
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminosilicate-mediated C(sp2)–H alkylation of furans using allylic alcohols†
Peter G. N. Neatea,
Liangliang Huang *b,
Margaret R. Jonesa,
Ruilian Wuc,
Nilusha M. Sudasinghea and
Xiaokun Yang*a
*b,
Margaret R. Jonesa,
Ruilian Wuc,
Nilusha M. Sudasinghea and
Xiaokun Yang*a
aChemistry Division, Los Alamos National Laboratory, Los Alamos NM 87544, USA. E-mail: xiaokuny@lanl.gov
bSchool of Sustainable Chemical, Biological & Materials Engineering, University of Oklahoma, Norman, OK 73019, USA. E-mail: hll@ou.edu
cBioscience Division, Los Alamos National Laboratory, Los Alamos NM 87544, USA
First published on 17th June 2025
Abstract
Alkyl furans have a variety of applications and therefore numerous approaches exist for their synthesis. While these methods can be effective, there are various drawbacks largely associated with the waste generated. Herein we report a novel method for the direct coupling of readily available furans with allylic alcohols. These include terpenoid alcohols such as geraniol and prenol, which are readily available from renewable, bio-derived feedstocks. Furthermore, the reaction is facilitated using aluminosilicates, including zeolites. It was found that Y-type zeolites were highly effective at mediating the coupling with furan, giving the 2-alkylated furan high yields. While various allylic alcohols were effective for this reaction, the identity of the furan proved to have a drastic impact on reactivity. For substituted furans, the reaction proved most effective with an amorphous aluminosilicate and calculations later revealed potential key factors in further developing and generalizing this methodology.
Introduction
Alkyl furans are used or have potential in various applications, including food additives/flavourings, surfactants, cosmetic additives, and insecticides.1–6 As such, a variety of methods exist for their synthesis.7–14 Three classical strategies dominate their synthesis and are summarized in Scheme 1: (a) Directed ortho-lithiation of furans followed by electrophilic quench7 generates 2-alkyl furans in good yields, but requires cryogenic temperatures and pyrophoric organolithium reagents, producing large quantities of salt waste. (b) Friedel–Crafts alkylation with alkyl halides (or alcohol-derived halides)8–11,13 employs stoichiometric Lewis or Brønsted acids. Although effective, these protocols suffer from corrosive reagents, over-alkylation, and difficult separation of metal salts or spent acid. (c) Transition-metal-catalyzed C–H activation couples furans with organohalides or boron reagents under Pd or Cu catalysis,12,14 offering excellent site-selectivity but relying on expensive ligands/metals and pre-functionalized coupling partners. These limitations motivate greener “direct-from-alcohol” methods. Dehydrative Friedel–Crafts reactions in which alcohol is protonated, loses water, and the resulting carbocation alkylates an arene provide an attractive alternative because water is the sole by-product and the alcohol feedstock can be bio-derived. However, literature examples are almost exclusively confined to benzylic or propargylic alcohols.8–11,13–19 Allylic alcohols, abundant in terpenoid biomass (prenol, geraniol, phytol),20 remain largely unexplored, and no general heterogeneous protocol exists for coupling them with furans. Here we report the first aluminosilicate-mediated dehydrative C(sp2)–H alkylation of furans with substituted allylic alcohols (Scheme 1, “Our approach”). Key features that distinguish our methodology from routes (a)–(c) include the direct use of alcohol thus avoiding the costly alkyl halides or organometallics; the mild reaction condition around 25–60 °C without external solvent when simple furans are used, or in benign solvents (toluene, DCE) for substituted substrates; the renewable feedstocks of biomass-based allylic alcohols and benign by-product of water; and the use of readily available Y-type or amorphous zeolites. Those attributes collectively offer a sustainable alternative to traditional furan alkylation chemistry and broaden the toolbox of zeolite-catalyzed C–C bond-forming reactions.15,21 | ||
Scheme 1 Approaches to alkylation of furan derivatives. In our approach, allyl alcohol (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2OH), corresponding to R2 = R3 = H, did not yield any product. CH–CH2OH), corresponding to R2 = R3 = H, did not yield any product. | ||
Results and discussion
Experimental
Our initial screening involved testing various commercially available zeolites for the coupling of furan 1 and phytol 2 (Table 1). Furan 1 was used in excess (10 equiv.) and the reaction performed neat, with excess furan acting as solvent due to its volatility facilitating easy removal and recovery after the reaction.
