
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-mode catalytic degradation of diclofenac by copper oxide-modified TiO2/MnOx composites: insights from dark and UV-A activation†
Gloria Issaa,
Sylvie Kříženecká c,
Petr Bezdičkab,
Daniela Popelkováb,
Martin Kormundad,
Jakub Edererc,
Daniel Bůžekbc,
Jan Čundrlebc,
Zdeněk Baďuraef,
Jiří Henychbc and
Martin Šťastný*b
c,
Petr Bezdičkab,
Daniela Popelkováb,
Martin Kormundad,
Jakub Edererc,
Daniel Bůžekbc,
Jan Čundrlebc,
Zdeněk Baďuraef,
Jiří Henychbc and
Martin Šťastný*b
aInstitute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Ak. G. Bontchev str., bl.9, 1113, Sofia, Bulgaria
bMaterials Chemistry Department, Institute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec-Řež, Czechia. E-mail: stastny@iic.cas.cz
cFaculty of Environment, Jan Evangelista Purkyně University in Ústí nad Labem, Pasteurova 3632/15, 400 96 Ústí nad Labem, Czechia
dFaculty of Science, Jan Evangelista Purkyně University in Ústí nad Labem, Pasteurova 3632/15, 400 96 Ústí nad Labem, Czechia
eRegional Centre of Advanced Technologies and Materials, Šlechtitelů 27, Olomouc 78371, Czech Republic
fNanotechnology Centre, VŠB – Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
First published on 10th June 2025
Abstract
Diclofenac sodium (DCF), a widely used nonsteroidal anti-inflammatory drug, is a persistent pharmaceutical contaminant that resists removal by conventional wastewater treatment. In this study, CuO-modified TiO2/MnOx composites were developed as multifunctional catalysts for DCF degradation under both dark and UV-A conditions. The materials exhibited dual-mode reactivity through distinct mechanisms: (i) non-radical oxidative degradation under dark conditions, and (ii) radical-mediated photocatalysis under UV-A irradiation. Under illumination, the formation of an interfacial p–n–p heterojunction between CuO, MnOx, and TiO2 generated internal electric fields that directed charge carrier migration—electrons flowing from the conduction band of TiO2 toward CuO and MnOx domains, and holes in the reverse direction. This spatial charge separation suppressed recombination and sustained redox cycling between Cu2+/Cu+ and Mn4+/Mn3+, promoting continuous ROS generation. In the absence of light, DCF degradation proceeded via non-radical oxidative pathways involving surface-bound reactive oxygen species and redox-active metal centers. Surface-sensitive XPS and hydroxyl quantification (TOTH) revealed elevated Mn3+/Mn4+ ratios, enriched surface-associated lattice oxygen, and high –OH group densities for the most active catalysts. These features collectively facilitated pollutant adsorption, oxygen activation, and sustained interfacial electron transfer. LC-MS/MS analysis confirmed a consistent degradation pathway across both regimes, involving hydroxylation, decarboxylation, and dechlorination of DCF. The Cu/5Ti5Mn-HT and Cu/8Ti2Mn-HT catalysts achieved exceptional dark-phase degradation efficiencies (∼99.8% and ∼99.4%, respectively), while Cu/TiO2 exhibited the highest UV-A photocatalytic performance (∼42%). These findings demonstrate the synergistic advantage of redox-active metal oxides and interfacial design, establishing CuO–MnOx–TiO2 composites as promising candidates for advanced pharmaceutical pollutant remediation.
Introduction
Diclofenac sodium (DCF), a widely prescribed non-steroidal anti-inflammatory drug (NSAID), is a persistent environmental pollutant due to its chemical stability and resistance to conventional wastewater treatment processes. Its widespread occurrence in aquatic ecosystems poses significant environmental and health risks, including endocrine disruption and toxicity to aquatic organisms and terrestrial mammals.1 The continuous discharge of DCF and similar pharmaceuticals into the environment necessitates the development of innovative and efficient methods for their removal, particularly under conditions relevant to real-world wastewater treatment.Traditional wastewater treatment plants, which rely predominantly on biological degradation and adsorption on activated carbon, often fail to achieve complete removal of pharmaceuticals such as DCF, leaving residual concentrations that persist in effluent streams.2
Advanced oxidation processes (AOPs), which employ reactive oxygen species (ROS) for the oxidative degradation of recalcitrant organic pollutants, have shown considerable promise as an alternative. AOPs exploit the high reactivity of species such as hydroxyl radicals (·OH), which are capable of non-selective attack on organic compounds, leading to their mineralization or transformation into less harmful products.3
Among the catalysts utilized in AOPs, titanium dioxide (TiO2) and manganese oxides (MnOx) are particularly interesting due to their unique photochemical and redox properties.
TiO2 has long been established as a highly effective photocatalyst. Upon UV irradiation, it generates electron–hole pairs, forming ROS, including ·OH, which can degrade a broad spectrum of organic pollutants.4,5
However, its activity is limited in the absence of light, restricting its applicability under dark conditions. In contrast, MnOx offers complementary advantages, leveraging its ability to transition between Mn2+, Mn3+, and Mn4+ oxidation states. This redox flexibility facilitates electron transfer processes and enables MnOx to catalytically degrade pollutants in both light and dark environments.6
These properties are strongly dependent on the surface characteristics of MnOx, including the density of redox-active sites, the presence of oxygen vacancies, and the availability of lattice oxygen species.7
Building on the established redox capabilities of MnOx, our prior research demonstrated their exceptional catalytic activity in the hydrolysis of bis(4-nitrophenyl)phosphate (BNPP), a model substrate mimicking phosphodiester bonds, and in the catalytic decomposition of methanol. This study highlighted the critical role of surface oxygen species (lattice oxygen and adsorbed species) and the interplay of Mn oxidation states in driving these reactions.8
Furthermore, the incorporation of CuO into heterogeneous catalysts9 and photocatalysts10 has been reported to enhance the generation of ROS under both dark and illuminated conditions, owing to the presence of the redox-active Cu2+/Cu+ couple, which facilitates electron transfer processes and promotes interfacial redox reactions.
While binary oxide-based photocatalysts, such as CuO/TiO2 and CuO/MnO2, have been extensively studied for the degradation of organic pollutants under both illuminated and dark conditions,11–14 tricomponent systems incorporating both Cu and Mn redox centers within a TiO2 matrix remain scarce and are predominantly investigated in non-photocatalytic applications, such as low-temperature NH3-SCR of NOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15–17 and CO-assisted NOx reduction.18
15–17 and CO-assisted NOx reduction.18
In this work, we address the existing gap by designing CuO-modified TiO2/MnOx composites capable of degrading diclofenac sodium (DCF) under both dark and UV-A conditions. The catalyst integrates redox-active Cu2+/Cu+ and Mn4+/Mn3+ couples, oxygen vacancies, and interfacial p–n-type junctions, enabling two distinct yet complementary reaction pathways. In the absence of light, degradation proceeds via a non-radical oxidative mechanism mediated by interfacial redox cycling and sustained electron transfer. Under UV-A irradiation, an internal electric field formed across the CuO–MnOx–TiO2 p–n–p heterojunction facilitates directional charge migration, promoting efficient separation of photogenerated carriers and continuous ROS formation.
Unlike previous studies focused exclusively on light-induced radical pathways, our approach combines both light-independent and photoactivated mechanisms within a single multifunctional material. Mechanistic validation via LC-MS/MS analysis, scavenger inhibition experiments, and radical identification confirms the presence of synergistic pathways responsible for DCF degradation. This dual-mode design expands the functional performance window of oxide-based catalysts and offers a robust strategy for treating persistent contaminants such as diclofenac in aqueous environments.
Experimental
Catalyst preparation
CuO-modified TiO2/MnOx composites were synthesized using a CTAB-assisted (N-hexadecyl-N,N,N-trimethylammonium bromide) method with minor modifications, followed by incipient wetness impregnation (IWI) and hydrothermal treatment (HT).19–21 Detailed experimental procedures are provided in ESI† (section S1).Catalyst characterization
Comprehensive catalyst characterization involved advanced techniques to detail textural properties, including Brunauer–Emmett–Teller (BET) surface area analysis and Barrett–Joyner–Halenda (BJH) pore size distribution. X-ray powder diffraction (XRPD) provided phase identification and crystallinity, while Raman and Fourier-transform infrared spectroscopy (FTIR) offered insights into structural features and vibrational modes. These techniques, along with hydrogen temperature-programmed reduction (H2-TPR) and X-ray photoelectron spectroscopy (XPS), were crucial in determining the catalysts' redox behaviors and elemental compositions, providing a robust understanding of their catalytic functionality. The potentiometric titration curves were used to calculate the pH point of zero charge (pHPZC), which is crucial for understanding the adsorption behavior of diclofenac on the catalyst surfaces under varying pH conditions. Additionally, the total titratable hydroxyl groups (TOTH) were evaluated, which provides insights into the surface acidity and basicity of the catalysts, directly influencing their catalytic performance in diclofenac degradation. Detailed experimental procedures, instrumentation, and additional conditions are provided in ESI,† sections S2 and S6, respectively.Experimental procedure for catalytic/photocatalytic degradation of DCF
In a typical catalytic/photocatalytic degradation experiment of DCF, 20 mg of catalyst was dispersed in 70 mL of 4 mg L−1 DCF solution. The reaction was carried out in a custom photoreactor with top illumination from three UV-A lamps (365 nm, 3.25 mW cm−2). The solution was equilibrated in the dark for 60 minutes, followed by 180 minutes of UV-A exposure. The pH of the reaction suspensions was maintained as-is during the experiments without any adjustments. The pH effect was tested at pH 4, 7, and 9 using acetate and Tris buffers. At specified intervals (5, 10, 20, 30, 50, and 60 minutes during the dark phase; and 70, 90, 120, 180, and 240 minutes during UV-A irradiation), 1 mL samples were taken, centrifuged (150 s, 18000 rpm), and analyzed for residual DCF and degradation by-products via high-performance liquid chromatography (see section S3 in the ESI†). The main experiments were performed in triplicate, with standard deviations shown as error bars.
Catalyst recycling procedure
To evaluate the reusability of the catalyst, recycling experiments were performed in four degradation cycles. After each experiment, the reaction mixture was transferred into 50 mL centrifuge tubes and centrifuged at 10000 rpm for 10 minutes to recover the catalyst. The separated solid was subsequently washed three times with deionized water to remove any adsorbed reaction intermediates or by-products, with each washing step followed by centrifugation under identical conditions. The wet catalyst was not dried between cycles but was immediately redispersed in deionized water using ultrasonic agitation to ensure uniform dispersion. All degradation experiments during recycling were conducted under the same conditions as the initial test, including catalyst loading, DCF concentration, volume, and irradiation parameters, to ensure a reliable comparison of catalytic performance.
