
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoacervates as enzymatic microreactors
Rif
Harris†
a,
Nofar
Berman†
a and
Ayala
Lampel
 *abcd
*abcd
aShmunis School of Biomedicine and Cancer Research, George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv, Israel. E-mail: ayalalampel@tauex.tau.ac.il
bCenter for Nanoscience and Nanotechnology Tel Aviv University, Tel Aviv, 69978, Israel
cSagol Center for Regenerative Biotechnology Tel Aviv University, Tel Aviv, 69978, Israel
dCenter for the Physics and Chemistry of Living Systems Tel Aviv University, Tel Aviv, 69978, Israel
First published on 14th March 2025
Abstract
Compartmentalization, a key aspect of biochemical regulation, naturally occurs in cellular organelles, including biomolecular condensates formed through liquid–liquid phase separation (LLPS). Inspired by biological compartments, synthetic coacervates have emerged as versatile microreactors, which can provide customed environments for enzymatic reactions. In this review, we explore recent advances in coacervate-based microreactors, while emphasizing the mechanisms by which coacervates accelerate enzymatic reactions, namely by enhancing substrate and enzyme concentrations, stabilizing intermediates, and providing molecular crowding. We discuss diverse coacervate systems, including those based on synthetic polymers, peptides, and nucleic acids, and describe the selection of enzymatic model systems, as well as strategies for enzyme recruitment and their impact on reaction kinetics. Furthermore, we discuss the challenges in monitoring reactions within coacervates and review the currently available techniques including fluorescence techniques, chromatography, and NMR spectroscopy. Altogether, this review offers a comprehensive perspective on recent progress and challenges in the design of coacervate microreactors, and addresses their potential in biocatalysis, synthetic biology, and nanotechnology.
 Rif Harris | Rif Harris is a PhD student in Professor Ayala Lampel's research group at the Shmunis School of Biotechnology and Cancer Research, Tel Aviv University. He holds a BSc in Chemistry and Biology and an MSc focused on developing LLPS-based bioreactors. In his PhD, Rif explores multicomponent phase-separating systems, studying molecular interactions that drive the formation of dynamic biomolecular structures with promising applications in biotechnology. |
 Nofar Berman | Nofar Berman received her BSc degree in Biotechnology from Tel Aviv University in 2024. She is currently a PhD student at the Shmunis School of Biotechnology and Cancer Research, Tel Aviv University under the supervision of Prof. Ayala Lampel. Her research focuses on the design of phase-separated materials and their applications in drug delivery systems for therapeutic antibodies. |
 Ayala Lampel | Ayala Lampel is an Associate Professor at the Shmunis School of Biomedicine and Cancer Research at Tel Aviv University (TAU). Her research group focuses on developing multicomponent biomolecular condensates from simple building blocks, including peptide- and nucleic acid-based polymers. The group investigates the potential of these condensates for various bionanotechnological applications, such as controlled catalysis, biocatalysis, molecular sensing, and the delivery of (macro)molecules. |
1. Introduction
Compartmentalization of biochemical reactions is essential for maintaining life and occurs mainly in cellular membrane-bound or membraneless organelles.1 The latter are dynamic assemblies, formed through liquid–liquid phase separation (LLPS) of various biomolecules including intrinsically disordered proteins (IDPs) and nucleic acids.2–5 Membraneless organelles, or biomolecular condensates, such as nucleoli, Cajal bodies, PML nuclear bodies, stress granules, and germ granules create distinct environments, which localize, organize, and concentrate enzymes and substrates and thus facilitate the efficiency of biochemical transformations.6,7 For instance, the mRNA decapping reaction occurs in P-bodies, where the Dcp1–Dcp2 decapping complex facilitates the degradation of mRNA.8 This process involves the removal of the protective 5′ cap from the mRNA molecule, marking it for degradation. PML protein nuclear bodies recruit and enrich substrates and enzymes of SUMOylation, a post transitional modification that covalently conjugates a small ubiquitin-like modifier protein (SUMO) to a target protein that is typically related to gene regulation.2,9 In stress granules, enzymes involved in protein translation regulation form condensates that modulate stress responses, without inducing harmful aggregation.7,10 Additionally, actin polymerization and cytoskeleton stabilization in neuronal synapses are often regulated by condensates composed of postsynaptic density proteins, which are responsible for communication between the postsynaptic neuronal activity and the spine cytoskeleton of the synapse.11Inspired by membrane-bound organelles and other hierarchical biological compartments such as viral capsids, a large variety of supramolecular nano- and micro-particles, including polymersomes,12–15 liposomes16,17 protein, peptide, or DNA cages have been examined as possible compartments for enzymatic reactions.17–20 These particles are relatively stable and can significantly enhance enzymatic reactions by mechanisms including enzyme immobilization.13,17,18,21–23 Unfortunately, due to their solid state, these particles may also have limited diffusivity, which hinders the recruitment of large and super charged macromolecules including enzymes present in oligomeric form.
In recent years and thanks to their liquid-like properties and spontaneous assembly,24 synthetic biomolecular condensates, or coacervates, have emerged as powerful tools for a wide range of nanobiotechnological applications, including drug delivery,25–28 biochemical sensing,29–31 tissue engineering,32 as adhesives33,34 and especially as microreactors.35–40 The interest in coacervate-based microreactor systems has grown rapidly, with a doubling in the number of studies in this field evidencing their growing importance and the increasing recognition of their potential.35,39,41 Coacervates offer several key advantages as microreactors, one being that they can provide a stable microenvironment that protects sensitive intermediates from degradation.42 This provision of a favourable microenvironment, by concentrating enzymes and substrates, providing molecular crowding, protecting against inhibitors and degradation, and stabilizing enzyme conformation, can greatly accelerate reaction kinetics. Thus, utilizing coacervates as reaction centers is especially attractive for applications where reaction efficiency, molecular stability, and precise regulation of biochemical processes are important.
The coacervates to be used as microreactors can be derived from synthetic polymers,43–48 or biomolecules including polypeptides, or nucleic acids,41,49–52 which are easy to produce by standard chemical synthesis methods and can be modified for specific applications. Typically, the polypeptide building blocks have intrinsic disordered regions, which gives them both flexibility and the ability to form the intermolecular interactions necessary for LLPS, while preventing the formation of more ordered secondary and tertiary structures.53 Common coacervate building blocks include simple polymers like polyethylene glycol and dextran (Dex), which form two distinct phases in an aqueous solution,54 but may also be polyelectrolytes and charged polymers like diethylaminoethyl-Dex,55,56 poly acrylic acid57,58 or polylysine59 which undergo complex coacervation through electrostatic interactions. Short peptides with an intrinsic disorder, derived from natural IDPs may also form condensates through multiple weak attractive forces.51,52,60 Importantly, coacervate microreactors can be designed to assemble/disassemble in response to environmental changes in pH, temperature, ionic strength, or molecular crowding. This dynamic responsiveness enables precise spatial and temporal control over the encapsulation and release of reactants or products, creating opportunities for targeted in situ production of molecules and their controlled delivery.61
In this review, we focus on recent advancements from the past 5–8 years in the field of coacervate-based microreactors. We begin by introducing various types of these microreactors and examining the criteria for selecting enzymatic models. Key factors include analytical considerations, enzymatic reactions that induce environmental changes (thereby affecting coacervate structure), reactions that model cellular biosynthetic pathways, and reactions with technological potential. We then review strategies for enzyme recruitment into coacervates, covering approaches such as spontaneous partitioning, enzyme conjugation to coacervate building blocks, and enzyme conjugation to ligands that bind to specific coacervate components. We discuss how these recruitment strategies influence reaction kinetics. Additionally, we explore the factors that impact reaction kinetics within coacervates, including molecular crowding and enzyme conformation stabilization by the condensed phase, highlighting cases where reactions are either accelerated or restricted. Finally, we review the methodologies currently available for monitoring enzymatic reactions in coacervates and discuss the analytical challenges they present. Overall, the aim of this review is to showcase recent progress in the emerging field of phase-separated microreactors based on designed coacervates, while exploring ongoing challenges and potential future directions.
2. Selection of enzyme model systems
The selection criteria for a model system providing enzymatic reactions in phase separated materials are reviewed below. In general, systems are selected because of their well-characterized properties, their complementary roles in enzymatic cascades, their relevance in mimicking cellular biochemical processes and studying spatial organization and reaction dynamics in synthetic coacervate systems, and for their potential in technological applications. Table 1 summarizes the enzyme model systems utilized in phase separated materials.| Enzyme | Substrates | Building blocks |
|---|---|---|
| HRP | Amplex red, H2O2;62–64o-phenylenediamine65 | Trans-azoTAB/Su-Amy;63 ELP–PEG;62 Q-Am/CM-Am;64 PDDA/ATP65 |
| Catalase | H2O266 | PDDA/PAA66 |
| GOx | Glucose43,65,67 | ATP/polylysine phase inside a GOx/DEAE-Dex phase;43 PDDA/dextran-sulfate and PLys/ADP;67 PDDA/ATP65 |
| Formate dehydrogenase | Formate and β-NAD+68,69 | PLys/ATP or CM-Dex;68 Plys/ATP or PDDA/CM-Dex69 |
| Nitric oxide synthase | Arginine70 | DEAE-Dex/PAA70 |
