
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium complexes supported on cross-linked polyacrylic acid as release-and-catch catalysts for continuous formic acid dehydrogenation†
Keito
Sawahara
ab,
Shinji
Tanaka
 b,
Ryota
Gemma
c,
Ryoichi
Kanega
d and
Hajime
Kawanami
*ab
b,
Ryota
Gemma
c,
Ryoichi
Kanega
d and
Hajime
Kawanami
*ab
aInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology, Tsukuba, 305-8565, Japan. E-mail: h-kawanami@aist.go.jp
bGraduate School of Pure and Applied Science, University of Tsukuba, Tsukuba, 305-8577, Japan
cDepartment of Applied Chemistry, School of Engineering, Tokai University, Kanagawa 259-1292, Japan
dResearch Institute for Energy Conservation, National Institute of Advanced Industrial Science and Technology, 305-8565, Japan
First published on 18th November 2024
Abstract
Although homogeneous catalysts outperform heterogenous catalysts for formic acid (FA) dehydrogenation, complicated energy-intensive processes are required for catalyst recovery. Herein, we developed an iridium complex immobilized on cross-linked polyacrylic acid as a “release-and-catch” catalyst. During catalysis, the immobilized iridium complex dissolved in the reaction solution and was subsequently recaptured by the support. The activity of this catalyst for FA dehydrogenation (turnover frequency: 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1; turnover number: 2180000) was comparable to that of homogeneous catalysts. Furthermore, the catalyst was recycled 10 times with near-perfect recovery in a flow-type reaction. These findings offer a promising approach for advancing formic acid-based hydrogen energy carriers.
000 h−1; turnover number: 2180000) was comparable to that of homogeneous catalysts. Furthermore, the catalyst was recycled 10 times with near-perfect recovery in a flow-type reaction. These findings offer a promising approach for advancing formic acid-based hydrogen energy carriers.
Introduction
The urgent challenge of global warming has intensified efforts to develop technologies that reduce dependence on fossil fuels and promote the use of clean hydrogen (H2) as an alternative energy source.1 However, H2 presents several challenges, including a low energy density per unit volume, leakage, and equipment design issues.2 To overcome these drawbacks, liquid organic hydrogen carriers (LOHCs)3,4 with high H2 densities, such as methyl cyclohexane (MCH)5–7 and ammonia,8–11 have been proposed. For H2 storage and production, formic acid (FA) is of particular interest owing to its low flammability, safe storage and transport characteristics, minimal environmental impact, high hydrogen content, and low reaction energy (eqn 1).12–15| HCOOH(liquid) → H2(gas) + CO2(gas) ΔH = +31 kJ mol−1, ΔG = −33 kJ mol−1 (T = 298 K) | (1) |
In recent years, numerous homogeneous catalysts based on Ir and Ru complexes have been reported for formic acid dehydrogenation (FA dehydrogenation).16–25 For example, we demonstrated the facile generation of high-pressure gas (H2 + CO2) exceeding 150 MPa through FA dehydrogenation using homogenous Ir complex catalysts26,27 and the subsequent easy separation of H2 and CO2.28 Advantageously, these homogeneous catalysts offer higher turnover frequencies (TOFs) and turnover numbers (TONs) than heterogenous catalysts.29 However, the use of costly and rare precious metals in highly active homogeneous catalysts requires post-reaction separation and recovery, which remains challenging due to the numerous steps and energy demands involved in the recycling process.30,31 Additionally, many processes use aqueous FA solutions instead of concentrated or neat FA (e.g., >98%), which complicates the separation, recovery, and reuse of dissolved catalysts.32 For example, 1,2,3,4,5-pentamethyl-2,4-cyclopentadienyl iridium (Cp*Ir) complex catalyst with 4,7-dihydroxy-1,10-phenanthroline ligand shows pH-dependent solubility in water, allowing catalyst recovery; however, a recovery rate of only 90% leads to inevitable catalyst loss.33
To solve these problems, immobilized catalysts that do not dissolve in water or other media are being investigated.34 However, immobilized catalysts face challenges such as decreased activity compared with homogeneous catalysts35–38 and metal complex leaching.38–41 Recently, we developed an immobilized catalyst suitable for FA dehydrogenation and successfully limited metal complex leaching to less than 5% by introducing vacant ligands into the support.37 Unfortunately, the activity of the immobilized catalyst was approximately 60% less than that of the free metal complex. As a result, enhancing catalyst activity and preventing leaching are conflicting goals, making it difficult to achieve both simultaneously.
To address both challenges, we focused on the concept of the “release-and-catch” catalytic systems42–51 to construct a new immobilized catalyst system for FA dehydrogenation (Fig. 1). When an aqueous FA solution is added, the catalyst immobilized on the support dissolves in the reaction solution and FA dehydrogenation proceeds to produce H2 and CO2 (Fig. 1a-(1)). Once FA is completely consumed, the homogeneous catalyst in the reaction solution is recaptured by the support (Fig. 1a-(2)). The supported catalyst is then separated by filtration for reuse (Fig. 1a-(3)).
 | ||
| Fig. 1 “Release-and-catch” catalytic system for FA dehydrogenation: a) concept of the release-and-catch type catalytic system, b) batchwise operation with a release-and-catch type catalyst, and c) flow type operation with a release-and-catch type catalyst. | ||
Thus, the system demonstrates high activity as a homogeneous catalyst during FA dehydrogenation, after which the catalyst is completely immobilized on the support, enabling solvent separation, catalyst recovery, and recycling via simple filtration. This system can be implemented in batch processes, as shown in Fig. 1b. Furthermore, this approach can be extended to construct semi-batch or flow catalytic reaction systems (Fig. 1c), thereby enhancing the catalyst recyclability, enabling solvent removal and catalyst recovery in a single step while maintaining the high activity of the complex catalyst. Based on this principle, we developed a release-and-catch catalytic system comprising a Cp*Ir complex immobilized on cross-linked polyacrylic acid (PAA). When adapted for catalyst recycling in both batch and flow processes, this system demonstrated efficient hydrogen production from FA, comparable to the performance of homogeneous catalysts.
