
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical engineering of zeolites: alleviating transport limitations through hierarchical design and shaping
Nibras Hijazi
 a,
Anastasiya Bavykina
a,
Irina Yarulina
a,
Tuiana Shoinkhorova
a,
Enrique V. Ramos-Fernandez
ab and
Jorge Gascon
*a
a,
Anastasiya Bavykina
a,
Irina Yarulina
a,
Tuiana Shoinkhorova
a,
Enrique V. Ramos-Fernandez
ab and
Jorge Gascon
*a
aAdvanced Catalytic Materials, KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa
bInorganic Chemistry Department, Laboratory of Advanced Materials, University Materials Institute of Alicante, University of Alicante, Alicante, 03080, Spain
First published on 29th May 2025
Abstract
Zeolites are indispensable catalysts in a wide range of industrial applications due to their well-defined microporous structures and exceptional shape-selective properties. However, their practical use is often constrained by diffusion limitations, which can hinder reactant accessibility, influence product selectivity, and accelerate catalyst deactivation. This review critically examines strategies to alleviate these diffusion constraints, focusing on hierarchical structuring, nanozeolite synthesis, and advanced shaping techniques. We discuss fundamental diffusion theories, experimental characterization methods, and emerging methodologies that enhance mass transport in zeolites. By bridging fundamental principles with industrial applications, this review provides a comprehensive overview of how tailored zeolite architectures can optimize catalytic performance, paving the way for more efficient and sustainable processes.
 Nibras Hijazi | Nibras Hijazi received his Bachelor of Chemical Engineering from the University of Minnesota, Twin Cities, in 2017 and his PhD in Chemical Engineering from KAUST in 2024. His thesis, entitled “Transport, Reactivity, and Deactivation in Zeolites: An Analysis of Alkene Oligomerization,” was supervised by Professor Jorge Gascon. From 2019 to 2020, Nibras worked in the High-Throughput Research Laboratory at Saudi Aramco, where he leveraged high-throughput experimentation for kinetic modeling studies. Prior to that, Nibras conducted undergraduate research under the supervision of Professor Michael Tsapatsis, working on zeolite synthesis and characterization. Nibras is currently working as Research Engineer in the Fuels & Chemicals R&D Division at Saudi Aramco. |
 Anastasiya Bavykina | Anastasiya Bavykina is a Research Scientist at KAUST Catalysis Center. Previously she was a postdoctoral researcher in the same group. She obtained two Master's degrees, the first at Novosibirsk State University in 2010 and the second jointly from the University of Barcelona and the Gdansk University of Technology in 2012, within the Erasmus Mundus program. She received her PhD at Delft University of Technology in 2017. |
 Irina Yarulina | Irina Yarulina holds a PhD in Chemical Engineering from Delft University of Technology, with a focus on heterogeneous catalysis. Over the past decade, she has developed catalysts for diverse applications across academia and industry. Now at Sulzer, she is dedicated to advancing renewable carbon initiatives through the novel polymer recycling technologies. |
 Tuiana Shoinkhorova | Dr Tuiana Shoinkhorova is a Senior Research Specialist in Dow's Core R&D organization, where she is the part of the high-throughput catalysis group, leading catalyst development efforts. She earned her PhD in Chemical Science at King Abdullah University of Science and Technology in 2022 under Prof. Jorge Gascon. Her dissertation, “Formulation of Zeolite-based Catalysts for Hydrocarbon Processing,” received the KAUST Physical Sciences & Engineering Best PhD Award 2022. Earlier, Tuiana completed a Specialist degree in Chemistry at Novosibirsk State University (2016). |
 Enrique V. Ramos-Fernandez | Enrique V. Ramos Fernández earned his PhD in Materials Science from the University of Alicante in 2008. Following this, he was awarded a Marie Curie fellowship to pursue scientific research at Delft University of Technology (TUDelft) in the Netherlands. He subsequently worked at the University of Amsterdam before joining the University of Alicante in 2014, where he served as an associate professor (on a leave of absence). He is now a Senior Research Scientist at King Abdullah University of Science and Technology (KAUST). His research focuses on the development of porous materials for applications ranging from adsorption to catalysis. He has published over 100 papers in leading scientific journals. |
 Jorge Gascon | Jorge Gascon was born in Huesca (Spain) in 1977, he received his MSc in Chemistry and his PhD in Chemical Engineering at the University of Zaragoza (Spain). Starting as post-doc in 2006 at TUDelft (NL), he went up the ranks to full Professor (2014) at the same institution. Since 2017 he is Professor of Chemical Engineering at King Abdullah University of Science and Technology. With an H-Index of >110 and over 50 |
1 Introduction
Zeolites, crystalline aluminosilicates with precisely defined pore structures at the molecular level, have revolutionized many industries due to their catalytic as well as molecular sieving properties.1 Their ability to discriminate molecules based on size and shape makes them indispensable in processes like oil refining, petrochemical synthesis, and environmental remediation.2,3 These shape-selective properties stem from their unique topologies, each defined by specific pore dimensions and connectivity, which govern both reactivity and diffusion.4,5While the unique pore structures of zeolites enable their remarkable properties, they are also responsible for potential diffusion limitations.5–9 Not only that these limitations restrict access to active sites, affecting the efficiency of catalysis, but also influence product selectivity and catalyst deactivation. This review aims to highlight the advancements made in overcoming diffusion limitations in zeolites, encompassing approaches from the molecular level to bulk-scale engineering. To establish an understanding of zeolites at the molecular level, the review begins with a general introduction to zeolites and shape selectivity in zeolites. Then, the basic theory of diffusion in zeolites is introduced, followed by an overview of the experimental methods used to study diffusion in zeolites.
The focus of Section 4 is on hierarchical and nanozeolites. These materials, owing to their reduced diffusion lengths, have emerged as effective strategies to alleviate diffusion limitations in zeolites. However, approaches to synthesize hierarchical and nanozeolites have trade-offs. Bottom-up synthesis methods, though precise, often rely on costly structure-directing agents and mesoporogens, while top-down methods, such as desilication, can suffer from reduced yields and structural imperfections. Balancing these pros and cons is vital for advancing hierarchical and nanozeolite design.
The industrial application of zeolites necessitates shaping strategies that retain their microporous volume while ensuring scalability and mechanical stability. Section 5 of this review addresses the challenges of translating zeolites from laboratory research to industrial catalysts. Extrudates for fixed-bed reactors and powders for fluidized beds are the dominant forms, with careful attention required to preserve macroporosity for optimal diffusion. Techniques like extrusion and spray drying are standard, but innovations such as 3D printing are opening new possibilities. For example, BASF's X3D catalyst platform showcases how additive manufacturing can precisely control particle geometry, potentially enhancing reactor performance.10 Patents in this area highlight a growing interest in leveraging advanced shaping technologies to meet industrial demands.11–16
Together, these perspectives illustrate a cohesive narrative: zeolite science begins with the synthesis of crystals possessing extraordinary molecular-level precision but must extend to the mesoscale and macroscale to ensure accessibility and industrial relevance. By addressing challenges at each scale, zeolite researchers and practitioners are paving the way for more efficient, sustainable, and scalable applications. This review captures this journey, offering insights into current practices and future directions for optimizing zeolites from the molecular to the reactor scale.
2 Prelude to zeolites and shape selectivity in zeolites
Zeolites are microporous aluminosilicate materials with channels and cavities of molecular dimensions. Their use as catalysts, sorbents, and molecular sieves has revolutionized oil refining and the petrochemical industry.17–20 The term “zeolite” derives from the Greek words zeo, meaning “boiling” and lithos, meaning “stone.” A. F. Cronstedt, a Swedish minerologist, coined this term in 1756 after observing that stilbite, a naturally occurring zeolite, releases steam when heated in a blowpipe flame.21Zeolites can be viewed as a three-dimensional network of TO4 (T = Si, Al) tetrahedra. Each oxygen atom in this network is shared between two tetrahedra. However, two AlO4 tetrahedra cannot be adjacent to each other, a rule in zeolite chemistry known as that of Löwenstein.22 The chemical formula of a given zeolite can be written as
| Mn+y/n[(SiO2)x·(AlO2−)y]·zH2O | (1) |
Zeolites come in a wide variety of structures. So far, about 265 different zeolite (and zeotype) topological structures have been recognized by the International Zeolite Association (IZA). Fig. 1 gives an overview of these structures and their void diameters. For simplicity, the structures are grouped by the number of tetrahedra in the largest ring circumscribing their voids. The void diameter in a given structure is defined by the maximum diameter of a sphere that can diffuse along. The Lennard-Jones diameters for some molecules of interest are also given for comparison.25,26
 | ||
| Fig. 1 Range of void diameters for the zeolites structures recognized by the IZA. The numbers above bars represent the number of zeolite structures with the given number of tetrahedra in largest ring. Source: the homepage of the IZA.27 Reproduced from ref. 23 with permission. Copyright 2000 Elsevier. | ||
What makes zeolites special is that their voids fall within the same size range as many small molecules (see Fig. 1). Therefore, zeolites are considered excellent molecular sieves. When used as catalysts, zeolites perform catalysis and molecular sieving in one step. This phenomenon is better known as “shape selective catalysis.” The concept was first introduced in 1960 by Weisz and Frilette,28 who observed that Ca-exchanged zeolite A shape selectively cracks n-paraffins to straight chain hydrocarbons. Since then, shape selective catalysis has turned from being a laboratory curiosity to a concept applied in various industrial processes.2,3
Weisz and Csicsery's29,30 work was fundamental in shaping the concept of shape selectivity. In principle, they classified shape selectivity in zeolites into (i) reactant shape selectivity, (ii) product shape selectivity, and (iii) transition state shape selectivity. Reactant shape selectivity occurs when part of the reactants is unable to access or diffuse through the zeolite. Therefore, these reactants end up excluded and unconverted. An example of this type of shape selectivity is the exclusion of branched paraffins such as iso-octane (2,2,4-trimethylpentane) in “selectoforming” gasoline over erionite.31 Product shape selectivity occurs when part of the products are unable to diffuse out of the zeolite. Thus, they are transformed into smaller or slimmer products or, in a worst case, transformed into coke deposits. An example of this type of shape selectivity is the retarded formation of ortho- and meta-ethyltoluene and hence the preferential formation of para-ethyltoluene in toluene ethylation over ZSM-5.32 Transition state shape selectivity refers to the situation in which a reaction is being suppressed due to the inability of the zeolite to accommodate the transition state complex. This type of shape selectivity can be exemplified by the inhibition of xylene disproportionation during xylene isomerization over ZSM-533 (Fig. 2).
 | ||
| Fig. 2 Examples of reactant, product, and transition state shape selectivity. Reproduced with permission from ref. 31. Copyright 1994 Elsevier. | ||
Reactant and product shape selectivity arise essentially from differences in the diffusional characteristics of reacting species or thus formed products. Therefore, these phenomena may be looked upon as manifestations of diffusion limitations. Haag et al.34 well demonstrated this by showing that reactant shape selectivity in paraffin cracking over ZSM-5 increases with increasing diffusion limitations. As shown in Fig. 3, the “effectiveness factor” for 2,2-dimethylbutane cracking decreased by virtue of reactant shape selectivity as the Thiele modulus increased, which is a measure for diffusion limitations based on physicochemical properties of the catalyst.35,36 Similarly, Olson and Haag37 demonstrated that product shape selectivity in toluene disproportionation over ZSM-5 increases through diffusion limitations, resulting in an enhanced selectivity towards the para-isomer of xylene at the expense of the other isomers. A strong relationship was found between para-xylene selectivity and a pseudo-Thiele modulus.
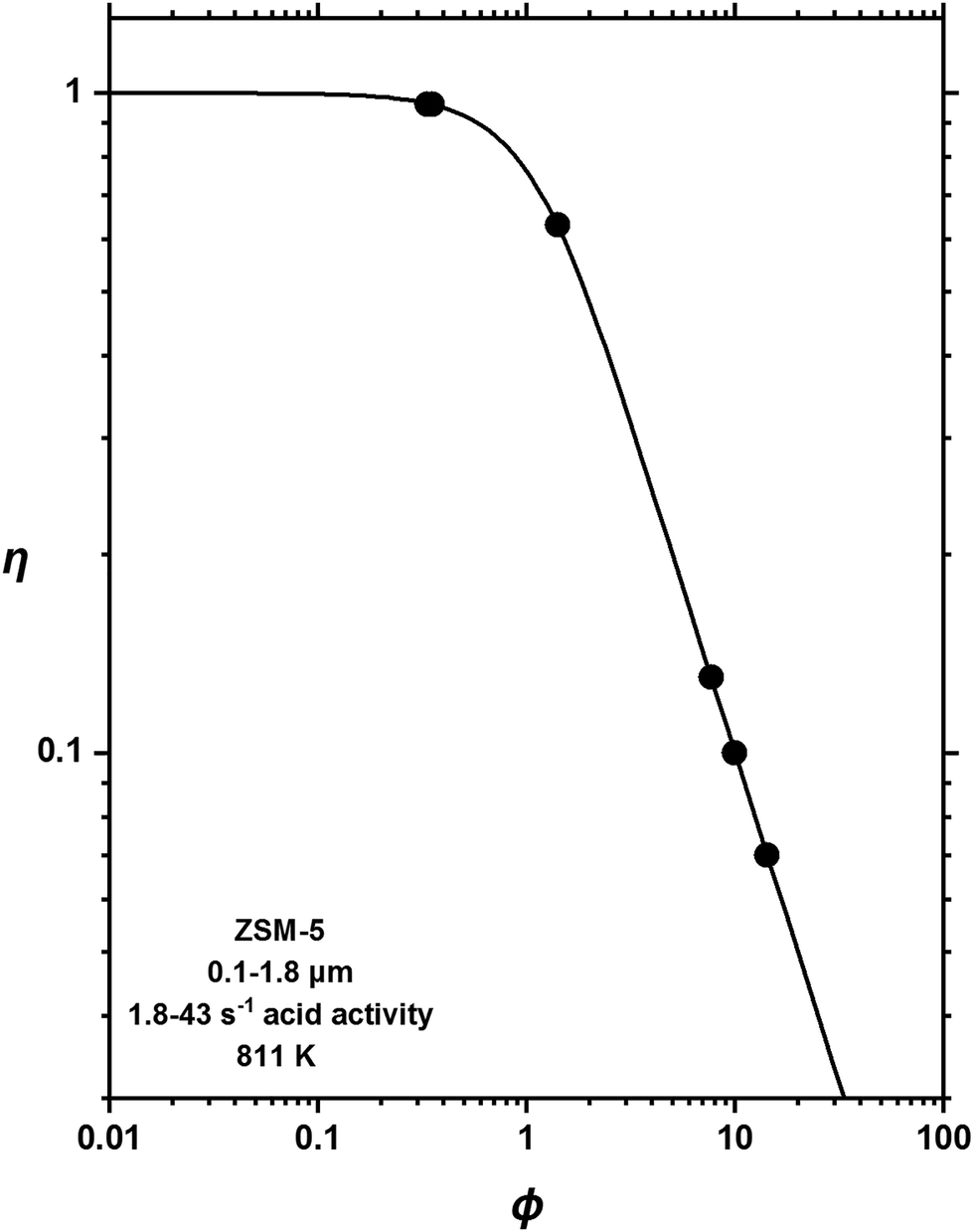 | ||
| Fig. 3 Effectiveness factors for 2,2-dimethylbutane cracking over ZSM-5 as a function of the Thiele modulus. The acid activity represents rate constants calculated for ZSM-5 samples based on the α-test.38 Reproduced with permission from ref. 34. Copyright 1981 Royal Society of Chemistry. | ||
Transition state shape selectivity differs from reactant and product shape selectivity in that it originates from spatial limitations rather than diffusional limitations. Thus, to probe this type of shape selectivity, one must consider the critical dimensions of the transition state as well as the effective pore dimensions. In this regard, the IZA reports parameters that can be of much utility to assess effective pore dimensions in zeolites such as the maximum diameter of a sphere that can diffuse along and the maximum diameter of a sphere that can be inscribed. Most of these parameters are adopted from work by Foster et al.,39 who computationally characterized pore dimensions in 165 zeolite structures. Descriptors developed by First et al.40 can be also useful to evaluate effective pore dimensions in zeolites. These descriptors include the pore limiting diameter and largest cavity diameter, computed for 202 zeolite structures.
Additionally, a number of experimental techniques can be utilized to probe effective pore dimensions in zeolites. One of these techniques is the constraint index (CI) test,41 which is based on cracking an equimolar mixture of n-hexane and 3-methylpentane and measuring their respective conversions. The CI is defined as follows:
 | (2) |

Another technique is the spaciousness index (SI) test,43–45 which has its basis on measuring the relative yield of isobutane and n-butane in hydrocracking of C10 naphthenes, preferably butylcyclohexane. Principally, the more spacious a zeolite, the greater its yield for isobutane and the higher its SI. Fig. 4b shows that the SI for selected zeolites increases linearly, with slight deviation, with the respective maximum diameter of a sphere that can be inscribed. This demonstrates the efficacy of the SI test to probe effective pore dimensions in zeolites.
Besides Weisz and Csicsery's concepts of shape selectivity, other concepts have been proposed such as molecular traffic control, the nest effect, the window effect, and secondary shape selectivity. Molecular traffic control is a concept pertaining to zeolites with intersecting channels of different widths. In such zeolites, the diffusion of differently sized molecules is directed so that small molecules diffuse through narrow channels while large molecules diffuse through wide channels. Derouane and Gabelica46 proposed this concept to explain the absence of counterdiffusional effects in methanol reactions over ZSM-5, a zeolite formed by intersecting straight (5.4 × 5.6 Å) and sinusoidal (5.1 × 5.6 Å) channels.
The concept of the nest effect was first introduced by Fraenkel et al.,47 who found that shape selectivity can take place not only in the internal surface of zeolites but also at their external surface. Their finding was based on the observation that ZSM-5 shape selectively methylates naphthalenes, molecules that are too bulky to enter and react in the intracrystalline channel system of ZSM-5. Later, Derouane48 offered an explanation for this peculiar phenomenon. He proposed that in such situations molecules conform themselves to optimize their van der Waals interactions with “nests” located at pore mouths or channel cuts. This conformation enhances the sorption of molecules and ultimately leads to shape selective reactions at nest locations. Other phenomena being conceptually similar to the nest effect are pore mouth and key-lock catalysis.49,50
The window or cage effect refers to the situation in which the size of a molecule approaches that of a cage, and this similarity in size induces shape selectivity against the molecule as it diffuses through the cage. This phenomenon was first observed by Gorring51 when studying the diffusion of n-alkanes over erionite. He found that the diffusion of n-octane is significantly reduced compared to that of lower or higher alkanes and explained that in terms of the proximity in size between n-octane (1.3 Å) and the erionite cage (1.3 × 0.6 Å). Chen and Garwood52 made similar observations as they studied the cracking of C4–C16 n-alkanes over erionite. They observed that the rate constant for cracking these alkanes goes through a minimum at C8 and attributed this observation to cage effects.
Inverse or secondary shape selectivity is a special phenomenon where the shape selectivity of a species is influenced by the presence of another species. The phenomenon was first observed by Namba et al.53 while studying n-octane cracking over ZSM-5. They found that the addition of 2,2-dimethylbutane, a species that diffuses slowly in ZSM-5, retards the cracking of n-octane. The authors argued that this retardation is a result of shape selectivity practiced against n-octane when slow diffusing 2,2-dimethylbutane was present.
3 Diffusion in zeolites
3.1 Theory
Diffusion in porous media falls in different regimes depending on pore dimensions. In wide pores, typically 0.1 μm or larger in diameter, diffusion is governed by molecule–molecule interactions and falls in the molecular regime (see Fig. 5b). In narrower pores, interactions between molecules and pore walls become predominant, and diffusion falls in the Knudsen regime. In even narrower pores, normally 9 Å or smaller in diameter, the size of molecules approaches pore dimensions, and diffusion falls in the configurational or zeolitic regime.54,55 | ||
| Fig. 5 Dependency of (a) diffusivity and (b) the diffusional activation energy on the pore diameter. Reproduced with permission from ref. 54. Copyright 1991 Elsevier. | ||
Zeolitic diffusion is considered far more complicated than molecular or Knudsen diffusion due to the fact that zeolitic diffusion is strictly dependent on how well molecules “fit” into the zeolite pore through which they diffuse. In other words, what determines diffusivity in the zeolitic regime is the continuous match in size and shape between molecules and zeolite pores.56 Besides, zeolitic diffusion is an activated process (see Fig. 5b), where activation arises mainly from steric hindrance imposed on molecules. This means that molecules have to overcome energy barriers in order to diffuse. Further, the fact that diffusing molecules interact among themselves and with the zeolite framework makes zeolitic diffusion strongly dependent on occupancy or loading. Kärger and Pfeifer57,58 demonstrated that there are five different dependencies for zeolitic diffusion on loading.
Zeolitic diffusion occurs by different mechanisms, the most prevalent of which is “transport diffusion”. Transport diffusion is a non-equilibrium process where the motion of molecules is driven by spatial gradients in concentration. Fick's first law gives a good representation of this diffusion mechanism:
 | (3) |
 | (4) |
μ = μ0 + RgT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnp lnp
| (5) |
 | (6) |
Here, Rg is the ideal gas constant, T is temperature, and p is the partial pressure of the adsorbed phase. By comparing eqn (3) and (6), the transport diffusivity can be defined as
 | (7) |
D0 is often referred to as the Maxwell–Stefan or corrected diffusivity and Γ is a thermodynamic correction factor which corrects for the non-linearity between the partial pressure and concentration of the adsorbed phase (Henry's law). The latter is directly related to loading through the following expression (for Langmuir isotherms):
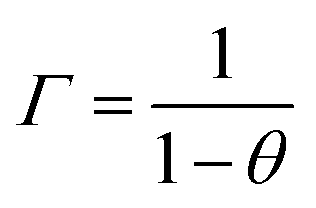 | (8) |
Another mechanism by which zeolitic diffusion occurs is self-diffusion. Unlike transport diffusion, self-diffusion is an equilibrium process and proceeds through Brownian motion. The flux in self-diffusion can be represented by
 | (9) |
 is the self-diffusivity. Although self-diffusion and transport diffusion represent two different diffusion mechanisms, Paschek and Krishna showed using the Maxwell–Stefan approach that a relationship between self-diffusivity and corrected diffusivity can be derived as follows:59
is the self-diffusivity. Although self-diffusion and transport diffusion represent two different diffusion mechanisms, Paschek and Krishna showed using the Maxwell–Stefan approach that a relationship between self-diffusivity and corrected diffusivity can be derived as follows:59
 | (10) |
 .
.
A special mechanism occurring in one-dimensional zeolites is single-file diffusion.60,61 This mechanism prevails when the mutual diffusion of molecules in zeolite channels is prohibited due to the narrowness of channels. A molecule can only pass through if the molecule in front of it is able to pass. Single-file diffusion can be described by the simple jump model where a molecule with occupancy θ jumps throughout a distance λ with a mean time between jumps of τ. The mean square displacement (a microscopic quantity) of the molecule can be expressed as a function of observation time t as follows:
 | (11) |
In the situation where the displacement of the molecule becomes uncorrelated with that of other molecules, which arises at long observation times, the mean square displacement can be written as
 | (12) |
 | (13) |
3.2 Measurement
Experimental methods used to measure zeolitic diffusion can be broadly categorized into microscopic and macroscopic methods. Microscopic methods rely on recording mean square displacements in subunits of zeolite crystals at known time intervals. Since microscopic methods study diffusion over short distances or time scales, they are well suited for measurements of self-diffusivity . Typically used microscopic methods are pulsed-field gradient nuclear magnetic resonance (PFG NMR) and quasi-elastic neutron scattering (QENS). Macroscopic methods, on the other hand, depend on measuring flux across well-defined concentration gradients or measuring rates of reaction under diffusion control. Unless extraneous factors such as surface resistance influence the diffusion process, transport diffusivity (Dt) is measured by macroscopic methods. Macroscopic methods include, but are not limited to, uptake rate measurements, membrane permeation and chromatography, the zero-length column (ZLC), temporal analysis of products (TAP), and effectiveness factor methods. Theoretical assessment of zeolitic diffusion by molecular dynamics calculations or Monte Carlo simulations is beyond the scope of this review and can be found in the work of Smit and Maesen.62
. Typically used microscopic methods are pulsed-field gradient nuclear magnetic resonance (PFG NMR) and quasi-elastic neutron scattering (QENS). Macroscopic methods, on the other hand, depend on measuring flux across well-defined concentration gradients or measuring rates of reaction under diffusion control. Unless extraneous factors such as surface resistance influence the diffusion process, transport diffusivity (Dt) is measured by macroscopic methods. Macroscopic methods include, but are not limited to, uptake rate measurements, membrane permeation and chromatography, the zero-length column (ZLC), temporal analysis of products (TAP), and effectiveness factor methods. Theoretical assessment of zeolitic diffusion by molecular dynamics calculations or Monte Carlo simulations is beyond the scope of this review and can be found in the work of Smit and Maesen.62
3.1.1.1 PFG NMR. Pulsed-field gradient nuclear magnetic resonance (PFG NMR) is a useful technique to measure self-diffusivity in zeolites. The technique is based on exciting NMR-active molecules loaded on a zeolite sample with a standard radio frequency echo pulse sequence and two space-dependent magnetic field gradient pulses (see Fig. 6).55 The two gradient pulses are applied for short time intervals (δ = 0.1–10 ms) and separated by relatively longer durations (Δ = 1–100 ms). When applied, gradient pulses mark nuclear spins that encode the position of the molecule in the axial direction of the gradient. Molecular migration causes phase incoherence between marked nuclear spins, resulting in an attenuation of the spin-echo signal. The extent of this attenuation is directly related to the mean square displacement of the molecule in the zeolite sample.
 | ||
| Fig. 6 Sequence of the gradient pulses in PFG NMR. Reproduced with permission from ref. 55. Copyright 2007 Elsevier. | ||
Self-diffusivity  is related to the mean square displacement through Einstein's equation:
is related to the mean square displacement through Einstein's equation:
 | (14) |
Using Einstein's equation, a relationship between self-diffusivity and the intensity of the spin-echo signal was derived as follows:
 | (15) |
PFG NMR, however, has limitations in the range of diffusivities that can be measured. For example, the lowest diffusivity that can be measured by PFG NMR is of the order of 10−13–10−12 m2 s−1. This lower bound exists because of limitations on the maximum duration between gradient pulses (Δ) that can be used. Furthermore, because the mean square displacement has to be smaller than the square of the average crystallite radius (R2), there exists an upper bound for diffusivities measured by PFG NMR. The upper bound is defined as follows:
 | (16) |
In traditional PFG NMR, the upper bound can be extended by using large crystallites or small durations between gradient pulses. Meanwhile, the lower bound can only be expanded though a modification of PFG NMR, namely by integrating the fast tracer desorption method.
