
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site blocking effects on P-modified Pd/Al2O3 catalysts for LOHC hydrogenation: an in situ DRIFTS study†
Yaoci
Sheng
 a,
Adrian
Seitz
b,
Thobani
Gambu
c,
Kailun
Zhang
a,
Patrick
Schühle
b and
Tanja
Retzer
*a
a,
Adrian
Seitz
b,
Thobani
Gambu
c,
Kailun
Zhang
a,
Patrick
Schühle
b and
Tanja
Retzer
*a
aInterface Research and Catalysis, ECRC, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: tanja.retzer@fau.de
bInstitute of Chemical Reaction Engineering, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany
cCatalysis Institute, Department of Chemical Engineering, University of Cape Town, Rondebosch, 7700, Cape Town, South Africa
First published on 23rd April 2025
Abstract
In this paper, we investigate structural changes of a P-modified Pd/Al2O3 catalyst for the hydrogenation of liquid organic hydrogen carriers (LOHCs) e.g., benzyltoluene (H0-BT). We elucidate the effect of P modification on the Pd nanoparticles (NPs) and the support material via a systematic combination of reactor studies, in situ diffuse reflectance FT-IR spectroscopy using CO as a probe molecule (CO-DRIFTS), and density functional theory (DFT). We find that P modification induces structural changes as well as site-blocking effects on the Pd NPs. In particular, phosphates on the Al2O3 support induce the formation of smaller Pd NPs. Phosphate species further reside on the terrace sites of Pd NPs, which leads to competitive adsorption between phosphates and CO/H2 especially at the Pd(111) facet. This demonstrates rearrangement of surface and adsorbate species on the P-modified Pd/Al2O3 catalysts under reactive gas atmosphere.
1 Introduction
Renewable hydrogen is a promising energy carrier to decarbonize industrial, energy, and mobility applications. Hydrogen can be stored and transported in liquid organic hydrogen carrier (LOHC) systems, allowing safe and loss-free H2-handling in the existing liquid fuel infrastructure.1 H2-storage is performed at locations with abundant energy supply via an exothermic catalytic hydrogenation of the LOHC.1 The corresponding endothermic dehydrogenation releases pure hydrogen at a time and place of H2-demand.1 Benzyltoluene (H0-BT) and its hydrogenated counterpart perhydro benzyltoluene (H12-BT) were recently introduced as a new LOHC system that combines the advantages of traditional systems.2,3 It offers a wide liquid temperature range, low toxicity, and simple production of high purity hydrogen, similar to the properties of the dibenzyltoluene (H0-DBT)/perhydro dibenzyltoluene (H18-DBT) system.3,4Pd-based catalysts are known to exhibit high productivity in the hydrogenation of H0-BT.5 However, Pd and precious metals are of limited availability and hence expensive. Therefore, we need strategies to apply them in a most efficient way, which in turn makes their use more sustainable and economical. To this aim, promotors such as P are often applied.6–9 With respect to the P/Pd system, different modes of interaction have been reported in the literature. On the one hand, incorporation of P into the Pd lattice produces distinct phosphide phases, among which eight are known according to the phase diagram.9 These palladium phosphides were shown to yield optimized catalytic performance, in particular regarding selectivity.7,10 This is due to the Pd site isolation effect (especially for high P contents7) and electronic effects on reactant molecules such as H atoms.10,11 On the other hand, formation of phosphates leads to distinct interactions with Al-based supports e.g. formation of AlPO4.12,13 Moreover, phosphates may potentially also be present directly at the surface of transition metals, where they can exert site blocking and electronic effects.14,15 We and others recently showed that P-modification of Al2O3-supported Pt and Pd catalyst is an effective strategy to increase activity and selectivity in the (de-)hydrogenation of carbohydrates and aromatic compounds such as LOHCs.15,16
In the current study, we systematically investigate the effect of P modification on Pd/Al2O3 catalysts and apply them for the hydrogenation of benzyltoluene (H0-BT). To this aim, we combine in situ IR spectroscopy and density functional theory (DFT) calculations with reaction studies. Our goal is to obtain a precise picture of the chemical state and location of the P promotor, its interaction with the catalytically active Pd nanoparticles (NP), and effects of reactive gases on the promoting species. A detailed understanding of the underlying processes will allow to further improve these systems in the future.
2 Experimental methods
2.1 Catalyst preparation
A commercial Pd/Al2O3 (4 wt% Pd) powder catalyst (Merck KGaA) was used as received. P modification of the Pd/Al2O3 catalyst was performed by impregnating the as-received catalyst with H3PO3 in aqueous solution for 16 h. This resulted in P-modified samples with a specific molar ratio x = nP/nPd (1.5/2.5). After solvent removal at 80 °C and 100 mbar, some of the catalysts were subjected to thermal, reductive treatment at Tred (500 °C/600 °C) in a tubular furnace for two hours under 10% H2/N2 (total flow 500 mL min−1). We indicate x and Tred in the identifier of the respective sample “Px–Tred”. For example, P1.5–500 represents the P-modified catalyst with nP/nPd = 1.5 molar ratio, reduced at 500 °C. P0-RT represents the unmodified, commercial Pd catalyst supported on Al2O3.2.2 Hydrogenation experiments and determination of productivity
Hydrogenation experiments were performed in a 300 mL batch autoclave (Parr Type 4566) equipped with cooling coil, electric heating mantle, four-blade gas-inducing stirrer, thermocouple (Type J), pressure recorder (Ashcroft Type G2), liquid sampling line with a filter and needle valve, temperature controller (Parr Type 4875), and cryostat (Huber Unichiller 022). The reactor was filled with 100 g H0-BT (Eastman Chemical Company) and the catalyst and was inertized with Ar. A constant nPd/nH0-BT molar ratio of 1.72 × 10−4 is present throughout all experiments. Once the target temperature (240 °C, stirred at 300 rpm) was reached, the liquid sample line was flushed with 1 mL of reaction liquid, and 0.1 mL was taken for gas chromatography (GC) analysis. During the experiment, we apply a constant pressure of 30 bar H2 and 1200 rpm stirrer speed. Samples collected throughout the reaction were analysed (30 μL sample in 1 mL acetone (Merck KGaA)) in a Shimadzu GC-2010 Plus equipped with a flame ionisation detector and a Restek Rxi-17Sil column. Qualitative and quantitative calibration of H0-, H6- and H12-BT peaks was described previously.3 Here, H6-BT represents the LOHC molecule where only one of the two aromatic rings is hydrogenated. The peak areas Ai of H0-, H6- and H12-BT were used to calculate the degree of hydrogenation (DoH) of the LOHC. | (1) |
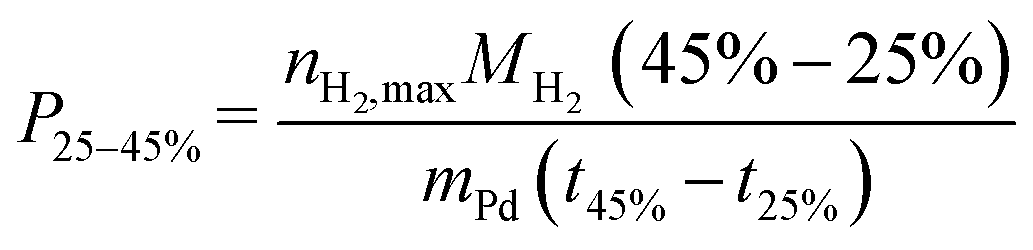 | (2) |
2.3 High-resolution transmission electron microscopy (HR-TEM)
HR-TEM images were acquired using a Philips CM30. The software ImageJ was used to measure particle diameters.17 The dispersion was determined according to Bergeret & Gallezot.182.4 X-ray diffraction (XRD)
XRD patterns were obtained using a Pananalytical X-Pert Pro-MD (Philips) equipped with a Cu-Kα radiation source with a wavelength of 0.154 nm. Measurements were conducted in a range of 10–90° (step size of 0.017°, time per step of 100 s).2.5 Inductively coupled plasma optical emission spectroscopy (ICP-OES)
The Pd and P content of the catalysts were determined by dissolving 100 mg of sample in a 10 mL mixture of HNO3/HCl (volumetric ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6) using microwave heating at 200 °C. The resulting solution was diluted to 100 mL with deionized water and analysed using a Ciros CCD from SPECTRO Analytical Instruments GmbH.