Scavenger experiments
The role of individual reactive species in the degradation mechanism was investigated through a series of scavenger experiments conducted under both dark and UV-A illuminated conditions. Prior to the start of the reaction, the appropriate scavenger was added directly into the reaction mixture containing 20 mg of catalyst dispersed in 70 mL of a 4 mg L−1 DCF solution. The mixture was then subjected to the standard procedure: a 60-minute equilibration period in the dark followed by 180 minutes of UV-A illumination (365 nm, 3.25 mW cm−2). The following scavengers were applied at a concentration of 1.0 mmol L−1: disodium ethylenediaminetetraacetate (EDTA-2Na) for hole (h+) quenching, isopropanol (IPA) as a scavenger of hydroxyl radicals (·OH), silver nitrate (AgNO3) for electron (e−) trapping, and p-benzoquinone (p-BQ) to scavenge superoxide radicals (O2·−). Additionally, terephthalic acid (TA) was employed as a selective probe for ·OH detection22 at a concentration of 4 mg L−1, equivalent to that of DCF. The formation of 2-hydroxyterephthalic acid (2-OHTA), the fluorescent product of TA oxidation by ·OH radicals, was monitored by high-performance liquid chromatography (HPLC), as detailed in section S3 of the ESI.† All scavenger tests were conducted in parallel with the reference experiments and evaluated comparatively to assess the contribution of each reactive species to the overall degradation process.Results and discussion
Materials characterization
In the CuO-modified TiO2/MnOx composites, various phases were identified: α-Mn2O3 (bixbyite, PDF# 41-1442, space group P21/c), Cu1.5Mn1.5O4 (cubic spinel-like copper–manganese oxide, PDF# 70-0260, space group C2/m), Mn5O8 also known as mixed-valence manganese oxide (PDF# 39-1218, space group C2/m), comprising both Mn3+ and Mn4+ oxidation states, and Ti0.9Mn0.1O2 (titanium manganese oxide, PDF# 04-022-6533, space group I41/amd). In contrast, the CuO-modified titanium oxide (Cu/TiO2-HT) exhibited the anatase phase of TiO2 (PDF# 00-021-1272, space group I41/amd) and monoclinic CuO (tenorite, PDF# 00-048-1548, space group C2/c). Similarly, Cu/MnOx-HT consisted of Bixbyite, Cu1.5Mn1.5O4, and Mn5O8.The formation of the body-centered cubic bixbyite phase (α-Mn2O3) indicates the presence of Mn3+ ions in [MnO6] octahedral sites, which enhances catalytic activity.23 Modification with CuO led to the formation of a cubic spinel-type Cu1.5Mn1.5O4 phase, indicating a strong interaction between CuO and MnOx and the development of Cu–O–Mn linkages. This spinel structure is considered the primary active phase in CuO-modified TiO2/MnOx catalysts, as it enables the redox cycle Cu2+ + Mn3+ ↔ Cu+ + Mn4+, which facilitates catalytic oxidation reactions.24,25
The formation of titanium manganese oxides (Ti0.9Mn0.1O2) in the composites suggests Mn substitution for Ti in the anatase structure, consistent with prior studies.26 The Mn5O8 phase, which has a layered birnessite-type structure consisting of [Mn34+O8]4− layers with vacant cationic sites surrounded by Mn2+ cations and oxygen anions, was also identified.27,28
In Cu/TiO2-HT, anatase TiO2 and tenorite (CuO) combination significantly influences catalytic and photocatalytic performance.29 Tenorite provides additional active sites, facilitating redox reactions, and its interaction with anatase creates p–n heterojunctions, enhancing charge separation and transfer.30
Fig. S1† presents the results of the quantitative phase analysis (QPA). Cu1.5Mn1.5O4 content was similar across all manganese-containing samples (∼9–12%), indicating good interaction between CuO and MnOx. The highest proportion of Ti0.9Mn0.1O2 (∼77.5%) was found in the Cu/8Ti2Mn-HT composite, which also exhibited the lowest Mn5O8 content (∼9%).
The phase composition and crystal parameters obtained from XRD via Rietveld refinement and the Scherrer method are summarized in Table S3.† The lattice parameters for various phases varied between samples, influenced by modification with CuO and differing Ti/Mn molar ratios.
For the α-Mn2O3 (bixbyite) phase, the lattice parameter “a” varied slightly from 9.4078 Å to 9.4134 Å, depending on Cu incorporation and Ti/Mn ratio variations. In contrast, the Cu1.5Mn1.5O4 phase showed minimal lattice parameter changes (“a” ranged from 8.2855 Å to 8.2874 Å), indicating a stable structure regardless of synthesis conditions. Mn5O8 displayed more significant fluctuations, with “a” ranging from 10.3640 Å to 10.3882 Å, “b” from 5.6757 Å to 5.7416 Å, and “c” from 4.8350 Å to 4.8825 Å, reflecting structural adjustments due to varying Mn content. The Ti0.9Mn0.1O2 phase exhibited lattice parameter variations, with “a” from 3.7860 Å to 3.7922 Å and “c” from 9.3059 Å to 9.4546 Å, indicating Mn incorporation's effect on the TiO2 lattice.
Modification with CuO impacted lattice parameters due to the larger Cu2+ ions (ionic radius ∼0.73 Å),31 compared to Ti4+ (ionic radius ∼0.605 Å)32 and Mn4+ (ionic radius ∼0.53 Å),33 causing lattice expansion. Higher Mn content caused more significant structural changes, which in turn may affect the catalytic and photocatalytic properties of the composites.
The N2-physisorption experiment determined the catalysts' surface area and pore size distribution. Fig. 2a displays the N2 adsorption–desorption isotherms of as-prepared catalysts. For most samples, the isotherms demonstrate a combination of type II and type IV behaviors, indicating macroporosity and mesoporosity.34 At lower relative pressures, the gradual increase in adsorption indicates multilayer adsorption on the external surface and larger pores, typical for type II isotherms. As the relative pressure increases, the isotherms begin to exhibit the hallmark of type IV behavior, characterized by capillary condensation within mesopores, particularly in the Cu/8Ti2Mn-HT and Cu/5Ti5Mn-HT samples, which point to significant mesoporous structures.
H3 hysteresis loops in these samples suggest the prevalence of slit-shaped pores. This type of hysteresis is typically associated with materials that have plate-like or layered particle structures, contributing to the formation of mesopores.35 However, the Cu/MnOx-HT sample exhibits a much smaller adsorption capacity, especially at higher P/P0, indicating a lower degree of mesoporosity and a more significant proportion of non-porous or macroporous surfaces, as evidenced by its low specific surface area and pore volume (Table 1).
| Sample | SSA, m2 g−1 | Vpore, cm3 g−1 |
|---|---|---|
| Cu/TiO2-HT | 54 | 0.10 |
| Cu/8Ti2Mn-HT | 77 | 0.31 |
| Cu/5Ti5Mn-HT | 82 | 0.26 |
| Cu/2Ti8Mn-HT | 47 | 0.19 |
| Cu/MnOx-HT | 19 | 0.04 |
The pore size distribution (PSD) curves, expressed as differential pore volume (dV/d(logd)) versus logarithmic pore diameter (Fig. 2b), provide valuable insight into the mesostructural organization of the CuO–modified TiO2–MnOx catalysts.36 This representation enhances the resolution of features within the mesoporous range and enables a more accurate comparison of pore architectures across samples with varying compositions.
The Cu/TiO2-HT sample exhibits a narrow, unimodal distribution centered at 4.6 nm, consistent with relatively uniform mesopores formed through the aggregation of TiO2 nanoparticles. In contrast, the Cu/MnOx-HT material shows a PSD maximum at approximately 3 nm, suggesting smaller mesopores resulting from densely packed manganese oxide domains with limited interstitial voids.
Compositional modification with both Ti and Mn leads to significantly broader PSD profiles. The Cu/5Ti5Mn-HT sample presents a single peak at ∼10.9 nm, reflecting the formation of more open mesopores within a mixed oxide matrix. Notably, both Cu/8Ti2Mn-HT and Cu/2Ti8Mn-HT samples show bimodal distributions, with primary peaks near ∼10.9 nm and secondary maxima at 15.9 nm and 16.1 nm, respectively. These bimodal profiles indicate the presence of hierarchical porosity, likely arising from the spatial heterogeneity of mixed oxide phases and variations in particle aggregation during synthesis.
The emergence of larger mesopores in Ti–Mn-containing systems suggests increased interparticle spacing and a less compact textural framework, which is beneficial for mass transport and reactant accessibility. The ability to tailor mesoporosity through compositional tuning is therefore a key structural feature contributing to the catalytic efficiency of these materials, which is consistent with the improved catalytic and photocatalytic degradation efficiencies observed in Fig. 9a.
X-ray photoelectron spectroscopy (XPS) was employed to evaluate the surface oxidation states and oxygen species in the copper oxide-modified titania-manganese oxide catalysts. High-resolution spectra for Cu 2p, Mn 2p3/2, Ti 2p, and O 1s are presented in Fig. S2–S5,† with fitted surface compositions summarized in Table 2.
| Samples | Mn2+ (%) | Mn3+ (%) | Mn4+ (%) | Mn3+/Mn4+ | Olatt-2/Ototal |
|---|---|---|---|---|---|
| Cu/TiO2-HT | 0 | 0 | 0 | 0 | 0.77 |
| Cu/MnOx-HT | 17.2 | 46.2 | 36.7 | 1.26 | 0.82 |
| Cu/2Ti8Mn-HT | 15.4 | 58.3 | 26.3 | 2.22 | 0.83 |
| Cu/5Ti5Mn-HT | 13.4 | 51.1 | 35.5 | 1.44 | 0.84 |
| Cu/8Ti2Mn-HT | 23.1 | 49.7 | 27.3 | 1.82 | 0.93 |
The Cu 2p spectra (Fig. S2†) display the expected spin–orbit doublets, with Cu 2p3/2 peaks between 933.2–934.2 eV and Cu 2p1/2 peaks at 952.5–954.3 eV, characteristic of Cu2+ in oxidized environments such as CuO.37
Following the method of Raja et al.,15 the Cu 2p3/2 peak was treated as a single feature without deconvolution. The presence of shake-up satellites at 942.8–943.5 eV further confirms the dominant Cu2+ character, while the absence of features around 932.0 eV and lack of satellites rule out a significant Cu+ contribution.38 Although the Cu LMM Auger signal provides valuable insight into oxidation state,39 it overlaps with Ti 2p under Al Kα excitation and was not included due to the strong Ti matrix signal.