| β-Galactosidase | 4-Methylumbelliferyl β-D-4-galactopyranoside.71,72 | Ionized synthetic polymers;72 positive peptide/negative peptide or RNA71 |
| Urease | Urea,73–75 | PEG/Dex/PAA;74 PEG/BSA;75 PDDA/CM-Dex73 |
| α-Chymotrypsin | Ala-Ala-Phe-7-amido-4-methylcoumarin76 | PEG/Dex76 |
| Adenylate kinase | ADP36,37,77 | Enzyme conjugated to LCD of LAF1, Dbp1, Dhh136,37,77 |
| Luciferase | Furimazine78 | Enzyme conjugated to LCD of LAF178 |
| Chlorocatechol 1,2-dioxygenase | 4-Chlorocatechol79 | Enzyme conjugated to LCD of Dhh179 |
| C45 | o-Phenylenediamine and H2O244 | DEAE-Dex/CM-Dex44 |
| Dextranase | Dex47,48 | PDDA/ATP, tetraethyleneglycol/Dex;47 ATP/PDDA condensates in PEG/Dex48 |
| GMP–AMP synthase | GTP and ATP80 | cGAS with dsDNA80 |
| L-Lactate dehydrogenase | Pyruvate,75 NADPH81 | PEG/BSA;75 NADPH/arginine-rich peptide81 |
| Proteinase K | Proteinosomes,67 FITC-BSA82 | Poly-D-lysine/ADP, PDDA/Dex sulfate;67 C-Am/Q-Am with Succinylated BSA, PEG-PCLgTMC and terpolymer membrane82 |
| Glucose-6-phosphate dehydrogenase | Glucose-6-phosphate81 | NADPH/arginine-rich peptide81 |
| Enzymatic cascades | ||
| GOx, HRP | Glucose and amplex red;48,82–84 Glucose and o-phenylenediamine65 | C-Am/Q-Am;82–84 PDDA/ATP;65 PEG/Dex48 |
| GOx and urease | Glucose, urea85 | Negatively charged sodium alginate and cationized silk fibroin85 |
| Hexokinase and G6PDH | ATP, glucose, glucose-6-phosphate, and NADP86 | Polylysine and anionic nucleotides (ATP, ADP, NADP, NADPH, NAD, and NADH)86 |
| Protein kinase A and protein phosphatase 1α | ATP, phosphorylated c-Raf pS233/pS259 peptide87 | Q-Am/Cm-Am87 |
| Uricase and catalase | Uric acid and H2O288 | Polylysine/DNA88 |
| Uricase and HRP | Uric acid and Amplex Red89 | PDDA/Succinylated Dex89 |
| Tryptophanase and flavin-containing monooxygenase | L-Tryptophan and NADPH90 | Q-Am/Cm-Am and NTA-amylose90 |
| ASL and ATIC | SAICAR, AICAR, 10-fTHF, FAICAR and IMP91 | PEG/Dex91 |
| SAE1/2 and Ubc9 | PML peptide, RanGAP peptide92 | PolySH3 with polyPRM92 |
2.1 Analytical considerations
One of the most critical criteria from a technological aspect is the analytical consideration, i.e. the ability to monitor reactions quantitatively. In most cases, this entails selection of a reaction that gives rise to a chromophore product. The most common enzyme model system in this category is the horseradish peroxidase (HRP)-catalyzed oxidation of Amplex Red in the presence of hydrogen peroxide (H2O2), which results in the formation of the fluorescent compound resorufin.62–64 Glucose oxidase (GOx) is commonly paired with HRP due to its ability to catalyze the oxidation of glucose into gluconic acid and hydrogen peroxide, forming a simple model cascade reaction.93 This pairing facilitates the spatial analysis of reaction dynamics within phase-separated materials, guiding the development of more accurate enzymatic models that mimic the complexity of cellular biochemical processes.86,91 Additional well-characterized enzymatic models that generate chromophores e.g. fluorescein and 4-methylumbelliferone, are β-galactosidase,25,71,94 cellulase and phosphatase-catalyzed hydrolysis reaction95 and formate dehydrogenase-catalyzed oxidation of formate, which donates electrons to NAD+ to form NADH.68,69 An indigo forming enzymatic cascade has also been used in synthetic coacervate-artificial protocells. The cascade includes the enzymes tryptophan anhydrase, which is responsible for the conversion of L-Trp to indole, and flavin-containing monooxygenase, which oxidizes indole by consuming nicotinamide adenine dinucleotide phosphate (NADPH). The two enzymes do not have a natural affinity for each other but serve as a synthetic cascade that provides a spectroscopic readout.902.2 Enzymatic reactions which regulate phase separation
Other enzyme model systems are selected based on the ability to modulate environmental changes and thereby regulate phase separation. For instance, the commonly used GOx catalyzes the oxidation of glucose into gluconic acid and H2O2, which lowers the pH.43,67 This approach was used by Mann and coworkers to construct pH-sensitive coacervates which disassemble in response to glucose oxidation by GOx.67 Another study by the same group employed GOx and urease and the corresponding substrates glucose and urea to modulate the pH within coacervates. Such changes induce reversible structural changes in the coacervate system, which mimic the effects of pH fluctuations seen in cellular processes and improve the functionality of synthetic protocells.85 In this study, coacervate formation and dissolution were regulated by the concentration of glucose, which controls the level of GOx activity, the subsequent pH change and the related assembly, stability, and dissolution of the coacervates.85 An alternative approach involves the use of glucose-6-phosphate dehydrogenase and lactate dehydrogenase to regulate the redox state of NADPH/NADP+, which is key to the formation and dissolution of the NADPH and peptide-based coacervates.812.3 Enzymatic reactions as models for cellular biosynthesis pathways
Additional considerations in the enzyme model selection process are the biological roles of the enzymes and their involvement in sequential biosynthetic pathways. For example, Xia and coworkers studied the impact of phase separation on cascade reactions by examining enzymes involved in menaquinone (vitamin K2) and terpene biosynthesis.96 Additionally, Liu and coworkers developed condensed systems to control the biosynthesis of 2′-fucosyllactose, an ingredient in infant formula.49The two-enzyme cascade from the SUMOylation pathway was monitored in phase-separated artificial cells. In this system, the dimer enzyme SAE1/2 activates the protein modifier SUMO by conjugating it to an AMP molecule using ATP as a substrate, while Ubc9 conjugates the activated SUMO to a protein or peptide with a SUMO-binding motif. Since no fluorescent product is formed, SDS-PAGE was used as a non-spectroscopic method to assess product formation.92
The effects of compartmentalization on reaction dynamics were also studied by sequestering hexokinase and glucose-6-phosphate dehydrogenase in liquid droplets. These enzymes were selected for their biological significance and the sequential relationship in metabolic pathways.86
2.4 The technological potential of the enzyme models
An important selection consideration for enzymatic reactions is the technological potential i.e. biotechnology- or biomedical-related applications. Enzymes that catalyze in vitro transcription–translation of proteins have been employed in phase-separated materials,49,97–100 while other enzymatic model systems have potential in biomedicine. For instance, the enzyme nitric oxide synthase (NOS), which catalyzes the formation of nitric oxide and plays a critical role in inducing apoptosis in cancer cells, was sequestered in coacervate microdroplets as a delivery system for cancer therapy.70 Another example involves the enzyme catalase (CAT), which catalyzes the decomposition of H2O2 into water and oxygen, and protects cells by significantly reducing oxidative stress and neutralizing reactive oxygen species (ROS).66 Therefore, CAT opens opportunities in biomedical applications with potential as a therapeutic approach to treat oxidative stress-related diseases including cardiovascular or neurodegenerative disorders and cancer.101 Integrating CAT in coacervates as delivery systems might improve the efficacy in mitigating ROS-related cellular dysfunctions.66 This concept is exemplified by coacervate protocells that combine uricase and CAT-catalyzed reactions. Uricase degrades uric acid, while CAT neutralizes the resulting H2O2, thereby reducing blood uric acid levels and preventing renal injuries in hyperuricemia mice. The combination of the two enzymes in the coacervate system addresses the dual challenge of hyperuricemia treatment, namely effective uric acid degradation and elimination of toxic by-products.883. Analytical techniques for monitoring enzymatic reactions in coacervates
Enzymatic reactions within coacervates present unique challenges and opportunities for analysis. Due to the distinct environment within coacervates, conventional techniques must often be adapted or combined with more advanced methods to analyze reactions in the complex setting of phase separated materials. In this section, we review the current analytical approaches for studying enzymatic activity, structure, and interactions within coacervate systems, focusing on methods tailored to these specialized environments.3.1 Spectroscopic and microscopic methods
Spectroscopic and microscopic techniques are among the most common methods for analyzing enzymatic activity in coacervates. These methods provide a straightforward and real-time measure of enzymatic kinetics by monitoring changes in the absorbance or fluorescence of the reaction product or substrate over time.37,38,42,48,62–64,67–69,72,73,79,82–87,89,93,97,102 Fluorescence-based methods are particularly advantageous in coacervate environments as they allow for direct spatial analysis of reaction products. The same fluorescent product can be analyzed both spectroscopically and microscopically, enabling spatial analysis of product localization. Notably, products may form in the dilute phase and subsequently partition into the dense phase.Fluorescence-activated cell sorting (FACS) is another useful technique that goes beyond simple fluorescence measurements by providing insights into individual coacervates. FACS measures not only fluorescence intensity but also the size and shape of each coacervate, allowing researchers to differentiate between various condensate populations and acquire more precise data.67,103 However, these spectroscopic methods have limitations, such as requiring substrates or products with fluorescent or absorbent properties. Certain reactions, such as post-transitional modifications of proteins, do not produce products with spectroscopic properties.92,104 In addition, cascade reactions with an intermediate product whose absorbance or fluorescence spectrum overlaps that of the final product cannot be monitored spectroscopically.91 A further issue is that light scattering within coacervates may either mask low absorbance signals in the visible light or alter fluorescence signals.38,44,71,95,105 These problems can be addressed by preparing calibration curves of the products or substrates in the presence of coacervates.38,44,71 Interestingly, fluorescence quenching due to compartmentalization in coacervates can be leveraged to monitor enzymatic reactions. Studies on the coronavirus main protease (Mpro) used this principle by employing fluorescent dye-labelled peptides that form coacervates. Upon cleavage by Mpro, the coacervates disassemble, increasing the fluorescence of the labelling dye in the solution and enabling accurate measurement of kinetic parameters.105