|
|||
|---|---|---|---|
| Entry | Catalyst (50 wt%) | Yield (%) | Catalyst details |
| a 25 wt% zeolite used. | |||
| 1 | CBV720CY | 92 | Y-type; SiO2/Al2O3 = 30 |
| 2 | CBV720CYa | 81 | Y-type; SiO2/Al2O3 = 30 |
| 3 | CBV760 | 93 | Y-type; SiO2/Al2O3 = 60 |
| 4 | CBV90A | 62 | Mordenite-type; SiO2/Al2O3 = 90 |
| 5 | CBV8014CY | ND | ZSM-5-type; SiO2/Al2O3 = 80 |
| 6 | CBV3024E | ND | ZSM-5-type; SiO2/Al2O3 = 30 |
| 7 | Perlkat 46–10 | 26 | Amorphous; SiO2 > 90 wt%; Al2O3 ∼10 wt% |
| 8 | SiO2 | ND | — |
| 9 | Al2O3 | ND | — |
| 10 | Amberlyst-15 | 78 | — |
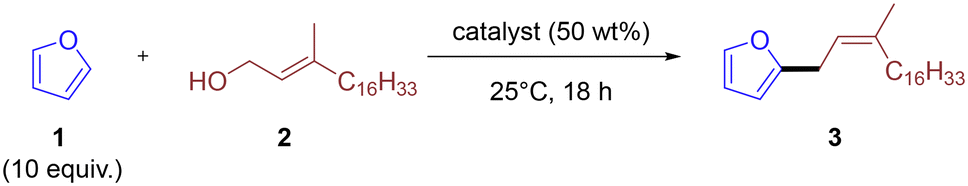
The reactions were conducted for 18 hours at 25 °C using 50 wt% of the heterogeneous catalyst. Y-type zeolites were found highly effective for this coupling, producing the allyl furan 3 in high yields in excess of 90% (Table 1, entries 1 and 3). Reducing zeolite loading to 25 wt% led to a modest drop in yield to 81% (Table 1, entry 2). Increasing the silica-to-alumina ratio, thereby enhancing the acidity of the Y-type zeolite, had no significant impact on the reaction (Table 1, entry 3). In contrast, other types of zeolites were much less effective. A mordenite-type zeolite resulted in a significantly lower yield (Table 1, entry 4), and ZSM-5-type zeolites did not yield any product, regardless of the silica-to-alumina ratio (Table 1, entries 5 and 6). Perhaps surprisingly, Perlkat 46–10, a commercially available amorphous aluminosilicate, did produce some product, albeit in low yield (Table 1, entry 7). Control reactions confirmed that silica or alumina alone did not yield any detectable product (Table 1, entries 8 and 9). Amberlyst-15, a more conventional heterogenous acid catalyst was also effective, although less so than Y-type zeolites (Table 1, entry 10). This suggests that the reaction likely proceeds via protonation of the alcohol and elimination of water to form a stabilized allylic cation. It should be noted, however, that reactions using Amberlyst-15 were found to be less regio-selective, with complete consumption of phytol and multiple alkene signals observed in the 1H-NMR spectrum of the crude reaction mixture.