The Mn 2p3/2 spectra (Fig. S3†) were deconvoluted into components assigned to Mn2+ (639.8–640.7 eV), Mn3+ (641.6–642.3 eV), and Mn4+ (643.4–643.9 eV), in agreement with values reported for mixed-valence manganese oxides.40,41 The peak assignment follows the approach adopted by Raja et al.,15 while the fitting parameters (binding energies and FWHM) were determined empirically based on the experimental spectra. Quantification of the oxidation states was performed using the integrated areas of the fitted peaks. The resulting analysis revealed that Mn3+ and Mn4+ are the dominant surface species, while Mn2+ (13.4–23.1%) likely arises from partial surface reduction during synthesis or Cu-induced redox shifts.42–44
The calculated Mn3+/Mn4+ ratio increases with Ti content (from 1.26 in Cu/MnOx-HT to 2.22 in Cu/2Ti8Mn-HT), suggesting that Ti incorporation promotes Mn3+ stabilization, potentially via structural distortion, changes in local coordination, or electronic interaction with the TiO2 matrix.43 However, it is important to note that quantification of Mn oxidation states based solely on the Mn 2p3/2 region is intrinsically limited due to significant peak overlap, multiplet splitting, and final-state screening effects.45 Although Mn 2p3/2 provides the main basis for oxidation state evaluation, we also examined the Mn 3s multiplet splitting, which can offer complementary insight into average Mn oxidation states. However, the signal was significantly attenuated in all samples due to the inherently low relative sensitivity of Mn 3s (approximately 20× lower than Mn 2p), resulting in insufficient spectral resolution. As such, Mn 3s data were excluded from the final analysis, and this limitation is now explicitly noted.
Accordingly, the Mn3+/Mn4+ ratios presented in Table 2 are interpreted semi-quantitatively and are used solely to track relative compositional trends across the sample series, rather than to establish absolute oxidation state distributions. This clarification is explicitly stated to ensure appropriate interpretation of the data.
A systematic shift in Mn 2p binding energies was also observed across the series. Compared to the Cu/MnOx-HT reference (Mn3+ at 641.6 eV), Mn3+ peaks in Ti-containing catalysts appeared progressively shifted to higher energies—641.8 eV in Cu/2Ti8Mn-HT, 642.1 eV in Cu/5Ti5Mn-HT, and 642.3 eV in Cu/8Ti2Mn-HT. Similar shifts were observed for Mn2+ and Mn4+ species. These positive shifts may reflect increased Mn–O–Ti coupling, localized electron withdrawal, enhanced final-state screening, or charge redistribution at the oxide interface.46,47
The Ti 2p spectra (Fig. S4†) confirmed Ti4+ in Cu/TiO2-HT with Ti 2p3/2 and Ti 2p1/2 peaks at 459.5 and 465.0 eV, respectively.48 In the Mn-containing composites, Ti 2p3/2 was shifted to lower BE (457.9–458.4 eV), indicative of partial Ti4+ → Ti3+ reduction, likely caused by oxygen vacancy formation and redox interaction with Cu or Mn.
Importantly, no asymmetry or low-BE shoulder was observed in the Ti 2p3/2 peaks of the Mn-containing samples. Therefore, the spectra were not deconvoluted into separate Ti3+ and Ti4+ components.
The presence of Ti3+ was inferred from the systematic negative shifts observed in the Ti 2p binding energies, which are commonly associated with substoichiometric titanium environments and the formation of oxygen vacancies. This interpretation aligns with the findings of Wang et al.,49 who demonstrated that Ti3+ species formed via vacancy generation exhibit distinctly lower Ti 2p binding energies due to local electronic redistribution and final-state effects. In our system, these features likely reflect interfacial Mn–O–Ti and Cu–O–Ti interactions, reinforcing the role of TiO2 as a redox-active support.
The introduction of Cu into the Ti–Mn composite matrix appears to influence the manganese redox state, with an elevated Mn3+/Mn4+ ratio observed in the ternary systems. This is consistent with a redox interplay involving equilibria such as:50
| Mn3+ + Cu2+ ⇌ Mn4+ + Cu |
Such interactions may help stabilize higher-valent Mn species by facilitating the faster reduction of Cu2+ to Cu+, thereby mitigating Mn4+ over-reduction and preserving redox functionality.
The O 1s XPS spectra (Fig. S5†) of the CuO–TiO2–MnOx catalysts were deconvoluted into three distinct components located at approximately 527.4–528.8 eV, 529.4–530.8 eV, and 531.6–532.9 eV. The lowest binding energy component is attributed to lattice oxygen species (Olatt-1), corresponding to oxygen in well-ordered bulk oxide environments. The intermediate peak (Olatt-2) is ascribed to surface-associated lattice oxygen, which is generally more labile and redox-active due to under-coordination or proximity to oxygen vacancies. The high binding energy signal is assigned to surface-adsorbed oxygen species (Oads), including hydroxyl groups, chemisorbed oxygen, and possibly trace amounts of molecular water or carbonates.
This three-component interpretation is consistent with prior studies on FeOx and perovskite-type La1−xCaxCoO3 systems, where O 1s signals at ≤529 eV are linked to lattice oxygen (O2−), while peaks near 530.6–531.6 eV are attributed to adsorbed oxygen forms such as O22−, O−, and OH−.51,52
Systematic shifts in binding energy across the sample series reflect changes in surface composition, defect density, and local electronic structure. For example, the Olatt-1 signal shifts from 528.8 eV in Cu/TiO2-HT to 527.4 eV in Cu/MnOx-HT, indicating increased lattice disorder and higher oxygen vacancy content in Mn-rich samples.53
To assess the relative abundance of redox-accessible lattice oxygen, the surface fraction of Olatt-2 was evaluated using the empirical ratio Olatt-2/Ototal, where Ototal = Olatt-1 + Olatt-2 + Oads. As summarized in Table 2, this ratio increases progressively across the catalyst series, reaching a maximum value of 0.93 for Cu/8Ti2Mn-HT. This suggests a greater availability of structurally labile oxygen species at the catalyst surface. The potential role of these species in facilitating non-radical oxidation pathways under dark conditions is further elaborated in the discussion on mechanistic insights into catalytic and photocatalytic oxidation of DCF.
The reduction properties were further investigated using hydrogen temperature-programmed reduction (H2-TPR), with the interpretation provided in the ESI† in section S4. The deconvoluted TPR profiles are presented in Fig. S6.† The H2-TPR analysis of CuO-modified TiO2/MnOx catalysts revealed distinct stepwise reduction behaviors, with lower reduction temperatures for Cu/8Ti2Mn-HT and Cu/5Ti5Mn-HT, suggesting strong interactions between CuO and the TiO2/MnOx support, which correlate with enhanced catalytic and photocatalytic activity due to the optimized redox properties of both Cu and Mn species.
The catalysts were characterized for morphology and elemental distribution using transmission electron microscopy (TEM) and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) coupled with an energy dispersive X-ray spectroscopy (EDS) detector. Fig. 3–7 present TEM, HAADF-STEM, and EDS images of the catalysts in their as-prepared states.
Fig. 3a and b illustrates the TEM micrograph and EDS spectrum for CuO-modified TiO2 (Cu/TiO2-HT). The TEM micrograph (Fig. 3a) reveals non-uniformly shaped TiO2 nanoparticles. The HAADF-STEM image (Fig. 3c) and EDS elemental maps (Fig. 3d and e), including the relative atomic number of elements, indicate uneven dispersion of Cu particles within TiO2 aggregates, averaging 12.3 at%. In contrast, CuO-modified MnOx catalyst (Cu/MnOx-HT) exhibits irregularly shaped aggregates with randomly dispersed Cu particles, as shown in Fig. 4a and e. The HAADF-STEM image (Fig. 4c), EDS elemental maps (Fig. 4d and e), and atomic number insert reveal an average Cu content of 2.7 at%.
Fig. 5a and b presents the TEM micrograph and EDS spectrum for CuO-modified mixed TiO2/MnOx catalyst with a Ti:Mn ratio 2:8 (Cu/2Ti8Mn-HT). The TEM micrograph (Fig. 5a) shows irregularly shaped aggregates with TiO2 nanoparticles distributed homogeneously on MnOx surfaces. The HAADF-STEM image (Fig. 5c), EDS elemental maps (Fig. 5d and e), and atomic number insert demonstrate an even distribution of Cu particles on the MnOx surface, averaging 4.9 at%. Similarly, Fig. 6a and b depict the TEM micrograph and EDS spectrum for CuO-modified mixed TiO2/MnOx catalyst with a Ti:Mn ratio of 1:1 (Cu/5Ti5Mn-HT), showing irregularly shaped aggregates with uneven distribution of Cu particles on MnOx surfaces. TiO2 nanoparticles are more regularly distributed. The average Cu content was 8 at%.
Vibrational spectroscopies
Raman and FTIR spectroscopies were employed to elucidate the phase composition, structural integrity, and local bonding environments within CuO-modified TiO2/MnOx composites.The Raman spectra (Fig. 8a) primarily probe the long-range order and symmetry of the TiO2 lattice. In Cu/TiO2-HT, well-defined bands at 147, 200, 390, 509, and 639 cm−1 are consistent with the Eg(1), Eg(2), B1g(1), A1g + B1g(2), and Eg(3) modes of crystalline anatase, respectively. These bands originate from symmetric and asymmetric stretching and bending vibrations of the O–Ti–O framework,54,55 confirming the preservation of a highly ordered anatase phase. Upon Mn incorporation, these modes become increasingly attenuated and broadened, indicating lattice distortion, diminished crystallinity, or the formation of local defects. Notably, the Ti–O stretching band around 600–640 cm−1 remains evident across all samples, with spectral deconvolution in Cu/8Ti2Mn-HT revealing a bimodal distribution attributable to differing coordination geometries of oxygen species at surface and bulk sites.56
FTIR spectroscopy (Fig. 8b) complements the Raman data by probing short-range interactions and surface-bound functionalities. The spectra exhibit three intense bands in the 2800–3000 cm−1 region, assigned to C–H stretching modes: symmetric methylene (νs CH2, ∼2850 cm−1), asymmetric methylene (νas CH2, ∼2925 cm−1), and asymmetric methyl (νas CH3, ∼2955–2960 cm−1) vibrations.57 These signals are indicative of residual organic species, likely originating from the CTAB surfactant or physisorbed hydrocarbons. The fingerprint region (500–800 cm−1) reveals characteristic metal–oxygen stretching modes: O–Mn–O (∼558 cm−1),58 Cu–O (∼596 cm−1),59 and Ti–O (∼722 cm−1),60 consistent with the formation of composite oxide linkages. Variations in peak intensity and position across the series reflect differences in the extent of Mn incorporation and the resulting modifications to the local bonding environment.