3.2 Advanced fluorescence techniques: FRET and AIE
Advanced fluorescence techniques, such as Förster resonance energy transfer (FRET), have been used to monitor enzymatic activity within coacervates. FRET occurs when energy is transferred from a donor fluorophore to an acceptor fluorophore in close proximity. In studies of Mpro, a fluorescent dye that serves as a donor was conjugated to a peptide containing the cleavage site of Mpro, while the counterpart acceptor of this FRET pair was conjugated to a second peptide with an opposite charge. When the two peptides undergo LLPS to form coacervates, the FRET signal increases. Following the cleavage of the donor-carrying peptide by Mpro, the donor emission increases and the FRET signal decreases, which allows a direct measurement of the protease activity.105 Another advanced fluorescence-based approach is aggregation-induced emission (AIE), where fluorescence intensifies in the aggregated state. In Mpro studies, a fluorescent dye with AIE properties termed PyTPE was attached to peptides within coacervates. The coacervate formation increases PyTPE fluorescence, which then decreases upon coacervate disassembly following Mpro-catalyzed peptide cleavage, thereby providing another method to monitor the enzymatic activity.1053.3 Luminescence-based assays
Luminescence assays, such as those involving luciferase, provide a means of detecting enzymatic reactions through light emission without the need for external excitation. Luciferase catalyzes the conversion of luciferin to luminescent oxyluciferin in the presence of ATP, oxygen, and magnesium. This approach is particularly valuable for real-time monitoring of reactions in systems that lack fluorescent substrates or products. Artificial cell lines, such as THP1-Lucia cells, can also be used to produce luciferase when specific pathways are activated, thereby supporting the indirect monitoring of enzymes like cyclic GMP-AMP synthase.78,803.4 Chromatography and mass spectrometry techniques
Chromatographic techniques like high-performance liquid chromatography (HPLC) separate and quantify substances based on hydrophobicity.49,86,91,96,106 However, due to the need for organic solvents, which may dissolve or damage coacervates, HPLC is typically limited to endpoint analysis after the reaction is terminated. Liquid chromatography quadrupole time-of-flight mass spectrometry (LC–MS Q-ToF) offers a more advanced option by combining separation with precise mass detection. This method has been used to provide detailed information about the reaction intermediates of enzymatic phosphorylation by protein kinase A (PKA).873.5 Sodium dodecyl–sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and western blot
Sodium dodecyl–sulfate polyacrylamide gel electrophoresis (SDS–PAGE) separates proteins on a polyacrylamide gel by mass after treating the proteins with SDS to eliminate their spatial structure and normalize the charge.107 This method can be used to analyze enzymatic reactions of post-transitional modifications like SUMOylation92 or phosphorylation.104 Western blots are an extension of SDS–PAGE, by which specific proteins are identified by specific antibody binding. This method can be useful when the enzymatic reaction is performed in vivo, because the specific protein of interest can be distinguished from the large variety of other proteins present in the cell.1043.6 Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance (NMR) spectroscopy provides insights into molecular structures and dynamics within coacervates. Specifically, 2D 1H–13C NMR is commonly employed to analyze protein structures by measuring the local magnetic environments of nuclei through chemical bonds and spatial interactions with surrounding atoms. This technique correlates protons with the carbon-13 atoms they are directly attached to, providing detailed structural information and provides valuable information about real-time conformational changes and reaction progress.108 Additionally, 2D 1H–15N NMR is commonly used to analyze protein structures and detect changes in the chemical shifts of the amide backbone. It can also be used to study post-translational modifications, such as phosphorylation, and to quantify the extent of phosphorylation at each residue, thereby enabling real-time kinetic analysis of phosphorylation reactions.104,1093.7 Fluorescence recovery after photobleaching (FRAP)
Fluorescence recovery after photobleaching (FRAP) is used to measure molecular diffusion within coacervates and provide information about the spatial distribution of reactants and products. By analyzing diffusion patterns, FRAP helps reveal how molecular mobility within coacervates influences reaction rates, enabling a deeper understanding of how these structures affect enzymatic efficiency.63,66,68,87,893.8 Red edge emission spectroscopy (REES) and fluorescence anisotropy (FA)
Red edge emission spectroscopy (REES) and fluorescence anisotropy (FA) provide additional means of analyzing changes in enzymatic conformation and binding within coacervates. REES takes advantage of the red shift in tryptophan fluorescence in a slowly relaxing solvent environment to provide information about changes in the tertiary structure and conformational alterations of proteins.44 While this method is relatively straightforward to perform using a spectrophotometer, it restricts the use of tryptophan-containing building blocks in the design of coacervate building blocks. When a fluorescently labelled peptide binds to a larger protein, its rotational motion slows, leading to an increase in anisotropy. These changes in FA can therefore be used to quantify the binding affinities and protein interactions within coacervates.87,954. Recruitment strategies of enzymes into coacervates: enzyme immobilization to coacervate building blocks vs. enzymes as client molecules
4.1 Spontaneous diffusion of enzyme clients
Enzymes can be used as client molecules that spontaneously partition into the coacervate dense phase, mimicking the intracellular conditions of living cells48,66,68,69,72,73,86,88,89,91,93,97,110–113 (Fig. 1(a)). The use of free and unconjugated enzymes in phase separated systems is ideal in terms of biomimicry because it better mimics the natural compartmentalization of enzymes in cellular condensates while retaining the native conformation of the enzyme binding and catalytic sites. Yet, it is important to note that protein conformations may change upon uptake into condensates, potentially influencing enzymatic activity and interactions within the dense phase. However, the recruitment and localization of enzymes in the coacervate dense phase depend heavily on the physicochemical properties of the coacervate system i.e. the polarity and hydrophobicity71,114 of the building blocks, the propensity for electrostatic interactions and charge stoichiometry,72,81,95 and the localization of counterions.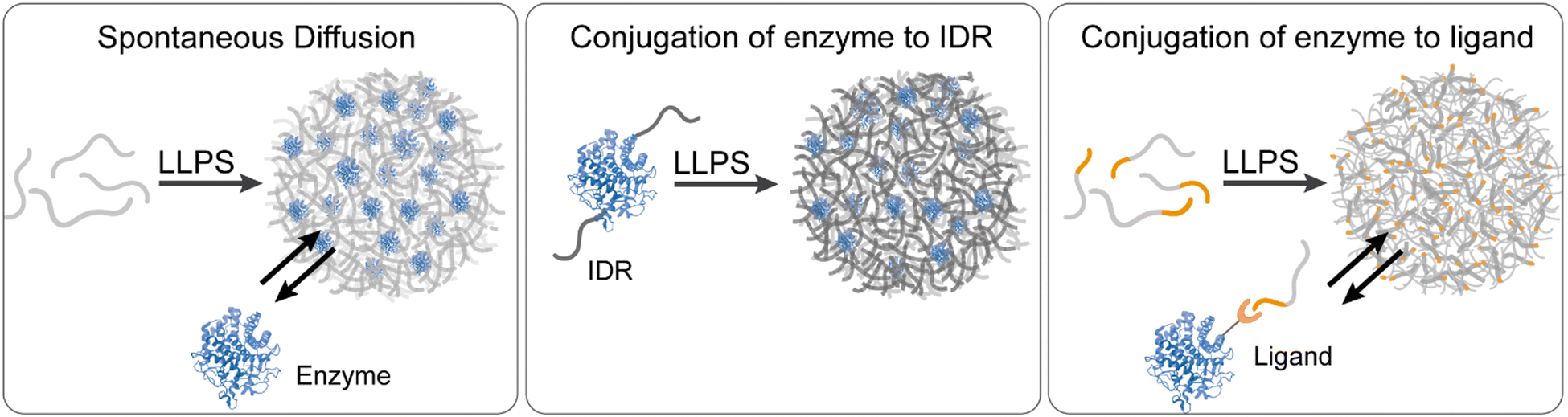 | ||
| Fig. 1 Enzymes recruitment strategies to coacervate reactors. The three central strategies for the recruitment of enzymes to the coacervate dense phase include spontaneous diffusion of enzymes into the condensates (left panel), conjugation of enzymes to IDRs which actively form the coacervates (middle panel), and conjugation of enzymes to ligands which bind specific motifs that are conjugated to the coacervate building block. Enzyme 3D structure is based on PDB 1IA7 (cellulase). | ||
4.2 Conjugation of enzymes to the coacervate building blocks
An alternative approach to the spontaneous diffusion of enzymes into coacervates as intact client molecules requires conjugation or fusion of enzymes to the coacervate-forming building blocks that undergo LLPS (Fig. 1(b)). This approach allows the recruitment of large enzymes, or enzymes which do not necessarily partition to the dense phase if added as intact proteins, due to a higher affinity for the dilute phase. The LLPS-promoting fusion domains are typically low complexity domains (LCDs) from IDRs or IDPs including LAF1, Ddh1, and Dbp1.36–38,79 These LCDs contain a more charged and polar amino acids than hydrophobic residues and are usually characterized by low sequence diversity.77,79 ELPs have been used to promote the recruitment of HRP through ELP–HRP bioconjugates. Under hyperosmotic conditions, the ELP–HRP bioconjugates undergo phase separation to form coacervates that concentrate the enzyme and substrates. This concept was further demonstrated by Arosio and coworkers in their study on NADH oxidase, where the enzyme was conjugated to various IDRs with varying net charges and the effect of IDR composition on the high local concentration of the enzyme, substrate and cofactor was studied.38 Another example involves fusing part of the enzyme β-galactosidase to recombinant intrinsically disordered polypeptides. When produced in bacteria cells, these polypeptides assemble into condensates that recruit the complementary part of β-galactosidase and restore the enzymatic activity.254.3 Recruitment of enzyme–ligand conjugates
In addition to direct conjugation to coacervate building blocks, enzymes can be recruited to the coacervate dense phase through conjugation to macromolecules which serve as ligands with specific affinity for the coacervate building block or associated domains (Fig. 1(c)). This approach was utilized by Rosen and Peeples92 to facilitate the inducible recruitment of enzymes of the SUMOylation enzymatic cascade into condensates that are formed by LLPS of the proteins PolySH33 and polyPRM5. For this purpose, a domain called FRB was attached to the N-terminus of polySH33, and the protein FKBP12 was attached to the SUMO-conjugating enzyme, Ubc9, and its substrates. Addition of the small molecule rapamycin triggers the binding of FRB and FKBP12, which results in the recruitment of the enzyme/substrate to polySH33 within the dense phase.92Another example, demonstrated by van Hest and coworkers, involved recruiting the enzymes tryptophanase and flavin-containing monooxygenase into amylose derivative coacervates. This was achieved using NTA-modified amylose, which binds Ni2+ ions and enables the recruitment of His-tagged enzymes through the formation of a strong coordination complex with the Ni2+ ions.90 This specific interaction facilitated the targeted and controlled sequestration of the enzymes into coacervate droplets.