Following the initial success with phytol, we examined other allylic alcohols (Fig. 1). The length of the alkyl chain did not significantly affect the reaction, with the terpenoids prenol and geraniol both proving effective, albeit with slightly lower yields compared to phytol (Fig. 1, 4, and 5). In contrast, allyl alcohol did not produce any product, with complete recovery of the starting materials. This may be due to the lack of substitution around the alkene, which would stabilize the carbocation intermediate. It is also worth noting that cinnamyl alcohol was also highly effective in coupling with furan, although it resulted in an inseparable mixture of the two products. The major component was the intended linear product (Fig. 1, 6), while the minor product was the branched isomer, where the furan ring reacted at the benzylic position (Fig. 1, 7). The mixture of products can be attributed to the allylic cation being more stable due to conjugation with the phenyl ring and less sterically hindered compared to previous examples.
 | ||
| Fig. 1 Scope of allylic alcohols tested for coupling with furan. Reported yields were determined by 1H-NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. Yields in parentheses correspond to isolated yields. | ||
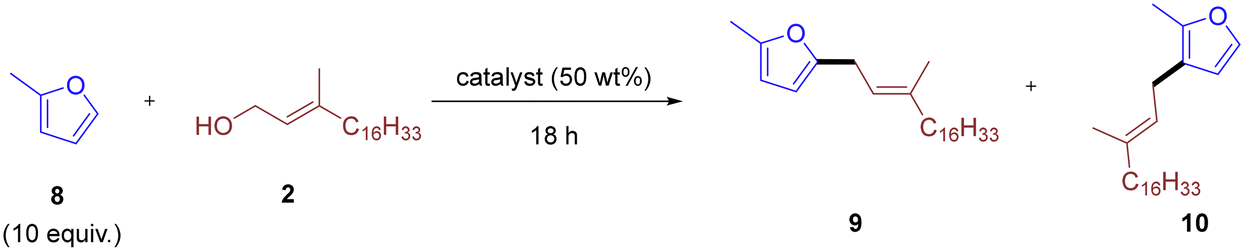
While the reaction was relatively insensitive to different allylic alcohols, substitution on the furan ring had a drastic effect. Extending the reaction protocol to 2-methylfuran resulted in negligible product formation. Consequently, we re-examined reaction parameters and catalysts using the reaction between 2-methylfuran 8 and phytol 2 as a platform (Table 2).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Temperature (°C) | Residual phytol (%) | 10 (%) | 9 (%) | 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 yield ratio :10 yield ratio |
| a 25 wt% used.b 10 wt% used. | ||||||
| 1 | CBV720CY | 25 | 95 | — | 3 | — |
| 2 | CBV760 | 25 | 97 | — | 2 | — |
| 3 | CBV90A | 25 | 80 | 5 | 7 | 1.4:1 |
| 4 | Perlkat 46–10 | 25 | 85 | 3 | 6 | 2.0:1 |
| 5 | CBV720CY | 40 | 91 | — | 5 | — |
| 6 | CBV760 | 40 | 85 | — | 8 | — |
| 7 | CBV90A | 40 | 44 | 11 | 18 | 1.6:1 |
| 8 | Perlkat 46–10 | 40 | 34 | 9 | 30 | 3.3:1 |
| 9 | CBV720CY | 60 | 75 | 4 | 9 | 2.3:1 |
| 10 | CBV760 | 60 | 19 | 8 | 30 | 3.8:1 |
| 11 | CBV90A | 60 | 11 | 19 | 35 | 1.8:1 |
| 12 | Perlkat 46–10 | 60 | — | 14 | 51 | 3.6:1 |
| 13 | Perlkat 46–10a | 60 | — | 14 | 50 | 3.6:1 |
| 14 | Perlkat 46–10b | 60 | 21 | 9 | 36 | 4.0:1 |
| 15 | Amberlyst-15b | 60 | — | 12 | 41 | 3.4:1 |

At 25 °C, a temperature effective for furan as the substrate, only trace amounts of product were observed with any aluminosilicates tested (Table 2, entries 1–4). Heating to 40 °C had little effect for Y-type and ZSM-5-type zeolites (Table 2, entries 5 and 6), while mordenite-type zeolite and amorphous Perlkat 46–10 showed improved reactivity with low but appreciable product formation (Table 2, entries 7 and 8). Further increasing the reaction temperature significantly improved product formation for all aluminosilicates except the Y-type zeolite (Table 2, entries 9–11). Using Perlkat 46–10 (Table 2, entry 12), the reaction provided a combined isolated yield of 65%, composed of 51% of the 5-substituted isomer 9 and 14% of the 3-substituted isomer 10. In all cases, the 5-substituted allyl furan 9 was the major product, with small amounts of the 3-substituted analog 10 also observed. Perlkat 46–10 (Table 2, entry 12) and CBV760 (Table 2, entry 10) gave comparable selectivity for the 5-substituted isomer, with product 9:10 yield ratios of 3.6:1 and 3.8:1, respectively. In contrast, mordenite CBV90A (entry 11) showed markedly lower selectivity for product 9, with a corresponding 9:10 yield ratio of 1.8:1. Amberlyst-15 was also effective, albeit with lower yields and significantly more side-product formation even at lower catalyst loadings (Table 2, entry 15). The loading of Perlkat 46–10 could be reduced to 25 wt% without a significant drop in yield, although any further reduction led to a marked decrease in yield (Table 2, entries 13 and 14).
