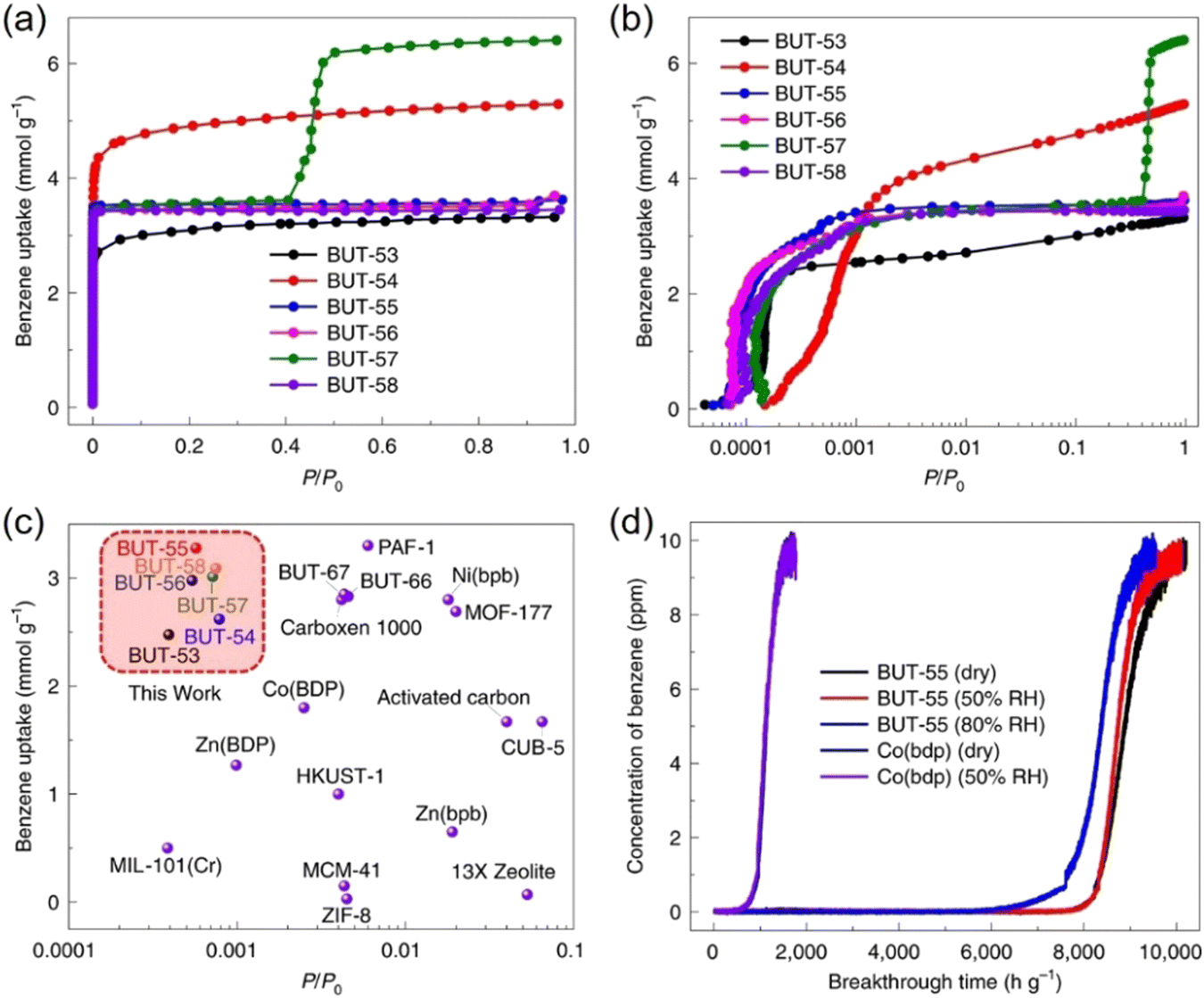 | ||
| Fig. 21 Benzene static isotherms of BUT-53 to BUT-58: (a) linear-scale plots and (b) logarithmic-scale plots of P/P0. (c) Comparison of benzene uptakes with various representative porous sorbents at low relative pressures. (d) Breakthrough curves for BUT-55 and Co(bdp).40 Reprinted with permission from Springer Nature 2022. | ||
To achieve high CH4 working capacities in MOFs for onboard storage applications, maximizing CH4 uptake at an adsorption pressure of 65 bar while minimizing uptake at desorption pressures of 5.0/5.8 bar is crucial. The Long group demonstrated high CH4 working capacity in two flexible MOFs, Fe(bdp) and Co(bdp), and effectively regulated internal heat during both adsorption and desorption processes.38 Specifically, these two flexible MOFs undergo a structural phase transition at certain CH4 pressures, resulting in adsorption and desorption isotherms with a distinct ‘step’. This behavior enables these materials to achieve greater storage capacities than traditional adsorbents. Notably, Co(bdp) has a record-high CH4 working capacity at 25 °C, with 155 cm3 STP cm−3 (v/v) at 35 bar and 197 v/v at 65 bar by then. This capacity is achieved by coupling with the heat offset resulting from the structural transformation, where the endothermic expansion reduces the heat released during adsorption and the exothermic contraction minimizes the cooling effect during desorption. In 2016, the same group further regulated the CH4-induced framework opening pressure by systematic ligand functionalization.61 The resulting frameworks, functionalized Co(bdp), are isoreticular to the parent structure, Co(bdp), and exhibit similar phase transition behaviors. Mechanism studies reveal that the fluorine atoms on the aryl ring disrupt edge-to-face π–π interactions, which help stabilize the collapsed phase and induce framework opening to occur at lower pressure, while deuterium atoms preserve these interactions and methyl groups strengthen them, resulting in framework opening at the highest pressure. This work provides a powerful tool to optimize the functionality of flexible MOFs for better CH4 storage performance.
The alkali cations of AxFe2(BDP)3 (A = Li+, Na+, K+; 0 < x ≤ 2) reside in proximity to the framework after activation, and the Long group studied how the surroundings affect the Lewis acidity of these cations using H2 as a probe molecule.113 Microcrystalline samples of LixFe2(BDP)3 (x = 1.18, 1.90), NaxFe2(BDP)3 (x = 1.14, 2.06), and KxFe2(BDP)3 (x = 0.68, 1.33) were synthesized through the stoichiometric reduction of Fe2(BDP)3 using lithium, sodium, or potassium naphthalenide. By varying the cation loadings, they assessed the effect of cation density on H2 uptake. The hydrogen adsorption results demonstrated that the reduced frameworks generally have lower adsorption capacities than Fe2(BDP)3, due to the partially quenched charge density by interactions with the linkers and iron–pyrazolate chains. Interestingly, Mg0.85Fe2(BDP)3 was obtained through reductive insertion with magnesium anthracene, incorporating charge-dense Mg2+ cations and endowing substantially improved H2 uptake and affinity compared to Fe2(BDP)3.
Meanwhile, they uncovered that AxFe2(BDP)3 (A = Na+, K+; 0 < x ≤ 2), which feature coordinatively saturated iron centers, could strongly and selectively adsorb O2 over N2 at ambient (25 °C) or even elevated (200 °C) temperatures, presenting a potential O2-selective adsorbent to produce high-purity oxygen from the air.114 A selective adsorption mechanism was proposed, involving outer-sphere electron transfer from the framework to generate superoxide species, which are stabilized by alkali metal cations within the 1D triangular channels. They also observed similar O2 adsorption behavior in an expanded-pore framework analogue, which supports the O2 adsorption mechanism. This approach involves the chemical reduction of a robust MOF to expose active metal sites, which enable O2 binding through an outer-sphere electron transfer mechanism, representing a promising and underexplored strategy for designing next-generation O2 adsorbents.