Fusion of ligands to enzymes has proven to be especially efficient in coacervate systems where the enzymes cannot be directly conjugated to the building blocks, such as in all-DNA coacervate systems where LLPS is triggered by heating–cooling cycles that can be damaging for enzyme conformation and stability.115 To address this challenge, Deng and Walther115 fused GOx and HRP to streptavidin. This construct can then be recruited to the dense phase when the streptavidin binds to coacervate-forming biotinylated ssDNA. Similarly, conjugation of DNAzyme to a ‘barcode’ ssDNA sequence enables recruitment to the dense phase of all-DNA coacervates by hybridization to a complementary ssDNA sequence conjugated to the coacervate building block.103,116
5. Design of microreactor coacervates
The need for stability, compatibility, and dynamic responsiveness drives the selection of building blocks for condensate formation in synthetic biology. Careful choice of components with specific properties, allows researchers to construct coacervate systems that mimic natural cellular processes and enhance our understanding of enzymatic regulation and compartmentalization. Several aspects should be considered when selecting the building blocks for a designed coacervate microreactor i.e. the selection of building blocks that provide a chemically favourable phase for partitioning of the enzyme and substrate.5.1 Coacervate reactors based on synthetic polymers
Synthetic polymers are frequently employed as building blocks for coacervate-microreactors (Fig. 2). These are typically, two immiscible polymers e.g. polyethylene glycol (PEG) and Dextran (Dex),48,54,76,91,117,118 which form distinct phases through dissociative LLPS. Also, conjugation of positively or negatively charged moieties e.g. dimethylaminomethyl (DEAE),43–45,93 or carboxymethyl (CM),44,93 respectively, to Dex, enables the construction of multiphase reactor systems. Examples include formation of complexes between DEAE-Dex and glucose oxidase (GOx),43 poly(acrylic acid) (PAA), or DNA,119 or construction of multicompartment reactor protocells through a layer-by-layer assembly of DEAE-Dex and CM-Dex.93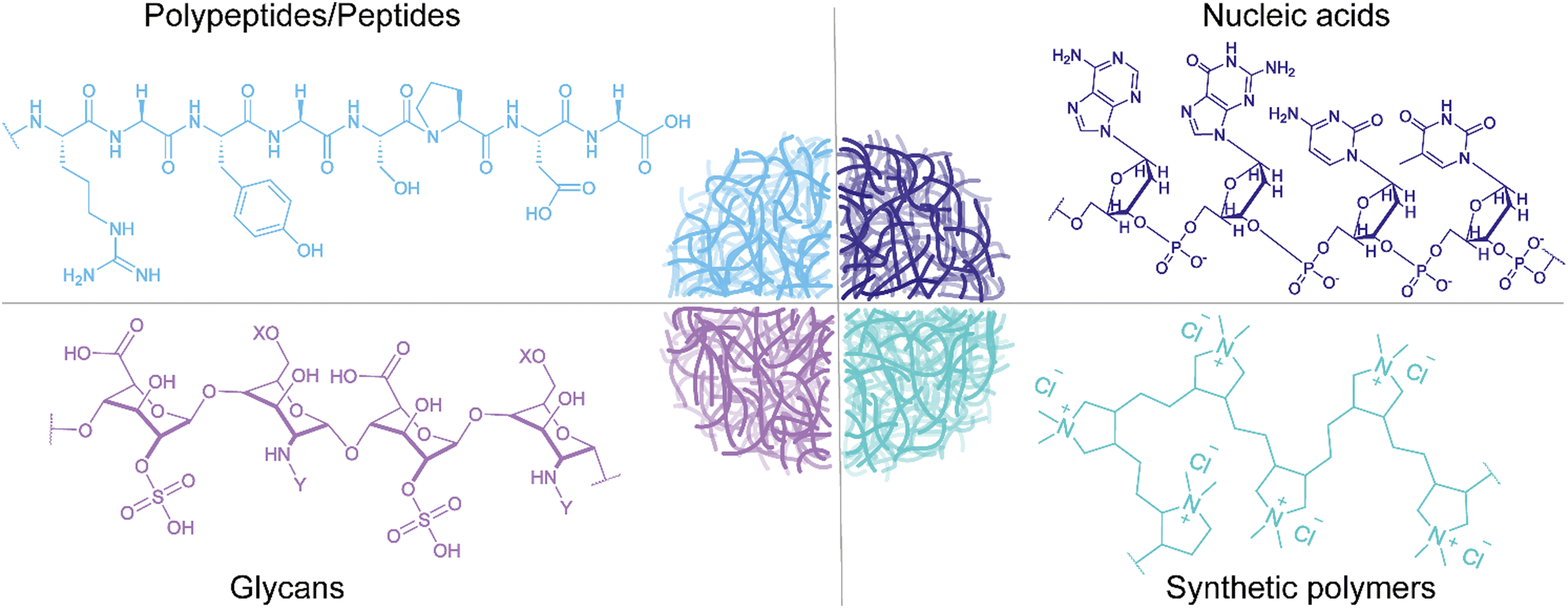 | ||
| Fig. 2 Schematic illustration of the four common building blocks of designed coacervate microreactors, including polypeptides/peptides, nucleic acid-based polymers, glycan-based polymers, and synthetic polymers. | ||
The cationic polymer PDDA has frequently been employed as a coacervate reactor building block,46–48,65,66,69,73,93 where the microreactor systems are based on the electrostatic complexation of PDDA and negatively charged polyelectrolytes including CM-Dex,69,73 PAA,66 ATP,65 DNA,93 and ATP in combination with PEG/Dex.48 Similarly, PDDA has also been used to form reactor coacervates by complexation with Dex sulfate (DS).67
5.2 Protein-based coacervate reactors
IDPs are the natural building blocks of the native cellular biomolecular condensates that host and concentrate enzymatic reactions physiologically. For this reason, both intact IDPs and their intrinsically disordered regions (IDRs) are frequently employed as building blocks of reactor coacervates (Fig. 2). Notably, the DEAD-box RNA helicase proteins LAF1, Dbp1, and Dhh1, which contain the LLPS-promoting arginine–glycine–glycine (RGG) repeating sequence are common sources of IDR motifs.36–38 Other proteins that also serve as coacervate reactor building blocks are involved in transcription–translation97 or are derived from the postsynaptic density (PSD) of neuronal synapses. The latter include the proteins GKAP, Shank, and Homer, which form cellular condensates that facilitate protein binding to actin bundles and actin polymerization.11,96 Additional proteins that naturally form cellular hubs for catalytic processes and were reported to undergo LLPS in vitro, include the conserved mRNA decapping complex, composed of the catalytic Dcp2 proteins and its corresponding activator Dcp1,8 and cyclic GMP-AMP synthetase (cGAS), which forms condensates with dsDNA through a disordered and positively charged N-terminal domain.80The elastin-like polypeptides (ELPs) and their conjugates, including ELP–polyethylene glycol (ELP–PEG) together with HRP linked to ELP (ELP–HRP) have been used as building blocks for a reactor coacervate encapsulated in lipid vesicles.62 When exposed to hyperosmotic stress, the ELP conjugates undergo phase separation where ELP–PEG acts as a stabilizer that supports compartmentalization and generates coacervates that increase the local concentration of HRP (Fig. 3(a)).62 Another example by Chilkoti and coworkers, mentioned in Section 4.2, involves in vivo formation of reactor condensates by 160–640 amino acid-long IDRs containing varying number of repeats of the sequence GRGDSPYS, derived from the Rec-1-resilin protein of Drosophilia melanogaster.25
 | ||
| Fig. 3 Types of coacervate microreactors. (a) Coacervate microreactors formed by ELP-b-PEG dense phase inside cytomimetic lipid compartments, which are formed in response to hyperosmotic stress. (b) Top: Chemical structure of a 14-mer cationic peptide building block. Bottom: The cationic peptide undergoes LLPS as a factor of ionic strength and pH to form simple coacervates that can recruit substrates and enzymes. (c) DNA-based coacervate reactors which are formed by sequence-controlled multiblock ssDNA polymers via rolling-circle amplification, leading to the formation of all-DNA coacervates with a liquid core and a crosslinked duplex shell. (d) Glycan-based coacervate reactors formed by mixing positively charged Q-Am and negatively charged Cm-Am and Ni-NTA-Am. His-tagged client proteins including phosphatase are recruited to the coacervate phase by Ni-His affinity. Client proteins are taken up based on their phosphorylation-dependent affinity and are released following the enzymatic dephosphorylation in the condensed phase. (a) was reproduced from ref. 62 with permission from Wiley-VCH GmbH; (b) was reproduced from ref. 52 with permission from Nature Publishing Group; (c) was reproduced from ref. 116 with permission from Nature Publishing Group; (d) was reproduced from ref. 87 with permission from Wiley-VCH GmbH. | ||
5.3 Peptide based coacervate reactors
Designed peptides are increasingly utilized as coacervate building blocks for various applications,26,27,29,31,51,60,120–123 including reactors49,71,95 (Fig. 2). The building blocks are frequently based on short IDR motifs with the incorporation of sticker amino acids. According to the stickers and spacers model, sticker amino acids promote associative forces including intermolecular interactions and are found in IDRs alongside spacer amino acids, which promote backbone flexibility.124 For instance, a 14-mer peptide composed of the LLPS promoting motif GRGRGR, three aromatic stickers, and an ELP domain52 was used to construct homotypic or heterotypic reactor coacervates (Fig. 3(b)).71,95 In addition, redox-responsive coacervate reactors were constructed by complex coacervation of short arginine (R)-rich peptides and NADPH. Incorporation of the fibronectin-derived RGD sequence improves selective binding to activated platelets at thrombus sites.815.4 Nucleic acid based coacervate reactors
Several studies have utilized DNA coacervates as microreactors (Fig. 2). These coacervates may be formed by electrostatic interactions through demixing of RNA, double stranded DNA (dsDNA) or single-stranded DNA (ssDNA) polymers with various cationic polyelectrolytes,88,93 polypeptides, or peptides.29,71 Alternatively, all-DNA coacervates may be formed through hydrogen-bonding of base pairs.103,115,125An all-DNA coacervate reactor developed by Walther and coworkers,103 involves base pairing of ssDNA sequences consisting of poly A20 and poly T20 strands, which contain barcode sequences. A DNAzyme was then immobilized within the dense phase through hybridization to the ssDNA barcode sequence (Fig. 3(c)).103
Another type of DNA-based reactor system is based on DNA nanostars.125 These are DNA assemblies with a secondary structure composed of several hybridized DNA strands with complementary sticky ends that hybridize to one another, creating a porous network of nanostar-like structures with liquid-like properties. The nanostar system described includes a 54 nucleic acid-long anchor strand, which hybridizes with ssRNA molecules and thereby becomes a substrate of RNase H, which degrades only hybridized RNA strands. Attaching different fluorophores to 14, 25 and 40 nucleic acid-long ssRNA strands, enabled monitoring the dynamics of RNAse H degradation of the differently sized materials. A further example of how the ssDNA sequences influence the properties of the coacervate reactor involves the light-controlled formation and disassembly of arylazopyrazole (AAP)-conjugated ssDNA-polylysine coacervates.126 Here, the results revealed that variations in the ssDNA sequences affect the efficiency of droplet formation under light exposure.
5.5 Glycan-based coacervate reactors
Amylose and its derivates may be employed as coacervate reactor building blocks (Fig. 2). Examples include coacervates formed by complexation of an anionic amylose derivative and synthetic cationic polymers, reinforced by a terpolymer membrane,72 or by the cationic quaternized amylose (Q-Am) and anionic carboxymethylated amylose (Cm-Am).87,90,127 In the latter coacervate system, amylose was functionalized with a nitrilotriacetic (NTA) group that coordinates Ni2+ and can bind his-tagged proteins. This approach was used to recruit his-tagged scaffold proteins and protein binders to study protein–protein interactions in the dense phase. Specifically, the study demonstrates that recruitment of proteins can be regulated by phosphorylation/dephosphorylation through the use of kinase and sequestration of phosphatase to the coacervate-artificial cell (Fig. 3(d)).87 As a further example, Huang and coworkers developed amylose-based coacervates with a light-controlled reactor. These coacervates are formed by hydrophobic and electrostatic interactions between succinylated amylose (Su-Amy) and azobenzene cation (trans-azoTAB). Under UV light, trans-azoTAB transitions to the more polar cis-azoTAB form, leading to the disassembly of the coacervate microdroplets. Under blue light, trans-azoTAB favours an interaction with Su-Amy, and the coacervate reassembles.63Additional example of a polysaccharide-based reactor system involves coacervates formed by electrostatic complexation of alginate and the positively charged cationized silk fibroin (CSF).85 The resulting membrane-like structures can be reversibly controlled by adjusting the pH within the protocells by adding the GOx/urease enzymatic system. Interestingly, the self-organization of CSF at the droplet surface under non-neutral charge conditions creates vesicles that resemble the cell membrane as they can selectively regulate the transport of solutes.