Results and discussion
Catalyst synthesis and structure
PAA is affordable, readily available, and has well-established physical properties that can be optimized. Furthermore, the chemical properties of PAA can be adjusted by tuning the content of carboxylic acid groups (–COOH or –COO–). Thus, PAA was chosen as a polymer support to facilitate Cp*Ir complex fixation via carboxylic groups. However, as PAA dissolves easily in pure water, cross-linking with N,N′-methylenebisacrylamide (MBA) was required to form an insoluble polymer support. Cp*Ir complexes (Cp*: 1,2,3,4,5-pentamethyl-2,4-cyclopentadienyl) with various ligands (2–6; Fig. 2a) were synthesized according to the previous reports,26,52 and Cp*Ir complex 1 and 7 was also synthesized same manner as shown in ESI.† All these Cp*Ir complexes 1–7 were considered for immobilization on PAA. The Cp*Ir complex 1 (Cp*Ir-HMDAbpy) with the N,N′-([2,2′-bipyridine]-4,4′-diyl)bis(hexane-1,6-diamine) (4-hmdabpy) ligand (Fig. S2–S4†), which exhibited the highest catalytic activity as a homogeneous catalysts (Fig. S43 and S44†), was selected for in-depth analysis. | ||
| Fig. 2 a) Cp*Ir complexes (1 to 7) for immobilization on cross-linked PAA. b) Synthetic scheme of release-and-catch catalyst consisting of a Cp*Ir complex 1 and cross-linked PAA. | ||
The immobilized 1, Cp*Ir-HMDAbpy, supported on PAA (Cp*Ir-HMDAbpy@PAA), was synthesized as shown in Fig. 2b. Briefly, acrylic acid (30.92 mL, 0.408 mol) and MBA (12.25 g, 0.0794 mol) were degassed in deionized water under N2 bubbling at 80 °C for 30 min. Subsequently, 2,2′-azobis(2-methylpropionitrille) (AIBN, 217 mg, 1.28 mmol) in MeOH was added under N2 bubbling at 80 °C and heated for 1 h to induce radical polymerization. After carefully washing three times with MeOH and deionized water, the obtained white powder was freeze-dried at −196 °C to obtain the polymer support (37.95 g). The polymer (860 mg) and 4-hmdabpy (70 mg, 0.182 mmol) were stirred together in dry CH2Cl2 under a N2 atmosphere at room temperature for 15 h. The resulting powder was filtered, washed three times with deionized water, and freeze-dried at −196 °C to obtain a white powder (909 mg). This white powder (216 mg) and [Cp*Ir(H2O)3]SO4 (7.7 mg, 16 μmol) were dispersed in deionized water and stirred at room temperature for 24 h to obtain Cp*Ir-HMDAbpy@PAA as a yellow solid. Finally, the yellow solid was filtered, washed three times with deionized water, and freeze-dried at −196 °C to obtain a pale-yellow powder (205 mg).
The successful synthesis of the Cp*Ir-HMDAbpy@PAA catalyst was confirmed using various characterization methods. First, the Ir loading was determined using inductively coupled plasma-atomic emission spectrometry (ICP-AES), and the presence of Ir was confirmed using energy dispersive X-ray spectroscopy (EDX, Fig. S11†). Scanning electron microscope (SEM) images of the obtained catalyst revealed particle sizes ranging from 10 μm to several hundred μm, with sub-micrometer-pores on the surfaces, which formed during freeze-drying (Fig. S12†).
Next, FT-IR spectra of cross-linked PAA, HMDAbpy@PAA, and Cp*Ir-HMDAbpy@PAA were compared (Fig. 3). Cross-linked PAA (Fig. 3a) exhibited a broad band at 2500–3300 cm−1 and a strong peak at 1703 cm−1 (a-1), which corresponded to OH and carbonyl stretching, respectively. The strong peak at 1634 cm−1 (a-2) was attributed to carbonyl stretching of the amide group, confirming that the PAA structure is cross-linked via amide bonds. Furthermore, strong adsorption bands resulting from hydrogen bonding between the carboxylic acids were observed at 1230 cm−1 (a-3) and 1171 cm−1 (a-4).53,54 The spectrum of HMDAbpy@PAA (Fig. 3b) contained peaks corresponding to alkyl chain CH stretching of hexamethylenediamine at 3000 cm−1 (b-1) and 2855 cm−1 (b-2) as well as strong sharp absorption peak at 1590 cm−1 (b-3) attributed to ring stretching of the bipyridine (bpy) framework. Additionally, an absorption band originating from pyridine ring breathing appeared at 986 cm−1 (b-4).55 Finally, in the Cp*Ir-HMDAbpy@PAA spectrum (Fig. 3c), the ring stretching vibration at 1590 cm-1 (b-3) shifted to 1620 cm−1 (c-1), which agrees well with the peak of homogeneous Cp*Ir-HMDAbpy (Fig. S15e†), confirming the formation of Cp*Ir-HMDAbpy and its incorporation into PAA.54
 | ||
| Fig. 3 FT-IR spectra of a) as prepared cross-linked PAA, b) HMDAbpy@PAA, and c) Cp*Ir-HMDAbpy@PAA. (a-1): 1730 cm−1; (a-2): 1634 cm−1; (a-3): 1230 cm−1; (a-4): 1171 cm−1; (b-1): 3000 cm−1; (b-2): 2855 cm−1; (b-3): 1590 cm−1; (b-4): 986−1; (c-1): 1620 cm−1. | ||
To obtain further details about the catalyst, 13C CPMAS solid-state NMR measurements were performed for cross-linked PAA, HMDAbpy@PAA, and Cp*Ir-HMDAbpy@PAA (Fig. 4a–c), respectively. For cross-linked PAA, peaks attributed to the methine group (–CH–) and methylene chains (–CH2–) of PAA were observed at 32.8 ppm and 40.1 ppm, respectively. Additionally, a peak corresponding to carboxylic acids (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) was observed at 180 ppm. Because the CO peak of dimerized carboxylic acid is observed around 183 ppm, thus the observed carboxylic acids appear at approximately 183 ppm, the observed peak was considered to correspond to free carboxylic acids. The peak at 177 ppm was estimated to be the amide carbon in the MBA crosslinker.53,56,57 For HMDAbpy@PAA, in addition to the peaks of PAA, peaks attributed to the methylene chains of the ligand (27.8, 30.1, 37.9, 41.9, and 43.4 ppm) and aromatic signals of bipyridine (102.6, 110.3, 148.1, 155.1, and 156.7 ppm) were observed. The sharpness of these peaks suggests that the ligand is freely supported. Cp*Ir-HMDAbpy@PAA exhibited peaks attributed to the C–
O) was observed at 180 ppm. Because the CO peak of dimerized carboxylic acid is observed around 183 ppm, thus the observed carboxylic acids appear at approximately 183 ppm, the observed peak was considered to correspond to free carboxylic acids. The peak at 177 ppm was estimated to be the amide carbon in the MBA crosslinker.53,56,57 For HMDAbpy@PAA, in addition to the peaks of PAA, peaks attributed to the methylene chains of the ligand (27.8, 30.1, 37.9, 41.9, and 43.4 ppm) and aromatic signals of bipyridine (102.6, 110.3, 148.1, 155.1, and 156.7 ppm) were observed. The sharpness of these peaks suggests that the ligand is freely supported. Cp*Ir-HMDAbpy@PAA exhibited peaks attributed to the C–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3 (methyl groups of Cp*) at 8.4 ppm and the –CH3 (Cp*) at 86.3 ppm in the 13C NMR spectrum. Additionally, the bipyridine signal at 148.1 ppm shifted to a higher magnetic field (140.4 ppm). Furthermore, 13C dipolar decoupling (DD) MAS NMR spectroscopy (Fig. S16†) revealed that the ratio of carboxylic acid to alkyl carbons in PAA was 29.2:70.8. The carbon ratio of the Cp* methyl group to the Cp* was 1:1, with Cp* carbons being introduced at approximately 1.5% of the PAA carbons. The signal of the Cp* methyl group was also observed at 1.66 ppm by 1H Solid-state NMR spectroscopy (Fig. S17†). Based on the above results, the structure of the synthesized immobilized catalyst is inferred to consists of a cross-linked PAA with carboxylic acid groups that immobilize the Ir complex through electrostatic or coordination interactions (Cp*Ir-HMDAbpy@PAA in Fig. 2b).