In 2013, Long and co-workers reported utilizing Fe2(BDP)3 featuring sharp triangular channels to separate hexane isomers.37 The static adsorption isotherms and isosteric heat of the isomers, n-hexane, 2-methylpentane, 3-methylpentane, 2,3-dimethylbutane and 2,2-dimethylbutane, indicate that a higher degree of branching leads to weaker binding with the framework. In breakthrough experiments conducted with an equimolar mixture of n-hexane, 2-methylpentane, 3-methylpentane, 2,3-dimethylbutane and 2,2-dimethylbutane, di-branched isomers were the first eluted, followed by the monobranched isomer, and the last linear isomer. Configurational-bias Monte Carlo simulations reveal the separation mechanism: the number of carbon atoms interacting with the pore surface of Fe2(BDP)3 decreases as the degree of branching increases. Therefore, dimethylbutane isomers exhibit the weakest interactions with the framework surface, allowing them to pass through the triangular channels quickly. In contrast, the linear n-hexane fits the corner of triangular channels with all six carbons, leading to the longest duration time.
In 2018, the Long group disclosed that the flexible Co(bdp) can achieve high CO2/CH4 selectivity across a broad range of pressures through a reversible CO2 templating mechanism (Fig. 22).60 At a CO2 partial pressure of 3.6 bar (7.2 bar total pressure), diffraction studies show that the selective CO2 adsorption is attributed to size exclusion: Co(bdp) forms a phase with pores large enough to permit the entrance of CO2, while being too narrow to allow CH4 to pass through. As pressure increases, Co(bdp) transfers to phases with expanded pores that can accommodate CH4, whereas CO2 clathrate formation remains energetically favorable, maintaining the exclusion of CH4. When the CO2/CH4 ratio heavily favors CH4 (6:94), the selectivity decreases, highlighting the need for comprehensive multicomponent equilibrium experiments to accurately assess the gas separation performance of flexible materials under varied conditions. In situ PXRD data further reveal that this selectivity resulted from a reversible guest-templating process, in which the framework expands to create a CO2 clathrate and returns to its original phase once desorption occurs. In addition, single-component adsorption isotherms for CO2, CH4, N2, and H2 suggest that Co(bdp) also has potentially high selectivities and capacities in the separation of other gas mixtures, such as CO2/N2, CO2/H2, CH4/N2, and CH4/H2. These findings position Co(bdp) as a promising candidate for CO2/CH4 separation and provide valuable references for further research on flexible adsorbents addressing significant gas separations.
 | ||
| Fig. 22 (a) CO2 adsorption over CH4 in a mixture induces a structural change in Co(bdp). (b) Multicomponent adsorption experiments for CO2/CH4 mixtures in Co(bdp) under equilibrium CO2/CH4 molar ratios of 46:54, 42:58, and 43:57, respectively and (c) in a CH4-rich atmosphere (with an equilibrium CO2/CH4 molar ratio of 6:94).60 Reprinted with permission from American Chemical Society 2018. | ||
Sulfur hexafluoride (SF6) is widely used in the power industry, but its emission is a major concern for the concentration of greenhouse gases.199 Efficient recovery of high-purity SF6 from industrial waste gases is challenging.200 In 2024, Li and co-workers demonstrated that BUT-53, with its dynamic molecular traps, can enrich SF6 from SF6/N2 mixtures, which can maintain performance under humid conditions (90% RH) over multiple cycles (Fig. 23).105 BUT-53 showed outstanding SF6 adsorption of 2.82 mmol g−1 at 0.1 bar and 298 K, and an exceptional SF6/N2 (10:90) selectivity of 2485. This high selectivity allows for the recovery of over 99.9% pure SF6 from a 10% SF6 mixture in breakthrough experiments.
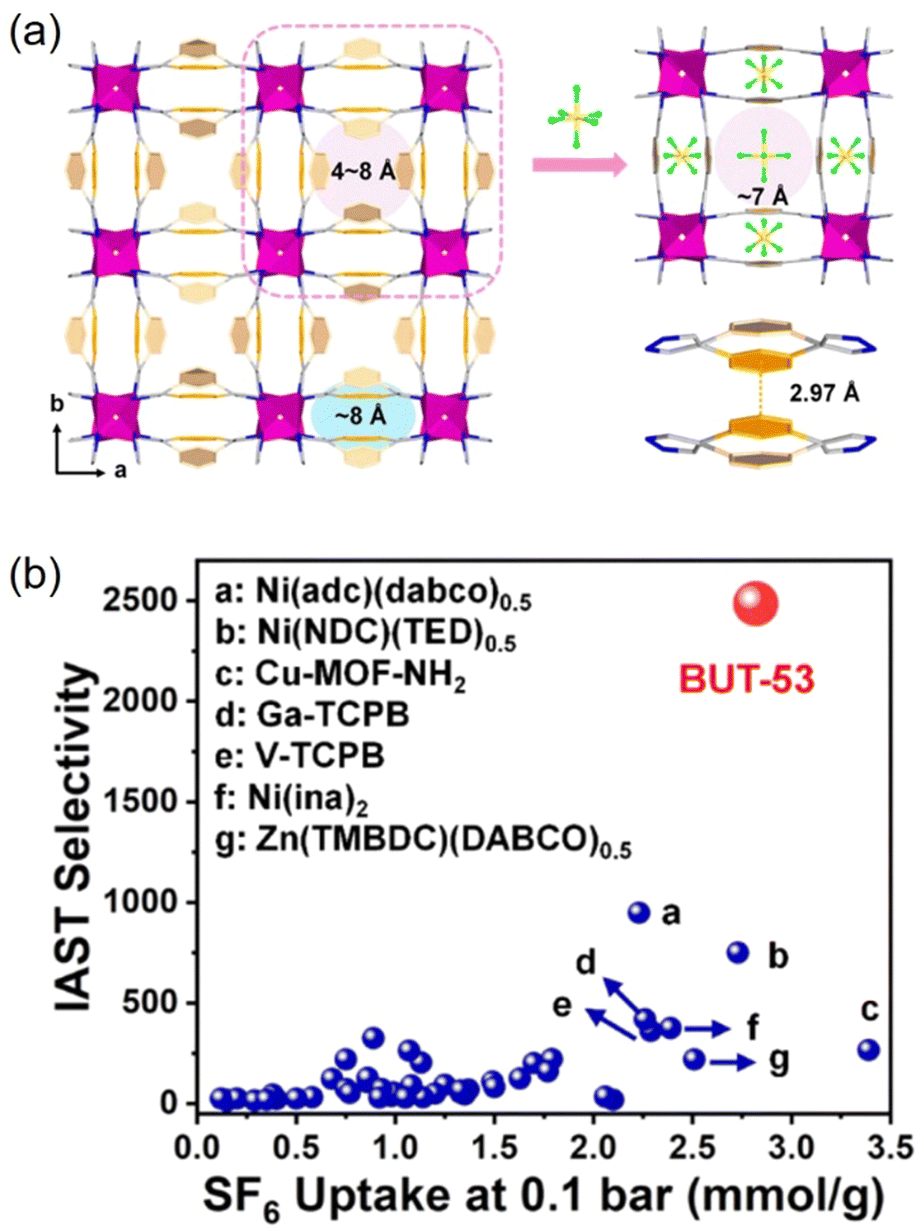 | ||
| Fig. 23 (a) Crystal structure of BUT-53 for trapping SF6 in the pore, and (b) comparison of selectivity and SF6 uptake of BUT-53 with reported materials.163 Reprinted with permission from American Chemical Society 2024. | ||
4.2. Heterogeneous catalysis
In addition to applications in gas capture, storage, and separation, MPFs are increasingly recognized for their potential in heterogeneous catalysis. These materials offer a solid framework that can incorporate functional molecular catalysts, such as porphyrins and salens, through rational design and modification of their ligands, and active metal species from the structural nodes or incoming sources.100,152,163 The robust, periodic structure of MPFs ensures that these active centers are securely immobilized within the framework, minimizing aggregation and thereby enhancing catalytic efficiency. In this section, we will introduce the progress made in leveraging MPFs as heterogeneous catalysts.Following this, Zhou and co-workers developed and characterized a new perfluorophenylene-functionalized metalloporphyrin MOF, PCN-624.150 This MPF, isostructural to PCN-602, features a pore surface decorated with perfluorophenylene groups, making it a highly efficient heterogeneous catalyst for the synthesis of fullerene–anthracene bisadducts. Thanks to its excellent chemical stability, PCN-624 can be reused more than five times with only a slight reduction in catalytic performance. In 2024, they also synthesized PCN-300, which has a lamellar structure with two distinct Cu centers and 1D open channels. PCN-300 demonstrated catalytic activity in the cross-dehydrogenative coupling reaction to form C–O bonds.163 In the same year, PCN-1004 was developed as a catalyst for cross-dehydrogenative coupling reactions, enabling the formation of C–O and C–S bonds without requiring directing groups.101 This system achieved impressive yields up to approximately 99%, exhibiting good cycling durability and broad substrate versatility.