6. The two-way effect of coacervates on reaction kinetics
6.1 The increase in local enzyme/substrate concentration in the condensed phase
Most studies on enzymatic reactions in phase-separated materials report accelerated reaction rate within the enzyme-active dense phase. This is because spatial compartmentalization of enzymes and the appropriate substrate in membraneless condensates improves reaction efficiency by increasing the local concentrations.48,62,69,86,88–90,92,128 As an example, encapsulation of CAT within PDDA and polyacrylic acid (PAA) coacervates significantly increases the global catalytic efficiency of H2O2 degradation, and enhances ROS scavenging.66 This concept is also exemplified by coacervate protocells that combine uricase and CAT-catalyzed reactions. Uricase degrades uric acid, while CAT neutralizes the resulting H2O2, thereby reducing blood uric acid levels and preventing renal injuries in hyperuricemia mice. The combination of the two enzymes in the coacervate system increases their local concentration, and in turn the global reaction rates, thus improving cells viability by addressing the dual challenge of hyperuricemia treatment, namely effective uric acid degradation and elimination of toxic by-products.88Similarly, the significant acceleration of Amplex Red oxidation to resorufin by the DNAzyme quadruplex-duplex-hemin in the photoswitchable coacervate system formed by AAP-conjugated ssDNA and polylysine can be attributed to the increase in local concentration of reactants within the droplets.126
In addition to the local concentration of reaction components, coacervates can also accelerate the rate of enzymatic cascades by reducing the diffusion distance of substrates and enzymes in the different steps of the cascade.115,129 For instance, co-encapsulation and increase in local concentration of GOx and HRP within hierarchical protocells facilitates the efficient transfer of intermediates, such as H2O2, and accelerates the global reaction rates. This is because the diffusion of H2O2 directly to HRP in the condensed phase of the co-encapsulated system is faster than the diffusion from the coacervate to the dilute phase.82 Moreover, enriching rate-limiting enzymes within condensates can enhance their activity and accelerate the overall reaction pathway. For example, enrichment of the enzymes MenH, MenF, and MenD from the menaquinone biosynthesis pathway within coacervates increases the global production rate of 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate (SHCHC) by 70%. Notably, MenD remains the rate-limiting step, because the partitioning does not increase significantly.96 Similarly, the global conversion of the reaction catalyzed by the enzymes Idi and IspA, involved in the terpene biosynthesis pathway, was increased by their sequestration in coacervates.96 The increase in the local concentration of enzymes and substrates in the condensed phase of coacervates not only facilitates standard enzyme–substrate reactions but also enables the interaction of enzymes with non-canonical substrates. Specifically, Holt, Zweckstetter and coworkers reported that the increased phosphorylation activity in coacervates composed of a polypeptide with small ubiquitin-like protein (SUMO) and small ubiquitin-like protein interacting motif (SIM), produces hyperphosphorylation and phosphorylation at unusual sites. Specifically, the kinases FUS3 and CDK1, which normally do not phosphorylate ELK1, were able to achieve a 3-10-fold increased phosphorylation within the coacervate environment. This indicates that coacervate encapsulation can expand the functional capabilities of enzymes beyond their typical substrate specificity.104
Multiphase coacervates in which the local concentrations of enzymes and substrates are increased in the different phases have also been used to enhance reaction kinetics.48,82 For instance, when coacervates composed of PDDA and succinylated Dex (Su-Dex) at different charge ratios were formed within protein cage hybrid microcompartments, the local uric acid oxidation was sixfold faster than that observed when uricase was dispersed in the protein cages.89
6.2 The effect of the condensed phase on enzyme conformation
Direct interactions between coacervate building blocks and enzymes can cause conformational changes in enzymes, which in turn can enhance or inhibit their catalytic efficiency. For instance, the encapsulation of the negatively charged C45 peroxidase in positively charged DEAE-Dex/CM-Dex coacervates through electrostatic interactions, not only enhances molecular crowding but also increases the rigidity and stability of the enzyme, which in turn improves the catalytic activity.44 As another example, the globular protein β-lactoglobulin, typically a transport protein not associated with cellular LLPS, forms coacervates in vitro in the presence of PEG as a molecular crowder. LLPS of the β-lactoglobulin leads to conformational changes, including a reduction in β-sheet content, increased structural flexibility, and prevention of aggregation and denaturation, which ultimately result in esterase-like properties (Fig. 4(a)–(c)).102 | ||
| Fig. 4 Acceleration of enzymatic reaction kinetics in coacervates. (a)–(c). Acceleration of hydrolysis by β-lactoglobulin coacervates. (a) Schematic illustration of β-lactoglobulin and PEG LLPS which results in the formation into liquid droplets. β-Lactoglobulin undergoes conformational changes in the coacervate phase which allows it to catalyze the hydrolysis of ester bonds. (b) and (c) Comparative analysis of β-lactoglobulin catalysis showing 4-nitrophenyl acetate hydrolysis rate (b) and kcat (c). (d) and (e) Acceleration of NADH oxidation in coacervates that are formed by NADH oxidase conjugated to IDRs. (d) Coacervate reactors are formed by IDRs (or LCDs) derived from the DEAD-box proteins Dbp1, Laf1, and Ddx4, that have varying net charge. The IDRs are fused to the N-terminus of the enzyme, creating chimeric proteins with different net charges. (e) Reaction kinetics in coacervates with varying net charge, monitored by a decrease in NADH concentration. (a)–(c) were reproduced from ref. 102 with permission from Royal Society of Chemistry and (d) and (e) were reproduced from ref. 38 with permission from Nature publishing group. | ||
The formation of coacervates by the decapping complex Dcp1/2, which marks mRNA for degradation by removing the 7-methylguanosine cap, can inhibit the mRNA decapping activity due to interactions between the C-terminal and catalytic domains of Dcp2. NMR analysis showed that when the C-terminal inhibitory domain is removed, the mRNA decapping rate in Dcp1/Dcp2(core) coacervates increases 30-fold. Additionally, adding the enhancer decapping protein Edc3 to Dcp1/Dcp2 coacervates induces a conformational change in the Dcp1/Dcp2 complex, which lowers the Kd for RNA by two orders of magnitude, leading to a 90-fold increase in mRNA decapping efficiency.8 Similarly, cyclic GMP–AMP synthetase, which catalyzes the conversion of GTP and ATP into cyclic GMP–AMP, has a disordered and positively charged N-terminal region that induces the formation of cellular condensates with dsDNA. Removing this N-terminal region prevents the LLPS of cyclic GMP–AMP synthetase with dsDNA and decreases the catalytic activity. Furthermore, while cyclic GMP–AMP synthetase can also form condensates with dsRNA, it only becomes catalytically active in condensates with dsDNA, indicating that binding to the dsDNA induces a conformational change and exposure of the catalytic site.80
6.3 Reaction restriction
Inhibition of enzymatic reactions by compartmentalization could be a result of several factors: (i) spatial segregation between an enzyme and substrate, (ii) electrostatic and hydrophobic interactions of the enzyme with the coacervate building block, which might hinder enzyme–substrate binding, and (iii) changes in the chemical environment within the coacervates. For instance, the activity of HRP is inhibited upon encapsulation in Q-Am/CM-Am coacervates. While the Michaelis–Menten constant (Km) of H2O2 is similar in the dense and dilute phases, the Vmax of HRP within the coacervates is 3-fold lower than that in bulk solution. These changes are attributed to coacervate–protein interactions and the excluded volume effect, which restricts movement of the enzyme active site. This limited flexibility hinders substrate binding, leading to a decreased Vmax. A similar decrease in Vmax is observed for Amplex Red, where it is thought to be due to interactions between Amplex Red and the coacervate matrix, which may limit accessibility to the enzyme active site. These findings were also observed for HRP when substrates interact with increasing concentrations of crowding agents, indicating that molecular crowding increases Km and decreases Vmax (Fig. 5(a)–(c)).64 Keating and coworkers reported that the inhibition of the two cascading enzymes from the biosynthesis of purine in a PEG/Dex coacervate system is due to spatial separation between the enzymes, which are recruited to the condensed phase, and their substrates, which are mostly found in the dilute phase.91 Partitioning of enzymes and substrates in the coacervate phase are also affected by charge stoichiometries. This can be demonstrated by comparing the enzymes cellulase and alkaline phosphatase whose reactivity can be monitored by the formation of the same fluorescent product produced from different substrates. The results of a comparative analysis indicated that while the substrates preferentially partition to the condensed phase in a charge-mediated manner, the enzymes primarily partition to the dilute phase, resulting in a spatial segregation that inhibits the reactions due to limited enzyme–substrate interactions within the condensed phase (Fig. 5(d)–(f)).95 Further evidence for spatial hindrance is given by the inhibition of amide bond formation by the enzyme α-chymotrypsin in PEG/Dex coacervates. In this case, while both the enzyme and substrate partition to the Dex condensed phase, the reaction is restricted due to the low water content in the Dex phase, which is crucial for hydrolysis.76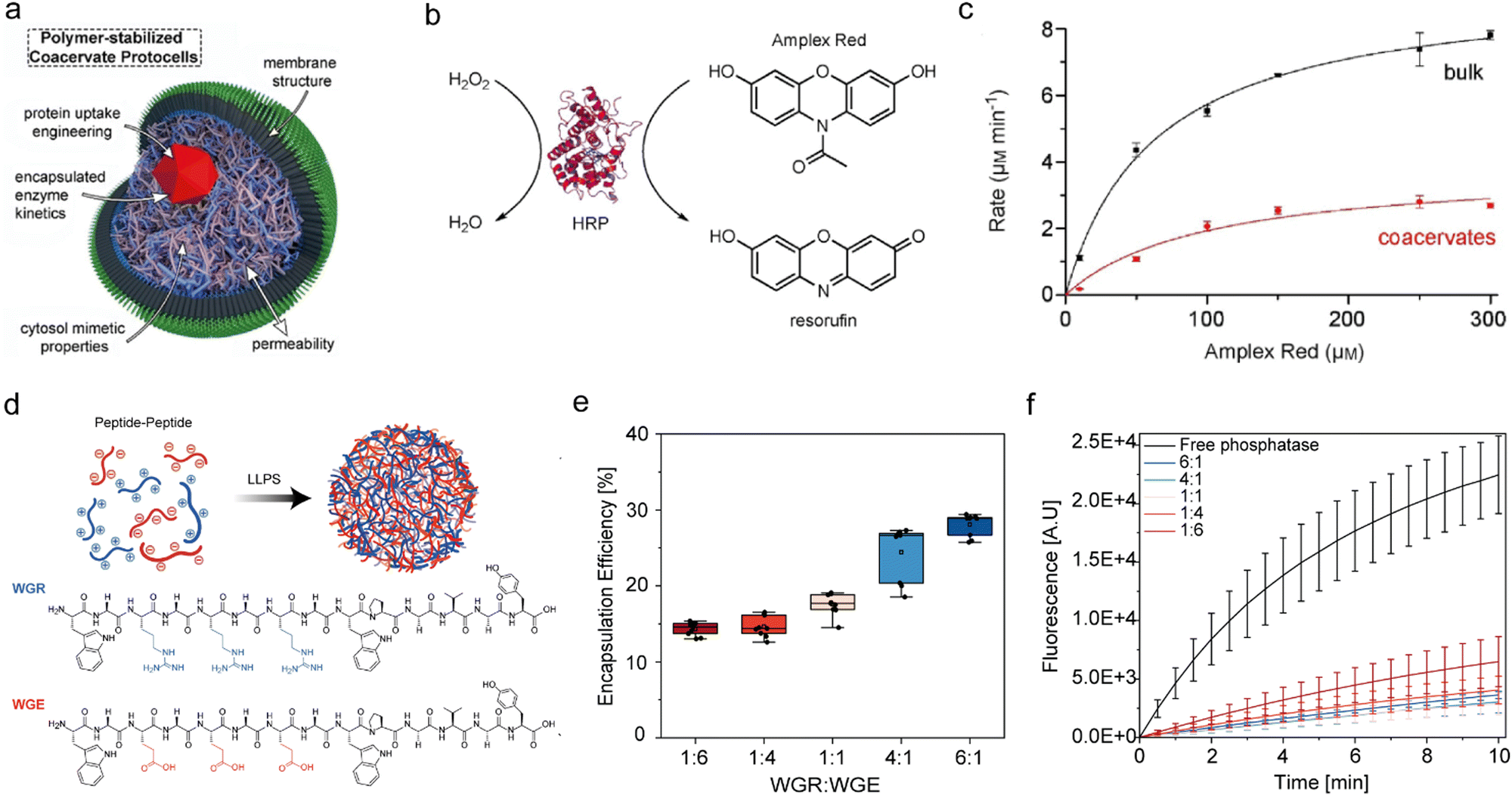 | ||