H3 (methyl groups of Cp*) at 8.4 ppm and the –CH3 (Cp*) at 86.3 ppm in the 13C NMR spectrum. Additionally, the bipyridine signal at 148.1 ppm shifted to a higher magnetic field (140.4 ppm). Furthermore, 13C dipolar decoupling (DD) MAS NMR spectroscopy (Fig. S16†) revealed that the ratio of carboxylic acid to alkyl carbons in PAA was 29.2:70.8. The carbon ratio of the Cp* methyl group to the Cp* was 1:1, with Cp* carbons being introduced at approximately 1.5% of the PAA carbons. The signal of the Cp* methyl group was also observed at 1.66 ppm by 1H Solid-state NMR spectroscopy (Fig. S17†). Based on the above results, the structure of the synthesized immobilized catalyst is inferred to consists of a cross-linked PAA with carboxylic acid groups that immobilize the Ir complex through electrostatic or coordination interactions (Cp*Ir-HMDAbpy@PAA in Fig. 2b).
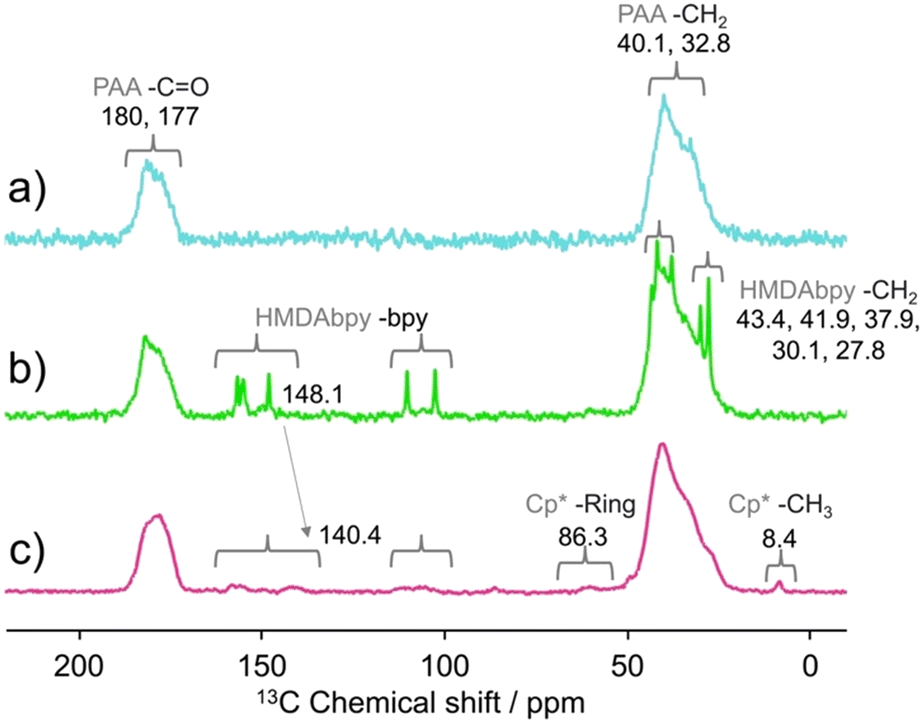 | ||
| Fig. 4 13C CPMAS NMR spectra of a) as prepared cross-linked PAA, b) HMDAbpy@PAA, and c) Cp*Ir-HMDAbpy@PAA. | ||
Catalyst activity for formic acid dehydrogenation
To optimize the catalytic activity of Cp*Ir-HMDAbpy@PAA, we adjusted the amount of ligand (4-hmdabpy) supported on the cross-linked PAA. After immobilizing 0 to 600 μmol g−1 of 4-hmdabpy on 1 g of cross-linked PAA to form HMDAbpy@PAA, 126 μmol g−1 of [Cp*Ir(H2O)3]SO4 was added to obtain Cp*Ir-HMDAbpy@PAA with a fixed Ir content of 2.5 μmol. FA dehydrogenation was performed using 20 mL of a 1 mol L−1 FA aqueous solution at 80 °C and the Turnover Frequency (TOF) was evaluated (Fig. 5a). In the absence of 4-hmdabpy (0 μmol g−1), [Cp*Ir(H2O)3]SO4 was directly supported on cross-linked PAA, but the catalytic activity was very low (TOF = 5600 h−1). However, upon increasing the amount of 4-hmdabpy supported on PAA from 0 to 212 μmol g−1, the TOF value increased linearly. In this case, [Cp*Ir(H2O)3]SO4 selectively reacts with 4-hmdabpy to form the Cp*Ir-HMDAbpy complex (with the plausible structure is [Cp*Ir(HMDAbpy)(H2O)]SO4) (Fig. S18†). As a result, the amount of the Cp*Ir complex increases proportionally with the amount of supported 4-hmdabpy, leading to enhanced catalytic activity. However, when the ligand content exceeds 212 μmol g−1, the TOF value stabilizes at 55000 h−1. When the ligand contents are higher than 212 μmol g−1, the amount of Cp*Ir complex formed was limited by the amount of added Ir, resulting in constant catalytic activity.
 | ||
| Fig. 5 Optimization of the ligand (4-hmdabpy) and Ir amounts in the Cp*Ir-HMDAbpy@PAA catalyst for FA dehydrogenation (reaction conditions: 1 mol L−1 aqueous FA, 2.5 μmol Ir; 80 °C reaction temperature, the TOF values were obtained from the gas evolution rate over the initial 5 min). Dependence of TOF on (a) the content of 4-hmdabpy with constant amounts of [Cp*Ir(H2O)3]SO4) and support (cross-linked PAA) and (b) content of Ir with constant amounts of 4-hmdabpy and the support. | ||
Next, we examined the effect of varying the amount of Ir support (0.3 to 5 wt%, determined by ICP-AES) on a carrier (cross-linked PAA, 1 g) with constant ligand content (4-hmdabpy, 212 μmol g−1) on the TOF for FA dehydrogenation (20 mL of 1 mol L−1, aqueous FA at 80 °C) (Fig. 5b). The highest TOF value (66000 h−1) was observed when the Ir content was 1.16 wt% (60 μmol g−1), which corresponds to approximately 30% relative to the ligand content. When the Ir content was 4.94 wt% (257 μmol g−1; 120% relative to the ligand content), the TOF value decreased to 21000 h−1. When the Ir content exceeded 30% relative to the ligand (0.3 mol of Ir per 1 mol of 4-hmdabpy), [Cp*Ir(H2O)3]SO4 that did not form a complex with the ligand was supported directly on cross-linked PAA, resulting in an increased amount of Ir that did not significantly contribute to the catalytic activity and thus lowering the activity of the immobilized catalyst. Therefore, the optimal immobilized catalyst contained 212 μmol g−1 of HMDAbpy@PAA and 1.16 wt% of Ir (60 μmol g−1), and further evaluations of the catalytic performance were conducted using this catalyst.
FA dehydrogenation was conducted using 36 mg of Cp*Ir-HMDAbpy@PAA (optimized immobilized catalyst, Ir: 413 μg, 2.15 μmol, as determined by ICP-AES) with 20 mL of a 1 mol L−1 FA aqueous solution at 80 °C (Fig. 6). The reaction proceeds as follows: 1) FA is added to the catalyst; 2) the Cp*Ir complex in the cross-linked PAA support is eluted, resulting in the solution turning yellow, and FA dehydrogenation begins; 3) as FA dehydrogenation progresses and FA concentration decreases, the reaction gradually slows; 4) upon further decreasing the FA concentration, the complex begins to be adsorbed onto the support; and 5) when FA is completely consumed, the Cp*Ir complex is fully adsorbed onto the support and the solution becomes transparent. The residual FA concentration after the reaction, determined using ion chromatography, was below 1.0 μmol L−1, with a conversion rate of nearly 100%. The generated gases were identified as H2 and CO2 in a 1:1 ratio (Fig. S19†). In addition, the TOF of the immobilized catalyst was 65000 h−1, which is comparable to or slightly higher than that of the free Cp*Ir-HMDAbpy complex ([Cp*Ir(HMDAbpy)(H2O)]SO4, 62000 h−1; Fig. S20†). These results suggest that the immobilized Cp*Ir complex dissolves in solution and functions as a homogeneous catalyst during FA dehydrogenation.
 | ||
| Fig. 6 Photographs during the FA dehydrogenation with the release-and-catch catalytic system (Cp*Ir-HMDAbpy@PAA). a) Before FA dehydrogenation, b) starting FA dehydrogenation, Cp*Ir complex is released from cross-linked PAA support into the solution, c) during FA dehydrogenation, d) ending FA dehydrogenation by consuming almost FA, Cp*Ir complex is caught by cross-linked PAA support, e) after FA dehydrogenation, Cp*Ir complex is completely caught by cross-linked PAA support. | ||
To confirm the dissolution of the catalyst during the reaction, the absorbance at 350 nm was monitored using UV-vis spectroscopy. Fig. 7a shows the time-dependent changes in the catalyst concentration, gas generation rate, and TON values determined from the UV-vis absorption spectra. As the 18 mg of immobilized catalyst contains 1 μmol of Ir, the catalyst concentration increased to 0.073 mmol L−1 within 0.1 h. This result shows that 73% of the immobilized Cp*Ir-HMDAbpy catalyst was dissolved in the solution. Subsequently, less than 0.015 ppm (by ICP-AES) of the immobilized Cp*Ir complex remained in the solution (Table S1†), indicating that almost 100% of the complex was re-adsorbed onto the support (Fig. 7a).
 | ||
| Fig. 7 a) Time course of TON values, gas evolution rate (L h−1), and Cp*Ir-HMDAbpy concentration (×10−1 mmol L−1) during FA dehydrogenation with Cp*Ir-HMDAbpy@PAA; b) pH dependence of UV-vis absorption at 350 nm of Cp*Ir-HMDAbpy in sulfuric acid solutions. The pH values were adjusted by the addition of CR-20 (basic ion exchange resin) in the following amounts, 1500 mg, 400 mg and 1500 mg; and left for 1 h. c) Cp*Ir-HMDAbpy concentration (mmol L−1) eluted from Cp*Ir-HMDAbpy@PAA (blue line) and pre-Cp*Ir-HMDAbpy@PAA (green line) during FA dehydrogenation. | ||
We further investigate the relationship between pH value and the absorption of the reaction solution. The Cp*Ir-HDMAbpy@PAA (18 mg containing 1 μmol Ir) was added to a sulfuric acid solution (20 mL) at 80 °C with a pH of 1.23 and an absorption of 0.54 at 350 nm, which is attributed to the Cp*Ir complex (Fig. 7b). With the addition of CR-20 (a basic ion-exchange resin) to the solution, the pH values increased while maintaining the solution concentration. This was accompanied by a decrease in absorption at 350 nm, indicating a reduction in the concentration of Cp*Ir-HMDAbpy. After 1 h at 5.37 of pH, almost the amount of Cp*Ir complex was absorbed by PAA support, not by CR-20 (Fig. S21†).
For comparison, we also investigated an immobilized catalyst (pre-Cp*Ir-HMDAbpy@PAA) synthesized via a different method, in which [Cp*Ir(4HMDAbpy)(H2O)]SO4 was prepared before immobilized on cross-linked PAA. Based on UV-vis spectroscopy, 0.025 mmol L−1 of Cp*Ir-4HMDAbpy was eluted from pre-Cp*Ir-HMDAbpy@PAA, which is only 34% of the amount eluted from Cp*Ir-HMDAbpy@PAA (0.073 mmol L−1) (Fig. 7c). Therefore, the TOF of pre-Cp*Ir-HMDAbpy@PAA (39000 h−1) was lower than that of Cp*Ir-HMDAbpy@PAA (65000 h−1). This indicates that, in the case of pre-Cp*Ir-HMDAbpy@PAA, a smaller amount of [Cp*Ir(HMDAbpy)(H2O)]2+ was eluted compared to the case of Cp*Ir-HMDAbpy@PAA, leading to a lower TOF.
The optimal FA concentration for the release-and-catch catalytic system was also determined. The maximum TOF value (110500 h−1) was achieved with 5 mol L−1 FA (Fig. S22a and b†). When a 20 mol L−1 (76 wt%) solution of FA was used, the initial reaction rate was slow. However, as FA was consumed and its concentration decreased to 5 mol L−1, the reaction rate increased (Fig. S23†). Regardless of the FA concentration used, the Ir concentration in the solution after the reaction was below 0.015 ppm by ICP-AES.
The activation energies of FA dehydrogenation in the presence of Cp*Ir-HMDAbpy and Cp*Ir-HMDAbpy@PAA were determined. The Arrhenius plots revealed activation energies as Ea = 75.6 kJ mol−1 for Cp*Ir-HMDAbpy and Ea = 74.2 kJ mol† for Cp*Ir-HMDAbpy@PAA, respectively (Fig. S24–S27†). These values are consistent with those previously reported for other Cp*Ir complexes.22,26,58,59 FA dehydrogenation is well documented as having the following reaction steps: (i) formation of an iridium formate as an intermediate, (ii) β-hydride elimination, direct transfer, or ligand assisted concerted transfer resulting in CO2 release and formation of an iridium hydride, and (iii) H2 generation via the reaction between the iridium hydride and a proton from the solvent.60,61 When using of Cp*Ir complexes as catalysts for FA dehydrogenation, the bpy ligand with strong electron donating groups is unlikely to dissociate from the Ir center. This reduces the formation of a vacant site on Ir for agnostic interaction, allowing the C–H bond of formate to be activated directly without the need for coordination in the step (ii). Kinetic isotope effects (KIEs) were evaluated to identify which of these processes – (i) intermediate IV to II, (ii) II to III, and (iii) III to IV in Scheme 1 – represents the rate-determining step (Table S2, Fig. S28†). When HCOOH was replaced with DCOOH (DCOOD + H2O), KIE value was obtained 2.15, whereas HCOOD (HCOOH + D2O) was used instead of HCOOH, KIE value was 1.40. From the obtained KIE values, it was determined that the rate-determining step is step (ii), which is more significantly affected than other steps. Additionally, the Eyring plot for Cp*Ir-HMDAbpy@PAA gave ΔS‡, ΔH‡, and ΔG‡ values of −124.37 kJ mol−1 K−1, 71.42 kJ mol−1, and 121.75 kJ mol−1, respectively.
 | ||
| Scheme 1 Plausible release-and-catch catalytic reaction mechanism for FA dehydrogenation with Cp*Ir-HMDAbpy@PAA. (I) is an iridium complex immobilized on a PAA support, (II) an iridium complex released from the PAA support by FA, (III) a hydride intermediate, (IV) a free Ir complex catalyst in water. | ||
To elucidate the release mechanism of Cp*Ir-HMDAbpy from the cross-linked PAA support, the Cp*Ir-HMDAbpy@PAA was added to sulfuric acid aqueous solution varying concentrations (0.01 to 1 mol L−1) and heated at 80 °C for 10 min while stirring (Fig. 8). The amount of eluted Cp*Ir complex increased with increasing sulfuric acid concentration. Additionally, when FA (pH 1) was used instead of sulfuric acid, ESI-MS analysis of the eluted solution revealed a signal at m/z 757 corresponding to [Cp*Ir-HMDAbpy-OCOH]+ (Fig. S30†). This result suggests that the Cp*Ir complex, which was immobilized by electrostatic or coordinating bonding between the carboxylic acid groups of PAA and the metal center of the Cp*Ir complex, was released as a Cp*Ir sulfonate or formate complex upon the addition of an acid stronger than the carboxylic acid of PAA, such as sulfuric acid or FA. To confirm this hypothesis, polystyrene sulfonic acid was used as the support instead of cross-linked PAA. Owning to the lower pKa of polystyrene sulfonic acid, Cp*Ir complex elution into the aqueous solution was not observed upon FA addition (Fig. S31†). Furthermore, even after heated at 80 °C for 24 h, no gas evolution resulting from FA dehydrogenation occurred.
 | ||
| Fig. 8 Effect of adding sulfuric acid and hearing at 80 °C for 10 min on the leaching amount of Cp*Ir-HMDAbpy, as measured by UV-vis absorption at 350 nm. | ||
The factors affecting the adsorption of the dissolved Cp*Ir complexes on various supports (activated carbon, PAA, α-Al2O3, and basic ion exchange resin (CR-20)) were investigated. In this experiment, 100 mg of each support was added to 0.1 mmol L−1 aqueous Cp*Ir-HMDAbpy solution. After stirring the mixtures at 80 °C for 1 h, the concentration of the Cp*Ir complex was determined using UV-vis absorption spectroscopy. No change in the complex concentration was observed with basic supports (α-Al2O3 and CR-20). In contrast, the acidic supports (activated carbon and PAA) adsorbed nearly 100% of the complex (Fig. S32†). Furthermore, after eluting Cp*Ir-HMDAbpy from Cp*Ir-HMDAbpy@PAA in a 0.01 mmol L−1 sulfuric acid solution, the solution was neutralized using CR-20, which caused Cp*Ir-HMDAbpy to be adsorbed onto the cross-linked PAA, but no Cp*Ir-HMDAbpy was adsorbed onto CR-20 (Fig. 7b and S21†). This behavior suggests that after dissolving Cp*Ir-HMDAbpy as a sulfonate salt, the sulfonate anion formed a complex with the cation in CR-20. Subsequently, the Cp*Ir-HMDAbpy cation would form a chelate complex with the carboxylic acid groups of PAA, resulting in PAA capturing the Cp*Ir complex.
Release-and-catch catalytic systems were also applied using Cp*Ir complexes with various bipyridine ligands (2 to 7 in Fig. 2a). Cross-linked PAA (100 mg) was added to aqueous solutions of Cp*Ir complexes 2 to 7 (0.1 mmol L−1, 20 mL), and the mixtures were stirred at room temperature for 1 h. Subsequently, the concentrations of the Cp*Ir complexes in solution were determined using UV-vis absorption spectroscopy. For all complexes nearly 100% adsorption by cross-linked PAA was observed, indicating that the ligand had a minimal impact on the release-and-catch catalytic system (Fig. S34–S40†). Next, the Cp*Ir complexes immobilized on cross-linked PAA were synthesized using Cp*Ir complexes 2 to 7, and these release-and-catch catalytic systems were applied to the FA dehydrogenation reaction (Fig. S41 and S42†). Similar to the case of homogeneous catalysts (Fig. S43 and S44†) the release-and-catch catalysts exhibited the highest activity when using a Cp*Ir complex containing a bipyridine ligand with HMDA as a substituent. Consistent with previous reports, the release-and-catch catalyst with a complex supported by an amino group at the para position also demonstrated high FA dehydrogenation activity.26 A Hammett plot analysis revealed that introducing an electron-donating substituent at the para position of the 2,2′-bipyridine ligands positively influences FA dehydrogenation, and a comparable effect was observed in the release-and-catch catalyst (Fig. S45†). The plot showed a slope of −1.3 (Fig. S45a†), based on the correlation between the Hammett constants of various substituents (NH2, OH, OMe, Me, and H) and the turnover frequency (TOF) values. In comparison, the homogeneous Cp*Ir complex exhibited a slope of −1.5 (Fig. S45b†). This suggests that the influence of PAA support on catalytic activity is minimal for the homogeneous catalysts, even in the case of the release-and-catch catalysts. Unlike complexes 1 to 4, which exhibited FA conversion rates of nearly 100%, Cp*Ir complexes 5, 6, and 7 achieved final conversion rates of 99.5%, 94.5%, and less than 1%, respectively, resulting in 0.017, 1.8 and 8.8 ppm of Cp*Ir complexes remaining in the solution after the reaction (Table S4†). Therefore, complexes 1 to 4 are suitable for use as release-and-catch catalytic systems.
For long-term continuous operation during practical applications, catalyst recycling is essential. Therefore, we first conducted recycling experiments using a batch operation. Cp*Ir-HMDAbpy@PAA (36 mg, Ir loading: 2 μmol) was placed in a 50 mL round-bottom glass flask, and FA (1 mol L−1, 20 mL per cycle) was added 5 times to perform FA dehydrogenation (Table 1, see ESI†). After each cycle, the catalyst was filtered, recovered, dried, and reused. Losses during the recovery procedure resulted in a slight decrease in the amount of catalyst used in each cycle. The TOF values remained between 63000 h−1 and 76000 h−1, showing no reduction (Fig. 9a). The amount of Ir remaining in the aqueous solution after the reaction was less than 0.1 ppm (0.002 ppm after the 5th cycle), indicating that almost 100% of the Ir was captured by the PAA support. Furthermore, solid-state NMR analyses of the catalyst after the reaction revealed no structural changes (Fig. S46 and S47†).
| Recycle run | TOF/h−1 | Catalyst/mg | Ir recovery rateb/% | Ir conc. After FA dehydrogenationb/ppm |
|---|---|---|---|---|
| a Reaction conditions: reaction temp.: 80 °C, FA: 1 mol L−1, 20 mL, Cp*Ir-HMDAbpy: 36 mg (including 2 μmol Ir). b The Ir concentrations in the residual solution were measured using ICP-AES analysis, and the recovery rate was calculated based on these concentrations. | ||||
| 1st | 65000 |
36.0 | 99.58 | 0.089 ± 0.001 |
| 2nd | 63000 |
34.0 | 99.78 | 0.047 ± 0.001 |
| 3rd | 64000 |
32.4 | 99.39 | 0.116 ± 0.001 |
| 4th | 70000 |
28.1 | 99.53 | 0.065 ± 0.003 |
| 5th | 76000 |
25.4 | 99.98 | 0.002 ± 0.001 |
 | ||
| Fig. 9 a) Time course of TON values for FA dehydrogenation during each cycle, and b) time course of the total TON value for FA dehydrogenation by the addition of a highly concentrated FA (>99 wt%, 26.2 mol L−1) into the solution (4.2 mol L−1, 38 mL) during batchwise recycling test of the release-and-catch catalytic system using Cp*Ir-HMDAbpy@PAA. | ||
Additionally, a comparable 5-cycle recycling test was performed by the addition of a highly concentrated FA (26.2 M, >99 wt%, approximately 38 mL per cycle) to the solution (4.2 mol L−1, 193 mL). With a total of 154 mL of FA solution (3.67 mol) used over 40 h, the TON value reached to 2185000, and 196 L of gas was generated through FA dehydrogenation (Fig. 9b). The conversion of FA in each cycle was greater than 99.3%, and the obtained gas had a stoichiometric ratio of H2 to CO2. After 40 h, no Ir was detected (<0.001 ppm by ICP-AES). To further investigate any changes in the Ir status during the reaction, we conducted XPS analyses of the Cp*Ir(HMDAbpy) complex immobilized on PAA after the first and fifth reactions. No peak shifts were observed compared to the Cp*Ir(HMDAbpy) complex (Fig. S49 and S50†). These results suggest that the Cp*Ir complex immobilized on PAA undergoes minimal degradation, even with the addition of a highly concentrated FA, thereby demonstrating suitable stability for practical applications.
Scheme 1 shows the plausible release-and-catch catalytic reaction mechanism for FA dehydrogenation. Initially, Cp*Ir-HMDAbpy (I) reacts with FA, leading to the formation of iridium formate (II) through the exchange of the carboxylic acid group of cross-linked PAA with FA. This reaction results in the release of iridium formate (II) into the solution. Upon dissolution of iridium formate (II) into the FA aq., β-hydride elimination or direct transformation occurs, producing CO2 and forming the iridium hydride complex (III). The iridium hydride complex (III) then reacts with a proton from water, releasing H2 and yielding the iridium complex (IV). Subsequently, iridium complex (IV) reacts with FA to generate iridium formate (II), thereby perpetuating the catalytic cycle, as many studies reported.58,61,62 After the complete consumption of FA by FA dehydrogenation, iridium complex (IV) reacts with the carboxylic acid of PAA and is captured by cross-linked PAA.
We adapted this release-and-catch catalytic system to a flow reaction system (Fig. 1c and S51 and S52†) and investigated its performance during continuous operation. The flow system offers the advantage of simultaneously removing residual water and recovering the catalyst. First, an aqueous FA solution (1.5 mol L−1,10 mL) heated to 80 °C was pumped into a column (Φ2.3 cm × 5.5 cm) packed with the catalyst, followed by an additional flow of 5 mL of pure water. Cp*Ir-HMDAbpy was eluted from the catalyst layer, and the FA solution containing the catalyst (15 mL) flowed into the reaction vessel. The FA dehydrogenation reaction proceeded in the reaction vessel (30 mL) heated to 80 °C, producing H2 and CO2. After completion of the reaction, when all FA was decomposed, the Cp*Ir-HMDAbpy aqueous solution remaining in the reaction vessel was passed through a column (Φ3 cm × 10 cm) packed with cross-linked PAA to capture Cp*Ir-HMDAbpy. Passing the aqueous solution (ca. 13 mL) through the column produced almost pure water. FA dehydrogenation was monitored using a gas meter to measure the gas production rate and volume, and the gas content after the reaction was analyzed using GC with a TCD detector.
FA dehydrogenation was repeated 10 times using the flow-type reaction system (Fig. 10). The gas production rate did not deteriorate over 10 cycles, demonstrating that the release-and-catch catalytic system could quantitatively generate gas through FA dehydrogenation in the flow-type system. Similar to the batch system, the generated gas consisted of H2 and CO2 in a 1:1 ratio, and the FA conversion rate after 10 cycles was nearly 100%. Additionally, the Ir content in the resulting aqueous solution was below the ICP-AES detection limit (Table S5†).
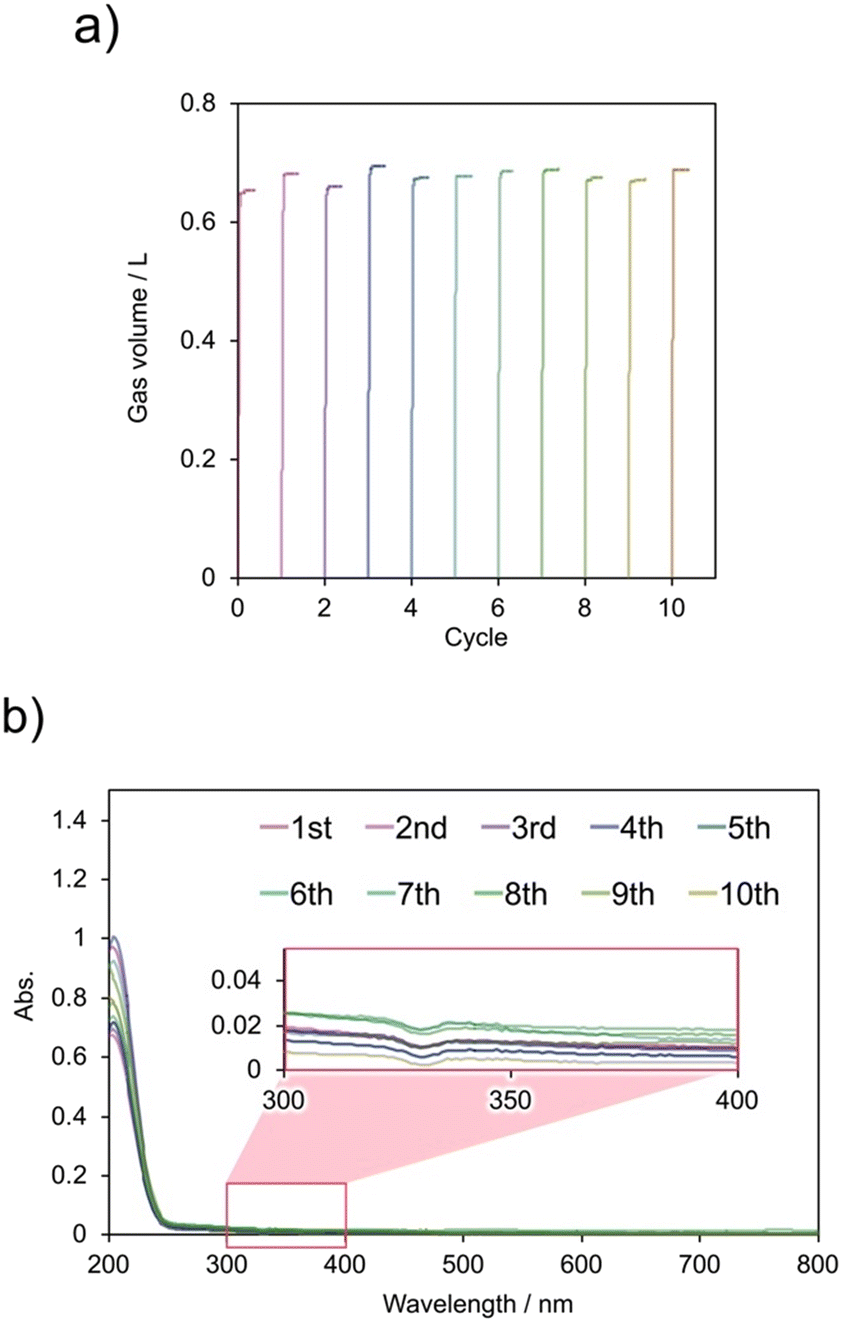 | ||
| Fig. 10 a) Flow-type recycling test for FA dehydrogenation with the release-and-catch catalytic system (Cp*Ir-HMDAbpy@PAA), and b) UV-vis spectra of the reaction solutions after each cycle. | ||
Conclusions