The use of solvents did not improve the reaction yields (see Table 3). Ethereal solvents such as THF and 1,2-dimethoxyethane resulted in significantly lower yields (Table 3, entries 1 and 2). While toluene and 1,2-dichloroethane did not significantly affect the overall yield or selectivity (Table 3, entries 3 and 4), they provided greater reproducibility. Under solvent-free conditions (Table 2, entry 12), approximately one in three reactions exhibited a roughly 15% lower yield, however, this issue was mitigated when using 1,2-dichloroethane or toluene. Additionally, reducing the loading of 2-methylfuran 8 when using 1,2-dichloroethane negatively impacted the yield (Table 3, entries 7 and 8). Although the decrease was not drastic, the yield dropped to 52% when using 3 equivalents of 2-methylfuran, and even lower yields were obtained with further reductions (Table 3, entries 5, 6, and 9). The higher reproducibility with toluene and 1,2-dichloroethane suggests that these solvents provide a more stable reaction environment, potentially by better solubilizing the reactants and intermediates. In contrast, the detrimental effect of ethereal solvents could indicate that these solvents interact unfavorably with the catalytic sites or intermediates.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Equivalents 2-methylfuran | Solvent volume (mL) | Residual phytol (%) | 10 (%) | 9 (%) | 9:10 yield ratio |
| 1 | THF | 10 | 2 | 66 | 6 | 15 | 2.5:1 |
| 2 | DME | 10 | 2 | 66 | 5 | 15 | 3.0:1 |
| 3 | Toluene | 10 | 2 | — | 13 | 48 | 3.7:1 |
| 4 | DCE | 10 | 2 | — | 15 | 50 | 3.3:1 |
| 5 | DCE | 5 | 2 | — | 12 | 44 | 3.7:1 |
| 6 | DCE | 3 | 2 | — | 10 | 42 | 4.2:1 |
| 7 | DCE | 3 | 1 | — | 10 | 40 | 4.0:1 |
| 8 | DCE | 3 | 0.5 | — | 9 | 39 | 4.3:1 |
| 9 | DCE | 1.5 | 2 | — | 10 | 33 | 3.3:1 |
With effective reaction conditions for 2-methylfuran 8 established, we assessed the general applicability of this system to different allylic alcohols and substituted furans (Fig. 2). Once again, the chain length of the allylic alcohol had little effect on the overall yield, with phytol, geraniol, and prenol giving similar yields (Fig. 2, 9/10, 11, and 12, respectively). Notably, prenol exhibited higher selectivity for the 5-substituted allyl furan product compared to phytol and geraniol. Cinnamyl alcohol, while effective, resulted in an inseparable mixture of isomers (Fig. 2, 13). Three isomers were obtained with the 5-substituted allyl furan the major product and 3-substituted the secondary product in similar ratios to reactions with phytol and geraniol. The minor component was the branched isomer, with the furan ring attached at the benzylic position, as was observed previously for the reaction between furan and cinnamyl alcohol (Fig. 1, 6 and 7). Perhaps unsurprisingly, the reaction between 2-ethylfuran and phytol 2 gave a similar yield and selectivity to the analogous reaction with 2-methylfuran 8 (Fig. 2, 14). Of note is that both methyl-2-furoate and benzofuran reacted to give only the 5-substituted furan, with no other isomers detected (Fig. 2, 15–17). These results demonstrate the versatility of the reaction conditions across various allylic alcohols and substituted furans. The higher selectivity observed with prenol, along with the mixture of isomers resulting from cinnamyl alcohol, suggest that steric and electronic effects on the allylic alcohol influence product distribution. As mentioned previously, allyl alcohol did not produce any observable product and remained unreacted (Fig. 2). Disubstituted furans were also ineffective in this reaction system, with both 2,3-dimethyl- and 2,5-dimethylfuran giving poor yields or no detectable product, respectively (Fig. 2, 18 and 19). Unlike methyl-2-furoate, both furfural and 2-acetylfuran resulted in poor yields of the intended product (Fig. 2, 20 and 21, respectively). This could be due to side reactions as all have similarly electron-withdrawing substituents. The lack of reactivity with allyl alcohol suggests that substitution around the alkene plays a crucial role, potentially in stabilizing the proposed carbocation intermediate. Understanding these limitations is essential for further optimizing the reaction conditions and expanding the range of compatible substrates.