| Fig. 5 Inhibition of enzymatic reaction kinetics in coacervates. (a)–(c). HRP-catalyzed reactions are restricted in coacervates that are formed by amylose derivatives, surrounded by a terpolymer membrane structure (a). (b) Chemical structures of H2O2 and Amplex red used as substrates to form the fluorescent product resorufin. (c) Michaelis–Menten curve which shows the rate of the reaction catalyzed by free and compartmentalized HRP. (d)–(f). Restricted phosphatase activity in peptide coacervates. (d) Schematic illustration of two oppositely charged peptide building blocks (WGR:WGE). (e) Encapsulation efficiency (EE%) of fluorescently-labelled phosphatase in peptide coacervates with varying charge ratio, achieved by varying the stoichiometry of WGR:WGE. (f) Reaction kinetics of phosphatase-catalyzed hydrolysis in coacervates with varying charge ratios, monitored by fluorescence spectroscopy of the MU product. (a)–(c) were reproduced from ref. 64 with permission from European Chemical Societies Publishing and (d)–(f) were reproduced from ref. 95 with permission from Wiley-VCH GmbH. | ||
6.4 Reaction can be either accelerated or inhibited
Understanding the effects of the spatial organization, concentration, and interaction dynamics of enzymes and substrates within confined environments can inform the design of coacervates to modulate enzyme reaction rates as desired. Specifically, SUMOylation activity can be increased by up to 36-fold if the coacervate concentrates the reaction components. For substrates with a high Km, LLPS accelerates SUMOylation, whereas for those with a low Km, it may lead to inhibition due to competitive binding and substrate saturation.92 In the case of β-galactosidase, lower charge-density polycations enhance enzyme activity, which is inhibited by higher charge-density polycations, such as those composed of p(TMAEMA) and p(AEMA). This inhibition is probably due to competitive interactions with the negatively charged Glu-537 residue on β-galactosidase, which hinder substrate binding (Fig. 6(a)–(c)).72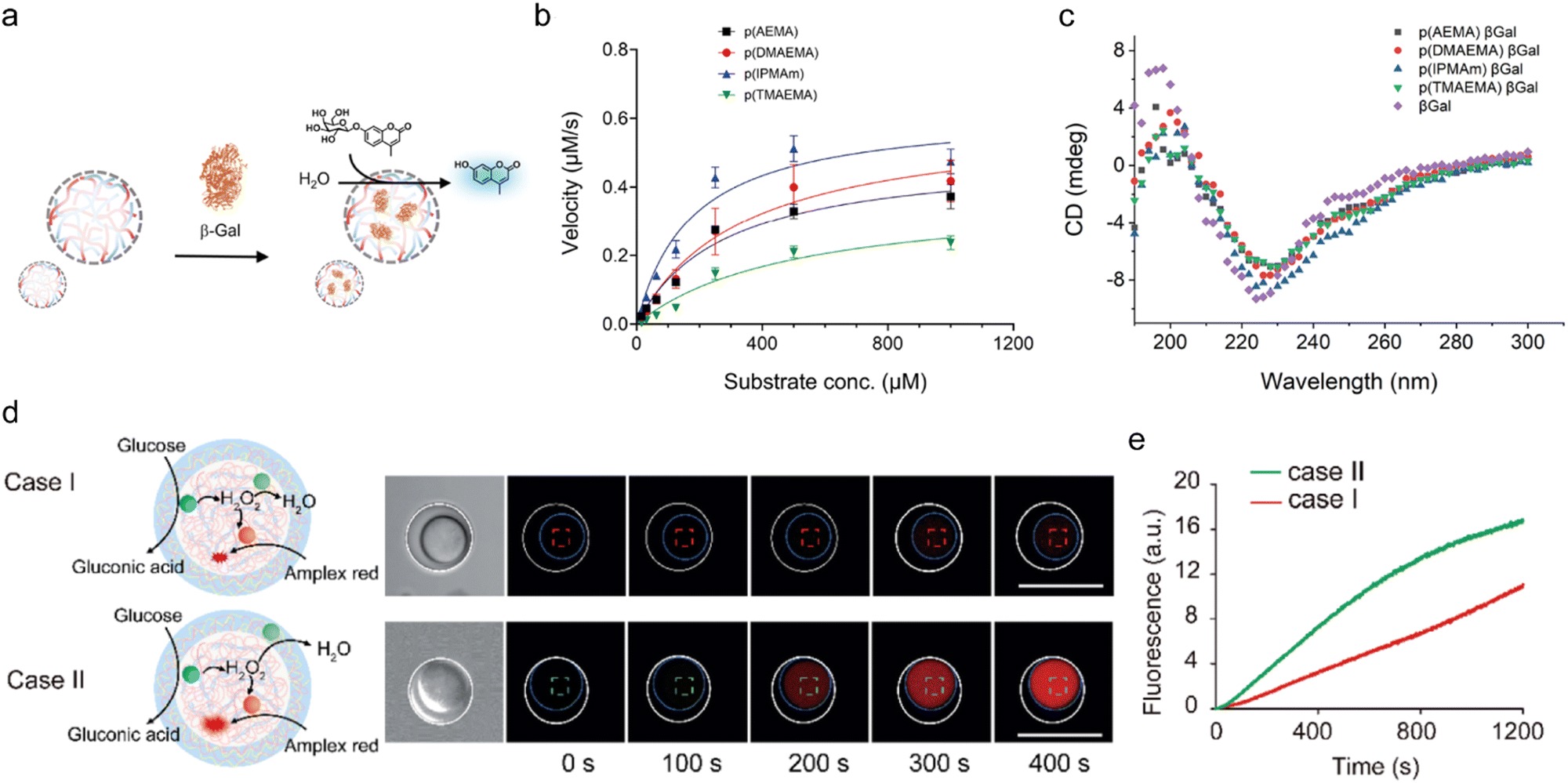 | ||
| Fig. 6 Acceleration vs. restriction of reaction kinetics in coacervates. (a)–(c) Inhibition of β-galactosidase activity in coacervates (a) that are formed by high charge-density polycations. (b) Michaelis–Menten kinetics plot of the reaction in coacervates that are formed by polycations with varying charge density. (c) CD spectroscopy of β-galactosidase in the different coacervates, showing secondary structure retention in the complex coacervates. (d), (e) Reaction acceleration/restriction by spatial organization of enzymatic cascade in a coacervate-in-coacervate multi-compartment system. (d) The reactions are restricted when GOx, HRP, and catalase are confined within the same compartment (case I) and accelerated when GOx and HRP are spatially separated from catalase (case II), as shown by bright-field and time-dependent fluorescence spectroscopy images upon glucose addition. The inner and outer coacervates were outlined with dashed blue and white circles, respectively. (e) Time profile of resorufin fluorescence following oxidation from Amplex Red by the cascade. (a)–(c) were reproduced from ref. 72 with permission from the American Chemical Society and (d), (e) were reproduced from ref. 93 with permission from the Royal Society of Chemistry. | ||
Another example of the effect of spatial organization is given by coacervate-in-coacervates constructed by a layer-by-layer assembly of PDDA and DNA, which form the inner coacervate microdroplets, and subsequent deposition of CM-Dex/DEAE-Dex, which forms the outer layer. Acceleration of the reaction is observed when GOx and HRP are separated from CAT within distinct compartments, allowing HRP to operate more efficiently with minimal competition for H2O2. Conversely, the reaction is inhibited when GOx, HRP, and CAT are confined within the same compartment, because the CAT consumes the available H2O2, thus preventing the HRP reaction from progressing effectively (Fig. 6(d) and (e)).93
7. Conclusions
Designing coacervates as efficient microreactors holds several challenges, particularly with respect to understanding how these environments enhance or restrict reaction kinetics. Coacervates typically accelerate enzymatic reactions by co-recruitment of enzymes and substrates in the dense phase, thereby increasing local concentrations and reducing diffusion distances48,66,126,128 or by reducing intermediate diffusion times and enhancing turnover rates by ensuring proximity between cascading enzymes.82 However, compartmentalization can also hinder reactions through spatial segregation of enzymes and substrates,91,95 unfavourable charge interactions, or restricted enzyme flexibility.72 Thus, it is still not trivial to predict which coacervate system will accelerate reaction kinetics. Designing microreactor coacervates requires a delicate balance between structural stability and the creation of a chemically favourable environment that allows both enzymes and substrates to partition effectively into the condensed phase. For instance, PEG and Dex are frequently used in synthetic polymer-based coacervates, where the more polar Dex preferentially attracts polar and charged macromolecules like enzymes. However, this poses challenges when apolar substrates, such as chromophores, are involved, as they tend not to co-partition with the enzymes.76,91 Fortunately, multiphase systems may be obtained by conjugating Dex to positively or negatively charged groups, such as DEAE or CM. Examples include coacervate systems composed of DEAE-Dex and GOx or DNA, which can facilitate enzyme localization and substrate compatibility,43,93 and coacervates that are formed by highly ionic polymers such as PDDA and Dex sulfate, which are stable at acidic pH.67 In amino acid-based coacervates, short peptides designed with aromatic or aliphatic stickers or short IDRs enhance coacervate stability and phase separation properties.50,130,131 For example, ELP conjugates can phase-separate under hyperosmotic stress, forming coacervates that localize and concentrate enzymes like HRP.62 Similarly, complexation of ssDNA or dsDNA polymers in nucleic acid-based coacervates with cationic polyelectrolytes like PDDA, creates microreactors that can host DNAzymes, and improve reaction kinetics within the dense phase.103 A key challenge in studying enzymatic reactions within coacervates lies in achieving high-resolution monitoring techniques that can accurately capture the dynamics and activity of enzymes in these complex environments. Current methods, such as spectroscopic and fluorescence-based approaches,42,48,62–64,67–69,72,73,82–87,89,93,97,102 often require specific fluorescent or absorbent substrates, thereby limiting their applicability. Additionally, the unique environment of coacervates can lead to significant light scattering and fluorescence quenching, which complicates data interpretation and reduces signal sensitivity. Techniques like FRET and AIE105 are useful but are restricted by the need for close fluorophore proximity and aggregation states, respectively. Chromatography and mass spectrometry49,86,87,91,96,106 provide detailed compositional data but are typically limited to endpoint analysis due to their reliance on organic solvents, which can dissolve or damage coacervates, making real-time monitoring challenging. NMR spectroscopy,8,104,108,109 while informative for structural analysis, requires high sample concentrations and specialized equipment, which limits its accessibility and utility for rapid kinetic studies. Furthermore, methods such as FRAP and REES44,63,66,68,87,89 are useful in analyzing the diffusion in the dense phase and enzyme conformation but do not directly quantify reaction rates or activity. When coacervates are engineered within cellular environments, indirect measurements of enzymatic activity, like cell growth or metabolite production,49 introduce additional complexity, as these indicators may be influenced by other cellular factors. These limitations highlight the need for developing advanced analytical tools and expanding the use of current high-resolution techniques such as NMR to achieve accurate monitoring of enzymatic activity and dynamics within the complex environment of coacervates. This will be required for more precise and comprehensive analysis in synthetic biology and biochemical research. Integrating computational models and high-resolution techniques132–138 will provide further information about coacervate dynamics and enable the rational design of coacervates with optimal reaction rates while avoiding inhibitory effects such as potential enzyme denaturation. These approaches will ultimately pave the way for leveraging coacervates for applications in biocatalysis, synthetic biology, and drug delivery.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Israel Science Foundation (ISF) grant no. 2589/21 and by the European Research Council (ERC) grant CORE 101162920. R. H. is funded by the ADAMA Center for Novel Delivery Systems in Crop Protection, Tel Aviv University. We thank the Chaoul Center for Nanoscale Systems of Tel Aviv University for the use of instruments and staff assistance.References
- Y. G. Zhao and H. Zhang, Dev. Cell, 2020, 55, 30–44 CrossRef CAS PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- W. Li, C. Jiang and E. Zhang, Transl. Cancer Res., 2021, 10, 4929–4946 CrossRef CAS PubMed.
- V. N. Uversky, Droplets of Life, Elsevier, 2023, pp. 101–132 Search PubMed.
- J. R. Espinosa, J. A. Joseph, I. Sanchez-Burgos, A. Garaizar, D. Frenkel and R. Collepardo-Guevara, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 13238–13247 CrossRef CAS PubMed.
- B. G. O’Flynn and T. Mittag, Curr. Opin. Cell Biol., 2021, 69, 70–79 CrossRef PubMed.
- T. Hirose, K. Ninomiya, S. Nakagawa and T. Yamazaki, Nat. Rev. Mol. Cell Biol., 2023, 24, 288–304 CrossRef CAS PubMed.
- R. W. Tibble, A. Depaix, J. Kowalska, J. Jemielity and J. D. Gross, Nat. Chem. Biol., 2021, 17, 615–623 CrossRef CAS PubMed.
- Y. Chen, Z. Jiang, Y. Yang, C. Zhang, H. Liu and J. Wan, Int. J. Biol. Macromol., 2023, 253, 126773 CrossRef CAS PubMed.
- P. Ivanov, N. Kedersha and P. Anderson, Cold Spring Harbor Perspect. Biol., 2019, 11, 1–18 Search PubMed.