3.1.1.2 QENS. The measuring principle behind quasi-elastic neutron scattering (QENS) is that neutrons interact with nuclei in atoms and this interaction causes scattering of neutrons. The scattering process involves small energy transfers (±2 meV), the measurement of which can give useful information about diffusion.60,63 Experimentally, one measures in Q–ω space a scattering function S(Q, ω), which is a four-dimensional Fourier-transform of the van Hove correlation function G(r, t). For incoherent scattering, the scattering function is defined as follows:
 | (17) |
 | (18) |
A temporal Fourier-transform of this intermediate incoherent scattering function gives an incoherent scattering function expressed solely in terms of Q and ω:
 | (19) |
The shape of this function is a Lorentzian. Therefore, by plotting the half-width at half-maximum (hwhm) of this function, which is  , against different values of Q2, self-diffusivity
, against different values of Q2, self-diffusivity  can be measured from the slope of the line. Sayeed et al., however, found from studying propane diffusion in NaY that the relationship between the hwhm and Q2 can be non-linaer (see Fig. 7).64 This led the authors to use other “jump diffusion” models for estimating (self-) diffusivity. The obtained diffusivities were in good agreement with diffusivities estimated by PFG NMR for a similar system.
can be measured from the slope of the line. Sayeed et al., however, found from studying propane diffusion in NaY that the relationship between the hwhm and Q2 can be non-linaer (see Fig. 7).64 This led the authors to use other “jump diffusion” models for estimating (self-) diffusivity. The obtained diffusivities were in good agreement with diffusivities estimated by PFG NMR for a similar system.
 | ||
| Fig. 7 Relationship between hwfm and Q2 for propane diffusion in NaY measured by QENS. Reprinted with permission from ref. 64. Copyright 2003 American Chemical Society. | ||
3.1.2.1 Uptake rate measurements. Uptake or sorption rate measurements are perhaps the most common method to measure transport diffusivity in zeolites. In a typical experiment, a zeolite sorbent is subjected to a step-change in sorbate pressure, and the weight of the sample or the pressure of the system is recorded as a function of time (see Fig. 8). To eliminate intrusion of surface resistance or heat transfer effects, small amounts of samples (10–30 mg) sieved to small particles or aggregates are typically used.55 The uptake of the sorbate by the sample can be modeled using Fick's second law (in radial coordinates):
 | (20) |
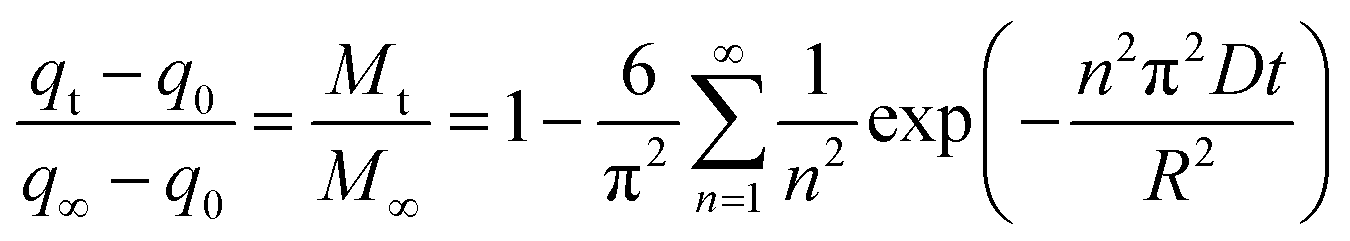 | (21) |
 | (22) |
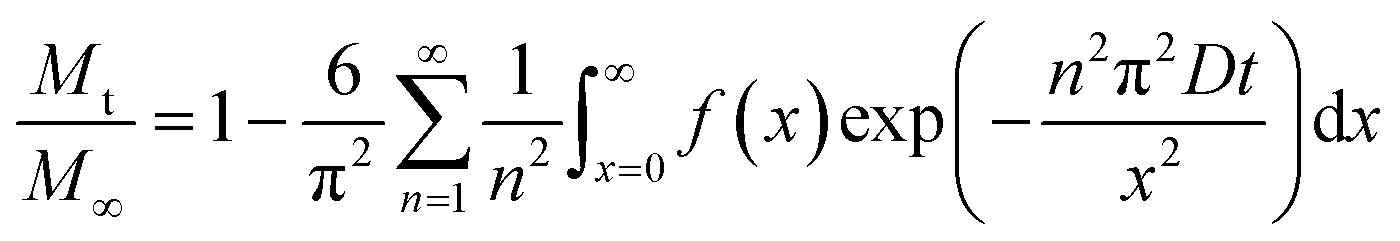 | (23) |
 | ||
| Fig. 8 Experimental setups for (left) gravimetric and (right) piezometric uptake rate measurements. Reproduced with permission from ref. 55. Copyright 2007 Elsevier. | ||
Fig. 9a shows theoretical uptake curves as a function of dimensionless time (Dt/x2) for different values of μ/σ. The uptake curves differ considerably, which implicates that the use of eqn (23) is implicit when there are broad size distributions (μ/σ < 5). Eqn (21) also entails that the uptake of the sorbate by the sample is small in comparison with the total amount of sorbate introduced. However, this is not always the case, as the uptake of the sorbate can be significant. In this situation, the solution of the uptake becomes67,68
 | (24) |
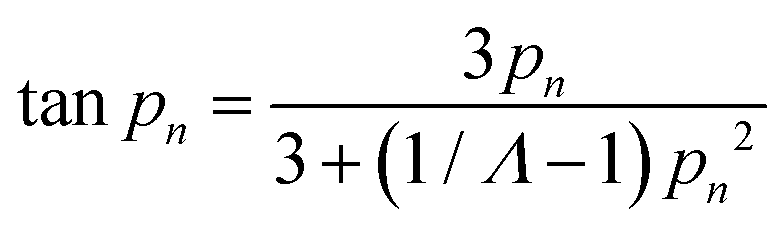 | (25) |
 | ||
| Fig. 9 (a) Effect of the size distribution on the uptake solution. Reproduced from ref. 66 with permission. Copyright 1971 Elsevier. (b) Effect of the amount of sorbate ultimately sorbed on the uptake solution. Reproduced from ref. 68 with permission. Copyright 2012 John Wiley and Sons. | ||
3.1.2.2 Frequency response. The measuring principle behind frequency response is that a sorbate–sorbent system brought into equilibrium is subjected to a periodic sine-wave perturbation. The perturbation leads, under limited diffusion of the sorbate, to different responses of volume and pressure (phase lag). Measurement of the Fourier-transformed volume and pressure wave forms gives quantitative information about sorbate diffusion.69 Starting with Fick's second law, Shen and Rees70 derived the following equations to describe diffusion in a sorbent subjected to a sinusoidal surface concentration modulation:
 | (26) |
 | (27) |
 | (28) |
 | (29) |
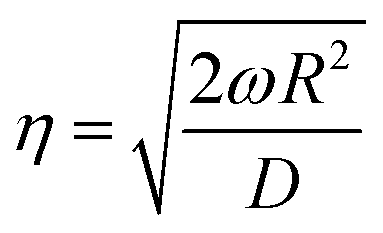 | (30) |
 | (31) |
 | (32) |
 | (33) |
A breakthrough in frequency response was made by Bourdin and co-workers,71–73 who integrated IR temperature detection to this method. They found that measuring phase differences between temperature and pressure gives more insightful results compared to phase differences between volume and pressure. With this “thermal frequency response” method, it was possible to delineate heat transport from mass transport and extract parameters related to both from specific windows in the angular frequency domain (see Fig. 10).
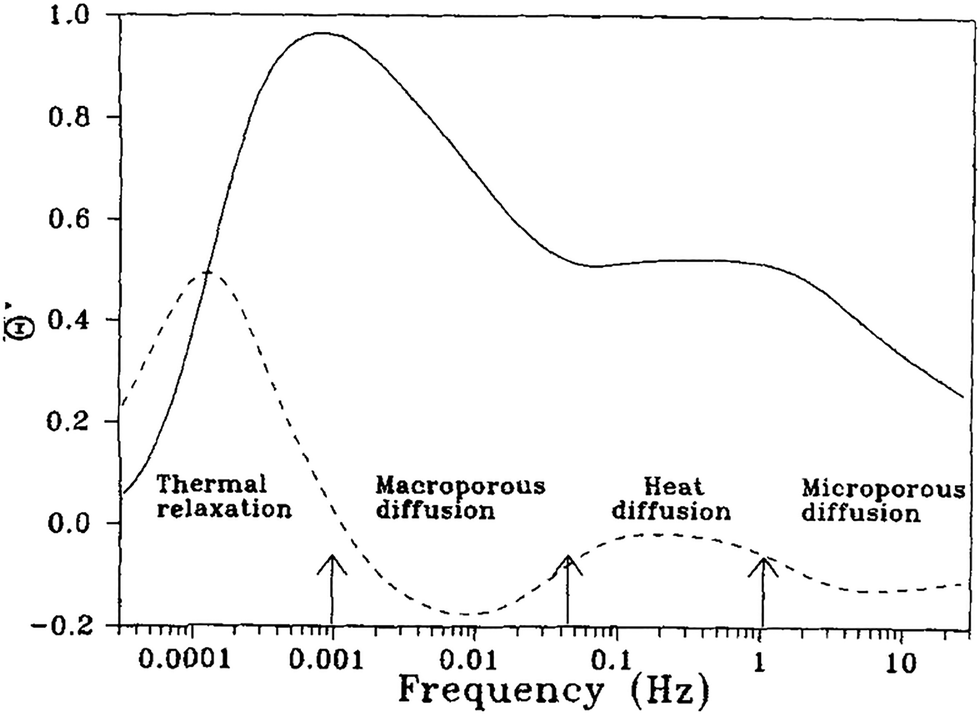 | ||
| Fig. 10 Decoupling of mass and heat transport effects from thermal frequency response characteristic functions. Reprinted with permission from ref. 72. Copyright 2004 John Wiley and Sons. | ||
3.1.2.3 Membrane permeation. Membrane permeation measurements can either be static or continuous.74 In the static mode, the gaseous component under study is fed from one side of the zeolite membrane (feed side), and in the other side (permeate side) the pressure change resulting from permeation is recorded as a function of time. Since the permeate side has fixed volume and temperature, the flux is directly proportional to the slope of pressure versus time and can be calculated using ideal gas law.
In the continuous mode, two gaseous components are fed from the feed side and a sweep gas is used in the permeate side to sweep away the permeated gases. Mass spectrometry or gas chromatography is used to determine the composition in the mixture of sweep and permeate gases. If no back diffusion occurs through the membrane, the flux of component i can be calculated as follows:
 | (34) |
The flux can be related to diffusivity starting from the dusty gas model originally developed by Maxwell and Stefan75,76 Kapteijn et al.77 were the first to apply this model to zeolite membranes. The dusty gas model considers that the force acting on a diffusing molecule is balanced by frictional forces caused by interactions with other diffusing molecules and the surface. The general form of the dusty gas model is given by
 | (35) |
 | (36) |
The partial pressure of the adsorbed phase (p) is related to occupancy through Langmuir isotherms:
 | (37) |
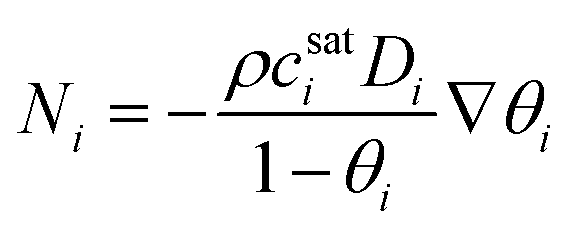 | (38) |
For two-component systems, the flux is expressed as
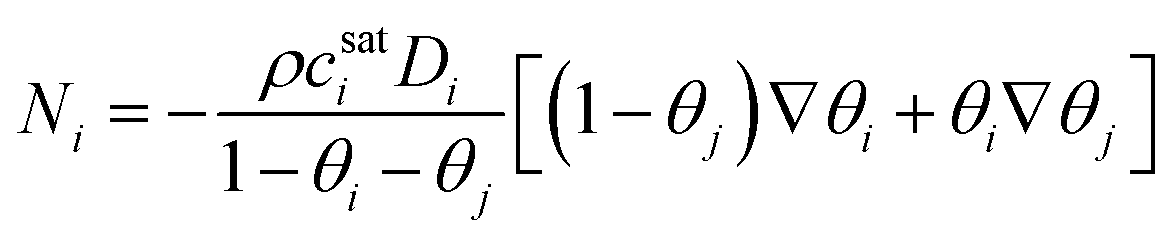 | (39) |
3.1.2.4 Chromatography. Chromatography relies on measuring the response of a chromatographic column packed with a zeolite sample to a pulse injection of sorbate. The response peak, if sufficiently broadened, reflects combined effects of axial dispersion, micropore (zeolitic) and macropore diffusion, and film resistance.54,60 The mean residence time (μ) and variance (σ) of the response peak are given by
 | (40) |
 | (41) |
 | ||
| Fig. 11 Illustration of the chromatography method. Reprinted with permission from ref. 54. Copyright 1991 Elsevier. | ||
Chromatography, just like many other techniques, has a limited range of (micropore) diffusivities that can be measured. An upper limit is reached when the influence of axial dispersion or macropore diffusion is significant compared to that of micropore diffusion. One way to increase the relative importance of micropore diffusion is to work with large crystallites.55 A lower limit is reached when micropore diffusion is so slow and rate-controlling that the response peak becomes excessively broad and peak tailing occurs.
3.1.2.5 The zero-length column (ZLC). Method may be looked upon as a flow variant of uptake rate measurements. In ZLC, a sorbate brought into equilibrium with a zeolite sorbent is purged, and the desorption of the sorbate is followed (see Fig. 12). The term “zero-length” derives from the use of only a few milligrams of sorbent, typically dispersed in a sinter as a monolayer. This use precludes artifacts such as heat transport from controlling the overall transport process. The use of purge gases also ensures that surface resistance is not rate-controlling. One must, however, perform measurements at various purge flow rates to confirm the latter. Ruthven and Eic well demonstrated this by showing that the diffusivity of o-xylene in NaX measured by ZLC is unaffected by changes in purge gas flow rates (30–140 cm3 min−1).79
 | ||
| Fig. 12 Experimental setup for ZLC measurements. Reproduced with permission from ref. 80. Copyright 1995 Elsevier. | ||
Starting with Fick's second law, Brandani and Ruthven81 derived the following expression for desorption from a sorbent with spherical crystallites:
 | (42) |
|
βncotβn + L − 1 = 0
| (43) |
The desorption from a sorbent with slab crystallites can be expressed as
 | (44) |
|
βntanβn = L
| (45) |
A noteworthy variant of the ZLC method is tracer ZLC. In this method, the sorbent is brought into equilibrium with an isotopically labeled sorbate. Then, the sorbent is purged with a gas stream containing the same sorbate but unlabeled, and desorption of the isotopically labeled sorbate is followed by a sensitive detection technique such as mass spectrometry. This procedure allows for the measurement of tracer or self-diffusivity, in contrast to traditional ZLC which measures transport diffusivity. Using tracer ZLC, Ruthven and co-workers80,82 reported self-diffusivities in excellent agreement with those reported using microscopic methods such as PFG NMR.
3.1.2.6 Temporal analysis of products (TAP). The concept of the temporal analysis of products (TAP) reactor was first introduced by Gleaves et al.83 and the application of TAP in diffusivity measurements was pioneered by Nijhuis et al.84,85 In TAP, a zeolite sample placed in an evacuated microreactor is pulsed with small amounts of sorbate (1014–1018 molecules per pulse; 100 μs pulse width) and the pulse response is measured. As in chromatography, the size and shape of the pulse response contain information about the physicochemical processes occurring in the microreactor such as adsorption and diffusion. The short residence time of the pulse, which is in the order of a few seconds, makes the extent of reaction negligible even at high measuring temperatures. The ultra-high vacuum maintained at the outlet of the microreactor makes the flow driven by Poiseuille flow and bulk diffusion of the Knudsen type.
Mass balances in the TAP reactor are written as follows:
 | (46) |
 | (47) |
 | (48) |
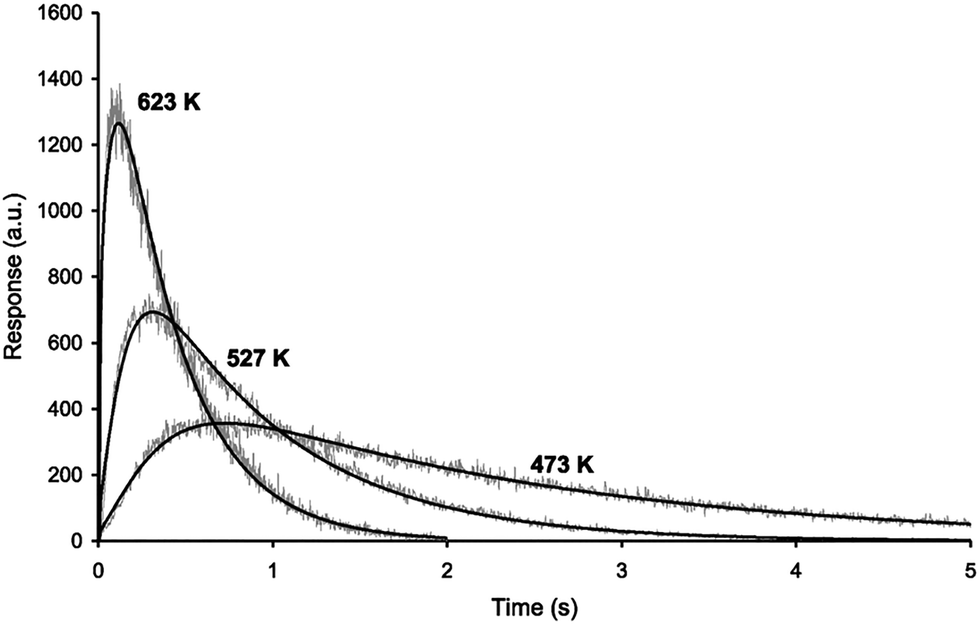 | ||
| Fig. 13 Measured and modeled pulse responses for TAP measurements of n-butane in silicalite-1 at different temperatures. Reprinted with permission from ref. 85. Copyright 1999 Elsevier. | ||
Notably, Keipert and Baerns,86 by studying diffusion using TAP, found that for a given crystallite size a narrow range of diffusivities (within three orders of magnitude) can be reliably measured. Outside this range, the simulated pulse response becomes insensitive to changes in diffusivity.
3.1.2.7 Effectiveness factor. The effectiveness factor method is based on measuring rates of reaction on zeolite samples similar in acidity but different in crystallize size. Under severe diffusion limitations, rates of reaction are strongly dependent on crystallite size because the diffusion length controls the accessibility of reactants to the acidic active sites. Having measured observed rate constants kobs,1 and kobs,2 for two zeolite samples with crystallite sizes R1 and R2, the ratio of effectiveness factors η1/η2 can be determined from the ratio of observed rate constants kobs,1/kobs,2, and the ratio of Thiele moduli ϕ1/ϕ2 can be calculated from the ratio of crystallite sizes R1/R2. The effectiveness factor is a strong function of the Thiele modulus (η = f(ϕ)), but the mathematical description varies depending on the reaction order and crystallite shape.87 However, a general relation can be derived as follows:
 | (49) |
From a double-logarithmic plot of eqn (49), the η1/η2 and ϕ1/ϕ2 ratios can give singular values of η1, η2, ϕ1 and ϕ2 using the method of triangulation (see Fig. 14).88 For a given sample, the intrinsic rate constant (k) can be determined from the effectiveness factor since they are related by kobs = ηk. Having known k, R, and ϕ for this sample, the diffusivity (D) can be calculated from the known definition of the Thiele modulus:35
 | (50) |
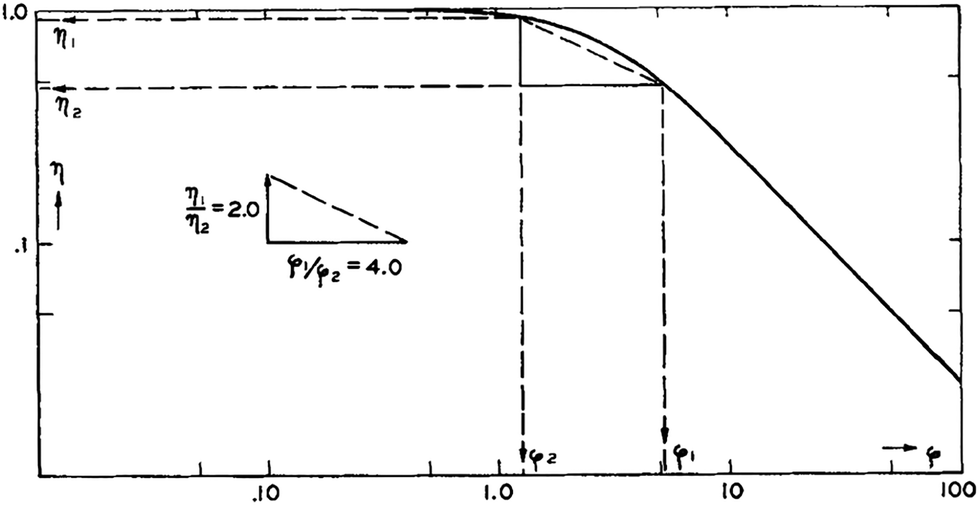 | ||
| Fig. 14 Application of the triangulation method for the determination of diffusivity from the effectiveness factor method. Reproduced with permission from ref. 88. Copyright 1954 Elsevier. | ||
Indeed, the diffusivity values attained from different samples are different. However, Haag et al.34 showed that they fall within the same order of magnitude. Post et al.89 confirmed the accuracy of diffusivity measurements by the effectiveness factor method by comparing them with measurements by (gravimetric) sorption and chromatography.
3.1.2.8 Fourier-transform infrared (FTIR). Diffusion measurements using Fourier-transform infrared spectroscopy are similar to uptake rate measurements in the sense that the uptake of a sorbate in a zeolite sorbent is recorded as a function of time. However, in FTIR, the uptake is followed using the absorbance of the IR band corresponding to the sorbate.90,91 Since different species absorb IR beams at different wavelengths, FTIR can be used to study diffusion of sorbates with more than one component. A typical apparatus for “macro”-FTIR diffusion measurements is shown in Fig. 15. The use of high gas flow rates (e.g., 800 mL min−1) in “macro”-FTIR for flowing the sorbate ensures isothermal operation and maintains constant bulk sorbate concentration.1 Therefore, the intrusion of heat transport or surface resistance is eliminated.
 | ||
| Fig. 15 Experimental setup for measuring diffusivity using FTIR. Reproduced with permission from ref. 90. Copyright 1991 Elsevier. | ||
Diffusivity is estimated by fitting the following uptake solution (for spherical crystallites) to experimental data:
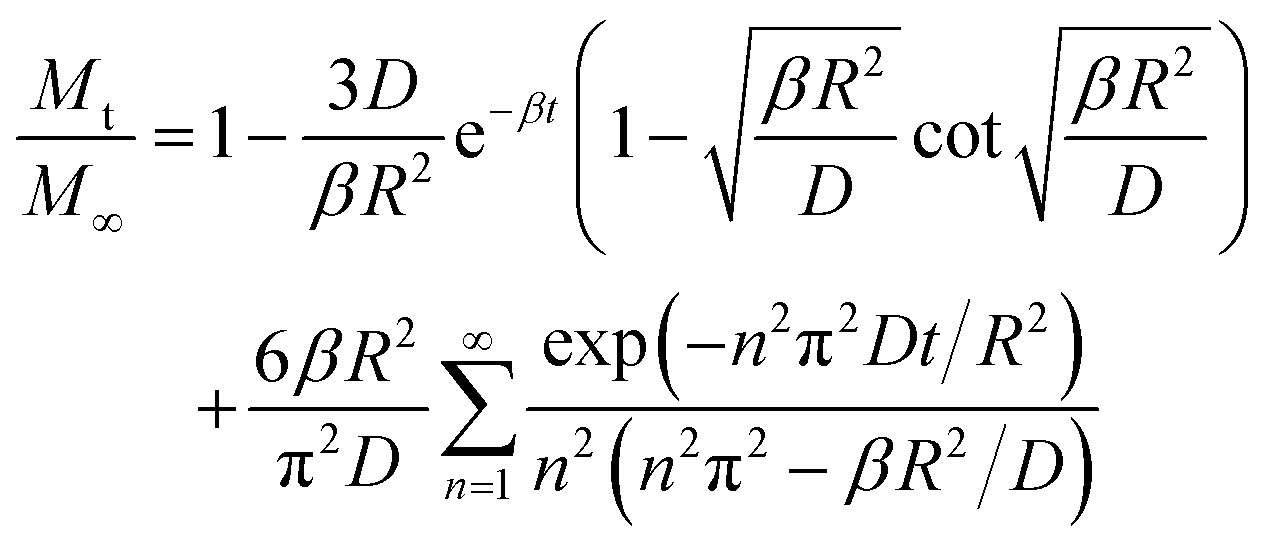 | (51) |
3.2.2.9 Interference microscopy. Interference microscopy is based on measuring the optical density in a loaded zeolite sample as a function of position. An interference pattern is generated by comparing with the optical density at a specific position inside a zeolite crystal with the optical density outside the zeolite crystal.92,93 Since the optical density is directly related to the concentration of loaded molecules, interference patterns generated at different positions can be used to construct concentration profiles throughout the zeolite crystal. Performing these measurements at different times gives concentration profiles in time and position, from which diffusivity can be determined using Fick's second law. Fig. 16 gives an example of 2D methanol concentration profiles measured inside a ferrierite crystal by interference microscopy.
 | ||
| Fig. 16 2D concentration profiles measured by interference microscopy for methanol sorption in a ferrierite crystal. Reproduced with permission from ref. 94. Copyright 2005 Springer Nature. | ||
4 Alleviating diffusion limitations through hierarchical and nanozeolite design
Diffusion in zeolites, whether driven by concentration gradients (transport diffusion) or Brownian motion (self-diffusion), can be limited by the narrowness of zeolite channels or presence of undulations within zeolite frameworks. As such, the diffusion length is a critical parameter that dictates the extent of diffusion limitations. In the context of zeolite-catalyzed reactions, diffusion can also be limited by the high activity or density of acid sites, provided that the rate of reaction is greater than that of diffusion. The Thiele modulus (eqn (50)) represents the best framework through which diffusion limitations in zeolites can be understood. The first term in the Thiele modulus, R, refers to the diffusion length within the zeolite crystallite or particle. Increasing this term by, for example, increasing crystallite size increases the value of the Thiele modulus, and hence, lowers the effectiveness factor because the diffusion time becomes longer than the reaction time. Therefore, designing zeolites with reduced diffusion length will allow more time for time reactants to react and thus increase the overall effectiveness factor of the catalyst. Tsapatsis and co-workers, in studying benzyl alcohol self-etherification, well demonstrated how reductions in diffusion lengths can enhance effectiveness factors.95 They prepared self-pillared MFI zeolites with crystals as small as the size of a few unit cells and compared their catalytic effectiveness with conventional, micron-sized MFI zeolites. The authors found that self-pillared MFI zeolites have effectiveness factors approaching unity, while the effectiveness factors for conventional MFI are well below unity (see Fig. 17a). | ||
| Fig. 17 (a) Effectiveness factors and Thiele moduli for benzyl alcohol self-esterification over differently sized MFI zeolites. Reprinted with permission from ref. 95. Copyright 2012 Science. (b) The ratio of trimers to dimers formed in propene oligomerization over MFI with samples different diffusion parameters (varying acid site densities). Reprinted with permission from ref. 96. Copyright 2016 American Chemical Society. (c) Effectiveness factors and Thiele moduli for cumene cracking over conventional (red triangle) and hierarchical (blue star) ZSM-5. Reprinted with permission from ref. 97. Copyright 2011 Royal Society of Chemistry. | ||
The second term in eqn (50) is k, which represents the reaction rate constant. Similar to the diffusion length, any increase in the reaction rate constant, or reactivity, increases the Thiele modulus and correspondingly decreases the effectiveness factor. Sarazen et al. demonstrated how increases in reactivity can increase the severity of diffusion limitations in propene oligomerization by comparing the extent of trimer formation from dimers, a reaction enhanced by diffusion limitations, on MFI zeolites with similar diffusion lengths but varying reactivities (i.e., acid site densities).96 They showed that the extent of the trimer formation from dimers increases monotonically with a diffusion parameter, φ, which scales proportionally with acid site density (see Fig. 17b).
The last term in eqn (50) is D, which represents the effective diffusivity of the reacting species in the zeolite. Increased values of this parameter reduce the Thiele modulus and thus enhance the effectiveness factor. Ways to increase the diffusivity of a given species include generating mesopores by desilication or dealumination, which cause partial destruction of the microporous framework and allow better overall accessibility. Zhou et al. studied the effect of mesopore formation on the catalytic effectiveness of cumene cracking over ZSM-5.97 They prepared conventional and hierarchical ZSM-5 samples and measured rates of cumene cracking on these samples. As shown in Fig. 17c, hierarchical ZSM-5 showed a higher effectiveness factor than conventional ZSM-5. The authors attributed this higher efficiency to a decreased “effective” diffusion length, induced by the higher diffusivity of cumene in the mesopores of hierarchical ZSM-5.
As the understanding of diffusion limitations in zeolites deepens, the need to overcome these barriers becomes increasingly evident. This need has driven the development of new zeolite designs, including hierarchical structures and nanozeolites, aimed at optimizing diffusion. In Section 4, we explore the strategies designed to create these advanced zeolite architectures, which focus on reducing diffusion distances within the material. Hierarchical zeolites, which incorporate both micropores and mesopores, are particularly promising in this respect. These modifications allow for better connectivity between different pore levels. This improvement in diffusion is essential to mitigate the limitations discussed earlier, particularly those associated with the restricted diffusion of molecules in traditional microporous structures.98–100
The importance of having efficient diffusion pathways within zeolites cannot be overstated. For instance, if we consider catalytic applications, as illustrated in Fig. 18, it is not possible to fully utilize the entire zeolite crystal unless intracrystalline mesopores are introduced.101 Without sufficient diffusion, only the outer regions of the zeolite are effectively engaged in the reaction, leaving a significant portion of the material underutilized. By incorporating mesopores, which enable faster diffusion, the entire zeolite crystal can be utilized for catalysis, enhancing both the efficiency and effectiveness of the catalyst. Therefore, ensuring proper diffusion pathways is a critical factor in the design and application of zeolite-based catalysts.
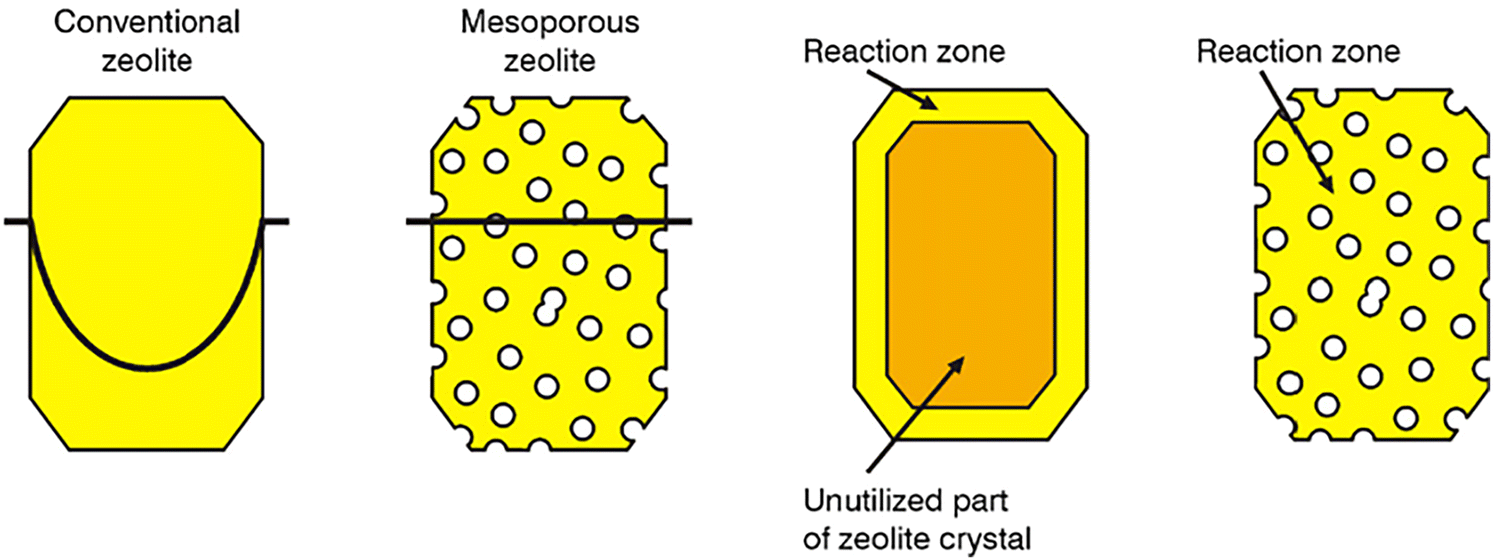 | ||
| Fig. 18 Schematic illustration of the concentration profile of a reactant through a conventional zeolite crystal when diffusion is limiting the catalyst's performance. In a mesoporous zeolite crystal, diffusion is fast enough to maintain the reactant concentration at the same level inside and outside the crystal during the reaction. This improvement in diffusion allows for the entire zeolite structure to be utilized in catalysis, optimizing its efficiency. Reproduced with permission from ref. 101 Copyright 1991 Elsevier. | ||
The microporous structure of zeolites also provides critical confinement effects, stabilizing reaction intermediates and transition states, which notably enhances catalytic selectivity and activity, making it vital for shape-selective catalysis. Introducing mesoporosity through functional mesopores, which have thin pore walls and active sites primarily on their surfaces, or auxiliary mesopores, which serve as internal shortcuts to microporous active sites, inevitably leads to a reduction in micropore volume. This reduction may partially diminish confinement effects, potentially lowering catalytic selectivity and precision. Additionally, the creation of mesopores might induce structural defects or alter the distribution of acidity, negatively impacting catalyst stability. To address this balance, strategies such as synthesizing zeolite nanosheets or carefully adjusting surface acidity have been developed, aiming to optimize mass transfer efficiency while preserving the beneficial microporous confinement.
