
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heteropolyacids and ruthenium on covalent triazine frameworks – a bifunctional, recyclable catalyst for bio-based tandem systems†
Lea
Hombach
a,
Fabian
Müller
b,
Fulvio
Varamo
c,
Charles Otieno
Ogolla
c,
Renée
Hoffmann
d,
Jonas
Frohne
c,
Holger
Schönherr
 d,
Regina
Palkovits
b,
Benjamin
Butz
c and
Anna Katharina
Beine
*ac
d,
Regina
Palkovits
b,
Benjamin
Butz
c and
Anna Katharina
Beine
*ac
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: katharina.beine@uni-siegen.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
cDepartment of Mechanical Engineering, University of Siegen, Paul-Bonatz-Str. 9-11, 57076, Siegen, Germany
dPhysical Chemistry I & Research Center of Micro and Nanochemistry and (Bio)Technology (Cμ), Department of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Straße 2, 57076 Siege, Germany
First published on 1st February 2025
Abstract
A recyclable catalytic system for the one-pot hydrolytic hydrogenation of xylan to xylitol has been developed. Phosphotungstic acid (PTA) and ruthenium were immobilized on covalent triazine frameworks (CTFs) to obtain a bifunctional catalyst which is capable of catalysing both hydrolysis and hydrogenation. PTA_Ru/CTF3 exhibits high activity with 88% xylan conversion and up to 80% selectivity to xylitol while its stability could be demonstrated over 6 recycling runs.
Utilizing lignocellulose as a feedstock for high value-chemicals is crucial for transitioning to a bio-based economy and reducing reliance on fossil resources. Within lignocellulosic biomass, the polysaccharides cellulose and hemicellulose, along with the aromatic macromolecule lignin, are key constituents. Among these, the acid-catalyzed hydrolysis of hemicellulose and cellulose yielding xylose and glucose, respectively, stands out as one of the key reactions for the production of bio-based platform chemicals, fuels, and materials. Moreover, there is increasing interest in the one-pot hydrolytic hydrogenation to sugar alcohols such as sorbitol and xylitol (Scheme 1).1–4 This approach is particularly desirable to mitigate side-reactions resulting from the high reactivity of sugars compared to sugar alcohols, thereby enhancing selectivity in product formation. It is commonly presumed that the acid-catalyzed hydrolytic depolymerization of polysaccharides is followed by a metal-catalyzed reductive hydrogenation producing the respective polyols. However, kinetic investigations by Negahdar et al. suggested a simultaneous occurrence of hydrolysis and hydrogenation.5 They found that hydrogenation is kinetically favored over hydrolysis, and depolymerization (i.e. hydrolysis) is accelerated when the terminal oligosaccharide group is pre-hydrogenated. Consequently, a sequential hydrogenation-hydrolysis pathway is proposed, wherein the terminal sugar unit is hydrogenated prior to its hydrolytic cleavage from the polysaccharide chain.
 | ||
| Scheme 1 Hydrolytic hydrogenation of xylan to xylitol. | ||
Among metals such as Ru, Rh, Ir, Pt, Pd and Ni, it is Ru that exhibits the highest activity for the hydrogenation to polyols.6–9 In catalyzing the hydrolysis, previous literature predominantly relies on homogeneous acids. This includes technical acids like HCl, H3PO4 and H2SO4, organic acids, enzymes, as well as strongly Brønsted acidic heteropolyacids (HPAs).10–16 HPAs possess a number of advantageous properties: they are more acidic yet less corrosive than mineral acids, and they exhibit superior thermal and oxidative stability, along with being non-toxic and easy to handle. Additionally, their high water tolerance renders them ideal for use as homogenous catalysts in biomass transformations. In the hydrolytic hydrogenation of cellulose commercially available heteropolyacids such as phosphotungsticacid H3PW12O40 (PTA) and silicotungsticacid H4SiW12O40 (STA) have been used in combination with Ru/C, yielding up to to 81% yield of sugar alcohols at carbon efficiencies above 90%.17,18 The use of insoluble salts of HPAs in combination with Ru/C or Ru supported on HPA salts for the same reaction facilitates catalyst recovery, yet catalyst activity is decreased by the heterogenisation.19–23 In other approaches PTA and ruthenium were immobilized on heterogenous support materials like ZrO2 and Nb2O5 or activated carbon (AC).24,25
While significant progress has been made in the valorisation of cellulose, studies on hemicellulose remain limited. Of particular interest in this context is the hemicellulose xylan, a polymer comprised of xylose units linked by β(1–4)-glycosidic bonds. In the hydrolytic hydrogenation of xylan homogenous PTA could be used in combination with Ru/C to yield xylitol with 82% under moderate reaction conditions.26 Although homogenous HPAs have demonstrated great potential in this context, it is essential to recycle the HPA catalysts in order to establish sustainable processes. Various approaches for HPA recycling have been explored in recent years, including assemblies, thermo-responsive systems, complexation into insoluble heteropolysalts as well as the immobilization on heterogenous supports.27,28 In previous investigations, we extensively examined the immobilisation of PTA onto AC and demonstrated its activity and stability in the hydrolysis of xylan.28 In this work we want to demonstrate the heterogeneous application of heteropolyacids in the tandem system of xylan to xylitol and investigate their stability under both hydrothermal and reductive conditions.