With rich active Ni(II) sites and excellent chemical stability, BUT-32 and BUT-33—extended versions of the sodalite-type Ni3(BTP)2—were employed as heterogeneous catalysts for C–C coupling reactions by Li and co-workers.122 The combination of superior chemical stability, large mesopores, and accessible Ni(II) sites makes BUT-33 particularly effective for a range of C–C coupling processes. While noble metal catalysts are known for their high activity, incorporating these metals into a MOF's skeleton can further improve their catalytic efficiency thanks to the isolation of active centers and the robust solid framework allows for catalyst recycling.203–205 In this regard, BUT-33(Pd), featuring high chemical stability and rich open Pd(II) sites within a mesoporous framework, is anticipated to perform well in cross-coupling reactions as a recyclable single-site catalyst.124 In model reactions including Suzuki and Heck coupling reactions, BUT-33(Pd) confirmed its potential as an effective catalyst with high conversion rates and yields, coupled with the ease of regeneration for consecutive reactions.
In 2021, Li and co-workers designed a pyrazole–benzothiadiazole–pyrazole organic molecule with a donor–acceptor–donor conjugated π-system for rapid charge separation, and its integration into MOFs resulted in the efficient heterogeneous photocatalyst JNU-204.106 Under visible-light irradiation, JNU-204 can catalyze three aerobic oxidation reactions (arylboronic acid oxidation, enamine oxidation, oxysulfonylation of alkynes) with different oxygenation pathways (Fig. 24). Recycling tests confirmed JNU-204's stability and reusability, demonstrating its robustness as a heterogeneous photocatalyst. Additionally, JNU-204 was employed for the straightforward synthesis of pyrrolo[2,1-a]isoquinoline heterocycles, which are core structures in various marine natural products. They also used this MPF for photocatalytic C–N and C–C coupling reactions.206 Based on this type of photosensitizer ligands (Ph-Pz, BT-Pz, BT-Ph-Pz, and BT-Th-Pz), they synthesized a series of porous organic polymers (JNU-208, -209, -210, and -211) for photocatalytic aerobic oxidation of benzylamines. Featuring a donor–π–acceptor–π–donor structure with a more suitable band gap for the generation of reactive oxygen species, JNU-211 was found to exhibit the highest photocatalytic activity.207
 | ||
| Fig. 24 Schematic illustration of JNU-204 as a photocatalyst for aerobic oxygenation under visible-light irradiation.106 Reprinted with permission from American Chemical Society 2021. | ||
Subsequently, they developed a series of isostructural fcu topology Ni8-MPFs (JNU-212, JNU-213, JNU-214, and JNU-215) using linear bipyrazolate ligands.145 Experimental results show that modifying the electron-accepting properties of the pyrazolate-bridging units enhances charge separation and transfer efficiency under visible-light irradiation. Among these, JNU-214, which includes a benzoselenadiazole unit, demonstrated the highest photocatalytic activity for the aerobic oxidation of benzylamines, achieving a 99% conversion rate in 24 hours. Recycling tests further confirmed JNU-214's stability and reusability as a robust heterogeneous catalyst. Tuning the electron-accepting capacity in donor–acceptor–donor MOFs provides a promising route to developing effective noble-metal-free photo-catalysts for aerobic oxidation reactions, with JNU series pyrazolate MOFs exemplifying this approach through its excellent photon absorption, suitable band gap, rapid charge separation, and exceptional chemical stability under visible light.
 | ||
| Fig. 25 Structure of (a) Ni8-TET, Ni-TPP, and PCN-601. (b) Potential active sites and (c) performance speculation of Ni8-TET, Ni-TPP, and PCN-601 applied in electrolytic cells.148 Reprinted with permission from Wiley-VCH 2022. | ||
Metalloporphyrin complexes are also among the most extensively investigated molecular catalysts for electrochemical CO2 reduction reactions (CO2RRs).208,209 While it is well recognized that the metal center plays a key role in determining the selectivity of CO2RR products, the influence of electrolyte composition has received comparatively less attention. Recent studies on a copper-porphyrin MPF catalyst, FICN-8, demonstrated that adjusting the electrolyte composition could systematically control the product distribution.100 Specifically, increasing the concentration of a proton source in an acetonitrile-based electrolyte shifted the primary CO2RR product from CO to HCOOH. A combination of experimental and computational approaches revealed that the formation of a ligand-bound hydride intermediate, facilitated by proton-coupled electron transfer, is a critical step in the generation of formic acid, whose formation is significantly influenced by the concentration of the proton source.
4.3. Conductivity
Developing efficient hydroxide-ion conducting materials is critical for advancing anion-exchange membranes.210,211 One strategy to enhance electronic mobility in poorly conductive MOFs is by introducing defects.212 Studies on the ionic conductivity of Ni8-MPFs [Ni8(OH)4(H2O)2(BDP-X)6] (X = H (1), OH (2), NH2 (3)) and their post-synthetically modified forms 1@KOH, 2@KOH, 3@KOH), which feature missing-linker defects, revealed a significant increase in conductivity-up to four orders of magnitude higher compared to the pristine 1–3 systems.131 For instance, at 313 K and 22% RH, 2 exhibits a conductivity of 5.86 × 10−9 Scm−1 with an activation energy Ea of 0.60 eV. When treated with KOH, the conductivity increases to 2.75 × 10−5 S cm−1, accompanied by a reduced activation energy of 0.40 eV, and further to 1.16 × 10−2 S cm−1 with Ea reduced to 0.20 eV at 100% RH. The improved performance of the KOH-modified materials is attributed to their increased porosity, basicity, and hydrophilicity compared to the unmodified systems.
Aubrey et al. recently reported the reductive doping of the poorly conducting Fe(III) MPF [Fe2(BDP)3] with potassium naphthalenide, resulting in mix-valence Fe(II)/Fe(III) systems of Kx[Fe2(BDP)3] (0 ≤ x ≤ 2).175 Such doping significantly enhances conductivity without compromising pore accessibility. Intermediate reduction levels show a characteristic inter-valence charge-transfer band at 0.52 eV, indicative of improved charge transport. Flash photolysis time-resolved microwave conductivity and single-crystal field-effect transistor measurements revealed an increase in charge mobility from 0.02 cm2 V−1 s−1 to 0.29 cm2 V−1 s−1 for K0.8[Fe2(BDP)3], with a further 10000-fold increase in conductivity to 7 × 102 S cm−1 for K0.98[Fe2(BDP)3], attributed to 1D conduction along the [Fe2(pyrazolate)6] chains.