Infrared bands located near 1378, 1465, and 1594 cm−1 are attributed to residual nitrate and carbonate species, retained from incomplete precursor decomposition.61,62 A distinct absorption near 1730 cm−1 corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, consistent with carboxylic or other carbonyl-containing functionalities, possibly originating from organic byproducts of synthesis.63
O stretching vibrations, consistent with carboxylic or other carbonyl-containing functionalities, possibly originating from organic byproducts of synthesis.63
The combined spectroscopic evidence confirms that Mn doping perturbs the anatase lattice, reduces crystallinity, and introduces surface heterogeneities. The emergence of Mn–O and Cu–O vibrational signatures, together with the attenuation of TiO2 phonon modes and the presence of surface-bound organic and inorganic residues, underscores the structural complexity of the composite catalysts. These findings corroborate XRD analysis (Fig. 1) and highlight the role of composition and synthetic history in determining local structure and surface chemistry.
 | ||
| Fig. 1 XRPD patterns of synthesized CuO-modified TiO2, MnOx, and their composites. | ||
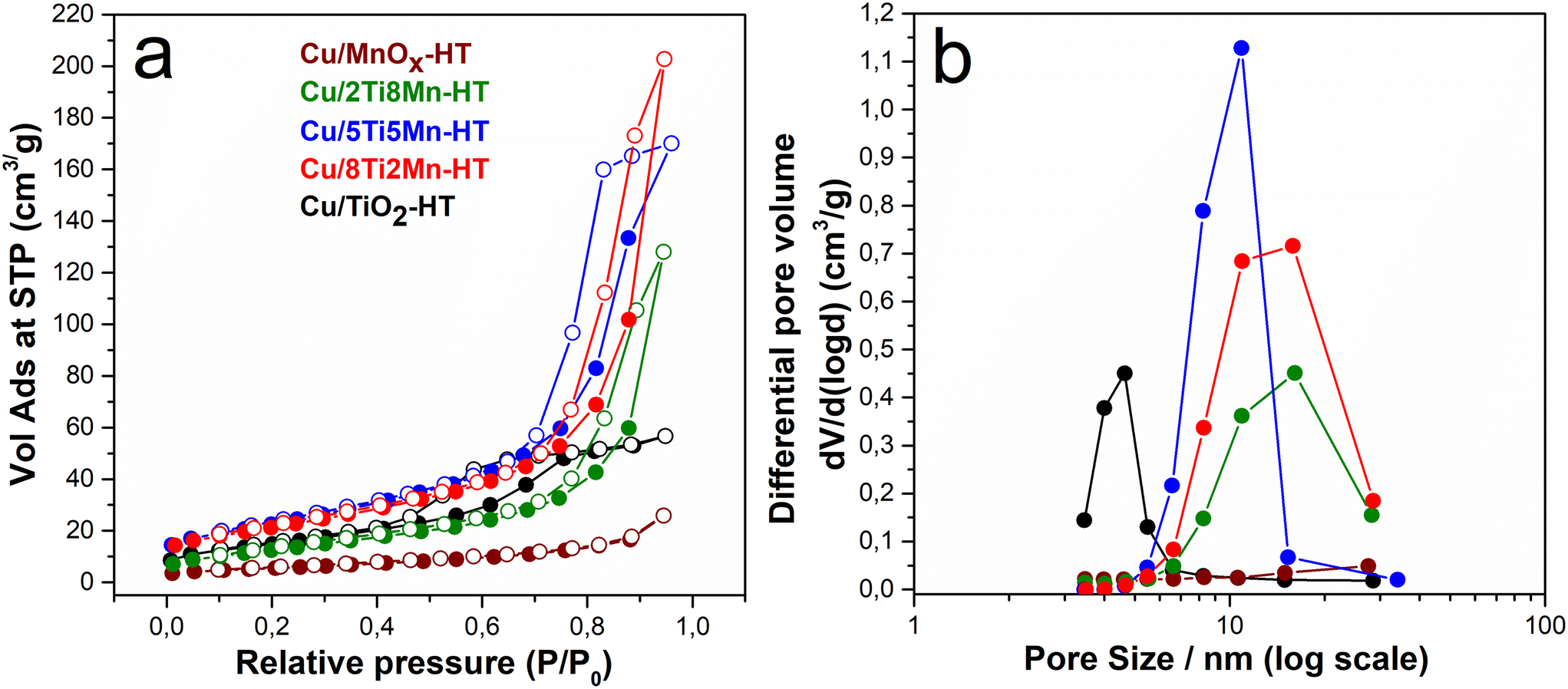 | ||
| Fig. 2 Sorption isotherms and pore size distribution of CuO-modified TiO2, MnOx, and their composites. | ||
 | ||
| Fig. 3 Morphology of Cu/TiO2-HT catalyst: (a) TEM micrograph, (b) EDS spectrum with elemental quantification, (c) HAADF-STEM micrograph, (d) EDS elemental distribution map for Ti, and (e) Ti/Cu systems. | ||
 | ||
| Fig. 4 Morphology of Cu/MnOx-HT catalyst: (a) TEM micrograph, (b) EDS spectrum with elemental quantification, (c) HAADF-STEM micrograph, (d) EDS elemental distribution map for Mn, and (e) Mn/Cu systems. | ||
 | ||
| Fig. 5 Morphology of Cu/2Ti8Mn-HT catalyst: (a) TEM micrograph, (b) EDS spectrum with elemental quantification, (c) HAADF-STEM micrograph, (d) EDS elemental distribution map for Ti/Mn, and (e) Mn/Cu systems. | ||
 | ||
| Fig. 6 Morphology of Cu/5Ti5Mn-HT catalyst: (a) TEM micrograph, (b) EDS spectrum with elemental quantification, (c) HAADF-STEM micrograph, (d) EDS elemental distribution map for Ti/Mn, and (e) Mn/Cu systems. | ||
 | ||
| Fig. 7 Morphology of Cu/8Ti2Mn-HT catalyst: (a) TEM micrograph, (b) EDS spectrum with elemental quantification, (c) HAADF-STEM micrograph, (d) EDS elemental distribution map for Ti/Mn, and (e) Mn/Cu systems. | ||
 | ||
| Fig. 8 Raman (a) and FTIR (b) spectra of CuO-modified TiO2, MnOx, and their composites. | ||
In contrast, the Cu-doped mixed catalyst with a higher TiO2 proportion (Cu/8Ti2Mn-HT) forms large aggregates with a bounded structure (Fig. 7a), where TiO2 nanoparticles are uniformly distributed. The average Cu content was 7.2 at%.
Catalytic performance in DCF degradation
The catalytic efficiency of CuO-modified TiO2/MnOx composites was assessed under both dark and UV-A conditions. Kinetic parameters, including dark-phase DCF conversion (DCFcat, %), total conversion (TC), and concentrations of the main degradation product, 5-hydroxy-diclofenac (5-OH-DCF), after 60 and 240 minutes, are summarized in Table S4.†Degradation rate constants (k) were calculated using a pseudo-first-order model: [DCF]t = [DCF]o * e(−k*t), where [DCF]t and [DCF]o represent the DCF concentrations at time t and initially, respectively, and k denotes the pseudo-first-order rate constant (min−1).
As shown in Fig. 9a, all CuO–TiO2–MnOx catalysts exhibited significant activity in both regimes, with pronounced degradation occurring even in the dark (within 60 minutes), followed by enhanced removal under UV-A. These effects are attributed to synergistic redox and photoinduced processes occurring at the newly formed oxide interfaces.
 | ||
| Fig. 9 (a) Normalized kinetic curves of catalytic/UV-A photocatalytic degradation of DCF on CuO-modified TiO2, MnOx, and their composites; (b) the course of the concentration of the main degradation product 5-OH-DCF over time; (c) dark/UV-A DCF normalized degradation curve and (d) formation/degradation of 5-OH-DCF product for Cu/2Ti8Mn-HT catalyst. | ||
Dark-phase activity followed the order: Cu/8Ti2Mn-HT (∼0.105 min−1) > Cu/5Ti5Mn-HT (∼0.079 min−1) > Cu/2Ti8Mn-HT (∼0.023 min−1) > Cu/MnOx-HT (∼0.017 min−1) > Cu/TiO2-HT (∼0.0053 min−1). This trend correlates with the increasing Ti content in the composite. Total DCF removal approached 100% for Cu/Ti–Mn composites, while individual oxides also showed notable conversion: Cu/MnOx-HT ∼87.9%, Cu/TiO2-HT ∼54%.
Although photocatalytic performance is partially masked by efficient dark-phase degradation, the UV-A rate constant of Cu/2Ti8Mn-HT (∼0.013 min−1) exceeded that of Cu/TiO2-HT and Cu/MnOx-HT (∼0.007 min−1). Cu/5Ti5Mn-HT and Cu/8Ti2Mn-HT exhibited the highest dark-phase conversion (∼99.8% and ∼99.4%), while Cu/TiO2 showed the strongest UV-A response (∼42%) despite limited dark activity (∼12%). The observed photocatalytic efficiency thus strongly depends on the extent of prior dark-phase degradation. No degradation occurred in control samples without catalyst under either dark or UV-A irradiation (Fig. S7†).
All catalysts promoted the formation of 5-OH-DCF, as confirmed by HPLC-DAD and LC-MS/MS (Fig. S11†). Concentration profiles in Fig. 9b highlight the dependence on catalyst composition, with Cu/5Ti5Mn-HT showing the highest 5-OH-DCF accumulation (∼145 μg L−1·60 min−1). UV-A further degraded 5-OH-DCF, particularly for Cu/5Ti5Mn-HT and Cu/8Ti2Mn-HT, where no residual product remained. In contrast, Cu/MnOx-HT generated 5-OH-DCF under both dark (∼59 μg L−1) and UV (∼73 μg L−1) conditions.
Fig. 9c and d illustrate DCF and 5-OH-DCF behavior under alternating dark and UV-A exposure for Cu/2Ti8Mn-HT. UV-A consistently accelerated degradation, confirming the composite's dual-mode catalytic function. Due to its balanced activity in both regimes, Cu/2Ti8Mn-HT was selected for further mechanistic investigation.
Influence of surface acid–base properties and initial pH on DCF degradation efficiency
The acid–base properties, including the point of zero charge (pHPZC) and total titratable hydroxyl groups (TOTH), were evaluated through potentiometric titration, as detailed in the ESI† in section S6.The differences in number of surface hydroxyl, as summarized in Table S7,† suggest that higher hydroxyl group content (e.g., in Cu/5Ti5Mn-HT) corresponds to increased catalytic activity, facilitating proton transfer reactions. For detailed calculations and TOTH curves, see ESI† eqn (S1) and (S2), and Fig. S17.
The pH values of the aqueous suspensions of these catalysts, detailed in Table S5,† reflect the varying surface chemistry of the catalysts, which influences their interaction with DCF and degradation products. Notably, the pH of the aqueous suspensions was not adjusted prior to measurement. The pH values indicate that Cu/TiO2-HT sample exhibited a slightly acidic pH of 5.65, while Cu/MnOx-HT was more neutral at 7.56. The pH values for the mixed CuO–TiO2–MnO2 composites ranged between 6.41 and 6.51, indicating their balanced surface properties contributing to their catalytic activity.