 | ||
| Fig. 2 Scope of coupling reaction between allylic alcohols and substituted furans. Product ratios are with respect to alkylation in the 5- and 3-position; *third isomer for 13 is substitution of furan ring onto α-aryl position. Reported yields were determined by 1H-NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. Yields in parentheses correspond to isolated yields. | ||
Calculations
To gain insight into the significant differences in reactivity observed among the zeolites, adsorption calculations were performed to compare faujasite (a Y-type zeolite), ZSM-5, and mordenite. During the optimization of reaction conditions for the coupling of furan 1 and phytol 2 (Table 1), these zeolites were highly effective, inactive, and moderately effective, respectively. Initially, the adsorption of phytol 2 was examined. Notably, adsorption calculations for ZSM-5 revealed that it could not accommodate phytol 2 due to the dimensions of its pores and channels. This finding is consistent with the observed lack of activity for ZSM-5 in the coupling of furan 1 and phytol 2. Both faujasite and mordenite could accommodate phytol 2 with adsorption energies of −117 and −119 kcal mol−1, respectively. The significantly more energetically favorable adsorption of phytol 2 for faujasite explains its greater reactivity compared to mordenite, as it better accommodates phytol 2 within its pores (Fig. 3). These initial computational results support the experimental observations by demonstrating that the physical properties of the zeolites, specifically pore size and adsorption energy, affect the reactivity. The inability of ZSM-5 to accommodate phytol 2 explains its inactivity, while the favorable adsorption in faujasite correlates with its high reactivity. | ||
| Fig. 3 One molecule of phytol 2 adsorbed in A) faujasite and B) mordenite. | ||
The adsorption energies of the product allylic furan 3 also provided insight into the high zeolite loading required for these reactions. The adsorption of the product was significantly more favorable than that of phytol. For faujasite and mordenite zeolites, adsorption energies for product 3 were −238 kcal mol−1 and −26 kcal mol−1, respectively, compared to −117 kcal mol−1 and −119 kcal mol−1 for phytol. These results suggest that the strong adsorption of the product allylic furan 3 within the zeolites might inhibit further catalytic activity by occupying active sites, thereby necessitating higher catalyst loadings to maintain reaction efficiency. Those computational results also reveal a potential limitation of the system, where strong product adsorption could lead to catalyst deactivation over time. As reviewed recently, tailoring the local properties of active sites in a zeolite to balance adsorption strengths and enhance the desorption of products for active site regeneration is critical for zeolite catalysis.22,23
Further supporting the superior reactivity of Y-type zeolite is its adsorption capacity for furan 1, which is 0.336 g g−1—more than twice that of ZSM-5 (0.142 g g−1) and over three times that of mordenite (0.0944 g g−1). Adsorption energies for furans, however, did not show significant differences among the three zeolites (Table 4). Notably, all substituted furans had higher adsorption energies than furan 1, especially those with carbonyl groups (Table 4, entries 4–6). This suggests that substituted furans, due to their excess and higher adsorption energies, outcompete phytol 2 for adsorption into the pores. Consequently, the reactivity of phytol 2 is hypothesized to occur on the outer surface, which, in return, explains why amorphous Perlkat 46–10 was the most effective aluminosilicate for substituted furans. Although this remains speculative, these observations underscore the complex interplay between adsorption capacities and reactivity. The higher adsorption capacity of Y-type zeolite for furan 1 likely contributes to its superior performance. However, the higher adsorption energies of substituted furans indicate that they dominate the adsorption sites, likely forcing reactions to occur on the outer surface of the zeolite catalyst.