:6) using microwave heating at 200 °C. The resulting solution was diluted to 100 mL with deionized water and analysed using a Ciros CCD from SPECTRO Analytical Instruments GmbH.
2.6 Transmission infrared spectroscopy (TIRS)
Transmission infrared spectroscopy (TIRS) was performed with a Vertex 80v FT-IR spectrometer (Bruker) in combination with a TIRS module, a KBr beam splitter, and a N2-cooled HgCdTe-detector. We recorded the spectrum of pure KBr (background, 2 min acquisition time) and the catalysts (diluted in KBr, 0.45 min acquisition time) in the form of compressed pellets. All spectra were recorded in vacuo (∼1 mbar), with a resolution of 2 cm−1 and a scanner velocity of 40 kHz. Post-data treatment included baseline correction.2.7 Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)
We performed in situ DRIFTS measurements with a Vertex 80v Fourier transform infrared (FTIR) spectrometer (Bruker) combined with a KBr beam splitter, a N2-cooled HgCdTe-detector, and a Praying Mantis DRIFTS accessory with a high temperature reaction chamber (both from Harrick). The latter is located in the home-built extension of the sample compartment of the spectrometer, which provides all electrical and gas dosing connections to the DRIFTS reactor and spectrometer. Evacuation of the complete beam path during the measurement leads to excellent long-term stability of the system. The reactor contains CaF2 windows, a sample cell for powder samples which is connected to the gas dosing system, and a K-type thermocouple in direct contact with the sample. The gas flow (up to five gases, each up to 20 mL min−1) and pressure (up to 20 bars) were regulated by different types of mass-flow and pressure controllers (both Bronkhorst). All parameters can be remote-controlled. In this work, H2 (Linde, >99.999%), Ar (Linde, >99.999%), CO (Linde, >99.997%) were used without further purification. We recorded the background spectrum in 1 bar Ar atmosphere (scan time: 10 min). We performed an identical gas dosing sequence for all samples, along with γ-Al2O3 as a reference. After filling of the sample powders into the sample holder and assembly of the reactor, we purged the reactor with Ar to remove residual traces of water and air. For the CO-DRIFTS experiments, we stepwise increased the pressure from 50 mbar, 100 mbar, 200 mbar, 500 mbar to 1000 mbar (scan time: 0.9 min). For the CO/H2 dosing experiments, we filled the reactor with 1 bar CO at a flow rate of 8 mLN min−1 and then gradually replaced it with H2, until H2:CO ratio reaching 8:1. The total pressure was maintained at 1 bar during the whole measurement. Post-data treatment includes normalization, baseline correction, and mathematical removal of the CO gas phase according to Blaumeiser et al.19
2.8 Density functional theory (DFT)
Spin-polarized density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP).20 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional21 was used with the projector augmented wave (PAW) method. Dispersion corrections were treated with the DFT-D3 method of Grimme22 with Becke-Johnson damping function.23A plane-wave basis set cutoff energy of 450 eV was used. The electronic and geometry optimization convergence criteria were 10−5 eV and 0.02 eV Å−1, respectively. The Pd(111)-p(3 × 3) and Pd(100)-p(3 × 3) slabs were cut from an optimized bulk structure, and each had five atomic layers and a vacuum gap of 12 Å between periodic slabs. A converged gamma centered k-point grid of 6 × 6 × 1 was used for all slabs. Only the top three and two layers were relaxed for the Pd(111) and Pd(100) slabs, respectively. Dipole correction was applied along the surface normal. The adsorption energy (Eads,POxHy) was calculated as follows,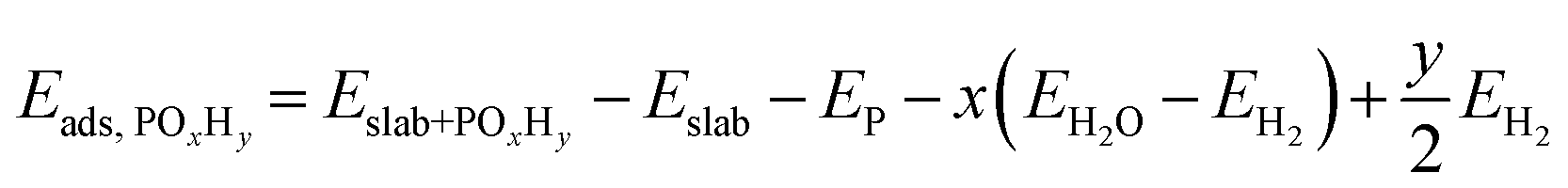 | (3) |
| EP = 0.25(EP4O10 − 10(EH2O − EH2)) | (4) |
| Eads,CO = Eslab+CO − Eslab − ECO | (5) |
3 Results and discussion
3.1 Catalytic testing of P-modified catalysts in hydrogenation of benzyltoluene (H0-BT)
We first investigate the effect of P modification of a commercial Pd/Al2O3 (P0-RT) in hydrogenation of H0-BT in a batch autoclave.