The graph in Fig. 10 illustrates the kinetics of DCF degradation using the Cu/2Ti8Mn-HT catalyst under various pH conditions, including without pH adjustment, at pH 4, pH 7, and pH 9. This catalyst was chosen for its optimal balance between catalytic oxidation and photocatalytic degradation of DCF, as in previous cases.
 | ||
| Fig. 10 Effect of pH on catalytic and photocatalytic degradation of diclofenac using Cu/2Ti8Mn-HT catalyst: (a) normalized kinetic curves of diclofenac degradation; (b) concentration trend of 5-OH-DCF, both in dark and light conditions. | ||
In graph in Fig. 10a, the catalytic oxidation of diclofenac in the dark phase varies with pH. At pH 4, the degradation rate is the highest (kdark ∼0.028 min−1), followed by the rate observed without pH adjustment (kdark ∼0.023 min−1). The efficiency decreases at pH 7 (kdark ∼0.016 min−1) and pH 9 (kdark ∼0.008 min−1). This is linked to the surface charge of the catalyst relative to its point of zero charge (pHPZC = 4.84).
Diclofenac has a pKa of 4.15, meaning it is predominantly ionized above this pH and non-ionized below it. At pH levels above its pKa, such as 7 and 9, diclofenac exists mainly in its anionic form, leading to repulsive interactions with the negatively charged catalyst surface at pH 9. In contrast, at pH 4, diclofenac is closer to its non-ionized form, favoring adsorption on the positively charged surface and enhancing degradation.64
Under UV-A light, photocatalytic activity increases degradation rates across all pH conditions. The highest efficiency is observed at pH 4, followed by unadjusted pH (pH ∼6.5, see Table S5†), pH 7, and pH 9.
Graph in Fig. 10b shows the formation and degradation of the 5-OH-DCF oxidation product. In the dark phase, 5-OH-DCF concentration increases as diclofenac is oxidized. Under UV-A light, the concentration trends vary with pH. At pH 4, the product concentration peaks and then decreases rapidly, indicating effective photocatalytic degradation of diclofenac and its oxidation product. Without pH adjustment, a similar but less pronounced trend was observed. At pH 7, the concentration remains relatively stable, reflecting moderate efficiency. At pH 9, the concentration initially increases and stabilizes, suggesting limited degradation due to repulsive interactions at the negatively charged surface.
Reusability of CuO-modified TiO2–MnOx catalysts
To evaluate the reusability and operational stability of the Cu/5Ti5Mn-HT catalyst, which exhibited the highest catalytic activity among the tested materials, four consecutive degradation cycles of DCF were performed under identical reaction conditions (see experimental section). After each cycle, the catalyst was recovered by centrifugation, washed thoroughly with deionized water, and directly reused without drying or thermal activation.The results of these recycling experiments are summarized in Fig. S8,† which displays both time-resolved kinetic curves (Fig. S8a†) and the corresponding degradation efficiencies after 60 minutes (inset). The apparent first-order rate constants under dark conditions decreased progressively from 0.078 to 0.056, 0.049, and finally 0.040 min−1, indicating a gradual decline in catalytic activity over the four cycles. This reduction can be primarily attributed to minor mechanical losses during the recovery and washing steps, experimentally determined to be approximately 5 wt%, rather than deactivation of the active sites.
The inset bar graph in Fig. S8a† provides a clear quantitative comparison of degradation efficiency at the 60-minute mark. The activity decreased only slightly, from 97.6% in the first cycle to 90.4% in the fourth, reflecting a net reduction of just ∼7.2%. Despite the observed decrease in reaction rate constants, the high overall DCF conversion values confirm that the catalyst retains excellent functional performance after multiple uses. This also suggests that mass transfer and surface reaction steps remain largely unaffected by recycling.
Furthermore, Fig. S8b† illustrates the evolution of the main transformation product, 5-hydroxy-diclofenac, across all four cycles. The consistent concentration-time profiles of this intermediate—both in terms of peak concentration and decay trend—indicate that the reaction mechanism and selectivity remain unaltered. This observation reinforces the conclusion that the redox-active sites responsible for oxygen activation and DCF transformation remain chemically stable throughout the recycling process.
Taken together, these results demonstrate that the Cu/5Ti5Mn-HT catalyst exhibits excellent recyclability, with high degradation efficiency, stable product formation behavior, and minimal structural or chemical degradation. This makes it a promising candidate for practical applications in water purification technologies. Further improvements in handling or immobilization may help minimize material loss and extend long-term performance.
Identification of reactive species
The ROS in the CuO-modified TiO2/MnOx system were investigated using scavenger experiments with Cu/2Ti8Mn-HT, chosen for its balanced performance in both catalytic and photocatalytic processes (Fig. 9a). Fig. 11 and Table S4† show the impact of various scavengers—p-benzoquinone (p-BQ) for superoxide anion radicals (O2·−),65 isopropanol (IPA) for hydroxyl radicals (·OH),66 silver nitrate (AgNO3) for electrons (e−), and EDTA-2Na for holes (h+)67—on the degradation of DCF. In dark conditions, the degradation rate (kdark) decreased most significantly with AgNO3 (to 0.0082 min−1), indicating that electrons play a crucial role. IPA also reduced kdark to 0.012 min−1, pointing to the involvement of hydroxyl radicals, though they appear less influential than electrons under dark conditions. Under UV-A illumination, the scavenger experiments highlighted the dominance of hydroxyl radicals (·OH), as the degradation rate dropped sharply from 0.013 min−1 to 0.0008 min−1 with the addition of IPA. The significant reduction with AgNO3 also suggested that electrons play an essential role under UV-A conditions, though hydroxyl radicals remain the primary reactive species. | ||
| Fig. 11 (a) Normalized kinetic curves of DCF degradation, and (b) formation of 5-hydroxy-diclofenac (5-OH-DCF) in the presence of scavengers using Cu/2Ti8Mn-HT catalyst. | ||
Terephthalic acid (TA) assays were employed to monitor hydroxyl radical (·OH) formation via the conversion of TA to 2-hydroxyterephthalic acid (2-OHTA) on the Cu/2Ti8Mn-HT sample.22
High-performance liquid chromatography (HPLC) was used to precisely quantify both TA and its hydroxylated product, 2-OHTA, in an aqueous solution. This method, described in detail in section S3 of the ESI,† offers a clear advantage over conventional fluorescence spectroscopy by eliminating interference and providing accurate measurements of TA and 2-OHTA, respectively.
As shown in Fig. S9a,† approximately 30% of TA is converted onto the surface of Cu/2Ti8Mn-HT during the dark phase (up to 60 minutes). Under UV-A illumination, a further 40% of TA is removed, suggesting continued photodegradation. Despite these significant decreases in TA concentration, the production of 2-OHTA remains very low, with only about 2.5% of TA converted to 2-OHTA, both in the dark and under UV-A conditions (Fig. S9b†).
This suggests that while there is a possibility of ·OH radicals forming even in the dark phase on Cu/2Ti8Mn-HT, other mechanisms are likely at play. Adsorption onto the catalyst surface or alternative degradation pathways appear to dominate, diverting ·OH radicals from significantly contributing to the hydroxylation of TA. The very low yield of 2-OHTA underscores the system's complexity, where competing processes, such as adsorption or non-radical degradation routes, limit the role of ·OH in TA conversion.
To elucidate the mechanisms of radical formation and their role in DCF degradation under dark and UV conditions, electron paramagnetic resonance (EPR) spectroscopy with spin trapping using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was employed. However, no free radicals were successfully trapped, possibly due to their extremely short lifetimes,68 leading to recombination before interacting with the spin trap, or their binding to the particle surface69 rather than being released into the environment. Phenyl-N-tert-butylnitrone (PBN) was used as an alternative scavenger to study radical formation. Data for the Cu/5Ti5Mn-HT sample, shown in Fig. S16a and b,† indicated the most significant signal after 15 minutes of UV irradiation. No adequate EPR signals were observed in the dark (data not shown).
Fig. S16a† displays the EPR spectrum at 250 K, revealing a complex resonance signal suggesting trapped adducts, likely PBN-OH and PBN-OOH. Due to low signal intensity and complexity, detailed analysis was challenging. An experiment at 113 K (Fig. S16b†) improved the signal-to-noise ratio, but the frozen state of the sample restricted tumbling effects, complicating detailed signal resolution and component analysis.
LC-MS/MS profiling of diclofenac transformation products
Time-resolved LC-MS/MS analysis was employed to identify intermediates and by-products generated during the catalytic degradation of DCF in both dark and under UV illumination. The Cu/2Ti8Mn-HT catalyst was selected for its optimal efficiency in DCF conversion across various conditions. Four distinct degradation products were identified during the dark phase of DCF degradation, with their concentrations (expressed as peak area) varying over time (Fig. S15†). These products are further degraded upon exposure to UV light, and their molecular structures and fragmentation patterns are summarized in Table S6.† A detailed interpretation of the mass spectra based on fragmentation analysis is provided in the ESI,† section S5.Diclofenac (DCF) degradation is initiated by hydroxylation, facilitated by surface–OH groups acting as active sites for adsorption and activation. Under dark conditions, non-radical oxygen species mediate the formation of 5-hydroxy-diclofenac (5-OH-DCF), followed by decarboxylation via intramolecular electron transfer to yield 2,6-dichloro-N-(o-tolyl)aniline (DP1). DP1 subsequently undergoes either hydroxylation to form 4-((2,6-dichlorophenyl)amino)-3-methylphenol (DP2) or dechlorination-cyclization, leading to 1-chloro-8-methyl-9H-carbazole (DP3). These transformation routes align with degradation mechanisms reported for DCF in manganese oxide systems.70–72 Upon UV-A irradiation, reactive oxygen species (primarily ·OH) accelerate the degradation of pre-formed intermediates (Fig. S15†), enhancing overall efficiency without altering the fundamental pathway. The proposed degradation mechanism is illustrated in Fig. 12.
 | ||
| Fig. 12 Proposed degradation mechanism of DCF by Cu/TiO2/MnOx catalysts. | ||
Mechanistic insights into catalytic and photocatalytic oxidation of DCF
The degradation of diclofenac (DCF) over CuO-modified TiO2/MnOx composites proceeds through distinct mechanisms under dark and UV-A illumination, driven by the tailored roles of each component—TiO2, MnOx, and CuO. These mechanisms were elucidated using kinetic studies, scavenger experiments, surface spectroscopy, and time-resolved LC-MS/MS analysis.Surface-sensitive XPS analysis (Table 2) provides a mechanistic basis for understanding the catalytic behavior of the CuO–TiO2–MnOx composites under dark conditions by elucidating the distribution of surface oxidation states and reactive oxygen species. The Mn3+/Mn4+ atomic ratios follow the order: Cu/5Ti5Mn-HT (1.44) < Cu/8Ti2Mn-HT (1.82) < Cu/2Ti8Mn-HT (2.22) < Cu/MnOx–HT (1.26), indicating compositional differences in the electronic structure and redox equilibria at the catalyst surface. The enrichment of Mn3+ species, which are commonly associated with the presence of oxygen vacancies and dynamic redox behavior, suggests enhanced electron transfer capability and increased capacity for oxygen activation.73
In parallel, the relative abundance of surface lattice oxygen—quantified by the Olatt-2/Ototal ratio—reaches its highest value in Cu/8Ti2Mn-HT (0.93), implying a greater availability of redox-accessible oxygen at or near the catalyst surface. This fraction reflects the relative amount of surface lattice oxygen, which is more reactive than the deeply embedded lattice oxygen (Olatt-1) and can participate in interfacial redox processes. Such surface lattice oxygen has been highlighted in recent literature as a key factor in catalytic oxidation reactions, where it promotes electron transfer and redox flexibility at defect-rich oxide interfaces.74,75
The convergence of elevated Mn3+ content and high surface lattice oxygen fraction reflects an electronically flexible surface environment that may facilitate non-radical oxidative pathways in the absence of photonic excitation.
Additional insight into the surface characteristics of the catalysts is provided by the total hydroxyl group content (TOTH, Table S7†), which reflects the relative abundance of surface –OH species formed via dissociative adsorption of water.76 The presence of surface hydroxyls may facilitate the initial adsorption and alignment of diclofenac at the solid–liquid interface, thereby supporting the subsequent activation steps involved in dark-phase transformation.
Fig. 13 illustrates the correlation between structural surface characteristics and catalytic performance under dark conditions. The degradation rate constant (kdark) exhibits a positive relationship with three surface-derived parameters: the Mn3+/Mn4+ atomic ratio, the relative fraction of surface-associated lattice oxygen (Olatt-2/Ototal), and the surface density of hydroxyl groups (q-OH, derived from TOTH). These descriptors collectively reflect the redox flexibility of the catalyst surface, the accessibility of reactive lattice oxygen, and the capacity for initial pollutant adsorption through hydroxyl-mediated interactions. The observed trends emphasize that the efficiency of non-radical oxidation in the dark is governed not by a single dominant factor, but rather by a synergistic interplay of redox-active metal centers, surface lattice oxygen, and surface –OH functionalities that together define the interfacial reactivity of the composite catalysts.
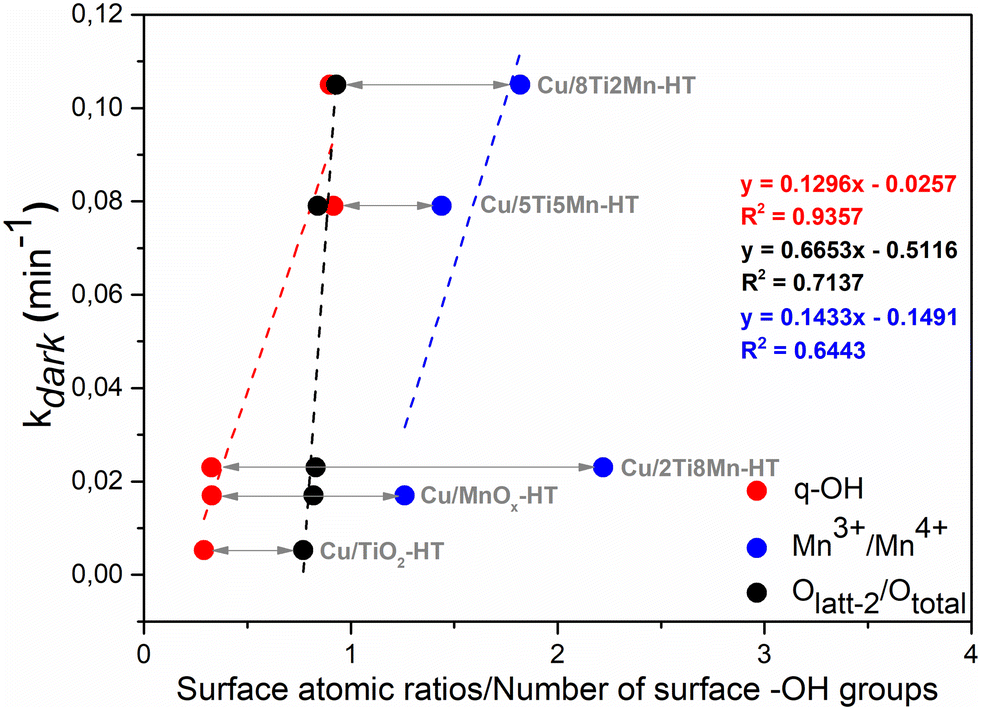 | ||
| Fig. 13 Correlations between surface atomic ratios and the number of surface hydroxyl groups per weight (q-OH) and DCF reaction rate under dark (kdark, min−1) for the individual phases and the composites. | ||
The oxidation mechanism was further confirmed by LC-MS/MS analysis, which identified several transformation products formed during DCF degradation in the dark. These include 5-hydroxy-diclofenac (5-OH-DCF) via hydroxylation, 2,6-dichloro-N-(o-tolyl)aniline (DP1) via decarboxylation, its hydroxylated derivative (DP2), and a dechlorinated and cyclized product, 1-chloro-8-methyl-9H-carbazole (DP3). These products (Table S6†) indicate that multiple oxidative pathways occur despite the absence of light and that they are consistent with previously reported Mn-based systems.70–72
While scavenger experiments showed partial inhibition in the presence of isopropanol (·OH scavenger), no significant formation of free radicals was observed by terephthalic acid probing (Fig. S9†) or EPR spectroscopy (Fig. S16†). These results confirm that the observed catalytic activity arises from non-radical oxygen species bound to the surface lattice oxygen, rather than from classical radical species such as ·OH or O2·−.
Based on a combination of spectroscopic, kinetic, and analytical observations, the most plausible mechanism for diclofenac (DCF) degradation under dark conditions has been proposed. In the absence of photonic excitation, the oxidation proceeds via a non-radical pathway involving redox-active surface species and reactive oxygen forms associated with the oxide lattice.
The process is initiated by the dissociative adsorption of water molecules on surface metal–oxygen bridges, generating terminal hydroxyl groups (Me–OH) on the catalyst surface.77
The abundance of these –OH groups, as quantified by TOTH analysis (Table S7†), indicates a high density of adsorption sites that may facilitate pollutant binding:
| H2O + Me−O−Me → 2Me−OH | (1) |
These hydroxyl groups interact with DCF via hydrogen bonding or electrostatic attraction, forming surface-bound complexes that facilitate interfacial electron transfer in the absence of light:
| DCF(aq) + Me−OH → DCF-OH(ads) | (2) |
This adsorption step enables pollutant pre-concentration and activation on the catalyst surface, which is critical for initiating the subsequent oxidation reactions without photonic excitation.
The catalytic activity is then sustained by reversible redox transitions within the MnOx and CuO domains. Manganese exists in a dynamic equilibrium between Mn3+ and Mn4+ oxidation states (Mn3+ → Mn4+ + e−), while CuO contributes through the Cu+ → Cu2+ + e− transition. These coupled redox cycles enable continuous electron donation to molecular oxygen adsorbed at oxygen vacancy sites (□), resulting in the formation of reactive non-radical oxygen species through the following cascade:78,79
| O2 + □ + e−→ O2−(ads) | (3) |
| O2−(ads) + e−→ O22−(ads) | (4) |
| O22−(ads) + e− → 2O−(ads) | (5) |
| O−(ads) + e− → O2−(latt) | (6) |
The resulting surface lattice oxygen species (O2−) serve as the primary oxidizing agents in the dark-phase process. In parallel, the TiO2 matrix stabilizes charge imbalances through the partial reduction of surface Ti4+ to Ti3+ (Ti4+ + e− → Ti3+), enabling temporary charge storage and local compensation during interfacial redox processes.32,80
Unlike conventional MnO2 systems, which rely solely on Mn4+ species and lack redox coupling with auxiliary metal oxides, the CuO–MnOx–TiO2 composite enables more effective electron flow and redox cycling. In this system, redox-active Cu2+/Cu+ couples bridge the Mn3+/Mn4+ redox system and enhance interfacial conductivity, resulting in significantly higher degradation rates even under light-free conditions. For example, Cu/8Ti2Mn-HT achieved a rate constant of kdark = 0.105 min−1, surpassing classical MnO2-based systems under similar conditions.81
Upon UV-A irradiation, the photocatalytic degradation of DCF proceeds through a synergistic mechanism enabled by the engineered p–n–p heterojunction architecture of the CuO–TiO2–MnOx composite. TiO2, with a wide bandgap (Ebg ≈ 3.0–3.2 eV),82 serves as the primary light-harvesting n-type semiconductor, while CuO (Ebg ≈ 1.4–1.7 eV)83 and MnOx (typically 1.3–1.8 eV)84 function as p-type components. The energetic coupling of these semiconductors creates interfacial electric fields upon Fermi-level equilibration, which facilitates directional charge carrier separation and suppresses electron–hole recombination.
Photoactivation occurs predominantly on TiO2, where band-gap excitation under UV-A light produces electron–hole pairs:85
| TiO2 + hν → e− + h+ | (7) |
These photogenerated carriers migrate across the heterojunction interfaces in a spatially resolved manner, initiating redox processes at the catalyst surface. Holes oxidize water or surface hydroxyls, generating hydroxyl radicals:86
| H+ + H2O → ·OH + H+ | (8) |
Simultaneously, electrons reduce adsorbed oxygen to form superoxide species:86
| e− + O2 → O2·− | (9) |
The involvement of both radical species in the degradation mechanism is corroborated by scavenger inhibition studies (Fig. 11), EPR spectroscopy (Fig. S16†), and TA probing (Fig. S9†).
Based on literature-supported band alignment models,87–89 electrons are transferred from the conduction band of TiO2 to adjacent CuO and MnOx domains, where they are transiently stabilized via redox cycling involving Cu2+/Cu+ and Mn4+/Mn3+ couples. In parallel, holes migrate toward the valence bands of MnOx and CuO, enabling sustained oxidation reactions.
In this mechanism, each component contributes distinct functionality: (i) CuO acts as a photoactive p-type semiconductor and efficient electron acceptor. Electrons reduce surface Cu2+ to Cu+, initiating a redox cycle that promotes superoxide generation:90,91
| e− + Cu2+ → Cu+ | (10) |
| Cu+ + O2 → Cu2+ + O2·− | (11) |
(ii) TiO2, as the central light absorber, mediates the photogeneration and directional transport of charge carriers.
(iii) MnOx serves as a redox-active domain and electron sink, supporting Mn4+/Mn3+ cycling and further superoxide formation:92
| e− + Mn4+ → Mn3+ | (12) |
| Mn3+ + O2 → Mn4+ + O2·− | (13) |
This redox interplay across Mn and Cu sites sustains ROS generation after initial charge separation. The pronounced photocatalytic activity of the Cu/2Ti8Mn-HT catalyst (kUV = 0.013 min−1, Fig. 9a) supports this mechanistic proposal. XPS analysis confirms the prevalence of surface Cu2+ and Mn4+ species (Table 2), as well as a high density of oxygen vacancies (O 1s ≈ 531.09 eV, Fig. S5†), both of which enhance O2 activation. Moreover, elevated surface hydroxylation (TOTH, Table S7†) facilitates ·OH generation.
Importantly, LC-MS/MS analysis (Fig. S15†) indicates that UV-A illumination does not induce the formation of new degradation products but accelerates the transformation of intermediates generated in the dark phase, along with residual DCF. This observation underscores the complementary relationship between both activation modes.
In summary, the enhanced photocatalytic degradation of diclofenac under UV-A light is governed by (i) efficient charge carrier generation on TiO2, (ii) spatially resolved charge separation via the CuO–MnOx–TiO2 p–n–p heterojunction, and (iii) redox-mediated oxygen activation through Cu2+/Cu+ and Mn4+/Mn3+ cycles. The integration of photophysical and redox functionalities enables persistent ROS formation and high photocatalytic efficiency.
Conclusion
In conclusion, CuO-modified TiO2/MnOx composites were successfully developed as multifunctional catalysts for the degradation of diclofenac sodium under both dark and UV-A irradiation. The materials exhibited dual-mode catalytic activity through two mechanistically distinct yet complementary pathways: (i) non-radical oxidation under dark conditions, enabled by redox-active metal centers, surface lattice oxygen, and surface hydroxyl functionalities, and (ii) radical-driven photocatalysis under UV-A light, initiated by photogenerated charge carriers.Under illumination, the formation of an interfacial p–n–p heterojunction between CuO, MnOx, and TiO2 established internal electric fields that facilitated directional charge separation—electrons migrating from the conduction band of TiO2 toward CuO and MnOx, and holes in the opposite direction. This charge carrier redistribution suppressed recombination and promoted continuous ROS generation via redox cycling involving Cu2+/Cu+ and Mn4+/Mn3+ pairs.
The mechanistic interpretation was supported by LC-MS/MS identification of transformation products, along with spectroscopic evidence of redox-active surface species and an increased abundance of surface hydroxyl groups.
The mechanistic interpretation was supported by LC-MS/MS identification of transformation products, as well as spectroscopic evidence of redox-active surface species, and enhanced hydroxylation.
Among the evaluated catalysts, Cu/5Ti5Mn-HT and Cu/8Ti2Mn-HT demonstrated the highest degradation efficiencies in the dark (∼99.8% and ∼99.4%, respectively), consistent with their elevated Mn3+/Mn4+ ratios, enriched surface lattice oxygen (Olatt-2/Ototal), and high hydroxyl group densities. Conversely, Cu/TiO2 exhibited superior photocatalytic performance under UV-A (∼42% removal), underscoring the influence of heterojunction architecture and interfacial conductivity on charge carrier utilization.
These findings underscore the versatility of CuO–MnOx–TiO2 composites as adaptable catalysts for pharmaceutical pollutant removal under variable environmental conditions and provide mechanistic insights to guide the design of next-generation oxide-based heterostructures with tunable redox and interfacial properties.
Data availability
The Institute of Inorganic Chemistry of the Czech Academy of Sciences (IIC) uses Fair Wizard (https://openscience.lib.cas.cz/fair-data/) to create the data management plan. Data are stored and archived at the digital repository of IIC, operated by the Institutional Data Repository of the Czech Academy of Sciences. We are willing to share research data with interested researchers. Data is available to all upon request; please contact the authors of this manuscript.Author contributions
Martin Šťastný: conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review and editing; Gloria Issa: conceptualization, data curation, formal analysis, funding acquisition, investigation, project administration, resources, visualization, writing – original draft, writing – review and editing; Sylvie Kříženecká: data curation, formal analysis, investigation, methodology, validation, writing – review and editing; Petr Bezdička: data curation, formal analysis, investigation, methodology, validation, writing – review and editing; Daniela Popelková: data curation, formal analysis, software, visualization, writing – review and editing; Martin Kormunda: data curation, formal analysis, investigation, visualization, writing – review and editing; Jakub Ederer: data curation, formal analysis, investigation, visualization, writing – review and editing; Daniel Bůžek: data curation, formal analysis, investigation, visualization, writing – review and editing; Jan Čundrle: data curation, formal analysis, investigation, methodology, validation, writing – original draft, writing – review and editing; Zdeněk Baďura: data curation, formal analysis, investigation, visualization, writing – review and editing; Jiří Henych: data curation, funding acquisition, project administration, resources, software, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the assistance provided by the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2023066. This article was funded by the Czech Academy of Sciences Strategy AV21 scheme as part of “The power of objects: Materiality between past and future” grant at the Institute of Inorganic Chemistry of the Czech Academy of Sciences. Additionally, support from the Bulgarian National Science Fund under Project No. KP-06-N59/12/12.11.2021 is gratefully acknowledged.References
- P. Sathishkumar, R. A. A. Meena, T. Palanisami, V. Ashokkumar, T. Palvannan and F. L. Gu, Sci. Total Environ., 2020, 698, 134057 CrossRef CAS PubMed.
- A. O. Oluwole, E. O. Omotola and O. S. Olatunji, BMC Chem., 2020, 14, 62 CrossRef CAS PubMed.
- P. Kumari and A. Kumar, Results in Surfaces and Interfaces, 2023, 11, 100122 CrossRef.
- E. Moctezuma, E. Leyva, C. Lara-Pérez, S. Noriega and A. Martínez-Richa, Top. Catal., 2020, 63, 601–615 CrossRef CAS.
- K. Fischer, M. Kühnert, R. Gläser and A. Schulze, RSC Adv., 2015, 5, 16340–16348 RSC.
- H. Li, B. Fu, H. Huang, S. Wu, J. Ge, J. Zhang, F. Li and P. Qu, Environ. Pollut. Bioavailability, 2022, 34, 395–406 CrossRef.
- I. J. De la Cruz, J. L. Rodríguez, S. I. Fuentes, H. Tiznado, J. L. Vazquez-Arce, I. Romero-Ibarra, J. I. Guzmán, C. and H. M. Gutiérrez, J. Environ. Chem. Eng., 2023, 11, 110753 CrossRef CAS.
- M. Šťastný, G. Issa, D. Popelková, J. Ederer, M. Kormunda, S. Kříženecká and J. Henych, Catal. Sci. Technol., 2021, 11, 1766–1779 RSC.
- Y. Ye, Q. Wang, H. Chen, L. Chen, L. Wang, L. Mo, L. Tan, Q. Wei and J. Xu, Res. Chem. Intermed., 2024, 50, 3351–3368 CrossRef CAS.
- M. Janczarek and E. Kowalska, Catalysts, 2017, 7, 317 CrossRef.
- D. A. Bopape, S. Mathobela, N. Matinise, D. E. Motaung and N. C. Hintsho-Mbita, Catalysts, 2023, 13, 163 CrossRef CAS.
- R. Solano, L. Mallarino, M. Mueses and A. Herrera, J. Environ. Chem. Eng., 2024, 12, 113576 CrossRef CAS.
- X. Luo, H. Liang, F. Qu, A. Ding, X. Cheng, C. Y. Tang and G. Li, Chemosphere, 2018, 200, 237–247 CrossRef CAS PubMed.
- T. Yu, Y. Sun, C. Zhe, W. Wang and P. Rao, J. Mater. Sci. Chem. Eng., 2017, 05, 12–25 CAS.
- S. Raja, M. S. Alphin, L. Sivachandiran, P. Singh, D. Damma and P. G. Smirniotis, Fuel, 2022, 307, 121886 CrossRef CAS.
- W. Luo, L. Yang, Z. Zhang, G. Cao, J. Li and B. Liu, ACS Appl. Nano Mater., 2024, 7, 6262–6272 CrossRef CAS.
- D. Fang, J. Xie, D. Mei, Y. Zhang, F. He, X. Liu and Y. Li, RSC Adv., 2014, 4, 25540 RSC.
- C. Sun, Y. Tang, F. Gao, J. Sun, K. Ma, C. Tang and L. Dong, Phys. Chem. Chem. Phys., 2015, 17, 15996–16006 RSC.
- T. Tsoncheva, A. Mileva, G. Issa, M. Dimitrov, D. Kovacheva, J. Henych, M. Kormunda, N. Scotti, M. Slušná, J. Tolasz and V. Štengl, J. Environ. Chem. Eng., 2018, 6, 2540–2550 CrossRef CAS.
- G. Issa, M. Dimitrov, R. Ivanova, M. Kormunda, D. Kovacheva and T. Tsoncheva, React. Kinet., Mech. Catal., 2022, 135, 105–121 CrossRef CAS.
- T. Tsoncheva, G. Issa, R. Ivanova, M. Dimitrov, D. Kovacheva, G. Atanasova and J. Henych, Symmetry, 2022, 14, 751 CrossRef CAS.
- T. Charbouillot, M. Brigante, G. Mailhot, P. R. Maddigapu, C. Minero and D. Vione, J. Photochem. Photobiol., A, 2011, 222, 70–76 CrossRef CAS.
- F. L. G. Silva, A. G. Veiga and N. M. F. Carvalho, Int. J. Hydrogen Energy, 2021, 46, 11677–11687 CrossRef CAS.
- X. Yang and Z. Zhang, ChemistrySelect, 2021, 6, 4837–4843 CrossRef CAS.
- R. Sun, S. Zhang, K. An, P. Song and Y. Liu, J. Fuel Chem. Technol., 2021, 49, 799–808 CrossRef CAS.
- S. Roy, B. Viswanath, M. S. Hegde and G. Madras, J. Phys. Chem. C, 2008, 112, 6002–6012 CrossRef CAS.
- Z. Zlatanova, D. Marinova, R. Kukeva, L. Mihaylov, D. Nihtianova and R. Stoyanova, J. Alloys Compd., 2021, 851, 156706 CrossRef CAS.
- P. Zhang, J. Liu, K. Page and A. Navrotsky, J. Am. Ceram. Soc., 2019, 102, 1394–1401 CrossRef CAS.
- G. Colón, M. Maicu, M. C. Hidalgo and J. A. Navío, Appl. Catal., B, 2006, 67, 41–51 CrossRef.
- A. I. M. A'srai, M. H. Razali, K. A. M. Amin and U. M. Osman, Dig. J. Nanomater. Biostruct., 2023, 18, 1005–1124 CrossRef.
- R. Kayestha, Sumati and K. Hajela, FEBS Lett., 1995, 368, 285–288 CrossRef CAS PubMed.