Similarly, the proton-conducting properties of MPFs can also be enhanced through post-synthetic modifications. He and co-workers prepared Ni8-MPFs, NiL1 and NiL2, and studied their proton conductivity in both pristine and imidazole-encapsulated forms (Im@NiL1 and Im@NiL2).142 The inclusion of imidazole increased the proton conductivity by 3 to 5 orders of magnitude, reaching up to 1.72 × 10−2 S cm−1 at 98% RH and 80 °C. Activation energy analyses indicated that NiL1, NiL2, and Im@NiL1 follow the Vehicle mechanism, while Im@NiL2 likely employs the Grotthuss mechanism due to the flexible alkyl side arms on L2 coordinating imidazole and water molecules.
4.4. Other applications
5. Conclusion and outlook
Under the guidance of crystal engineering/reticular chemistry principles, researchers have developed plenty of MPFs with great structure diversity and base resistance. Various classic MOF families such as Co(bdp), Fe2(BDP)3, Ni8(L)6, PCN-601, and BUT-55 demonstrated fascinating architectures and record-breaking performances over the past decade. This review comprehensively introduces MPFs, including the superiority and significance of construction through rational design, synthetic strategies, structural features, and stability. Potential applications have also been explored in energy and environmental fields, with insights gained into the mechanisms to shed light on underlying structure–property relationships.Although significant progress has been made, there are still a few issues pending in the development of this field. Strong metal–pyrazolate bonds, despite being beneficial for stability, pose significant synthetic challenges, such as kinetic trapping and reduced crystallinity. The difficulty in obtaining high-quality single crystals further inhibits detailed structure analysis and new materials discovery, impeding a deeper understanding of their structure–property relationships. Due to the coordination constraints of pyrazolate ligands, and the relatively small library of inorganic chains and clusters, the number of matched topologies remains limited. Targeting at easy access to advanced functional materials, general design principles and further structure diversification are still actively sought after.
In addition to these scientific limitations, the scalability of MPFs presents another critical challenge for industrial applications. The synthesis and post-synthetic treatments (washing and activation) of MPFs, usually performed under controlled laboratory conditions, requires adaptation to scalable, cost-effective, and reproducible procedures suitable for mass production while ensuring the maintenance of structures and properties. Key considerations include the development of efficient, solvent-free or green synthetic routes (for both ligands and MFPs), as well as the optimization of reaction parameters to reduce energy and resource consumption. Furthermore, the design of synthetic workflows that enable continuous or modular manufacturing of MPFs, rather than batch processes, will be essential for meeting industrial demands. The integration of MPFs into industrial applications also necessitates the evaluation of their performance under real-world conditions, such as chemical robustness, thermal stability, possibility of shaping and processing, and reusability over extended operational cycles. Addressing these issues will be vital for their use in areas like gas storage and separation, catalysis, and environmental remediation.
Substantial interdisciplinary efforts are on demand to develop versatile synthetic strategies, advanced characterization/analysis techniques, computational/theoretical modeling, and artificial intelligence/machine learning-assisted materials design and screening, to unveil the full palette of MPFs. Bridging the gap between laboratory-scale synthesis and large-scale production will further rely on close collaborations between academia, industry, and government initiatives to foster standardization. Continued innovation in this area will not only advance the chemistry of porous frameworks but also inspire the development of next-generation materials to tackle global challenges in a more sustainable manner. By addressing both the fundamental scientific limitations and the practical challenges of scalability, MPFs have the potential to play a transformative role in shaping a greener future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 22225803, 22038001 and 22401168), the Beijing Natural Science Foundation (No. Z230023), and the Beijing Outstanding Young Scientist Program (Project No. JWZQ20240102008).Notes and references
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H. C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- S. S. Chui, S. M. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS PubMed.
- C. Gropp, S. Canossa, S. Wuttke, F. Gandara, Q. Li, L. Gagliardi and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1255–1273 CrossRef CAS PubMed.
- O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15507–15509 CrossRef CAS PubMed.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Woll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CAS.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- H. Y. Li, X. J. Kong, S. D. Han, J. Pang, T. He, G. M. Wang and X. H. Bu, Chem. Soc. Rev., 2024, 53, 5626–5676 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- T. He, X.-J. Kong and J.-R. Li, Acc. Chem. Res., 2021, 54, 3083–3094 CrossRef CAS PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- C. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549–554 CrossRef CAS PubMed.
- C. Jia, T. He and G.-M. Wang, Coord. Chem. Rev., 2023, 476, 214930 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729–4756 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- X. F. Lu, P. Q. Liao, J. W. Wang, J. X. Wu, X. W. Chen, C. T. He, J. P. Zhang, G. R. Li and X. M. Chen, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS PubMed.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC.
- Z. Wang, A. Bilegsaikhan, R. T. Jerozal, T. A. Pitt and P. J. Milner, ACS Appl. Mater. Interfaces, 2021, 13, 17517–17531 CrossRef CAS PubMed.
- H. J. Choi, M. Dinca and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS PubMed.
- N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. Lopez, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef CAS PubMed.
- Z. R. Herm, B. M. Wiers, J. A. Mason, J. M. van Baten, M. R. Hudson, P. Zajdel, C. M. Brown, N. Masciocchi, R. Krishna and J. R. Long, Science, 2013, 340, 960–964 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS PubMed.
- T. He, X. J. Kong, Z. X. Bian, Y. Z. Zhang, G. R. Si, L. H. Xie, X. Q. Wu, H. Huang, Z. Chang, X. H. Bu, M. J. Zaworotko, Z. R. Nie and J. R. Li, Nat. Mater., 2022, 21, 689–695 CrossRef CAS PubMed.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS PubMed.
- R. Kumar Gupta, P. Kallem, G.-G. Luo, P. Cui, Z. Wang, F. Banat, C.-H. Tung and D. Sun, Coord. Chem. Rev., 2023, 497, 215436 CrossRef CAS.
- C. Pettinari, A. Tăbăcaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS.
- M. Parshad, D. Kumar and V. Verma, Inorg. Chim. Acta, 2024, 560, 121789 CrossRef CAS.
- J. A. R. Navarro, Isr. J. Chem., 2018, 58, 1112–1118 CrossRef CAS.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef CAS.
- K. Wang, Y. Li, L. H. Xie, X. Li and J. R. Li, Chem. Soc. Rev., 2022, 51, 6417–6441 RSC.
- J. Klingele, S. Dechert and F. Meyer, Coord. Chem. Rev., 2009, 253, 2698–2741 CrossRef CAS.
- J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203–240 CrossRef.
- N. Masciocchi, M. Moret, P. Cairati, A. Sironi, G. A. Ardizzoia and G. La Monica, J. Am. Chem. Soc., 1994, 116, 7668–7676 CrossRef CAS.
- N. Masciocchi, G. A. Ardizzoia, A. Maspero, G. LaMonica and A. Sironi, Inorg. Chem., 1999, 38, 3657–3664 CrossRef CAS PubMed.
- N. Masciocchi, G. A. Ardizzoia, S. Brenna, G. LaMonica, A. Maspero, S. Galli and A. Sironi, Inorg. Chem., 2002, 41, 6080–6089 CrossRef CAS PubMed.
- A. Cingolani, S. Galli, N. Masciocchi, L. Pandolfo, C. Pettinari and A. Sironi, J. Am. Chem. Soc., 2005, 127, 6144–6145 CrossRef CAS PubMed.
- M. A. Omary, O. Elbjeirami, C. S. Gamage, K. M. Sherman and H. V. Dias, Inorg. Chem., 2009, 48, 1784–1786 CrossRef CAS PubMed.