Results and discussion
Initially, the AC supported PTA was tested in the hydrolytic hydrogenation of xylan to xylitol in combination with commercially available Ru/C as a hydrogenation catalyst. Reactions were typically performed in 10 mL autoclaves at 130 °C under 50 bar H2 in water. In the first reaction run a yield of 55% xylitol at high xylan conversion of 87% was achieved. Apart from xylitol, no additional products were identified by HPLC analysis of the reaction solution, suggesting complete hydrogenation of xylose. However, the discrepancy in carbon balance (32%) indicates either side-reactions like e.g. polymerization or substrate adsorption onto the AC-support, the latter of which has already been observed previously.29 The catalyst was re-cycled over 4 runs and therefore separated by centrifugation and washed three times after each run. After the first run the activity decreased significantly to 26%, followed by further catalyst deactivation to only 8% conversion in the 4th run 8 (Fig. 1a). While Ru-leaching was found to be <0.1 ppm, 53% leaching of PTA was observed by XRF in the first run. Consequently, the catalyst deactivation can be attributed to the loss of the active acid centres. When the catalyst was recycled only in the presence of either hydrogen or Ru/C (see Fig. S1†), the found leaching was significantly lower (11 and 8%, respectively). This suggests that the combination of both, thus reductive conditions, favours PTA leaching and thus lowers the catalyst stability. In our previous study we found that the attachment of the HPA to the support is facilitated by the presence of oxygen functional groups on the carbon surface.29 When the AC support is treated under reductive conditions, a decrease in the oxygen content from 7% to 3% can be observed by elemental analysis, suggesting that the oxygen groups are reduced under reductive conditions and therefore cannot efficiently bind the HPA on the surface anymore.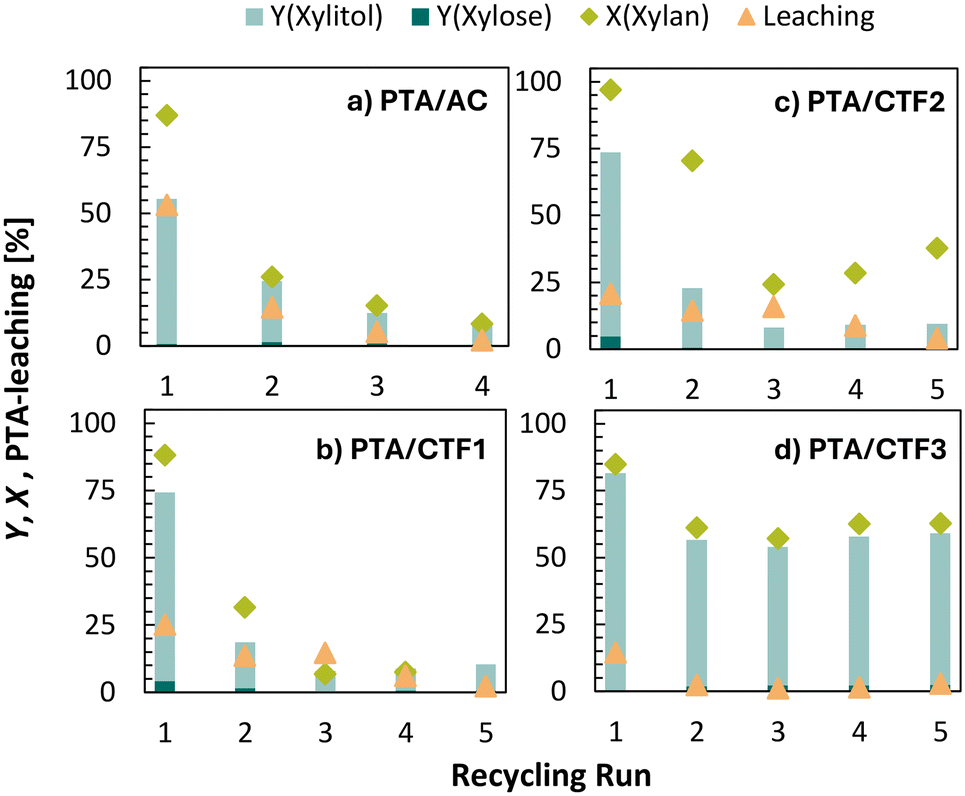 | ||
| Fig. 1 Xylan conversion X, xylitol yield Y and PTA-leaching in the recycling of PTA/AC, PTA/CTF1, PTA/CTF2 and PTA/CTF3 in the hydrolytic hydrogenation of xylan (conditions: 130 °C, 2 h, 50 bar H2, 750 rpm, m(xylan) = 0.115 g, c(HPA) = 4.35 mmol L−1, m(Ru/C 5 wt%) = 0.0115 g, V(H2O) = 5 mL). | ||