Fig. 1 Shows the productivities of the prepared catalysts in H0-BT hydrogenation. The as-received commercial P0-RT catalyst exhibits a productivity of 2.64 gH2 gPd−1 min−1 (highlighted in blue). In the first set of experiments, we investigate the effect of the temperature treatment after H3PO3 impregnation at a fixed molar P:Pd ratio of 1.5. The catalyst that was only impregnated and not thermally treated (P1.5-RT) shows a slight productivity increase as compared to the unmodified benchmark. This increase is even more pronounced after reductive treatment at 500 °C (P1.5–500). Reduction at 600 °C (P1.5–600) results in the highest productivity of 3.10 gH2 gPd−1 min−1, i.e., a relative productivity boost of approximately 17%. This relative increase is comparable to what has been observed for Pt–P/Al2O3 catalysts for perhydro benzyltoluene dehydrogenation which we previously studied.15
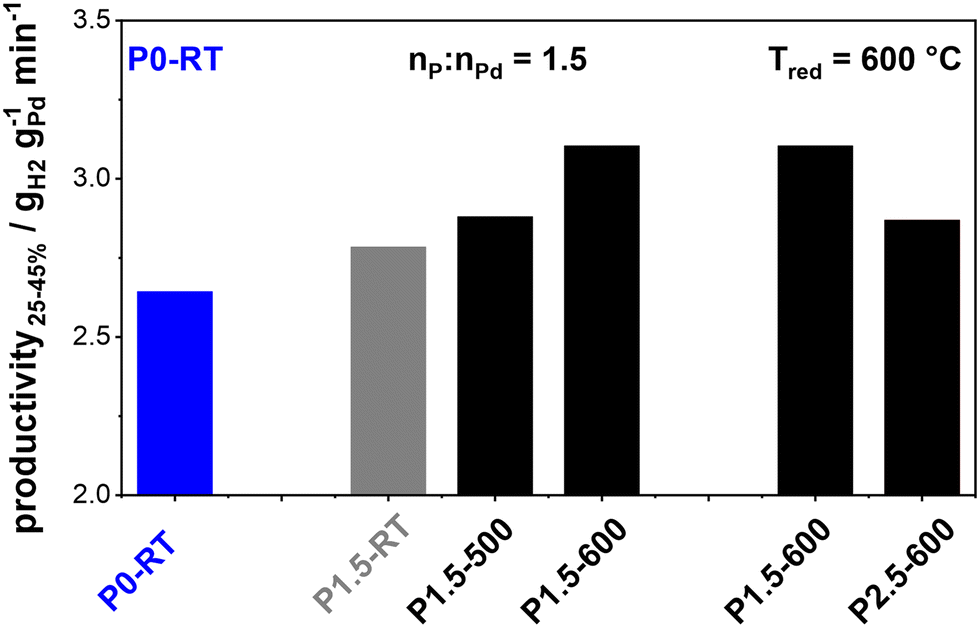 | ||
| Fig. 1 Productivity25–45% of P0-RT and P-modified catalysts during hydrogenation of H0-BT at 240 °C, 30 bar H2 pressure, and nPd/nH0-BT = 1.72 × 10−4. “Px–Tred” indicates the molar ratio (x = nP/nPd) and reduction temperature Tred during pretreatment of the catalyst. | ||
In a second set of experiments, we compare catalysts with different molar P:Pd ratios (1.5/2.5) subjected to the optimum reductive treatment at 600 °C. As shown in Fig. 1, P2.5–600 yields lower productivity as compared to P1.5–600. This is consistent with our previous study where a 1.5 nP/nPt molar ratio was indeed an ideal value for the Pt/Al2O3 system.15 Higher P:Pt ratios were shown to lead to blocking of active Pt sites and consequently reduced activity.15 Thus, our results clearly show that the performance of the catalysts depends on both, the specific molar ratio x = nP:nPd and the reduction temperature Tred with best results found for the combination applied in P1.5–600.
3.2 Structural analysis of P-modified catalysts
We proceed to investigate and identify the state of P on the surface of the as-prepared catalysts. To this aim we combine TIRS and XRD on an extended set of samples, in particular including a sample with a higher P-loading (P5.1–600).Fig. 2a displays the XRD patterns obtained from P0-RT, P1.6–600, and P5.1–600. The position of the characteristic reflections of Al2O3, Pd, and AlPO4 are included at the bottom of the graph and indicated with triangles (black, blue, and green, respectively) in the experimentally obtained patterns. All XRD patterns show clear reflections corresponding to metallic Pd0 and γ-Al2O3. Most importantly, no reflections of palladium phosphide phases are observed. Thus, we exclude a complete reduction of the H3PO3 precursor during preparation of the catalysts. The XRD patterns of P0-RT and P1.6–600 look virtually identical. In contrast, the XRD pattern of P5.1–600 displays new reflections at position 21.4 and 35.2° which are consistent with the reflections of AlPO4.
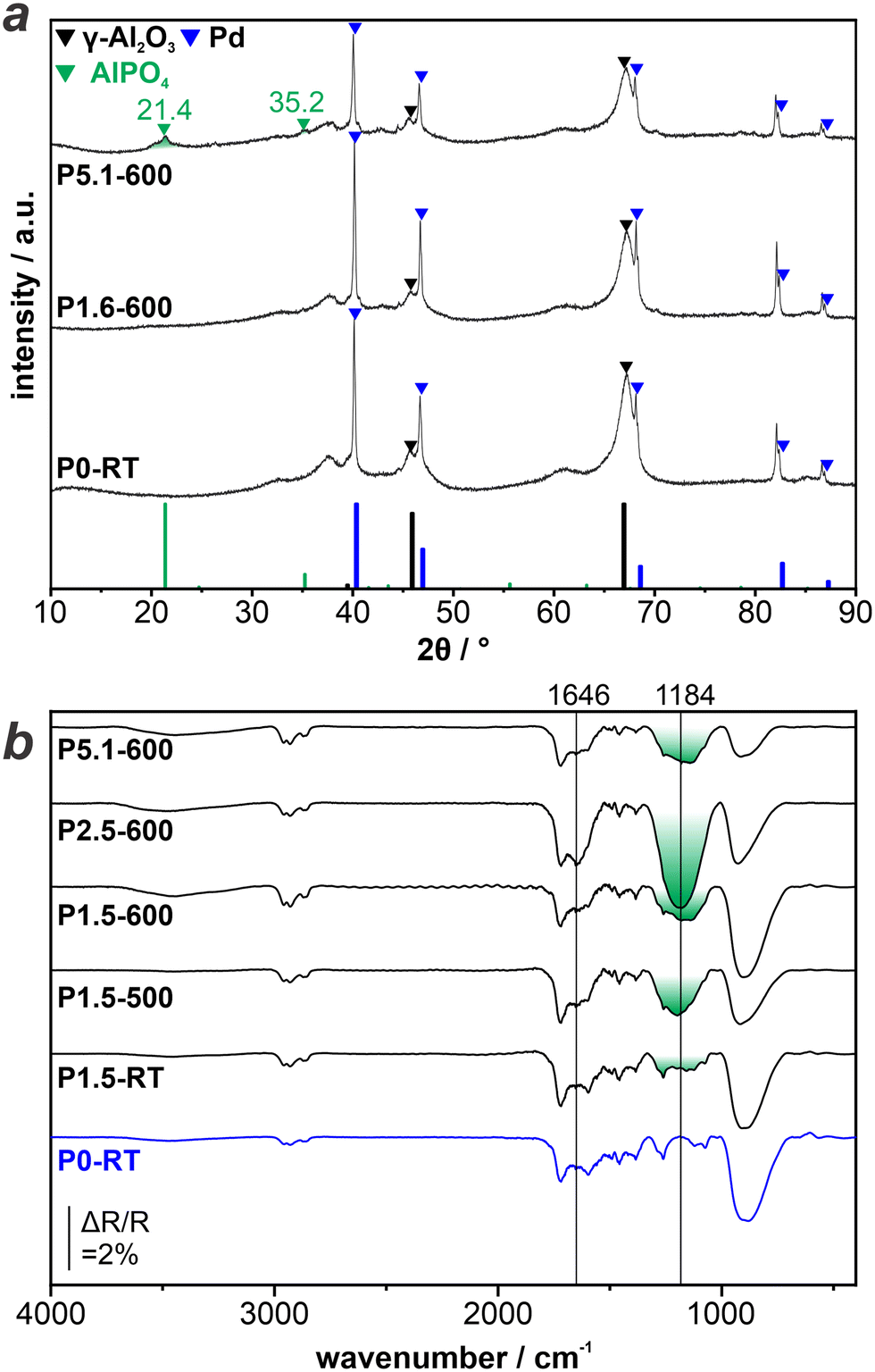 | ||
| Fig. 2 Ex situ characterization of LOHC catalysts: a) XRD patterns of P-modified catalysts with varying P-loading. b) TIRS reference spectra of P-modified catalysts measured under ambient conditions. | ||
The XRD results prove that the pretreatment with H3PO3 and subsequent reduction modifies the Al2O3 support material via the formation of AlPO4. This is in agreement with previous reports in the literature.12,24 In this study, the observation of the phosphate species via XRD is limited to high P-loadings, due to the fact that sufficiently large and crystalline phases need to be present. Thus, we apply TIRS to investigate catalysts with lower amounts of P, in particular samples used during catalytic testing.