- M. A. Omary, M. A. Rawashdeh-Omary, M. W. Gonser, O. Elbjeirami, T. Grimes, T. R. Cundari, H. V. Diyabalanage, C. S. Gamage and H. V. Dias, Inorg. Chem., 2005, 44, 8200–8210 CrossRef CAS PubMed.
- Q. Liu, Y. Song, Y. Ma, Y. Zhou, H. Cong, C. Wang, J. Wu, G. Hu, M. O'Keeffe and H. Deng, J. Am. Chem. Soc., 2019, 141, 488–496 CrossRef CAS PubMed.
- G. Hu, Q. Liu, Y. Zhou, W. Yan, Y. Sun, S. Peng, C. Zhao, X. Zhou and H. Deng, J. Am. Chem. Soc., 2023, 145, 13181–13194 CrossRef CAS PubMed.
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knofel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and G. Ferey, J. Am. Chem. Soc., 2010, 132, 13782–13788 CrossRef CAS PubMed.
- Y. Lu, M. Tonigold, B. Bredenkötter, D. Volkmer, J. Hitzbleck and G. Langstein, Z. Anorg. Allg. Chem., 2008, 634, 2411–2417 CrossRef CAS.
- M. K. Taylor, T. Runcevski, J. Oktawiec, J. E. Bachman, R. L. Siegelman, H. Jiang, J. A. Mason, J. D. Tarver and J. R. Long, J. Am. Chem. Soc., 2018, 140, 10324–10331 CrossRef CAS PubMed.
- M. K. Taylor, T. Runcevski, J. Oktawiec, M. I. Gonzalez, R. L. Siegelman, J. A. Mason, J. Ye, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2016, 138, 15019–15026 CrossRef CAS PubMed.
- S. Galli, N. Masciocchi, V. Colombo, A. Maspero, G. Palmisano, F. J. López-Garzón, M. Domingo-García, I. Fernández-Morales, E. Barea and J. A. R. Navarro, Chem. Mater., 2010, 22, 1664–1672 CrossRef CAS.
- C. Lucarelli, S. Galli, A. Maspero, A. Cimino, C. Bandinelli, A. Lolli, J. Velasquez Ochoa, A. Vaccari, F. Cavani and S. Albonetti, J. Phys. Chem. C, 2016, 120, 15310–15321 CrossRef CAS.
- G. Huang, L. Yang, Q. Yin, Z. B. Fang, X. J. Hu, A. A. Zhang, J. Jiang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 4385–4390 CrossRef CAS PubMed.
- Y. Ye, Y. Xie, Y. Shi, L. Gong, J. Phipps, A. M. Al-Enizi, A. Nafady, B. Chen and S. Ma, Angew. Chem., Int. Ed., 2023, 62, e202302564 CrossRef CAS PubMed.
- Q. Q. Huang, Z. B. Fang, K. Pang, W. K. Qin, T. F. Liu and R. Cao, Adv. Funct. Mater., 2022, 32, 2205147 CrossRef CAS.
- F. G. Cirujano, E. Lopez-Maya, N. Almora-Barrios, A. Rubio-Gaspar, N. Martin, J. A. R. Navarro and C. Marti-Gastaldo, Inorg. Chem., 2020, 59, 18168–18173 CrossRef CAS PubMed.
- H. J. Choi, M. Dincă, A. Dailly and J. R. Long, Energy Environ. Sci., 2010, 3, 117–123 RSC.
- Y. S. Wei, L. Sun, M. Wang, J. Hong, L. Zou, H. Liu, Y. Wang, M. Zhang, Z. Liu, Y. Li, S. Horike, K. Suenaga and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 16013–16022 CrossRef CAS PubMed.
- X.-L. Liu, Q. Yin, G. Huang and T.-F. Liu, Inorg. Chem. Commun., 2018, 94, 21–26 CrossRef CAS.
- Q. Xie, W. Si, Y. Shen, Z. Wang and H. Uyama, Nanoscale, 2021, 13, 16296–16306 RSC.
- H. Babaei, K. R. Meihaus and J. R. Long, Chem. Mater., 2023, 35, 6220–6226 CrossRef CAS PubMed.
- V. Colombo, C. Montoro, A. Maspero, G. Palmisano, N. Masciocchi, S. Galli, E. Barea and J. A. Navarro, J. Am. Chem. Soc., 2012, 134, 12830–12843 CrossRef CAS PubMed.
- S. Rojas, F. J. Carmona, C. R. Maldonado, P. Horcajada, T. Hidalgo, C. Serre, J. A. R. Navarro and E. Barea, Inorg. Chem., 2016, 55, 2650–2663 CrossRef CAS PubMed.
- Z. Wang, Q. Xie, Y. Wang, Y. Shu, C. Li and Y. Shen, New J. Chem., 2020, 44, 18319–18325 RSC.
- T. He, Y. Z. Zhang, B. Wang, X. L. Lv, L. H. Xie and J. R. Li, ChemPlusChem, 2016, 81, 864–871 CrossRef CAS PubMed.
- M. Tonigold, Y. Lu, A. Mavrandonakis, A. Puls, R. Staudt, J. Mollmer, J. Sauer and D. Volkmer, Chem. – Eur. J., 2011, 17, 8671–8695 CrossRef CAS PubMed.
- M. Moroni, L. Nardo, A. Maspero, G. Vesco, M. Lamperti, L. Scapinello, R. Vismara, J. A. R. Navarro, D. Monticelli, A. Penoni, M. Mella and S. Galli, Chem. Mater., 2023, 35, 2892–2903 CrossRef CAS PubMed.
- S. Galli, A. Maspero, C. Giacobbe, G. Palmisano, L. Nardo, A. Comotti, I. Bassanetti, P. Sozzani and N. Masciocchi, J. Mater. Chem. A, 2014, 2, 12208–12221 RSC.
- Z. Xue, J. J. Zheng, Y. Nishiyama, M. S. Yao, Y. Aoyama, Z. Fan, P. Wang, T. Kajiwara, Y. Kubota, S. Horike, K. I. Otake and S. Kitagawa, Angew. Chem., Int. Ed., 2023, 62, e202215234 CrossRef CAS PubMed.
- H. C. Wentz, G. Skorupskii, A. B. Bonfim, J. L. Mancuso, C. H. Hendon, E. H. Oriel, G. T. Sazama and M. G. Campbell, Chem. Sci., 2019, 11, 1342–1346 RSC.
- J. Li, A. Kumar, B. A. Johnson and S. Ott, Nat. Commun., 2023, 14, 4388 CrossRef CAS PubMed.
- A. Kumar, J. Li, A. K. Inge and S. Ott, ACS Nano, 2023, 17, 21595–21603 CrossRef PubMed.
- J. Li, A. Kumar and S. Ott, J. Am. Chem. Soc., 2024, 146, 12000–12010 CrossRef CAS PubMed.
- S. Chong, S. M. J. Rogge and J. Kim, Chem. Mater., 2021, 34, 254–265 CrossRef.
- C. R. Wade, M. Li and M. Dinca, Angew. Chem., Int. Ed., 2013, 52, 13377–13381 CrossRef CAS PubMed.
- C. R. Wade, T. Corrales-Sanchez, T. C. Narayan and M. Dincă, Energy Environ. Sci., 2013, 6, 2172–2177 RSC.
- K. Nakamoto, J. Bai, M. Zhao, R. Sakamoto, L. Zhao, M. Ito, S. Okada, E. Yamamoto, H. Murayama and M. Tokunaga, RSC Adv., 2023, 13, 22070–22078 RSC.
- V. Monnier, F. Odobel and S. Diring, J. Am. Chem. Soc., 2023, 145, 19232–19242 CrossRef CAS PubMed.