Additionally, designing such materials involves significant challenges, as it is necessary to balance pore size and structural integrity of the zeolite. Modifying the porous architecture, such as introducing mesopores, can enhance the accessibility of molecules to active sites, but it may also affect the stability and mechanical strength of the material.102 Therefore, the selection of synthesis methods, whether top-down or bottom-up, plays a pivotal role in determining the final performance of these advanced zeolites. Each of these approaches presents its own set of advantages and disadvantages: while bottom-up methods allow for more precise control over structural properties, top-down methods offer greater simplicity and scalability, though often with some sacrifice in structural perfection. Consequently, the choice of synthesis method must be based on the specific requirements of the application, balancing the enhancement of diffusion with the preservation of the desired zeolite characteristics.103–105
Before discussing the various methodologies used to design zeolites with improved molecular diffusion, it is essential to describe the most general method for synthesizing zeolites. Zeolite synthesis is a complex process influenced by numerous factors, each of which can significantly impact the resulting structure and properties of the material. A typical synthesis of zeolites involves a hydrothermal process, which occurs under autogenous pressure and elevated temperatures. The process begins with a gel preparation, where a mixture of silica (SiO2) and alumina (Al2O3) sources is combined with water, alkali (typically OH− or F−), and a structure-directing agent (SDA) to form a homogeneous gel. The molar ratios of these components as well as concentration are crucial as they dictate the final zeolite framework, influencing properties such as pore size, shape, and acidity.104,106–113
Once prepared, the gel is transferred into a Teflon-lined stainless-steel autoclave, where it undergoes hydrothermal treatment at a temperature ranging from 100 °C to 200 °C. During this process, nucleation and crystal growth occur, forming the zeolitic framework. The time of crystallization can vary, from a few hours to several days, depending on the type of zeolite being synthesized. After the hydrothermal reaction, the resulting solid product is separated from the liquid by filtration, washed thoroughly with deionized water to remove residual alkaline components, and then dried at a moderate temperature (typically 100 °C). Following drying, the zeolite material is often subjected to calcination at high temperatures (500 °C to 600 °C) to remove the organic SDA, which burns off, leaving the zeolite's porous framework.
4.1 Nanozeolites
One of the initial approaches to improving the diffusion of reactants and products through the zeolite crystal is to reduce the crystal size.110,114,115 This practice significantly decreases the distance between the crystal edge and the center in other words the diffusion length and generating auxiliary mesoporous by aggregation of the nanocristal. Additionally, the surface-to-volume ratio increases substantially, providing a larger external surface that can be utilized when molecules are too large to diffuse through the intrinsic microporosity of the zeolite. Another advantage of reducing the zeolite size is that it can be synthesized with the desired Si/Al ratio, as no post-synthetic treatment is required that might alter its composition.113 By organizing synthesized nanozeolites into larger, ordered structures, a hierarchical framework with inter(nano)crystalline mesopores can be achieved. The size of these mesopores can be adjusted based on the dimensions of the nanocrystals. The assembled nanozeolites exhibit improved characteristics compared to traditional zeolites, combining the benefits of nanozeolites with the additional advantage of mesopores. These mesopores facilitate faster transport of reactants and products in and out of the zeolite system, enhancing overall efficiency.Nucleation is the first step in crystal formation, where atoms or molecules gather into small clusters, called nuclei, within a solution. These nuclei can form a stable crystalline structure under proper conditions but may dissolve if the conditions are unfavorable. Supersaturation drives nucleation, occurring when the concentration of precursors exceeds their solubility. Increasing temperature can enhance nucleation but may also promote undesired phases.
Before discussing nanozeolites, it is important to establish the criteria that define the size of zeolite crystals to be considered as nanozeolites. In the pioneering work by Valtchev et al.,115,116 it was established that a material can be classified as a nanozeolite if it has at least one dimension smaller than 100 nm.117 This size threshold distinguishes nanozeolites from conventional zeolites, allowing for enhanced diffusion and accessibility to active sites. The small size of nanozeolites makes them difficult to handle, and while traditional zeolites often need to be structured into pellets or monoliths, this becomes even more important and critical for nanozeolites. Structuring is essential to improve their mechanical stability and facilitate their use in practical applications, as the extremely small particle size can lead to challenges in processing and handling without such structuring.110
Within bottom-up techniques, some of the most utilized methods include the control of nucleation to promote the growth of nanosized materials and the design of organic structure-directing agents (SDAs), which guide the formation of specific frameworks. These methods help to control the size, shape, and porosity of the final product. A more recent addition to this toolkit involves data analytics and machine learning,118,119 which offer innovative ways to predict and optimize synthesis conditions, potentially transitioning zeolite synthesis away from empirical, trial-and-error methods.
4.1.1.1 Control of nucleation. Rimer et al.110,120,121 and other authors,116 in successive studies, have demonstrated that the nucleation and precipitation of nanozeolites differ from the classical nucleation theory, in which the monomers or zeolite precursors arrange themselves to form initial nuclei that subsequently grow. These studies reveal that amorphous solids are first formed, which then follow various pathways before crystallizing into zeolite structures.122 Controlling these processes is crucial for the synthesis of nanozeolites with precisely controlled structure and morphology at the nanometric level.
There are several approaches to controlling the nucleation and growth of zeolite crystals, even though this is a topic that is not yet fully understood and requires further research. In fact, there is not a clear correlation between crystal size and concentration of precussors that can be applied to all zeolites. A deeper understanding is essential to develop effective synthesis protocols that offer high yields and are environmentally friendly.110
The size of crystals is determined by the relationship between the nucleation rate and the growth rate. To synthesize nanozeolites, two key factors must be controlled: (i) ensuring a high nucleation rate and (ii) stabilizing the nanoscale nuclei (either amorphous or crystalline). The first factor is influenced by various parameters such as temperature, alkalinity, aging, and concentration and solubility of the reactant while the second is primarily governed by the influence of SDAs. A summary of these parameters is provided below.
4.1.1.1.1 Temperature. Temperature is crucial in regulating both nucleation and crystal growth. While higher temperatures generally promote nucleation, they can also accelerate growth (Ostwald repining), which requires careful adjustment to maintain small crystal sizes. Crystallization is often carried out at relatively low temperatures (80–100 °C) because, under these conditions, the growth rate exceeds the nucleation rate. However, it is important to note that very low temperatures can lead to poor crystallinity, reduced efficiency, and extended crystallization times. Li et al.123 investigated the impact of temperature on the nucleation and crystallization kinetics of nanosized faujasite zeolites using a two-stage, varying-temperature technique. Their study demonstrated that both nucleation and crystal growth of zeolite Y occur simultaneously, in contrast to the sequential process seen in other systems like TPA-silicalite-1.124 At 100 °C, the nucleation period for zeolite Y spanned 6.5 days, nearly overlapping with the entire crystal growth phase. The authors compared crystallization at 100 °C and 130 °C and showed that higher temperatures reduce the crystallization time from 7 to 3 days and increase crystal size from 75 nm to 137 nm, but at the cost of lowering the crystal yield. This highlights how temperature influences not only the kinetics but also the size and yield of the final product. Their findings emphasized that precise temperature control is crucial for optimizing the nucleation process.
4.1.1.1.2 Alkalinity. As previously mentioned, there is no general rule for such a complex process as zeolite synthesis. However, certain trends can be identified. Cardoso et al.125 explored a method to synthesize nanosized faujasite crystals without using organic structure-directing agents (SDAs). The focus of the study is to examine the effect of alkalinity and aging in controlling the size of the crystals, as well as the influence of the hydrothermal treatment process on the properties of the resulting material. The authors found that increasing the alkalinity of the synthesis gel—achieved by varying the Na2O/Al2O3 ratio—led to a significant reduction in particle size. Samples synthesized with higher alkalinity ratios exhibited smaller, irregular particles compared to those with lower ratios, which had more well-defined octahedral morphology. However, at very high alkalinity, the formation of secondary phases, such as NaP1 (GIS framework) zeolite, was observed, suggesting a limit to the alkalinity that could be used to synthesize pure faujasite.
The introduction of an aging step, during which the synthesis gel was aged at 25 °C for up to 72 hours before hydrothermal treatment, had a substantial effect on both the crystal size and the material's structural organization. Aging the gel increased the number of nuclei formed, resulting in smaller, more uniform crystals and higher microporosity. This aging step also prevented the contamination of the product by secondary phases, ensuring a purer faujasite phase. Crystallinity and micropore volume were measured and showed that the highest alkalinity sample (Na2O/Al2O3 ratio of 8.5) had the lowest crystallinity and micropore volume, likely due to the dissolution of the zeolite during synthesis. However, the introduction of aging improved the microporosity and led to the formation of a more organized, mesoporous structure.
4.1.1.1.3 Reactant concentration. The concentration of reactant significantly influences the system's saturation level. At lower supersaturation, crystal growth is promoted over nucleation, and dilution of the solution tends to result in the formation of larger crystals. Therefore, maintaining a high concentration is essential to favor nucleation and control crystal size.
4.1.1.2 Inhibitors for crystal growth. The synthesis of nanozeolites using a growth inhibitor involves introducing an organic additive, distinct from the structure-directing agent (SDA), to hinder the growth process and produce smaller zeolite crystals. The success of this method relies on two key factors: the reactivity of the additive and its concentration in the synthesis mixture. The additive must adsorb onto or interact with the surface of silicate particles, preventing further aggregation. However, if the additive concentration is too high, zeolite formation may be inhibited due to a lack of free aluminosilicate species required for building the structure. On the other hand, if the concentration is too low, the inhibition effect will be insufficient.
For instance, Zhang et al.126 developed a synthetic strategy for producing high-quality single-crystalline MFI-type nanozeolites with diameters between 10 and 55 nm. This method employed a two-step crystallization process in combination with L-lysine, which acted as a growth inhibitor to limit crystal growth. Initially, at 80 °C, metastable nanoparticles with irregular morphology were formed. In the second step, these nanoparticles were further crystallized at 170 °C, resulting in well-defined crystals without excessive growth, thanks to the inhibition effect of L-lysine. The use of L-lysine not only stabilized the crystal size but also prevented aggregation by forming hydrogen bonds with silanol groups on the zeolite surface. The two-step process and the introduction of L-lysine allowed for the synthesis of nanozeolites with high monodispersity, crystallinity, and yield. This approach was also effective in creating highly stable ZSM-5 catalysts, which demonstrated superior performance in methanol-to-propylene (MTP) reactions, with prolonged catalytic lifetimes and improved selectivity compared to conventional ZSM-5 crystals.
In another example, Wen et al.127 investigated the synthesis of hierarchical porous ZSM-5 (HP-ZSM-5) using organosilane-assisted methods to interfere with the normal crystallization process of the zeolite. Four different organosilanes were employed as growth inhibitors: (3-aminopropyl) triethoxysilane (AMEO), γ-chloropropyltriethoxysilane (CPTEO), triethoxyvinylsilane (VTES), and 3-triethoxysilypropylmercaptan (CPTES). These organosilanes were grafted onto the surface of zeolite precursors through covalent Si–O–Si and hydrogen bonds, hindering crystal growth. The study demonstrated that the use of organosilane significantly altered the morphology, particle size, and porosity of the ZSM-5. The resulting HP-ZSM-5 samples exhibited spherical aggregates of smaller crystals with rough surfaces, enhancing diffusion and mass transfer compared to conventional ZSM-5. The introduction of mesopores also improved the catalyst's stability and selectivity in light-olefin production from chloromethane, reducing side reactions and coke formation.
Similarly, Yang et al.128 explored the synthesis of nano-sized ZK-5 zeolites using β-cyclodextrin as a growth inhibitor to control the crystal size. By regulating the amount of β-cyclodextrin in the synthesis process, the authors were able to reduce the size of ZK-5 crystals from 3 μm to 50–100 nm. The hydrogen bonds formed between the hydroxyl groups of β-cyclodextrin and the silicon hydroxyl groups on the surface of ZK-5 nuclei helped to slow down the crystal growth, while the nano-cavity of β-cyclodextrin provided spatial confinement, further limiting crystal expansion. The optimized nano-ZK-5 sample, designated as S-0.005, exhibited superior properties, including a high specific surface area of 370 m2 g−1 and a pore volume of 0.22 cm3 g−1, compared to the conventional ZK-5 sample with a surface area of 150 m2 g−1. These improvements resulted in enhanced methane (CH4) adsorption capacity, with the nano-ZK-5 achieving a 64% increase in capacity compared to the micron-sized counterpart. Additionally, the nano-ZK-5 demonstrated faster gas diffusion and mass transfer rates, confirmed through adsorption kinetics experiments.
Other organic compounds, including 1,2,3-hexanetriol,129 polyethylene glycol,130 polyoxyethylene lauryl ether,130 and methylene blue,130 have also been employed to reduce zeolite crystal size. Despite these efforts, most of the zeolites synthesized remain in the sub-micron range, and reports of true nanosized zeolites are still rare.
4.1.1.3 Confined space synthesis. In 2000, Schmidt et al.131 introduced the concept of synthesizing zeolites within confined spaces (CSS). This method is based on three key principles: (i) preparing the synthesis gel within the confined space of inert matrices, (ii) crystallizing the zeolite within the pore networks of these matrices, and (iii) preventing the migration of zeolite growth species to the external surface of the inert matrix during crystallization. The CSS approach allows for precise control over the crystal size and morphology by restricting crystal growth to the internal pores, resulting in hierarchical zeolite structures. This technique enhances porosity and accessibility to active sites, making it highly effective for improving catalytic and adsorptive properties.
There are multiple methods for preparing the synthesis gel within an inert matrix. The most common approach is through incipient wetness impregnation, where the structure-directing agent (SDA) and the silica and alumina sources are dissolved in a solvent and then added to the porous matrix. The amount of solution used matches the pore volume of the inert matrix. Capillary forces allow the solution to be absorbed and confined within the matrix pores. This process can be carried out in one or multiple steps. Another method involves impregnating the inert matrix with the zeolite synthesis components through an alternative approach. In some cases, clear solution synthesis is employed, where a transparent solution containing zeolite precursors, typically in nanometer-scale colloidal form, is prepared. When these precursors are smaller than the pore size of the inert matrix, they can be introduced into the matrix, where crystallization occurs. The use of clear solutions often leads to the formation of zeolite nanocrystals, as the precursors remain colloidally stable throughout the process. A third method for introducing synthesis components into an inert matrix involves producing a composite matrix consisting of silica and the inert material in a single step. For example, mesoporous silica has been employed as a hard template to create mesoporous carbon and polymer-based materials. These hard templates are typically formed through a multi-step process, where a carbon resin is infused into the silica template and then carbonized. Additionally, the silica template can act as a growth framework for the development of hierarchical zeolites. Utilizing silica/carbon composites as starting materials not only enables the synthesis gel to be confined within the matrix but also streamlines the synthesis process.132 Another method recently published introduced the reactant in the solid matrix by ball milling.133
Once the matrix is loaded with the zeolite precursors, crystallization must occur. There are similar used methods for this process are steam-assisted crystallization (SAC), vapor-phase transport (VPT). In the SAC method, the precursor-loaded matrix, including the structure-directing agent (SDA), is exposed to water vapor, initiating the crystallization process. In the VPT method, if the vapor consists of a mixture of water and a volatile SDA, the matrix only needs to contain the silicon and aluminum sources. In this case, the SDA is supplied through the vapor phase, facilitating crystallization.
An example of this is the one published by Ogura et al.,132 who explores the synthesis of hierarchical zeolite-mesoporous composites (ZMM-n) by employing the vapor-phase transport (VPT) method, using SBA-15 as the inert matrix. Their approach emphasized the role of carbon filling to preserve the mesostructure of SBA-15 during the crystallization process. The study demonstrated that without carbon, the mesoporous structure of SBA-15 tends to collapse during the VPT synthesis, whereas carbon-filled mesopores help maintain structural stability. The synthesis process involved impregnating SBA-15 with aluminum to create Al-SBA-15, followed by the introduction of carbon precursors such as sucrose or furfuryl alcohol into the mesopores. This composite is then subjected to VPT, where structure-directing agents (SDAs) in vapor form facilitate the crystallization of nanosized ZSM-5 along the mesopore walls. The carbon filling controls the diffusion of SDAs, slowing down the nucleation process and ensuring the formation of uniform zeolitic building units within the mesoporous framework.
Chen et al.134 explored the synthesis of nanozeolites within the confined space of a three-dimensionally ordered mesoporous (3DOm) carbon template using a hydrothermal method. The study demonstrates how this confined space approach allows for precise control over crystal size and morphology, producing hierarchical zeolites with both micro- and mesoporosity. The 3DOm carbon, used as a hard template, was filled with a synthesis gel via incipient wetness impregnation, ensuring that nucleation and crystal growth occurred exclusively within the mesopores of the carbon framework. The authors synthesized several types of zeolites, including BEA, LTA, FAU, and LTL, using this method. The process involved multiple synthesis cycles, with each step reinforcing the crystal formation within the mesopores. The zeolite crystals produced exhibited interconnected spherical elements limited by the size of the mesopores in the 3DOm carbon template, confirming the effectiveness of the confined growth strategy.
Ma et al.133 presented an innovative method for the synthesis of nanosized hierarchical Beta zeolites through a steam-assisted crystallization (SAC) process. This approach employs solid raw materials that are ball-milled to ensure thorough mixing before crystallization. By leveraging the SAC method, the authors achieved highly dispersed nanosized zeolites with interconnected mesopores, which enhance diffusion and accessibility to active catalytic sites. The study demonstrated that Beta zeolites synthesized via this method, with Si/Al ratios ranging from 10 to 40, possess superior structural characteristics compared to those produced through conventional hydrothermal methods. Crystallization occurs within mulberry-like aggregates composed of uniform nanoparticles (∼15 nm), promoting both high crystallinity and excellent dispersion. In this method, the steam atmosphere facilitates the migration of raw solid components during crystallization, preventing agglomeration and ensuring uniform particle distribution.
Wakihara et al.135 presented a top-down approach for the synthesis of nanozeolites using bead milling followed by a post-milling recrystallization process. In this study, commercially available zeolite A (LTA type) was initially subjected to bead milling to reduce its size, resulting in a powder with significantly smaller particles. However, the milling process also led to partial amorphization of the zeolite, damaging its crystalline structure. To restore the crystallinity and improve the quality of the nanozeolites, the authors employed a recrystallization step. This involved treating the milled zeolite in a dilute aluminosilicate solution, which facilitated the selective recrystallization of the damaged parts of the zeolite. During this process, the amorphous regions dissolved and recrystallized onto the remaining crystalline framework, while maintaining the nano-scale size achieved during the milling step. The combination of bead milling and post-milling recrystallization yielded nanozeolites. The obtained crystals had an average size of approximately 50 nm and high crystallinity.
Liu et al.136 improved the top-down methodology for nanosized zeolite production by focusing on the post-milling recrystallization process. Unlike previous studies where recrystallization took several hours, this work introduces a much faster recrystallization step, taking only a few minutes (10 minutes for SSZ-13 and 5 minutes for AlPO4-5) using a tubular reactor. Anand et al.137 introduced a significant advancement in the top-down synthesis of zeolites by developing an in situ recrystallization method during bead milling. This method addresses the limitations of traditional top-down approaches, where zeolite crystals are mechanically broken down, resulting in partial amorphization, which previously required a separate post-milling recrystallization step. In their study, the authors combined milling and recrystallization into a single process by modifying the milling apparatus to operate under high alkalinity and controlled temperatures. Using this method, zeolite A crystals were reduced from approximately 3 μm to 66 nm in just 30 minutes, a much faster and more efficient process compared to prior techniques. During milling, three simultaneous effects—miniaturization, amorphization, and recrystallization—occur, allowing for the formation of highly crystalline nanoparticles. The recrystallization process happens in situ within the milling apparatus, driven by the thermal and alkaline conditions, eliminating the need for a post-milling step.
The advantages of nanozeolites are not limited to light olefin synthesis. In hydrocracking and hydroprocessing of heavy oils (which use zeolites like Beta or Y), diffusion within micropores often limits the cracking of bulky feed molecules. Nanoscale Beta or Y zeolites can dramatically improve the conversion of heavy hydrocarbons by giving molecules better access to active sites. For instance, a recent study on a Beta zeolite for crude oil hydrocracking found that a catalyst containing <80 nm Beta crystals with added mesoporosity achieved “much higher heavy oil conversion activity and increased naphtha selectivity” than a catalyst with conventional Beta.142 This means more of the heavy fraction was cracked into lighter products (naphtha range) when using the nano-Beta, owing to enhanced diffusion of large feed molecules into, and products out of, the zeolite. In the fluid catalytic cracking (FCC) process, it is also well known that smaller Y zeolite crystallites or desilicated (mesoporous) Y lead to higher gasoline yields and lower coke yields.139,142
In hydroisomerization of long-chain alkanes, researchers observed that nano-sized Pt/ZSM-5 and Pt/Beta catalysts exhibited higher turnover and a narrower product distribution, consistent with fewer diffusion bottlenecks that would otherwise cause over-cracking of intermediates.143 Moreover, catalyst deactivation by coking is universally mitigated in nanozeolites. Thick crystals tend to trap polyaromatic coke in their cores, rapidly closing off channels. When those same frameworks are nano-sized, coke tends to form on the external surface or in non-critical locations, allowing the catalyst to function longer. Hedlund et al. demonstrated a strong correlation between ZSM-5 crystal thickness and deactivation rate: 400 nm thick ZSM-5 crystals deactivated much faster than 35 nm crystals, due to intracrystalline coke blocking the larger crystals’ pores.144 At equal Si/Al and identical reaction conditions, the 35 nm ZSM-5 retained activity far better, confirming that diffusion path length was the key differentiator in coking behavior. Additionally, they found defect-free nano-ZSM-5 was more resistant to deactivation than defect-rich nano-ZSM-5, again implying that an ideal nanozeolite should combine small size with good crystallinity to maximize lifetime.
4.2 2D zeolites
2D zeolites have gained significant attention due to their unique structural properties, which offer advantages over conventional three-dimensional zeolites. The reduced thickness of 2D zeolites enhances the accessibility of their active sites, facilitating faster diffusion of reactants and products. This increased surface area and improved mass transfer make 2D zeolites highly effective in catalysis, adsorption, and separation processes. Additionally, their tunable pore structures allow for greater control over reaction pathways, making them ideal for advanced applications in energy conversion, environmental remediation, and chemical synthesis.These materials, with thicknesses typically on the scale of a few unit cells, can be produced through three main approaches: layered precursors, the ADOR (assembly-disassembly-organization-reassembly) mechanism, and direct synthesis. In the layered precursor method, stacked zeolite layers are exfoliated or delaminated into individual 2D sheets. The ADOR mechanism involves breaking down the zeolite framework into its building units and then reorganizing and reassembling them into 2D structures. Direct synthesis, on the other hand, forms 2D zeolites directly during crystallization, often guided by surfactants or organic structure-directing agents, which promote the formation of thin, plate-like layers.
The first example of this is the one published by Roth et al.,153 who focused on the development of the ADOR process by preparing layered materials. The study began with the zeolite UTL, which contains germanium-rich double four-ring (D4R) units. By selectively removing these D4R units through hydrolysis, the researchers were able to disassemble the three-dimensional framework of UTL and obtain a layered intermediate material, termed IPC-1P. This layered precursor was key to the ADOR strategy, as it served as the basis for further manipulation. The disassembly step was carefully controlled to ensure that the germanium-rich units were removed while preserving the integrity of the remaining layers, resulting in stable and well-defined sheets (see Fig. 19). This layered material could then be reorganized and reassembled into new zeolitic frameworks by introducing different linkers between the layers, such as oxygen bridges or new silicate units. The ability to maintain the layered structure during disassembly was a crucial aspect of the ADOR process, allowing for the subsequent modification and reconstruction into new zeolites with varying pore architectures, such as IPC-2 and IPC-4. However, the method is somewhat limited by the high cost of germanium, which makes large-scale applications challenging.
 | ||
| Fig. 19 Schematic of the ADOR method. A is the assembly step (in blue is the germanium rich areas and in red is the rest of the zeolite components) and B is the disassembly step by hydrolysis of the germanium rich areas. Reproduced with permission from ref. 153 Copyright 2013 Nature Publishing Group. | ||
Surfactants can also be used in the synthesis of 2D zeolites. In this process, the surfactant plays a dual role: the “head” of the surfactant acts as a structure-directing agent (SDA), guiding the formation of the zeolite framework, while the “tail” introduces steric hindrance, preventing the zeolite from growing in all directions. This steric barrier restricts the growth of the material, encouraging the formation of layered or two-dimensional structures. By carefully selecting and designing surfactants with these properties, it is possible to tailor the synthesis of 2D zeolites with specific structural features. This approach was first demonstrated by Choi et al.,155 who introduced a novel method for synthesizing single-unit-cell nanosheets of MFI zeolite using a bifunctional surfactant. The surfactant not only directed the crystallization of the MFI framework but also limited the growth in one dimension, resulting in ultrathin zeolite layers with a thickness of just 2 nm. The diammonium head of the surfactant served as a structure-directing agent (SDA), while the long alkyl tail provided steric hindrance, preventing further crystal growth and enabling the formation of two-dimensional nanosheets. This breakthrough demonstrated the potential of using surfactants to precisely control the structure and dimensions of zeolites (Fig. 20). An excellent illustration of the control achievable in this study is the ability to adjust the thickness of the nanosheets by modifying the number of quaternary ammonium groups in the surfactant. Increasing the number of cationic units in the surfactant head from two to three or four results in progressively thicker zeolite nanosheets. This demonstrates that by altering the composition of the surfactant, the thickness of the zeolite layers can be finely tuned.
 | ||
| Fig. 20 (a) Suggested structural model for a single MFI nanosheet. The surfactant molecules are oriented along the straight channels of the MFI framework, with two quaternary ammonium groups (represented by red spheres) positioned at the channel intersections. One ammonium group is embedded within the framework, while the other is situated at the pore opening on the external surface. These nanosheets can form either multilamellar stacks along the b-axis (b), or assemble in a disordered manner as individual unilamellar sheets (c). Reproduced with permission from ref. 155. Copyright 2009 Nature Publishing Group. | ||
In addition to higher initial activity, 2D zeolites resist deactivation by coke deposition far better than bulk analogues. Choi et al.155 reported that single-unit-cell MFI nanosheets maintained methanol conversion much longer than normal ZSM-5, with significantly slower coke accumulation. These ultrathin ZSM-5 layers gave higher conversion in heavy hydrocarbon cracking (e.g. polyethylene) and showed delayed catalyst decay in methanol-to-gasoline trials relative to conventional ZSM-5. Nanoscale zeolites in general benefit from short diffusion lengths – for instance, 10–50 nm ZSM-5 crystals in the methanol-to-propylene reaction achieved 54 h stable run time (at 470 °C, WHSV 7.2 h−1) with 74% light olefins selectivity (49% propylene).
Crucially, the benefits of 2D zeolite catalysts have been realized across a wide range of chemistries – from refining and petrochemical processes to biomass conversions. In reactions involving larger molecules (alkylation, acylation, hydroisomerization, aromatization, hydrodesulfurization, etc.), hierarchically structured and 2D-form zeolites consistently show higher effectiveness than purely microporous equivalents.158
4.3 Hierarchical zeolites
4.3.1.1 Nanozeolite self-assembly. It is important to note that nanozeolite powders are not considered mesoporous zeolites, as their mesopores are only present temporarily when the powder is compacted. For a material to be classified as truly mesoporous, the mesoporosity must be a permanent characteristic of the material. Additionally, nanozeolite powders are typically produced in low yields, and their isolation is both time-intensive and costly. In contrast, mesoporous zeolites should be synthesized with minimal reaction steps and preferably using affordable templates to make them viable for industrial applications. Considering these points, the self-assembly of nanozeolites into stable, mesoporous aggregates appears promising. These structures offer several benefits: they provide an interconnected secondary pore network that facilitates mass transport, they have large external surface areas, and they enable short intraparticle diffusion paths.159
One of the first approaches to assembly crystal nanoparticles was published by Gu et al.160 They developed a novel strategy for synthesizing hierarchical mesoporous zeolites by employing a mixed-template system. In their approach, they used a cationic surfactant, cetyltrimethylammonium bromide (CTAB), with the co-solvent tert-butyl alcohol (TBA) and a swelling agent, 1,3,5-trimethylbenzene (TMB). This methodology enabled the formation of hierarchical structures by condensing zeolite nanocrystals onto meso-scaled surfactant micelles (Fig. 21). The hierarchical mesoporous zeolites synthesized, including zeolite Y or sodalite fragments, demonstrated the characteristic diffraction peaks of zeolites, confirming the preservation of zeolitic structure within the mesoporous framework. The addition of TBA and TMB played critical roles in the self-assembly process. TBA increased the charge density on the surfactant micelle surface, promoting stronger interactions with the aluminosilicate species, while TMB acted as a swelling agent, expanding the micelle size to match the zeolite fragments. This assembly method produced materials with large surface areas, high mesoporous volumes, and improved stability. Testing showed that these hierarchical zeolites, exhibited enhanced adsorption capacities, particularly for large molecules like N-nitrosonornicotine (NNN), outperforming conventional NaY zeolite in certain adsorption applications.
 | ||
| Fig. 21 Proposed route for the synthesis of hierarchical mesoporous zeolites by nanozeolite self-assembly. Reproduced with permission from ref. 160. Copyright 2010 American Chemical Society. | ||
Similarly, Wei et al.161 described a method for synthesizing ordered mesoporous silicalite-1 zeolites through the self-assembly of nanosized silicalite-1 seeds. The process employs a two-step procedure where silicalite-1 seeds of varying sizes are produced by controlling the heating time of an alkali precursor solution. These nanocrystals are then assembled into mesoporous materials using the triblock copolymer P123 as a template in acidic conditions. This assembly method, inspired by a bottom-up approach, leads to the formation of materials with well-defined micro- and mesopores without phase separation, producing a stable hierarchical structure. The synthesized mesoporous zeolite exhibits a large surface area (>700 m2 g−1) and high mesopore volume, with pore sizes around 3.8 nm. Detailed analyses, including X-ray diffraction, electron microscopy, and nitrogen adsorption, confirm the hierarchical arrangement of the microporous zeolitic framework within a mesoporous matrix.