Fig. 2b shows the TIR spectra obtained from P0-RT, P1.5–RT, P1.5–500, P1.5–600, P2.5–600, and P5.1–600. All TIR spectra show an intense band at <1000 cm−1, which is due to strong absorption of the Al2O3 support. The unmodified P0-RT sample shows additional small features at 1000–1750 cm−1. In consequence, these bands are also present in the spectra obtained from the P-modified samples, but will not be discussed further. In addition, the spectrum obtained from P2.5–600 shows a band centered at 1646 cm−1, which is due to δ(OH) in H2O.25,26 After the modification of the catalysts with H3PO3, a broad band at 1184 cm−1 is present with varying intensity. For P1.5–RT, it is barely visible, whereas it is more pronounced after the temperature treatment. However, the peak intensity does not correlate linearly with the P loading. Several researchers have attributed this band to ν(P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond in phosphates, which can arise from both aluminum phosphate and palladium-phosphate (surface) species.13,24,26–28
O) bond in phosphates, which can arise from both aluminum phosphate and palladium-phosphate (surface) species.13,24,26–28
While XRD detected crystalline AlPO4 only at higher P-loadings (P5.1–600), the absence of XRD signals at lower P-loading (P1.5-RT, P1.5–500, P1.5–600) does not exclude the presence of highly dispersed or amorphous phosphate species. The trend of the peak intensity of ν(PO) is in line with this finding. The formation of phosphates occurs during the thermal pretreatment of the catalysts. Therefore, the corresponding band is barely visible in case of P1.5–RT.
Previous studies e.g. by Dong et al., showed that modification with phosphates stabilizes the Al2O3 support and inhibits sintering of Pd at elevated temperatures.29 Thus, it is reasonable to assume that the improved catalytic performance of the P-modified catalysts in this work is at least in part due to this effect. In the next step, we evaluate the effect of the reductive thermal treatment and phosphates on the structure of the catalytically active Pd NPs.
3.3 Influence of reductive thermal treatment
In order to investigate the influence of thermal treatment and phosphate modification on the Al2O3-supported Pd catalysts, we perform DRIFTS with CO as a probe molecule (in the following referred to as CO-DRIFTS experiment). This approach is commonly used in the context of characterizing heterogeneous catalysts via IR spectroscopy. The stretching frequency of CO adsorbates ν(CO) on metal surfaces is sensitive to the binding site, adsorption motif, electronic properties of the metal binding partner, and the presence of co-adsorbates.30–32 The latter includes other CO molecules, i.e., there is a distinct coverage-dependence of ν(CO) due to dipolar coupling effects.33,34 To account for this effect, we record CO-DRIFTS spectra at a constant pressure and remove the interfering CO gas phase signals during post-data treatment. First, we investigate the impact of reductive, thermal treatment via pressure-dependent CO-DRIFTS of samples without phosphate modification (P0-RT, P0–500, P0–600). During the experiment, we step-wise increase the CO pressure (50 mbar, 100 mbar, 200 mbar, 500 mbar, and 1 bar). Fig. 3a depicts the corresponding spectra in the wavenumber range of 1840 to 2140 cm−1. | ||
| Fig. 3 Pressure-dependent CO-DRIFTS on Pd/Al2O3 catalysts: a) DRIFTS spectra recorded under increasing CO pressure from 50 mbar to 1 bar with P0-RT, P0–500, and P0–600; b) integrated peak area of COtop (solid lines) and CObr+h (dashed lines). | ||
On all samples, we observe a single peak at ∼2100 cm−1 with a broad shoulder at ∼2075 cm1 and pronounced peaks at ∼1984 cm−1 and ∼1940 cm−1. We assign all features based on a surface science approach with the help of literature. Here, metal NPs are regarded as entities with a regular shape that expose distinct crystal facets along with edges and corners. The most stable, i.e., most abundant crystal facets in case of Pd are the (111) and (100) facets.
At complete saturation at 300 K, CO adsorbs at bridge (CObr) and three-fold hollow (COh) sites on the Pd(111) facet, resulting in a single band at ∼1930 cm−1.24,35–37 On Pd(100) terraces, CObr shows a band at 1990 cm−1.34,38,39 CObr at particle edges cause a pronounced band between 1950–1980 cm−1, i.e., at a similar position as compared to CObr on Pd(100).40 Bands arising from CO in on-top configuration (COtop) on low-coordinated Pd atoms are located at ∼2090 to 2100 cm−1, while COtop on Pd(111) facet is located at ∼2075 to 2080 cm−1.39–41 Based on these findings, we assign the bands obtained from the (P-modified) Pd-catalysts as follows:
• 1939 cm−1: CObr/COh on Pd(111) terraces
• 1984 cm−1: CObr on particle edges with a minor contribution of CObr on Pd(100)
• 2075 cm−1: COtop on Pd(111) with contribution of Pd(100)
• 2100 cm−1: COtop on edge and defect sites
The spectrum obtained from P0-RT exhibits distinct features for CO adsorbed on Pd(111) terraces (1939 cm−1) and on particles edges (1984 cm−1). The high temperature pretreatment at 500 °C and 600 °C did not induce significant structural changes to the catalyst, and the signal peaks for COtop and CObr/COh remain at high intensity. Please note that the peak intensities for P0–500 and P0–600 is significantly higher as compared to P0-RT, which is due to a significantly higher overall signal intensity during the measurement. Fig. 3b visualizes changes in the cumulated peak areas of the COtop and CObr+h bands. In particular, we find an 20 ± 4% increase of the CObr+h band going from 50 mbar to 1 bar. The COtop area decreases accordingly. We attribute these changes to restructuring of the CO adsorbate layers at higher CO pressures. These observations serve as a reference for pressure-dependent CO-DRIFTS studies on the P-modified catalysts presented in section 3.5. Beforehand, we take a turn to compare the distribution of catalytically active Pd sites in case of the pristine Pd/Al2O3 samples and the P-modified counterparts.