In this study, we developed Cp*Ir-HMDAbpy@PAA, which consisted of a Cp*Ir complex supported on cross-linked PAA, as a release-and-catch catalytic system for FA dehydrogenation. This system achieved catalytic performance and durability comparable to those of homogeneous complex catalysts while enabling 100% recovery of the catalyst after the reaction. The mechanism of the release-and-catch catalytic system is as follows: 1) initially, the Cp*Ir complex is supported by electrostatic or coordination interactions with the cross-linked PAA; 2) when an acid stronger than PAA, such as FA or sulfonic acid, is added, the Cp*Ir complex forms salts with the acid and is released from PAA support; and 3) in the presence of FA, the released Cp*Ir complex becomes a free complex through FA dehydrogenation and is recaptured by forming electrostatic or coordination bonds with the PAA support. Notably, various Cp*Ir complexes can be employed as release-and-catch catalytic systems.The Cp*Ir-HMDAbpy@PAA catalysts showed a TOF of 110000 h−1 and a TON of 2180000, demonstrating catalytic activity and durability comparable to homogeneous complex catalysts. Even after more than 40 h of reaction time, no decline in the catalytic performance was observed. Furthermore, when applied to FA dehydrogenation in a continuous flow system, the catalyst showed no deterioration, even after 10 recycles. Additionally, the catalyst and water were separated from the reaction solution after FA dehydrogenation, allowing quantitative recovery of the Cp*Ir complex. This technology holds great promise for developing a practical catalytic dehydrogenation system for hydrogen production from aqueous FA solutions.
Data availability
The data for this article are available in the ESI† and from the corresponding author upon reasonable request.Author contributions
K. Sawahara: conceptualization, data curation, formal analysis, investigation, methodology, funding acquisition and writing – original draft. S. Tanaka: resources, data curation, formal analysis, investigation. R. Gemma: data curation. R. Kanega: resources, data curation, investigation. H. Kawanami: conceptualization, resources, funding acquisition, investigation, project administration, supervision, validation, visualization, methodology, writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors also acknowledge the financial support by the Canon Foundation, Sasakawa Scientific Research Grant from The Japan Science Society, Support for Pioneering Research Initiated by the Next Generation (SPRING), Grant Number JPMJSP2124 from Japan Science and Technology Agency (JST), and Grants-in-Aid for Scientific Research (KAKENHI), Grant Number 24K01619 from Japan Society for the Promotion of Science (JSPS). RG thanks the Technology Joint Management Office at Tokai University for their support in conducting the SEM observations. The authors also acknowledge Dr Koichiro Masuda in Interdisciplinary Research Center for Catalytic Chemistry in AIST for his support with XPS observations.References
- C. C. Elam, C. E. G. Padró, G. Sandrock, A. Luzzi, P. Lindblad and E. F. Hagen, Int. J. Hydrogen Energy, 2003, 28, 601–607 CrossRef CAS.
- H. T. Hwang and A. Varma, Curr. Opin. Chem. Eng., 2014, 5, 42–48 CrossRef.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC.
- P. Preuster, A. Alekseev and P. Wasserscheid, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 445–471 CrossRef CAS PubMed.
- Y. Saito and Y. Okada, in Energy Technology Roadmaps of Japan: Future Energy Systems Based on Feasible Technologies Beyond 2030, ed. Y. Kato, M. Koyama, Y. Fukushima and T. Nakagaki, Springer Japan, Tokyo, 2016, pp. 459–462, DOI:10.1007/978-4-431-55951-1_34.
- X. Cui, M. Ishii, T. Tsujimura, T. Taniguchi, Y. Hashimoto and T. Nanba, J. Jpn. Pet. Inst., 2019, 62, 67–73 CrossRef CAS.
- J. Meng, F. Zhou, H. Ma, X. Yuan, Y. Wang and J. Zhang, Top. Catal., 2021, 64, 509–520 CrossRef CAS.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- A. T. Wijayanta, T. Oda, C. W. Purnomo, T. Kashiwagi and M. Aziz, Int. J. Hydrogen Energy, 2019, 44, 15026–15044 CrossRef CAS.
- I. Dutta, R. K. Parsapur, S. Chatterjee, A. M. Hengne, D. Tan, K. Peramaiah, T. I. Solling, O. J. Nielsen and K.-W. Huang, ACS Energy Lett., 2023, 8, 3251–3257 CrossRef CAS.
- H. Kawanami, Y. Himeda and G. Laurenczy, in Inorganic Reaction Mechanisms, 2017, pp. 395–427, DOI:10.1016/bs.adioch.2017.04.002.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- H. Zhong, M. Iguchi, M. Chatterjee, Y. Himeda, Q. Xu and H. Kawanami, Adv. Sustainable Syst., 2018, 2, 1700161 CrossRef.
- S. Kushwaha, J. Parthiban and S. K. Singh, ACS Omega, 2023, 8, 38773–38793 CrossRef CAS.
- S. Kushwaha, J. Parthiban and S. K. Singh, Organometallics, 2023, 42, 3066–3076 CrossRef CAS.
- C. Wang, M. Li, Q. Fan, C. Yang, G. Wang, X. Kong and Q. Zhu, Int. J. Hydrogen Energy, 2024, 66, 148–155 CrossRef CAS.
- J. Guo, M. Li, C. Yin, X. Li, Y. Wang, J. Yuan and T. Qi, Dalton Trans., 2023, 52, 4856–4861 RSC.
- L. Piccirilli, B. Rabell, R. Padilla, A. Riisager, S. Das and M. Nielsen, J. Am. Chem. Soc., 2023, 145, 5655–5663 CrossRef CAS PubMed.
- B. Maji, A. Kumar, A. Bhattacherya, J. K. Bera and J. Choudhury, Organometallics, 2022, 41, 3589–3599 CrossRef CAS.