Wang et al.162 developed a method for synthesizing hierarchical ZSM-22 hollow spheres through a two-stage hydrothermal process, resulting in structures with mesoporosity and enhanced surface area. In the first stage, ZSM-22 nanorods were formed, which later self-assembled into hollow spheres in the second stage with the assistance of cetyltrimethylammonium bromide (CTAB) and potassium fluoride (KF). The process leverages the developed grain boundaries of ZSM-22 nanorods, facilitating their self-assembly into stable hollow spheres. Characterization techniques, including X-ray diffraction (XRD), scanning electron microscopy (SEM), and nitrogen adsorption–desorption, confirmed the hierarchical structure with both micropores and intercrystalline mesopores. The nanorods created during the first crystallization stage provided the necessary structural rigidity and allowed for mesoporous channels to form between them.
Wang et al.163 reported a method for the direct, single-step synthesis of hierarchical ZSM-5 zeolites without the need for secondary templating. This approach, which is both cost-effective and efficient, leverages controlled nucleation and self-assembly of zeolite precursor particles. By carefully managing the formation and organization of nanoparticles early in the crystallization process, the researchers were able to create a hierarchical structure featuring a micro-mesoporous core and a thin crystalline shell. This structure consists of aggregated MFI nanocrystals approximately 30–50 nm in size, with interstitial mesopores that enhance mass transport and catalytic efficiency, particularly for reactions involving bulky molecules. The study demonstrates how altering synthesis conditions such as silica source, Si/Al ratio, NaOH concentration, and aging time influences the formation of precursor particles and the resulting hierarchical structures. Using methods like SEM and TEM and argon physisorption, the authors observed that mesoporosity develops as the crystallization time increases, resulting in a broadened mesopore size distribution from 3 to 15 nm. The mesoporosity and relatively short diffusion pathways of approximately 25 nm within the sub-micrometer spherical particles significantly improve mass transport properties, making the synthesized zeolite particularly effective for catalytic applications.
4.3.1.2 Seeded growth. Incorporating seeds in zeolite synthesis has emerged as a promising approach to develop hierarchical structures with enhanced mass transport properties. The addition of nanometric seeds into the synthesis mixture serves as a nucleation site, facilitating anisotropic crystal growth. The seeds used during synthesis can remain in the final structure if they are not consumed as a nutrient source, or they may be depleted if used as a nutrient. In some cases, this consumption of the seed leads to the formation of hollow structures, as described by certain studies.164
A very nice example of seed assisted growth where the seeds is not consumed, it was reported by the group of Rimer, the researchers developed a novel method to enhance the mass-transport properties of ZSM-11 and ZSM-5 zeolites by growing nanometric fin-like protrusions on the surfaces of seed crystals.165 These “fins” increased the external surface area and reduced coke formation by enhancing access to active sites and reducing diffusion limitations. In this example the seed is no dissolved during the synthesis, so it is preserved in the final structure. They demonstrated that these finned zeolites significantly outperformed conventional zeolites in catalytic tests, exhibiting improved selectivity and catalyst lifespan in methanol-to-hydrocarbons reactions. Building on the concept of finned zeolites, Dai et al.166 extended this approach to ferrierite, a two-dimensional zeolite with restrictive pore networks. By using seed crystals, they synthesized finned ferrierite structures with nanometric fin-like protrusions on the surface, enhancing mass transport and catalytic performance. Notably, the seeds remained intact during synthesis, and the fins did not alter the internal pore network but acted as pseudo-nanoparticles on the surface. Catalytic tests in 1-butene isomerization showed a three-fold increase in catalyst lifespan and a 12% improvement in isobutene selectivity compared to conventional ferrierite.
Zhang et al.167 presented an intriguing example of hierarchical zeolite synthesis using a nonclassical crystallization approach. In this study, the authors employ a salt-assisted, seed-induced synthesis method to create MFI zeolite mesocrystals in the form of bundled nanorods. Through anisotropic growth, precursor particles assemble and crystallize in an oriented fashion on the surface of silicalite-1 seeds. This oriented attachment of nanoparticles enables the formation of a unique core–shell–shell hierarchical structure. By optimizing anisotropic crystallization, the researchers enhanced the physical and chemical properties of the zeolites, leading to improved catalytic performance in adsorption and large-molecule conversion applications, contrast to the previous example here the seed is dissolved and used as nutrient for the crystal growth of the secondary structures.
Sun et al.140 explored the synthesis of hierarchical, nano-sized SAPO-34 zeolites using a seed-assisted method. The process involves using triethylamine (TEA) as the sole template and seeds with a nanosheet-like morphology to induce the growth of SAPO-34 crystals. Over time, the seeds undergo partial dissolution, which allows the formation of voids within the growing SAPO-34 structure, leading to a hollow morphology. The resulting zeolites not only exhibit meso- and macroporosity, but also maintain high crystallinity and yield. This hollow, hierarchical structure contributes to a significantly enhanced catalytic performance in the methanol-to-olefin (MTO) reaction, with up to a four-fold increase in catalytic lifetime and an improved selectivity for ethylene and propylene compared to conventional SAPO-34 zeolites.
Jain et al.168 demonstrate the formation of self-pillared pentasil (SPP) zeolites through a seed-assisted synthesis method that does not require SDA. The authors used MEL- and MFI-type zeolite seeds to initiate the spontaneous formation of nanosheets, which resulted in a hierarchical structure. During synthesis, the seeds partially dissolved, facilitating the formation of “pillars” or nanosheets that branch out from amorphous precursors. This approach generated a structure with high surface area and an elevated concentration of external acid sites, which enhanced the catalytic performance of the zeolite in methanol-to-hydrocarbons and Friedel–Crafts alkylation reactions.
4.3.1.3 Hard templating. The hard templating method uses solid, nonporous or porous materials with a rigid structure as sacrificial templates to introduce additional porosity during zeolite crystallization. This approach is versatile and applicable across various zeolite structures, as the solid template is chemically inert and does not interfere with the formation of the zeolite framework. Typically, the process involves adding solid templates to the zeolite synthesis mixture, allowing the zeolite to crystallize around the template, and then removing the template through calcination or leaching. Solid templates can be integrated into zeolite crystals during dry-gel conversion or throughout the crystallization process, resulting in highly crystalline zeolites with uniform and customizable porosity. Examples of materials used as hard templates include carbon-based substances, polymers, aerogels, resins, inorganic solids, and biological materials.
4.3.1.3.1 Carbon-based templates. The use of carbon-based hard templates is a widely applied strategy for synthesizing hierarchical zeolites. These templates include activated carbon,169–174 carbon nanotubes (CNTs),175–179 and mesoporous carbon,175,180,181 which act as sacrificial scaffolds to introduce additional porosity in the zeolite structure. One of the main advantages of using carbon materials as a hard template in zeolite synthesis is its easy removal by calcination. In this process, carbon is burned off at high temperatures in a controlled environment, typically in the presence of air or oxygen, without compromising the crystalline structure of the zeolite. Moreover, using carbon templates allows precise tuning of the Si/Al ratio in zeolite synthesis, which is essential for customizing the acid–base properties and catalytic activity of the material. There are two main approaches to using carbon materials as hard templates in zeolite synthesis. The first involves allowing the zeolite to grow around carbon nanoparticles, nanofibers, or nanotubes. When the carbon is later burned off, the zeolite is left with pores that match the size of these carbon structures. The second approach takes advantage of the internal porosity of activated or mesoporous carbons to grow the zeolite within these pores. This method results in a zeolite structure that mirrors the negative of the carbon's porous network.100,182
4.3.1.3.1.1 Zeolites around carbon materials. Jacobsen et al.183 used carbon nanoparticles as hard templates to synthesize large mesoporous ZSM-5 zeolite single crystals. They impregnated carbon black particles with a zeolite precursor solution containing tetrapropylammonium hydroxide, sodium aluminate, and tetraethyl orthosilicate. After hydrothermal crystallization at 180 °C for 72 hours, the carbon was removed by calcination at 550 °C, resulting in mesoporous ZSM-5 crystals.184 This approach was further extended to nanotubes. Schmidt et al.185 demonstrated a novel approach for synthesizing mesoporous zeolite single crystals using multiwall carbon nanotubes (MWNTs) as templates. The MWNTs, purified by hydrochloric acid reflux, were sequentially impregnated with zeolite precursors, including tetrapropylammonium hydroxide (TPAOH) and tetraethyl orthosilicate (TEOS). This method allowed silicalite-1 crystals to grow around the nanotubes, with hydrothermal crystallization occurring at 175 °C for 24 hours. After crystallization, the carbon nanotubes were removed by calcination at 600 °C, creating straight mesopores within the zeolite. Transmission electron microscopy (TEM) images revealed that the mesopores retained the dimensions and orientation of the original nanotubes, effectively penetrating the zeolite crystal. Selected area electron diffraction confirmed the high crystallinity of the silicalite-1 single crystals, with mesopores evenly distributed throughout. The resulting silicalite-1 zeolite exhibited high crystallinity and uniform mesopores that mirrored the structure of the original carbon nanotubes. This technique enabled the creation of straight and interconnected mesopores, improving mass transport and accessibility to catalytic sites. TEM analysis confirmed the presence of mesopores that penetrated the zeolite crystals, showcasing the method's ability to create single-crystal zeolites with enhanced mesoporosity tailored by the dimensions of the carbon nanotubes.
In recent studies, carbon materials have been used not only as hard templates but also as carriers for inorganic components. Chang et al.179 explored the use of carbon nanotubes (CNTs) containing gallium oxide (GaOx) nanoparticles as hard templates to synthesize Ga-supported ZSM-5 zeolites for methanol-to-aromatics (MTA) conversion. The CNTs were either encapsulated with GaOx inside their channels or decorated with GaOx nanoparticles on their surfaces. During the zeolite synthesis, these CNTs facilitated the crystallization of mesoporous ZSM-5 around the GaOx-loaded CNTs. The subsequent calcination removed the CNT templates, embedding GaOx within the ZSM-5 matrix. Two types of catalysts were produced for comparison: GaOx located on the outer CNT surfaces and GaOx confined within the CNT channels. The encapsulated GaOx provided stronger Lewis acid sites and preserved the Brønsted acid concentration, leading to improved methanol to aromatics (MTA) performance.
4.3.1.3.1.2 Zeolites inside carbon matrices. This method leverages the internal porosity of activated or mesoporous carbons to allow zeolite growth within these pores. One drawback of using carbon templates is the challenge in tightly confining zeolite crystallization within the template structure. This limitation arises from the incompatibility between the hydrophobic nature of the carbon template and the aqueous zeolitic precursor solution. As a result, some of the precursor gel can migrate away from the template, leading to crystallization outside the desired template boundaries.
One method to prevent unwanted crystallization outside the carbon template is to confine the zeolite precursors within the pores of the carbon matrix and then dry them to solidify. This is followed by exposure to a water vapor-rich atmosphere (SAC or VPT), initiating the conversion of the dried precursors into zeolite. The drying step forms a powder, significantly reducing the mobility of the precursor within the reaction environment, thereby keeping the zeolite precursor in controlled contact with the carbon template.174
Following the strategy of confining zeolite precursors within the pores of a carbon matrix, Chen et al.134 demonstrated a hydrothermal synthesis method to create zeolites with a three-dimensionally ordered mesoporous-imprinted (3DOm-i) structure. They used mesoporous carbon as a hard template, impregnating it with a zeolite precursor solution. During synthesis by SAC, the zeolite crystals grew within the carbon matrix's pores. After synthesis, the carbon was removed by calcination, leaving behind a well-defined, hierarchical mesoporous zeolite structure. This 3DOm-i structure enhanced mass transport and improved accessibility to catalytic sites, with potential applications in catalysis and separations. Chen's approach allowed for precise control over mesopore size and connectivity, producing hierarchical zeolites like BEA, FAU, and LTL, which retained both microporosity and uniform mesoporosity imprinted from the carbon template (see Fig. 22).
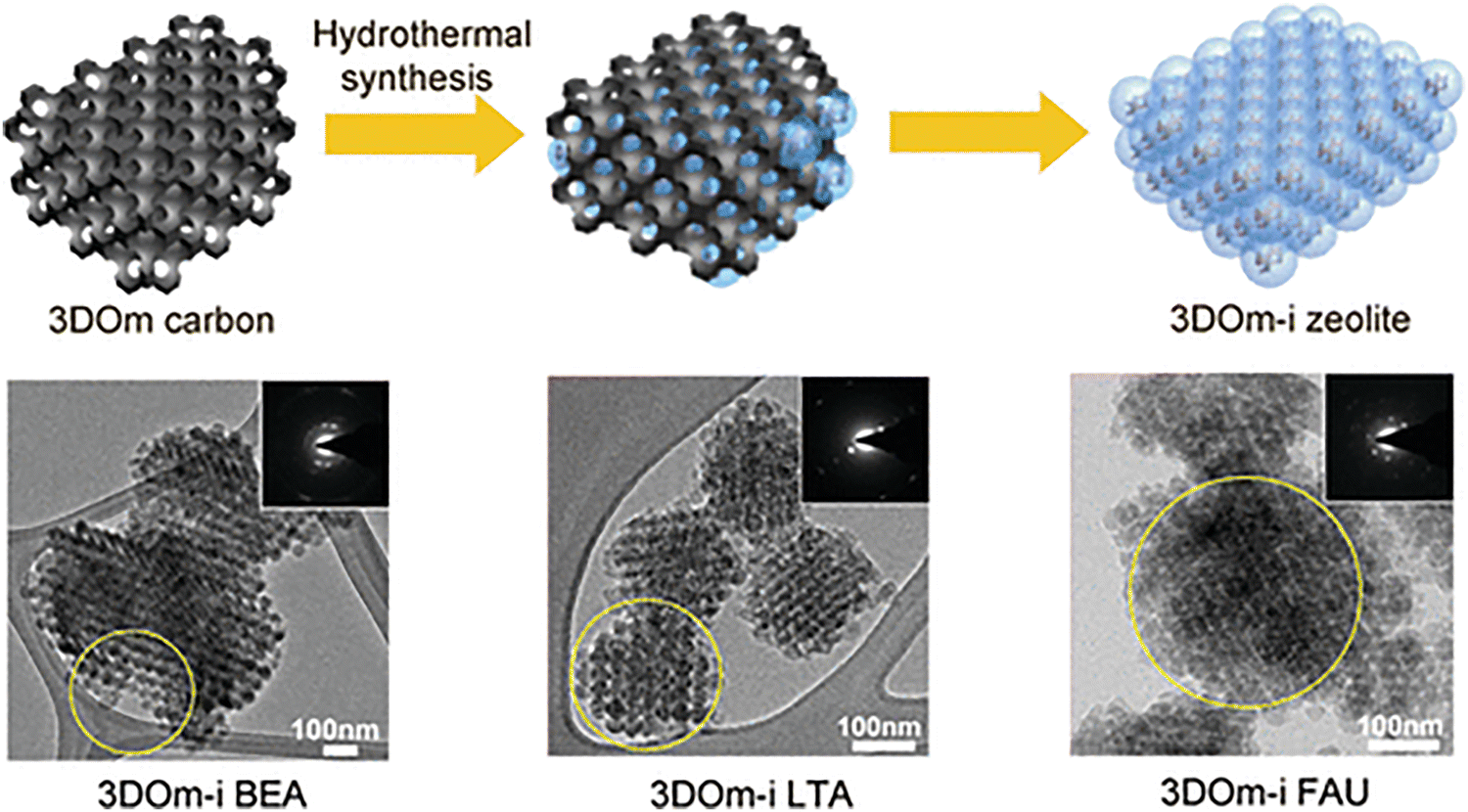 | ||
| Fig. 22 Synthesis of 3DOm-i zeolite from 3DOm carbon. Reproduced with permission from ref. 134. Copyright 2011 American Chemical Society. | ||
4.3.1.3.2 Polymers. Resins and polymers can also be used as hard templates for synthesizing hierarchical zeolites. Polymers, including polymer aerogels,186,187 polymer microspheres,188 and resins,108,189–191 are widely employed to create zeolite monoliths with bimodal, trimodal, or even tetramodal porosity. These hierarchical structures integrate multiple pore sizes, combining the advantages of each type to enhance mass transport and catalytic properties. Typically, the process involves coating a rigid porous matrix with a zeolite film or growing the zeolite within the porous structure of the polymer. Once the zeolite structure is formed, the polymer can be removed through calcination, leaving a well-interconnected pore system that retains the zeolite's intrinsic microporosity along with the introduced mesopores or macropores.
In addition to growing zeolites within the interstices formed by resins or polymers, zeolite nanocrystals can also self-assemble on the surface of polymers. This method involves modifying the polymer to possess specific surface charges that promote strong interactions between the zeolite nanocrystals and the polymer surface. By introducing functional groups or coatings that alter the polymer's surface charge, electrostatic attractions are enhanced, facilitating the uniform deposition and assembly of zeolite nanocrystals onto the polymer.192–194
4.3.1.3.3 Mesoporous silica/alumina spheres. It has been reported that the use and removal of polymer templates often lead to environmental pollution. Additionally, the calcination process used to remove the polymer cores can cause significant shrinkage (over 20%) or even fractures in the final products. As an alternative, mesoporous silica or aluminosilicate spheres can be used as hard templates to eliminate the need for a separate template removal step. This approach is similar to the seeding method but utilizes mesoporous silicas or aluminosilicate. These spheres serve both as meso- or macropore templates and as a source of silica nutrients for zeolite growth, allowing for continuous digestion of the spheres during the crystallization of the zeolites.195–200
Pashkova et al.201 developed an eco-friendly, and cost-effective approach for synthesizing hollow ZSM-5 zeolite spheres using a “self templating” method that bypasses expensive structure-directing agents (SDAs). This method relies on creating a spray-dried aluminosilicate precursor, prepared by mixing a colloidal silicic acid solution with aluminum butoxide. This precursor acts as both the silica and alumina source for the zeolite framework and as a shape-directing agent to form spherical particles. During the synthesis, the precursor is crystallized into ZSM-5 by adding seeds of ZSM-5 crystals. In the alkaline solution, the amorphous aluminosilicate spheres undergo swelling, acting as internal templates that eventually form hollow structures. The result is a stable, hollow ZSM-5 zeolite sphere with a well-defined shell structure composed of intergrown zeolite crystals. This process eliminates the need for template removal, simplifying the synthesis and avoiding potential environmental impacts associated with template disposal. The produced hollow spheres exhibit good mechanical stability and high aluminum content, making them highly suitable for applications in catalysis and adsorption.
Another highly significant example is the work published by Song et al.,202 which details a method for synthesizing hexagonal, hollow, aluminum-containing ZSM-5 zeolite tubes using mesoporous silica (MS) as a template. In this approach, the MS template, characterized by a worm-like morphology, functions both as a structural scaffold and as a silica source for the zeolite formation. The process begins by coating the MS template with nanocrystalline silicalite-1 seeds. Aluminum is then incorporated by impregnating the seeded MS with an aluminum nitrate solution, which provides the aluminum necessary for the zeolite framework. During hydrothermal synthesis, the MS template gradually dissolves, leading to the formation of hollow, hexagonal ZSM-5 tubes. This method effectively removes the need for an additional template removal step, as the MS is consumed during the synthesis, resulting in a streamlined production process for these hollow zeolite tubes.
4.3.1.3.4 Other types of templates. Biological templates offer an abundant, cost-effective, eco-friendly, readily available, and renewable option for zeolite synthesis. Biotemplating methods that use natural or biological tissues and materials as templates can lead to bioinspired zeolites with unique shapes, intricate functional patterns, and hierarchical porosity. These characteristics hold significant potential for advancing zeolite designs with enhanced performance in various applications.203,204
4.3.1.4 Soft templating. In zeolite synthesis, both a structure-directing agent (SDA) and a mesoporogen can be introduced, classifying this as a dual-template method. The SDA is responsible for generating micropores within the zeolite structure, while the mesoporogen facilitates the formation of mesopores. Common mesoporogens, such as surfactants, polymers, or organosilanes, function as soft templates. Alternatively, hard templates may be employed, which were covered in previous sections. Soft templates can serve as a physical scaffold, often by forming micelles around which mesopores develop, and they may also chemically interact with the zeolite phase as it forms. After synthesis, mesoporogens can be easily removed through calcination, creating mesopores in their former locations. A diverse range of soft templates can be utilized, and because their properties can be adjusted, mesopore size can be precisely controlled However, it is essential to select mesoporogens that are hydrothermally stable under synthesis conditions. Care should be taken, as the formation of amorphous mesoporous materials instead of crystalline hierarchical zeolites can occur, and defect sites may form, potentially reducing the zeolite's hydrothermal stability. Despite the advantages and control over pore architecture offered by soft templates, their use incurs higher costs (as these templates are often expensive), making them less suitable for industrial applications. Additionally, toxic gases may be generated during the template removal process.103,159,170,188
Chen et al.100 classified soft templating methods into two categories: primary methods, where the mesoporogen is added along with the other synthesis components, and secondary methods, where the mesoporogen is introduced in a second stage after the zeolite precursors are mixed and the gel is formed.
4.3.1.4.1 Primary methods. 4.3.1.4.1.1 Macromolecular structures. The most extensive category of soft templates consists of macromolecular structures, including soluble nonsurfactant polymers. These templates are widely accessible and can be adjusted in terms of their chemical composition, molecular weight, and surface charge. For instance, nonsurfactants like silylated polymers205,206 or cationic polymers207 have demonstrated effectiveness as primary templates.
One of the earliest examples of using silylated polymers in zeolite synthesis was reported by Wang and Pinnavaia.189,205 They developed a method to prepare ZSM-5 zeolites with small and uniform intracrystal mesopores by employing a silane-functionalized polyethylenimine as a mesoporogen. This approach involved the integration of the polymer into the zeolite matrix through covalent Si–O–Si linkages, which allowed the formation of an intracrystal polymer network. Upon calcination, the polymer was removed, creating uniform mesopores within the zeolite structure, demonstrating the effective use of silylated polymers as templates for hierarchical pore development. Most recently, Guoqiang Song et al.208 synthesized and catalytically characterized mesoporous ZSM-5 zeolites using a novel organosiloxane as a mesopore-directing template. This approach allowed for the formation of intracrystalline mesopores of 3–6 nm within the zeolite structure, while maintaining high hydrothermal stability.
In the synthesis of zeolites, nonsurfactant cationic polymers like polydiallyldimethylammonium chloride (PDADMAC) serve as a dual-function template, facilitating the creation of both micropores and mesopores within the zeolite structure. These polymers are effective because their quaternary ammonium groups strongly interact with negatively charged aluminosilicate species, guiding the formation of the microporous zeolite framework while also acting as a porogen for mesopores. Unlike surfactant-based templates, which self-assemble into ordered structures, nonsurfactant polymers do not form regular micelles or ordered structures due to the lack of hydrophobic segments. This absence allows for the crystallization of zeolite without disruption from template self-assembly, promoting the formation of a thermodynamically stable single-crystal structure.207,209
4.3.1.4.1.2 Organosilanes. The use of organosilanes, such as 3-(trimethoxysilyl)propylhexadecyldimethylammonium chloride (TPHAC), as templates in zeolite synthesis involves creating mesoporosity within the zeolite framework.210,211 Organosilanes are amphiphilic molecules that possess both hydrophilic (silane) and hydrophobic (alkyl chain) parts. In the synthesis process, these molecules help form mesoporous structures within the zeolite by acting as mesopore-generating agents. When added to the synthesis gel, the organosilanes arrange into micelles, creating a network that guides the formation of mesopores. During the hydrothermal crystallization of zeolites, these micelles serve as templates around which the silica and alumina species condense, eventually forming a crystalline zeolite structure with interconnected mesopores. The organosilanes' hydrolysable silane groups bond with the framework, stabilizing the mesostructure. After calcination, the organosilanes are removed, leaving a hierarchical zeolite with both microporous and mesoporous networks.
4.3.1.4.1.3 Surfactants. Amphiphilic organosilane surfactants and nonsurfactant polymers provide precise control over mesoporosity; however, they are challenging to synthesize and quite costly. Therefore, there remains a strong interest in using conventional cationic surfactants, as they are affordable and easily scalable porogens for introducing mesopores into zeolites. The initial attempts to create mesoporous and microporous structures within a single zeolite material employed a mix of monovalent surfactant templates, such as cetyltrimethylammonium bromide (CTABr) for mesopores, and small organic ions like tetrapropylammonium (TPA+) for micropores. However, the interaction between the zeolite and monofunctional surfactants like CTABr is not strong enough to compete with the zeolite's structure-directing agent (e.g., TPA+ for MFI zeolite). As a result, in most cases, only a physical mixture of bulk zeolite and amorphous mesoporous material is obtained due to phase separation caused by competition between the two templating systems.
To address this challenge, researchers have explored dual-template strategies that employ both micropore-directing agents and mesopore-directing agents simultaneously. For instance, a study synthesized hierarchical ZSM-5 catalysts using a combination of templates: tetrapropylammonium bromide (TPABr) as the micropore template, and nonionic copolymer F127 along with cationic surfactant CTAB as mesoporogens. This approach successfully integrated mesoporous and microporous structures within the zeolite, enhancing its catalytic performance.211,212 Another example involves the synthesis of mesoporous SSZ-13 zeolite using a dual-template method with trimethyl-adamantanammonium hydroxide and a diquaternary-ammonium mesoporogen. This strategy induced significant mesoporosity without compromising the zeolite's intrinsic microporosity, resulting in improved accessibility of Brønsted acid sites and enhanced catalytic stability.212 Another nice example is the incorporation of carboxyl-ended organosilanes with CTAB in ZSM-5 synthesis has demonstrated that carboxyl groups strengthen the interaction between zeolite seeds and the surfactant, forming well-integrated mesoporous structures. This approach, involving organosilanes like, avoids amorphous phases and yields highly crystalline hierarchical zeolites, enhancing accessibility to active sites and stability in catalytic applications.213
Another strategy to prevent phase segregation when mixing a template with a mesogen is the use of multifunctional templates that can act as both structure-directing agents and mesogens. These surfactants direct a periodically ordered mesostructure through the self-assembly of supramolecular micelles, while micropores are generated by the individual surfactant head groups. Ryoo et al. described the synthesis of zeolite nanocrystals using multivalent surfactants as capping agents to prevent unwanted growth, enabling the formation of nanostructures with mesoporosity. The multivalent surfactants, featuring multiple ammonium head groups, enhance electrostatic interactions with the zeolite framework, facilitating the formation of nanocrystals with stable mesostructures. The synthesis yielded various morphologies, including nanorods, nanosheets, and agglomerates, depending on the surfactant used. This approach improved catalytic performance by reducing diffusion limitations and expanding the external surface area, proving effective across multiple zeolite types, including MOR, FAU, CHA, and MFI.214
4.3.1.4.2 Secondary methods. Secondary methods follow a two-step synthesis approach: in the initial step, all ingredients except the surfactant are introduced. In the subsequent step, the soft template either facilitates the arrangement of zeolite seeds into hierarchically porous structures or creates microemulsions/reverse micelles for “confined space synthesis” or “steam-assisted conversion” of hierarchically porous zeolites.
4.3.1.4.2.1 Soft template assembly of zeolite seeds. One of the earliest examples of this approach was published by Serrano et al.215 This study introduces a novel method for synthesizing hierarchical zeolites by employing silanized zeolite seeds, which improve both the textural and catalytic properties of the final material. The process begins with a pre-crystallization step to form zeolite nuclei, followed by the addition of a silanizing agent, phenylaminopropyl-trimethoxysilane (PHAPTMS), which anchors to the surface of the zeolite seeds. This silanization step inhibits seed aggregation during the hydrothermal crystallization phase, resulting in a structure with interconnected mesopores and micropores. The hierarchical ZSM-5 and beta zeolites produced through this method exhibited a higher external surface area, additional secondary porosity, and enhanced catalytic performance, particularly in polypropylene cracking.
Zhu et al.216 developed a method for synthesizing hierarchical mesoporous ZSM-5 zeolites using hexadecyl trimethyl ammonium bromide (CTAB) as a soft template with kinetic control over zeolite seed formation. By aging the precursor at 100 °C for 2–3 days, subnanocrystal-type zeolite seeds with a high degree of polymerization were formed, which then assembled with CTAB to create mesostructures without phase separation. The resulting ZSM-5 zeolites exhibited improved catalytic performance, particularly in aldol condensation reactions, due to the enhanced accessibility provided by the mesoporosity alongside traditional microporosity.
4.3.1.4.2.2 Microemulsions as nanoreactors. A further approach to soft templating involves synthesizing hierarchically porous zeolites within microemulsions that act as nanoreactors, facilitating zeolite growth. This technique achieves similar effectiveness to confined voids found in porous carbons. In essence, the process relies on a phase-separation mechanism between an organic and an aqueous phase during zeolite synthesis, forming a stable biphasic emulsion in which the aqueous phase contains the zeolite mixture, while the organic solvent contains the porogen.217,218
4.3.2.1 Dealumination. Dealumination is widely regarded as one of the most effective methods for demetallation, frequently applied to produce zeolites with higher Si/Al ratios, enhanced stability, and increased mesoporosity. Common techniques include steaming, calcination, and acid leaching to hydrolyze Al–O–Si bonds within the zeolite framework. Originally, dealumination was used to increase the Si/Al ratio for greater stability, but it was later discovered that this process also unexpectedly generates mesopores.
4.3.2.1.1 Steaming. Steaming, typically performed above 500 °C, breaks Si–O–Al bonds, causing aluminum loss and creating structural defects. These defects allow less stable silicon atoms to move into former aluminum sites, forming silanol-rich domains. This process can partially restore the structure and produce mesopores.