| Entry | EAds (kcal mol−1) | ZSM-5 | FAU | MOR |
|---|---|---|---|---|
| 1 |  |
−89.1 | −81.6 | −86.1 |
| 2 |  |
−96.2 | −89.5 | −93.2 |
| 3 |  |
−101.7 | −96.4 | −100.8 |
| 4 |  |
−170.9 | −165.0 | −168.7 |
| 5 |  |
−137.9 | −131.4 | −136.8 |
| 6 |  |
−167.9 | −158.3 | −164.1 |
We examined the rates of self-diffusion for both allyl alcohol and prenol within a faujasite cell containing 15 molecules of furan 1. Extremely high diffusion rates can lead to a lack of reactivity, as the substrate may not interact effectively with the solid catalyst due to rapid movement.24 The self-diffusion rates for allyl alcohol and prenol were 5.271 × 10−9 m2 s−1 and 1.307 × 10−9 m2 s−1, respectively. Similarly, the self-diffusion rates of furan did not significantly change in the presence of either allyl alcohol or prenol, with rates of 4.789 × 10−9 m2 s−1 and 2.054 × 10−9 m2 s−1, respectively. The self-diffusion rates highlight that physical movement within the zeolite pores does not significantly affect the reaction. Instead, the electronic properties of the reactants play a crucial role. The greater substitution of prenol likely stabilizes the cationic intermediate more effectively than allyl alcohol, enhancing reactivity.
Proposed strategies to mitigate catalyst deactivation
Catalyst deactivation, particularly due to strong product adsorption within zeolite pores, represents a significant challenge for industrial heterogeneous catalysis. To enhance catalyst durability, efficiency, and selectivity, several tailored mitigation strategies25–27 can be pursued: (a) Adjustment of Zeolite Acidity zeolite acidity profoundly influences adsorption–desorption equilibrium, as strong acid sites often lead to excessively strong interactions with product molecules, resulting in deactivation through blocked active sites. Reducing or fine-tuning the Brønsted and Lewis acidity by altering the SiO2/Al2O3 ratio (Si/Al ratio) effectively moderates these interactions. For example, increasing the Si/Al ratio decreases zeolite acidity, weakening adsorption interactions and facilitating easier product desorption. Our experimental results indicated comparable yields across different Si/Al ratios, suggesting acidity tuning could be implemented without compromising overall catalytic activity. Precise control over the distribution and density of acidic sites through controlled zeolite synthesis can be particularly advantageous for achieving an optimal balance between catalytic activity and ease of regeneration. (b) Surface Hydrophobicity Modification zeolites are inherently hydrophilic due to their aluminol and silanol groups. The hydrophilic nature of zeolites can lead to strong binding and retention of polar or charged products, severely limiting catalyst turnover. Surface modification techniques, such as silylation, introduce hydrophobic alkylsilyl groups onto zeolite surfaces, dramatically reducing surface polarity and enhancing hydrophobic character. This treatment reduces the adsorption strength of polar or charged products, promoting easier desorption and regeneration. (c) Alteration of Pore Structures pore structure is a critical determinant of adsorption–desorption dynamics in zeolites. Conventional microporous zeolites often suffer from slow mass transport and pore blockage by strongly adsorbed bulky products. Introducing hierarchical pore structures, which combine micropores with meso- or macropores, significantly improves diffusion pathways and facilitates efficient transport of reactants and products. Such hierarchical structures can be achieved through various methods, including templating, etching, or steam-assisted crystallization. In our computational analyses, Y-type zeolite (faujasite) demonstrated strong product adsorption due to its large pore cavities. Hierarchical modifications would substantially reduce diffusion resistance, facilitating product escape from catalytic sites and mitigating rapid deactivation.For practical industrial applications, an integrated approach combining several of these strategies might prove most effective. For example, initially synthesizing or choosing zeolites with controlled acidity and hierarchical porosity followed by targeted surface modifications (e.g., hydrophobic surface silylation) could achieve an optimal balance between catalytic efficiency and long-term stability. Periodic regeneration steps should also be integrated into reaction protocols, informed by continuous catalyst performance monitoring.
Conclusions
In summary, we have established a novel, operationally simple protocol for the dehydrative coupling of furan and furan derivatives with allylic alcohols using environmentally benign and readily available aluminosilicates. The identity of the aluminosilicate catalyst proved crucial, especially depending upon the furan derivative used. Initial computational efforts have identified key aspects of zeolites and their interactions with substrates, providing valuable insights into the reaction system. The identification of key zeolite properties and their interactions with substrates provides a foundation for further optimization. Future efforts will focus on identifying and mitigating side reactions and exploring catalysts or conditions that can accommodate a wider variety of substituted furans. Additionally, deeper mechanistic studies will aid in the refinement of our understanding and guide the development of even more efficient and selective catalytic processes.Data availability
The data supporting the findings of this study are available within the manuscript and its ESI.† Additional raw data, including experimental procedures, characterization data, and computational details, are available from the corresponding author upon reasonable request.Author contributions
Conceptualization: Peter Neate and Xiaokun Yang, methodology: Peter Neate, Xiaokun Yang, and Ruilian Wu, validation: Margaret R. Jones, formal analysis: Liangliang Huang, investigation: Peter Neate and Margaret Jones, resources: Xiaokun Yang and Nilusha Sudasinghe, data curation: Margaret Jones, writing – original draft: Peter Neate, writing – review & editing: Ruilian Wu, Margaret Jones, Liangliang Huang, Xiaokun Yang, and Nilusha Sudasinghe, supervision: Xiaokun Yang and Nilusha Sudasinghe, project administration: Xiaokun Yang and Nilusha Sudasinghe, funding acquisition: Xiaokun Yang and Nilusha Sudasinghe.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Laboratory Directed Research and Development program of Los Alamos National Laboratory under Project Grant No. 20230161ER. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract No. 89233218CNA000001). We acknowledge funding provided by Los Alamos National Laboratory, Laboratory Directed Research and Development (LDRD) Program Office under 20230161ER. Cameron Moore is acknowledged for his previous discussions on related chemical reactions. Yulu Ge is acknowledged for her help on chemical structure confirmation and edits for IUPAC names in the ESI.†References
- M. Younes, G. Aquilina, L. Castle, K.-H. Engel, P. Fowler, M. J. Frutos Fernandez, P. Fürst, U. Gundert-Remy, R. Gürtler, T. Husøy, M. Manco, P. Moldeus, S. Passamonti, R. Shah, I. Waalkens-Berendsen, D. Wölfle, M. Wright, R. Benigni, C. Bolognesi, K. Chipman, E. Cordelli, G. Degen, D. Marzin, C. Svendsen, M. Carfì, G. Vianello and W. Mennes, EFSA J., 2021, 19, e06386 CAS.
- H. P. Surburgh, Common Fragrance and Flavor Materials, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 177–238 Search PubMed.