- Z. Xue, M. S. Yao, K. I. Otake, Y. Nishiyama, Y. Aoyama, J. J. Zheng, S. Zhang, T. Kajiwara, S. Horike and S. Kitagawa, Angew. Chem., Int. Ed., 2024, 63, e202401005 CrossRef CAS PubMed.
- C. Pettinari, A. Tabacaru, I. Boldog, K. V. Domasevitch, S. Galli and N. Masciocchi, Inorg. Chem., 2012, 51, 5235–5245 CrossRef CAS PubMed.
- Y.-L. Zhao, Y. Xie, X. Zhang, X.-Y. Li, X. Bai and J.-R. Li, Green Chem. Eng., 2025 DOI:10.1016/j.gce.2025.01.004.
- Q. Fan, J. Yao, S. Zhao, X. Wu, J. Huang, H. Luo and Q. Xia, Small, 2025, e2409215 CrossRef PubMed.
- S. Spirkl, M. Grzywa, C. S. Zehe, J. Senker, S. Demeshko, F. Meyer, S. Riegg and D. Volkmer, CrystEngComm, 2015, 17, 313–322 RSC.
- R. Vismara, G. Tuci, A. Tombesi, K. V. Domasevitch, C. Di Nicola, G. Giambastiani, M. R. Chierotti, S. Bordignon, R. Gobetto, C. Pettinari, A. Rossin and S. Galli, ACS Appl. Mater. Interfaces, 2019, 11, 26956–26969 CrossRef CAS PubMed.
- N. Mosca, R. Vismara, J. A. Fernandes, G. Tuci, C. Di Nicola, K. V. Domasevitch, C. Giacobbe, G. Giambastiani, C. Pettinari, M. Aragones-Anglada, P. Z. Moghadam, D. Fairen-Jimenez, A. Rossin and S. Galli, Chem. – Eur. J., 2018, 24, 13170–13180 CrossRef CAS PubMed.
- A. Nowacka, R. Vismara, G. Mercuri, M. Moroni, M. Palomino, K. V. Domasevitch, C. Di Nicola, C. Pettinari, G. Giambastiani, I. X. F. X. Llabres, S. Galli and A. Rossin, Inorg. Chem., 2020, 59, 8161–8172 CrossRef CAS PubMed.
- R. Vismara, G. Tuci, N. Mosca, K. V. Domasevitch, C. Di Nicola, C. Pettinari, G. Giambastiani, S. Galli and A. Rossin, Inorg. Chem. Front., 2019, 6, 533–545 RSC.
- P. Campitelli, A. Tombesi, C. Di Nicola, C. Pettinari, A. Mauri, S. Galli, T. Yan, D. Liu, J. D. Duan, S. Goswami, G. Tuci, G. Giambastiani, J. T. Hupp and A. Rossin, ACS Appl. Energy Mater., 2023, 6, 9231–9242 CrossRef CAS.
- S. H. Pu, T. Huang, D. H. Si, M. J. Sun, W. W. Wang, T. Zhang and R. Cao, Angew. Chem., Int. Ed., 2024, e202411766 CAS.
- R. R. Liang, Z. Liu, Z. Han, Y. Yang, J. Rushlow and H. C. Zhou, Angew. Chem., Int. Ed., 2024, e202414271 Search PubMed.
- S. Millan, B. Gil-Hernández, E. Hastürk, A. Schmitz and C. Janiak, Z. Anorg. Allg. Chem., 2018, 644, 1311–1316 CrossRef CAS.
- S. Millan, G. Makhloufi and C. Janiak, Z. Anorg. Allg. Chem., 2019, 645, 893–899 CrossRef CAS.
- G. Berkbigler, Q. Liu, N. Hoefer, Y. Xie, J. S. Hilliard, D. W. McComb and C. R. Wade, Eur. J. Inorg. Chem., 2024, e202300548 CrossRef CAS.
- X. Zhang, Y. L. Zhao, X. Y. Li, X. Bai, Q. Chen and J. R. Li, J. Am. Chem. Soc., 2024, 146, 19303–19309 CrossRef CAS PubMed.
- J. K. Jin, K. Wu, X. Y. Liu, G. Q. Huang, Y. L. Huang, D. Luo, M. Xie, Y. Zhao, W. Lu, X. P. Zhou, J. He and D. Li, J. Am. Chem. Soc., 2021, 143, 21340–21349 CrossRef CAS PubMed.
- H. L. Xia, K. Zhou, S. Wu, D. Ren, K. Xing, J. Guo, X. Wang, X. Y. Liu and J. Li, Chem. Sci., 2022, 13, 8036–8044 RSC.
- J. Liu, J. Miao, L. Ma, J. Li, J. Li and H. Wang, AIChE J., 2024, e18656 Search PubMed.
- Z. Cai, W. Tao, C. E. Moore, S. Zhang and C. R. Wade, Angew. Chem., Int. Ed., 2021, 60, 21221–21225 CrossRef CAS PubMed.
- Q. Gao, A.-L. Li, X. Chen, N. Lu, Y.-M. Zhang and L.-Z. Chen, Sep. Purif. Technol., 2021, 276, 119424 CrossRef CAS.
- R. Vismara, S. Terruzzi, A. Maspero, T. Grell, F. Bossola, A. Sironi, S. Galli, J. A. R. Navarro and V. Colombo, Adv. Mater., 2023, e2209907 Search PubMed.
- C. Giacobbe, E. Lavigna, A. Maspero and S. Galli, J. Mater. Chem. A, 2017, 5, 16964–16975 RSC.
- N. Biggins, M. E. Ziebel, M. I. Gonzalez and J. R. Long, Chem. Sci., 2020, 11, 9173–9180 RSC.
- A. Jaffe, M. E. Ziebel, D. M. Halat, N. Biggins, R. A. Murphy, K. Chakarawet, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2020, 142, 14627–14637 CrossRef CAS PubMed.
- J. Perego, C. X. Bezuidenhout, A. Pedrini, S. Bracco, M. Negroni, A. Comotti and P. Sozzani, J. Mater. Chem. A, 2020, 8, 11406–11413 RSC.
- M. Grzywa, C. Gessner, D. Denysenko, B. Bredenkotter, F. Gschwind, K. M. Fromm, W. Nitek, E. Klemm and D. Volkmer, Dalton Trans., 2013, 42, 6909–6921 RSC.
- A. Tăbăcaru, S. Galli, C. Pettinari, N. Masciocchi, T. M. McDonald and J. R. Long, CrystEngComm, 2015, 17, 4992–5001 RSC.
- Z. Li, Z. Zhang, Y. Ye, K. Cai, F. Du, H. Zeng, J. Tao, Q. Lin, Y. Zheng and S. Xiang, J. Mater. Chem. A, 2017, 5, 7816–7824 RSC.
- M. C. de Koning, L. Dadon, L. C. M. Rozing, M. van Grol and R. Bross, ACS Appl. Mater. Interfaces, 2023, 15, 55877–55884 CrossRef CAS PubMed.
- H. Xue, J.-R. Huang, Z.-S. Wang, Z.-H. Zhao, W. Shi, P.-Q. Liao and X.-M. Chen, Chin. J. Catal., 2023, 53, 102–108 CrossRef CAS.
- X. Li, Z. Yan-Long, S.-N. Chen, K. Wang, S. Wang, L.-H. Xie and J.-R. Li, Chem. Sci., 2024, 15, 14425–14430 RSC.
- T. He, Z. Huang, S. Yuan, X.-L. Lv, X.-J. Kong, X. Zou, H.-C. Zhou and J.-R. Li, J. Am. Chem. Soc., 2020, 142, 13491–13499 CrossRef CAS PubMed.
- T. Xue, T. He, L. Peng, O. A. Syzgantseva, R. Li, C. Liu, D. T. Sun, G. Xu, R. Qiu, Y. Wang, S. Yang, J. Li, J. R. Li and W. L. Queen, Sci. Adv., 2023, 9, eadg4923 CrossRef CAS PubMed.