- J. Guo, M. Li, C. Yin, D. Zhong, Y. Zhang, X. Li, Y. Wang, J. Yuan, H. Xie and T. Qi, Inorg. Chem., 2023, 62, 18982–18989 CrossRef CAS PubMed.
- L. Guo, Z. Li, M. Cordier, R. Marchal, B. Le Guennic and C. Fischmeister, ACS Catal., 2023, 13, 13626–13637 CrossRef CAS.
- S. Ge, L. Gong, P. Yi, X. Mo, C. Liu, X. Y. Yi and P. He, Inorg. Chem., 2023, 62, 18375–18383 CrossRef CAS PubMed.
- A. Léval, H. Junge and M. Beller, Eur. J. Inorg. Chem., 2020, 2020, 1293–1299 CrossRef.
- R. Kanega, N. Onishi, L. Wang, K. Murata, J. T. Muckerman, E. Fujita and Y. Himeda, Chem. – Eur. J., 2018, 24, 18389–18392 CrossRef CAS PubMed.
- H. Kawanami, M. Iguchi and Y. Himeda, Inorg. Chem., 2020, 59, 4191–4199 CrossRef CAS.
- M. Iguchi, M. Chatterjee, N. Onishi, Y. Himeda and H. Kawanami, Sustainable Energy Fuels, 2018, 2, 1719–1725 RSC.
- S. Ono, D. C. Santra, R. Kanega and H. Kawanami, Sustainable Energy Research, 2024, 11, 9 CrossRef.
- H. Zhong, M. Iguchi, F.-Z. Song, M. Chatterjee, T. Ishizaka, I. Nagao, Q. Xu and H. Kawanami, Sustainable Energy Fuels, 2017, 1, 1049–1055 RSC.
- V. S. Shende, V. B. Saptal and B. M. Bhanage, Chem. Rec., 2019, 19, 2022–2043 CrossRef CAS.
- T. A. Fassbach, J.-M. Ji, A. J. Vorholt and W. Leitner, ACS Catal., 2024, 14, 7289–7298 CrossRef CAS.
- S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS.
- M. Iguchi, Y. Himeda, Y. Manaka and H. Kawanami, ChemSusChem, 2016, 9, 2749–2753 CrossRef CAS.
- L. Alrais, S. S. Gholap, I. Dutta, E. Abou-Hamad, B. W. J. Chen, J. Zhang, M. N. Hedhili, J.-M. Basset and K.-W. Huang, Appl. Catal., B, 2024, 342, 123439 CrossRef CAS.
- J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed.
- P. Knörr, N. Lentz and M. Albrecht, Eur. J. Inorg. Chem., 2023, 26, e202300300 CrossRef.
- K. Sawahara, S. Tanaka, T. Kodaira, R. Kanega and H. Kawanami, ChemSusChem, 2023, 17, e202301282 CrossRef PubMed.
- L. Tensi, A. V. Yakimov, C. Trotta, C. Domestici, J. De Jesus Silva, S. R. Docherty, C. Zuccaccia, C. Copéret and A. Macchioni, Inorg. Chem., 2022, 61, 10575–10586 CrossRef CAS PubMed.
- H. Liu, H. Zou, D. Wang, C. Wang, F. Li, H. Dai, T. Song, M. Wang, Y. Ji and L. Duan, Angew. Chem., Int. Ed., 2023, 62, e202216739 CrossRef CAS PubMed.
- C. Trotta, G. Menendez Rodriguez, L. Tensi, A. V. Yakimov, L. Rocchigiani, A. Donnadio, C. Zuccaccia, C. Copéret and A. Macchioni, Eur. J. Inorg. Chem., 2023, 26, e202300211 CrossRef CAS.
- C. Broicher, S. R. Foit, M. Rose, P. J. C. Hausoul and R. Palkovits, ACS Catal., 2017, 7, 8413–8419 CrossRef CAS.
- J. S. Kingsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 4510–4517 CrossRef CAS PubMed.
- M. Gruttadauria, F. Giacalone and R. Noto, Green Chem., 2013, 15, 2608–2618 RSC.
- D. B. Eremin and V. P. Ananikov, Coord. Chem. Rev., 2017, 346, 2–19 CrossRef CAS.
- M. Genelot, V. Dufaud and L. Djakovitch, Adv. Synth. Catal., 2013, 355, 2604–2616 CrossRef CAS.
- C. Vriamont, M. Devillers, O. Riant and S. Hermans, Chem. – Eur. J., 2013, 19, 12009–12017 CrossRef CAS PubMed.
- S. Vásquez-Céspedes, K. M. Chepiga, N. Möller, A. H. Schäfer and F. Glorius, ACS Catal., 2016, 6, 5954–5961 CrossRef.
- A. M. Kluwer, C. Simons, Q. Knijnenburg, J. I. Van Der Vlugt, B. De Bruin and J. N. H. Reek, Dalton Trans., 2013, 42, 3609 RSC.
- A. Ohtaka, E. Sakaguchi, T. Yamaguchi, G. Hamasaka, Y. Uozumi, O. Shimomura and R. Nomura, ChemCatChem, 2013, 5, 2167–2169 CrossRef CAS.
- M. Ahmed, A. G. M. Barrett, D. C. Braddock, S. M. Cramp and P. A. Procopiou, Tetrahedron Lett., 1999, 40, 8657–8662 CrossRef CAS.
- S. Gámez, E. de la Torre and E. M. Gaigneaux, Chem. Eng. J., 2022, 427, 131820 CrossRef.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- J. Dong, Y. Ozaki and K. Nakashima, Macromolecules, 1997, 30, 1111–1117 CrossRef CAS.
- T. P. Gerasimova and S. A. Katsyuba, Dalton Trans., 2013, 42, 1787–1797 RSC.
- S. Maeda, Y. Fujiwara, C. Sasaki and K.-K. Kunimoto, Polym. J., 2012, 44, 200–203 CrossRef CAS.
- H. Cruz, B. Laycock, E. Strounina, T. Seviour, A. Oehmen and I. Pikaar, Environ. Sci. Technol., 2020, 54, 9573–9583 CrossRef CAS PubMed.
- T. Miyoshi, K. Takegoshi and K. Hikichi, Polymer, 1997, 38, 2315–2320 CrossRef CAS.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chem. – Eur. J., 2017, 23, 17017–17021 CrossRef CAS PubMed.
- C. Trotta, V. Langellotti, I. Manco, G. M. Rodriguez, L. Rocchigiani, C. Zuccaccia, F. Ruffo and A. Macchioni, ChemSusChem, 2024, e202400612 CrossRef CAS.
- M. Iguchi, H. Zhong, Y. Himeda and H. Kawanami, Chemistry, 2017, 23, 17788–17793 CrossRef CAS PubMed.
- G. Menendez Rodriguez, F. Zaccaria, L. Tensi, C. Zuccaccia, P. Belanzoni and A. Macchioni, Chem. – Eur. J., 2021, 27, 2050–2064 CrossRef CAS PubMed.
- K. Sawahara and H. Kawanami, J. Jpn. Pet. Inst., 2024, 67, 147–157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01067a |
| This journal is © The Royal Society of Chemistry 2025 |