In this regard nitrogen containing covalent triazine frameworks (CTFs) have been considered as a more reduction-stable alternative. CTFs represent a class of highly cross-linked porous polymers which are formed by the cyclotrimerization of dicyano-(hetero)aryl building blocks.30,31 In this work, three different CTF-materials have been prepared based on the monomers 4,5-dicyanoimidazole (CTF1), 1,4-benzenedicarbonitrile (CTF2) and 4,4′-biphenyl-dicarbonitrile (CTF3) under ionothermal conditions in molten ZnCl2 at 400 °C for 10 h and 600 °C for 10 h. After ball milling CTF1–3 have been isolated as amorphous and partially carbonized frameworks, featuring high specific surface areas between 883 and 1675 m2 g−1 which are consistent with previous reports (Table 1 and Fig. S2†).32,33 The N2-physisorption-isotherms for CTF1 and CTF2 can be assigned to microporous materials according to IUPAC type I (Fig. S2†). This is also reflected in the high micropore volumes of 0.18 and 0.20 cm3 g−1 respectively (Table 1). In contrast, a type IV isotherm is obtained for CTF3, indicative of a mesoporous material (Fig. S2†). The N-content was determined by elemental analysis and varied from 17.5 to 46.8 wt% depending on the monomer used (Table 1).
| # | CTF-monomer | Name | S BET [m2 g−1] | V total [cm3 g−1] | V micro [cm3 g−1] | N-content [wt%] | PTA-loading [μmol g−1] | PTA-loading per surface [μmol m−2] | S BET (PTA/CTF) [m2 g−1] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4,5-Dicyanoimidazole | CTF1 | 883 | 0.39 | 0.18 | 46.8 | 433 | 0.49 | 309 |
| 2 | 1,4-Benzenedicarbonitrile | CTF2 | 1075 | 0.47 | 0.20 | 26.3 | 473 | 0.44 | 346 |
| 3 | 4,4′-Biphenyldicarbonitrile | CTF3 | 1575 | 0.66 | 0.00 | 17.5 | 448 | 0.28 | 611 |
PTA was immobilized on CTF1–3 by equilibrium wet impregnation for 16 h at 60 °C after which the catalyst was filtered off and washed until the washing solution reached pH 7. For all materials ICP-OES revealed a high PTA loading of 433–473 μmol g−1 resulting in a significant decrease in the specific surface area (Table 1). In dependence of the specific surface area an increased PTA-loading was found with increased N-content of the CTF. The same trend was already observed for oxygen rich activated carbon materials, indicating that the PTA is bound by the functional groups on the surface.29 In the FT-IR-spectrum of CTF1–3 the characteristic Keggin stretching bands of H2O (1633 cm−1), P–Oa (1078 cm−1), W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od (976 cm−1), W–Ob–W (899 cm−1), and W–Oc–W (817 cm−1) can be found, confirming the integrity of PTA after immobilization (see Fig. S3†).
Od (976 cm−1), W–Ob–W (899 cm−1), and W–Oc–W (817 cm−1) can be found, confirming the integrity of PTA after immobilization (see Fig. S3†).