- T. He, X.-J. Kong, J. Zhou, C. Zhao, K. Wang, X.-Q. Wu, X.-L. Lv, G.-R. Si, J.-R. Li and Z.-R. Nie, J. Am. Chem. Soc., 2021, 143, 9901–9911 CrossRef CAS PubMed.
- G. R. Si, X. J. Kong, T. He, Z. Zhang and J. R. Li, Nat. Commun., 2024, 15, 7220 Search PubMed.
- M. Tonigold, Y. Lu, B. Bredenkotter, B. Rieger, S. Bahnmuller, J. Hitzbleck, G. Langstein and D. Volkmer, Angew. Chem., Int. Ed., 2009, 48, 7546–7550 Search PubMed.
- N. Masciocchi, S. Galli, V. Colombo, A. Maspero, G. Palmisano, B. Seyyedi, C. Lamberti and S. Bordiga, J. Am. Chem. Soc., 2010, 132, 7902–7904 CrossRef CAS PubMed.
- J. L. Obeso, K. Gopalsamy, M. Wahiduzzaman, E. Martínez-Ahumada, D. Fan, H. A. Lara-García, F. J. Carmona, G. Maurin, I. A. Ibarra and J. A. R. Navarro, J. Mater. Chem. A, 2024, 12, 10157–10165 RSC.
- R. Freund, A. Kalytta-Mewes, M. Kraft and D. Volkmer, Chem. Commun., 2022, 58, 9349–9352 Search PubMed.
- E. López-Maya, C. Montoro, V. Colombo, E. Barea and J. A. R. Navarro, Adv. Funct. Mater., 2014, 6130–6135 CrossRef.
- C. Montoro, P. Ocon, F. Zamora and J. A. R. Navarro, Chem. – Eur. J., 2016, 22, 1646–1651 CrossRef CAS PubMed.
- L. Marleny Rodriguez-Albelo, E. Lopez-Maya, S. Hamad, A. Rabdel Ruiz-Salvador, S. Calero and J. A. R. Navarro, Nat. Commun., 2017, 8, 14457 CrossRef PubMed.
- F. G. Cirujano, E. López-Maya, J. A. R. Navarro and D. E. De Vos, Top. Catal., 2018, 61, 1414–1423 CrossRef CAS.
- F. G. Cirujano, E. Lopez-Maya, M. Rodriguez-Albelo, E. Barea, J. A. R. Navarro and D. E. De Vos, ChemCatChem, 2017, 9, 4019–4023 Search PubMed.
- N. Martin, F. G. Cirujano, E. Garcia-Verdugo, J. Llorca, E. Del Rio, I. Jimenez-Morales, A. Bogeat-Barroso, E. Lopez-Maya and M. G. Alvarez, ChemPlusChem, 2023, 88, e202300447 CrossRef CAS PubMed.
- T. He, Y.-Z. Zhang, H. Wu, X.-J. Kong, X.-M. Liu, L.-H. Xie, Y. Dou and J.-R. Li, ChemPhysChem, 2017, 18, 3245–3252 CrossRef CAS PubMed.
- H. Yang, X. Zhang, G. Zhang and H. Fei, Chem. Commun., 2018, 54, 4469–4472 RSC.
- S. Tarnowicz-Ligus, A. Augustyniak and A. M. Trzeciak, Eur. J. Inorg. Chem., 2019, 4282–4288 Search PubMed.
- Y.-Z. Zhang, T. He, X.-J. Kong, Z.-X. Bian, X.-Q. Wu and J.-R. Li, ACS Mater. Lett., 2019, 1, 20–24 Search PubMed.
- F. Afshariazar, A. Morsali, S. Sorbara, N. M. Padial, E. Roldan-Molina, J. E. Oltra, V. Colombo and J. A. R. Navarro, Chem. – Eur. J., 2021, 27, 11837–11844 CrossRef CAS PubMed.
- J. Hu, X. Deng, H. Zhang, Y. Diao, S. Cheng, S. L. Zheng, W. M. Liao, J. He and Z. Xu, Inorg. Chem., 2021, 60, 161–166 CrossRef CAS PubMed.
- J. Hu, H. Zhang, Z. Feng, Q.-R. Luo, C.-M. Wu, Y.-H. Zhong, J.-R. Li, L.-H. Chung, W.-M. Liao and J. He, Chin. Chem. Lett., 2022, 33, 3227–3230 CrossRef CAS.
- Q. R. Luo, J. Hu, Z. Jiang, S. Chen, X. Ye, J. Li, L. H. Chung, L. Yu and J. He, Inorg. Chem., 2023, 62, 5229–5236 Search PubMed.
- K. Li, J. Hu, Q. Gu, J. He, Y. K. Peng and Z. Xu, ACS Appl. Mater. Interfaces, 2023, 15, 35107–35116 CrossRef CAS PubMed.
- K. Wu, X. Y. Liu, P. W. Cheng, Y. L. Huang, J. Zheng, M. Xie, W. Lu and D. Li, J. Am. Chem. Soc., 2023, 145, 18931–18938 Search PubMed.
- R. Freund, A. Schulz, P. Lunkenheimer, M. Kraft, T. Bergler, H. Oberhofer and D. Volkmer, Adv. Funct. Mater., 2024, 2415376 CrossRef.
- Z. B. Fang, T. T. Liu, J. Liu, S. Jin, X. P. Wu, X. Q. Gong, K. Wang, Q. Yin, T. F. Liu, R. Cao and H. C. Zhou, J. Am. Chem. Soc., 2020, 142, 12515–12523 CrossRef CAS PubMed.
- S. N. Sun, L. Z. Dong, J. R. Li, J. W. Shi, J. Liu, Y. R. Wang, Q. Huang and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202207282 Search PubMed.
- X.-L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 211–217 CrossRef CAS PubMed.
- N. Huang, K. Wang, H. Drake, P. Cai, J. Pang, J. Li, S. Che, L. Huang, Q. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 6383–6390 Search PubMed.
- Y. Chen, C. Li, X. Wang, L. Fan, Y. Zhang, X. Zhao, Q. Y. Li and X. J. Wang, Inorg. Chem., 2024, 63, 19924–19930 CrossRef CAS PubMed.
- F. X. Ma, L. Y. Lyu, J. Chen, T. Huang, T. Zhang and R. Cao, Chem. Commun., 2024, 60, 1293–1296 RSC.
- X.-J. Kong, T. He, A. A. Bezrukov, S. Darwish, G.-R. Si, Y. Z. Zhang, W. Wu, Y. Wang, X. Li, N. Kumar, J.-R. Li and M. J. Zaworotko, J. Am. Chem. Soc., 2024, 146, 28320–28328 CAS.
- F. X. Ma, F. Q. Mi, M. J. Sun, T. Huang, Z. A. Wang, T. Zhang and R. Cao, Inorg. Chem. Front., 2022, 9, 1812–1818 RSC.
- Y.-Z. Zhang, T. He, X.-L. Lv, B. Wang, L.-H. Xie, X.-M. Liu and J.-R. Li, J. Coord. Chem., 2016, 69, 3242–3249 CrossRef CAS.
- Z. Li, Y. Ye, Z. Yao, J. Guo, Q. Lin, J. Zhang, Z. Zhang, F. Wei and S. Xiang, J. Mater. Chem. A, 2018, 6, 19681–19688 RSC.
- D. Zhang, P. Du, J. Chen, H. Guo and X. Lu, Sens. Actuators, B, 2021, 341, 130000 Search PubMed.
- L. Z. Dong, Y. Zhang, Y. F. Lu, L. Zhang, X. Huang, J. H. Wang, J. Liu, S. L. Li and Y. Q. Lan, Chem. Commun., 2021, 57, 8937–8940 RSC.