3.4 Availability of catalytically active Pd-sites on (P-modified) catalysts
Fig. 4a depicts CO-DRIFTS spectra of the unmodified P0-RT, P0–500, and P0–600 sample (in blue) combined with the P-modified samples P1.5-RT, P1.5–500, P1.5–600, and P2.5–600 (in black) all recorded at 1 bar CO. For discussion of the unmodified samples, we refer the reader to the previous section. Direct comparison shows that in case of the P-modified catalysts, the peak for COtop at ∼2100 cm−1 remains and the CObr+h band at 1984 cm−1 becomes more pronounced. The features at 1939 cm−1 and 2070 cm−1 are not clearly resolved anymore but rather contribute to the bands at ∼1984 and ∼2100 cm−1 which are asymmetric towards the low-wavenumber side. It is important to note that the features which are less intense on the P-modified catalysts originate from CO adsorbates on the Pd(111) and Pd(100) terraces. This indicates that treatment with H3PO3 induces the formation of comparatively smaller Pd NPs that naturally expose much less extended facets.41,42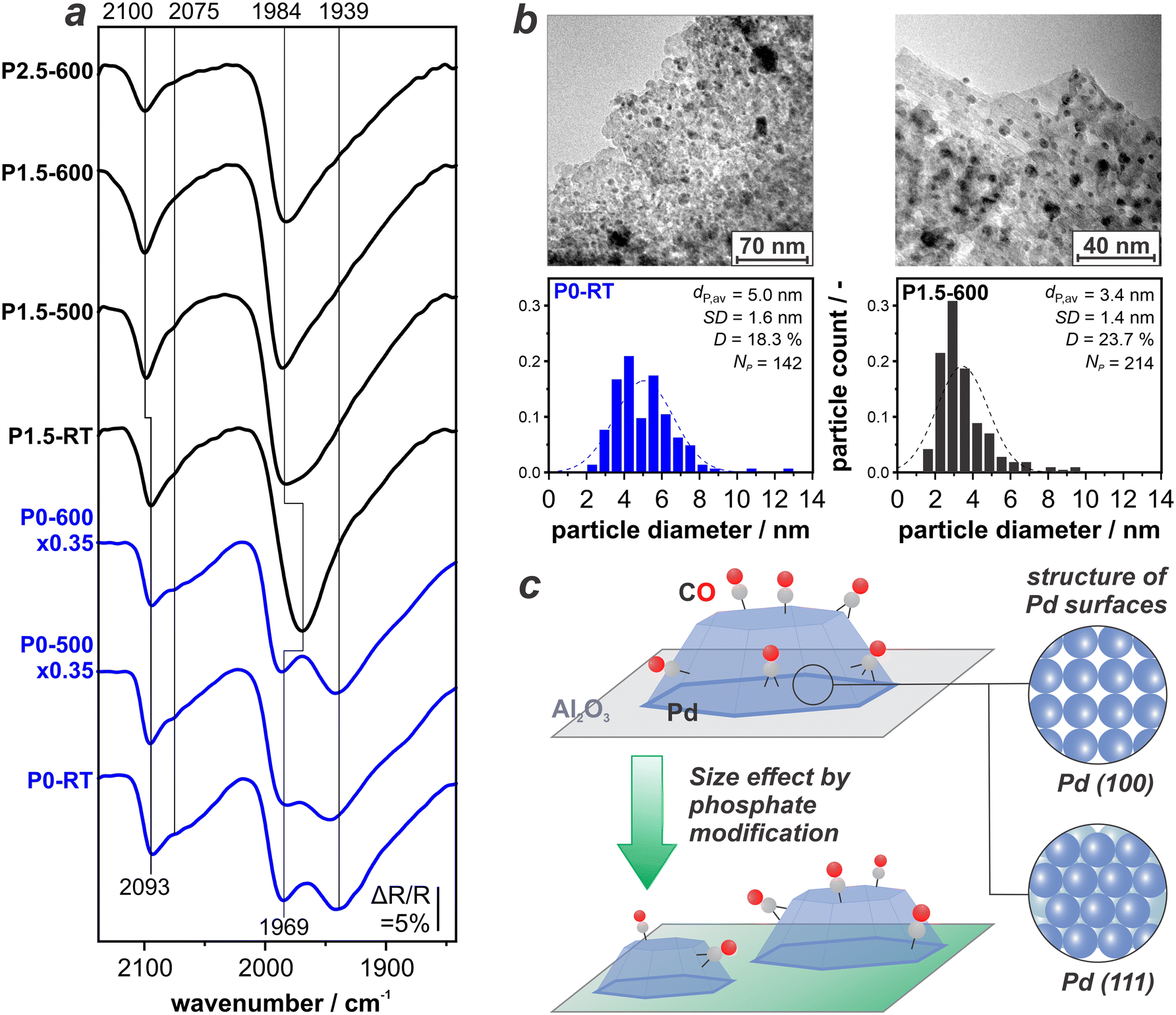 | ||
| Fig. 4 Site-distribution on (P-modified) Pd/Al2O3 catalysts: a) CO-DRIFTS spectra after the subtraction of CO gas phase, recorded at RT under 1 bar CO; b) HR-TEM pictures and particle size distribution of P0-RT and P1.5–600; c) sketch of CO adsorption on Pd before and after P modification. | ||
To confirm this observation, we perform HR-TEM on selected samples. Fig. 4b depicts the resulting images as well as the particle size distribution for P0-RT and P1.5–600. Indeed, we find that P modification of the support decreases the average diameter of Pd NPs from 5.0 nm to 3.4 nm. This corresponds to a theoretical dispersion increase from 18.3% for P0-RT to 23.7% for P1.5–600. Thermal treatment at such high temperatures of 600 °C usually leads to active metal sintering due to an increased mobility of the metal.43 The opposite behaviour and therefore efficient stabilization of small NPs against sintering is found for the P-modified catalysts. Note that the DRIFTS data suggests that re-dispersion of the Pd NPs occurs even without the thermal, reductive treatment (see P1.5-RT in Fig. 4a). This is, however, not reflected by the catalytic performance of the respective sample. Our combined findings show that the formation of phosphates on Al2O3 does not only impact the structure and thermal stability of the support, but also the size of the catalytically active Pd NP. Fig. 4c schematically depicts this geometric effect. In terms of catalytic performance, the availability of smaller Pd NPs on a phosphate-modified alumina is highly beneficial. A similar effect has been observed for P-modified Pt/Al2O3.15 Here, we found maximum productivity after P-modification and thermal treatment at 600 °C. Indeed, we found a higher Pt dispersion which is stable up to temperatures of 900 °C.15
At this point, the question remains whether the P-treatment has further effects on the Pd NPs. An additional contribution to the trend observed in the DRIFT spectra could be selective site blocking, i.e., phosphates occupying the Pd terrace sites but not particle edges or defect sites. This would similarly lead to a suppression of the band at 1939 cm−1.