The CTF supported catalysts were then tested in the hydrolytic hydrogenation of xylan using Ru/C as hydrogenation catalyst. In the first run high conversions of 88 to 97% and xylitol yields of 68 to 70% were observed for all catalysts (Fig. 1b–d). The found PTA-leaching in XRF ranges between 21–25% for CTF1–3 while Ru-leaching was below 0.2 ppm in all experiments. Although comparable leaching values were found for CTF1–3, a significant deactivation to <34% conversion was observed for PTA/CTF1 and PTA/CTF2 in the second reaction, followed by full deactivation in the following recycling runs (Fig. 1b–c). On the other hand, PTA/CTF3 showed less deactivation to 61% xylan conversion and only minor decrease in the xylitol yield to 54% in run 2 (Fig. 1d). In subsequent recycling runs the catalyst exhibits high stability achieving constant yields and conversions with low PTA leaching of 1–3%. To clarify possible deactivation mechanisms of PTA/CTF1 and PTA/CTF2, FT-IR-spectroscopy of the recycled catalysts was performed (see Fig. S3†). However, the intactness of PTA could be confirmed for all recycled catalysts. Since the CTF supported catalysts cannot be separated from Ru/C after the reaction, comparable experiments were carried out under the same reaction conditions without the addition of Ru/C. This enabled to independently investigate the influence of the reaction conditions on the surface properties of CTF1–3. N2-physisorption of once recycled PTA/CTF1 and PTA/CTF2 revealed a drastic decrease of the specific surface area to 121 m2 g−1 and 12 m2 g−1, respectively, whereas CTF3 still exhibit a high specific surface area of 585 m2 g−1 (see Fig. S4†). This suggests that the deactivation in the catalytic activity results from pore blocking and minimisation of the catalytically active surface. This suggests that the deactivation in the catalytic activity results from pore blocking and minimisation of the catalytically active surface. In contrast to CTF3, CTF 1 and CTF2 exhibit a lower intrinsic specific surface area on the one hand and a high degree of microporosity on the other hand, whereby the latter could promote irreversible adsorption of the substrate. To gain further insight into the substrate adsorption, the catalysts were subjected to thermal gravimetric analysis (TGA) before and after the reaction (see Fig. S5†). Notwithstanding the observed leaching of HPW, HPW/CTF1 and HPW/CTF2 exhibited an increased relative weight loss in comparison to that observed prior to the reaction. In the case of HPW/CTF3, the weight loss is decreased after the adsorption study, which can be attributed to the leaching of the immobilised HPW. These results confirm an increased adsorption of xylan, xylose or possible by-products on CTF1 and CTF2, which leads to the deactivation of the catalyst.
Given the potential for enhanced mass transfer of intermediates between catalytic centres in a bifunctional catalyst, the additional loading of CTF3 with ruthenium was investigated in the following. Therefore, PTA/CTF3 was impregnated with Ru by coordination with RuCl3·xH2O in ethanol. In order to keep up the molar ratio between the two catalysts Ru and PTA, a Ru-loading of 0.82 wt% was desired. ICP-OES of the prepared catalyst confirmed a reached Ru-loading of 0.83 wt%. The catalyst was reduced under hydrogen at a moderate temperature of 250 °C to prevent PTA decomposition. The preservation of the Keggin structure was confirmed by FT-IR spectroscopy of Ru_PTA/CTF3 after the reduction procedure (see Fig. S6†). XPS analysis of the bifunctional catalyst confirms the presence of C, N, W, O and Ru on the catalyst surface (see Fig. S7†). The Ru 3d5/2 signal at a binding energy of 281.1 eV corresponds to a partially oxidised Ru as the sample was not stored in inert gas atmosphere. The N element spectrum confirms the co-existence of two N-species; pyridinic nitrogen (398.4 eV) and nitrile functionality (400.6 eV). W is present as one single species corresponding to the WO6 octahedra (binding energy of W 4f7/2 = 35.7 eV).34
In the hydrolytic hydrogenation of xylan under 50 bar H2, Ru_PTA/CTF3 showed high activity with 88% xylan conversion (Fig. 2 run 1). After 2 h a product mixture of xylose (31% yield) and xylitol (40% yield) is obtained, which indicates that hydrogenation is not yet complete at this point. The mechanism seems therefore to proceed via a first hydrolysis of the polysaccharide to the sugar and a subsequent hydrogenation to the sugar alcohol. However, 70% of xylitol is yielded with prolonged reaction time of 4 h (Fig. 2 run 6). XRF revealed moderate PTA leaching of 13% in the first run which decreased to 5% in the subsequent runs. In addition, the Ru-leaching found in XRF was <0.2 ppm in all runs. The catalyst stability could be demonstrated over 6 recycling runs reaching constant conversions of 83–87%. In addition, the selectivity was maintained, confirming the stability of both acidic and reduction catalytic sites. With prolonged reaction time of 4 h in the 6th run 70% of xylitol, which is comparable to studies with 5 wt% Ru/C and indicates that the hydrogenation with is Ru_PTA/CTF3 is merely slower.