- D. S. Park, K. E. Joseph, M. Koehle, C. Krumm, L. Ren, J. N. Damen, M. H. Shete, H. S. Lee, X. Zuo, B. Lee, W. Fan, D. G. Vlachos, R. F. Lobo, M. Tsapatsis and P. J. Dauenhauer, ACS Cent. Sci., 2016, 2, 820–824 CrossRef CAS PubMed.
- A. Velty, S. Iborra and A. Corma, ChemSusChem, 2022, 15, e202200181 CrossRef CAS PubMed.
- C. R. Rodriguez-Saona and J. T. Trumble, Entomol. Exp. Appl., 1999, 90, 131–140 CrossRef CAS.
- B. Gabriele and G. Salerno, Chem. Commun., 1997, 1083–1084 RSC.
- M. Silvi, R. Schrof, A. Noble and V. K. Aggarwal, Chem. – Eur. J., 2018, 24, 4279–4282 CrossRef CAS PubMed.
- Y. S. Cho, S. T. Kim and D. H. Ryu, Org. Lett., 2022, 24, 1732–1736 CrossRef CAS PubMed.
- J. Li, C. Yue, P. Chen, Y. Xiao and Y. Chen, Angew. Chem., Int. Ed., 2014, 53, 5449–5452 CrossRef CAS PubMed.
- A. S. Makarov, A. E. Kekhvaeva, C. J. J. Hall, D. R. Price, I. V. Trushkov and M. G. Uchuskin, Tetrahedron, 2017, 73, 7042–7053 CrossRef CAS.
- A. A. Merkushev, D. A. Eshmemet'eva and M. G. Uchuskin, Tetrahedron, 2023, 145, 133627 CrossRef CAS.
- M. Peña-López, L. A. Sarandeses and J. Pérez Sestelo, Eur. J. Org. Chem., 2013, 2013, 2545–2554 CrossRef.
- G. Yang, D. Dilixiati, T. Yang, D. Liu, B. Yu and C. Hu, Appl. Organomet. Chem., 2018, 32, e4450 CrossRef.
- J. Yuan, X. Zhang and C. Yang, RSC Adv., 2021, 11, 13832–13838 RSC.
- J. R. Cabrero-Antonino, A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 5658–5661 CrossRef CAS PubMed.
- M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 48, 935–959 CrossRef CAS.
- S. Estopiñá-Durán and J. E. Taylor, Chem. – Eur. J., 2021, 27, 106–120 CrossRef PubMed.
- Y. Tao, B. Wang, J. Zhao, Y. Song, L. Qu and J. Qu, J. Org. Chem., 2012, 77, 2942–2946 CrossRef CAS PubMed.
- S. Zhang, M. Vayer, F. Noël, V. D. Vuković, A. Golushko, N. Rezajooei, C. N. Rowley, D. Lebœuf and J. Moran, Chem, 2021, 7, 3425–3441 CAS.
- S. M. Rowland, S. Van Wychen and L. M. L. Laurens, Identification and Quantification of Photosynthetic Pigments in Algae (Laboratory Analytical Procedure (LAP)), National Renewable Energy Laboratory (NREL), Golden, CO, Report No. NREL/TP-5100-84170, 2022 Search PubMed.
- M. Tejeda-Serrano, S. Sanz-Navarro, F. Blake and A. Leyva-Pérez, Synthesis, 2020, 52, 2031–2037 CrossRef CAS.
- E. Pérez-Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647–17695 CrossRef PubMed.
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- D. F. Plant, G. Maurin and R. G. Bell, J. Phys. Chem. B, 2007, 111, 2836–2844 CrossRef CAS PubMed.
- F. Lin, M. Xu, K. K. Ramasamy, Z. Li, J. L. Klinger, J. A. Schaidle and H. Wang, ACS Catal., 2022, 12, 13555–13599 CrossRef CAS.
- T. Ennaert, J. V. Aelst, J. Dijkmans, R. D. Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- M. Guisnet and F. R. Ribeiro, Deactivation and Regeneration of Zeolite Catalysts, Imperial College Press, London, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01320d |
| This journal is © The Royal Society of Chemistry 2025 |