3.5 Site blocking effects on P-modified catalysts
To elucidate this possibility, we revert to evaluate changes in the CO-DRIFT spectra upon varying the CO pressure. Panels in Fig. 5a show CO-DRIFT spectra recorded at 50 mbar, 100 mbar, 200 mbar, 500 mbar, and 1 bar on P0-RT (in blue, for sake of comparison), P1.5-RT, P1.5–500, P1.5–600, and P2.5–500. Fig. 5b depicts the resulting integrated peak areas of COtop (∼2100 cm−1) and CObr+h (∼1990–1930 cm−1). As stated above, the area of the COtop and CObr+h peaks on P0-RT basically remain unchanged with increasing CO pressure. In contrast, the total peak area of CObr+h increases significantly with increasing CO pressure to 1 bar for all P-modified catalysts (peak area highlighted in purple in Fig. 5a). The relative increase of the peak area is 120±40% as compared to 20±4% in case of the non-modified samples (see Fig. S1 in ESI†). This proves that indeed the modification with H3PO3 causes the observed changes. Further increase of the CO pressure does not lead to further changes (see Fig. S2 in ESI†). Changes in the morphology of the Pd NPs are highly unlikely under the conditions applied throughout the experiment, i.e., CO dosing at RT. On closer inspection, the DRIFT spectra show an increase of the low wavenumber side of the CObr+h band. In particular spectra recorded on the P1.5–500 catalyst show an increase of the CObr+h band at 1939 cm−1 at elevated CO pressure. We hypothesize that phosphate species block adsorption sites at the Pd surface until they are replaced by stronger-binding CO at elevated CO pressure. This site blocking is depicted in Fig. 5c.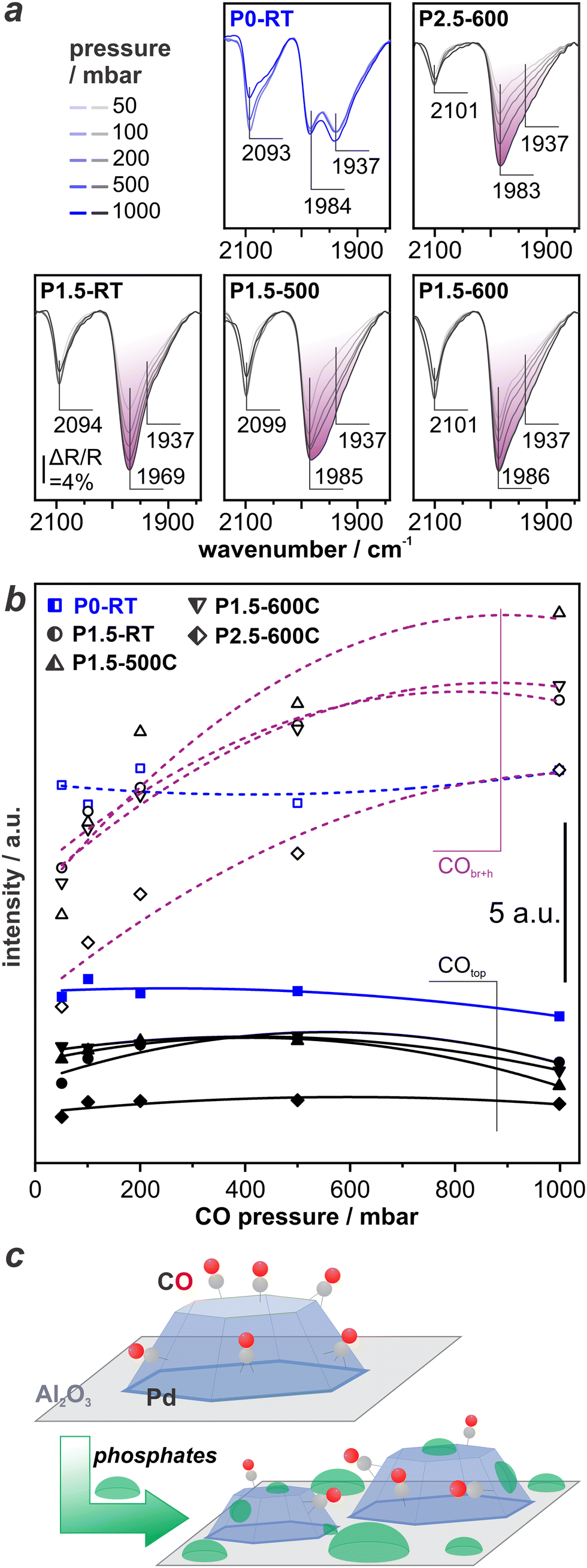 | ||
| Fig. 5 Pressure-dependent CO-DRIFTS on P-modified Pd/Al2O3 catalysts: a) DRIFTS spectra recorded under increasing CO pressure from 50 mbar to 1 bar with P0-RT, P1.5-RT, P1.5–500, P1.5–600, P2.5–600; b) integrated peak area of COtop (solid lines) and CObr+h (dashed lines); c) sketch for CO adsorption before and after P modification. | ||
To validate this assumption, we perform density functional theory (DFT) calculations. We explore different adsorption configurations of CO and POxHy-species (PH, P, PO and POH) on Pd(111) and Pd(100), i.e., the most stable and abundant crystal facets according to the literature.40,44Table 1 shows the lowest calculated adsorption energies at a 1/9 monolayer coverage and at 0 K.
| Adsorption energy/kJ mol−1 | |||||
|---|---|---|---|---|---|
| Surface | CO | PH | P | PO | POH |
| Pd(111)-p(3 × 3) | −214 | −134 | −142 | −160 | −170 |
| Pd(100)-p(3 × 3) | −216 | −254 | −239 | −230 | −283 |
The employed exchange-correlation functional leads to the correct prediction of the CO site preference. However, the absolute adsorption energy is overestimated by the applied exchange-correlation functional compared to experimental data.45 CO has a nearly isoenergetic adsorption behavior on both surfaces (Table 1, CO adsorption energy on different sites of Pd including vibrational frequency please see Table S1 in the ESI†). All POxHy-species adsorb significantly stronger on Pd(100) than on Pd(111) resulting in the following adsorption strength order: POxHy/Pd(111) < CO/Pd(111) ∼ CO/Pd(100) < POxHy/Pd(100). Thus, increasing the chemical potential of CO (achieved experimentally by increasing the CO partial pressure), CO can likely displace POxHy-species adsorbed on Pd(111), but can hardly replace POxHy-species on Pd(100) facets. Competitive adsorption on the Pd(111) facet is also reflected by the coverage-dependent adsorptions energies (see Fig. 6). The differential adsorption energy of each POxHy adsorbate decreases with increasing coverage and becomes positive above ca. 0.44 ML. This suggests that further adsorption beyond this coverage is thermodynamically not feasible, making 0.44 ML the saturation coverage. On the other hand, our calculations show a saturation coverage greater than 0.44 ML for CO on Pd(111)-p(3 × 3). Please note adjusting the adsorption strength of CO to match the experimental data in the literature (140 kJ mol−1, “CO-adjusted” in Fig. 6)22 will further enhance the competitiveness of CO and POxHy adsorption on Pd(111). Overall, with increasing CO partial pressure, CO adsorption will dominate on the Pd(111) facet. Bader charge analyses, shown in Fig. S4,† indicate that the adsorption of CO differs significantly to that of POxHy. While the former draws more electrons from the topmost surface layer, the latter mostly donates electrons to this layer. The effect of adsorption on the 2nd and 3rd layers is comparable across all adsorbates. However, the second layer appears to have an accumulation of charge compared to a clean slab. This suggests that POH pre-covered surfaces are more negatively charged and able to donate more electrons to adsorbing CO molecule, leading to a stronger adsorption and a weaker C–O bond.46 Table S2 and Fig. S5† show the latter where CO–CO interactions lead to weaker adsorption and poor activation of the C–O bond while co-adsorption of CO with POH is seen to increase the adsorption strength and elongate the C–O bond. Please note that the changes in the CO geometry are too small to lead to spectral changes observable in the DRIFTS experiment. However, the trend concerning the replacement of phosphate species is perfectly in line with our experimentally obtained DRIFTS spectra, where we observe competitive adsorption between POxHy-species and CO on the Pd(111) terraces via changes in the low wavenumber side of the CObr+h band at ∼1940 cm−1. CO dosing at ambient pressure is sufficient to make the respective surface sites available for CO. It is important to note that according to our DFT data, phosphate species also block the Pd(100) terraces, but as the site occupation does not change, we do not observe the respective changes during increase of the CO partial pressure. However, the peak area obtained for COtop and CObr+h on P2.5–600 (see Fig. 5b) is remarkably lower as compared to the samples with a lower P-loading. This shows that blocking of Pd surface sites by phosphates is indeed not limited to the Pd(111) surface and occurs to a higher level at increased P-loading. Please note that this is also in good agreement with the results from the catalytic tests, which showed that the optimum P–Pd molar ratio lies at an intermediate value of 1.5.
 | ||
| Fig. 6 Calculated differential adsorption energy of CO and POxHy species on Pd(111) surfaces plotted against coverage; sketch of CO and POxHy species adsorption on Pd(111). The energies are at 0 K (without zero-point correction) and are relative to CO, H2, H2O and P4O10 in the gas phase (see eqn (3)–(5) in the experimental part). | ||
Possible rearrangement of the phosphate adsorbates to free up sites for CO adsorption includes changes in the adsorption mode (from bidentate to monodentate) or migration to the support. Moreover, the coverage-dependent adsorption energies (see Fig. 6) show that the POxHy species interact more strongly with each other as compared to the CO–CO interactions at the same coverage. Thus, agglomeration/vertical growth of the phosphate species is another possible scenario. Note that the presence of phosphates on the Pd surface does apparently not induce electronic effects on co-adsorbates, as ν(CO) is identical for all samples and throughout the experimental procedure. Similarly, Chen et al. reported competitive adsorption of phosphates and CO on Pd/Al2O3 without further electronic effects visible in the IR spectrum.24
As the presence of gases alters the availability of the catalytically active Pd surface sites already at mild conditions, we proceed to investigate the effect of phosphates closer to reaction conditions. In a first step, we elucidate the effect of H2. To follow potential changes spectroscopically, we start with pure CO gas, and gradually exchange it with H2 (up to a H2:CO ratio of 8:1) while maintaining a constant total pressure of 1000 mbar. It is important to note that maximum H2 partial pressure (880 mbar) applied during this procedure exceeds the one used during the reductive pretreatment (100 mbar). Fig. 7 displays the time-resolved spectra acquired during the continuous gas change for P0-RT, P1.5–600, and P2.5–600. For spectra of other P-modified samples, please refer to Fig. S6 and S7 in the ESI.† In case of all samples, we observe the characteristic bands assigned above, in short: ∼1930 cm−1 (CObr and COh on Pd(111)), ∼1980 cm−1 (CObr on particle edges), and 2100 cm−1 (COtop). In case of the unmodified P0-RT sample (spectra shown in blue in Fig. 7a), the initial spectrum is in perfect agreement with the spectra obtained during pressure-dependent CO dosing (compare with Fig. 3a and 5a) with all CO peaks being clearly recognizable and resolved. Upon introducing H2, the peak intensity and band position remain virtually unchanged. Notably, the presence of H2 does not change ν(CO), although it is known to interact with Pd via dissociative adsorption and diffusion to sub-surface regimes.47 This also holds for the non-modified P0–500 and P0–600 samples (see Fig. S8 in ESI†) On the contrary, we observe spectral changes immediately after H2 was added for the P-modified samples (spectra shown in Fig. 7b and c). In particular, the band at 1936 cm−1 (CObr and COh on Pd(111), highlighted in purple) starts to appear and even dominates the spectrum at a H2:CO ratio of 8:1. The band at 1980 cm−1 (CObr on particle edges) then becomes a higher frequency shoulder. This trend is in line with our previous finding: phosphates block the Pd facets, but the Pd(111) surface sites can be recovered at mild conditions in the presence of reactive gases. For comparison between the ability of CO and H2 to lift this site-specific blocking, it is important to keep in mind that during the H2 dosing experiment the partial pressure of CO indeed decreases.
 | ||
| Fig. 7 Hydrogen dosing DRIFTS results using different samples: a) P0-RT; b) P1.5–600; c) P2.5–600. Spectra of other P-modified samples are included in Fig. S6 and S7 in the ESI.† | ||
Together with the fact that the band for CObr and COh at Pd(111) terraces become significantly more pronounced during H2 dosing, the combined results prove that H2 has a greater ability than CO to remove P-blocking on the (111) facets. Thus, the ability to lift the blocking of surface sites by phosphates increases with greater reduction strength of the reactive gas. We checked the reversibility of the changes induced by H2 for the P1.5–500C sample. Fig. 7b depicts spectra acquired during changing from pure CO atmosphere to a H2:CO ratio of 8:1 and back to pure CO atmosphere in the system. The spectra show that the dominating peak at 1939 cm−1 (highlighted in purple) remains present. This observation proves that the changes induced by H2 persist after its removal. Please note that the P-loading of the catalyst did not change during the treatment with H2 (see Table S3 in ESI†). Given the fact that LOHC hydrogenation occurs under even higher H2 partial pressures, it is fair to assume that similar lifting of site blocking effects occur.
In summary, our study proves that P-modified LOHC catalysts are very likely subjected to (structural) changes during the hydrogenation reaction. Our study shows that phosphate-modified Pd/Al2O3 catalysts experience site-blocking, which is sensitive to the applied reaction parameters. Therefore, the site-blocking effects must be taken into account when interpreting the catalytic results. This is especially true for impure gas feeds (e.g. with traces of CO), where the interactions between all components (LOHC, H2, CO and phosphate species) under reaction conditions must be taken into account. Therefore, we will investigate the effects of (model) LOHC reactants and products on the (modified) catalyst systems in the future.
4 Conclusions
In this study, we applied TIRS and in situ DRIFTS to elucidate the nature and location of phosphorus species on Pd/Al2O3 catalysts for LOHC hydrogenation. In particular, we elucidate how the phosphorous modification affects the catalytically active Pd NPs at different pretreatment protocols. Furthermore, we investigate how the presence of reactive gases such as CO and H2 affects the state of the catalyst. Here, we summarize our most important findings:1) Treatment of Pd/Al2O3 catalysts with H3PO3 and subsequent heating under reductive conditions increases their performance in LOHC hydrogenation. A P loading of 1.5 molar ratio and thermal pretreatment at 600 °C leads to a 17% increase in activity.
2) P modification leads to the formation of phosphates on the support (e.g., AlPO4) and induces structural changes of the Pd NPs: the phosphate-modified support exposes and stabilizes smaller Pd NPs with a narrower size distribution.
3) Phosphate species also adsorb on the facets of the Pd NPs and bind more strongly on Pd(100) as compared to the Pd(111). Comparatively weak binding on Pd(111) leads to competitive adsorption with other adsorbates (e.g., CO). Treatment with reactive gases under mild conditions lifts the selective site blocking of Pd(111) facets caused by phosphates. This effect is more pronounced for H2 as compared to CO.
4) Coadsorbates such as CO do not experience any electronic effects caused by the phosphates that can be seen in the IR spectra.
Data availability
The data that support the findings of this study are openly available in Zenodo at https://doi.org/10.5281/zenodo.14258340.Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used DeepL in order to improve the language. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation: Project-ID 431791331), SFB 1452 (CLINT Catalysis at Liquid Interfaces). This work is based on the research supported in part by the National Research Foundation of South Africa (Ref Numbers TTK23031482384). The authors further acknowledge the Centre for High Performance Computing (CHPC), South Africa, for providing computational resources to this research project. The authors further gratefully acknowledge the Federal Ministry of Education and Research for funding of the BMBF Junior Research Group FAIR-H2 (Grant number (FKZ): 03SF0730).References
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2016, 50, 74–85 CrossRef PubMed.
- N. Brückner, K. Obesser, A. Bösmann, D. Teichmann, W. Arlt, J. Dungs and P. Wasserscheid, ChemSusChem, 2014, 7, 229–235 CrossRef PubMed.
- T. Rüde, S. Dürr, P. Preuster, M. Wolf and P. Wasserscheid, Sustainable Energy Fuels, 2022, 6, 1541–1553 RSC.
- Y. Kwak, J. Kirk, S. Moon, T. Ohm, Y. J. Lee, M. Jang, L. H. Park, C. i. Ahn, H. Jeong, H. Sohn, S. W. Nam, C. W. Yoon, Y. S. Jo and Y. Kim, Energy Convers. Manage., 2021, 239, 114124 CrossRef CAS.