 | ||
| Fig. 2 Xylan conversion X, xylitol yield Y and PTA-leaching in the recycling of Ru_PTA/CTF3 hydrolytic hydrogenation of xylan. *4 h reaction time in 6th run (conditions: 130 °C, 2 h, 50 bar H2, 750 rpm, m(xylan) = 0.115 g, m(Ru_PTA/CTF-2) = 140 mg, V(H2O) = 5 mL). | ||
Additionally, cellobiose as model for cellulose was tested as substrate for the tandem reaction to broaden the substrate scope (see Fig. S8†). The Ru_PTA/CTF3 catalyst is capable of converting cellobiose to glucose (Y = 75%) and the sugar alcohol sorbitol (Y = 23%). A further acid catalysed dehydration reaction to sorbitan or isosorbide was not observed.
Since the molar ratio between Ru and PTA is comparable in both systems the influence of the Ru-particle-size on the catalyst activity was investigated by means of transmission electron microscopy (TEM) of 5 wt% Ru/C catalyst (Fig. 3a and b) and Ru_PTA/CTF3 (Fig. 3c and d). The atomic number-dependent contrast in the representative high-angle annular dark-field scanning TEM (HAADF-STEM) micrographs in Fig. 3a and c shows bright Ru particles on the darker (porous) substrate particles. This interpretation is corroborated by energy-dispersive X-ray spectroscopic analyses, which confirm the bright particles to be Ru (Fig. 4). The atomic tungsten species of PTA only contribute a homogeneous intensity background. For both Ru/C and Ru_PTA/CTF3 a homogeneous distribution of the Ru particles on the CTF substrate (Fig. 3a and c) with a narrow particle size distribution (PSD) and a comparable diameter of approximately 1 nm (Fig. 3d) was found. These results indicate that the comparatively diminished hydrogenation activity of Ru_PTA/CTF3 is not attributable to an increase in particle size and correspondingly reduced catalytic surface area. Prior research has suggested that the electronic structure of the Ru surface is influenced by the donating properties of nitrogen present in the CTF support.33 Consequently, the activity may also be subject to influence from the electronic effects associated with different supports of Ru/C and Ru_PTA/CTF3.
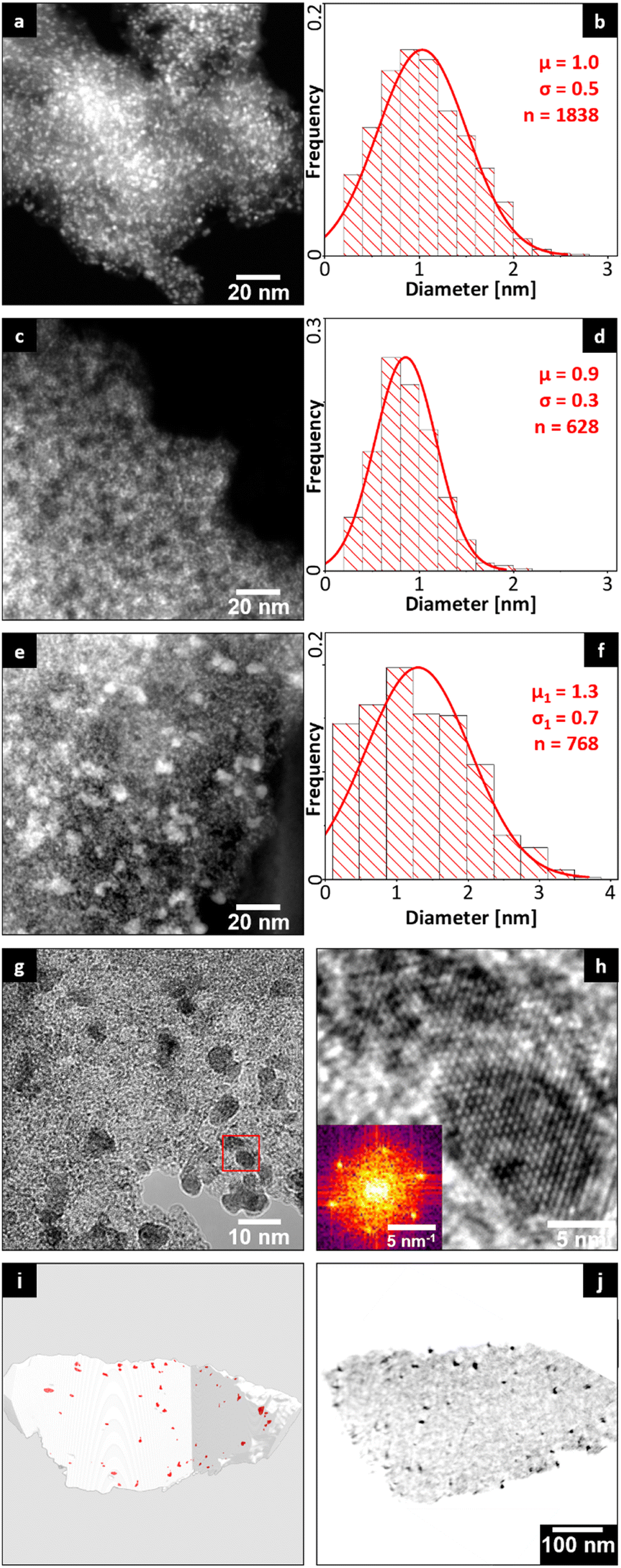 | ||
| Fig. 3 HAADF-STEM images with bright Ru nanoparticles and corresponding PSDs of a) commercial 5 wt% Ru/AC catalyst (PSD in b), c) Ru_PTA/CTF3 (PSD in d), and e) Ru_PTA/CTF3 after 6th recycling run, f) bimodal PSD of individual (background plot) and coalesced (inset) Ru particles from e), g) and h) HRTEM of coarsened Ru particles with dark crystallographic contrast, i) STEM tomography reconstruction: iso-surfaces of coarsened Ru particles (red) embedded within the open-porous CTF support (transparent grey), j) orthoslice (cross-section) of reconstruction with dark Ru agglomerates. | ||