- X. Chen, B. Jia, S. Zhu and M. Zhang, eLife, 2023, 12, 1–22 CrossRef CAS PubMed.
- Y. Wang, Q. Zhao, R. Haag and C. Wu, Angew. Chem., Int. Ed., 2022, 61, e202213974 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- L. Ruiz-Pérez, J. Madsen, E. Themistou, J. Gaitzsch, L. Messager, S. P. Armes and G. Battaglia, Polym. Chem., 2015, 6, 2065–2068 RSC.
- J. van Hest and H. Che, ChemNanoMat, 2019, 5, 1092–1109 CrossRef.
- B. C. Buddingh’ and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef PubMed.
- M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Curr. Opin. Biotechnol, 2014, 28, 10–16 CrossRef CAS PubMed.
- F. Xu, Q. Xia and P. Wang, Front. Chem., 2020, 8, 751 CrossRef CAS PubMed.
- Q. Chi, Z. Yang, K. Xu, C. Wang and H. Liang, Front. Pharmacol., 2019, 10, 1585 CrossRef CAS PubMed.
- N. A. Yewdall, A. F. Mason and J. C. M. van Hest, Interface Focus, 2018, 8, 20180023 CrossRef PubMed.
- S. B. P. E. Timmermans and J. C. M. van Hest, Curr. Opin. Colloid Interface Sci., 2018, 35, 26–35 CrossRef CAS.
- H. Seo and H. Lee, Biomicrofluidics, 2021, 15, 021301 CrossRef CAS PubMed.
- R. J. Wheeler and A. A. Hyman, Philos. Trans. R. Soc., B, 2018, 373, 20170193 CrossRef PubMed.
- M. Dzuricky, B. A. Rogers, A. Shahid, P. S. Cremer and A. Chilkoti, Nat. Chem., 2020, 12, 814–825 CrossRef CAS PubMed.
- I. Katzir, E. Haimov and A. Lampel, Adv. Mater., 2022, 34, e2206371 CrossRef PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- Y. Sun, X. Xu, L. Chen, W. L. Chew, Y. Ping and A. Miserez, ACS Nano, 2023, 17, 16597–16606 CrossRef CAS PubMed.
- A. Netzer, I. Katzir, A. Baruch Leshem, M. Weitman and A. Lampel, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2310569120 CrossRef CAS PubMed.
- C. M. Green, D. Sementa, D. Mathur, J. S. Melinger, P. Deshpande, S. Elbaum-Garfinkle, I. L. Medintz, R. V. Ulijn and S. A. Díaz, Commun. Chem., 2024, 7, 49 CrossRef CAS PubMed.
- D. Gaash, S. Dewan, A. B. Leshem, K. S. Jaiswal, R. Jelinek and A. Lampel, Chem. Commun., 2023, 59, 12298–12301 RSC.
- S. Roberts, T. S. Harmon, J. L. Schaal, V. Miao, K. J. Li, A. Hunt, Y. Wen, T. G. Oas, J. H. Collier, R. V. Pappu and A. Chilkoti, Nat. Mater., 2018, 17, 1154–1163 CrossRef CAS PubMed.
- J. Sun, L. Xiao, B. Li, K. Zhao, Z. Wang, Y. Zhou, C. Ma, J. Li, H. Zhang, A. Herrmann and K. Liu, Angew. Chem., Int. Ed., 2021, 60, 23687–23694 CrossRef CAS PubMed.
- B. K. Ahn, S. Das, R. Linstadt, Y. Kaufman, N. R. Martinez-Rodriguez, R. Mirshafian, E. Kesselman, Y. Talmon, B. H. Lipshutz, J. N. Israelachvili and J. H. Waite, Nat. Commun., 2015, 6, 8663 CrossRef CAS PubMed.
- A. Chatterjee, A. Reja, S. Pal and D. Das, Chem. Soc. Rev., 2022, 51, 3047–3070 RSC.
- U. Capasso Palmiero, A. M. Küffner, F. Krumeich, L. Faltova and P. Arosio, Angew. Chem., Int. Ed., 2020, 59, 8138–8142 CrossRef CAS PubMed.
- A. M. Küffner, M. Prodan, R. Zuccarini, U. Capasso Palmiero, L. Faltova and P. Arosio, ChemSystemsChem, 2020, 2, e2000001 CrossRef.
- M. Gil-Garcia, A. I. Benítez-Mateos, M. Papp, F. Stoffel, C. Morelli, K. Normak, K. Makasewicz, L. Faltova, F. Paradisi and P. Arosio, Nat. Commun., 2024, 15, 3322 CrossRef CAS PubMed.
- I. B. A. Smokers, B. S. Visser, A. D. Slootbeek, W. T. S. Huck and E. Spruijt, Acc. Chem. Res., 2024, 57, 1885–1895 CrossRef CAS PubMed.
- S. Ghosh, M. G. Baltussen, N. M. Ivanov, R. Haije, M. Jakštaitė, T. Zhou and W. T. S. Huck, Chem. Rev., 2024, 124, 2553–2582 CrossRef CAS PubMed.
- Y. Dai, L. You and A. Chilkoti, Nat. Rev. Bioeng., 2023, 1–15 Search PubMed.
- B. Saini and T. K. Mukherjee, J. Phys. Chem. B, 2023, 127, 180–193 CrossRef CAS PubMed.
- H. Karoui, M. J. Seck and N. Martin, Chem. Sci., 2021, 12, 2794–2802 RSC.
- E. Kluczka, V. Rinaldo, A. Coutable-Pennarun, C. Stines-Chaumeil, J. L. R. Anderson and N. Martin, ChemCatChem, 2024, 16, e202400558 CrossRef CAS.
- T. Ivanov, S. Cao, N. Bohra, M. de Souza Melchiors, L. Caire da Silva and K. Landfester, ACS Appl. Mater. Interfaces, 2023, 15, 50755–50764 CrossRef CAS PubMed.
- A. A. D. T. Abeysinghe, E. J. Young, A. T. Rowland, L. C. Dunshee, S. Urandur, M. O. Sullivan, C. A. Kerfeld and C. D. Keating, Small, 2024, 20, e2308390 CrossRef PubMed.
- N. G. Moreau, N. Martin, P. Gobbo, T.-Y. D. Tang and S. Mann, Chem. Commun., 2020, 56, 12717–12720 RSC.
- T. Kojima and S. Takayama, ACS Appl. Mater. Interfaces, 2018, 10, 32782–32791 CrossRef CAS PubMed.
- W. Yu, K. Jin, D. Wang, N. Wang, Y. Li, Y. Liu, J. Li, G. Du, X. Lv, J. Chen, R. Ledesma-Amaro and L. Liu, Nat. Commun., 2024, 15, 7989 CrossRef CAS PubMed.
- M. Abbas, W. P. Lipiński, K. K. Nakashima, W. T. S. Huck and E. Spruijt, Nat. Chem., 2021, 13, 1046–1054 CrossRef CAS PubMed.
- C. Yuan, Q. Li, R. Xing, J. Li and X. Yan, Chem, 2023, 9, 2425–2445 CAS.
- A. Baruch Leshem, S. Sloan-Dennison, T. Massarano, S. Ben-David, D. Graham, K. Faulds, H. E. Gottlieb, J. H. Chill and A. Lampel, Nat. Commun., 2023, 14, 421 CrossRef CAS PubMed.
- W. Borcherds, A. Bremer, M. B. Borgia and T. Mittag, Curr. Opin. Struct. Biol., 2021, 67, 41–50 CrossRef CAS PubMed.
- T. Massarano, A. Baruch Leshem, M. Weitman and A. Lampel, ACS Appl. Mater. Interfaces, 2022, 14, 20520–20527 CrossRef CAS PubMed.
- W. Mu, Z. Ji, M. Zhou, J. Wu, Y. Lin and Y. Qiao, Sci. Adv., 2021, 7, eabf9000 CrossRef CAS PubMed.
- H. V. Le, V. Dulong, L. Picton and D. Le Cerf, Colloids Surf., A, 2021, 629, 127485 CrossRef CAS.
- E. N. Durmaz, J. D. Willott, A. Fatima and W. M. de Vos, Eur. Polym. J., 2020, 139, 110015 CrossRef CAS.
- A. Rizvi and J. P. Patterson, Soft Matter, 2024, 20, 1978–1982 RSC.
- T. Nobeyama, T. Furuki and K. Shiraki, Langmuir, 2023, 39, 17043–17049 CrossRef CAS PubMed.
- M. Abbas, W. P. Lipiński, J. Wang and E. Spruijt, Chem. Soc. Rev., 2021, 50, 3690–3705 RSC.
- N. Martin, ChemBioChem, 2019, 20, 2553–2568 CrossRef CAS PubMed.
- C. Schvartzman, H. Zhao, E. Ibarboure, V. Ibrahimova, E. Garanger and S. Lecommandoux, Adv. Mater., 2023, 35, e2301856 CrossRef PubMed.
- Y. Huang, X. Wang, J. Li, Y. Lin, H. Chen, X. Liu and X. Huang, ChemSystemsChem, 2021, 3, e2100006 CrossRef CAS.
- N. A. Yewdall, B. C. Buddingh, W. J. Altenburg, S. B. P. E. Timmermans, D. F. M. Vervoort, L. K. E. A. Abdelmohsen, A. F. Mason and J. C. M. van Hest, ChemBioChem, 2019, 20, 2643–2652 CrossRef CAS PubMed.
- L. Tian, M. Li, J. Liu, A. J. Patil, B. W. Drinkwater and S. Mann, ACS Cent. Sci., 2018, 4, 1551–1558 CrossRef CAS PubMed.
- Y. Chen, M. Yuan, Y. Zhang, S. Zhou, K. Wang, Z. Wu and J. Liu, Biomater. Sci., 2022, 10, 4588–4595 RSC.
- Y. Qiao, M. Li, D. Qiu and S. Mann, Angew. Chem., Int. Ed., 2019, 58, 17758–17763 CrossRef CAS PubMed.
- C. Love, J. Steinkühler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T.-Y. D. Tang, Angew. Chem., 2020, 132, 6006–6013 CrossRef.
- T. Beneyton, C. Love, M. Girault, T.-Y. D. Tang and J. Baret, ChemSystemsChem, 2020, 2, e2000022 CrossRef CAS.
- Y. Zhang, Y. Yao, S. Liu, Y. Chen, S. Zhou, K. Wang, X. Yang and J. Liu, J. Mater. Chem. B, 2021, 9, 9784–9793 RSC.
- R. Harris, S. Veretnik, S. Dewan, A. Baruch Leshem and A. Lampel, Commun. Chem., 2024, 7, 90 CrossRef CAS PubMed.
- A. B. Cook, B. D. Gonzalez and J. C. M. van Hest, Biomacromolecules, 2024, 25, 425–435 CrossRef CAS PubMed.
- G. B. Perin, S. Moreno, Y. Zhou, M. Günther, S. Boye, B. Voit, M. I. Felisberti and D. Appelhans, Biomacromolecules, 2023, 24, 5807–5822 CrossRef CAS PubMed.
- A. T. Rowland, D. N. Cacace, N. Pulati, M. L. Gulley and C. D. Keating, Chem. Mater., 2019, 31, 10243–10255 CrossRef CAS.
- A. Testa, M. Dindo, A. A. Rebane, B. Nasouri, R. W. Style, R. Golestanian, E. R. Dufresne and P. Laurino, Nat. Commun., 2021, 12, 6293 CrossRef CAS PubMed.