- K. McColl and F. Corà, J. Mater. Chem. A, 2019, 7, 3704–3713 RSC.
- Y. Zhu, S. Zhao, J. Wang and N. Zhang, J. Solid State Chem., 2023, 320, 123854 CrossRef CAS.
- K. S. W. Sing and R. T. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- P. Seekhiaw, S. Jantasee, P. Praserthdam and B. Jongsomjit, ACS Omega, 2023, 8, 32775–32783 CrossRef CAS PubMed.
- T. Asefa and V. Dubovoy, in Comprehensive Supramolecular Chemistry II, Elsevier, 2017, pp. 157–192 Search PubMed.
- P. V. F. de Sousa, A. F. de Oliveira, A. A. da Silva and R. P. Lopes, Environ. Sci. Pollut. Res., 2019, 26, 14883–14903 CrossRef CAS PubMed.
- M. Khan, M. I. Abdullah, A. Samad, Z. Shao, T. Mushiana, A. Akhtar, A. Hameed, N. Zhang, U. Schwingenschlögl and M. Ma, Small, 2023, 19(27), 2205499 CrossRef CAS PubMed.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- L. Han, W. Song, J. Ji, W. Zou, J. Sun, C. Tang, B. Xu and L. Dong, Chem. Phys. Impact, 2023, 6, 100227 CrossRef.
- G. Mitran, S. Chen and D. K. Seo, Mol. Catal., 2020, 483, 110704 CrossRef CAS.
- G. Koch, M. Hävecker, D. Teschner, S. J. Carey, Y. Wang, P. Kube, W. Hetaba, T. Lunkenbein, G. Auffermann, O. Timpe, F. Rosowski, R. Schlögl and A. Trunschke, ACS Catal., 2020, 10, 7007–7020 CrossRef CAS.
- L. G. Bousiakou, P. J. Dobson, T. Jurkin, I. Marić, O. Aldossary and M. Ivanda, J. King Saud Univ., Sci., 2022, 34, 101818 CrossRef.
- F. Cheng, J. Yang, L. Yan, J. Zhao, H. Zhao, H. Song and L. Chou, Eur. J. Inorg. Chem., 2019, 2019, 1236–1242 CrossRef CAS.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- M. Tsega, Indian J. Phys., 2025, 99, 177–189 CrossRef CAS.
- A. Sathe, M. A. Peck, C. Balasanthiran, M. A. Langell, R. M. Rioux and J. D. Hoefelmeyer, Inorg. Chim. Acta, 2014, 422, 8–13 CrossRef CAS.
- T. Jedsukontorn, T. Ueno, N. Saito and M. Hunsom, J. Alloys Compd., 2017, 726, 567–577 CrossRef CAS.
- J. Wang, D. N. Mueller and E. J. Crumlin, J. Eur. Ceram. Soc., 2024, 44, 116709 CrossRef CAS.
- D. L. Cocke and S. Vepřek, Solid State Commun., 1986, 57, 745–748 CrossRef CAS.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS.
- Y. Wei, B. Wang, R. Ren and R. Wang, Aerosol Air Qual. Res., 2023, 23, 220438 CrossRef CAS.
- S. Liu, C. Sun, J. Chen, J. Xiao and J.-L. Luo, ACS Catal., 2020, 10, 13437–13444 CrossRef CAS.
- S. K. Mukherjee and D. Mergel, J. Appl. Phys., 2013, 114, 013501 CrossRef.
- Y. Zhang, C. X. Harris, P. Wallenmeyer, J. Murowchick and X. Chen, J. Phys. Chem. C, 2013, 117, 24015–24022 CrossRef CAS.
- B. Taudul, F. Tielens and M. Calatayud, Nanomaterials, 2023, 13, 1856 CrossRef CAS PubMed.
- E. Calabrò and S. Magazù, Adv. Phys. Chem., 2013, 2013, 1–8 CrossRef.
- A. T. Mekuria, J. Nanotechnol. Nanomater., 2024, 5, 31–45 Search PubMed.
- N. Kumar, S. S. Parui, S. Limbu, D. K. Mahato, N. Tiwari and R. N. Chauhan, Mater. Today: Proc., 2021, 41, 237–241 CAS.
- D. Huang, S. Liao, S. Quan, L. Liu, Z. He, J. Wan and W. Zhou, J. Mater. Res., 2007, 22, 2389–2397 CrossRef CAS.
- R. L. Frost and S. Bahfenne, J. Raman Spectrosc., 2009, 40, 360–365 CrossRef CAS.
- A. Akbari, M. Khammar, D. Taherzadeh, A. Rajabian, A. Khorsand Zak and M. Darroudi, J. Mol. Struct., 2017, 1149, 771–776 CrossRef CAS.
- P. Wang, J. Zhao, Q. Zhao, X. Ma, X. Du, X. Hao, B. Tang, A. Abudula and G. Guan, J. Colloid Interface Sci., 2022, 607, 100–110 CrossRef CAS PubMed.
- A. Zaka, T. H. Ibrahim and M. Khamis, Desalin. Water Treat., 2021, 212, 401–414 CrossRef CAS.
- M. Zhu, J. Lu, Y. Hu, Y. Liu, S. Hu and C. Zhu, Environ. Sci. Pollut. Res., 2020, 27, 31289–31299 CrossRef CAS PubMed.
- D. Ewing and H. L. Walton, Radiat. Res., 1991, 126, 187 CrossRef CAS PubMed.
- M. Fakhrul Ridhwan Samsudin, L. T. Siang, S. Sufian, R. Bashiri, N. M. Mohamed and R. M. Ramli, Mater. Today: Proc., 2018, 5, 21703–21709 CAS.
- E. Braxton, D. J. Fox, B. G. Breeze, J. J. Tully, K. J. Levey, M. E. Newton and J. V. Macpherson, ACS Meas. Sci. Au, 2023, 3, 21–31 CrossRef CAS PubMed.
- Y. Nosaka and A. Nosaka, ACS Energy Lett., 2016, 1, 356–359 CrossRef CAS.
- H. Shi, G. Zhou, Y. Liu, Y. Fu, H. Wang and P. Wu, RSC Adv., 2019, 9, 31370–31377 RSC.
- V. Muelas-Ramos, M. J. Sampaio, C. G. Silva, J. Bedia, J. J. Rodriguez, J. L. Faria and C. Belver, J. Hazard. Mater., 2021, 416, 126199 CrossRef CAS PubMed.
- Y. Peng, H. Shi, Z. Wang, Y. Fu and Y. Liu, RSC Adv., 2021, 11, 6804–6817 RSC.
- K. Yang, C. Li, Q. Zhu, H. Wang and J. Qi, Nanomaterials, 2025, 15, 524 CrossRef CAS PubMed.
- H. Du, H. Luo, M. Jiang, X. Yan, F. Jiang and H. Chen, Appl. Catal., A, 2023, 664, 119348 CrossRef CAS.
- M. Li, H. Zhang, Z. Liu, Y. Su and C. Du, Chem. Eng. J., 2022, 450, 138147 CrossRef CAS.
- H. Tamura, K. Mita, A. Tanaka and M. Ito, J. Colloid Interface Sci., 2001, 243(1), 202–207 CrossRef CAS.
- T. Hanawa, in Titanium in Medical and Dental Applications, Elsevier, 2018, pp. 95–113 Search PubMed.
- Z. Ye, G. Wang, J. Giraudon, A. Nikiforov, J. Chen, L. Zhao, X. Zhang and J. Wang, J. Hazard. Mater., 2022, 424, 127321 CrossRef CAS PubMed.
- M. Anpo, G. Costentin, E. Giamello, H. Lauron-Pernot and Z. Sojka, J. Catal., 2021, 393, 259–280 CrossRef CAS.
- C. S. Chen, T. C. Chen, C. C. Chen, Y. T. Lai, J. H. You, T. M. Chou, C. H. Chen and J.-F. Lee, Langmuir, 2012, 28, 9996–10006 CrossRef CAS PubMed.
- M. Huguet, M. Deborde, S. Papot and H. Gallard, Water Res., 2013, 47, 5400–5408 CrossRef CAS PubMed.
- A. Bumajdad and M. Madkour, Phys. Chem. Chem. Phys., 2014, 16, 7146 RSC.
- C. Liu, F. Meng, L. Zhang, D. Zhang, S. Wei, K. Qi, J. Fan, H. Zhang and X. Cui, Appl. Surf. Sci., 2019, 469, 276–282 CrossRef CAS.
- E. Cockayne and L. Li, Chem. Phys. Lett., 2012, 544, 53–58 CrossRef CAS.
- D. R. Eddy, M. D. Permana, L. K. Sakti, G. A. N. Sheha, Solihudin, S. Hidayat, T. Takei, N. Kumada and I. Rahayu, Nanomaterials, 2023, 13, 704 CrossRef CAS PubMed.
- N. Ramesh, C. W. Lai, M. R. Bin Johan, S. M. Mousavi, I. A. Badruddin, A. Kumar, G. Sharma and F. Gapsari, Heliyon, 2024, 10, e40998 CrossRef CAS PubMed.
- E. F. Mohamed and G. Awad, J. Ind. Eng. Chem., 2021, 98, 366–374 CrossRef.
- A. Kubiak, Z. Bielan, M. Kubacka, E. Gabała, A. Zgoła-Grześkowiak, M. Janczarek, M. Zalas, A. Zielińska-Jurek, K. Siwińska-Ciesielczyk and T. Jesionowski, Appl. Surf. Sci., 2020, 520, 146344 CrossRef CAS.
- A. Balapure, J. Ray Dutta and R. Ganesan, RSC Appl. Interfaces, 2024, 1, 43–69 RSC.
- Y. T. Prabhu, V. Navakoteswara Rao, M. V. Shankar, B. Sreedhar and U. Pal, New J. Chem., 2019, 43, 6794–6805 RSC.
- X. Zhou, H. Luo, B. Sheng, X. Chen, Y. Wang, Q. Chen and J. Zhou, J. Hazard. Mater., 2021, 411, 125050 CrossRef CAS PubMed.
- M. Malhotra, K. Poonia, P. Singh, A. A. P. Khan, P. Thakur, Q. Van Le, E. T. Helmy, T. Ahamad, V.-H. Nguyen, S. Thakur and P. Raizada, J. Taiwan Inst. Chem. Eng., 2024, 158, 104945 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI provides comprehensive experimental details, including the preparation of CuO-modified TiO2/MnOx composites, catalyst characterization (XRD, XPS, H2-TPR), and in-depth analysis of the degradation mechanisms of diclofenac. Fragmentation pathways and corresponding kinetic data are summarized in tables, along with mass spectra confirming the identification of intermediates and products, further supporting the catalytic performance discussed in the main manuscript. See DOI: https://doi.org/10.1039/d4cy01400f |
| This journal is © The Royal Society of Chemistry 2025 |