 | ||
| Fig. 4 HAADF-STEM images and corresponding energy-dispersive X-ray spectroscopy (EDXS) analyses (net intensity distributions of Ru, W, P and Ru + W color map) of a), b) Ru_PTA/CTF3 and c, d) after recycling. | ||
Further it has to be investigated whether the decreased reduction temperature in the Ru_PTA/CTF3 preparation has resulted in incomplete reduction of Ru, which could diminish the catalyst's hydrogenation activity.
TEM images of the spent catalyst after 6 recycling runs show a progressive coarsening of part of the ruthenium particles in Ru_PTA/CTF3 (Fig. 3e and S10†). The localized Ru coalescence is indicated by the bimodal size distribution of the ruthenium particles with the majority still as small as 1.3 nm (similar to the original state) and sparse but significantly larger particles with sizes even up to more than 10 nm (Fig. 4e–h and S9,† details about particle-size evaluation in Fig. S9†). Both, the primary Ru particles as well as the coarsened ones are crystalline as confirmed by high-resolution TEM (HRTEM) (Fig. 3g and h). STEM tomography revealed that the observed coarsening counterintuitively occurs in the whole volume of the open-porous CTF support. Consequently, a large fraction of the primary Ru particles remains accessible and active, which explains the sustained performance even after six recycling runs. The observed coarsening may be attributed to coalescence of migrating Ru nanoparticles within larger voids of the mesoporous support (Fig. 3i and j).
Energy dispersive X-ray spectroscopy (EDXS) complements STEM imaging and proves the localized nanoparticles to be Ru, whereas the PTA appears to be more homogeneously distributed within the pore channels of the CTF support (Fig. 4). Even after recycling (Fig. 4c and d), both, tungsten and phosphorous from the HPA are still homogenously distributed on the CTF support indicating successful inclusion, immobility, and long-term stability of the acid.
Although local Ru coalescence occurs, the detailed TEM analyses confirmed that a major fraction of the primary Ru nanoparticles as well as the PTA are long-term stable under operational conditions and facilitate multiple recycling of Ru_PTA/CTF3.
XPS analysis of the spent catalyst shows again the presence of C, N, O, W and Ru on the catalyst surface (see Fig. S13†). The Ru 3d5/2 signal at a binding energy of 280.4 eV corresponds to a reduced Ru-species. The N element spectrum remains unchanged displaying again the two N-species pyridinic N and nitrile functionality. W is still present as one single species corresponding to the WO6 octahedra of the Keggin structure. XPS therefore confirms the stability of the catalyst.
Conclusions
In conclusion, PTA/CTF3 presents a highly active and selective acidic catalyst for the hydrolytic hydrogenation of xylan and yielded up to 85% of xylitol in combination with commercial Ru/C as hydrogenation catalyst. Whereas for AC support a high PTA leaching of >50% and consequently deactivation in activity was found under the reductive reaction conditions, PTA/CTF3 demonstrated a very good stability and reusability over 5 cycles. Furthermore, the additional loading of CTF3 with Ru nanoparticles was demonstrated. The resulting bifunctional catalyst Ru_PTA/CTF3 exhibited excellent stability in both its acidic and hydrogenation activity over 6 runs with up to 80% xylitol selectivity. TEM analysis revealed local Ru coalescence but confirmed that the major fraction remains its small particle size and accessibility. Further EDXS shows that the homogeneous distribution of P, W and Ru remained after recycling, emphasising the stability of the catalyst.Data availability
Supporting data are given in the uploaded ESI† of the article. Additionally, data for this article, including raw data of FT-IR, N2-physisorption and TEM/SEM, are available at Zenodo under the following DOI: https://doi.org/10.5281/zenodo.13682057.Author contributions
L. H. performed experimental work, participated in the data interpretation and the conceptual planning of experimental work flow and wrote the manuscript draft. F. V. supported in the catalytic testing of the synthesized catalysts and participated in data evaluation and interpretation. F. M. synthesized and characterized the CTF-materials, participated in the data interpretation and reviewed the manuscript. C. O. O. and B. B. measured and interpreted the TEM data and contributed to the manuscript draft and revision. J. F. collected tomography data and did the 3D reconstructions. R. H. and H. S. measured, plotted and interpreted the XPS data. R. P. contributed to the success of the project through a variety of scientific discussions and manuscript revisions. A. K. B. participated in the basic project definition, the conceptual planning of the experimental work flow, the data interpretation, supervised the experimental work and reviewed the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Hombach gratefully acknowledges the networking programme ‘Sustainable Chemical Synthesis 2.0’ for support and discussions across disciplines. The authors thank N. Avraham, A. Jakubowski and A. Gurowski for analytical support. The research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under the project number 501103334. Open access funding was enabled and organized by Project DEAL. C. Ogolla, J. Frohne and B. Butz acknowledge the use of the DFG-funded Micro-and Nanoanalytics Facility (MNaF) at the University of Siegen (INST 221/131-1) allowing the use of its major TEM instrument FEI Talos F200X (DFG INST 221/93-1, DFG INST 221/126-1) and sample preparation equipment. C. Ogolla and B. Butz further acknowledge Thermo Fisher Scientific specifically A. Meledin for UltraX EDX spectral imaging.Notes and references
- L. S. Ribeiro, J. J. M. Órfão and M. F. R. Pereira, Mater. Today Sustain., 2021, 11–12, 100058 Search PubMed.
- E. Redina, O. Tkachenko and T. Salmi, Molecules, 2022, 27, 1353 CrossRef CAS PubMed.
- O. V. Manaenkov, O. V. Kislitsa, V. G. Matveeva, E. M. Sulman, M. G. Sulman and L. M. Bronstein, Front. Chem., 2019, 7, 834 CrossRef CAS PubMed.
- Y. Liu, L. Chen, W. Zhang and H. Liu, CCS Chem., 2022, 4, 3162–3169 CrossRef CAS.
- L. Negahdar, P. J. C. Hausoul, S. Sibirtsev, S. Palkovits and R. Palkovits, Int. J. Chem. Kinet., 2018, 50, 325 CrossRef CAS.
- Y. Li, Y. Liao, X. Cao, T. Wang, L. Ma, J. Long, Q. Liu and Y. Xua, Biomass Bioenergy, 2015, 74, 148–161 CrossRef CAS.
- X. Zhao, J. Xu, A. Wang and T. Zhang, Chin. J. Catal., 2015, 36, 1419–1427 CrossRef CAS.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- M. Yang, H. Qi, F. Liu, Y. Ren, X. Pan, L. Zhang, X. Liu, H. Wang, J. Pang, M. Zheng, A. Wang and T. Zhang, Joule, 2019, 3, 1937–1948 CrossRef CAS.
- G. Hilpmann, N. Becher, F. A. Pahner, B. Kusema, P. Mäki-Arvela, R. Lange, D. Y. Murzin and T. Salmi, Catal. Today, 2016, 259, 376–380 CrossRef CAS.
- G. Hilpmann, P. Kurzhals, T. Reuter and M. M. Ayubi, Front. Chem. Eng., 2020, 2, 1–12 CrossRef.
- T. Marzialetti, M. B. V. Olarte, C. Sievers, T. J. C. Hoskins, P. K. Agrawal and C. W. Jones, Ind. Eng. Chem. Res., 2008, 47, 7131–7140 CrossRef CAS.
- P. Mäki-Arvela, T. Salmi, B. Holmbom, S. Willför and D. Y. Murzin, Chem. Rev., 2011, 111, 5638–5666 CrossRef PubMed.
- J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962 RSC.
- M. M. Ayubi, A. Werner, S. Steudler, S. Haase, R. Lange, T. Walther and G. Hilpmann, Catal. Today, 2021, 367, 137–144 CrossRef CAS.
- G. Hilpmann, S. Steudler, M. M. Ayubi, A. Pospiech, T. Walther, T. Bley and R. Lange, Catal. Lett., 2018, 149, 69–76 CrossRef.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 2167–2174 RSC.
- X. Xie, J. Han, H. Wang, X. Zhu, X. Liu, Y. Niu, Z. Song and Q. Ge, Catal. Today, 2014, 233, 70–76 CrossRef CAS.
- N. V. Gromov, T. B. Medvedeva, O. P. Taran, M. N. Timofeeva, O. Said-Aizpuru, V. N. Panchenko, E. Y. Gerasimov, I. V. Kozhevnikov and V. N. Parmon, Appl. Catal., A, 2020, 595, 117489 CrossRef CAS.
- M. Liu, W. Deng, Q. Zhang, Y. Wang and Y. Wang, Chem. Commun., 2011, 47, 9717–9719 RSC.
- N. V. Gromov, T. B. Medvedeva, V. N. Panchenko, O. P. Taran, M. N. Timofeeva and V. N. Parmon, Catal. Ind., 2023, 15, 87–98 CrossRef.
- N. V. Gromov, T. B. Medvedeva, Y. A. Rodikova, M. N. Timofeeva, V. N. Panchenko, O. P. Taran, I. V. Kozhevnikov and V. N. Parmon, Bioresour. Technol., 2021, 319, 124122 CrossRef CAS PubMed.
- M. Almohalla, I. Rodríguez-Ramos, L. S. Ribeiro, J. J. M. Órfão, M. F. R. Pereira and A. Guerrero-Ruiz, Catal. Today, 2018, 301, 65–71 CrossRef CAS.
- K. Dietrich, C. Hernandez-Mejia, P. Verschuren, G. Rothenberg and N. R. Shiju, Org. Process Res. Dev., 2017, 21, 165–170 CrossRef CAS.
- L. Hombach, N. Simitsis, J. T. Vossen, A. J. Vorholt and A. K. Beine, ChemCatChem, 2022, 14, e202101838 CrossRef CAS.
- E. Rafiee and S. Eavani, RSC Adv., 2016, 6, 46433–46466 RSC.
- L. Hombach, N. Hausen, A. G. Manjón, C. Scheu, H. Kraffczyk, M. Rose, J. Albert and A. K. Beine, Appl. Catal., A, 2023, 666, 119392 CrossRef CAS.
- J. Artz, ChemCatChem, 2018, 10, 1753–1771 CrossRef CAS.
- L. Liao, M. Li, Y. Yin, J. Chen, Q. Zhong, R. Du, S. Liu, Y. He, W. Fu and F. Zeng, ACS Omega, 2023, 8, 4527–4542 CrossRef CAS PubMed.
- A. K. Beine, A. J. D. Krüger, J. Artz, C. Weidenthaler, C. Glotzbach, P. J. C. Hausoul and R. Palkovits, Green Chem., 2018, 20, 1316 RSC.
- G. Tuci, A. Iemhoff, H. Ba, L. Luconi, A. Rossin, V. Papaefthimiou, R. Palkovits, J. Artz, C. Pham-Huu and G. Giambastiani, Beilstein J. Nanotechnol., 2019, 10, 1217 CrossRef CAS PubMed.
- N. Lopes da Costa, L. Guedes Pereira, J. V. Mendes Resende, C. A. Diaz Mendoza, K. Kaiser Ferreira, C. Detoni, M. M. V. M. Souza and F. N. D. C. Gomes, Mol. Catal., 2021, 500, 111334 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01071j |
| This journal is © The Royal Society of Chemistry 2025 |