Sheng et al.220 investigated the impact of steam treatment on the catalytic performance of HZSM-5 zeolite for ethanol dehydration to ethylene. The study focused on understanding how steam treatment alters the acidity, pore structure, and stability of HZSM-5, a zeolite known for its strong acidic properties, which are crucial for catalytic applications but can lead to rapid deactivation due to coke deposition. The authors treated commercial HZSM-5 zeolite with steam at different temperatures (400 °C, 450 °C, 500 °C, and 550 °C) to examine how these conditions affect the catalyst's acidity and porosity. They found that steam treatment led to the partial dealumination of the zeolite framework, which increased the Si/Al ratio and generated mesopores. This mesoporosity, alongside the original microporosity, created a hierarchical structure that improved the diffusion of reactants and products, reducing the risk of coke formation. Characterization techniques, including XRD, nitrogen adsorption–desorption, and NH3-TPD, revealed that steam-treated HZSM-5 exhibited lower total acidity and a reduced Brønsted/Lewis acid site ratio. These changes were more pronounced at higher steam temperatures, with significant decreases in strong Brønsted acid sites, which are associated with coke formation. The optimal treatment temperature identified was 500 °C, where the catalyst showed a balance between maintaining crystallinity and enhancing mesoporosity. The catalytic testing results showed that the steam-treated HZSM-5, particularly the sample treated at 500 °C, exhibited high stability and selectivity for ethylene production during ethanol dehydration. The treated catalyst displayed reduced coke deposition and better long-term performance compared to untreated HZSM-5. This improvement in stability was attributed to the presence of mesopores, which allowed coke to deposit in less obstructive locations, preventing the blockage of micropores and maintaining access to active sites.221
The steaming method is often followed by acid treatment to remove debris generated during the process, which can potentially accumulate on the surface of the zeolite particles or within the channels, causing blockages. Common acids used for debris removal include nitric acid, hydrochloric acid, and organic acids such as oxalic, acetic, and tartaric acid. Since the primary goal of dealumination is to extract aluminium, the feedstock zeolites typically have a low Si/Al ratio.222,223
4.3.2.1.2 Calcination. Dealumination can also be performed through a calcination process. Müller et al.170,224 conducted a study comparing the dealumination effects of calcination on different zeolites, including beta, mordenite, ZSM-5, and ferrierite. Similar to the steaming method, a mild acid treatment, such as with oxalic acid, is required after calcination to remove any residual debris. NMR analysis indicated that the degree of dealumination increased with a higher number of Brønsted acid sites interacting with the zeolite framework, leading to the formation of structural defects. These changes made the zeolite framework more flexible, facilitating interactions between Brønsted acid sites and oxygen atoms within the network. Zhang et al.225 also found that mesoporous ZSM-5 created via thermal treatment maintained its crystallinity up to temperatures of 1000 °C. For beta zeolite, the effects of steam treatment at 500 °C and calcination at 750 °C were examined. Results showed that Lewis acid sites increased while Brønsted acid sites decreased. Additionally, beta zeolites treated with steam and calcination demonstrated HMF selectivity of 51% and 55%, respectively, in glucose isomerization and dehydration reactions.223
4.3.2.1.3 Acid leaching. Acid leaching is also used to remove aluminum from the zeolite framework. Mild acids can promote hydrolysis of Si–O–Al bonds in concentrated solutions, facilitating aluminum extraction and mesopore formation. The efficiency of dealumination depends on the zeolite type, acid characteristics, and pH. The commonly used inorganic acids for chemical etching of zeolites, such as hydrochloric acid (HCl), hydrofluoric acid (HF) or chromic acid (H2CrO4), are reviewed below.
4.3.2.1.3.1 Hydrochloric acid. An interesting example was reported by Alver et al.,226 where they treated clinoptilolite, a natural zeolite, with hydrochloric acid (HCl) to examine changes in its structural and adsorption properties. Acid treatments with HCl concentrations of 0.1, 0.5, 1.0, and 2.0 M increased the specific surface area, micropore volume, and mesoporosity by removing extra-framework cations and causing partial dealumination. XRD analysis showed that higher acid concentrations, particularly from 1.0 M upwards, reduced crystallinity. The treated samples exhibited enhanced methane (CH4) adsorption, with the highest uptake observed in samples treated with 0.1 M HCl. This increase in adsorption capacity is attributed to the exchange of metal cations with protons (H+), which opened the channels within the zeolite structure and enhanced its adsorption potential.
Quiao et al.227 presented a method for creating hollow SAPO-34 zeolite crystals via selective acid etching with hydrochloric acid (HCl), achieving hollow structures while maintaining crystal integrity. By treating SAPO-34 precursor crystals in a 0.58 M HCl solution at 80 °C for three hours, the process selectively removes aluminum from the framework, resulting in substantial internal voids and producing a hollow morphology with shell thicknesses of around 30 nm. This acid etching technique dissolves aluminum-rich regions within the crystal, generating mesopores and preserving the external crystal structure. The resulting hollow structure significantly enhances the external surface area and pore volume, which is advantageous for catalytic applications as it improves accessibility to active sites within the crystal (Fig. 23).
 | ||
| Fig. 23 SAPO-34 samples before and after acid tradesman. Reproduced with permission from ref. 227. Copyright 2016 Royal Society of Chemistry. | ||
A study on beta, mordenite, and ZSM-5 using microwave irradiation in acidic environments revealed that microwave treatments accelerate dealumination compared to conventional methods, with beta zeolite showing the highest ease of dealumination and ZSM-5 displaying notable resistance. Microwave-assisted treatment of mordenite with HCl increased mesoporosity, surface area, and Brønsted acidity, while beta zeolite showed minimal changes.
4.3.2.1.3.2 Hydrofluoric acid. Hydrofluoric acid (HF) is another commonly used inorganic acid for chemically etching zeolites. Unlike other inorganic acids, HF not only functions as an etching agent but can also act as a mineralizer.228
Wang et al.229 investigated the effects of modifying HZSM-5 zeolite with hydrofluoric acid (HF) and nickel (Ni) on catalytic fast pyrolysis (CFP) of cellulose The study demonstrated that HF treatment partially removes aluminum from the framework, enhancing mesoporosity and reducing Brønsted acid sites, which facilitates better diffusion of reactants and mitigates coke formation. Additionally, HF slightly extracts silicon, balancing the acidic properties of the zeolite and further increasing its mesoporosity. The authors found that HF-treated HZSM-5 (designated as F-Z5) produces a hierarchical structure that significantly boosts selectivity for light aromatics in CFP applications. The dealumination process is optimized at an HF concentration of 0.5 M, which maximizes mesopore formation without compromising crystallinity. When Ni is introduced, it further enhances light aromatic yields due to improved deoxygenation and hydrogenation capabilities. The combined modification with HF and Ni results in a catalyst with superior activity, offering improved access to active sites and reducing diffusion limitations, which are critical for efficient biomass conversion.
Another example was published by Xi et al.230 that investigates the synthesis of hierarchically macroporous SAPO-34 zeolite using an in situ growth-etching method facilitated by hydrofluoric acid (HF). The HF serves as both a mineralizer and an etching agent, enabling the creation of macropores within the SAPO-34 crystals. This approach removes framework aluminum and silicon, forming hollow structures with macroporous channels, as seen in the resulting rhombohedral crystal morphology with central voids. The macroporous structure significantly enhances the diffusion of reactants, improving mass transfer and extending the catalyst's lifespan in methanol-to-olefin (MTO) reactions. SEM and TEM images reveal that HF treatment creates internal parallel macrochannels within the crystals, facilitating access to active sites. The SAPO-34 synthesized with HF displays superior catalytic performance, with higher ethylene and propylene selectivity and increased resistance to deactivation due to reduced coke formation.
4.3.2.1.3.3 Chromic acid. Although uncommon, chromic acid can also be used as a dealuminating agent. There are very few studies on this topic, with perhaps the most notable being the work published by Babic et al.231 that explores the use of chromic acid (H2CrO4) as a novel dealuminating agent for various zeolite structures, including CHA, MFI, and LTL. Chromic acid, known for its strong oxidizing properties, is applied in different concentrations (0.1% to 10%) to examine its effect on the framework stability, crystallinity, porosity, and acidity of the treated zeolites. The study demonstrated that chromic acid can selectively remove aluminum atoms from the zeolite framework, thereby increasing the Si/Al ratio and creating defects that enhance mesoporosity. This process is particularly effective for the LTL framework, which is more susceptible to dealumination due to its larger pore openings, compared to the more resistant CHA and MFI structures. Through techniques such as XRD, SEM, TEM, and nitrogen physisorption, the authors observed that chromic acid treatment preserves the crystallinity of CHA and MFI zeolites at lower concentrations but causes partial amorphization in LTL at higher concentrations. Nitrogen adsorption data indicate that treated samples exhibit increased micropore and mesopore volumes, contributing to enhanced accessibility for reactants in catalytic applications. Additionally, infrared spectroscopy with pyridine and deuterated acetonitrile probes reveals changes in the acidic properties of the zeolites, with chromic acid treatment increasing the accessibility of Brønsted acid sites and enhancing the formation of Lewis acid sites. This increase in accessible acid sites, along with the enhanced porosity, suggests that chromic acid-treated zeolites could offer improved performance in catalytic processes, particularly in reactions that benefit from hierarchical porosity and optimized acidity.
4.3.2.2 Desilication. Desilication is a well-established post-synthetic modification process that selectively removes silicon atoms from the framework of zeolites, introducing mesoporosity while preserving the crystalline structure to a significant extent. This hierarchical modification enhances molecular diffusion and increases access to active sites, addressing the intrinsic diffusion limitations of purely microporous zeolites. The process has gained significant attention due to its simplicity, scalability, and its application across various zeolite frameworks such as ZSM-5, beta, and mordenite.
The concept of desilication dates back to the 1960s, with Dean Arthur Young's pioneering work demonstrating enhanced benzene adsorption and hydrocracking performance in alkaline-treated mordenite. Subsequent studies in the 1990s provided a deeper scientific understanding of silicon dissolution mechanisms.
Dessau et al.232 identified the anisotropic dissolution of ZSM-5 crystals and the role of aluminum in inhibiting local dissolution, while Groen et al.109,233–244 later established the importance of specific Si/Al ratios for achieving optimal mesoporosity.108,245
A good example of the benefits of desilication is the study published by Groen et al. in their work on mesoporous ZSM-5 zeolites.244 In their study, they achieved a two-order-of-magnitude increase in gas transport by introducing intracrystalline mesoporosity into ZSM-5 crystals via a controlled desilication process. The results indicated that this mesoporosity led to a dramatic reduction in the characteristic diffusion path length, which in turn enhanced the diffusion rates of probe molecules, such as neopentane. The diffusion time was reduced from 120 minutes in the conventional calcined zeolite to just 2 minutes in the desilicated sample, highlighting the critical role of mesopores in improving gas transport within zeolite frameworks (Fig. 24). This enhancement demonstrates the importance of creating interconnected mesopore networks for efficient catalyst utilization in industrial applications where diffusional limitations are a concern.
 | ||
| Fig. 24 Neopentane uptake curves for calcined and alkaline-treated ZSM-5 (desilicated) crystals measured using the TEOM (Tapered Element Oscillating Microbalance) at 393 K. The TEOM is a high-precision device that measures real-time changes in mass by detecting variations in the oscillation frequency of a tapered element when molecules are adsorbed or desorbed from the sample. Reproduced with permission from ref. 244. Copyright 2007 American Chemical Society. | ||
4.3.2.2.1 Mechanisms of desilication. Desilication is primarily conducted in an alkaline environment, using bases such as sodium hydroxide (NaOH) or potassium hydroxide (KOH). The silicon extraction mechanism involves the hydrolysis of Si–O–Si bonds in the framework, leaving defects that develop into mesopores. Framework aluminum plays a critical role in the dissolution process, as it modulates silicon extraction. This aluminum-based regulation has led to the concept of aluminum as a “pore-directing agent” (PDA).233
4.3.2.2.1.1 Role of Si/Al ratio. Desilication is most effective in ZSM-5 zeolites with Si/Al ratios between 25 and 50. Within this range, controlled silicon removal occurs, leading to the formation of mesopores while maintaining the structural integrity of the zeolite. This balance enhances catalytic performance by improving accessibility to active sites without compromising the framework's stability.234 At lower Si/Al ratios, the increased presence of aluminum introduces more negatively charged AlO4− tetrahedra into the framework. These units stabilize the structure and resist silicon extraction during alkaline treatment, resulting in limited mesopore formation. The protective effect of aluminum at these ratios hinders the desilication process, making it less effective. In contrast, ZSM-5 zeolites with high Si/Al ratios are more susceptible to uncontrolled silicon extraction during desilication. The reduced aluminum content means fewer AlO4− units are available to stabilize the framework, leading to excessive dissolution of silicon. This uncontrolled process can cause the formation of irregular and broad pore-size distributions, structural collapse, and significant loss of material yield.234–238
4.3.2.2.1.2 Effect of morphology. The morphology of the parent zeolite significantly affects the desilication process. Zeolites composed of intergrown particles with higher external surface areas have more grain boundaries and structural defects compared to large single crystals. These defects act as preferential sites for attack during alkaline desilication, promoting silicon extraction and leading to increased mesoporosity. However, this improvement in mesoporosity often comes at the cost of reduced microporosity and crystallinity, potentially compromising the material's catalytic performance. Svelle et al.246 delves into how the morphology and defects of ZSM-5 zeolites influence the desilication process. Morphological differences play a critical role: zeolites composed of intergrown crystallites with pronounced defects are more prone to directed mesopore formation compared to larger, uniform crystals. This is attributed to the higher susceptibility of boundaries and defect sites to alkaline attack. The study confirms this through extensive characterization techniques such as SEM, TEM, and FTIR, demonstrating the presence of radial channels and distinct mesoporous networks in particles with complex morphologies. While desilication improves the diffusion properties by creating mesopores, it can also reduce the microporosity and crystallinity, especially in zeolites with high defect densities. The process also leads to changes in acidity, as indicated by FTIR and NH3-TPD analyses, with a reduction in strong Brønsted acid sites and an increase in weak acid sites. Despite these trade-offs, the study highlights the potential of mesoporous zeolites in catalytic applications, particularly in reactions such as methanol-to-hydrocarbons conversion.
4.3.2.2.2 Alternative reagents. While the desilication method using NaOH etching is straightforward and effective, its strong alkalinity can partially damage the zeolite structure, potentially leading to amorphization in some cases.247 To address this limitation, alternative bases have been explored to minimize structural degradation while achieving mesoporosity.
Bases such as potassium hydroxide (KOH),248,249 sodium carbonate (Na2CO3),250 and ammonium hydroxide (NH4OH)251 have been employed as gentler alternatives. These bases offer milder dissolution effects on the silicon framework, reducing the risk of amorphization. For instance, KOH tends to create mesoporosity with less aggressive framework damage due to the larger size of the potassium ion, which limits penetration into the zeolite lattice. Similarly, Na2CO3 provides controlled silicon extraction while preserving crystallinity, as it promotes selective dissolution of the framework. NH4OH,251 on the other hand, is known for its mildness and has been used to protect the zeolite structure during mesopore generation, though it may result in slower desilication processes.
4.3.2.2.3 Desilication of partially detemplated zeolites. Desilication may lreed to the amorphization of the zeolite. Using partially detemplated zeolites in desilication has proven effective to protect the framework from aggressive base attack. The residual organic templates act as barriers, regulating the extent of silicon dissolution. Pérez-Ramírez et al.252 highlighted the benefits of partial detemplation in conjunction with desilication for the controlled creation of mesoporosity in beta zeolites, effectively preserving structural integrity. The research demonstrated that partial calcination of as-synthesized zeolites removes only a fraction of the organic template, leaving certain micropores protected from NaOH attack during desilication. These template-protected regions safeguard the zeolite framework from amorphization, allowing selective mesopore formation in unprotected areas. Fig. 25 illustrates this approach. By varying the calcination temperature (230 °C, 330 °C, or 550 °C), the degree of detemplation can be tailored, resulting in progressively increasing mesoporosity (20–230 m2 g−1) upon alkaline treatment. Higher calcination temperatures and complete template removal led to enhanced mesoporosity but with greater micropore loss.
 | ||
| Fig. 25 Schematic representation of the desilication treatment for templated, partially detemplated, and template-free zeolites to tailor mesoporosity development. Reproduced with permission from ref. 252. Copyright 2009 Wiley-VCH. | ||
van Laak et al.253 explored the alkaline treatment of template-containing zeolites as a strategy to introduce mesoporosity while preserving the intrinsic properties of the material. Using NaOH solutions under controlled conditions, the method selectively dissolves silicon at the external surfaces and intercrystalline boundaries. Three zeolites—ZSM-5, ZSM-12, and Beta—were studied. The presence of organic templates during treatment played a protective role, safeguarding the microporous framework from excessive dissolution. Smaller crystallites were found to produce higher mesoporosity due to their increased surface area and defect sites. Importantly, NH3-TPD analysis showed that the acidic properties, including Brønsted acid sites, were well-preserved, Surfactant-assisted desilication.
4.3.2.2.4 Surfactant-assisted desilication. External PDAs represent a significant advancement in controlling alkaline treatment during desilication to generate mesoporosity in zeolites. These agents, such as tetraalkylammonium cations (TAA+), alkylammonium hydroxides, and metal hydroxides like Al(OH)4− and Ga(OH)4−, interact specifically with the zeolite surface under alkaline conditions.254 This interaction provides partial protection against excessive dissolution, enabling the controlled formation of intracrystalline mesopores. The protective effect of PDAs minimizes micropore loss, preserves crystallinity, and increases the yield of mesoporous zeolites compared to standard alkaline treatments. PDAs provide several advantages in desilication. PDAs act as “pore-growth moderators,” selectively regulating silicon dissolution and enabling precise control over mesopore size and distribution. They also offer versatility by allowing desilication of zeolites with Si/Al ratios ranging from 25 to infinity, thus overcoming the traditional limitations of aluminum-dependent processes. Furthermore, PDAs, such as tetraalkylammonium cations (TAA+), enhance desilication efficiency by generating comparable mesopore surface areas while preserving microporosity and achieving higher material yields, making them an effective tool for tailoring hierarchical zeolite structures.238,255
Gackowski et al.256,257 explored the use of tetrabutylammonium hydroxide (TBAOH) as a PDA in the desilication of zeolite Y to achieve hierarchical porosity. The addition of TBAOH to the NaOH solution was found to significantly improve the desilication process by mitigating the risk of amorphization, a common issue with conventional alkaline treatments. TBAOH acts by interacting with the silica framework, moderating the dissolution process and enabling the formation of mesopores while preserving the microporous structure and crystallinity. The optimized desilication conditions, using a mixture of 10 mol% TBAOH at 353 K, yielded a zeolite with enhanced mesoporosity and well-preserved acidity. The treated zeolite exhibited a controlled mesopore size distribution and higher material yield compared to samples treated with pure NaOH. These hierarchical properties resulted in improved catalytic performance, as demonstrated in α-pinene isomerization tests, where the modified zeolite outperformed the untreated material due to better diffusion and accessibility to active sites.
4.3.3.1 Methanol-to-hydrocarbons (MTH) reactions. Perhaps the most striking improvements are observed in methanol-to-hydrocarbons conversion, a process notoriously limited by intracrystalline coke deposition in microporous zeolites.
4.3.3.1.1 Catalyst lifetime extension. Hierarchical ZSM-5 zeolites—featuring combined micro- and mesoporosity—demonstrate significantly longer catalytic lifetimes in methanol-to-hydrocarbons (MTH) conversion compared to conventional microporous ZSM-5. For instance, Zhao et al. reported that introducing mesoporosity into ZSM-5 (Si/Al = 100) extended the time-on-stream from ∼10 hours (for conventional ZSM-5) to ∼83 hours before deactivation.258 Likewise, Asghari et al. found that a hierarchical ZSM-5 catalyst maintained >90% methanol conversion for 100 hours, whereas the conventional sample deactivated after ∼50 hours.259 In another study, Weissenberger et al. showed that a hierarchical ZSM-5 with both mesopores and macropores had a lifetime nearly 10 times longer than its purely microporous counterpart.260 These improvements are directly linked to enhanced mass transport through the hierarchical pore network, which delays deactivation.
4.3.3.1.2 Reduced coke formation. Hierarchical structures also lead to lower coke formation rates, a critical advantage in MTH. Compared to microporous ZSM-5, hierarchical catalysts accumulate carbonaceous residues more slowly. Asghari et al. reported that their hierarchical sample showed a lower coke deposition rate and sustained longer operation before deactivation due to coke.259 Weissenberger et al. observed that while macropores reduced the coke formation rate, mesopores improved the catalyst's coke tolerance, allowing more carbon to accumulate without performance loss.260 Li et al. confirmed that the enhanced diffusion pathways in hierarchical ZSM-5 suppress rapid coke buildup, improving overall stability.
4.3.3.1.3 Enhanced light olefin selectivity. Hierarchical ZSM-5 catalysts also demonstrate higher selectivity to light olefins, particularly propylene, compared to conventional ZSM-5. In Zhao et al.'s study, mesoporous ZSM-5 exhibited a propylene selectivity of ∼54% and reduced ethylene formation (∼6%) compared to the conventional sample (∼47% propylene, ∼13% ethylene).258 These results suggest that hierarchical porosity favors the olefin-based reaction cycle and suppresses the aromatics-based cycle that typically leads to ethylene and coke. Similarly, Li et al. found that hierarchical macro–microporous ZSM-5 increased selectivity toward C3–C4 olefins, shifting the product slate toward more valuable light olefins.
4.3.3.1.4 Hydrocraking of long chain alkenes. Hierarchical zeolites have significantly improved the performance of zeolite-based hydrocracking catalysts, especially for processing long-chain alkanes and heavy oil fractions. In conventional microporous zeolites like USY (ultrastable Y, FAU) and Beta (BEA), the diffusion of bulky molecules is severely restricted, resulting in underutilization of internal acid sites and catalytic activity concentrated near the external surface. Introducing mesoporosity into these materials enhances intracrystalline diffusion and accessibility, enabling more complete cracking and isomerization reactions throughout the zeolite particle.
Introducing intracrystalline mesoporosity alleviates these mass transport issues. For instance, desilicated USY zeolites with mesopores (∼10–20 nm) have shown improved VGO conversion of up to 83 wt%, compared to ∼68–70 wt% in their purely microporous counterparts under identical conditions. Furthermore, even after deactivation, mesoporous USY retained higher activity: one study reported 60 wt% VGO conversion versus only 50 wt% for the unmodified catalyst.261
In top-down methods, dealumination (via steaming or acid leaching) removes framework aluminum, increasing the Si/Al ratio and reducing total Brønsted acid site density. The extracted aluminum may remain as extra-framework alumina (EFAl), introducing new Lewis acid sites. In contrast, desilication (mild alkaline treatment) selectively removes silicon, preserving the framework aluminum and often maintaining or slightly increasing total acidity by exposing previously inaccessible acid sites. The key distinction is that dealumination typically decreases total Brønsted acidity while increasing acid strength, whereas desilication maintains or enhances acid site density while introducing mild Lewis acidity.
Bottom-up methods allow precise control over Si/Al ratio and acid site distribution, with most aluminum incorporated directly into the framework. This results in materials with a high proportion of framework Brønsted acid sites, few defects, and uniform acid distribution. The final acidity profile depends on the synthesis gel composition and crystallization conditions rather than post-synthetic treatment.
In terms of porosity and texture, top-down methods produce mesopores via etching of the parent zeolite, often resulting in non-uniform pore sizes and partially closed mesopores if treatment is not well-controlled. In contrast, bottom-up approaches (e.g., surfactant templating) yield uniform, interconnected mesopores formed concurrently with the microporous framework, enabling better integration of micro-mesoporosity and minimizing loss of crystallinity or micropore volume. Nanocrystal assembly (e.g., nano-Beta) can also introduce interparticle mesoporosity with preserved intrinsic acidity and full microporosity.
Regarding catalytic performance, both approaches can yield highly active and stable catalysts if appropriately optimized. Top-down methods may slightly compromise crystallinity or acid site distribution but are generally more cost-effective and scalable. Bottom-up strategies offer greater design flexibility and superior textural properties but require complex synthesis procedures and costly organic templates, limiting industrial adoption.
Bottom-up methods, in contrast, allow for more tailored pore architectures and precise control of composition, but are inherently more complex. These routes involve dual or hard templates and long crystallization times, and they often use expensive organics that must be removed, increasing both environmental burden and cost. Additionally, the resulting materials may require binders for shaping, which can dilute the overall acidity.
The main trade-off is one of “acid site retention versus synthetic complexity.” While top-down methods may diminish total acidity, they often yield stronger and more accessible acid sites—especially in USY zeolites—ultimately enhancing heavy-feed performance. Bottom-up synthesis retains Brønsted acid sites and crystallinity but demands sophisticated protocols. For example, templated mesoporous Beta can outperform its conventional counterpart in hydrocracking, but industrial uptake is hindered by the requirement for long syntheses with costly surfactants.
Nevertheless, recent developments, such as ultrafast bottom-up synthesis strategies and scalable routes for hierarchical Beta, are bridging this gap. A notable example is the pilot-scale production of 100 kg batches of hierarchical Beta with demonstrated improvements in VGO hydrocracking performance. In summary, top-down methods are practical and tunable, but must be controlled to prevent degradation. Bottom-up approaches provide excellent design flexibility and acid retention, though they are currently less viable at scale. Often, the most effective strategy combines both—synthesizing nanozeolite particles bottom-up and applying mild post-treatment to optimize acidity and accessibility.
5 Powder shaping
While hierarchical and nanozeolites show significant improvements in catalytic performance, their practical application in industrial settings requires overcoming challenges related to material stability, scalability, and the preservation of mesoporosity. The transition from laboratory-scale synthesis to industrial-scale production is not straightforward, as it involves maintaining the delicate balance of structural integrity while scaling up production. At industrial scales, it is essential to preserve the zeolites' mesoporosity, which plays a crucial role in facilitating efficient reactant transport and improving catalytic performance. Without this, the benefits of these advanced zeolite designs might be compromised in large-scale operations, where mechanical stresses and harsh conditions can often lead to deactivation or loss of efficiency.In Section 5, we address the strategies employed to shape zeolites for industrial applications, particularly focusing on powder shaping and the design of structured reactors. Techniques such as extrusion, spray drying, and 3D printing have become essential tools for creating zeolite-based monoliths and extrudates that can maintain the material's structural integrity while enhancing its mechanical stability. These approaches not only help preserve mesoporosity but also enable the integration of zeolites into large-scale catalytic reactors. By improving scalability and reactor performance, these shaping methods bridge the gap between fundamental zeolite research and industrial applications, ensuring that the advancements in zeolite structure can be effectively utilized in real-world catalytic processes.
Zeolites are typically synthesized as insoluble powders, leading researchers to often treat their processability as a secondary concern. However, for these porous materials to be practically useful, they must be shaped into larger, more manageable forms such as pellets, films, membranes, or microreactors.262,263 While much of the literature focuses on the high surface area of zeolites, their effective application relies on structuring them into meso- or macroscopic forms to fully utilize their microporous volume. These hierarchical structures must meet several critical requirements, including (i) chemical stability, (ii) mechanical strength, and (iii) resistance to attrition, all while retaining the material's intrinsic microscopic properties.263,264
The development of industrial catalysts involves careful consideration of their shapes, sizes, and compositions to optimize factors like pressure drop, hydrodynamics, and mass and heat transfer. In industrial catalysis and adsorption, zeolites are commonly processed into forms such as beads, granules, or extrudates through methods like spray drying, extrusion, pelletization, and granulation.265 These techniques are favored for their cost-effectiveness and their ability to achieve high reactor loading. During preparation, various additives such as binders, fillers, and modifiers are incorporated. These additives are essential for enhancing the mechanical and chemical stability of the catalyst and for adjusting the rheological properties of the precursor feed.266
Although shaping is primarily undertaken to render zeolite powders into mechanically robust, usable forms, it also profoundly influences mass transport characteristics at multiple length scales. The macroscopic geometry established by different shaping techniques – whether extruded pellets, monoliths, foams, or emerging 3D-printed shapes – governs parameters such as pressure drop and flow distribution through the catalyst.109,267,268 For instance, an extruded catalyst pellet must be sized and structured to minimize internal diffusion resistance while avoiding excessive pressure drop in a packed bed; very small pellets or those with roughened surfaces shorten diffusion distances and thin the external boundary layer for mass transfer, but they can cause high bed pressure drops.269 In contrast, monolithic and foam structures with large, open channels offer superb flow permeability (virtually eliminating macroscopic pressure drop) and provide extensive surface area, yet the active material is confined to thin walls or coatings to maintain short diffusion path lengths within the catalyst. At the microscale, the way a catalyst is shaped determines pore connectivity and available porosity inside the formed body. Binders, additives, and sacrificial pore formers used in extrusion or other shaping methods can either impede or enhance pore connectivity – an optimally formulated body will retain hierarchical porosity. In essence, the transport properties of a zeolite catalyst are an emergent feature of its shape and internal architecture. Understanding and tuning this shaping-transport relationship is critical for translating the intrinsic performance of powder catalysts to industrial reactors.270 By carefully balancing geometric design (for convective flow and low pressure drop) with internal pore structure (for high diffusivity and accessibility), engineers can bridge the gap between lab-scale powder performance and practical catalytic functionality at scale, ensuring that shaped zeolite catalysts achieve maximal activity and selectivity under industrial process conditions.
Traditionally, the formulation and fine-tuning of catalyst preparation have been managed by industry due to the process's complexity and empirical nature. The success of shaping catalysts depends on the type and quantity of additives as well as the order in which they are added, all of which can greatly influence the catalyst's final properties. An in-depth review by Perez-Ramirez et al.263,266 discussed the primary challenges associated with scaling up catalyst production and highlighted how commonly used additives can significantly impact catalytic performance.
On the other hand, structured supports and reactors have been more extensively studied in academic settings, where their relatively straightforward fabrication and advantages-such as open structures, uniform flow patterns, and enhanced mass transfer–are emphasized.271 The concept of structured reactors revolves around highly porous, hierarchical designs that serve as catalyst supports or frameworks for loading granules. These designs merge the characteristics of catalyst particles and reactor structures, highlighting the importance of multi-scale interactions. This section will provide examples of such applications, including monoliths, pellets, membranes, microreactors, and more, detailing their fabrication methods with zeolites as the active phase.