- R. Oliva, S. Banerjee, H. Cinar and R. Winter, Chem. Commun., 2020, 56, 395–398 RSC.
- L. Faltova, A. M. Küffner, M. Hondele, K. Weis and P. Arosio, ACS Nano, 2018, 12, 9991–9999 CrossRef CAS PubMed.
- M. Guan, M. V. Garabedian, M. Leutenegger, B. S. Schuster, M. C. Good and D. A. Hammer, Biochemistry, 2021, 60, 3137–3151 CrossRef CAS PubMed.
- N. N. Evangelista, M. C. Micheletto, E. Kava, L. F. S. Mendes and A. J. Costa-Filho, Int. J. Biol. Macromol., 2024, 270, 132294 CrossRef CAS PubMed.
- M. Du and Z. J. Chen, Science, 2018, 361, 704–709 CrossRef CAS PubMed.
- J. Wang, M. Abbas, Y. Huang, J. Wang and Y. Li, Commun. Chem., 2023, 6, 243 CrossRef CAS PubMed.
- A. F. Mason, N. A. Yewdall, P. L. W. Welzen, J. Shao, M. van Stevendaal, J. C. M. van Hest, D. S. Williams and L. K. E. A. Abdelmohsen, ACS Cent. Sci., 2019, 5, 1360–1365 CrossRef CAS PubMed.
- A. Jobdeedamrong, S. Cao, I. Harley, D. Crespy, K. Landfester and L. Caire da Silva, Nanoscale, 2023, 15, 2561–2566 RSC.
- A. F. Mason, B. C. Buddingh’, D. S. Williams and J. C. M. van Hest, J. Am. Chem. Soc., 2017, 139, 17309–17312 CrossRef CAS PubMed.
- Z. Yin, L. Tian, A. J. Patil, M. Li and S. Mann, Angew. Chem., Int. Ed., 2022, 61, e202202302 CrossRef CAS PubMed.
- T. Ura, S. Tomita and K. Shiraki, Chem. Commun., 2021, 57, 12544–12547 RSC.
- T. W. van Veldhuisen, W. J. Altenburg, M. A. M. Verwiel, L. J. M. Lemmens, A. F. Mason, M. Merkx, L. Brunsveld and J. C. M. van Hest, Adv. Mater., 2023, 35, e2300947 CrossRef PubMed.
- Q. Hu, H. Lan, Y. Tian, X. Li, M. Wang, J. Zhang, Y. Yu, W. Chen, L. Kong, Y. Guo and Z. Zhang, J. Control. Release, 2024, 365, 176–192 CrossRef CAS PubMed.
- J. Li, M. Zhu, S. Wang, Z. Tao, X. Liu and X. Huang, Chem. Commun., 2021, 57, 11713–11716 RSC.
- W. J. Altenburg, N. A. Yewdall, D. F. M. Vervoort, M. H. M. E. van Stevendaal, A. F. Mason and J. C. M. van Hest, Nat. Commun., 2020, 11, 6282 CrossRef CAS PubMed.
- B. W. Davis, W. M. Aumiller, N. Hashemian, S. An, A. Armaou and C. D. Keating, Biophys. J., 2015, 109, 2182–2194 CrossRef CAS PubMed.
- W. Peeples and M. K. Rosen, Nat. Chem. Biol., 2021, 17, 693–702 CrossRef CAS PubMed.
- Y. Chen, M. Yuan, Y. Zhang, S. Liu, X. Yang, K. Wang and J. Liu, Chem. Sci., 2020, 11, 8617–8625 RSC.
- M. Abbas, J. O. Law, S. N. Grellscheid, W. T. S. Huck and E. Spruijt, Adv. Mater., 2022, 34, e2202913 CrossRef PubMed.
- A. Lampel, R. Harris and N. Berman, ChemSystemsChem, 2025, 1, e202400055 Search PubMed.
- M. Liu, S. He, L. Cheng, J. Qu and J. Xia, Biomacromolecules, 2020, 21, 2391–2399 CrossRef CAS PubMed.
- L. L. J. Schoenmakers, N. A. Yewdall, T. Lu, A. A. M. André, F. H. T. Nelissen, E. Spruijt and W. T. S. Huck, ACS Synth. Biol., 2023, 12, 2004–2014 CrossRef CAS PubMed.
- A. A. M. André and E. Spruijt, Int. J. Mol. Sci., 2020, 21, 5908 CrossRef PubMed.
- C. D. Reinkemeier, G. E. Girona and E. A. Lemke, Science, 2019, 363, 6434 CrossRef PubMed.
- C. D. Reinkemeier and E. A. Lemke, Cell, 2021, 184, 4886–4903 CrossRef CAS PubMed.
- K. Jomova, R. Raptova, S. Y. Alomar, S. H. Alwasel, E. Nepovimova, K. Kuca and M. Valko, Arch. Toxicol., 2023, 97, 2499–2574 CrossRef CAS PubMed.
- S. Rai, S. Pramanik and S. Mukherjee, Chem. Sci., 2024, 15, 3936–3948 RSC.
- A. Samanta, M. Hörner, W. Liu, W. Weber and A. Walther, Nat. Commun., 2022, 13, 3968 CrossRef CAS PubMed.
- D. Sang, T. Shu, C. F. Pantoja, A. Ibáñez de Opakua, M. Zweckstetter and L. J. Holt, Mol. Cell, 2022, 82, 3693–3711 CrossRef CAS PubMed.
- Z. Jin, C. Ling, W. Yim, Y.-C. Chang, T. He, K. Li, J. Zhou, Y. Cheng, Y. Li, J. Yeung, R. Wang, P. Fajtová, L. Amer, H. Mattoussi, A. J. O’Donoghue and J. V. Jokerst, ACS Nano, 2023, 17, 16980–16992 CrossRef CAS PubMed.
- A. Bhattacharya, H. Niederholtmeyer, K. A. Podolsky, R. Bhattacharya, J.-J. Song, R. J. Brea, C.-H. Tsai, S. K. Sinha and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18206–18215 CrossRef CAS PubMed.
- A. B. Nowakowski, W. J. Wobig and D. H. Petering, Metallomics, 2014, 6, 1068–1078 CrossRef CAS PubMed.
- T.-L. Hwang, D. Batabyal, N. Knutson and M. Wikström, Molecules, 2014, 6, 1068–1078 CrossRef PubMed.
- P. Chakraborty, A. Ibáñez de Opakua, J. A. Purslow, S. A. Fromm, D. Chatterjee, M. Zachrdla, S. Zhuang, S. Puri, B. Wolozin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2414176121 CrossRef CAS PubMed.
- N. Modi, S. Chen, I. N. A. Adjei, B. L. Franco, K. J. M. Bishop and A. C. Obermeyer, Chem. Sci., 2023, 14, 4735–4744 RSC.
- C. E. Mills, A. Obermeyer, X. Dong, J. Walker and B. D. Olsen, Langmuir, 2016, 32, 13367–13376 CrossRef CAS PubMed.
- H. V. Sureka, A. C. Obermeyer, R. J. Flores and B. D. Olsen, ACS Appl. Mater. Interfaces, 2019, 11, 32354–32365 CrossRef CAS PubMed.
- H. Tjo, N. Zervoudis and A. Obermeyer, Protein-Polymer Complex Coacervates as Synthetic Membrane-less Organelles, Columbia University, 2020, vol. 14, pp. 21–25 Search PubMed.
- Y. Wang, Y. Dong, H. Liu, W. Yin, T. Guo, H. Yuan and T. Meng, ACS Appl. Mater. Interfaces, 2022, 14, 5009–5016 CrossRef CAS PubMed.
- J. Deng and A. Walther, Chem, 2020, 6, 3329–3343 CAS.
- A. Samanta, V. Sabatino, T. R. Ward and A. Walther, Nat. Nanotechnol., 2020, 15, 914–921 CrossRef CAS PubMed.
- A. T. Rowland and C. D. Keating, Soft Matter, 2021, 17, 3688–3699 RSC.
- H. Wang, X. Zhou, J. Wang, X. Zhang, M. Zhu and H. Wang, Bioact. Mater., 2022, 17, 261–275 CAS.
- Y. Zhang, S. Liu, Y. Yao, Y. Chen, S. Zhou, X. Yang, K. Wang and J. Liu, Small, 2020, 16, e2002073 CrossRef PubMed.
- J. Liu, E. Spruijt, A. Miserez and R. Langer, Nat. Rev. Mater., 2023, 8, 139–141 CrossRef CAS.
- R. V. Ulijn and A. Lampel, Isr. J. Chem., 2020, 60, 1129–1140 CrossRef CAS.
- A. Lampel, Chem, 2020, 6, 1222–1236 CAS.
- S. Song, T. Ivanov, D. Yuan, J. Wang, L. C. da Silva, J. Xie and S. Cao, Biomacromolecules, 2024, 25, 5468–5488 CrossRef CAS PubMed.
- J. Wang, J.-M. Choi, A. S. Holehouse, H. O. Lee, X. Zhang, M. Jahnel, S. Maharana, R. Lemaitre, A. Pozniakovsky, D. Drechsel, I. Poser, R. V. Pappu, S. Alberti and A. A. Hyman, Cell, 2018, 174, 688–699 CrossRef CAS PubMed.
- J. Bucci, L. Malouf, D. A. Tanase, N. Farag, J. R. Lamb, R. Rubio-Sánchez, S. Gentile, E. Del Grosso, C. F. Kaminski, L. Di Michele and F. Ricci, J. Am. Chem. Soc., 2024, 146, 31529–31537 CrossRef CAS PubMed.
- W. A. Wee, H. Sugiyama and S. Park, iScience, 2021, 24, 103455 CrossRef CAS PubMed.
- S. Song, A. F. Mason, R. A. J. Post, M. De Corato, R. Mestre, N. A. Yewdall, S. Cao, R. W. van der Hofstad, S. Sanchez, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Nat. Commun., 2021, 12, 6897 CrossRef CAS PubMed.
- C. Love, J. Steinkühler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T.-Y. D. Tang, Angew. Chem., Int. Ed., 2020, 59, 5950–5957 CrossRef CAS PubMed.
- B. Saini and T. K. Mukherjee, ACS Appl. Mater. Interfaces, 2022, 14, 53462–53474 CrossRef CAS PubMed.
- A. Netzer, A. Baruch Leshem, S. Veretnik, I. Edelstein and A. Lampel, Small, 2024, 20, e2401665 CrossRef PubMed.
- S. Cao, T. Ivanov, J. Heuer, C. T. J. Ferguson, K. Landfester and L. Caire da Silva, Nat. Commun., 2024, 15, 39 CrossRef CAS PubMed.
- S. Rekhi, C. G. Garcia, M. Barai, A. Rizuan, B. S. Schuster, K. L. Kiick and J. Mittal, Nat. Chem., 2024, 16, 1113–1124 CrossRef CAS PubMed.
- S. Shi, L. Zhao and Z.-Y. Lu, J. Phys. Chem. Lett., 2024, 15, 7280–7287 CrossRef CAS PubMed.
- T. Yamashita, in Phase separation in living cells: benefits and risks, ed. R. Kurokawa, Springer Nature, Singapore, 2023, pp. 21–38 Search PubMed.
- J. E. Bramham and A. P. Golovanov, Nat. Commun., 2022, 13, 1767 CrossRef CAS PubMed.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed.
- N. Galvanetto, M. T. Ivanović, A. Chowdhury, A. Sottini, M. F. Nüesch, D. Nettels, R. B. Best and B. Schuler, Nature, 2023, 619, 876–883 CrossRef CAS PubMed.
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS PubMed.
Footnote |
| † Authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |