
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the low-temperature performance of Pt-based three-way catalysts using CeO2(core)@ZrO2(shell) supports†‡
Chih-Han
Liu
a,
Junjie
Chen§
 *a,
Patrick R.
Raffaelle
b,
Michael J.
Lance
c,
Jacob
Concolino
a,
Prateek
Khatri
a,
Tala
Mon
a,
Todd J.
Toops
c,
Alexander A.
Shestopalov
b and
Eleni A.
Kyriakidou
*a
*a,
Patrick R.
Raffaelle
b,
Michael J.
Lance
c,
Jacob
Concolino
a,
Prateek
Khatri
a,
Tala
Mon
a,
Todd J.
Toops
c,
Alexander A.
Shestopalov
b and
Eleni A.
Kyriakidou
*a
aDepartment of Chemical and Biological Engineering, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA. E-mail: jchen299@buffalo.edu; elenikyr@buffalo.edu
bHajim School of Engineering and Applied Sciences, University of Rochester, Rochester, New York 14627, USA
cOak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 17th April 2025
Abstract
Developing robust Pt/CeO2-based three-way catalysts (TWCs) with enhanced oxygen buffering capability and low-temperature activity is highly desirable. In this study, a new TWC family, Pt/(1 − x)CeO2(core)@xZrO2(shell) (where x = 0–0.5), was prepared and evaluated at degreened (DG) and hydrothermally aged (HTA) states. Incorporation of 0.1 molar concentration of ZrO2 resulted in a decreased temperature that 50% (T50) (CO: 167 °C, THCs: 218 °C, NO: 228 °C) and 90% (T90) (CO: 207 °C, THCs: 237 °C, NO: 244 °C) conversions achieved over HTA 1.8 wt% Pt/0.9CeO2@0.1ZrO2 compared to the HTA 1.8 wt% Pt/CeO2 sphere (CO: T50,90 = 179, 222 °C, THCs: 234, 252 °C, NOx: 240, 260 °C). An enhanced oxygen storage capacity and oxygen release rate were observed over Pt/0.9CeO2@0.1ZrO2 compared to the Pt/CeO2 sphere. Increasing the ZrO2 molar concentration to values greater than 0.2 resulted in increased T50s (224, 265![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 274 °C) and T90s (251, 289, 292 °C) for CO, THCs, and NOx, respectively, over 1.8 wt.% Pt/0.5CeO2@0.5ZrO2. Overall, this work highlights the potential of forming a ZrO2 shell on CeO2 spheres as a support for TWC applications.
274 °C) and T90s (251, 289, 292 °C) for CO, THCs, and NOx, respectively, over 1.8 wt.% Pt/0.5CeO2@0.5ZrO2. Overall, this work highlights the potential of forming a ZrO2 shell on CeO2 spheres as a support for TWC applications.
1. Introduction
Three-way catalysts (TWCs) can simultaneously convert carbon monoxide (CO), hydrocarbons (HCs), and nitrogen oxides (NOx) to less harmful products (CO2, H2O, N2) and they are the key components for the control of stoichiometric spark ignition engine emissions.1 TWCs are currently highly efficient when the vehicle emission temperature is higher than 350 °C. However, approximately 80% of vehicle emissions are emitted to the atmosphere during the cold start period (first 1–3 min of vehicle operation) when the TWCs are not warm enough.2 Moreover, the improved engine efficiency of vehicles operating on stoichiometric feed has led to low engine operating temperatures and lower exhaust temperatures. Low exhaust temperatures in gasoline aftertreatment systems necessitate the development of TWCs that are active at low temperatures.3–5 Specifically, the “150 °C challenge”6 aimed to achieve 90% conversion of CO, HCs, and nitrogen NOx at temperatures as low as 150 °C. The most common commercial TWCs utilize platinum group metals (PGMs), such as Pd,7 Pd/Rh,8,9 Pt/Rh,10,11 and Pt/Pd/Rh,12,13 which can simultaneously convert CO, HCs, and NOx to CO2, H2O, and N2, respectively, via the following reaction routes:14

Pt-based TWCs have attracted significant attention due to the lower cost of Pt ($956 per troy ounce; Jan. 2025) compared to Rh ($4675 per troy ounce; Jan. 2025).15

The active metals are often dispersed on a support to maximize their active surface area. The rare earth metal oxide, ceria (CeO2), is of particular interest in TWCs because Ce exists in two oxidation states (Ce4+/Ce3+) under operating conditions.1 The Ce4+/Ce3+ redox couple allows CeO2 to store/release O2 through a redox reaction (2CeO2 ↔ Ce2O3 + 0.5 O2)16,17 under oxygen-rich/deficient conditions, respectively.18–20 The oxygen storage capacity (OSC) of CeO2 is important in TWCs as it can buffer the oxygen-rich/deficient conditions resulting from the imprecise control of fuel injection in gasoline vehicles, leading to 1% oscillations for an air/fuel ratio of around 14.7 during vehicle operation.21,22 Oxygen-rich/deficient conditions can significantly impact the efficiency of TWCs to mitigate CO, HCs, and NOx.12,23,24 Specifically, oxygen can be stored in CeO2 under lean conditions and can be supplied to the oxidation reactions under rich conditions. Zirconium is typically incorporated into CeO2 to increase its lattice oxygen mobility to further improve the TWC performance.25–27 For example, a more than an order of magnitude increase in OSC (from 25 to 350 μmolO g−1) was reported by increasing the Ce:Zr molar ratio from 0 (CeO2) to 1 (Ce0.5Zr0.5O2).25 Moreover, the CO and NOx catalytic activity over Pd/CeO2–ZrO2–Pr2O3 with various Ce:Zr ratios revealed that a higher OSC (488.4 molO g−1) was responsible for a wider operational window (100% CO conversion at λ ≥ 0.925 and 100% NO conversion at λ ≤ 1.075).26
Although CeO2 possesses high OSC, it can suffer from sintering when exposed to elevated temperatures. For instance, TWCs can exposed to 900–950 °C during deceleration fuel cut-offs, scavenging, and disabled fuel enrichment at high engine loads.28 Polyhedral CeO2, commonly used in catalytic reactions,29–31 losses the majority of its surface area when exposed to elevated temperatures. Specifically, the surface area of polyhedral CeO2 is decreased from 89 to 21 m2 g−1 when increasing the calcination temperature from 450 to 600 °C.32 Therefore, CeO2 supports that can maintain their surface area after exposure to elevated temperatures attract a lot of interest. Several strategies have been adopted to enhance the thermal stability of CeO2. In a study by Chen et al., CeO2 islands were anchored onto the surface of penta-site rich Al2O3 leading to smaller CeO2 crystalline sizes (∼11–17 nm) than the bare CeO2 sample (∼22 nm) after hydrothermal aging.1 Other methods include the incorporation of ZrO2 to the CeO2 lattice to form ceria–zirconia solid solutions.32 Herein, an approach of coating CeO2 nanospheres with a ZrO2 shell is studied by synthesizing (1 − x)CeO2@xZrO2 (x = 0, 0.1, 0.2, 0.3, 0.5) supports for TWC applications. The TWC activity and hydrothermal stability of Pt/(1 − x)CeO2@xZrO2 catalysts were evaluated using the U.S. DRIVE test protocol.33 High-resolution transmission electron microscopy (HRTEM), scanning transmission electron microscopy (STEM), and energy dispersive X-ray spectroscopy (EDS) were conducted to reveal the morphology of (1 − x)CeO2@xZrO2. Moreover, CO-pulse chemisorption and CO diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFTS) were conducted to identify the Pt dispersion. X-ray photoelectron spectroscopy (XPS) was used to determine the Ce oxidation states. Finally, complete oxygen storage capacity (OSCC) and oxygen release rate (ORR) measurements were conducted to elucidate their effect on the TWC performance.
2. Experimental
2.1. Reagents and materials
Zirconium(IV) butoxide (Zr(BuO)4) solution (80 wt% in 1-butanol, Sigma-Aldrich), anhydrous ethanol (EtOH, 200 proof, Decon), ethylene glycol (C2H6O2) (Fisher Scientific, 99.99%), ammonium hydroxide (NH4OH, 28–30 wt% solution of NH3 in water, Sigma-Aldrich), 2,2,4-trimethylpentane (i-C8H18) (Sigma Aldrich, 99.8%), and silicon carbide (SiC) (Alfa Aesar, ≥98%) were used as received. A Brij30 solution (100 wt%, Acros Organics, purity 99.79%) was diluted with D.I. water to a 3.8 wt% Brij30 solution prior to usage. Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O) (Sigma Aldrich, 99.999%), zirconium(IV) oxynitrate hydrate (ZrO(NO3)2·xH2O) (Sigma Aldrich, 99%), and tetraammineplatinum nitrate (Pt(NH3)4(NO3)2) (STREM Chemicals, 99%) were diluted with D.I. water to form 0.1 M, 0.5 M Ce(NO3)3, 0.1 M ZrO(NO3)2, and 0.015 M Pt(NH3)4(NO3)2 solutions prior to usage. D.I. water with a resistivity of 18.2 MΩ cm was used for all the syntheses in this study.2.2. Catalyst synthesis
CeO2 spheres were prepared by a hydrothermal synthesis method (Scheme 1).34 Briefly, CeO2 spheres were synthesized by mixing 5.2 mL of 0.5 M Ce(NO3)3 solution with 78 mL ethylene glycol and the obtained solution was stirred for 4 h. The mixture was then transferred to an autoclave fitted with a Teflon liner and heated to 180 °C for 8 h, followed by cooling down to room temperature. The obtained mixture was centrifuged at 5000 rpm for 20 min and the solid phase was redispersed in D.I. water and centrifuged three times. The solid phase was subsequently redispersed in anhydrous EtOH and centrifuged three times. The retrieved solid phase was then dried (110 °C/overnight) and calcined (500 °C/2 h) to obtain CeO2 spheres. | ||
| Scheme 1 Synthesis procedure of CeO2@ZrO2 supports. | ||
(1 − x)CeO2@xZrO2 supports were synthesized using CeO2 spheres (dispersed in anhydrous EtOH). Briefly, CeO2 spheres were dispersed in 39 g anhydrous EtOH, followed by the addition of 0.13 mL of 3.8 wt% Brij 30 solution under constant stirring at room temperature. The solution was stirred for an hour, followed by the addition of 0.132, 0.297, 0.509, and 1.189 mL of 0.1 M Zr(BuO)4, followed by overnight stirring to synthesize (1 − x)CeO2@xZrO2 with x = 0.1, 0.2, 0.3, 0.5, respectively. The solution was centrifuged and redispersed in 50 mL D.I. water. This step was repeated four times. The solution was then aged for 3 days at room temperature. The solid phase was separated, followed by drying (110 °C/overnight) and calcination (500 °C/2 h).
The reference Ce0.9Zr0.1O2 solid solution support was synthesized by co-precipitation as reported previously32 for an immediate comparison with 0.9CeO2@0.1ZrO2. Briefly, 72 mL of 0.1 M Ce(NO3)3 solution and 8 mL of 0.1 M ZrO(NO3)2 solution were mixed, resulting in a Ce/Zr precursor solution. This Ce/Zr precursor solution was added dropwise into 48 mL of NH4OH under stirring. The solution was stirred for 30 min. The formed precipitant was washed with 50 mL D.I. water followed by vacuum filtation. The obtained solids were dried (110 °C/overnight) and calcined (500 °C/2 h).
1.8 wt% Pt was deposited to (1 − x)CeO2@xZrO2 (x = 0.1, 0.2, 0.3, 0.5) and Ce0.9Zr0.1O2 by wet-impregnation. Specifically, 6.3 mL of 0.015 M Pt(NH3)4(NO3)2 solution was added to 1 g of (1 − x)CeO2@xZrO2 and Ce0.9Zr0.1O2 supports. The liquid was evaporated under stirring at room temperature, followed by drying (110 °C/overnight) and calcination (500 °C/2 h).
2.3. Material characterization
The Brunauer–Emmett–Teller (BET) surface areas of as-synthesized supports and catalysts were determined by nitrogen physisorption with a Micromeritics Tri-Star II surface area analyzer.The morphology of (1 − x)CeO2@xZrO2 (x = 0 and 0.3) supports was determined by HRTEM, STEM, and EDS techniques. HRTEM images were obtained at an accelerating voltage of 200 kV using a JEM-2010 equipment. The powdered samples were dispersed in anhydrous EtOH (HRTEM) and isopropyl alcohol (STEM, EDS) followed by ultrasonication. The obtained mixture was then added dropwise onto carbon-coated copper grids (Electron Microscopy Sciences, CF300-CU) followed by drying in air at room temperature. STEM images and EDS elemental maps of individual particles were collected using an FEI Talos F200X.
X-ray diffraction (XRD) patterns of (1 − x)CeO2@xZrO2 (x = 0, 0.1, 0.2, 0.3, 0.5) supports were collected using a Rigaku Ultima IV with a Cu Kα X-ray source. The data were collected from 2θ = 10 to 90° with a step size of 0.02° and scan speed of 2° min−1.
CO-pulse chemisorption experiments were conducted using a Micromeritics AutoChem II 2920 equipped with a thermal conductivity detector. An approximately 50 mg sample was loaded in a U-shaped quartz tube reactor. The catalysts were initially oxidized with 20% O2/Ar at 500 °C for 30 min. The catalysts were then cooled down to 250 °C in Ar, followed by purging with Ar for 30 min to remove physisorbed O2. The catalysts were then reduced with 10% H2/Ar for 30 min at 250 °C followed by purging with Ar for another 30 min to remove physisorbed H2. The reactor was immersed in a dry ice–ethanol mixture and cooled to −78 °C under a He flow to prevent CO adsorption on CeO2.35 CO pulses (10% CO/He, 0.5 cm3) were injected every 5 min until no CO consumption was observed. The total flow rate for the CO-pulse chemisorption experiment was maintained at 50 sccm (cm3 min−1 (STP)). The Pt particle size was calculated assuming all particles were equally sized hemispheres and a stoichiometry of CO/Pt = 1 was assumed.36
CO-DRIFTS experiments were performed as reported previously37 using a Nicolet iS50 FTIR spectrometer (Thermo Fisher) equipped with a high temperature reaction chamber (Harrick Praying Mantis). An approximately 25 mg sample was loaded in a sample holder and it was initially pretreated in Ar at 200 °C for 30 min. The sample was then cooled down to 25 °C in Ar, where background spectra were collected at a resolution of 4 cm−1 and 32 scans. Samples were then exposed to 1% CO/Ar for 30 min, followed by purging with Ar for another 30 min to remove gas phase CO. The total flow rate for the CO-DRIFTS experiments was 100 sccm.
The OSCC and ORR were measured using the low-temperature TWC test protocol defined by U.S. DRIVE in a customized reactor setup described in section 2.4.33 OSCC measurements on ∼25 mg of degreened catalyst were performed isothermally from 550 °C to 350 °C to 150 °C. The catalysts were initially exposed to 1.5% O2/Ar for 10 min, followed by switching to 0.2% CO/Ar for another 10 min. The switch from one gas stream to another was facilitated with an automated four-way valve for a smoother transition. Before exposure to O2 at each steady-state temperature, the catalyst surface was purged with Ar for 30 min. The OSCC was calculated by integrating the CO consumption from 0 to 10 min and the ORR was calculated from the slope of the CO concentration vs. time plot between 6 and 9 s after 0.2% CO/Ar was introduced to the feed stream. CO (m/z = 29) and Ar (m/z = 40) signals were recorded with a Pfeiffer Omnistar GSD 320 mass spectrometer with a 200 ms interval.
XPS spectra were recorded on a Kratos Axis Ultra XPS spectrometer equipped with an Al Kα (1486.6 eV) X-ray source at 200 W power and a pressure of 3.0 × 10−8 mbar. Survey scans were obtained between 0 and 1200 eV with a step size of 1 eV, a dwell time of 200 ms, and a pass energy of 140 eV averaged over 5 scans. Core-level region scans for Ce 3d, Zr 3d, C 1s, and O 1s were obtained at the corresponding binding energy ranges with a step size of 0.1 eV, an average dwell time of 260 ms, and a pass energy of 20 eV averaged over 5 scans. Data processing was performed using CasaXPS software employing Shirley-routine background subtraction and instrument-specific atomic sensitivity factors.
2.4. Catalyst evaluation
Catalyst evaluation was performed in a customized U-shaped quartz reactor, as reported previously.38 Briefly, 100 mg of catalyst powder diluted with 200 mg SiC was placed in the reactor tube stabilized by two loosely packed quartz wool plugs. The reactor was then placed in a cylindrical furnace surrounded by quartz wool to eliminate heat gradients. A K-type thermocouple was inserted into the top quartz wool plug in the U-shaped reactor to record the inlet gas temperature. Another K-type thermocouple was attached outside the reactor at the same height as the inlet temperature thermocouple to allow the PID temperature controller to control the furnace temperature. The gases used to simulate the TWC performance were purchased from Airgas and were the following: 99.999% CO2, 99.999% O2 (UHP), 10.01% H2/Ar, 10.1% CO/Ar, 1% NO/Ar, 5% C2H4/Ar, 5% C3H6/Ar, 1% C3H8/Ar, and 99.999% Ar (UHP). All gases were regulated by a set of MKS mass flow controllers. Liquid i-C8H18 was placed in a bubbler immersed in a controlled temperature (0 °C) water bath and gas phase i-C8H18 was carried by Ar to the reactor. The Clausius–Clapeyron equation and the vapor pressure of i-C8H18 (1.78 kPa at 0 °C)39 were used to calculate the Ar flow required to carry i-C8H18 from the bubbler to the reactor. Water was supplied to the reactor (0.027 mL min−1) by a D-series pump (Teledyne Isco) and steam was generated in a tube furnace set at 200 °C that was carried by Ar to the reactor. The catalytic performance experiments were conducted from 100 to 500 °C at a heating rate of 2 °C min−1. The temperature was recorded every second using a LabView program. The total flow rate was maintained to 333 sccm, corresponding to a gas hourly space velocity of 142500 h−1. The reactor outlet gas was diluted with 1332 sccm Ar and analyzed using an MKS MultiGas 2030 FTIR gas analyzer operating at 191 °C. The reactor gas lines were heated at 170 °C to avoid water condensation.
Catalysts were evaluated after degreening (DG) and redox hydrothermal aging (HTA). Degreening of the catalysts was conducted at 700 °C for 4 h under 10% H2O, 10% CO2, Ar balance. Redox HTA was conducted at 800 °C for 10 h at a switching frequency of 0.1 Hz between a lean (5% O2, 10% H2O, 10% CO2, Ar balance) and a rich (3% CO, 1% H2, 10% H2O, 10% CO2, Ar balance) gas stream. Prior to evaluating the DG and HTA catalysts, the catalysts were pretreated at 600 °C (10% H2O, 13% CO2, Ar balance) for 20 min. Simulated TWC oxidation experiments were performed using the low temperature oxidation catalyst test protocol stoichiometric gasoline direct injection (S-GDI) gas composition defined by U.S. DRIVE33 (13% CO2, 10% H2O, stoichiometric O2, 1670 ppm H2, 5000 ppm CO, 1000 ppm NO, 700 ppm C2H4, 1000 ppm C3H6, 300 ppm C3H8, 1000 ppm i-C8H18, hydrocarbons in C1 basis, Ar balance).
3. Results and discussion
3.1. Surface characterization
The BET surface areas and N2 adsorption/desorption isotherms of (1 − x)CeO2@xZrO2 (where x = 0, 0.1, 0.2, 0.3, and 0.5) and Ce0.9Zr0.1O2 solid solution supports are summarized in Table S1 and Fig. S1,† respectively. All (1 − x)CeO2@xZrO2 supports have surface areas of 69.3–95 m2 g−1 and type II isotherms, characteristic of non-porous or macroporous materials.40 The solid solution Ce0.9Zr0.1O2 has a surface area of 77.2 m2 g−1 and a type IV isotherm, characteristic of mesoporous materials.The morphology of synthesized CeO2 is spherical (Fig. S2†) with a uniform diameter of ∼160 nm. Fig. 1(a, b and d) show the HRTEM image and EDS maps of the 0.7CeO2@0.3ZrO2 support. The EDS elemental maps of Ce and Zr suggest that the ZrO2 overlayer covers the surface of CeO2 uniformly. Fig. 1(c) shows that ZrO2 is enriched in the shell layer of 0.7CeO2@0.3ZrO2, while the Ce signal is more dominant in the core and decreases when approaching the shell, suggesting that a core@shell structure was prepared. However, an uneven ZrO2 coating was formed (Fig. S3†) when the Zr molar concentration increased from 0.3 (0.7CeO2@0.3ZrO2) to 0.5 (0.5CeO2@0.5ZrO2), attributed to excess ZrO2 that cannot be coated evenly.
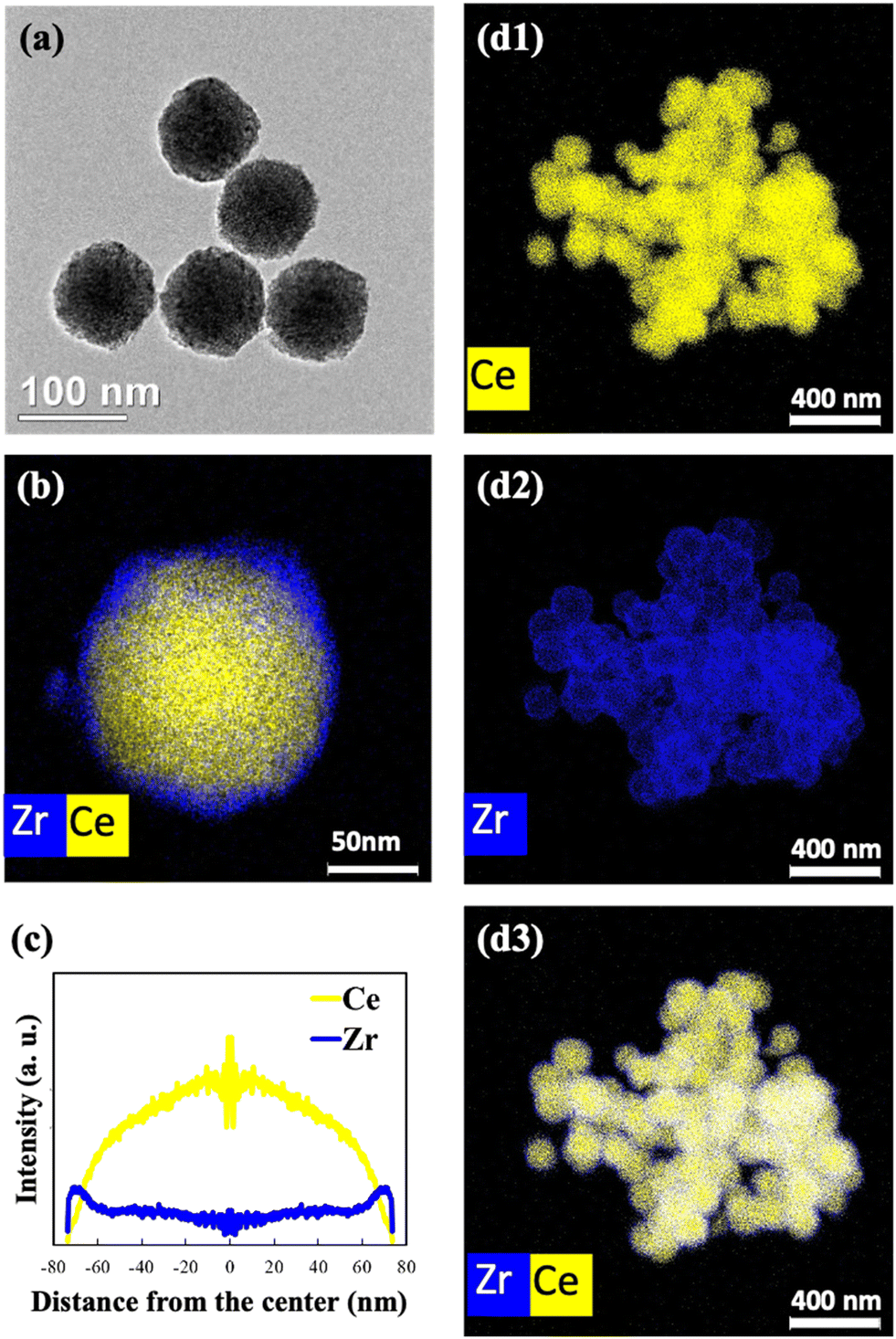 | ||
| Fig. 1 (a) HRTEM image, (b and d) EDS elemental maps of Ce and Zr of the 0.7CeO2@0.3ZrO2 support and (c) Ce and Zr elemental distributions obtained by converting the brightness in intensity from the EDS elemental map in (b). | ||
XRD spectra of the (1 − x)CeO2@xZrO2 supports (Fig. 2) show that CeO2 spheres possess peaks at 2θ of 28.5° and 33.1° that correspond to the (111) and (200) planes of cubic CeO2, respectively.41 Increasing the Zr molar concentration to values ≥0.2 led to the appearance of additional peaks at 30.2 and 35.2°, which correspond to the (111) and (200) planes of tetragonal ZrO2, respectively.42,43
 | ||
| Fig. 2 XRD patterns of (1 − x)CeO2@xZrO2 (x = 0, 0.1, 0.2, 0.3, 0.5) supports (t: tetragonal). | ||
The average Pt particle size and dispersion of 1.8 wt% Pt/(1 − x)CeO2@xZrO2 were obtained by CO-pulse chemisorption (Fig. 3). The Pt/CeO2 sphere showed an average Pt particle size of 7.7 nm, corresponding to a Pt dispersion of 13.5%. Similar average Pt particle sizes and dispersions were observed over Pt/(1 − x)CeO2@xZrO2 with x ≤ 0.2. Specifically, the average Pt particle size of Pt/0.9CeO2@0.1ZrO2 and Pt/0.8CeO2@0.2ZrO2 was 6.8 nm (Pt dispersion: 15.4%) and 7.2 nm (Pt dispersion: 15.7%), respectively. The similar Pt particle size and dispersion observed at low Zr molar concentrations suggest the existence of strong metal–support interactions between Pt and CeO2.44 An increase in Zr molar concentration to 0.3 and 0.5 in Pt/(1 − x)CeO2@xZrO2 led to an increase in the average Pt particle size to 9.9 nm (Pt dispersion: 11.4%) and 14.3 nm (Pt dispersion: 7.9%), respectively.
 | ||
| Fig. 3 Average Pt particle size and dispersion of 1.8 wt% Pt/(1 − x)CeO2@xZrO2 catalysts obtained by CO-pulse chemisorption. | ||
CO-DRIFTS experiments conducted over Pt/(1 − x)CeO2@xZrO2 catalysts (Fig. 4) showed that Pt/(1 − x)CeO2@xZrO2 contains two types of Pt species. The high wavenumber peak (2097 cm−1) is attributed to CO adsorbed on ionic Pt2+ strongly interacting with CeO2.45,46 The low wavenumber peak (2087 cm−1) is attributed to CO adsorbed on Pt0 particles.47,48 The 2097 cm−1 and 2087 cm−1 peaks are more pronounced at low (x ≤ 0.1) and high (x > 0.1) Zr molar concentrations, respectively. This observation suggests that increasing the molar concentration of Zr can hinder the occurrence of Pt2+ species that can strongly interact with CeO2. Instead, Pt0 particles are formed, consistent with the CO-pulse chemisorption results that show a decreased Pt dispersion with increasing Zr molar concentration.
 | ||
| Fig. 4 CO-DRIFTS over fresh 1.8 wt% Pt/(1 − x)CeO2@xZrO2 (x = 0, 0.1, 0.2, 0.3, 0.5) catalysts. | ||
The XPS survey of 0.9CeO2@0.1ZrO2 (Fig. S4†) shows the presence of 16.6 atomic% of Zr, suggesting the successful deposition of ZrO2 on the surface of 0.9CeO2@0.1ZrO2. The XPS results for Ce 3d of the CeO2 spheres and 0.9CeO2@0.1ZrO2 supports are shown in Fig. 5. The peaks labeled Vo, V′, u0, and u′ correspond to Ce3+ and peaks labeled u, u′′, u′′′, v, v′′, and v′′′ correspond to Ce4+.49 The peak area that belongs to Ce3+ is 13.0% and 17.2 % for CeO2 spheres and 0.9CeO2@0.1ZrO2, respectively, suggesting that Zr incorporation leads to a higher Ce3+/Ce4+ ratio. It was reported that oxygen vacancies from nano-sized CeO2 are associated with the presence of Ce3+ on the surface resulting in a higher Ce3+/Ce4+ ratio compared to bulk CeO2.50 Therefore, the increase of Ce3+/Ce4+ ratio in 0.9CeO2@0.1ZrO2 may result in a higher oxygen storage capacity and oxygen release rate (discussed in section 3.2).
 | ||
| Fig. 5 XPS results for Ce 3d of (a) CeO2 and (b) 0.9CeO2@0.1ZrO2. | ||
3.2. TWC performance
The temperatures that 50% and 90% CO, total HCs (THCs) and NOx conversions achieved (T50 and T90, respectively) over Pt/(1 ∼ x)CeO2@xZrO2 (DG) catalysts are summarized in the bar chart shown in Fig. 6 and the light off curves shown in Fig. S5.†T90s for CO/THCs/NOx were achieved at 207 °C/244 °C/256 °C over Pt/CeO2 spheres (DG), respectively. The performance improved with the incorporation of 0.1 molar concentration of Zr. Specifically, Pt/0.9CeO2@0.1ZrO2 (DG) achieved T90s for CO (188 °C), THCs (229 °C), and NOx (240 °C) at a lower temperature compared to Pt/CeO2 spheres (DG). Further, Pt/0.8CeO2@0.2ZrO2 (DG) showed a slightly improved performance compared to Pt/0.9CeO2@0.1ZrO2 (DG) with T90s achieved at 185 °C, 223 °C, and 234 °C for CO, THCs, and NOx, respectively. However, a further increase in Zr molar concentration to ≥0.3 led to a decrease in the catalytic performance with a shift in T90s to higher temperatures. For instance, the T90s achieved over Pt/0.7CeO2@0.3ZrO2 (DG) and Pt/0.5CeO2@0.5ZrO2 (DG) were 206 °C/245 °C/254 °C and 216 °C/255 °C/268 °C for CO/THCs/NOx, respectively. The decreased performance of the catalysts with Zr loadings ≥0.3 can be attributed to sintering of Pt particles on crystallized ZrO2. Furthermore, the activity of 1.8 wt% Pt/Ce0.9Zr0.1O2 (DG) was compared to 1.8 wt% Pt/0.9CeO2@0.1ZrO2 (DG) (Fig. S6a†). The results indicate that Pt/0.9CeO2@0.1ZrO2 (DG) outperformed Pt/Ce0.9Zr0.1O2 (DG), with the latter achieving T90s of CO/THCs/NOx at 205 °C/248 °C/260 °C, respectively. | ||
| Fig. 6 Comparison of T50,90s of CO, THCs and NOx over DG 1.8 wt% Pt/(1 ∼ x)CeO2@xZrO2 (x = 0, 0.1, 0.2, 0.3, 0.5) catalysts; each bar represents the average T50,90s of three different batches of the catalyst and the error bars represent the standard deviation of the T50,90s. | ||
The oxygen storage/release properties of the catalyst are crucial in near stoichiometric TWC reactions. Therefore, the OSCC and ORR of Pt/(1 − x)CeO2@xZrO2 (DG) were measured and correlated with the catalytic performance results (Fig. 7 and S7†). The Pt/CeO2 sphere had an OSCC and ORR of 155 μmol g−1 and 4.7 μmol g−1 s−1, respectively. The OSCC and ORR were improved when a small amount of ZrO2 (x = 0.1, 0.2) was deposited on CeO2 spheres. Specifically, Pt/0.9CeO2@0.1ZrO2 and Pt/0.8CeO2@0.2ZrO2 had a greater OSCC (259 and 246 μmol g−1, respectively) and ORR (5.2 and 5.1 μmol g−1 s−1, respectively) compared to Pt/CeO2 spheres. However, further addition of Zr (molar concentrations >0.2) decreased the OSCC and ORR. Specifically, the OSCC decreased to 211 and 185 μmol g−1 and the ORR decreased to 4.6 and 4.1 μmol g−1 s−1 for Zr molar concentrations of 0.3 and 0.5, respectively. Fig. 7 shows that an increase in OSCC and ORR was accompanied by a decrease in T50s and vice versa, implying that optimization of the OSCC and ORR of Pt/(1 − x)CeO2@xZrO2 (DG) by varying the Zr molar concentrations can tune the TWC performance. The ORR of bare supports was measured at three different temperatures (150, 350, and 550 °C). 0.7CeO2@0.3ZrO2 showed an enhanced ORR compared to CeO2 spheres at 150 °C and a similar ORR compared to CeO2 spheres at 350 and 550 °C (Fig. S8†). Moreover, 0.7CeO2@0.3ZrO2 and the Ce0.7Zr0.3O2 solid solution showed a similar ORR at 150 °C, while 0.7CeO2@0.3ZrO2 showed a greater ORR than the Ce0.7Zr0.3O2 solid solution at 350 and 550 °C.
 | ||
| Fig. 7 T 50s of CO, THCs, NOx (left) and OSCC and ORR (right) of 1.8 wt% Pt/(1 − x)CeO2@xZrO2 (DG) catalysts. | ||
The hydrothermal stability of Pt/(1 − x)CeO2@xZrO2 was assessed after redox HTA was conducted, followed by a TWC performance evaluation. The evaluation results of Pt/(1 − x)CeO2@xZrO2 (HTA) catalysts are summarized in Fig. 8 and Fig. S9.† All catalysts showed higher T50,90s compared to their DG states (Fig. 6), suggesting that the catalysts deactivate after redox HTA. For example, the Pt/CeO2 sphere (HTA) reached T90s for CO/THCs/NOx at 222 °C/252 °C/258 °C, which are 15 °C/8 °C/3 °C higher than the T90s achieved over Pt/CeO2 sphere (DG), respectively. Deposition of the Zr shell with a Zr molar concentration <0.2 alleviated the deactivation caused by redox HTA and improved the catalytic performance compared to the Pt/CeO2 sphere (HTA). For instance, Pt/0.9CeO2@0.1ZrO2 (HTA) achieved lower T90s for CO (207 °C), THCs (237 °C), and NOx (244 °C) compared to Pt/CeO2 (HTA) with T90s for CO/THCs/NOx achieved at 222 °C/252 °C/258 °C, respectively. Moreover, Pt/0.9CeO2@0.1ZrO2 (HTA) outperformed Pt/0.8CeO2@0.2ZrO2 (HTA) (CO/THCs/NOxT90s achieved at 223 °C/251 °C/258 °C, respectively). A further increase in Zr molar concentrations to 0.3 and 0.5 led to higher T90s for Pt/0.7CeO2@0.3ZrO2 (HTA) (239 °C (CO)/270 °C (THCs)/279 °C (NOx)) and Pt/0.5CeO2@0.5ZrO2 (HTA) (252 °C (CO)/289 °C (THCs)/292 °C (NOx)). Finally, Pt/0.9CeO2@0.1ZrO2 (HTA) outperformed the Pt/Ce0.9Zr0.1O2 (HTA) (Fig. S6†) solid solution catalyst, with the latter achieving T90s at 220 °C/262 °C/263 °C for CO/THCs/NOx, respectively. This behavior indicates the advantage of the formation of the (1 − x)CeO2@xZrO2 structured support compared to the bare CeO2 support. TEM images of redox HTA Pt/0.9CeO2@0.1ZrO2 (Fig. S10†) showed mild sintering of the spherical support, while no large Pt nanoparticles were identified, which is consistent with the CO-DRIFTS results (Fig. 4). Pt/CeO2 and Pt/0.9CeO2@0.1ZrO2 showed the lowest ΔT50s (= T50,HTA – T50,DG) (Fig. S11†), suggesting the least deactivation after redox HTA compared to the rest of the studied catalysts. The ΔT50s increased when the Zr molar concentration increased to values ≥0.2, indicating that a thick ZrO2 coating layer does not favor stability.
 | ||
| Fig. 8 Comparison of T50,90s of CO, THCs and NOx for 1.8 wt% Pt/(1 − x)CeO2@xZrO2 (HTA) (x = 0, 0.1, 0.2, 0.3, 0.5) catalysts; each bar represents the average T50,90s of the three different batches of the catalyst and the error bars represent the standard deviation of the T50,90s. | ||
4. Conclusions
A simulated S-GDI gas mixture was used for the TWC evaluation of a series of Pt/(1 − x)CeO2@xZrO2 catalysts. Pt/CeO2 spheres (DG) achieved T90s at 207, 244, and 256 °C for CO, THCs, and NOx, respectively. However, deactivation was observed after HTA with the T90s of the Pt/CeO2 sphere (HTA) increased to 222, 252, and 258 °C for CO, THCs, and NOx, respectively. With the incorporation of 0.1 molar concentration of Zr, Pt/0.9CeO2@0.1ZrO2 showed the most promising performance considering both DG and HTA evaluation results by achieving the lowest T90s for CO, THCs, and NOx at 207, 237 and 244 °C (HTA), respectively, compared to all studied Pt/(1 − x)CeO2@xZrO2 catalysts. Tuning the Zr molar concentration and thus the Ce:Zr molar ratio of the Pt/(1 − x)CeO2@xZrO2 TWCs affected their low temperature catalytic activity by altering their OSCC and ORR. Specifically, improved OSCC (259 μmol g−1) and ORR (5.2 μmol g−1 s−1) values were observed over Pt/0.9CeO2@0.1ZrO2 (DG), while increasing the Zr molar concentration further led to a decrease in OSCC and ORR. Furthermore, sintering of Pt can be minimized (average Pt particle size of 6.8 nm) over Pt/0.9CeO2@0.1ZrO2 compared to an excess amount of zirconium (x > 0.2), which contains thick ZrO2 shell that interacts weakly with Pt. Overall, this work highlights the potential of Pt-only catalysts supported on (1 − x)CeO2@xZrO2 supports as TWCs and reveals that an improved TWC performance can be achieved by incorporating moderate amounts of zirconium (Zr molar concentration = 0.1).
Data availability
The data supporting this article have been included as part of the ESI.† Any additional data are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by start-up funding from the Department of Chemical and Biological Engineering, University at Buffalo (UB), The State University of New York (SUNY). A portion of this research was sponsored by the U. S. Department of Energy's Vehicle Technology Office, with particular thanks to Gurpreet Singh, Siddiq Khan, and Nicholas Hansford of the Offroad, Rail, Marine and Aviation (ORMA). Additionally, the FEI Talos F200X STEM was provided by the Department of Energy, Office of Nuclear Energy, Fuel Cycle R&D Program and the Nuclear Science User Facilities.References
- J. Chen, C.-H. Liu, H. N. Pham, T. J. Toops, A. K. Datye and E. A. Kyriakidou, Designing ultrastable Pt/CeO2-Al2O3 nanosheet catalysts for three-way catalysts applications, Chem. Eng. J., 2023, 477, 147086 CrossRef CAS.
- M. Weilenmann, J.-Y. Favez and R. Alvarez, Cold-start emissions of modern passenger cars at different low ambient temperatures and their evolution over vehicle legislation categories, Atmos. Environ., 2009, 43(15), 2419–2429 CrossRef CAS.
- J. Lee, J. R. Theis and E. A. Kyriakidou, Vehicle emissions trapping materials: Successes, challenges, and the path forward, Appl. Catal., B, 2019, 243, 397–414, DOI:10.1016/j.apcatb.2018.10.069.
- A. P. Wong, E. A. Kyriakidou, T. J. Toops and J. R. Regalbuto, The catalytic behavior of precisely synthesized Pt–Pd bimetallic catalysts for use as diesel oxidation catalysts, Catal. Today, 2016, 267, 145–156 CrossRef CAS.
- E. Kyriakidou, T. J. Toops, J.-S. Choi, M. J. Lance and J. E. Parks II Exhaust treatment catalysts with enhanced hydrothermal stability and low-temperature activity, US Pat., 1042137 B2 (October 1, 2019) Search PubMed.
- M. Zammit, C. L. DiMaggio, C. H. Kim, C. Lambert, G. G. Muntean, C. H. Peden, J. E. Parks and K. Howden, Future automotive aftertreatment solutions: The 150 C challenge workshop report (US Drive Workshop), 2012, https://www.pnnl.gov/main/publications/external/technical_Reports/PNNL-22815.pdf Search PubMed.
- S. B. Kang, J. B. Lim, D. Jo, I.-S. Nam, B. K. Cho, S. B. Hong, C. H. Kim and S. H. Oh, Ostwald-ripening sintering kinetics of Pd-based three-way catalyst: Importance of initial particle size of Pd, Chem. Eng. J., 2017, 316, 631–644 CrossRef CAS.
- C. Wang, J. Tan, G. Harle, H. Gong, W. Xia, T. Zheng, D. Yang, Y. Ge and Y. Zhao, Ammonia formation over Pd/Rh three-way catalysts during lean-to-rich fluctuations: The Effect of the catalyst aging, exhaust temperature, lambda, and duration in rich conditions, Environ. Sci. Technol., 2019, 53(21), 12621–12628 CrossRef CAS PubMed.
- A. A. Vedyagin, R. M. Kenzhin, M. Y. Tashlanov, E. A. Alikin, V. O. Stoyanovskii, P. E. Plyusnin, Y. V. Shubin, I. V. Mishakov, M. Y. Smirnov and A. V. Kalinkin, Effect of La addition on the performance of three-way catalysts containing palladium and rhodium, Top. Catal., 2020, 63, 152–165 CrossRef CAS.
- J. A. Anderson, R. A. Daley, S. Y. Christou and A. M. Efstathiou, Regeneration of thermally aged Pt-Rh/CexZr1− xO2-Al2O3 model three-way catalysts by oxychlorination treatments, Appl. Catal., B, 2006, 64(3–4), 189–200 CrossRef CAS.
- J. G. Nunan, H. J. Robota, M. J. Cohn and S. A. Bradley, Physicochemical properties of Ce-containing three-way catalysts and the effect of Ce on catalyst activity, J. Catal., 1992, 133(2), 309–324 CrossRef CAS.
- J. Wang, H. Chen, Z. Hu, M. Yao and Y. Li, A review on the Pd-based three-way catalyst, Catal. Rev.: Sci. Eng., 2015, 57(1), 79–144 CrossRef CAS.
- V. V. Eskina, O. A. Dalnova, D. G. Filatova, V. B. Baranovskaya and Y. A. Karpov, Direct precise determination of Pd, Pt and Rh in spent automobile catalysts solution by high-resolution continuum source graphite furnace atomic absorption spectrometry, Spectrochim. Acta, Part B, 2020, 165, 105784 CrossRef CAS.
- I. Mejía-Centeno, A. Martínez-Hernández and G. A. Fuentes, Effect of low-sulfur fuels upon NH3 and N2O emission during operation of commercial three-way catalytic converters, Top. Catal., 2007, 42(1), 381–385 CrossRef.
- Metals Daily, https://www.metalsdaily.com/accessed Search PubMed.
- Y. Malyukin, P. Maksimchuk, V. Seminko, E. Okrushko and N. Spivak, Limitations of self-regenerative antioxidant ability of nanoceria imposed by oxygen diffusion, J. Phys. Chem. C, 2018, 122(28), 16406–16411 CrossRef CAS.
- L. Sun, W. Xiao, X. Hao, Q. Meng and M. Zhou, A first-principles study on the structural, thermal and electronic properties of cerium oxides by using different functionals, Electron. Struct., 2018, 1(1), 015003 CrossRef.
- P. Li, X. Chen, Y. Li and J. W. Schwank, A review on oxygen storage capacity of CeO2-based materials: Influence factors, measurement techniques, and applications in reactions related to catalytic automotive emissions control, Catal. Today, 2019, 327, 90–115 CrossRef CAS.
- F. Dong, T. Tanabe, N. Takahashi and H. Shinjoh, Investigation of the effective oxygen storage and release performances on the Pt/CeO2-ZrO2 catalysts by breakthrough method, Catal. Today, 2019, 332, 259–266 CrossRef CAS.
- C. Paun, O. V. Safonova, J. Szlachetko, P. M. Abdala, M. Nachtegaal, J. Sa, E. Kleymenov, A. Cervellino, F. Krumeich and J. A. van Bokhoven, Polyhedral CeO2 nanoparticles: size-dependent geometrical and electronic structure, J. Phys. Chem. C, 2012, 116(13), 7312–7317 CrossRef CAS.
- M. V. Twigg, Roles of catalytic oxidation in control of vehicle exhaust emissions, Catal. Today, 2006, 117(4), 407–418 CrossRef CAS.
- T. Suzuki and M. Ichiyanagi, Robust control design for air-fuel ratio fluctuation of gasoline engine (1st report: Development of feed-forward controller with heat transfer model at intake), Journal of Japan Society for Design Engineering, 2018, 53(5), 377–390 Search PubMed.
- J. A. Cook, I. V. Kolmanovsky, D. McNamara, E. C. Nelson and K. V. Prasad, Control, computing and communications: technologies for the twenty-first century model T, Proc. IEEE, 2007, 95(2), 334–355 Search PubMed.
- O. Barbarisi, A. Gaeta and L. Glielmo, An extended kalman observer for the in-cylinder air mass flow estimation, in Proceedings of MECA02 International Workshop on Diagnostics in Automotive Engines and Vehicles, Fisciano SA, Italy, 2002 Search PubMed.
- Q. Dong, S. Yin, C. Guo and T. Sato, Aluminum-doped ceria-zirconia solid solutions with enhanced thermal stability and high oxygen storage capacity, Nanoscale Res. Lett., 2012, 7(1), 1–5 CrossRef PubMed.
- X. Yang, L. Yang, S. Lin and R. Zhou, Investigation on properties of Pd/CeO2–ZrO2–Pr2O3 catalysts with different Ce/Zr molar ratios and its application for automotive emission control, J. Hazard. Mater., 2015, 285, 182–189 CrossRef CAS PubMed.
- R. Wang, L. Lan, M.-C. Gong and Y.-Q. Chen, Catalytic Combustion of Gasoline Particulate Soot over CeO2-ZrO2 Catalysts, Acta Phys. Chim. Sin., 2016, 32(7), 1747–1757 CAS.
- J. Schoenhaber, J. M. Richter, J. Despres, M. Schmidt, S. Spiess and M. Roesch, Advanced TWC Technology to Cover Future Emission Legislations, SAE Tech. Pap., 2015 Search PubMed.
- C. Larese, M. L. Granados, F. C. Galisteo, R. Mariscal and J. Fierro, TWC deactivation by lead: a study of the Rh/CeO2 system, Appl. Catal., B, 2006, 62(1–2), 132–143 CrossRef CAS.
- L. S. Kibis, D. A. Svintsitskiy, E. A. Derevyannikova, T. Y. Kardash, E. M. Slavinskaya, O. A. Stonkus, V. A. Svetlichnyi and A. I. Boronin, From highly dispersed Rh3+ to nanoclusters and nanoparticles: Probing the low-temperature NO+CO activity of Rh-doped CeO2 catalysts, Appl. Surf. Sci., 2019, 493, 1055–1066 CrossRef CAS.
- M. M. Natile and A. Glisenti, Nanostructured CeO2 powders by XPS, Surf. Sci. Spectra, 2006, 13(1), 17–30 CrossRef CAS.
- J. Chen, B. D. Carlson, T. J. Toops, Z. Li, M. J. Lance, S. Karakalos, J.-S. Choi and E. Kyriakidou, Methane combustion over Ni/CexZr1-xO2 catalysts: impact of ceria/zirconia ratio, ChemCatChem, 2020, 12(21), 5558–5568 CrossRef CAS.
- K. G. Rappé, C. DiMaggio, J. A. Pihl, J. R. Theis, S. H. Oh, G. B. Fisher, J. Parks, V. G. Easterling, M. Yang and M. L. Stewart, Aftertreatment Protocols for Catalyst Characterization and Performance Evaluation: Low-Temperature Oxidation, Storage, Three-Way, and NH3-SCR Catalyst Test Protocols, Emiss. Control Sci. Technol., 2019, 5(2), 183–214 CrossRef.
- Q. Fang and X. Liang, CeO2–Al2O3, CeO2–SiO2, CeO2–TiO2 core-shell spheres: formation mechanisms and UV absorption, RSC Adv., 2012, 2(12), 5370–5375 RSC.
- M. Li, J. Deng, X. Yin, W. Wang, Y. Zhao, H. Xu, J. Wang and Y. Chen, Effect of lauric acid on the grain growth of CeO2-ZrO2-Y2O3-La2O3 in different periods, J. Alloys Compd., 2022, 894, 162301 CrossRef CAS.
- H. Nassiri, K. E. Lee, Y. Hu, R. E. Hayes, R. W. Scott and N. Semagina, Platinum Inhibits Low-Temperature Dry Lean Methane Combustion through Palladium Reduction in Pd− Pt/Al2O3: An In Situ X-ray Absorption Study, ChemPhysChem, 2017, 18(2), 238–244 CrossRef CAS PubMed.
- J. Chen, K. Giewont, E. A. Walker, J. Lee, Y. Niu and E. A. Kyriakidou, Cobalt-Induced PdO Formation in Low-Loading Pd/BEA Catalysts for CH4 Oxidation, ACS Catal., 2021, 11(21), 13066–13076 CrossRef CAS.
- C.-H. Liu, J. Chen, T. J. Toops, J.-S. Choi, C. Thomas, M. J. Lance and E. A. Kyriakidou, Hydrothermally stable Pd/SiO2@Zr Core@Shell catalysts for diesel oxidation applications, Chem. Eng. J., 2021, 130637 CrossRef CAS.
- R. Oktavian, V. Amidelsi, A. Rasmito and G. Wibawa, Vapor pressure measurements of ethanol–isooctane and 1-butanol–isooctane systems using a new ebulliometer, Fuel, 2013, 107, 47–51 CrossRef CAS.
- K. S. Sing, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984), Pure Appl. Chem., 1985, 57(4), 603–619 CrossRef CAS.
- J. Xu, M. Li, J. Qiu, X.-F. Zhang, Y. Feng and J. Yao, PEGylated deep eutectic solvent-assisted synthesis of CdS@CeO2 composites with enhanced visible light photocatalytic ability, Chem. Eng. J., 2020, 383, 123135 CrossRef CAS.
- R. Zhang, H. Liu and D. He, Pure monoclinic ZrO2 prepared by hydrothermal method for isosynthesis, Catal. Commun., 2012, 26, 244–247 CrossRef CAS.
- M. R. Loghman-Estark, R. S. Razavi and H. Edris, Synthesis and thermal stability of nontransformable tetragonal (ZrO2) 0.96 (REO1. 5) 0.04 (RE= Sc3+, Y3+) nanocrystals, in Defect and Diffusion Forum, Trans Tech Publ, 2013, vol. 334, pp. 60–64 Search PubMed.
- Q. Yang, L. Li, X. Wang and Y. Ma, Tunable metal-support interaction of Pt/CeO2 catalyst via surfactant-assisted strategy: Insight into the total oxidation of CO and toluene, J. Hazard. Mater., 2022, 424, 127601 CrossRef CAS PubMed.
- W. Tan, H. Alsenani, S. Xie, Y. Cai, P. Xu, A. Liu, J. Ji, F. Gao, L. Dong and E. Chukwu, Tuning Single-atom Pt1−CeO2 Catalyst for Efficient CO and C3H6 Oxidation: Size Effect of Ceria on Pt Structural Evolution, ChemNanoMat, 2020, 6(12), 1797–1805 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard and L. Kovarik, Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation, Science, 2017, 358(6369), 1419–1423 CrossRef CAS PubMed.
- W. Ruettinger, X. Liu, X. Xu and R. J. Farrauto, Effect of Mo and Re Promoters on the Activity and Stability of a Pt/ZrO2 Water-Gas Shift Catalyst (Part 1), Top. Catal., 2008, 51(1–4), 60–67 CrossRef CAS.
- A. Litke, H. Frei, E. J. Hensen and J. P. Hofmann, Interfacial charge transfer in Pt-loaded TiO2 P25 photocatalysts studied by in-situ diffuse reflectance FTIR spectroscopy of adsorbed CO, J. Photochem. Photobiol., A, 2019, 370, 84–88 CrossRef CAS.
- K. Reed, A. Cormack, A. Kulkarni, M. Mayton, D. Sayle, F. Klaessig and B. Stadler, Exploring the properties and applications of nanoceria: is there still plenty of room at the bottom?, Environ. Sci.: Nano, 2014, 1(5), 390–405 RSC.
- V. Nicolini, E. Gambuzzi, G. Malavasi, L. Menabue, M. C. Menziani, G. Lusvardi, A. Pedone, F. Benedetti, P. Luches and S. D'Addato, Evidence of catalase mimetic activity in Ce3+/Ce4+ doped bioactive glasses, J. Phys. Chem. B, 2015, 119(10), 4009–4019 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00921e |
| ‡ This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| § Current address: SUNCAT Center for Interface Science and Catalysis, Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA |
| This journal is © The Royal Society of Chemistry 2025 |