5.1 Monoliths, foams, and meshes
Monolithic structures, typically composed of parallel, straight channels, are constructed from materials such as ceramics (e.g., cordierite, mullite) or metals (e.g., steel, copper). These channels, which can be configured into shapes like squares, circles, or hexagons, vary widely in density, commonly ranging from a few dozen to several hundred channels per square inch (cpsi).272 Both ceramic and metal monoliths provide notable advantages, including low pressure drop, high porosity, and extensive geometric surface area. Metallic monoliths, however, offer superior heat transfer properties when compared to their ceramic counterparts. Monoliths serve as versatile catalytic supports that enable efficient catalyst loading due to their short diffusion paths, while also exhibiting robust thermal, chemical, and mechanical stability, contributing to long-term reactor performance. Their cost-effectiveness stems from straightforward production processes and scalability.Foam structures also serve as effective supports and feature an open-cell architecture with interconnected pores, resulting in high void fractions, fine pore sizes, and considerable specific surface areas with minimal pressure drop.273 These foams can be made from materials such as ceramics, metals, carbon, silicon carbide, and polymers. Their intricate geometries promote complex flow paths that regenerate hydrodynamic and thermal boundary layers. Pore sizes in foam structures range from a few to hundreds of pores per linear inch (ppi), similar to other structured materials used in packed beds.274–277
Meshes are also commonly used as support structures in catalytic and separation applications. Constructed from metals like stainless steel, they provide a durable framework with high permeability and minimal pressure drop. The surface can be treated to enhance the adhesion of catalytic or adsorbent coatings. Meshes are especially valued for their mechanical strength and suitability for processes like filtration, oil/water separation, and gas-phase reactions.
Despite the variety of support types, the strategies used in the manufacture of these systems are similar. Therefore, the primary focus will be on incorporating the active phase into monoliths. This topic has been explored in several reviews over the past few years.277–280 There are four general methods for preparing monolithic catalysts: (i) extrusion, (ii) in situ synthesis, (iii) coating, and (iv) deposition of an active phase. Additional methods involve combinations of these techniques. It is also worth noting that additive manufacturing, such as 3D printing, has recently entered the field of chemical engineering, offering a range of shapes and structures for various support materials.
The standard preparation process for integral monoliths involves several key steps: (i) mixing the monolithic precursors, (ii) adding water and selected additives followed by thorough kneading, (iii) extrusion of the mixture into the desired shape, (iv) drying, and (v) calcination. Aranzabal et al.283 highlighted the importance of optimizing each step, particularly the drying phase. Rapid heating during calcination can lead to defects or cracks in the structure, whereas a controlled, gradual heating rate (below 0.15 °C min−1) promotes uniform shrinkage and minimizes deformation (Fig. 26).
 | ||
| Fig. 26 Comparative image of two monoliths dried at different heating rate. Reproduced with permission from ref. 283. Copyright 2010 Elsevier. | ||
Integral monoliths are used in various applications, such as the selective catalytic reduction (SCR) of NOx.284 Nonetheless, supported monoliths—where the active phase is applied to pre-formed structures—are more widely adopted due to their adaptability and broader range of potential uses.
Mosca et al.287 utilized the seeded method to grow NaX coatings on cordierite monoliths. The coatings were produced either through a single four-hour hydrothermal synthesis using gel or clear solutions as precursors, or through a multi-step synthesis process carried out in five steps (1 hour and 20 minutes each) with fresh synthesis solution applied at each step. The thickness and extent of crystal intergrowth were dependent on the precursor mixture composition and preparation method, with the highest quality films achieved via the multi-step approach. Well-intergrown crystals were also obtained in the one-step synthesis using a gel precursor, though a significant amount of sedimented zeolite crystals was observed on top of the film.
Li et al.288 synthesized Linde A, Linde Y, mordenite, and ZSM-5 zeolites on cordierite supports by in situ hydrothermal synthesis, without the use of templates or zeolite seeds. They proposed that under basic hydrothermal conditions, Al atoms from the cordierite are activated and react with Si atoms from the precursor solution to form the zeolite framework. In this mechanism, Al atoms from the cordierite act as a bridge between the monolith and the zeolite, enhancing adhesion properties.
From this perspective, in situ methods that involve partial dissolution of the support to supply Si and/or Al atoms for zeolite synthesis are particularly noteworthy. These methods not only improve zeolite adhesion but also reduce the difference in thermal expansion coefficients between the zeolite and the coating.289 For instance, Ocampo,290 following the approach of Louis,291 obtained ZSM-5 coatings on glass monoliths via hydrothermal synthesis. Under the strong alkaline conditions used, the glass monoliths were partially dissolved and converted into ZSM-5, resulting in a system with a trimodal pore distribution.
The fundamental process of these methods involves immersing the monolith in a solution or slurry of the active phase, allowing the macropores to absorb the liquid. After immersion, the monolith is removed, excess liquid is drained, and the structure is calcined to stabilize the coating.272 The stability of the resulting monolith is often evaluated using ultrasonic vibration and thermal shock tests.293 Despite these techniques, the adhesion of the active phase can be weaker compared to direct hydrothermal synthesis, which often necessitates the use of binders. However, the adhesion properties of the active phase are generally inferior compared to those achieved by direct hydrothermal synthesis, often necessitating the use of a binder.
Efforts to enhance the adhesion and stability of coated monoliths while reducing binder usage have led to innovative techniques. Zamaro et al.294 introduced a binderless zeolite-coated monolith by integrating hydrothermal synthesis with washcoating. In their method, a cordierite honeycomb structure was first washcoated with an aqueous suspension containing 30 wt% mordenite powder in its ammonium form. This was followed by a hydrothermal treatment using a zeolite precursor mixture. The resulting monolith exhibited high catalytic activity and selectivity, particularly for SCR applications. Mechanical stability tests involving ultrasonic vibration demonstrated minimal weight loss, while SEM analysis revealed a uniform, well-adhered coating.
Building on recent advances in coating and synthesis techniques, several innovative methods have been developed to enhance the functionality and application range of structured supports. One example involves the incorporation of magnetic Fe3O4-ZSM-5 nanozeolites into a dual-layer nanofiber system composed of chitosan as the bottom layer and polyacrylonitrile as the top layer. This nanofiber composite was coated onto polyurethane sponges to boost their oil absorption capabilities. Performance tests revealed high absorption capacities for various oils, with the sponges absorbing up to 99.4 g g−1 of lubricating oil, 95.3 g g−1 of motor oil, and 88.1 g g−1 of pump oil, achieved using Fe3O4-ZSM-5 at 2 wt% and a nanofiber thickness of 12 μm. Importantly, regeneration tests demonstrated that these sponges could be reused across multiple cycles without significant loss in absorption efficiency, highlighting their potential for sustainable oil spill remediation.295
The work of Li et al. exemplified a streamlined approach for enhancing oil/water separation capabilities through a one-step coating method. Stainless steel mesh (SSM) was coated with pure-silica zeolite beta (PSZB) using a secondary seeded-growth technique. The incorporation of polydopamine facilitated complete seed layer coverage, creating a micro/nanoscale hierarchical structure that endowed the SSM with superhydrophobic and superoleophilic properties. The PSZB coating demonstrated robust adhesion and corrosion resistance, maintaining a separation efficiency above 97.5% even after 90 cycles. This high performance indicates the PSZB-coated SSMs promise for real-world oil/water separation applications.296
Bonaccorsi et al. explored the application of copper foam coated with zeolite 4A for use in adsorption-based heat pumps. The fabrication process involved foaming a mixture of epoxy resin, copper powder, and a foaming agent, followed by sintering to create a porous metal structure. The foam was then seeded with colloidal zeolite particles and subjected to two hydrothermal synthesis steps, producing a multilayer zeolite coating approximately 10 μm thick and accounting for 17% of the total foam weight. XRD analysis confirmed the crystalline quality of the zeolite layer, while SEM imaging showed continuous, intergrown crystals. This method enhanced heat transfer properties and maintained a high adsorption surface area comparable to commercial zeolite 4A, making it a strong candidate for improving the efficiency of heat pump systems.297
A range of 3D printing techniques have been applied to the creation of monolithic reactors, with extrusion-based printing being particularly prominent in fabricating zeolite-based monoliths.298 The preparation of extrusion pastes, incorporating the zeolite material, binders like bentonite, dispersants, and water, is a critical first step.299–301 The rheological properties of these pastes determine the mechanical stability and performance of the printed monoliths. For instance, pastes designed for CO2 capture often use zeolite 13X or 5A, with additives such as methyl cellulose and poly(vinyl) alcohol, enabling precise layer-by-layer construction through robocasting printers.299 The selection of binders plays a pivotal role; research has demonstrated that binary binder systems, such as those combining bentonite with aluminophosphate, can enhance mechanical strength, minimize shrinkage during calcination, and maintain a high active material content-up to 65%-compared to the 33.5% typically achieved through surface coating alone (Fig. 27).301
 | ||
| Fig. 27 Image of the robocasting process by extrusion of zeolite/binary binder paste through the 0.9 mm nozzle. Reproduced with permission from ref. 301. Copyright 2017 Elsevier. | ||
In the study by Li et al., 3D printing was employed to fabricate HZSM-5 and HY zeolite monoliths, which were subsequently evaluated for their efficacy in the catalytic cracking of n-hexane to produce light olefins. The monoliths were further enhanced with SAPO-34 coatings applied via a secondary growth method, resulting in modifications to surface area, porosity, and acidity.302
Another strategy involves using 3D printing to create the desired monolithic architecture followed by active phase deposition through washcoating. Lefevere et al. utilized 3D fiber deposition (3DFD) to create monoliths with zigzag or straight channels, followed by sintering at 1300 °C. A ZSM-5 slurry was then applied using a peristaltic pump to perfuse the structure.303 These 3D-printed monoliths showed enhanced performance in methanol-to-olefins conversion compared to ZSM-5 powders304 and exhibited slightly reduced performance in CO2/N2 and CO2/CH4 separations.305
3D printing allows for the fabrication of different monolith geometries. Lawson et al. explored the fabrication of zeolite 13X monoliths with different cell densities (200, 400, and 600 cpsi) and porosities using 3D printing. The designs were created in AutoCAD 2017 and converted to gcode via Simplify 3D software. The printing paste, composed of zeolite 13X, bentonite, and methylcellulose, was extruded using a Nordson precision tip (0.25 mm for standard and 0.61 mm for macroporous formulations). The printed monoliths were dried at ambient conditions overnight, followed by calcination at 550 °C for six hours to remove organic components and solidify the structure. Adjustments in porosity were made by varying the methylcellulose content or replacing bentonite with macroporous kaolin, enhancing mass transfer through increased pore space.306
Core–shell structures via 3D printing were achieved by Wang et al. Their method used a uniform printing ink composed of ZSM-5 powders, sepiolite nanofibers (SNFs), colloidal silica, hydroxypropyl methylcellulose (HPMC), and deionized water. The ink was extruded layer by layer and subsequently freeze-dried and calcined at 650 °C to produce a robust, macroporous structure. Post-hydrothermal crystallization was employed to grow a silicalite-1 layer (100–200 nm thick) on the ZSM-5 core, creating a hydrophobic shell. This core–shell design enhanced toluene adsorption capacity and water resistance, making the monoliths particularly effective for capturing volatile organic compounds under humid conditions.307
Khalil et al.308 demonstrated a novel application of 3D printing for shaping zeolite-Y into monolithic structures. Using direct ink writing (DIW) technology, the authors developed a printable zeolite-Y paste with nanoclay and polymeric binders to ensure structural integrity and rheological consistency. Bragina et al.309 developed an innovative method using 3D printing to fabricate block zeolite catalysts with and without binders, showcasing the versatility of additive manufacturing for zeolite shaping. The process involved printing polylactic acid (PLA) templates with predefined void geometries, which were filled with zeolite paste or precursor gels. For binder-free blocks, steam-assisted crystallization was employed to form nanozeolite crystals directly within the template channels, resulting in highly crystalline, binder-free MFI-type zeolite blocks. This approach allowed for the precise replication of template voids and facilitated the production of monolithic structures with enhanced mechanical stability and hierarchical porosity.
5.2 Membranes and films
Zeolite membranes play a crucial role in a wide range of separation and catalytic processes due to their distinctive structural properties. These membranes are broadly categorized into organic–inorganic composite membranes and purely inorganic membranes with zeolite films. Organic–inorganic composites integrate zeolites with polymer matrices, enhancing their mechanical flexibility. In contrast, inorganic membranes, often constructed entirely from zeolitic films on porous supports, offer high selectivity and uniform pore size distribution.Initial research into the use of zeolite membranes for separation processes revealed the inherent fragility of self-supported zeolite structures.310 To address this limitation, robust porous supports are frequently employed, onto which zeolite films are grown. The resulting composite membranes must meet stringent criteria: continuity with minimal defects, appropriate thickness, and controlled pore orientation.311 Two primary methods for fabricating zeolite films are direct hydrothermal synthesis and secondary growth.312
In the direct hydrothermal method, a porous support is immersed in a precursor solution containing a structure-directing agent, silica and alumina sources, a base, and water. This setup undergoes hydrothermal treatment, promoting nucleation and intergrowth of the zeolite on the support surface. For example, Güntner et al.313 combined zeolite MFI membranes on Al2O3 supports with Pd-doped SnO2 gas sensors for selective formaldehyde detection. Although effective in enhancing selectivity, the membrane increased sensor response times, highlighting a need for reduced thickness. Similarly, Abruzzi et al.314 utilized zeolite 13X to enhance the methane selectivity of SnO2-based CO2 sensors, demonstrating moisture resistance.
Alternative approaches include spin-coating and nanoporous layer deposition. Pullano et al. used a mixture of zeolite and high iodine value oil applied via spin-coating, followed by low-temperature annealing, to create thin, stable zeolite films on metal-coated glass. This method enabled precise thickness control and nanoporous film formation.315 Direct synthesis advancements are illustrated by Jeon et al.,316 who employed bottom-up seeded growth to fabricate ultra-thin, defect-free MFI zeolite membranes, achieving superior molecular sieving for xylene isomers. Chen et al.317 improved mordenite membrane performance on Al2O3 hollow fibers by ball milling zeolite seeds for uniform coatings, yielding high flux and selectivity for solvent dehydration in industrial applications.
Surface-modified membranes have also been explored for specific separations. Crick et al. developed membranes tailored for oil–water separation by coating glass filter membranes with silicalite-1 zeolite crystals using hydrothermal treatment and hydrophobic modification with hexamethyldisilazane. This dual-step approach achieved complete oil–water separation efficiency, enabling customizable suction pressures based on oil type.318 Similarly, Botoman's work on zeolite-embedded fabric sheets demonstrated the process that involved embedding LTA zeolite powder between non-woven fabric sheets, pressing the layers, calcining the composite, and washing it to eliminate excess powder. These sheets removed over 99% of lead from contaminated water within 30 seconds without the need for centrifugation or complex separation, showcasing a simple yet effective method for potable water treatment.319
HZSM-5 micromembranes with various Si/Al ratios (∞ to 30) were prepared by microfabricating zeolite films on silicon substrates. This involved seeding with TPA-Sil-1 nanocrystals, followed by hydrothermal growth and etching to form freestanding membranes. Micromembranes with higher aluminum content demonstrated strong proton conductivity and fuel cell performance comparable to Nafion MEAs.320
Aside from gas separation and proton exchange, zeolites films are used as drug delivery system. Zeolites have been shown to be biocompatible molecules and find their place in the production of microneedles. Such microneedles are used for transdermal delivery of drugs. In these methods, a pre-shaped template, often composed of a biocompatible material, serves as the foundation for zeolite crystal growth. Through controlled synthesis conditions, zeolite crystals form on the surface of the template, creating a structured film that maintains the desired microneedle morphology. The process can produce composite structures if additional materials are integrated during growth, enhancing mechanical properties and functional capabilities. However, the film-based approach ensures a uniform coating and facilitates specific applications in targeted delivery systems, particularly for biomedical uses such as transdermal drug delivery. This templating strategy supports the customization of zeolite needle characteristics, such as porosity and structural stability, enabling a wide range of functional applications.321
Both the organic and inorganic components can synergize effectively. For instance, siliceous zeolites, while valuable for many uses, generally show poor sensitivity at low humidity due to the lack of hydrophilic groups. This limitation can be overcome by incorporating hydrophilic polymers, enhancing performance in humidity sensing. In 2018, MCM-41/polypyrrole (PPy) humidity sensors were developed through an in situ gaseous polymerization method, where pyrrole vapor was polymerized within the MCM-41 film. This approach ensured precise polymerization control and uniform PPy deposition, achieving high sensitivity across the humidity spectrum, albeit with longer response and recovery times compared to metal oxide-based sensors.324
Dorosti et al. prepared asymmetric mixed matrix membranes (MMMs) by embedding ZSM-5 and MIL-53 particles into a Matrimid® 5218 matrix using a dry–wet phase inversion method. The polymer was dissolved in NMP, and particles were dispersed with stirring and sonication. The casting solution was degassed, cast on glass, partially dried, and immersed in water, ethanol, and hexane baths to solidify. Post-processing included coating with a PMHS solution and vacuum drying.325 Amooghina et al. enhanced the compatibility of zeolites with the by incorporating aminosilane-grafted NaY zeolite into a Matrimid® 5218 matrix for CO2/CH4 separation. The NaY zeolite was modified with 3-aminopropyl(diethoxy)methylsilane (APDEMS), resulting in membranes with improved CO2 permeability and selectivity due to superior interfacial adhesion.326
Zarshenas et al. explored MMMs incorporating nano-zeolite NaX into a Pebax-1657 matrix. A phase inversion technique via solvent evaporation on a polyethersulfone (PES) support was employed. The microwave-assisted synthesis of NaX particles ensured their uniform dispersion, boosting CO2/N2 and O2/N2 selectivity despite decreased gas permeability.327 Sancho et al. developed zeolite–polymer composites with NaA zeolite, mordenite, ETS-10, and umbite, mixed with PVDF as a binder for enhanced mechanical strength. These membranes demonstrated greater thermal stability than Nafion® 117, maintaining high conductivity at temperatures up to 150 °C.328
The method applies not only for gas separation. High-temperature proton exchange membranes (HTPEMs) have been constructed by integrating ionic liquids, such as 2-hydroxyethyl trimethylammonium dimethyl phosphate (IL1) and other similar compounds, into large-pore zeolites like NH4BEA and NaY, which were then mixed into a PBI casting solution.329 Nanocomposite proton exchange membranes featuring Nafion and sulfonic acid-functionalized zeolite beta were produced using a casting technique to mitigate methanol crossover, a common issue at elevated temperatures. These composite membranes retained Nafion's swelling behavior but significantly reduced methanol permeability, resulting in increased power output. Selectivity ratios (proton conductivity/methanol permeability) were enhanced by up to 93% at 21 °C and 63% at 80 °C, demonstrating marked performance improvements over commercial Nafion 117.330 Zeolite fillers have also been added to Nafion composite membranes using mordenite and analcime, prepared through spray-coating. These membranes showed improvements in properties such as mechanical strength, water uptake, ion-exchange capacity, and direct methanol fuel cell (DMFC) performance.331–333
Though starting with a typical composite fabrication approach, binder-free ZSM-5@silica beads were developed for the removal of organic pollutants using an innovative OSDA-free method. The process began with using an anion exchange resin as a macro-template, which was charged with silica through an ion exchange step. The resin template was then removed via high-temperature combustion, resulting in silica beads that retained the shape and structure imparted by the resin. These silica beads underwent hydrothermal treatment in an OSDA-free system to partially convert the silica into ZSM-5 while maintaining the macrostructure. The synthesis temperature was a key factor in controlling the final zeolite content and mesopore system within the beads.334
Zeolite-based composite structures can be created without incorporating organic polymers, broadening their range of applications. Narang et al. developed an electrochemical microfluidic paper-based analytical device (EμPAD) specifically for ketamine sensing. This device integrated nanocrystals of zeolites and graphene oxide nanoflakes, which were deposited onto the circular working electrode and dried at room temperature. The resulting device offered a cost-effective alternative to conventional metal-based electrodes, demonstrating a low detection limit of 0.001 nM mL−1 and a rapid response time of just two seconds, positioning it as a promising tool for scalable industrial production.335 Additionally, sensors can be enhanced using zeolite-based composites. A study developed a modified carbon paste electrode (CPE) by incorporating zeolite material with multi-walled carbon nanotubes (MWCNTs). This composite sensor was effective for quantifying caffeine in raw materials, commercial beverages, and pharmaceutical samples. The inclusion of an anionic surfactant, sodium dodecyl sulfate (SDS), further improved the performance by promoting hydrophobic interactions that facilitated stronger caffeine adsorption and prolonged its presence near the electrode, thus enhancing the signal.336 Chen et al. introduced a practical application of zeolite composites in water purification. They designed a low-cost flow-through filter composed of mesoporous single-crystal chabazite zeolite bound to cotton fibers, fabricated without a template. This zeolite-cotton filter effectively reduced heavy metal concentrations, such as Cu2+, Cd2+, and Pb2+, to below 5 ppb in a simulated purification of 8 liters of contaminated water. For disinfection, Ag-exchanged zeolite-cotton was employed to remove bacteria like E. coli without releasing harmful levels of silver ions (below 0.05 ppm). This modification allowed for the safe treatment of bacteria-contaminated water, leaving no residual pathogens. The simplicity, affordability, and effectiveness of the zeolite-cotton filter highlight its potential for global household water treatment applications.337
5.3 Spray drying
Spray drying represents an essential and highly efficient method for shaping zeolites into forms suitable for industrial applications. This process involves atomizing a zeolite-containing slurry into fine droplets, which are rapidly dried in a stream of heated gas, yielding spherical particles or powders with controlled size and uniform morphology.338 Unlike extrusion, which is favored for producing structured catalysts like monoliths, spray drying excels in producing granulated materials suitable for fluidized beds, adsorption systems, and catalytic cracking units. A key advantage of spray drying lies in its capacity to integrate binders, fillers, or other functional additives directly into the zeolite slurry prior to drying, thereby tailoring the mechanical and catalytic properties of the resulting particles. This method ensures control over particle size distribution and enhances packing density, minimizing pressure drops in reactor systems. Additionally, spray drying imposes minimal mechanical stress on frameworks, making it particularly suitable for hierarchical or nanozeolites, where the preservation of pore structure is critical.Liu et al.339 developed an approach for shaping SAPO-34 into macroporous microspheres using spray drying combined with polystyrene spheres as hard templates. This method involved blending pre-crystallized SAPO-34 with kaolin, silica sol, and other precursors, followed by spray drying to form 30–50 μm microspheres. The use of polystyrene spheres introduced macroporosity, which improved diffusion and accessibility to active sites compared to non-macroporous counterparts. Post-spray drying, hydrothermal treatment and calcination were employed to enhance crystallinity and acid site strength. The resulting macroporous SAPO-34 demonstrated superior catalytic performance and longer inactivation times in methanol-to-olefins (MTO) reactions, highlighting the critical role of shaping in overcoming diffusion limitations and optimizing catalytic efficiency.
A year later, Majano et al.340 employed a one-step spray drying technique to assemble hierarchical zeolite-silica composites with multimodal porosity. This approach integrated a surfactant-templated silica matrix with mesoporous ZSM-5 zeolite, creating composites that combined micro-, meso-, and macropores. The spray drying process enabled precise control of particle morphology and porosity by adjusting variables such as slurry composition, nozzle size, and drying temperature. Notably, the silica matrix functioned as a binder without compromising the zeolite's structural or acidic properties, ensuring the retention of its catalytic activity. The method was further validated with other zeolite types, including USY and mordenite, demonstrating its versatility and scalability.
Shang et al.341 went further and introduced a novel approach combining spray drying and steam-assisted crystallization (SAC) to create robust, binder-free hierarchical zeolite microspheres. Unlike conventional methods, this approach integrates TPAOH encapsulation during spray drying, enabling precise control of particle size, morphology, and hierarchical porosity. The use of SAC further transforms these precursors into fully crystalline zeolites with enhanced catalytic activity and mechanical stability. The novelty lies in achieving binder-free, uniformly porous microspheres while maintaining high crystallinity and scalability—addressing key challenges in zeolite shaping for industrial catalysis.
5.4 Structured reactors
Structured microreactors, known for their improved mass and heat transfer and straightforward scalability, have increasingly integrated zeolite coatings for enhanced catalytic performance. Various strategies for fabricating zeolite-coated microreactors have been explored, each addressing challenges such as catalyst incorporation within microchannels.342One notable approach was demonstrated by Rebrov et al., who created Ce-ZSM-5-coated stainless steel microreactors for the selective catalytic reduction of NO with NH3. Through a two-step synthesis involving the generation of nucleation centers and controlled crystal growth, uniform zeolite coatings were achieved. These microreactors maintained stable performance without delamination for over 24 hours and were successfully scaled to 72 plates with consistent crystal coverage and minimal deviation.343,344
Navascués et al. contributed to this field by applying seeded liquid-phase hydrothermal synthesis to coat microreactor channels with Pt/ZSM-5 and Pt/Zeolite Y films. The ion-exchange-incorporated Pt enhanced the microreactor's performance in VOC combustion, demonstrating superior results compared to fixed-bed reactors under identical conditions.345 Truter et al. showcased another method involving in situ transformation of a mesoporous silica precoating into titanium silicate-1 (TS-1) zeolite, carefully balancing silica dissolution, nucleation, and growth. The TS-1 microreactor demonstrated high stability over 500 hours and achieved a propylene oxide selectivity of over 90% during propene epoxidation.346,347
Glass microreactors, prized for their chemical inertness and resistance to corrosion, present a challenge in achieving surface roughness for zeolite adherence. One innovative method involved sealing glass plates to form a chip microreactor, followed by a combination of fluoride-based pretreatment and in situ hydrothermal synthesis. This method yielded ZSM-5-coated glass reactors with optimal crystal properties and Brønsted acidity, which performed well in the synthesis of methyl isobutyl ketone and the hydrogenation of 3-methyl-1-pentyn-3-ol, though deactivation due to palladium leaching was observed over time.347
A microfabrication process proposed by Almazánin incorporated a siliceous MFI-type zeolite layer into microconcentrators for VOC sensing. This integration, completed before the anodic bonding step, emphasized the importance of incorporating the zeolite layer via hydrothermal synthesis for functional performance.348
6. Emerging trends and opportunities for hierarchical zeolites
Hierarchical zeolites – zeolites featuring both micropores and larger meso/macropores – are at the forefront of innovative catalysis and adsorption research. Their multi-scale porosity alleviates diffusion limitations and coking in ways traditional zeolites cannot, opening new avenues in sustainable processing and advanced applications. Below we discuss four promising future directions: sustainable and scalable synthesis, plastic waste valorization, CO2 capture/conversion, and AI-guided zeolite design.349,3506.1 Sustainability and scalable fabrication
Recent efforts focus on greener, scalable routes to synthesize hierarchical zeolites, reducing environmental footprint while maintaining performance. Key advances include:Crucially, researchers are evaluating these green methods using metrics like the E-factor (mass waste per product) and life-cycle assessments. One example reported a solvent-free, surfactant-templated route to hierarchical zeolites that achieved an 82% yield in a target fine chemical synthesis (2-aminochromene) with double the catalytic activity of a conventional zeolite, all under solvent-free conditions.349
6.2 AI-Assisted zeolite design and discovery
Artificial intelligence and machine learning (ML) are revolutionizing how we design and optimize zeolites, including hierarchical variants. Traditionally, zeolite discovery (finding new frameworks or improved synthesis routes) was largely trial-and-error. Now, data-driven models can predict synthesis outcomes, structure–property relationships, and even suggest new zeolite structures or templates. In the realm of hierarchical zeolites, AI tools are being used to navigate the complex interplay between synthesis conditions and multi-scale structure, accelerating innovation.AI is also being harnessed to discover novel zeolite structures (including those with inherent hierarchical porosity). Millions of hypothetical zeolite frameworks have been proposed by algorithmic enumeration; the challenge is to find which are synthesizable and useful. Machine learning is helping screen these candidates for stability and guiding synthetic targets. Notably, a recent study applied ML techniques to forecast low-framework-density zeolites – those with extra-large pores – by learning from known synthesis data. The ML model identified patterns in the database that hint at how to crystallize frameworks with very large cages (which could inherently be hierarchical due to large voids).360 This has led to predictions of specific composition and condition “recipes” that might yield new ultra-mesoporous zeolites. In parallel, deep generative models (like inverse design algorithms) are being explored to propose entirely new framework topologies optimized for certain functions (e.g. a target pore size distribution for a given reaction).
Looking ahead, AI-assisted design may allow “on-demand” hierarchical zeolites: given a desired application (e.g. a bulky biomass molecule conversion), one could input the required pore sizes and acid site density, and an AI algorithm could propose a feasible framework and synthesis route to achieve it. Early steps toward this include an ML model that suggests synthesis conditions for hypothetical zeolites not yet made, effectively bridging the gap between computationally predicted structures and lab realization.363 By integrating synthesis data with crystal chemistry, such models can point to combinations of inorganic/organic structure-directing agents and parameters that might crystallize a novel hierarchical structure.
In summary, AI and machine learning are becoming indispensable in zeolite research. They are accelerating the discovery of new hierarchical zeolite structures, optimizing synthesis protocols (improving success rates and scalability), and predicting performance (allowing virtual testing of designs). This data-driven revolution complements experimental efforts: for example, an ML classifier can sort through thousands of possible synthesis mixtures to find a few likely to yield a mesoporous zeolite, which chemists can then try in the. As these tools mature, we can expect a faster pace of innovation – new zeolite frameworks, bespoke pore architectures, and tuned active sites – all achieved with fewer resources by guiding experiments with predictive models. The fusion of AI with traditional zeolite science thus represents a powerful opportunity to realize the next generation of hierarchical zeolites tailored for sustainability and performance.
7. Conclusions
Zeolitic materials are both celebrated and challenged by their well-defined pores and molecular dimensions. On one hand, their uniform microporous structure enables molecular sieving, high surface area, and shape-selective catalysis, making them indispensable in applications such as petrochemical refining, gas separation, and environmental remediation. However, these same features also impose diffusion limitations, particularly for larger molecules, which can struggle to access or exit the active sites within the narrow pore network. Such constraints can lead to reduced catalytic efficiency, mass transport bottlenecks, and deactivation due to pore blockage. Addressing these limitations through hierarchical structuring, mesoporous incorporation, or post-synthetic modifications is crucial for optimizing the performance of zeolites in practical applications.In this comprehensive review, after introducing the governing equations of transport phenomena in zeolites along with the different experimental techniques used to measure diffusion, we have outlined the different strategies applied in practice to address these limitations, from the microlevel to the macroscale.
To overcome diffusion limitations, various synthetic strategies are employed, each addressing different terms in the governing equations. For example, to reduce the diffusion pathlength, thereby increasing the overall mass transport rate, nano-zeolites are synthesized, ensuring shorter distances for molecules to travel. Additionally, hierarchical zeolites introduce mesopores and macropores, effectively increasing Deff by providing additional pathways for molecular transport and mitigating pore-blockage effects. Post-synthetic modifications, such as desilication and dealumination, can tune pore connectivity and improve accessibility, further enhancing diffusion rates. These strategies collectively optimize the interplay between pore architecture and transport properties at the micro and mesoscale, ensuring that the intrinsic catalytic potential of zeolites is not hindered by mass transfer limitations.
At the macro-scale, commercial zeolite bodies encounter additional diffusion limitations due to their use in pelletized, extruded, or monolithic forms. The governing equations of diffusion extend beyond intracrystalline transport and must also account for macroscopic mass transport within catalyst bodies, often described by the Thiele modulus and Weisz-Prater criterion. To address diffusion constraints at this scale, several strategies are implemented in commercial zeolite formulations.
Pelletization techniques such as spray drying allow control over the macroscopic porosity and pellet size. By reducing pellet diameter, external diffusion limitations can be minimized, however, too small a pellet size increases pressure drop in packed-bed reactors, necessitating an optimal balance between transport and process efficiency. Monolithic and 3D-printed zeolite structures provide another emerging solution by incorporating ordered macroporous networks, significantly reducing external diffusion limitations while maintaining structural integrity. These hierarchical macrostructures facilitate convective transport, effectively increasing the apparent diffusivity and enhancing overall performance in catalytic and adsorption applications. Together, these macro-scale engineering approaches complement nano- and meso-structural modifications, ensuring that commercial zeolite catalysts achieve both high accessibility and durability while maintaining practical applicability in large-scale industrial processes.
Despite significant advancements in mitigating diffusion limitations in zeolites across multiple length scales, several challenges remain for future development. A key hurdle lies in the precise control of hierarchical porosity without compromising zeolite crystallinity, stability, or catalytic activity. Current synthesis methods often involve trade-offs between mesoporosity and framework integrity, necessitating novel bottom-up and post-synthetic strategies that offer greater structural control. Additionally, in commercial zeolite bodies, optimizing mass transport while maintaining mechanical strength and hydrothermal stability remains a complex engineering challenge, particularly in high-pressure and high-temperature industrial environments. The integration of advanced manufacturing techniques, such as 3D printing and templated synthesis, holds promise for designing zeolite catalysts with tailored microporous architectures, yet scalability and cost-efficiency remain key concerns. Furthermore, computational modeling and machine learning approaches offer new opportunities to predict and design optimal pore structures, but experimental validation and translation into industrial-scale production are still in their infancy. Addressing these challenges will require an interdisciplinary approach, combining advances in material science, catalysis, and process engineering to push the boundaries of zeolite performance and applicability in next-generation industrial processes.
Data availability
No new data were generated or analyzed in this study. All data supporting the findings of this review are available in the referenced literature.Conflicts of interest
There are no conflicts to declare.References
- W. Vermeiren and J. P. Gilson, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- N. Y. Chen, W. E. Garwood and F. G. Dywer, Shape Selective Catalysis in Industrial Applications, Marcel Dekker, New York, NY, 2 edn, 1996 Search PubMed.
- T. F. Degnan, J. Catal., 2003, 216, 32–46 CrossRef CAS.
- S. Ezenwa and R. Gounder, Chem. Commun., 2024, 60, 12118–12143 RSC.
- X. Liu, C. Wang, J. Zhou, C. Liu, Z. Liu, J. Shi, Y. Wang, J. Teng and Z. Xie, Chem. Soc. Rev., 2022, 51, 8174–8200 RSC.
- J. van den Bergh, J. Gascon and F. Kapteijn, in Zeolites and Catalysis: Synthesis, Reactions and Applications, ed. J. Čejka, A. Corma and S. Zones, Wiley, 2010, ch. 13, pp. 361–387 Search PubMed.
- P. Peng, X.-H. Gao, Z.-F. Yan and S. Mintova, Natl. Sci. Rev., 2020, 7, 1726–1742 CrossRef PubMed.
- G. Noh and M. L. Sarazen, J. Catal., 2021, 404, 679–686 CrossRef CAS.
- C. W. Hullfish, J. Z. Tan, H. I. Adawi and M. L. Sarazen, ACS Catal., 2023, 13, 13140–13150 CrossRef CAS.
- X3D®: Shaping tomorrow's catalysts with today's technology, https://chemical-catalysts-and-adsorbents.basf.com/global/en/sustainability-process-catalysts/X3D, 20th May 2025.
- A. K. Ghosh, C. Mihut and M. Simmons, US Pat., 20140080696A1, 2014 Search PubMed.
- D. Bazer-Bachi, B. Harbuzaru and E. Lecolier, EP Pat., 3071324B1, 2014.
- S. H. Jones, US Pat., 20160297685A1, 2016 Search PubMed.
- S. Hirano and T. Kawamoto, CN Pat., 102177093A, 2011 Search PubMed.
- F. Rezaei and H. V. Thakkar, US Pat., 20190083954A1, 2019 Search PubMed.
- W. Liu, X. S. Li and N. L. Canfield, US Pat., 20150265975A1, 2015 Search PubMed.
- P. M. M. Blauwhoff, J. W. Gosselink, E. P. Kieffer, S. T. Sie and W. H. J. Stork, in Catalysis and Zeolites: Fundamentals and Applications, ed. J. Weitkamp and L. Puppe, Springer, Berlin, Heidelberg, 1999, pp. 437–538 Search PubMed.
- T. F. Degnan, Top. Catal., 2000, 13, 349–356 CrossRef CAS.
- R. Gläser and J. Weitkamp, in Basic Principles in Applied Catalysis, ed. M. Baerns, Springer, Berlin, Heidelberg, 2004, pp. 161–212 Search PubMed.
- E. T. C. Vogt, G. T. Whiting, A. D. Chowdhury and B. M. Weckhuysen, Adv. Catal., 2015, 58, 143–314 Search PubMed.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- W. Loewenstein, Am. Mineral., 1954, 39, 92–96 CAS.
- J. Weitkamp, Solid State Ionics, 2000, 131, 175–188 CrossRef CAS.
- R. A. van Santen and G. J. Kramer, Chem. Rev., 1995, 95, 637–660 CrossRef CAS.
- R. B. Bird, D. J. Klingenberg, E. N. Lightfoot and W. E. Stewart, Introductory Transport Phenomena, Wiley, 2014 Search PubMed.
- B. E. Poling, J. M. Prausnitz and J. P. O'Connell, The Properties of Gases and Liquids, McGraw-Hill, New York, NY, 5 edn, 2001 Search PubMed.
- International Zeolite Association, https://www.iza-online.org, 20th May 2025.
- P. B. Weisz and V. J. Frilette, J. Phys. Chem. C, 1960, 64, 382 CrossRef CAS.
- P. B. Weisz, Stud. Surf. Sci. Catal., 1981, 7, 3–20 CrossRef CAS.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- J. Weitkamp and S. Ernst, Catal. Today, 1994, 19, 107–150 CrossRef CAS.
- W. W. Kaeding, L. B. Young and C.-C. Chu, J. Catal., 1984, 89, 267–273 CrossRef CAS.
- L. B. Young, S. A. Butter and W. W. Kaeding, J. Catal., 1982, 76, 418–432 Search PubMed.
- W. O. Haag, R. M. Lago and P. B. Weisz, Faraday Discuss. Chem. Soc., 1981, 72, 317–330 RSC.
- E. W. Thiele, Ind. Eng. Chem., 1939, 31, 916–920 CrossRef CAS.
- P. B. Weisz, Science, 1973, 179, 433–440 CrossRef CAS PubMed.
- D. H. Olson and W. O. Haag, in Catalytic Materials: Relationship Between Structure and Reactivity, ed. T. E. Whyte, R. A. Dalla Betta, E. G. Derouane and R. T. K. Baker, American Chemical Society, 1984, pp. 275–310 Search PubMed.
- J. N. Miale, N. Y. Chen and P. B. Weisz, J. Catal., 1966, 6, 278–287 CrossRef CAS.
- M. D. Foster, I. Rivin, M. M. J. Treacy and O. Delgado Friedrichs, Microporous Mesoporous Mater., 2006, 90, 32–38 CrossRef CAS.
- E. L. First, C. E. Gounaris, J. Wei and C. A. Floudas, Phys. Chem. Chem. Phys., 2011, 13, 17339–17358 RSC.
- V. J. Frilette, W. O. Haag and R. M. Lago, J. Catal., 1981, 67, 218–222 CrossRef.
- J. Weitkamp, S. Ernst and L. Puppe, in Catalysis and Zeolites: Fundamentals and Applications, ed. J. Weitkamp and L. Puppe, Springer, 1999, pp. 327–376 Search PubMed.
- J. Weitkamp, S. Ernst and R. Kumar, Appl. Catal., 1986, 27, 207–210 CrossRef CAS.
- J. Weitkamp, S. Ernst and C. Y. Chen, Stud. Surf. Sci. Catal., 1989, 49, 1115–1129 CrossRef.
- J. Weitkamp, C. Y. Chen and S. Ernst, Stud. Surf. Sci. Catal., 1989, 44, 343–350 CrossRef CAS.
- E. G. Derouane and Z. Gabelica, J. Catal., 1980, 65, 486–489 CrossRef CAS.
- D. Fraenkel, M. Cherniavsky, B. Ittah and M. Levy, J. Catal., 1986, 101, 273–283 CrossRef CAS.
- E. G. Derouane, J. Catal., 1986, 100, 541–544 CrossRef CAS.
- J. A. Martens, W. Souverijns, W. Verrelst, R. Parton, G. F. Froment and P. A. Jacobs, Angew. Chem., 1995, 34, 2528–2530 CrossRef CAS.
- J. A. Martens, G. Vanbutsele, P. A. Jacobs, J. Denayer, R. Ocakoglu, G. Baron, J. A. M. Arroyo, J. Thybaut and G. B. Marin, Catal. Today, 2001, 65, 111–116 CrossRef CAS.
- R. L. Gorring, J. Catal., 1973, 31, 13–26 CrossRef CAS.
- N. Y. Chen and W. E. Garwood, in Molecular Sieves, ed. W. M. Meier and J. B. Uytterhoeven, American Chemical Society, 1973, pp. 575–582 Search PubMed.
- S. Namba, K. Sato, K. Fujita, J. H. Kim and T. Yashima, Stud. Surf. Sci. Catal., 1986, 28, 661–668 CrossRef CAS.
- M. F. M. Post, Stud. Surf. Sci. Catal., 1991, 58, 391–443 CrossRef CAS.
- D. M. Ruthven, Stud. Surf. Sci. Catal., 2007, 168, 737–785 CrossRef.
- E. G. Derouane, Stud. Surf. Sci. Catal., 1980, 5, 5–18 CrossRef CAS.
- J. Kärger and J. Pfeifer, Zeolites, 1987, 7, 90–107 CrossRef.
- H. Pfeifer, J. Kärger, A. Germanus, W. Schirmer, M. Bülow and J. Caro, Adsorpt. Sci. Technol., 1985, 2, 229–239 CrossRef.
- D. Paschek and R. Krishna, Chem. Phys. Lett., 2001, 333, 278–284 CrossRef CAS.
- H. G. Karge and J. Weitkamp, Adsorption and Diffusion, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in Nanoporous Materials, 2012, pp. 111–142 Search PubMed.
- B. Smit and T. L. M. Maesen, Chem. Rev., 2008, 108, 4125–4184 CrossRef CAS PubMed.
- H. Jobic, Membr. Sci. Technol., 2000, 6, 109–137 CAS.
- A. Sayeed, S. Mitra, A. V. Anil Kumar, R. Mukhopadhyay, S. Yashonath and S. L. Chaplot, J. Phys. Chem. B, 2003, 107, 527–533 CrossRef CAS.
- J. Crank, The Mathematics of Diffusion, Clarendon Press, Ely House, London, 2 edn, 1975 Search PubMed.
- D. M. Ruthven and K. F. Loughlin, Chem. Eng. Sci., 1971, 26, 577–584 CrossRef CAS.
- D. M. Ruthven, Principles of Adsorption and Adsorption Processes, John Wiley & Sons, New York, NY, 1984 Search PubMed.
- J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in Nanoporous Materials, Wiley-VCH, Weinheim, Germany, 2012, vol. 1, pp. 143–190 Search PubMed.
- N. G. van-den-Begin and L. V. C. Rees, Stud. Surf. Sci. Catal., 1989, 49, 915–924 CrossRef.
- D. Shen and L. V. C. Rees, J. Chem. Soc., Faraday Trans., 1994, 90, 3011–3015 RSC.
- V. Bourdin, A. Germanus, P. Grenier and J. Kärger, Adsorption, 1996, 2, 205–216 CrossRef CAS.
- V. Bourdin, P. Grenier, F. Meunier and L. M. Sun, AIChE J., 1996, 42, 700–712 Search PubMed.
- P. Grenier, A. Malka-Edery and V. Bourdin, Adsorption, 1999, 5, 135–143 CrossRef CAS.
- M. J. den Exter, J. C. Jansen, J. M. van de Graaf, F. Kapteijn, J. A. Moulijn and H. van Bekkum, Stud. Surf. Sci. Catal., 1996, 102, 413–454 Search PubMed.
- R. Krishna, Gas Sep. Purif., 1993, 7, 91–104 CrossRef CAS.
- R. Krishna and J. A. Wesselingh, Chem. Eng. Sci., 1997, 52, 862–911 CrossRef.
- F. Kapteijn, W. J. W. Bakker, G. Zheng, J. Poppe and J. A. Moulijn, Chem. Eng. J., 1995, 57, 145–153 CAS.
- D. B. Shah and D. M. Ruthven, AIChE J., 1977, 23, 804–809 CrossRef CAS.
- D. M. Ruthven and M. Eic, Perspectives in Molecular Sieve Science, American Chemical Society, 1988, ch. 22, vol. 368, pp. 362–375 Search PubMed.
- S. Brandani, J. Hufton and D. Ruthven, Zeolites, 1995, 15, 624–631 Search PubMed.
- S. Brandani and D. M. Ruthven, Adsorption, 1996, 2, 133–143 CrossRef CAS.
- D. M. Ruthven and S. Brandani, Membr. Sci. Technol., 2000, 6, 187–212 Search PubMed.
- J. T. Gleaves, J. R. Ebner and T. C. Kuechler, Catal. Rev.: Sci. Eng., 1988, 30, 49–116 CrossRef CAS.
- T. A. Nijhuis, L. J. P. van den Broeke, J. M. van de Graaf, F. Kapteijn, M. Makkee and J. A. Moulijn, Chem. Eng. Sci., 1997, 52, 3401–3404 CrossRef CAS.
- T. A. Nijhuis, L. J. P. van den Broeke, M. J. G. Linders, J. M. van de Graaf, F. Kapteijn, M. Makkee and J. A. Moulijn, Chem. Eng. Sci., 1999, 54, 4423–4436 CrossRef CAS.
- O. Keipert and M. Baerns, Chem. Eng. Sci., 1998, 53, 3623–3634 Search PubMed.
- R. Aris, Chem. Eng. Sci., 1957, 6, 262–268 Search PubMed.
- P. B. Weisz and C. D. Prater, Adv. Catal., 1954, 6, 143–196 CAS.
- M. F. M. Post, J. van Amstel and H. W. Kouwenhoven, Diffusion and Catalytic Reaction of 2,2-Dimethylbutane in ZSM-5 Zeolite, The 6th International Zeolite Conference, Reno, NV, 1983 Search PubMed.
- H. G. Karge and W. Nießen, Catal. Today, 1991, 8, 451–465 CrossRef CAS.
- H. G. Karge, C. R. Chim, 2005, 8, 303–319 CrossRef CAS.
- U. Schemmert, J. Kärger, C. Krause, A. Rákoczy and J. Weitkamp, Europhys. Lett., 1999, 46, 204–210 Search PubMed.
- U. Schemmert, J. Kärger and J. Weitkamp, Microporous Mesoporous Mater., 1999, 32, 101–110 Search PubMed.
- P. Kortunov, C. Chmelik, J. Kärger, R. A. Rakoczy, D. M. Ruthven, Y. Traa, S. Vasenkov and J. Weitkamp, Adsorption, 2005, 11, 235–244 CrossRef CAS.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed.
- M. L. Sarazen, E. Doskocil and E. Iglesia, ACS Catal., 2016, 6, 7059–7070 CrossRef CAS.
- J. Zhou, Z. Liu, Y. Wang, H. Gao, L. Li, W. Yang, Z. Xie and Y. Tang, RSC Adv., 2014, 4, 43752–43755 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 Search PubMed.
- E. Andres-Garcia, A. Dikhtiarenko, F. Fauth, J. Silvestre-Albero, E. V. Ramos-Fernández, J. Gascon, A. Corma and F. Kapteijn, Chem. Eng. J., 2019, 360, 569–576 Search PubMed.
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- C. H. Christensen, K. Johannsen, E. Törnqvist, I. Schmidt, H. Topsøe and C. H. Christensen, Catal. Today, 2007, 128, 117–122 CrossRef CAS.
- Z. Li, J. Martínez-Triguero, P. Concepción, J. Yu and A. Corma, Phys. Chem. Chem. Phys., 2013, 15, 14670–14680 RSC.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- G. Aguirre-Cruz, F. Legorreta-Garcia, G. Aguirre-Cruz, L. Stanciu and G. Aguirre-Alvarez, Microporous Mesoporous Mater., 2022, 345, 112274 CrossRef CAS.
- L. Tan, N. Jiao, X. Bai, H. Wang, J. Wang, H. Wang and X. Zhang, Eur. J. Inorg. Chem., 2023, e202300314 CrossRef CAS.
- S. Ghojavand, E. Dib and S. Mintova, Chem. Sci., 2023, 14, 12430–12446 RSC.
- S. Mitchell, A. B. Pinar, J. Kenvin, P. Crivelli, J. Kärger and J. Pérez-Ramírez, Nat. Commun., 2015, 6, 8633 Search PubMed.
- K. Li, J. Valla and J. Garcia-Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS.
- J. Pérez-Ramírez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- A. J. Mallette, S. Seo and J. D. Rimer, Nat. Synth., 2022, 1, 521–534 Search PubMed.
- R. Simancas, A. Chokkalingam, S. P. Elangovan, Z. Liu, T. Sano, K. Iyoki, T. Wakihara and T. Okubo, Chem. Sci., 2021, 12, 7677–7695 RSC.
- T. Weissenberger, A. G. F. Machoke, B. Reiprich and W. Schwieger, Adv. Mater. Interfaces, 2021, 8, 2001653 CrossRef CAS.
- J. Li, M. Gao, W. Yan and J. Yu, Chem. Sci., 2023, 14, 1935–1959 RSC.
- L. Hong, J. Zang, B. Li, G. Liu, Y. Wang and L. Wu, Inorganics, 2023, 11, 214 CrossRef CAS.
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494–2513 Search PubMed.
- W. Schwieger, A. G. Machoke, T. Weissenberger, A. Inayat, T. Selvam, M. Klumpp and A. Inayat, Chem. Soc. Rev., 2016, 45, 3353–3376 RSC.
- Z. Jensen, E. Kim, S. Kwon, T. Z. H. Gani, Y. Román-Leshkov, M. Moliner, A. Corma and E. Olivetti, ACS Cent. Sci., 2019, 5, 892–899 CrossRef CAS PubMed.
- X. Li, H. Han, N. Evangelou, N. J. Wichrowski, P. Lu, W. Xu, S.-J. Hwang, W. Zhao, C. Song, X. Guo, A. Bhan, I. G. Kevrekidis and M. Tsapatsis, Nat. Commun., 2023, 14, 3152 Search PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- A. I. Lupulescu and J. D. Rimer, Science, 2014, 344, 729–732 CrossRef CAS PubMed.
- R. Li, N. Linares, J. G. Sutjianto, A. Chawla, J. Garcia-Martinez and J. D. Rimer, Angew. Chem., Int. Ed., 2018, 57, 11283–11288 CrossRef CAS PubMed.
- Q. Li, D. Creaser and J. Sterte, Chem. Mater., 2002, 14, 1319–1324 Search PubMed.
- Q. Li, B. Mihailova, D. Creaser and J. Sterte, Microporous Mesoporous Mater., 2000, 40, 53–62 Search PubMed.
- T. F. Chaves, H. O. Pastore and D. Cardoso, Microporous Mesoporous Mater., 2012, 161, 67–75 CrossRef CAS.
- Q. Zhang, G. Chen, Y. Wang, M. Chen, G. Guo, J. Shi, J. Luo and J. Yu, Chem. Mater., 2018, 30, 2750–2758 CrossRef CAS.
- D. Wen, Q. Liu, Z. Fei, Y. Yang, Z. Zhang, X. Chen, J. Tang, M. Cui and X. Qiao, Ind. Eng. Chem. Res., 2018, 57, 446–455 CrossRef CAS.
- J. Yang, X. Tang, J. Liu, J. Wang, H. Shang, L. Wu, J. Li and S. Deng, Chem. Eng. J., 2021, 406, 126599 CrossRef CAS.
- J. Zheng, W. Zhang, Z. Liu, Q. Huo, K. Zhu, X. Zhou and W. Yuan, Microporous Mesoporous Mater., 2016, 225, 74–87 Search PubMed.
- S. R. Venna and M. A. Carreon, J. Phys. Chem. B, 2008, 112, 16261–16265 CrossRef CAS PubMed.
- I. Schmidt, C. Madsen and C. J. H. Jacobsen, Inorg. Chem., 2000, 39, 2279–2283 Search PubMed.
- M. Ogura, Y. Zhang, S. P. Elangovan and T. Okubo, Microporous Mesoporous Mater., 2007, 101, 224–230 Search PubMed.
- Z. Ma, Q. Zhang, L. Li, M. Chen, J. Li and J. Yu, Chem. Sci., 2022, 13, 8052–8059 RSC.
- H. Chen, J. Wydra, X. Zhang, P.-S. Lee, Z. Wang, W. Fan and M. Tsapatsis, J. Am. Chem. Soc., 2011, 133, 12390–12393 CrossRef CAS PubMed.
- T. Wakihara, R. Ichikawa, J. Tatami, A. Endo, K. Yoshida, Y. Sasaki, K. Komeya and T. Meguro, Cryst. Growth Des., 2011, 11, 955–958 CrossRef CAS.
- Z. Liu, N. Nomura, D. Nishioka, Y. Hotta, T. Matsuo, K. Oshima, Y. Yanaba, T. Yoshikawa, K. Ohara, S. Kohara, T. Takewaki, T. Okubo and T. Wakihara, Chem. Commun., 2015, 51, 12567–12570 Search PubMed.
- C. Anand, Y. Yamaguchi, Z. Liu, S. Ibe, S. P. Elangovan, T. Ishii, T. Ishikawa, A. Endo, T. Okubo and T. Wakihara, Sci. Rep., 2016, 6, 29210 Search PubMed.
- I. L. Motta, J. G. P. Vicente and D. Cardoso, Mol. Catal., 2018, 458, 127–138 CrossRef CAS.
- C. Manrique, T. Botero, R. Solano, C. Mendoza and A. Echavarría-Isaza, J. Porous Mater., 2025 DOI:10.1007/s10934-024-01703-8.
- Q. Sun, N. Wang, R. Bai, X. Chen and J. Yu, J. Mater. Chem. A, 2016, 4, 14978–14982 RSC.
- C. Zhang, K. Fan, G. Ma, C. Lei, W. Xu, J. Jiang, B. Sun, H. Zhang, Y. Zhu and S. Wen, Crystals, 2021, 11, 1247 CrossRef CAS.
- F. M. Alotaibi, L. Ding, H. Sitepu, Q. Saleem, D. A. Alsewdan, A. S. Ali, S. I. Bin Hamad, M. H. Alotaibi and M. F. Aljishi, Catal. Commun., 2024, 187, 106871 CrossRef CAS.
- M. V. Landau, L. Vradman, V. Valtchev, J. Lezervant, E. Liubich and M. Talianker, Ind. Eng. Chem. Res., 2003, 42, 2773–2782 Search PubMed.
- J. Hedlund, M. Zhou, A. Faisal, O. G. W. Öhrman, V. Finelli, M. Signorile, V. Crocellà and M. Grahn, J. Catal., 2022, 410, 320–332 Search PubMed.
- A. Corma, C. Corell and J. Pérez-Pariente, Zeolites, 1995, 15, 2–8 CrossRef CAS.
- L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 1996, 6, 259–271 CrossRef CAS.
- K. Na, W. Park, Y. Seo and R. Ryoo, Chem. Mater., 2011, 23, 1273–1279 CrossRef CAS.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Čejka, Chem. Rev., 2014, 114, 4807–4837 CrossRef CAS PubMed.
- L. Zhang, N. Wang and Y. Li, Chem. Sci., 2023, 14, 5266–5290 Search PubMed.
- S. Zanardi, A. Alberti, G. Cruciani, A. Corma, V. Fornés and M. Brunelli, Angew. Chem., Int. Ed., 2004, 43, 4933–4937 CrossRef CAS PubMed.
- P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris and J. Čejka, Chem. Soc. Rev., 2015, 44, 7177–7206 Search PubMed.
- D. N. Rainer, C. M. Rice, S. J. Warrender, S. E. Ashbrook and R. E. Morris, Chem. Sci., 2020, 11, 7060–7069 Search PubMed.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. Čejka, Nat. Chem., 2013, 5, 628–633 Search PubMed.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 Search PubMed.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 Search PubMed.
- D. Mehlhorn, A. Inayat, W. Schwieger, R. Valiullin and J. Kärger, ChemPhysChem, 2014, 15, 1681–1686 Search PubMed.
- J. Aguilar, S. B. C. Pergher, C. Detoni, A. Corma, F. V. Melo and E. Sastre, Catal. Today, 2008, 133–135, 667–672 Search PubMed.
- X. Zhu, M. G. Goesten, A. J. J. Koekkoek, B. Mezari, N. Kosinov, G. Filonenko, H. Friedrich, R. Rohling, B. M. Szyja, J. Gascon, F. Kapteijn and E. J. M. Hensen, Chem. Sci., 2016, 7, 6506–6513 RSC.
- K. Möller and T. Bein, Chem. Soc. Rev., 2013, 42, 3689–3707 RSC.
- F. N. Gu, F. Wei, J. Y. Yang, N. Lin, W. G. Lin, Y. Wang and J. H. Zhu, Chem. Mater., 2010, 22, 2442–2450 CrossRef CAS.
- F. Wei, W. Song, F. Wei and C. Cao, Chin. J. Catal., 2015, 36, 838–844 CrossRef CAS.
- X. Wang, X. Zhang and Q. Wang, Mater. Lett., 2019, 244, 96–99 Search PubMed.
- Z. Wang, C. Li, H. J. Cho, S.-C. Kung, M. A. Snyder and W. Fan, J. Mater. Chem. A, 2015, 3, 1298–1305 Search PubMed.
- K. Iyoki, K. Itabashi and T. Okubo, Microporous Mesoporous Mater., 2014, 189, 22–30 Search PubMed.
- H. Dai, Y. Shen, T. Yang, C. Lee, D. Fu, A. Agarwal, T. T. Le, M. Tsapatsis, J. C. Palmer, B. M. Weckhuysen, P. J. Dauenhauer, X. Zou and J. D. Rimer, Nat. Mater., 2020, 19, 1074–1080 CrossRef CAS PubMed.
- H. Dai, C. Lee, W. Liu, T. Yang, J. Claret, X. Zou, P. J. Dauenhauer, X. Li and J. D. Rimer, Angew. Chem., Int. Ed., 2022, 61, e202113077 Search PubMed.
- H. Zhang, H. Zhang, Y. Zhao, Z. Shi, Y. Zhang and Y. Tang, Chem. Mater., 2017, 29, 9247–9255 CrossRef CAS.
- R. Jain, A. Chawla, N. Linares, J. García Martínez and J. D. Rimer, Adv. Mater., 2021, 33, 2100897 Search PubMed.
- J. Du, Q. Wang, Y. Wang, Y. Guo and R. Li, Mater. Lett., 2019, 250, 139–142 Search PubMed.
- X. Jia, W. Khan, Z. Wu, J. Choi and A. C. K. Yip, Adv. Powder Technol., 2019, 30, 467–484 Search PubMed.
- H. Qu, Y. Ma, B. Li and L. Wang, Emerg. Mater., 2020, 3, 225–245 CrossRef CAS.
- L. Liu, M. Zhang, L. Wang, X. Zhang and G. Li, Appl. Catal., A, 2020, 602, 117664 Search PubMed.
- H. Wang, G. Du, J. Jia, S. Chen, Z. Su, R. Chen and T. Chen, Front. Chem. Sci. Eng., 2021, 15, 1444–1461 CrossRef CAS.
- H. S. Cho and R. Ryoo, Microporous Mesoporous Mater., 2012, 151, 107–112 Search PubMed.
- S. Abdulridha, Y. Jiao, S. Xu, R. Zhang, Z. Ren, A. A. Garforth and X. Fan, Appl. Catal., A, 2021, 612, 117986 Search PubMed.
- Z. Asgar Pour, D. G. Boer, S. Fang, Z. Tang and P. P. Pescarmona, Catalysts, 2021, 11, 1346 Search PubMed.
- S. Soltanali and J. T. Darian, Microporous Mesoporous Mater., 2019, 286, 169–175 CrossRef CAS.
- R. Khoshbin, S. Oruji and R. Karimzadeh, Adv. Powder Technol., 2018, 29, 2176–2187 CrossRef CAS.
- C.-J. Chang, C.-H. Chen, J.-F. Lee, T. Sooknoi and Y.-C. Lin, Ind. Eng. Chem. Res., 2020, 59, 11177–11186 Search PubMed.
- K. Zhu, K. Egeblad and C. H. Christensen, Eur. J. Inorg. Chem., 2007, 3955–3960 Search PubMed.
- Y. Wang, T. Li, Y. Ouyang, J. Zhong, Y. Zhang, X. Xiong, Q. Hu, J. Deng, H. Sun and Z. Yan, Catal. Sci. Technol., 2024, 14, 2461–2469 RSC.
- M. Hartmann, M. Thommes and W. Schwieger, Adv. Mater. Interfaces, 2021, 8, 2001841 Search PubMed.
- C. J. H. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116–7117 CrossRef CAS.
- Y. Hou, X. Li, M. Sun, C. Li, S. U. H. Bakhtiar, K. Lei, S. Yu, Z. Wang, Z. Hu, L. Chen and B.-L. Su, Front. Chem. Sci. Eng., 2021, 15, 269–278 CrossRef CAS.
- I. Schmidt, A. Boisen, E. Gustavsson, K. Ståhl, S. Pehrson, S. Dahl, A. Carlsson and C. J. H. Jacobsen, Chem. Mater., 2001, 13, 4416–4418 CrossRef CAS.
- Y. Tao, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 6044–6045 CrossRef CAS PubMed.
- Y. Tao, H. Kanoh and K. Kaneko, J. Phys. Chem. B, 2003, 107, 10974–10976 CrossRef CAS.
- K. Na, M. Choi and R. Ryoo, Microporous Mesoporous Mater., 2013, 166, 3–19 CrossRef CAS.
- H. Wang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2006, 45, 7603–7606 Search PubMed.
- R. W. Pekala, C. T. Alviso, F. M. Kong and S. S. Hulsey, J. Non-Cryst. Solids, 1992, 145, 90–98 CrossRef CAS.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- X. D. Wang, W. L. Yang, Y. Tang, Y. J. Wang, S. K. Fu and Z. Gao, Chem. Commun., 2000, 2161–2162, 10.1039/B006539K.
- V. Valtchev and S. Mintova, Microporous Mesoporous Mater., 2001, 43, 41–49 CrossRef CAS.
- Y.-B. Zhang, X.-F. Qian, H.-A. Xi, J. Yin and Z.-K. Zhu, Mater. Lett., 2004, 58, 222–225 CrossRef CAS.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC.
- S. Lopez-Orozco, A. Inayat, A. Schwab, T. Selvam and W. Schwieger, Adv. Mater., 2011, 23, 2602–2615 CrossRef CAS PubMed.
- H. Awala, J.-P. Gilson, R. Retoux, P. Boullay, J.-M. Goupil, V. Valtchev and S. Mintova, Nat. Mater., 2015, 14, 447–451 CrossRef CAS PubMed.
- L. Huang, W. Guo, P. Deng, Z. Xue and Q. Li, J. Phys. Chem. B, 2000, 104, 2817–2823 CrossRef CAS.
- V. Valtchev, G. Majano, S. Mintova and J. Pérez-Ramírez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- C. Mei, P. Wen, Z. Liu, H. Liu, Y. Wang, W. Yang, Z. Xie, W. Hua and Z. Gao, J. Catal., 2008, 258, 243–249 Search PubMed.
- V. Pashkova, V. Tokarova, L. Brabec and J. Dedecek, Microporous Mesoporous Mater., 2016, 228, 59–63 Search PubMed.
- W. Song, R. Kanthasamy, V. H. Grassian and S. C. Larsen, Chem. Commun., 2004, 1920–1921, 10.1039/B406753C.
- F. Ocampo, J. A. Cunha, M. R. de Lima Santos, J. P. Tessonnier, M. M. Pereira and B. Louis, Appl. Catal., A, 2010, 390, 102–109 CrossRef CAS.
- V. Valtchev, F. Gao and L. Tosheva, New J. Chem., 2008, 32, 1331–1337 Search PubMed.
- D. H. Park, S. S. Kim, H. Wang, T. J. Pinnavaia, M. C. Papapetrou, A. A. Lappas and K. S. Triantafyllidis, Angew. Chem., Int. Ed., 2009, 48, 7645–7648 CrossRef CAS PubMed.
- D. P. Serrano, T. J. Pinnavaia, J. Aguado, J. M. Escola, A. Peral and L. Villalba, Catal. Today, 2014, 227, 15–25 CrossRef CAS.
- J. Zhu, Y. Zhu, L. Zhu, M. Rigutto, A. van der Made, C. Yang, S. Pan, L. Wang, L. Zhu, Y. Jin, Q. Sun, Q. Wu, X. Meng, D. Zhang, Y. Han, J. Li, Y. Chu, A. Zheng, S. Qiu, X. Zheng and F.-S. Xiao, J. Am. Chem. Soc., 2014, 136, 2503–2510 CrossRef CAS PubMed.
- G. Song, D. Xue, J. Xue and F. Li, Microporous Mesoporous Mater., 2017, 248, 192–203 CrossRef CAS.
- L. Wang, Z. Zhang, C. Yin, Z. Shan and F.-S. Xiao, Microporous Mesoporous Mater., 2010, 131, 58–67 CrossRef CAS.
- K. Cho, H. S. Cho, L.-C. de Ménorval and R. Ryoo, Chem. Mater., 2009, 21, 5664–5673 CrossRef CAS.
- N. Kalantari, A. Farzi, N. Çaylak Delibaş, A. Niaei and D. Salari, Res. Chem. Intermed., 2021, 47, 4957–4984 CrossRef CAS.
- L. Wu, V. Degirmenci, P. C. M. M. Magusin, B. M. Szyja and E. J. M. Hensen, Chem. Commun., 2012, 48, 9492–9494 RSC.
- Y. Zhang, K. Zhu, X. Duan, P. Li, X. Zhou and W. Yuan, RSC Adv., 2014, 4, 14471–14474 Search PubMed.
- C. Jo, J. Jung, H. S. Shin, J. Kim and R. Ryoo, Angew. Chem., Int. Ed., 2013, 52, 10014–10017 CrossRef CAS PubMed.
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodríguez and Á. Peral, Chem. Mater., 2006, 18, 2462–2464 CrossRef CAS.
- Y. Zhu, Z. Hua, J. Zhou, L. Wang, J. Zhao, Y. Gong, W. Wu, M. Ruan and J. Shi, Chem. – Eur. J., 2011, 17, 14618–14627 CrossRef CAS PubMed.
- J. Sun, C. Wang, L. Wang, L. Liang, X. Liu, L. Chen and F.-S. Xiao, Catal. Today, 2010, 158, 273–278 CrossRef CAS.
- M. A. Malik, M. Y. Wani and M. A. Hashim, Arabian J. Chem., 2012, 5, 397–417 CrossRef CAS.
- L. Figueroa-Quintero, D. Villalgordo-Hernández, J. J. Delgado-Marín, J. Narciso, V. K. Velisoju, P. Castaño, J. Gascón and E. V. Ramos-Fernández, Small Methods, 2023, 7, 2201413 CrossRef CAS PubMed.
- Q. Sheng, K. Ling, Z. Li and L. Zhao, Fuel Process. Technol., 2013, 110, 73–78 CrossRef CAS.
- Y. Fan, X. Bao, X. Lin, G. Shi and H. Liu, J. Phys. Chem. B, 2006, 110, 15411–15416 Search PubMed.
- D. Barthomeuf, Zeolites, 1990, 10, 131–133 Search PubMed.
- R. Otomo, T. Yokoi, J. N. Kondo and T. Tatsumi, Appl. Catal., A, 2014, 470, 318–326 CrossRef CAS.
- M. Müller, G. Harvey and R. Prins, Microporous Mesoporous Mater., 2000, 34, 135–147 CrossRef.
- C. Zhang, Q. Liu, Z. Xu and K. Wan, Microporous Mesoporous Mater., 2003, 62, 157–163 CrossRef CAS.
- B. E. Alver and M. Sakizci, Adsorption, 2015, 21, 391–399 CrossRef CAS.
- Y. Qiao, M. Yang, B. Gao, L. Wang, P. Tian, S. Xu and Z. Liu, Chem. Commun., 2016, 52, 5718–5721 RSC.
- Z. Qin, J.-P. Gilson and V. Valtchev, Curr. Opin. Chem. Eng., 2015, 8, 1–6 CrossRef.
- J.-X. Wang, J.-P. Cao, X.-Y. Zhao, S.-N. Liu, X.-Y. Ren, M. Zhao, X. Cui, Q. Chen and X.-Y. Wei, Bioresour. Technol., 2019, 278, 116–123 Search PubMed.
- D. Xi, Q. Sun, X. Chen, N. Wang and J. Yu, Chem. Commun., 2015, 51, 11987–11989 Search PubMed.
- V. Babić, S. Koneti, S. Moldovan, M. Debost, J.-P. Gilson and V. Valtchev, Microporous Mesoporous Mater., 2022, 329, 111513 Search PubMed.
- R. M. Dessau, E. W. Valyocsik and N. H. Goeke, Zeolites, 1992, 12, 776–779 Search PubMed.
- J. C. Groen and J. Pérez-Ramírez, Appl. Catal., A, 2004, 268, 121–125 CrossRef CAS.
- J. C. Groen, T. Sano, J. A. Moulijn and J. Pérez-Ramírez, J. Catal., 2007, 251, 21–27 Search PubMed.
- J. C. Groen, J. A. Moulijn and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2005, 87, 153–161 Search PubMed.
- J. C. Groen, S. Abelló, L. A. Villaescusa and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2008, 114, 93–102 CrossRef CAS.
- D. Verboekend, S. Mitchell, M. Milina, J. C. Groen and J. Pérez-Ramírez, J. Phys. Chem. C, 2011, 115, 14193–14203 CrossRef CAS.
- D. Verboekend, N. Nuttens, R. Locus, J. Van Aelst, P. Verolme, J. C. Groen, J. Pérez-Ramírez and B. F. Sels, Chem. Soc. Rev., 2016, 45, 3331–3352 RSC.
- J. C. Groen, L. A. A. Peffer, J. A. Moulijn and J. Pérez-Ramírez, Colloids Surf., A, 2004, 241, 53–58 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer, J. A. Moulijn and J. Pérez-Ramírez, Chem. – Eur. J., 2005, 11, 4983–4994 CrossRef CAS PubMed.
- J. C. Groen, T. Bach, U. Ziese, A. M. Paulaime-van Donk, K. P. de Jong, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2005, 127, 10792–10793 CrossRef CAS PubMed.
- J. C. Groen, J. A. Moulijn and J. Pérez-Ramírez, J. Mater. Chem., 2006, 16, 2121–2131 RSC.
- J. C. Groen, J. C. Jansen, J. A. Moulijn and J. Pérez-Ramírez, J. Phys. Chem. B, 2004, 108, 13062–13065 Search PubMed.
- J. C. Groen, W. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef CAS PubMed.
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
- S. Svelle, L. Sommer, K. Barbera, P. N. R. Vennestrøm, U. Olsbye, K. P. Lillerud, S. Bordiga, Y.-H. Pan and P. Beato, Catal. Today, 2011, 168, 38–47 CrossRef CAS.
- D. Verboekend, G. Vilé and J. Pérez-Ramírez, Cryst. Growth Des., 2012, 12, 3123–3132 CrossRef CAS.
- L. Bonetto, C. Fermanelli and C. Saux, Waste Biomass Valorization, 2024, 15, 6599–6613 Search PubMed.
- E. Diguilio, E. D. Galarza, M. E. Domine, L. B. Pierella and M. S. Renzini, New J. Chem., 2020, 44, 4363–4375 RSC.
- R. Le Van Mao, S. T. Le, D. Ohayon, F. Caillibot, L. Gelebart and G. Denes, Zeolites, 1997, 19, 270–278 CrossRef CAS.
- Z. Hasan, J. W. Jun, C.-U. Kim, K.-E. Jeong, S.-Y. Jeong and S. H. Jhung, Mater. Res. Bull., 2015, 61, 469–474 CrossRef CAS.
- J. Pérez-Ramírez, S. Abelló, A. Bonilla and J. C. Groen, Adv. Funct. Mater., 2009, 19, 164–172 CrossRef.
- A. N. C. van Laak, L. Zhang, A. N. Parvulescu, P. C. A. Bruijnincx, B. M. Weckhuysen, K. P. de Jong and P. E. de Jongh, Catal. Today, 2011, 168, 48–56 CrossRef CAS.
- K. Tarach, K. Góra-Marek, J. Tekla, K. Brylewska, J. Datka, K. Mlekodaj, W. Makowski, M. C. Igualada López, J. Martínez Triguero and F. Rey, J. Catal., 2014, 312, 46–57 CrossRef CAS.
- D. Verboekend and J. Pérez-Ramírez, Chem. – Eur. J., 2011, 17, 1137–1147 CrossRef CAS PubMed.
- M. Gackowski, K. Tarach, Ł. Kuterasiński, J. Podobiński, B. Sulikowski and J. Datka, Microporous Mesoporous Mater., 2019, 281, 134–141 CrossRef CAS.
- M. Gackowski, K. Tarach, Ł. Kuterasiński, J. Podobiński, S. Jarczewski, P. Kuśtrowski and J. Datka, Microporous Mesoporous Mater., 2018, 263, 282–288 Search PubMed.
- X. Zhao, J. He and J. Li, Catalysts, 2022, 12, 1216 CrossRef CAS.
- A. Asghari, M. K. Khorrami and S. H. Kazemi, Sci. Rep., 2019, 9, 17526 CrossRef PubMed.
- T. Weissenberger, A. G. F. Machoke, J. Bauer, R. Dotzel, J. L. Casci, M. Hartmann and W. Schwieger, ChemCatChem, 2020, 12, 2461–2468 CrossRef CAS.
- J. Fals, J. F. Garcia-Valencia, E. Puello-Polo, F. Tuler and E. Márquez, Molecules, 2024, 29, 3085 CrossRef CAS PubMed.
- F. Akhtar, L. Andersson, S. Ogunwumi, N. Hedin and L. Bergström, J. Eur. Ceram. Soc., 2014, 34, 1643–1666 CrossRef CAS.
- S. Mitchell, N.-L. Michels and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 6094–6112 Search PubMed.
- F. Akhtar, L. Andersson, S. Ogunwumi, N. Hedin and L. Bergstrom, J. Eur. Ceram. Soc., 2014, 34, 1643–1666 Search PubMed.
- J. Gascon, J. R. van Ommen, J. A. Moulijn and F. Kapteijn, Catal.: Sci. Technol., 2015, 5, 807–817 Search PubMed.
- N.-L. Michels, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2014, 4, 2409–2417 CrossRef CAS.
- P. Forzatti, D. Ballardini and L. Sighicelli, Catal. Today, 1998, 41, 87–94 CrossRef CAS.
- Z. Asgar Pour, M. M. Abduljawad, Y. A. Alassmy, L. Cardon, P. H. M. Van Steenberge and K. O. Sebakhy, Catalysts, 2023, 13, 656 Search PubMed.
- J. Zhu, P. Wu, Y. Chao, J. Yu, W. Zhu, Z. Liu and C. Xu, Chem. Eng. J., 2022, 433, 134341 CrossRef CAS.
- R. Bingre, B. Louis and P. Nguyen, Catalysts, 2018, 8, 163 CrossRef CAS.
- J. Ding, P. Chen, G. Zhao, Y. Liu and Y. Lu, J. Catal., 2018, 360, 40–50 CrossRef CAS.
- T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I. Hoek, F. Kapteijn and J. A. Moulijn, Catal. Rev.: Sci. Eng., 2001, 43, 345–380 CrossRef CAS.
- M. V. Twigg and J. T. Richardson, Chem. Eng. Res. Des., 2002, 80, 183–189 CrossRef CAS.
- J. A. Moulijn and F. Kapteijn, Curr. Opin. Chem. Eng., 2013, 2, 346–353 CrossRef.
- K. Pangarkar, T. J. Schildhauer, J. R. van Ommen, J. Nijenhuis, F. Kapteijn and J. A. Moulijn, Ind. Eng. Chem. Res., 2008, 47, 3720–3751 CrossRef CAS.
- C. P. Stemmet, J. N. Jongmans, J. van der Schaaf, B. F. M. Kuster and J. C. Schouten, Chem. Eng. Sci., 2005, 60, 6422–6429 CrossRef CAS.
- V. Tomasic and F. Jovic, Appl. Catal., A, 2006, 311, 112–121 CrossRef CAS.
- A. Cybulski and J. A. Moulijn, Catal. Rev.: Sci. Eng., 1994, 36, 179–270 CrossRef CAS.
- P. Avila, M. Montes and E. E. Miro, Chem. Eng. J., 2005, 109, 11–36 CrossRef CAS.
- V. Meille, Appl. Catal., A, 2006, 315, 1–17 CrossRef CAS.
- K. Shams and S. J. Mirmohammadi, Microporous Mesoporous Mater., 2007, 106, 268–277 CrossRef CAS.
- S. Govender and H. Friedrich, Catalysts, 2017, 7, 62 CrossRef.
- A. Aranzabal, D. Iturbe, M. Romero-Sáez, M. P. González-Marcos, J. R. González-Velasco and J. A. González-Marcos, Chem. Eng. J., 2010, 162, 415–423 Search PubMed.
- I. M. Lachman and J. L. Williams, Catal. Today, 1992, 14, 317–329 CrossRef CAS.
- L. Li, B. Xue, J. Chen, N. Guan, F. Zhang, D. Liu and H. Feng, Appl. Catal., A, 2005, 292, 312–321 CrossRef CAS.
- J. Jänchen, T. H. Herzog, K. Gleichmann, B. Unger, A. Brandt, G. Fischer and H. Richter, Microporous Mesoporous Mater., 2015, 207, 179–184 CrossRef.
- A. Mosca, J. Hedlund, P. A. Webley, M. Grahn and F. Rezaei, Microporous Mesoporous Mater., 2010, 130, 38–48 Search PubMed.
- L. D. Li, B. Xue, J. X. Chen, N. J. Guan, F. X. Zhang, D. X. Liu and H. Q. Feng, Appl. Catal., A, 2005, 292, 312–321 CrossRef CAS.
- P. Colombo, C. Vakifahmetoglu and S. Costacurta, J. Mater. Sci., 2010, 45, 5425–5455 CrossRef CAS.
- F. Ocampo, H. S. Yun, M. M. Pereira, J. P. Tessonnier and B. Louis, Cryst. Growth Des., 2009, 9, 3721–3729 CrossRef CAS.
- B. Louis, C. Tezel, L. Kiwi-Minsker and A. Renken, Catal. Today, 2001, 69, 365–370 CrossRef CAS.
- J. Gascon, J. R. van Ommen, J. A. Moulijn and F. Kapteijn, Catal. Sci. Technol., 2015, 5, 807–817 RSC.
- D. F. Wu and H. Zhang, Ind. Eng. Chem. Res., 2013, 52, 14713–14721 CrossRef CAS.
- J. M. Zamaro and E. E. Miro, Chem. Eng. J., 2010, 165, 701–708 CrossRef CAS.
- S. Samadi, S. S. Yazd, H. Abdoli, P. Jafari and M. Aliabadi, Int. J. Biol. Macromol., 2017, 105, 370–376 CrossRef CAS PubMed.
- Y. Li, X. Shang and B. Zhang, Ind. Eng. Chem. Res., 2018, 57, 17409–17416 CrossRef CAS.
- L. Bonaccorsi, A. Freni, E. Proverbio, G. Restuccia and F. Russo, Microporous Mesoporous Mater., 2006, 91, 7–14 CrossRef CAS.
- X. Zhou and C. j Liu, Adv. Funct. Mater., 2017, 27, 1701134 CrossRef.
- H. Thakkar, S. Eastman, A. Hajari, A. A. Rownaghi, J. C. Knox and F. Rezaei, ACS Appl. Mater. Interfaces, 2016, 8, 27753–27761 CrossRef CAS PubMed.
- S. Couck, J. Cousin-Saint-Remi, S. Van der Perre, G. V. Baron, C. Minas, P. Ruch and J. F. M. Denayer, Microporous Mesoporous Mater., 2018, 255, 185–191 Search PubMed.
- J. Lefevere, L. Protasova, S. Mullens and V. Meynen, Mater. Des., 2017, 134, 331–341 Search PubMed.
- X. Li, W. Li, F. Rezaei and A. Rownaghi, Chem. Eng. J., 2018, 333, 545–553 Search PubMed.
- J. Lefevere, M. Gysen, S. Mullens, V. Meynen and J. Van Noyen, Catal. Today, 2013, 216, 18–23 CrossRef CAS.
- J. Lefevere, S. Mullens, V. Meynen and J. Van Noyen, Chem. Pap., 2014, 68, 1143–1153 CAS.
- S. Couck, J. Lefevere, S. Mullens, L. Protasova, V. Meynen, G. Desmet, G. V. Baron and J. F. M. Denayer, Chem. Eng. J., 2017, 308, 719–726 Search PubMed.
- S. Lawson, B. Adebayo, C. Robinson, Q. Al-Naddaf, A. A. Rownaghi and F. Rezaei, Chem. Eng. Sci., 2020, 218, 115564 CrossRef CAS.
- S. Wang, P. Bai, Y. Wei, W. Liu, X. Ren, J. Bai, Z. Lu, W. Yan and J. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 38955–38963 CrossRef CAS PubMed.
- A. Khalil, R. Hashaikeh and N. Hilal, J. Water Process Eng., 2021, 42, 102187 Search PubMed.
- A. A. Bragina, A. I. Lysikov and E. V. Parkhomchuk, Pet. Chem., 2022, 62, 853–861 CrossRef CAS.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastián, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829–2844 CrossRef CAS.
- M. A. Snyder and M. Tsapatsis, Angew. Chem., Int. Ed., 2007, 46, 7560–7573 CrossRef CAS PubMed.
- N. Kosinov, J. Gascon, F. Kapteijn and E. J. M. Hensen, J. Membr. Sci., 2016, 499, 65–79 Search PubMed.
- A. T. Güntner, S. Abegg, K. Wegner and S. E. Pratsinis, Sens. Actuators, B, 2018, 257, 916–923 CrossRef.
- R. C. Abruzzi, M. J. R. Pires, B. A. Dedavid and C. F. Galli, Mater. Res., 2019, 22, e20180818 CrossRef CAS.
- S. A. Pullano, F. Falcone, D. C. Critello, M. G. Bianco, M. Menniti and A. S. Fiorillo, Sensors, 2020, 20, 2143 CrossRef CAS PubMed.
- M. Y. Jeon, D. Kim, P. Kumar, P. S. Lee, N. Rangnekar, P. Bai, M. Shete, B. Elyassi, H. S. Lee, K. Narasimharao, S. N. Basahel, S. Al-Thabaiti, W. Xu, H. J. Cho, E. O. Fetisov, R. Thyagarajan, R. F. Dejaco, W. Fan, K. A. Mkhoyan, J. I. Siepmann and M. Tsapatsis, Nature, 2017, 543, 690–694 CrossRef CAS PubMed.
- C. Chen, Y. Cheng, L. Peng, C. Zhang, Z. Wu, X. Gu, X. Wang and S. Murad, Microporous Mesoporous Mater., 2019, 274, 347–355 CrossRef CAS.
- C. R. Crick, F. T. Ozkan and I. P. Parkin, Sci. Technol. Adv. Mater., 2015, 16, 055006 CrossRef PubMed.
- L. Botoman, E. Shukla, E. Johan, S. Mitsunobu and N. Matsue, J. Water Health, 2017, 16, 159–163 CrossRef PubMed.
- W. Han, S. M. Kwan and K. L. Yeung, Chem. Eng. J., 2012, 187, 367–371 CrossRef CAS.
- A. Ramović, Z. Zorlak, Đ. Husić and S. Ramić, Zeolite microneedles: recent advancements and implications in the delivery of collagen, Proceedings of the International Conference on Medical and Biological Engineering, Banja Luka, Bosnia and Herzegovina, 2020 Search PubMed.
- N. S. Hassan, A. A. Jalil, M. B. Bahari, N. F. Khusnun, E. M. S. Aldeen, R. S. Mim, M. L. Firmansyah, S. Rajendran, R. R. Mukti, R. Andika and H. Devianto, Chemosphere, 2023, 314, 137709 CrossRef CAS PubMed.
- N. Rangnekar, N. Mittal, B. Elyassi, J. Caro and M. Tsapatsis, Chem. Soc. Rev., 2015, 44, 7128–7154 RSC.
- R. Qi, X. Lin, J. Dai, H. Zhao, S. Liu, T. Fei and T. Zhang, Sens. Actuators, B, 2018, 277, 584–590 CrossRef CAS.
- F. Dorosti, M. Omidkhah and R. Abedini, J. Nat. Gas Sci. Eng., 2015, 25, 88–102 CrossRef CAS.
- A. Ebadi Amooghin, M. Omidkhah and A. Kargari, J. Membr. Sci., 2015, 490, 364–379 CrossRef CAS.
- K. Zarshenas, A. Raisi and A. Aroujalian, J. Membr. Sci., 2016, 510, 270–283 CrossRef CAS.
- T. Sancho, J. Soler and M. P. Pina, J. Power Sources, 2007, 169, 92–97 CrossRef CAS.
- A. Eguizábal, J. Lemus and M. P. Pina, J. Power Sources, 2013, 222, 483–492 CrossRef.
- B. A. Holmberg, X. Wang and Y. Yan, J. Membr. Sci., 2008, 320, 86–92 CrossRef CAS.
- P. Prapainainar, Z. Du, P. Kongkachuichay, S. M. Holmes and C. Prapainainar, Appl. Surf. Sci., 2017, 421, 24–41 CrossRef CAS.
- P. Prapainainar, S. Maliwan, K. Sarakham, Z. Du, C. Prapainainar, S. M. Holmes and P. Kongkachuichay, Int. J. Hydrogen Energy, 2018, 43, 14675–14690 CrossRef CAS.
- P. Prapainainar, Z. Du, A. Theampetch, C. Prapainainar, P. Kongkachuichay and S. M. Holmes, Energy, 2020, 190, 116451 CrossRef CAS.
- P. He, J. Ding, Z. Qin, L. Tang, K. G. Haw, Y. Zhang, Q. Fang, S. Qiu and V. Valtchev, Inorg. Chem. Front., 2020, 7, 2080–2088 RSC.
- J. Narang, N. Malhotra, C. Singhal, A. Mathur, D. Chakraborty, A. Anil, A. Ingle and C. S. Pundir, Biosens. Bioelectron., 2017, 88, 249–257 CrossRef CAS PubMed.
- S. M. Azab, M. Shehata and A. M. Fekry, New J. Chem., 2019, 43, 15359–15367 RSC.
- X. Chen, L. Yu, S. Zou, L. Xiao and J. Fan, Sci. Rep., 2020, 10, 4719 CrossRef CAS PubMed.
- D. P. Debecker, S. Le Bras, C. Boissière, A. Chaumonnot and C. Sanchez, Chem. Soc. Rev., 2018, 47, 4112–4155 RSC.
- Y. Liu, L. Wang, J. Zhang, F. Chen and M. Anpo, Res. Chem. Intermed., 2011, 37, 949–959 CrossRef CAS.
- G. Majano, A. Restuccia, M. Santiago and J. Pérez-Ramírez, CrystEngComm, 2012, 14, 5985–5991 RSC.
- C. Shang, Z. Wu, W. Duo Wu and X. Dong Chen, Chem. Eng. Sci., 2021, 229, 116080 CrossRef CAS.
- Y. S. S. Wan, J. L. H. Chau, A. Gavriilidis and K. L. Yeung, Microporous Mesoporous Mater., 2001, 42, 157–175 CrossRef CAS.
- E. V. Rebrov, G. B. F. Seijger, H. P. A. Calis, M. H. J. M. de Croon, C. M. van den Bleek and J. C. Schouten, Appl. Catal., A, 2001, 206, 125–143 CrossRef CAS.
- M. J. M. Mies, J. L. P. van den Bosch, E. V. Rebrov, J. C. Jansen, M. H. J. M. de Croon and J. C. Schouten, Catal. Today, 2005, 110, 38–46 CrossRef CAS.
- N. Navascués, M. Escuin, Y. Rodas, S. Irusta, R. Mallada and J. Santamaría, Ind. Eng. Chem. Res., 2010, 49, 6941–6947 CrossRef.
- L. A. Truter, V. Ordomsky, J. C. Schouten and T. A. Nijhuis, Microporous Mesoporous Mater., 2016, 226, 424–432 CrossRef CAS.
- L. A. Truter, V. Ordomsky, J. C. Schouten and T. A. Nijhuis, Appl. Catal., A, 2016, 515, 72–82 CrossRef CAS.
- F. Almazán, I. Pellejero, A. Morales, M. A. Urbiztondo, J. Sesé, M. P. Pina and J. Santamaría, J. Micromech. Microeng., 2016, 26, 084010 CrossRef.
- M. J. Mendoza-Castro, N. Linares and J. García-Martínez, Catal. Today, 2023, 419, 114152 CrossRef CAS.
- T. Pan, Z. Wu and A. C. K. Yip, Catalysts, 2019, 9, 274 CrossRef CAS.
- A. S. F. Nanda and G. T. M. Kadja, J. Porous Mater., 2024, 31, 1155–1173 CrossRef CAS.
- H. Xie, W. Xu, S. Ran, X. Li, B. Li, Y. Li and L. Wang, Microporous Mesoporous Mater., 2022, 345, 112238 CrossRef CAS.
- K. Zhang, S. Fernandez, J. T. O’Brien, T. Pilyugina, S. Kobaslija and M. L. Ostraat, Catal. Today, 2018, 316, 26–30 CrossRef CAS.
- Y. Jia, W. Han, G. Xiong and W. Yang, Mater. Lett., 2008, 62, 2400–2403 CrossRef CAS.
- C. Zhang, S. Li and S. Bao, Waste Biomass Valorization, 2019, 10, 2825–2835 Search PubMed.
- F. Pan, X. Lu, Y. Wang, S. Chen, T. Wang and Y. Yan, Mater. Lett., 2014, 115, 5–8 Search PubMed.
- X. Zeng, X. Hu, H. Song, G. Xia, Z.-Y. Shen, R. Yu and M. Moskovits, Microporous Mesoporous Mater., 2021, 323, 111262 CrossRef CAS.
- J. Zhu, Z. Liu, S. Sukenaga, M. Ando, H. Shibata, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2018, 268, 1–8 Search PubMed.
- E. Pan, S. Kwon, Z. Jensen, M. Xie, R. Gómez-Bombarelli, M. Moliner, Y. Román-Leshkov and E. Olivetti, ACS Cent. Sci., 2024, 10, 729–743 Search PubMed.
- G. Raman, J. Solid State Chem., 2023, 327, 124290 Search PubMed.
- F. Daeyaert, F. Ye and M. W. Deem, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3413–3418 Search PubMed.
- M. Wu, S. Zhang and J. Ren, APL Mater., 2025, 13, 020601 Search PubMed.
- D. Schwalbe-Koda, D. E. Widdowson, T. A. Pham and V. A. Kurlin, Digital Discovery, 2023, 2, 1911–1924 RSC.
| This journal is © The Royal Society of Chemistry 2025 |