- H. Jorschick, M. Vogl, P. Preuster, A. Bösmann and P. Wasserscheid, Int. J. Hydrogen Energy, 2019, 44, 31172–31182 CrossRef CAS.
- J. W. Bae, S.-M. Kim, Y.-J. Lee, M.-J. Lee and K.-W. Jun, Catal. Commun., 2009, 10, 1358–1362 CrossRef CAS.
- Y. Liu, A. J. McCue, C. Miao, J. Feng, D. Li and J. A. Anderson, J. Catal., 2018, 364, 406–414 CrossRef CAS.
- N. I. Skripov, L. B. Belykh, T. P. Sterenchuk, K. L. Gvozdovskaya, V. V. Zherdev, T. M. Dashabylova and F. K. Schmidt, Kinet. Catal., 2020, 61, 575–588 CrossRef CAS.
- T. Xie and R. M. Rioux, Catal. Today, 2021, 371, 29–39 CrossRef CAS.
- A. Neyyathala, F. Flecken and S. Hanf, ChemPlusChem, 2023, 88, e202200431 CrossRef CAS PubMed.
- A. Sampath and D. W. Flaherty, Catal. Sci. Technol., 2020, 10, 993–1005 RSC.
- T. Roy, D. Wisser, M. Rivallan, M. C. Valero, T. Corre, O. Delpoux, G. D. Pirngruber and G. Lefèvre, J. Phys. Chem. C, 2021, 125, 10909–10918 CrossRef CAS.
- H. A. Khalaf, G. A. Mekhemer, A. K. Nohman and S. A. A. Mansour, Monatsh. fur Chem., 2007, 138, 641–648 CrossRef CAS.
- P. Ferrari, L. M. Molina, V. E. Kaydashev, J. A. Alonso, P. Lievens and E. Janssens, Angew. Chem., Int. Ed., 2016, 55, 11059–11063 CrossRef CAS PubMed.
- A. Ellert, F. Herold, M. Rønning, A. Hutzler, L. Piccirilli, T. V. W. Janssens, P. N. R. Vennestrøm, P. Wasserscheid and P. Schühle, J. Catal., 2024, 436, 1–10 CrossRef.
- J. Oh, N. Jeon, I. Chung, O. Seo, J. Park, A. Tayal and Y. Yun, Appl. Catal., A, 2024, 681, 119783 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- G. Bergeret and P. Gallezot, Handb. Heterog. Catal., 2008, 738–765 CAS.
- D. Blaumeiser, C. Schuschke, L. Fromm, N. Taccardi, S. Schötz, R. Eschenbacher, H. Bühlmeyer, T. Xu, T. Bauer, P. Wasserscheid, A. Görling and J. Libuda, J. Phys. Chem. C, 2021, 125, 15301–15315 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- K. Li, L. Luo, Y. Zhang, W. Li and Y. Hou, ACS Appl. Mater. Interfaces, 2018, 10, 41525–41534 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Chen, G. Zhang, Y. Wu, W. Hu, P. Qu, Y. Wang, L. Zhong and Y. Chen, Ind. Eng. Chem. Res., 2020, 59, 6497–6505 CrossRef CAS.
- V. Siva Kumar, A. H. Padmasri, C. V. V. Satyanarayana, I. A. K. Reddy, B. D. Raju and K. S. R. Rao, Catal. Commun., 2006, 7, 745–751 CrossRef CAS.
- K. Lertjiamratn, P. Praserthdam, M. Arai and J. Panpranot, Appl. Catal., A, 2010, 378, 119–123 CrossRef CAS.
- A. John, D. Philip, K. R. Morgan and S. Devanarayanan, Spectrochim. Acta, Part A, 2000, 56, 2715–2723 CrossRef CAS PubMed.
- L. C. Thomas and R. A. Chittenden, Spectrochim. Acta, 1965, 21, 1905–1914 CrossRef CAS.
- J. Dong, J. Wang, J. Wang, M. Yang, W. Li and M. Shen, Catal. Sci. Technol., 2017, 7, 5038–5048 RSC.
- P. Jakob and A. Schiffer, Surf. Sci., 2009, 603, 1135–1144 CrossRef CAS.
- E. Ozensoy and E. I. Vovk, Top. Catal., 2013, 56, 1569–1592 CrossRef CAS.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51–94 CrossRef CAS.
- T. Risse, A. Carlsson, M. Bäumer, T. Klüner and H. J. Freund, Surf. Sci., 2003, 546, L829–L835 CrossRef CAS.
- A. M. Bradshaw and F. M. Hoffmann, Surf. Sci., 1978, 72, 513–535 CrossRef CAS.
- N. M. Martin, M. Van Den Bossche, H. Grönbeck, C. Hakanoglu, F. Zhang, T. Li, J. Gustafson, J. F. Weaver and E. Lundgren, J. Phys. Chem. C, 2014, 118, 1118–1128 CrossRef CAS.
- S. Surnev, M. Sock, M. G. G. Ramsey, F. P. P. Netzer, M. Wiklund, M. Borg and J. N. N. Andersen, Surf. Sci., 2000, 470, 171–185 CrossRef CAS.
- T. Bauer, S. Mehl, O. Brummel, K. Pohako-Esko, P. Wasserscheid and J. Libuda, J. Phys. Chem. C, 2016, 120, 4453–4465 CrossRef CAS.
- A. R. Head, O. Karslıoǧlu, T. Gerber, Y. Yu, L. Trotochaud, J. Raso, P. Kerger and H. Bluhm, Surf. Sci., 2017, 665, 51–55 CrossRef CAS.
- S. Bertarione, D. Scarano, A. Zecchina, V. Johánek, J. Hoffmann, S. Schauermann, J. Libuda, G. Rupprechter, H. J. Freund, M. M. Frank, J. Libuda, G. Rupprechter and H. J. Freund, J. Catal., 2004, 108, 64–73 CrossRef.
- M. Kettner, C. Stumm, M. Schwarz, C. Schuschke and J. Libuda, Surf. Sci., 2019, 679, 64–73 CrossRef CAS.
- I. V. Yudanov, R. Sahnoun, K. M. Neyman, N. Rösch, J. Hoffmann, S. Schauermann, V. Johánek, H. Unterhalt, G. Rupprechter, J. Libuda and H.-J. J. Freund, J. Phys. Chem. B, 2003, 107, 255–264 CrossRef CAS.
- G. Rupprechter, H. Unterhalt, M. Morkel, P. Galletto, L. Hu and H.-J. Freund, Surf. Sci., 2002, 502–503, 109–122 CrossRef CAS.
- R. J. Liu, P. A. Crozier, C. M. Smith, D. A. Hucul, J. Blackson and G. Salaita, Appl. Catal., A, 2005, 282, 111–121 CrossRef CAS.
- C. R. Henry, Prog. Surf. Sci., 2005, 80, 92–116 CrossRef CAS.
- A. Patra, H. Peng, J. Sun and J. P. Perdew, Phys. Rev. B, 2019, 100, 035442 CrossRef CAS.
- G. T. K. Kalhara Gunasooriya and M. Saeys, ACS Catal., 2018, 8, 3770–3774 CrossRef CAS.
- F. D. Manchester, A. San-Martin and J. M. Pitre, J. Phase Equilib., 1994, 15, 62–83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01456a |
| This journal is © The Royal Society of Chemistry 2025 |