- J. He, J. Duan, H. Shi, J. Huang, J. Huang, L. Yu, M. Zeller, A. D. Hunter and Z. Xu, Inorg. Chem., 2014, 53, 6837–6843 Search PubMed.
- A. Tăbăcaru, C. Pettinari, F. Marchetti, S. Galli and N. Masciocchi, Cryst. Growth Des., 2014, 14, 3142–3152 Search PubMed.
- Z. Xu, L. L. Han, G. L. Zhuang, J. Bai and D. Sun, Inorg. Chem., 2015, 54, 4737–4743 CrossRef CAS PubMed.
- J. Fritzsche, M. Grzywa, D. Denysenko, V. Bon, I. Senkovska, S. Kaskel and D. Volkmer, Dalton Trans., 2017, 46, 6745–6755 RSC.
- R.-R. Liang, Z. Han, P. Cai, Y. Yang, J. Rushlow, Z. Liu, K.-Y. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 14174–14181 CrossRef CAS PubMed.
- R.-R. Liang, Y. Fu, Z. Han, Y. Yang, V. I. Bakhmutov, Z. Liu, J. Rushlow and H.-C. Zhou, Nat. Water, 2024, 2, 1218–1225 CrossRef CAS.
- V. V. Ponomarova, V. V. Komarchuk, I. Boldog, H. Krautscheid and K. V. Domasevitch, CrystEngComm, 2013, 15, 8280–8287 Search PubMed.
- J. Ren, W. Zeng, Y. Chen, X. Fu and Q. Yang, Sep. Purif. Technol., 2022, 295, 121286 Search PubMed.
- S. Mukherjee, N. Sikdar, D. O'Nolan, D. M. Franz, V. Gascon, A. Kumar, N. Kumar, H. S. Scott, D. G. Madden, P. E. Kruger, B. Space and M. J. Zaworotko, Sci. Adv., 2019, 5, eaax9171 CrossRef CAS PubMed.
- M. K. Ehlert, S. J. Rettig, A. Storr, R. C. Thompson and J. Trotter, Can. J. Chem., 1993, 71, 1425–1436 Search PubMed.
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 Search PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Ferey, J. Am. Chem. Soc., 2002, 124, 13519–13526 Search PubMed.
- E. Albanese, B. Civalleri, M. Ferrabone, F. Bonino, S. Galli, A. Maspero and C. Pettinari, J. Mater. Chem., 2012, 22, 22592–22602 RSC.
- V. Monnier, F. Odobel and S. Diring, Chem. Commun., 2022, 58, 9429–9432 RSC.
- S. Spirkl, M. Grzywa and D. Volkmer, Dalton Trans., 2018, 47, 8779–8786 Search PubMed.
- S. R. Miller, P. A. Wright, C. Serre, T. Loiseau, J. Marrot and G. Ferey, Chem. Commun., 2005, 3850–3852 Search PubMed.
- M. L. Aubrey, B. M. Wiers, S. C. Andrews, T. Sakurai, S. E. Reyes-Lillo, S. M. Hamed, C.-J. Yu, L. E. Darago, J. A. Mason, J.-O. Baeg, F. Grandjean, G. J. Long, S. Seki, J. B. Neaton, P. Yang and J. R. Long, Nat. Mater., 2018, 17, 625–632 CrossRef CAS PubMed.
- L. Hou, Y. Y. Lin and X. M. Chen, Inorg. Chem., 2008, 47, 1346–1351 Search PubMed.
- L. Pandolfo and C. Pettinari, CrystEngComm, 2017, 19, 1701–1720 Search PubMed.
- H. V. Dias, H. V. Diyabalanage, M. G. Eldabaja, O. Elbjeirami, M. A. Rawashdeh-Omary and M. A. Omary, J. Am. Chem. Soc., 2005, 127, 7489–7501 CrossRef CAS PubMed.
- G. Yang, J. R. Martínez and R. G. Raptis, Inorg. Chim. Acta, 2009, 362, 1546–1552 Search PubMed.
- M. Grzywa, D. Denysenko, A. Schaller, A. Kalytta-Mewes and D. Volkmer, CrystEngComm, 2016, 18, 7883–7893 Search PubMed.
- M. K. Ehlert, A. Storr, D. A. Summers and R. C. Thompson, Can. J. Chem., 1997, 75, 491–498 Search PubMed.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- M. Dinca, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876–16883 CrossRef CAS PubMed.
- J. Y. Xu, X. Qiao, H. B. Song, S. P. Yan, D. Z. Liao, S. Gao, Y. Journaux and J. Cano, Chem. Commun., 2008, 6414–6416 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- T. J. Azbell, T. A. Pitt, M. M. Bollmeyer, C. Cong, K. M. Lancaster and P. Milner, Angew. Chem., Int. Ed., 2023, e202218252 CAS.
- T. Yang, T. Willhammar, H. Xu, X. Zou and Z. Huang, Nat. Protoc., 2022, 17, 2389–2413 CrossRef CAS PubMed.
- W. Xiang, Y. Zhang, Y. Chen, C.-J. Liu and X. Tu, J. Mater. Chem. A, 2020, 8, 21526–21546 RSC.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- W. Morris, B. Volosskiy, S. Demir, F. Gandara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- D. Feng, H.-L. Jiang, Y.-P. Chen, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Inorg. Chem., 2013, 52, 12661–12667 CrossRef CAS PubMed.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- V. Charra, P. de Frémont and P. Braunstein, Coord. Chem. Rev., 2017, 341, 53–176 Search PubMed.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- M. Woellner, S. Hausdorf, N. Klein, P. Mueller, M. W. Smith and S. Kaskel, Adv. Mater., 2018, 30, 1704679 CrossRef PubMed.
- J. E. Szulejko and K.-H. Kim, Sep. Purif. Technol., 2019, 228, 115729 CrossRef CAS.
- D. Zhang, J. Liu, M. Liu, L. Liu and D. D. Do, Sep. Purif. Technol., 2020, 251, 117306 CrossRef CAS.
- F. Illuzzi and H. Thewissen, J. Integr. Environ. Sci., 2010, 7, 201–210 Search PubMed.
- C. Y. Chuah, Y. Lee and T.-H. Bae, Chem. Eng. J., 2021, 404, 126577 Search PubMed.
- W. Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 Search PubMed.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 Search PubMed.
- S. Parshamoni, R. Nasani, A. Paul and S. Konar, Inorg. Chem. Front., 2021, 8, 693–699 RSC.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Havecker, A. Knop-Gericke and R. Schlogl, ACS Catal., 2015, 5, 2740–2753 Search PubMed.
- C.-H. Wang, W.-Y. Gao, Q. Ma and D. C. Powers, Chem. Sci., 2019, 10, 1823–1830 RSC.
- K. Wu, J.-K. Jin, X.-Y. Liu, Y.-L. Huang, P.-W. Cheng, M. Xie, J. Zheng, W. Lu and D. Li, J. Mater. Chem. C, 2022, 10, 11967–11974 RSC.
- K. Wu, X.-Y. Liu, M. Xie, P.-W. Cheng, J. Zheng, W. Lu and D. Li, Appl. Catal., B, 2023, 334, 122847 CrossRef CAS.
- T. Yan, J. H. Guo, Z. Q. Liu and W. Y. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 25937–25945 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- J. Thomas, M. E. Thomas, S. Thomas, A. Schechter and F. Grynszpan, Mater. Today Chem., 2024, 35, 101866 CrossRef CAS.
- M. Sadakiyo, H. Kasai, K. Kato, M. Takata and M. Yamauchi, J. Am. Chem. Soc., 2014, 136, 1702–1705 CrossRef CAS PubMed.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |