
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidation of the factors governing the oxygen evolution reaction in Ba0.5Sr0.5CoxFe1−xO3−δ catalysts via operando hard and soft X-ray absorption spectroscopy†
Weijie
Cao‡
 a,
Mukesh
Kumar‡
*a,
Neha
Thakur
a,
Mitsuhiro
Matsumoto
b,
Yoichiro
Tsuji
b,
Toshiki
Watanabe
a,
Toshiyuki
Matsunaga
a,
Yoshiyuki
Kuroda
c,
Shigenori
Mitsushima
cd and
Yoshiharu
Uchimoto
a
a,
Mukesh
Kumar‡
*a,
Neha
Thakur
a,
Mitsuhiro
Matsumoto
b,
Yoichiro
Tsuji
b,
Toshiki
Watanabe
a,
Toshiyuki
Matsunaga
a,
Yoshiyuki
Kuroda
c,
Shigenori
Mitsushima
cd and
Yoshiharu
Uchimoto
a
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida Nihonmatsu-cho, Sakyo, Kyoto 606-8501, Japan. E-mail: kumar.mukesh.5x@kyoto-u.ac.jp
bOffice of Institutional Advancement and Communications, Kyoto University, Yoshida Honmachi, Kyoto 606-8501, Japan
cGraduate School of Engineering Science, Yokohama National University, 79-5, Tokiwadai, Hodogaya-ku, Yokohama, Kanagawa 240-8501, Japan
dInstitute of Advanced Sciences, Yokohama National University, 79-5, Tokiwadai, Hodogaya-ku, Yokohama, Kanagawa 240-8501, Japan
First published on 16th May 2025
Abstract
Ba0.5Sr0.5CoxFe1−xO3−δ (BSCF) has garnered considerable attention as a promising catalyst for the alkaline oxygen evolution reaction (OER). However, the intrinsic roles of Co and Fe at the active sites during the OER process remain inadequately understood due to the limited studies integrating bulk-sensitive and surface-sensitive operando techniques. Prior studies on BSCF catalysts have either focused on surface transformations or employed bulk-sensitive techniques to probe redox dynamics. Our work uniquely integrates both bulk-sensitive (Co/Fe K-edge XAS) and surface-sensitive (Co/Fe L-edge) operando spectroscopy, which provides surface-sensitive and 3d orbital-specific information, and O K-edge XAS, which captures the dynamics of oxygen-containing active species. This comprehensive multi-edge approach directly correlates the bulk redox behavior with the surface electronic structure. The electrochemical performance is systematically evaluated for different Ba0.5Sr0.5CoxFe1−xO3−δ. Further, intrinsic OER activity was evaluated by eliminating the bubbles' influence during OER by using the forced flow method. Our combined soft and hard XAS revealed that the superior OER activity of Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF5582) is attributed to the structural and electronic features of the electrochemically active layer formed at the interface. Minor Fe doping (x = 0.2) predominantly occupies octahedral sites, modulating the electronic states of Co. Under increasing potentials, Co2+ in tetrahedral sites (Td) transitions to Co3+ in octahedral sites (Oh), promoting the formation of the catalytically active CoOOH phase. In contrast, Ba0.5Sr0.5Co0.2Fe0.8O3−δ (BSCF5528), with higher Fe doping, stabilizes Co3+ in Oh sites without substantial changes in oxidation state, leading to the predominance of FeOOH as the active phase and diminished catalytic performance. Operando O K-edge XAS results align closely with the observed behavior of the 3d transition metals, revealing that the formation of reducible Fe3+/Co3+–O(H) sites, particularly μ2-O(H) bridges, significantly enhances OER activity. These findings provide a mechanical insight highlighting the critical role of Fe doping in optimizing the structural and electronic properties of BSCF catalysts to achieve superior OER performance.
Introduction
Hydrogen energy, widely recognized as a clean and sustainable fuel, is crucial for transitioning the global energy landscape away from reliance on fossil fuels.1–3 Among various hydrogen production methods, large-scale alkaline water electrolysis has garnered significant attention due to its environmental compatibility and cost-effectiveness.4–7 Within this framework, perovskite oxides, particularly Ba0.5Sr0.5CoxFe1−xO3−δ (BSCF), have emerged as highly promising catalysts for the oxygen evolution reaction (OER) in alkaline media.8–12Shao-Horn et al. demonstrated a volcano-shaped relationship between the eg orbital filling of perovskite oxides and their intrinsic OER activity, establishing BSCF as a benchmark material due to its optimal eg orbital filling of approximately 1.2.10,13 Furthermore, studies have revealed that during OER, dynamic surface reconstruction occurs at B-site cations (Co/Fe), facilitated by lattice oxygen evolution reactions:8,14
 | (1) |
These findings underscore the critical role of BSCF's flexible perovskite structure and high oxygen vacancy content in achieving superior OER performance. Recent advancements, including doping strategies and hybrid material designs, have further optimized BSCF derivatives, achieving overpotentials as low as 260 mV in 0.1 M KOH (Table S2†).9 These achievements demonstrate the competitiveness of BSCF-based materials.
Despite extensive studies on BSCF, significant uncertainties remain regarding the exact structure of the active Co/FeOOH layer, the potential synergistic effect between Co and Fe, and the involvement of Co and/or Fe at active sites in BSCF catalysts with different Co/Fe ratios,15 particularly those with high Fe content, such as Ba0.5Sr0.5Co0.2Fe0.8O3−δ (BSCF5528). Clarifying this issue is essential for further improving the activity of this widely researched BSCF, and for this purpose, there is a strong desire to apply operando measurement techniques that clarify the active sites of the catalysts during the reaction.
X-ray absorption spectroscopy (XAS) has been widely used to investigate the local electronic and geometric structures of BSCF and to determine the active Co/FeOOH layer based on oxidation states and coordination environments.8,16–18 While hard X-ray XAS (e.g., Co/Fe K-edge) provides bulk-sensitive, average structural information such as oxidation state transitions and coordination changes,19–22 OER is predominantly governed by the surface/electronic structure of the catalyst, surface-sensitive operando analysis methods are essential to acquire information about the catalyst surface during OER.
Soft X-ray XAS in total-electron-yield (TEY) mode at the Co/Fe L-edge, with a probing depth of approximately 5 nm, has emerged as a powerful tool for providing detailed surface-sensitive information.23,24 Recent developments in operando O K-edge TEY-XAS have also enabled in-depth studies of catalytic interfaces, particularly in Ir-based catalysts under acidic OER conditions.22,25 However, such techniques remain underexplored in studying BSCF for alkaline OER. To bridge this gap, the combination of bulk-sensitive and surface-sensitive operando techniques and the use of information on oxygen-containing species, which are crucial for the elementary reaction processes, and not just information on 3d transition metals, is essential. A comprehensive understanding of the dynamic structural and electronic changes that occur in the BSCF catalyst during the OER process is necessary to resolve the ambiguity, but this has not been achieved through integrated operations.
In this study, we synthesized four different catalysts: Ba0.5Sr0.5CoO3−δ (BSC), Ba0.5Sr0.5FeO3−δ (BSF), Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF5582), and Ba0.5Sr0.5Co0.2Fe0.8O3−δ (BSCF5528). To mitigate the influence of bubbles during OER activity testing using the three-electrode method, a new electrochemical measurement technique, the “time-zero method”, was introduced. This approach allows us to estimate kinetic currents under conditions of no supersaturation or bubbles on the catalyst surface. Additionally, operando Co/Fe K-edge, Co/Fe L-edge, and O K-edge XAS were employed to investigate the dynamic changes in the surface structure of BSCF during OER. The relationship between the Fe content and the dynamic structural changes of Co and Fe, which promote the formation of active sites, was clarified, and the formation of oxygen-containing species corresponding to the dynamic changes in 3d transition metals was quantitatively clarified by operando O K-edge measurement. Through this analysis, we attempted to explain the critical role of minor Fe doping in optimizing BSCF's surface structure and improving OER performance. This is expected to provide a solid foundation for the advanced design of perovskite-type OER catalysts.
Experimental
Materials
Barium nitrate (Ba(NO3)2, 99.0%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, 99.0%), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99.0%), citric acid (98.0%), ethylenediaminetetraacetic acid (EDTA, 99.5%), ammonia solution (NH3·H2O, 28.0%), perchloric acid (HCl, 35.0%), strontium nitrate (Sr(NO3)2, 99.9%), ethanol (99.5%) were acquired from Fujifilm. All chemical purities are reported as weight percentages (wt%) and used as received without further purification. The electrolyte was prepared from ultra-pure water (Milli-Q, 18.2 MΩ) and perchloric acid (ultra-pure, Kanto Chemical Co. Inc.).Synthesis of Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.5, 0.8, 1)
The four catalysts, Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.8, 1), were synthesized using an ethylenediaminetetraacetic acid–citric acid (EDTA–CA) complexation sol–gel method. For Ba0.5Sr0.5Co0.8Fe0.2O3−δ as an example, stoichiometric amounts of 0.26 g Ba(NO3)2, 0.21 g Sr(NO3)2, 0.46 g Co(NO3)2·6H2O, and 0.16 g Fe(NO3)3·9H2O were dissolved in 20 mL deionized water. 1.17 g EDTA and 1.54 g citric acid were added sequentially as complexing agents in a mole ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 for total metal ions:EDTA:citric acid. The pH of the solution was adjusted to 6–7 using NH3·H2O to ensure complete complexation. The solution was heated to 90 °C and stirred continuously until a gel formed. This gel was dried in a furnace at 250 °C for 5 hours to produce a solid precursor. The solid precursor was milled and sintered at 1000 °C for 5 hours in an air atmosphere to form the BSCF powder.
:1:2 for total metal ions:EDTA:citric acid. The pH of the solution was adjusted to 6–7 using NH3·H2O to ensure complete complexation. The solution was heated to 90 °C and stirred continuously until a gel formed. This gel was dried in a furnace at 250 °C for 5 hours to produce a solid precursor. The solid precursor was milled and sintered at 1000 °C for 5 hours in an air atmosphere to form the BSCF powder.
The synthesis of the other catalysts followed the same procedure as Ba0.5Sr0.5Co0.8Fe0.2O3−δ, with the only difference being the molar amounts of Co(NO3)2·6H2O and Fe(NO3)3·9H2O used.
Characterization
X-ray diffraction (XRD) analyses were performed using a Rigaku Ultima IV with Cu Kα radiation at a scanning rate of 2° per minute to characterize the crystalline structure of the samples. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) were conducted using a Hitachi S-3400N microscope at an accelerating voltage of 20 kV. For these analyses, a 0.2 mg sample was dispersed in anhydrous ethanol, dropped onto tape attached to the sample holder, and allowed to dry before observation. Sample morphologies were examined using a JEM-2200FS transmission electron microscope (JEOL, Ltd.) at an accelerating voltage of 200 kV. Surface chemical compositions were analyzed using X-ray photoelectron spectroscopy (XPS) on a PHI5000 VersaProbe II, utilizing monochromatic Al Kα radiation for high precision. Ex situ Fe and Co K-edge XAS were conducted in transmission mode using a Si (111) monochromator on BL14B2 at SPring-8, Japan. High-energy-resolution fluorescence-detected XANES (HERFD-XANES) of the Fe and Co K-edge were acquired at the BL36XU and BL39XU at SPring-8, Japan. X-ray emission spectroscopy (XES) was used to monochromatize the fluorescent X-ray before acquiring HERFD-XANES signal. Incident X-ray were generated using a pair of Si (111) monochromators and a total-reflection Rh mirror (4.5 mrad). For energy alignment, Co and Fe metal foils were used as references. The first peak of the first derivative of the foil spectra was assigned to 7708.9 eV (Co K-edge) and 7110.75 eV (Fe K-edge). The surface area of the nanosized material was calculated using the Brunauer–Emmett–Teller (BET) method. N2 adsorption–desorption experiments were conducted at 77.00 K using an ASAP 2020 physisorption analyzer (Micromeritics Instrument Corporation).RDE measurements
The synthesized Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.8, 1) catalysts were evaluated for OER activity using electrochemical measurements in rotating disk electrode (RDE) mode. Catalyst inks were prepared by dispersing 11.76 mg of catalyst powder in 973 μL of ethanol and 26 μL of 5 wt% Nafion solution (Sigma-Aldrich), followed by sonication in an ice bath for 20 minutes. A total of 5 μL of the ink was then dropped onto a polished glassy carbon (GC) electrode (Hokuto Denko, 5 mm diameter, 0.196 cm2).OER performance in a 0.1 M KOH solution was assessed using a standard three-electrode electrochemical cell (Naito Rika) controlled by an HZ-7000 electrochemistry workstation (Hokuto Denko Ltd.). The GC cast with the catalyst served as the working electrode, with Pt mesh as the counter electrode and a reversible hydrogen electrode (RHE) as the reference electrode prepared using 5% H2 gas. The electrode was equilibrated for a minimum of 30 minutes with 5% H2 prior to measurement to ensure a stable and reliable reference potential. A continuous gas flow was maintained during the electrochemical performance. To calibrate our potential to the standard 100% RHE scale, the recorded potentials were corrected by adding 0.0385 V to account for the Nernst equation, ensuring accurate conversion to the RHE scale.
Cyclic voltammetry (CV) was performed at 50 mV s−1 from 1.0 V to 1.6 V vs. RHE for 50 cycles for pre-treatment, followed by 5 cycles at 10 mV s−1 from 0 V to 1.6 V vs. RHE. This activation cycle is essential for inducing surface reconstruction and generating the catalytically active phase.8 Linear sweep voltammetry (LSV) was conducted at 10 mV s−1 from 1.0 V to 1.8 V vs. RHE, with the RDE rotating at 1600 rpm.
Forced-flow cell measurement
The anode catalysts and cathode catalyst used were Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.8, 1) and Pt/C (Pt 28.4%, Tanaka Kikinzoku Kogyo, Japan), respectively. The Pt/C was dispersed in ethanol uniformly sprayed onto a 1 × 1 cm2 carbon paper (SGL Carbon Japan Ltd.) with a loading amount of approximately 0.5–0.6 mg cmPt−2 and served as the cathode material. The BSCF catalyst ink (ethanol:H2O = 1:2) was ultrasonically homogenized for less than 5 minutes, then applied to Zirfon® (Perl UTP 500, Agfa, Belgium) using a masking tape with five rectangular holes (1 × 8 mm2 each), resulting in an approximate catalyst loading of 1.2 mg cm−2, and was dried before electrochemical testing.
In the cell setup (Fig. S1†), interdigitated and parallel flow fields were applied to the anode and cathode, respectively. The membrane pressure was set at 4 MPa, with both the anode and cathode chambers circulating 7 M KOH solution at 30 °C. The anode flow rate was 50 mL min−1, while the cathode flow rate was 30 mL min−1.
OER performance in 7 M KOH solution was evaluated using a forced-flow cell controlled by a Bio-Logic MPG-205 electrochemistry workstation. The electrochemical measurement protocol included pre-treatment with chronopotentiometry (CP) at 400 mA cm−2 for 2 hours. The electrochemical impedance spectroscopy (EIS) with an AC amplitude of 10 mV and frequencies from 100 kHz to 0.1 Hz at 1.5 V, 1.6 V, and 1.7 V vs. RHE. Pulsed potential step measurements were performed with a constant increasing potential step applied for 10 seconds per 0.01 V from 1.5 V to 1.7 V vs. RHE. Chronoamperometry (CA) intervals of 360 s were set to remove bubbles generated within the catalyst layer, applying a lower potential during these intervals to avoid phase transitions in the catalyst (Fig. S2a†).
Time-zero analysis details are provided in the ESI† (Fig. S2b).
Operando XAS measurement
Operando soft XAS was conducted at BL27 in SPring-8 using total electron yield (TEY) mode. The working electrode material was prepared by spraying the ink onto an anion exchange membrane (Sustainion® X37-50 Grade RT), achieving a catalyst loading of approximately 0.1 mg cm−2. The flow cell, assembled as shown in the Fig. S3,† utilized a Pt wire wrapped around the RHE as the counter electrode, a handmade RHE (H2 100% purity) as the reference electrode, and a glassy carbon with the catalyst membrane as the working electrode. A high-purity H2 gas for RHE was locally generated via electrolysis using a 9 V battery on the platinum wire. The electrolyte was 7 M KOH, flowing at 10 mL min−1 during the operando soft XAS tests. Monochromatic X-ray were generated using a line-spacing plane grating monochromator. Electrode polarization ranged from 0.5 V to 1.9 V vs. RHE in a vacuum environment. XAS signals were recorded after stabilizing each potential for 10 minutes to ensure steady current conditions. Co L3-edge spectra was normalized by dividing the intensity by the averaged maximum intensities of the A and B peaks. Fe L3-edge spectra was normalized by dividing the intensity by the averaged maximum intensities of the D and E peaks.Operando hard XAS was performed at BL37 in SPring-8 using partial fluorescence yield (PFY) mode. During the test, a pair of Si (111) monochromators and a 4 mrad total-reflection Rh mirror were used to obtain collimated and monochromatic X-ray. The electrode was polarized from 0.5 V to 1.9 V vs. RHE in an air atmosphere. The XAS signal was collected after maintaining polarization at each potential for 10 minutes to achieve stable current conditions.
Results and discussion
Characterization
Various Fe-doped Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.8, 1) catalysts were synthesized using the ethylenediaminetetraacetic acid–citric acid (EDTA–CA) complex sol–gel method. The perovskite oxides have a general formula ABO3, where the A-site cations (typically Ba2+ and Sr2+) occupy the corners of the unit cell, and the smaller B-site cations (Co and Fe) are located at the body center. The oxygen anions reside at the face centers, forming a three-dimensional network of corner-sharing BO6 octahedra. The high flexibility of A and B site substitutions facilitates a wide range of electronic, structural, and catalytic properties. The XRD patterns of various catalysts are shown in (Fig. 1a). For Ba0.5Sr0.5FeO3−δ (BSF) the characteristic crystalline long range order diffraction peaks are observed at approximately at 23°, 32°, 39°, 46°, 52°, 58°, and 68°, corresponding to the (100), (110), (111), (200), (210), (220), and (300) planes, respectively. These reflections match well with those of a cubic perovskite structure (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/i_char_0033_0304.gif) m), in agreement with previously reported work.9,26 As the Co/Fe atomic ratio increases from BSF to Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.5 and 0.8), the diffraction peaks are slightly shifted due to the partial replacement of larger Fe3+ ions with smaller Co3+ ions.27 These changes could alter Co–O and Fe–O bond lengths, distorting the BO6 octahedra (detailed in the following section). However, it is noteworthy to point out that from BSF to Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.5 and 0.8) the cubic structure remains well maintained. In contrast, Ba0.5Sr0.5CoO3−δ (BSC), without Fe, exhibits an XRD pattern with a distinctly different set of peaks that can be indexed to a hexagonal structure (space group P63/mmc), indicating a structural phase transition from cubic to hexagonal symmetry. This phenomenon has also been confirmed in previous studies, where increasing the Co content (x = 0.9, 0.95, 1) induces a phase transition (Fig. S4†), attributed to the additional effects of the A-site Ba/Sr.28,29
m), in agreement with previously reported work.9,26 As the Co/Fe atomic ratio increases from BSF to Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.5 and 0.8), the diffraction peaks are slightly shifted due to the partial replacement of larger Fe3+ ions with smaller Co3+ ions.27 These changes could alter Co–O and Fe–O bond lengths, distorting the BO6 octahedra (detailed in the following section). However, it is noteworthy to point out that from BSF to Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.5 and 0.8) the cubic structure remains well maintained. In contrast, Ba0.5Sr0.5CoO3−δ (BSC), without Fe, exhibits an XRD pattern with a distinctly different set of peaks that can be indexed to a hexagonal structure (space group P63/mmc), indicating a structural phase transition from cubic to hexagonal symmetry. This phenomenon has also been confirmed in previous studies, where increasing the Co content (x = 0.9, 0.95, 1) induces a phase transition (Fig. S4†), attributed to the additional effects of the A-site Ba/Sr.28,29
 | ||
| Fig. 1 (a) XRD patterns and (b) TEM images of Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.5, 0.8, 1). | ||
The particle distribution and surface morphology of the synthesized samples were analyzed using SEM and TEM (Fig. 1b and S5†). The results show that the average particle size of the four catalysts ranges from 200 nm to 400 nm, with BSC and BSF having smaller average particle sizes around 200 nm. Fig. S6† presents the EDS spectrum and corresponding elemental mapping images of as-prepared BSCF5582. The metal atom ratios in the catalysts are roughly consistent with the calculated values.
The BET-specific surface areas of the catalysts were determined from N2 adsorption–desorption isotherms (Fig. S7 and Table S3†). The N2 adsorption–desorption isotherms of all samples exhibit type III characteristics and show a rapid decrease in N2 adsorption with a decrease in pressure. This suggests weak adsorbent–absorbate interactions could be due to a less porous structure.30 Compared to pure BSC (0.73 m2 g−1) and BSF (0.56 m2 g−1), the BET specific surface areas of BSCF catalysts are slightly smaller, corresponding to the larger nanoparticle sizes observed in SEM and TEM images. Among the BSCF catalysts, BSCF5582 has a BET surface area of 0.41 m2 g−1, similar to BSCF5528 (0.49 m2 g−1) and BSCF5555 (0.49 m2 g−1). This indicates that the B-site metal ratio does not significantly affect the BET surface area of perovskite oxide catalysts.
To evaluate the catalysts' structural characteristics in an alkaline electrolyte, they were immersed in 0.1 M KOH for two hours before XAS measurement.11 HERFD-XANES measurements were performed, offering enhanced spectral resolution over conventional XANES by selectively detecting fluorescence from a specific emission line and minimizing core-hole lifetime broadening effects. This improved resolution allows detection of subtle energy shifts and fine structural features critical for distinguishing small variations in oxidation states.31,32
The oxidation states of the B-site metals (Co and Fe) were quantitatively assigned using the first derivative of the HERFD-XANES spectra (Fig. 2a, b, and S8†). The average bulk oxidation states were approximately +2.57 for Co and +2.90 for Fe across all samples. The oxidation state of Co followed the order: BSC > BSCF5528 > BSCF5582, indicating that Fe doping reduces Co oxidation and increases oxygen vacancy concentration.17 However, excessive Fe doping in BSCF5528 leads to further oxidation of Co. In Fig. S9b,† BSCF5582 exhibits the highest pre-edge (1s to 3d) intensity in the Co K-edge XANES spectra and the lowest oxidation state among all samples. This is due to the sensitivity of the pre-edge peak to the metal's oxidation state and coordination symmetry, with tetrahedral (Td) sites showing more intense pre-edge features than octahedral (Oh) sites discussed in detail in the following paragraph with FT-EXAFS analysis (Fig. S9a†).33
 | ||
| Fig. 2 (a) The normalized Co K-edge HERFD-XANES spectra and (c) the R-space Fourier-transformed of the Co K-edge EXAFS spectra for Co3O4, Co(OH)2, CoOOH, BSC, BSCF5528 and BSCF5582. (b) The normalized Fe K-edge HERFD-XANES spectra and (d) Fourier-transformed of the Fe K-edge EXAFS spectra for Fe3O4, FeOOH, BSF, BSCF5528 and BSCF5582. Peaks A1/A2 correspond to the single-scattering path from metal ions to the nearest oxygen atoms (Co–O and Fe–O). The peaks at B1 (Co–Co(Oh))/B2 (Fe–Fe(Oh)) and C1 (Co–Co(Td)/Co–Fe(Td)) represent the scattering from adjacent metal ions in octahedral or tetrahedral sites. | ||
Comparing BSC, it is observed that with a Fe doping of 0.2 (BSCF5582), Co2+(Td) occupancy increases, leading to the reduction in an average oxidation state of Co as Fe ions preferentially occupy Oh sites.11 However, when Fe doping is increased to 0.8 (BSCF5528), there is a noticeable decrease in the pre-edge region, and the spectral energy shifts positively due to Fe3+ increasingly occupying Td sites, reducing the presence of Co2+(Td).34 FT-EXAFS analysis validated the distances and structural deformations within the crystals. As shown in Fig. 2c and d, the first peak at position A corresponds to the single-scattering path from metal ions to the nearest oxygen atoms (Co–O and Fe–O). The second and third peaks at positions B1 (Co–Co(Oh))/B2 (Fe–Fe(Oh)) and C1 (Co–Co(Td)/Co–Fe(Td)) represent the scattering from adjacent metal ions in octahedral or tetrahedral sites, depending on the initial position of the metal ions.35 In the Co K-edge FT-EXAFS spectra (Fig. 2c), a small amount of Fe doping (BSCF5582) leads to Co ions preferentially occupying Td sites, resulting in higher signal intensity at position C1. Under heavy doping conditions (BSCF5528), most Co ions occupy Oh sites, consistent with XANES observations. Additionally, since the Co3+(Oh)–O distance is greater than the Co2+(Td)–O distance, the signal at position A1 in BSCF5582 shifts to a shorter scattering path. Further, the Fe K-edge XANES spectra comparison in Fig. S9c† also revealed distinct pre-edge features. Peaks around ∼7109 eV and ∼7110 eV correspond to tetrahedral Fe3+(Td) and octahedral Fe3+(Oh), respectively.36 All BSCF samples showed positive energy shifts compared to Fe3O4, suggesting dominant Fe3+(Oh) coordination in BSCF. Among them, BSCF5582 exhibited stronger pre-edge intensity and a slight positive energy shift, reinforcing that Fe preferentially occupies Oh sites, stabilizing Co in Td coordination and suppressing Co oxidation.
From the Fe K-edge FT-EXAFS spectra (Fig. 2d), it is evident that in all catalysts, most Fe ions reside in Oh sites. Despite the presence of Fe3+(Td) in BSCF5528, it still primarily occupies Oh positions due to its lower formation energy, consistent with previous studies.35
O K-edge XAS provides critical insights into the density of unoccupied states around the Fermi level of transition metals. As shown in Fig. S10,† the peaks at 529.1 eV and 530.5 eV correspond to the hybridization of O 2p with the transition metal 3d t2g and eg orbitals, respectively. The lower intensity at approximately 529.1 eV in the BSCF5528 sample indicates more occupied states in the eg orbitals, suggesting a higher spin state because of the heavy Fe doping.22,37
XPS spectra further confirm the surface oxygen states in the BSCF catalysts. Fig. S11† shows the O 1s XPS spectra of BSCF5528 and BSCF5582, deconvoluted into three peaks: lattice oxygen (O2−) at 528.6 eV, highly oxidative species (O2−/O−) at 529.9 eV, and hydroxyl groups or surface-absorbed oxygen (OH− or O2) at 531.2 eV.38 BSCF5582 has a significantly higher O2− content than BSCF5528, enhancing lattice oxygen oxidation during the OER process.39,40
Electrochemical measurement
The OER performance of the synthesized catalysts in 0.1 M KOH solution was evaluated using a standard three-electrode electrochemical cell. To minimize the adverse effects of bubble formation and facilitate bubble detachment during OER, all electrochemical OER measurements were performed under continuous electrode rotation at 1600 rpm. Fig. 3b presents the LSV polarization curves for BSC, BSF, BSCF5582, and BSCF5528. Among these BSCF5582 catalyst demonstrates a lower overpotential (η) at each current density compared to the other catalysts, indicating superior OER electrocatalytic activity. The Tafel plot in Fig. 3c further reveals that BSCF5582 has the lowest Tafel slope (111.0 mV dec−1), suggesting faster OER kinetics than other catalysts.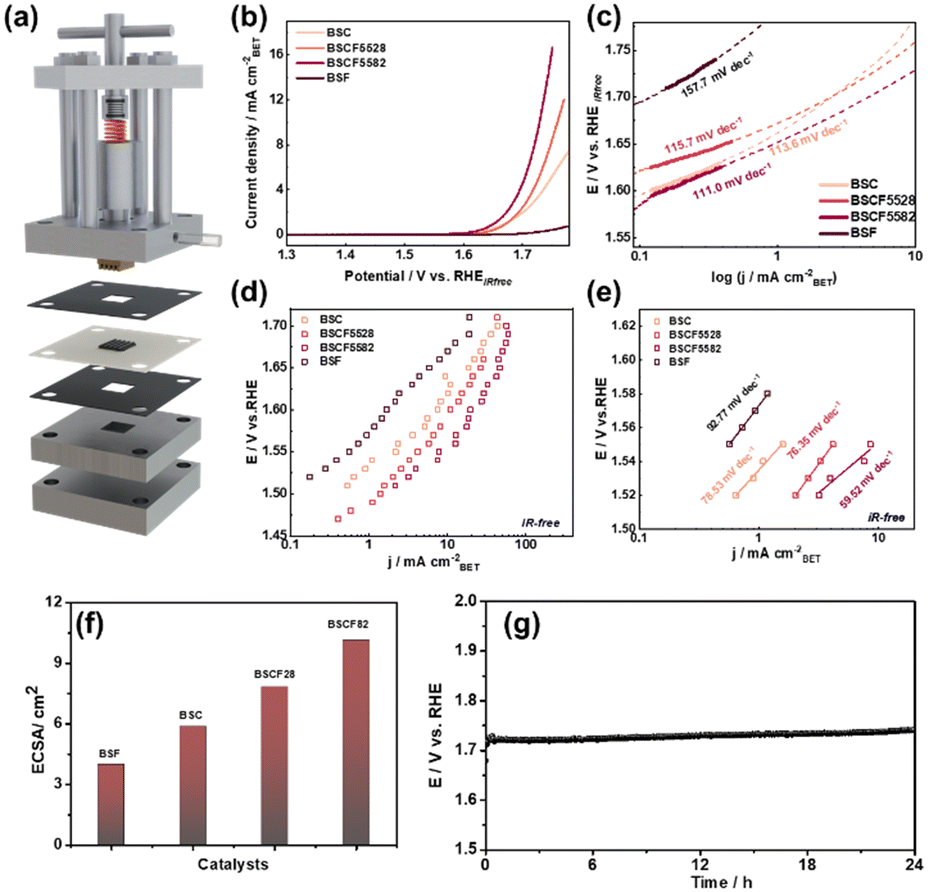 | ||
| Fig. 3 (a) Schematic illustration of the forced-flow cell. (b) LSVs curves and (c) tafel slope of BSC, BSF, BSCF5582 and BSCF5528 by RDE method. (d) i–e curves and (e) tafel slope of BSC, BSF, BSCF5582 and BSCF5528 using the forced-flow cell method and time zero analysis. (f) ECSA, (g) stability of BSCF5582 with forced flow cell at 100 mA cm−2 for 24 h. | ||
However, persistent bubble adhesion was observed at higher OER potential despite using electrode rotation. Oxygen gas evolution leads to the formation of microbubbles on the electrode surface, which progressively nucleate and grow into larger bubbles. These gas bubbles can block active catalytic sites, increase charge transfer resistance, and limit mass transport by impeding ion diffusion between the electrode and the electrolyte, which can still affect the apparent OER activity and hinder the accurate evaluation of reaction kinetics and intrinsic OER activity. To more precise OER activity measurement, a new “time-zero method” using a forced-flow cell was introduced to address limitations of standard electrochemical cells, allowing for accurate OER activity measurements by preventing catalyst detachment and eliminating bubble interference (Fig. 3a).41,42 CA tests using a forced-flow cell were performed to precondition and activate the catalysts by maintaining a current density of 400 mA cm−2 for two hours with forced flow cell. This process accelerated the dissolution of surface Ba/Sr and structural transformation, resulting in the formation of a Co/Fe-rich surface layer, which lowered the overpotential and enhanced OER activity (Fig. S12 and S13†). EIS was also conducted under the operating conditions for OER, with BSCF5582 exhibiting the lowest charge transfer resistance (0.7 Ω at 1.7 V vs. RHE), signifying excellent conductivity and rapid charge transfer rates (Fig. S14†).
CV measurements further confirmed the impact of the Co/Fe-rich surface on the OER activity of BSCF catalysts. Fig. S15† shows the CV curves for the four catalysts at different cycle numbers, with BSCF5582 exhibiting significant surface reconstruction, resulting in increased activity. The redox peak observed around 1.4 V vs. RHE is associated with the Co2+ to Co3+ transition in Co3O4 spinel and the formation of surface hydroxide Co3+–OHad.43,44 Compared to BSC, the anodic shift of BSCF catalysts indicates that Fe doping suppresses Co oxidation and promotes its transition to hydroxides, confirming the presence of a Co/Fe-rich surface.11,34 For the BSF catalyst, the peak around 1.3 V vs. RHE corresponds to the Fe2+/Fe3+ transition, contributing minimally to OER activity.45
Fig. 3d and e present the OER activity for all four BSCF catalysts measured using the forced-flow cell and time-zero method, normalized by BET surface area. Within the potential range of 1.45 V to 1.75 V vs. RHE, BSCF5582 demonstrated the highest current density and the lowest Tafel slope among the catalysts, consistent with results from the RDE method. The ECSA provides a good measure of OER active sites. The OER catalytic activity and ECSA of the BSCF series catalysts follow the order: BSCF5582 > BSCF5528 > BSC > BSF, which clearly demonstrates that more active sites are generated after adding a smaller amount of Fe (Fig. 3f and S16B†).
Under accelerated degradation conditions, the OER durability of BSCF5528 and BSCF5582 was evaluated at 100 mA cm−2 over different cycle numbers (Fig. S16†). After 600 cycles, BSCF5528 exhibited a significant decrease in OER activity (∼81 mV), while BSCF5582 showed excellent stability with only a ∼58 mV increase. This superior stability of BSCF5582 is attributed to the reversible surface reconstruction between Co/Fe spinel and Co/Fe oxyhydroxide under accelerated degradation conditions.11 Further, we have performed the stability@100 mA cm−2 fixed current density under forced flow cell conditions for 24 h (Fig. 3g). The catalysts maintained a stable response at higher current density, demonstrating their good stability.
At the same applied potential, the current densities measured using the time-zero method were higher for all four samples compared to those obtained using the RDE method. This indicates that the time-zero method accurately assesses the intrinsic OER activity at the initial time of bubble generation, demonstrating its superiority in electrochemical measurement. The catalytic OER activity measured by both the RDE and time-zero methods exhibits a volcano-type relationship with the Fe doping level (denoted as 1-X), as illustrated in Fig. S17.† This suggests that slight Fe doping (1-X = 0.2) enhances OER activity to an optimal level, beyond which the activity decreases. Thus, varying Fe doping levels result in different surface structures and electronic states, affecting the catalytic activity.
Operando Co/Fe K and L-edge XAS measurement
To elucidate the superior performance of BSCF5582 and the relationship between Fe substitution and OER activity, operando XAS measurements with a custom-built electrochemical cell were used, highlighting the essential role of in situ/operando techniques in identifying actual active sites due to changes in the electronic structure of Fe/Co surface under OER conditions.8,9,20 The CV curves of BSCF5582 obtained with the operando cell and a three-electrode RDE setup demonstrate consistent redox potentials and reliable current-potential relationships, confirming the operando method's effectiveness (Fig. S18†).The operando Co K-edge spectra of BSCF5582, shown in Fig. 4a and S19a,† indicate a consistent shift to higher energy levels with increasing OER potential, suggesting a continuous increase in the average oxidation state of Co from 0.8 V to 2.0 V vs. RHE (ΔValence = 0.13).46 The rapid changes in Co oxidation states occur around 1.3 V vs. RHE, correlating with the onset of OER and the surface structural reconstruction. This oxidation is more pronounced in BSCF5582 than in BSC (Fig. S20a†), correlating with higher OER activity attributed to minor Fe doping. The pre-edge peaks of BSCF5582 show a slight decrease, implying a transition of Co ions from Td to Oh sites, further supported by FT-EXAFS spectra in Fig. 4e. Fig. 4e reveals two overlapping peaks at ∼2.8 Å and ∼3.0 Å in the Co–Co/Fe coordination shells of BSCF5582, corresponding to Co3+(Oh) in Co/Fe oxyhydroxide and Co2+(Td) in Co/Fe spinel-like structures.11 During anodic polarization, a transition from Co2+(Td) to Co3+(Oh) peaks was observed, indicating the growth of a self-assembled Co hydroxide layer, forming the actual active surface.8 This conclusion is further supported by the FT-EXAFS fitting results in Table S5.† In contrast, BSC showed minimal growth at the ∼2.8 Å scattering peak due to side oxide phase formation, inhibiting surface layer reconstruction (Fig. S20c†).17 Therefore, minor Fe doping promotes structural transformation of Co ions, and enhances the OER process with the formation of the active CoOOH surface layer.
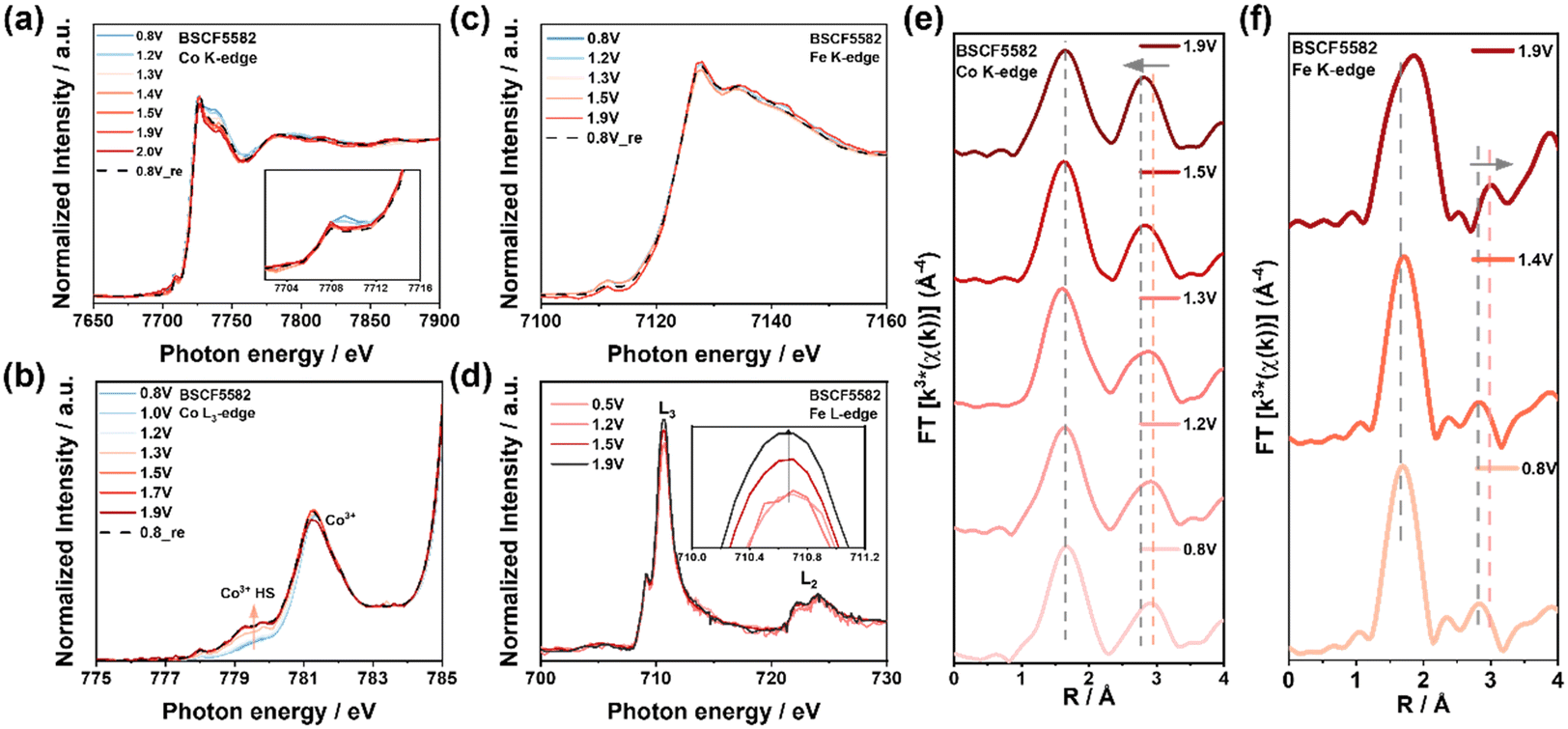 | ||
| Fig. 4 Operando XAS for investigating the origin of OER activity of BSCF5582 under application of various potentials from 0.8 V to 1.9 V vs. RHE. (a) Co K-edge XANES, inset showing the enlargement of pre-edge peaks of BSCF5582. (b) Co L3-edge XANES spectra show low and high spin states. (c) Fe K-edge XANES and (d) Fe L-edge XANES spectra. (e) Fourier transform of the Co K-edge EXAFS spectrum showing transition from Co2+(Td) to Co3+(Oh) peaks, (f) Fe K-edge EXAFS spectra showing an increase in the average Fe-O bond length. Aand changes in the position of the Fe–Fe(Oh) scattering peak. | ||
Operando Fe K-edge measurements on BSCF5582 revealed a slight positive shift in the XANES energy position during increasing potential, indicating a minor increase in the average oxidation state of Fe (ΔValence = 0.02) (Fig. 4c and S19c†). While many studies suggest that Fe enhances the activity of other transition metals without inherent changes,18,34,47 research by Boettcher et al. demonstrated that Fe can act as an active site in Co(Fe)OxHy catalysts during OER.48 Therefore, further investigation is required to understand Fe's role in BSCF catalysts during the OER process. The FT-EXAFS spectra of BSCF5582 at different potentials showed a positive shift in the scattering paths at ∼1.8 Å (Fe–O) and ∼2.8 Å (Fe–Fe(Oh)) at 1.9 V vs. RHE, indicating an increase in average Fe–O bond length and structural transformation (Fig. 4f). Studies have shown that average bond lengths in metal oxides during OER consist of metal sites coordinated with lattice oxygen (O2−) and adsorbed OH− species (M–OH).9,49 When Fe participates in the reaction, it forms Fe–OH intermediates. Since O2− carries more negative charge than OH−, the Fe–OH bond length is relatively longer, leading to an increase in the average Fe–O bond length. Additionally, changes in the position of the Fe–Fe(Oh) scattering peak were observed by comparing the FT-EXAFS spectra of FeOOH and Fe3O4 (Fig. S21a†). At high OER potentials, Fe undergoes a configuration change, resulting in the formation of FeOOH active sites. This suggests that while Fe serves an auxiliary role, it also actively participates in the reaction. However, due to the dominant role of Co sites, Fe's contribution is less pronounced at lower OER potentials.
To gain deeper insights into the exceptional OER performance of BSCF5582, operando soft XAS was conducted using the surface-sensitive TEY mode, which enabled a detailed characterization of the catalyst's surface electronic structure evolution.22,50 As shown in Fig. 4b and S22,† the Co L-edge spectra, dominated by the Co 2p to 3d transition, split into L3 and L2 edges due to spin-orbit coupling. The shape and normalized intensity of the L3 edge depend on the Co 3d–3d interaction and the hybridization of Co 3d orbitals with O 2p orbitals, reflecting the symmetry and spin state.51 Compared to the reference CoOOH L3-edges, the absorption peaks of BSCF5582 and BSC are attributed to the Co3+ states (Fig. S23a†). In Fig. 4b, the decreasing intensity of the low-spin (LS, t2g6 eg0, S = 0) Co3+ peak (781.5 eV) with increasing potential indicates partial filling of the eg orbitals, associated with the formation of Co3+ in high-spin (HS, t2g4 eg2, S = 2) states.40,52–54 Additionally, the increasing intensity of the peak at approximately 780.0 eV with increasing potential confirms the formation of the HS-Co3+ (Fig. S24†),51,55–57 a change not observed in BSC (Fig. S20b†). It has also been reported that the peak around 780.0 eV can be attributed to twofold metal-coordinated oxygen atoms (μ2-OH) bridging pairs of oxygen-coordinated HS-Co3+ and Co2+ ions,58 which will be further discussed in the operando O K-edge results. Research suggests that HS-Co3+ enhances electron transfer and facilitates the adsorption of OH and OOH intermediates. Fe heterojunction doping has been shown to adjust Co materials to the high-spin state,59,60 thereby contributing to the superior OER activity of BSCF5582 compared to BSC.
To elucidate the behavior of BSCF5582 under operando conditions, the peak intensity ratio IB/IA for Co L-edge spectra was calculated (Fig. S25†). At 0.8 V vs. RHE, BSCF5582 exhibited an IB/IA value of approximately 1.55, indicating a low HS-Co3+ content. With increasing potential, BSCF5582 showed a notable decrease in IB/IA (Δ ≈ 0.20), correlating with the Co2+(Td) to Co3+(Oh) transition. This rapid high-spin state increase near the OER potential (∼1.3 V vs. RHE) aligns with the observed operando Co K-edge oxidation state increase.
The shoulder at ∼778 eV in Fig. S23a† suggests the potential formation of Co2+, though the primary process involves the oxidation of Co2+ to Co3+. Mechanisms such dissolution of A-site cations (e.g., Ba, Sr) lattice oxygen loss, and intermediate species like Co(OH)2 during OER could contribute to Co2+ formation.61 However, our overall analysis supports the dominance of Co3+ as the primary active species driving the OER process.
Fig. 4d and S23b† displays the Fe L3,2-edge spectra of BSCF5582 at different potentials. The two split peaks near ∼710 eV correspond to the transitions of 2p electrons to 3d t2g orbitals (∼709 eV, D peak) and 3d eg orbitals (∼711 eV, E peak) in an octahedral site.62 The dominance of the E peak in all spectra, compared to the reference Fe3O4 and FeOOH, indicates the typical Fe3+ oxidation state. According to Cramer's research, an L3/(L3 + L2) value above 0.75 indicates a high spin state for Fe.63 All calculated L3/(L3 + L2) values in BSCF catalyst exceeding 0.8 suggest that Fe3+ is in a high spin state (t2g3 eg2). Although no significant energy shift in the Fe L-edge was observed, the IE/ID peak intensity ratio varied with different potentials, reflecting the number of holes in the t2g and eg orbitals.64 In high spin Fe3+, electrons are preferentially removed from the eg state during oxidation, leading to a stronger E peak transition.22 The increasing IE/ID ratio with potential indicates minor oxidation of Fe during OER (Fig. 4d), further confirming that Fe not only aids in enhancing Co activity but also directly participates in the reaction.
The exceptional OER activity of BSCF5582 can be attributed to several key factors related to its structure and electronic structure: minor Fe doping preferentially occupies Oh sites, forming Co2+(Td)–O–Fe3+(Oh), which modulates the electronic state of Co3+(Oh).38 As the potential increases, Co2+(Td) transitions to Co3+(Oh) with forming CoOOH, serving as active sites during the OER process. The formation of HS-Co3+ also promotes OER. Fe in BSCF5582 is in a high-spin state and undergoes slight oxidation during the OER process. Fe supports the catalytic process by forming FeOOH active sites and enhancing the activity of Co sites.
Operando XAS analysis on BSCF5528 revealed a distinct OER reaction mechanism compared to BSCF5582. Operando Co K-edge and FT-EXAFS analysis indicated that Co in BSCF5528 did not experience significant changes in oxidation state or configuration (Fig. 5a, e, S26a and Table S6†). This stability is due to heavy Fe doping, where Fe3+ occupies both Td and Oh sites, keeping Co3+ primarily in Oh sites.38 Additionally, the lower electron density of Fe3+(Oh) in the t2g orbitals reduces electron–electron repulsion between Fe3+(Oh) and O 2p πu orbitals, lowering the energy level and inhibiting further oxidation of Co3+.34Operando Co L3-edge results showed that Co3+ in BSCF5528 consistently exhibited a lower IB/IA (≈1.03), with the proportion of HS-Co3+ increasing from 0.8 to 1.9 V vs. RHE ((Δ ≈ 0.04)), thus providing more active sites for OER (Fig. 5b and S27†). Fe serves as a critical reaction site in BSCF5528. Operando Fe K-edge and L-edge analysis revealed that Fe3+ undergoes further oxidation at high potentials, forming a Fe3+δ oxidation state as indicated by standard FeOOH spectra (Fig. 5c, d, S23b and S26c†).
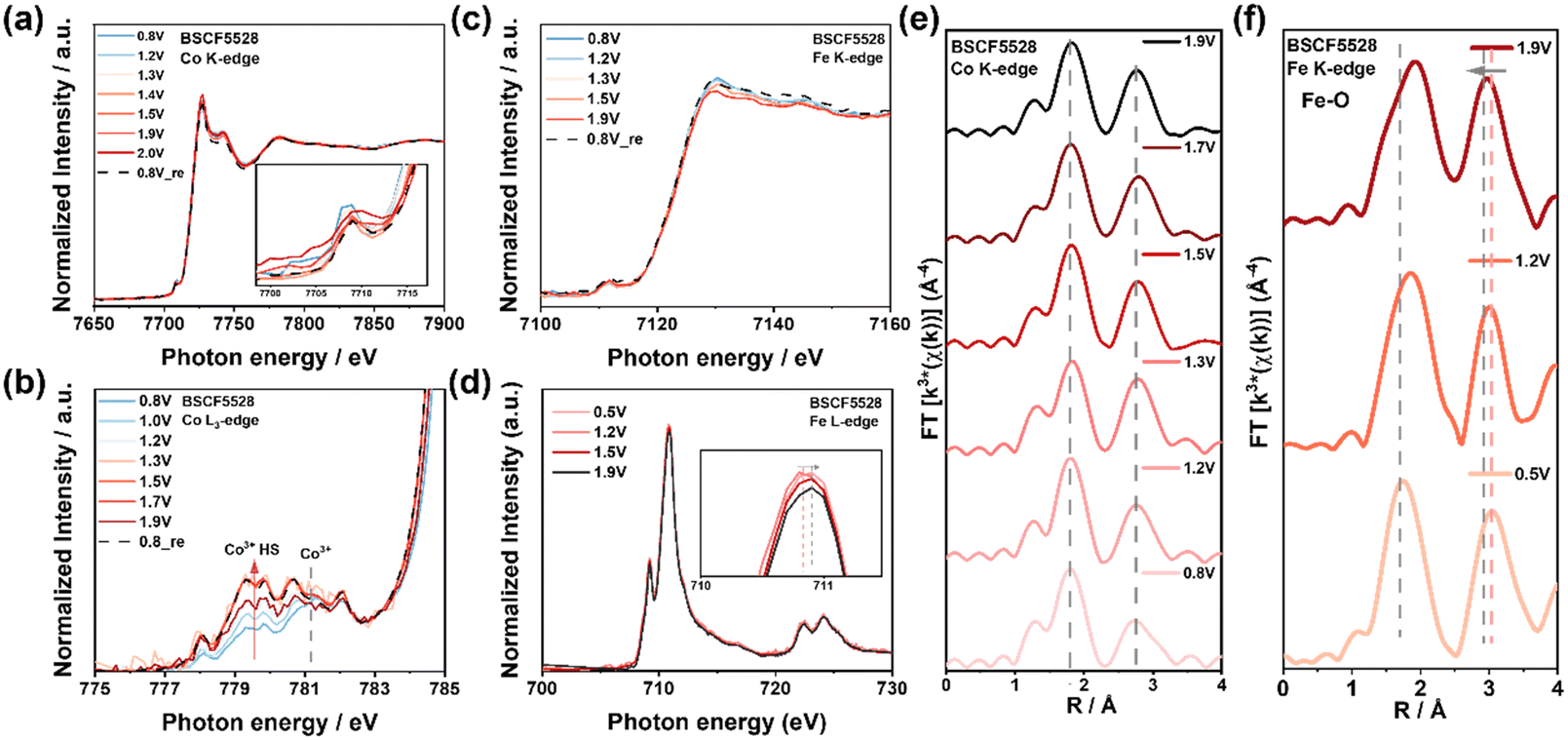 | ||
| Fig. 5 Operando XAS for investigating the origin of OER activity of BSCF5528 under application of various potentials from 0.8 V to 1.9 V vs. RHE. (a) Co K-edge XANES inset showing the enlargement of pre-edge peaks and (b) Co L3-edge XANES spectra showing low and high spin states. (c) Fe K-edge XANES and (d) Fe L-edge XANES spectra. (e) Fourier-transformed of the Co K-edge EXAFS spectra shows no significant changes, and (f) Fe K-edge EXAFS spectra shows Fe3+(Td) transitioning to Fe3+(Oh) at high potentials. | ||
FT-EXAFS spectra showed that Fe3+ also experiences configuration changes during OER (Fig. 5f), with initial Fe3+(Td) transitioning to Fe3+(Oh) at high potentials, contributing to FeOOH formation and causing a slight negative shift in the scattering peak around ∼3.0 Å (Fig. S21b†). Additionally, the Fe–O peak (∼1.8 Å) shifts positively at lower potentials (1.2 V vs. RHE), indicating the formation of Fe–OH intermediates, a phenomenon observed only near 1.9 V vs. RHE in BSCF5582 (Fig. 4f). Thus, Fe in BSCF5528 acts as a significant reaction site, actively participating in the OER process alongside HS-Co3+.
During the OER process in BSCF5528, Co3+ remains primarily in Oh sites without significant oxidation state or configuration changes due to heavy Fe doping, stabilizing Co3+ in a high-spin state and increasing the number of active sites as the potential rises. Meanwhile, Fe3+ transitions from Td to Oh sites at high potentials, forming FeOOH and further oxidizing to Fe3+δ. These changes enhance electron transfer and the formation of reactive intermediates, significantly promoting OER activity. However, BSCF5528 exhibits lower activity compared to BSCF5582, as Co(Fe)OOH in BSCF5582 generally has higher OER activity than Fe(Co)OOH in BSCF5528. CoOOH has better electronic conductivity and a more favorable electronic structure, facilitating the adsorption and evolution of oxygen intermediates during OER.65 Additionally, although BSCF5528 has more HS-Co3+ activity sites ratio, the overall lower Co content limits the number of active sites, reducing its effectiveness compared to BSCF5582.
Operando O K-edge XAS measurement
Most OER reaction mechanisms involve the formation of O–O bonds through bulk water nucleophilic attack (BWNA), occurring at the terminal or bridging oxygen (marked μ-O) sites.66,67 Arno Bergmann et al. identified a unified chemical structure where μ2-OH bridged Co2+/3+ ion clusters form on three-dimensional cross-linked and layered CoOx(OH)y precursors.58 Therefore, operando O K-edge spectroscopy is essential for understanding the reaction mechanisms, identifying active sites, and examining the structural response of the catalyst during the OER process.Under OER conditions, hybridization between O 2p and TM 3d orbitals is observed in the pre-edge of the O K-edge XANES spectra below 534.0 eV (Fig. 6). The experimental O K-edge XANES spectra of BSCF catalysts, recorded in TEY mode at different OER potentials, show energy resonance around 530.5 eV attributed to Fe3+/Co3+–O hybridized states with oxygen ions three- or fourfold metal-coordinated (blue: μ3/4-O). The resonance around 531.5 eV corresponds to protonated, double metal-coordinated oxygen atoms (pink: μ2-OH) bridging oxygen-coordinated high-spin Fe3+, Co3+, or Co2+ ions, typically found at edges, corners, and metal vacancies in the Co(Fe)Ox(OH)y domains.68–70 The resonance around 532.5 eV represent a combination of μ2-O (resulting from the deprotonation of μ2-OH) and μ3-O(H), an irreversible species formed during OER.63
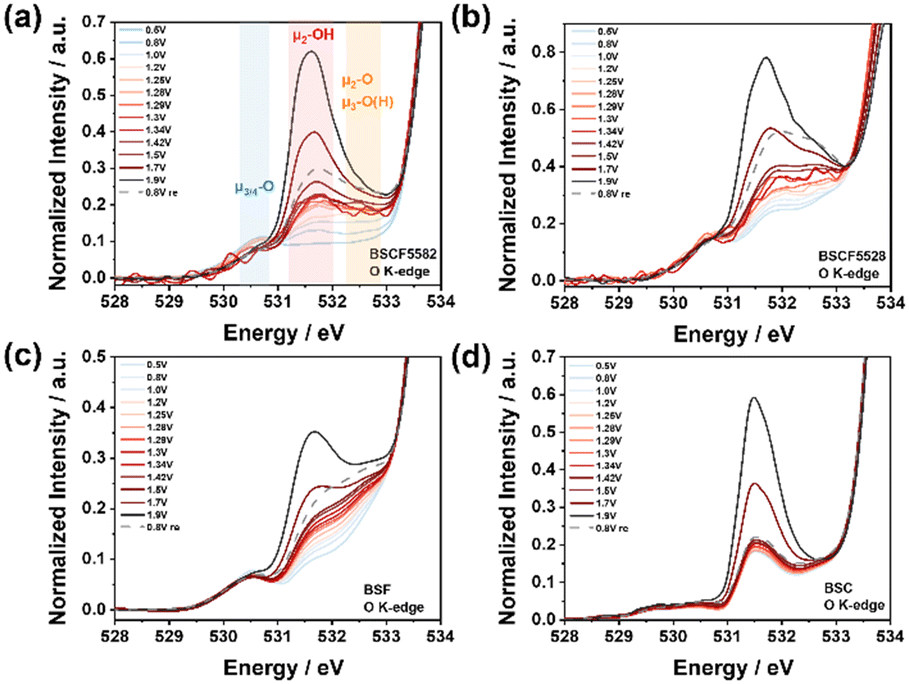 | ||
| Fig. 6 Operando O K-edge XAS spectra of (a) BSCF5582, (b) BSCF5528, (c) BSF and (d) BSC. The spectra were recorded using the TEY detection mode. | ||
As the OER potential increases, the peak intensity at 530.5 eV remains relatively unchanged, indicating that these electrochemically non-reducible Fe3+/Co3+–O sites have a limited contribution to OER activity (Fig. 7a). However, a slight decrease in peak intensity was observed in BSCF5582 and BSCF5528 (Fig. 6a and b), which is associated with a reduction in LS-Co3+,58 consistent with operando Co L-edge results. During the formation of O–O bonds and the eventual generation of molecular oxygen, both terminal oxygen sites and μ2-O(H) ligands are crucial for catalytic activity, enabling the deprotonation of water molecules during BWNA. As the potential increases, the peak intensities at 531.5 eV and 533.0 eV increase significantly A simultaneous increase in the HS Co3+ signal at the Co L3-edge peak and the intensities at 531.5 eV and 533.0 eV in the O K-edge during the OER further confirms that lower Fe doping in BSCF5582 enhances electron transfer and facilitates the adsorption of OH and OOH intermediates. This leads to forming more active CoOOH sites, which is consistent with the Co K-edge results. In contrast, for higher Fe doping, there is a lesser increase in the HS Co3+ signal and a more pronounced shift in the Fe L-edge, indicating that FeOOH species act as the active sites in this case.
 | ||
| Fig. 7 (a) Structure model of Co(Fe)OOH. The Co/Fe atoms, O atoms and H atoms are shown in green, red and gray. (b) The relationship between growth of normalized intensity and OER activity in forced-flow cell. (c) Pre-edge of ex situ O K-edge XAS spectra of CoOOH and FeOOH, as well as pre-edge of operando O K-edge XAS spectra of Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.8, 1) at 1.9 V vs. RHE. | ||
Further integrating the peak areas reveals a volcano-shaped trend from 0.5 V to 1.9 V vs. RHE, consistent with OER activity trends (Fig. 7b and S28†). Interestingly, although BSCF5582 exhibits significantly higher catalytic activity than BSCF5528, their integrated areas are remarkably similar, primarily due to the formation of μ3-O(H). In BSCF5528, the μ3-O(H) peak reflects irreversible oxidation at Fe-rich sites, reducing catalytic activity by limiting surface dynamics. In contrast, BSCF5582, with lower Fe content, minimizes μ3-O(H) contributions and promotes Co2+(Td) to Co3+(Oh) transitions, enhancing OER activity through synergistic Co and Fe interactions. The reducible Fe3+/Co3+–O sites, attributed to μ2-OH bridges, are highly favorable for fast OER process. Furthermore, O K-edge XANES spectra of the four BSCF catalysts at 1.9 V vs. RHE demonstrate that Fe doping effectively adjusts the coordination environment and bonding state of Co/Fe–O, causing a slight shift in the μ2-OH peak position, which directly influences OER activity (Fig. 7c).
Many research has investigated the reversible structural transformation between spinel and oxyhydroxide phases in BSCF catalysts during the OER process.8,11 Chen et al. observed that the ratio of coordination numbers between Oh and Td sites increases in the oxidation region and decreases during reduction. This indicates that Co3+(Oh) in the oxyhydroxide phase can revert to either Co2+(Td) (active site) or Co3+(Oh) (inactive site) in the initial cobalt oxides.34 This multipath conversion can interfere with the overall activation process, as the random migration of Co ions may render the original active sites inactive. Most previous reports have focused primarily on cobalt (Co) as the active site, with iron (Fe) playing a role in enhancing the oxygen evolution reaction (OER) activity of Co. However, the true role of Fe at various Co–Fe compositions remains elusive. In contrast to previous studies, the present work utilizes operando Co/Fe K-edge, L-edge, and O K-edge measurements to gain a deeper understanding of the catalyst behavior. Our findings reveal that minor Fe doping significantly suppresses the conversion of Co3+ to inactive sites, likely due to the geometric confinement effect of Fe ions. This effect forces the restoration of Co2+ in a tetrahedral (Td) coordination environment, thus maintaining continuous activation of these sites. This conclusion is further supported by a comparison of the O 1s X-ray photoelectron spectroscopy (XPS) spectra before and after the OER (Fig. S29†).
On the other hand, when higher amounts of Fe are present, Fe occupies the octahedral sites, and during the OER, FeOOH remains the active site. To the best of our knowledge, this is the first report to integrate both bulk- and surface-sensitive operando techniques to clearly specify the role of Fe in the catalyst. Our operando XAS data reveal that bulk oxidation state variations (e.g., Co and Fe valence shifts) are clearly observable across the composition series. Our soft operando measurements reveal that surface-specific transformations dominate catalytic activity, particularly forming an amorphous, Co/Fe oxyhydroxide-rich layer under OER conditions. However, the characteristics of the active surface layer are strongly influenced by the Fe content, leading to compositional and structural variations in the reconstructed oxyhydroxide phase that ultimately lead to different OER activity. While the bulk composition serves as a critical reservoir, providing the structural integrity and electronic environment necessary to support surface transformations, it is ultimately the surface reconstruction into an OER-active oxyhydroxide phase that dictates the catalytic performance. Furthermore, the dynamic structural and electronic changes of the catalysts during the OER were thoroughly investigated. Besides, we have compared the key findings of our study with reported literature (Table S2B†). We believe that this fundamental understanding will guide researchers in designing highly active OER catalysts.
Conclusion
The study synthesized and characterized four different Ba0.5Sr0.5CoxFe1−xO3−δ (x = 0, 0.2, 0.8, 1) catalysts with varying Fe doping levels to evaluate their OER performance using RDE and forced-flow cell techniques. To the best of our knowledge, this is the first comprehensive study on BSCF-based perovskites employing operando Co/Fe K-edge, Co/Fe L-edge, and O K-edge XAS simultaneously to unravel the site-specific redox evolution and formation of catalytically active oxygen species under OER conditions. The results demonstrate that the outstanding OER performance of BSCF catalysts, particularly BSCF5582, is primarily due to the unique interplay of their local and electronic structure Minor Fe doping preferentially occupies Oh sites, influencing the electronic state of Co3+. As the applied potential increases, Co2+(Td) transitions to Co3+(Oh), forming CoOOH and creating active sites for the OER process. In contrast, BSCF5528, with significant Fe doping, stabilizes Co3+ in Oh sites without notable changes in oxidation state, leading to lower overall activity due to a less favourable electronic structure and reduced Co content. Fe3+ transitions from Td to Oh sites at high potentials, forming FeOOH and further oxidizing to Fe3+δ, acting as a primary reaction site for OER. Operando O K-edge XAS analysis reveals that the formation of reducible Fe3+/Co3+–O sites, particularly μ2-O(H) bridges between high-spin Co/Fe ions, significantly enhances OER electrocatalysis, with Fe doping effectively regulating the coordination environment and bonding states, thereby affecting overall activity. Overall, this work establishes the importance of Fe doping in tuning the redox behavior and coordination structure of BSCF catalysts, combining the multiple operando XAS methodology for a mechanistic understanding of complex perovskite OER systems. Further, the Fe content plays a crucial role in shaping the properties of the active surface layer, inducing compositional and structural modifications in the reconstructed oxyhydroxide phase, which leads to different OER activity, which is not thoroughly investigated in prior studies combining various operando techniques. These findings offer a new framework for designing advanced electrocatalysts with optimized transition metal–oxygen interactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
W. Cao developed a plan for the experiment, collected the data, performed the analyses, and wrote the manuscript. M. Kumar validated the data, supervised the project, and revised the manuscript. N. Thakur, M. Matsumoto and Y. Tsuji revised the manuscript. T. Watanabe performed the XAS analysis. T. Matsunaga helped XRD analysis. Y. Kuroda and S. Mitsushima supported the electrochemical measurements Y. Uchimoto validated the data, supervised the project, provided the resources, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was performed by the commissioned research fund provided by F-REI (JPFR 24030301). The synchrotron radiation experiments were performed at the beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal 2021B1041, 2021B1042, 2022A1037, 2022A1484, 2022A1535, 2022A1555, 2022A1811, 2022A1820, 2022A1475, 2022A1038, 2022B1438, and 2022B1436). We would like to thank Dr. T. Uchiyama for his help with the synchrotron radiation experiments.References
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- M.-I. Jamesh, D. Hu, J. Wang, F. Naz, J. Feng, L. Yu, Z. Cai, J. C. Colmenares, D.-J. Lee, P. K. Chu and H.-Y. Hsu, J. Mater. Chem. A, 2024, 12, 11771–11820 RSC.
- L. Magnier, G. Cossard, V. Martin, C. Pascal, V. Roche, E. Sibert, I. Shchedrina, R. Bousquet, V. Parry and M. Chatenet, Nat. Mater., 2024, 23, 252–261 CrossRef CAS.
- X. Ren, Y. Dai, X. Wen, B. Guo, C. Shi, X. Huang, Y. Guo and S. Li, Adv. Sustainable Syst., 2024, 8, 2300379 CrossRef CAS.
- H. Tüysüz, Acc. Chem. Res., 2024, 57, 558–567 Search PubMed.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384–391 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- D. Guan, G. Ryu, Z. Hu, J. Zhou, C.-L. Dong, Y.-C. Huang, K. Zhang, Y. Zhong, A. C. Komarek, M. Zhu, X. Wu, C.-W. Pao, C.-K. Chang, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 3376 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- T.-H. Shen, L. Spillane, J. Vavra, T. H. M. Pham, J. Peng, Y. Shao-Horn and V. Tileli, J. Am. Chem. Soc., 2020, 142, 15876–15883 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, X. Cheng and T. J. Schmidt, Adv. Energy Mater., 2015, 5, 1402033 CrossRef.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Kötz and T. J. Schmidt, Sci. Rep., 2015, 5, 12167 CrossRef CAS.
- Z. Hu, Q. Yan and Y. Wang, Mater. Today Chem., 2023, 34, 101800 CrossRef CAS.
- J. Timoshenko and B. R. Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- B.-J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, J. Am. Chem. Soc., 2019, 141, 5231–5240 CrossRef CAS.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628–8635 CrossRef CAS.
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, J. Am. Chem. Soc., 2017, 139, 8960–8970 CrossRef CAS PubMed.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef.
- D. Wang, J. Zhou, Y. Hu, J. Yang, N. Han, Y. Li and T.-K. Sham, J. Phys. Chem. C, 2015, 119, 19573–19583 CrossRef CAS.
- D. Aegerter, E. Fabbri, N. S. Yüzbasi, N. Diklić, A. H. Clark, M. Nachtegaal, C. Piamonteze, J. Dreiser, T. Huthwelker, T. Graule and T. J. Schmidt, ACS Catal., 2023, 13, 15899–15909 CrossRef CAS.
- J. Huang, C. N. Borca, T. Huthwelker, N. S. Yüzbasi, D. Baster, M. El Kazzi, C. W. Schneider, T. J. Schmidt and E. Fabbri, Nat. Commun., 2024, 15, 3067 CrossRef CAS PubMed.
- R. V. Mom, L. J. Falling, O. Kasian, G. Algara-Siller, D. Teschner, R. H. Crabtree, A. Knop-Gericke, K. J. J. Mayrhofer, J.-J. Velasco-Vélez and T. E. Jones, ACS Catal., 2022, 12, 5174–5184 CrossRef CAS.
- A. Soltanizade, A. Babaei, A. Ataie and S. V. Seyed-Vakili, J. Appl. Electrochem., 2019, 49, 1113–1122 CrossRef CAS.
- P. S. Mahadik, A. N. Shirsat, B. Saha, N. Sitapure, D. Tyagi, S. Varma, B. N. Wani and S. R. Bharadwaj, J. Therm. Anal. Calorim., 2019, 137, 1857–1866 CrossRef CAS.
- D. N. Mueller, R. A. De Souza, T. E. Weirich, D. Roehrens, J. Mayer and M. Martin, Phys. Chem. Chem. Phys., 2010, 12, 10320–10328 RSC.
- Z. Zhang, L. Andre and S. Abanades, Sol. Energy, 2016, 134, 494–502 CrossRef CAS.
- F. Ambroz, T. J. Macdonald, V. Martis and I. P. Parkin, Small Methods, 2018, 2, 1800173 CrossRef.
- W. Cao, M. Kumar, N. Thakur, T. Uchiyama, Y. Gao, S. Tominaka, A. Machida, T. Watanabe, R. Sato, T. Teranishi, M. Matsumoto, H. Imai, Y. Sakurai and Y. Uchimoto, ACS Appl. Energy Mater., 2024, 7, 8515–8525 CrossRef CAS.
- W. Cao, N. Thakur, M. Kumar, T. Uchiyama, Y. Gao, S. Tominaka, A. Machida, T. Watanabe, R. Sato, T. Teranishi, M. Matsumoto, H. Imai, Y. Sakurai and Y. Uchimoto, J. Mater. Chem. A, 2024, 12, 29843–29853.32 RSC.
- Y. Long, X. Zhu, C. Gao, W. Si, J. Li and Y. Peng, Nat. Commun., 2025, 16, 1048 CrossRef CAS.
- S.-F. Hung, Y.-Y. Hsu, C.-J. Chang, C.-S. Hsu, N.-T. Suen, T.-S. Chan and H. M. Chen, Adv. Energy Mater., 2018, 8, 1701686 CrossRef.
- X. Gao, J. Liu, Y. Sun, X. Wang, Z. Geng, F. Shi, X. Wang, W. Zhang, S. Feng, Y. Wang and K. Huang, Inorg. Chem. Front., 2019, 6, 3295–3301 RSC.
- J.-S. Lee, W.-B. Wu, J. Chen, C.-L. Chen, H.-W. Kuo, C.-R. Lin, H.-J. Lin and C.-T. Chen, Appl. Phys. Lett., 2021, 118, 072403 CrossRef CAS.
- X. Zhang, H. Zhong, Q. Zhang, Q. Zhang, C. Wu, J. Yu, Y. Ma, H. An, H. Wang, Y. Zou, C. Diao, J. Chen, Z. G. Yu, S. Xi, X. Wang and J. Xue, Nat. Commun., 2024, 15, 1383 CrossRef CAS.
- X. Xu, Y. Chen, W. Zhou, Z. Zhu, C. Su, M. Liu and Z. Shao, Adv. Mater., 2016, 28, 6442–6448 CrossRef CAS PubMed.
- S. Wu, J. Li, J. Hu, Y. Huang, H.-S. Xu and K. Tang, ACS Appl. Energy Mater., 2023, 6, 6289–6298 CrossRef CAS.
- C. Liu, D. Ji, H. Shi, Z. Wu, H. Huang, Z. Kang and Z. Chen, J. Mater. Chem. A, 2022, 10, 1336–1342 RSC.
- K. Nagasawa, I. Matsuura, Y. Kuroda and S. Mitsushima, Electrochemistry, 2022, 90, 017012 CrossRef CAS.
- K. Nagasawa, L. Kunpeng, Y. Takenaga, Y. Kuroda and S. Mitsushima, Electrochemistry, 2022, 90, 047004–047004 CrossRef CAS.
- F. Reikowski, F. Maroun, I. Pacheco, T. Wiegmann, P. Allongue, J. Stettner and O. M. Magnussen, ACS Catal., 2019, 9, 3811–3821 CrossRef CAS.
- T.-H. Shen, L. Spillane, J. Peng, Y. Shao-Horn and V. Tileli, Nat. Catal., 2022, 5, 30–36 CrossRef CAS.
- E. M. Davis, A. Bergmann, C. Zhan, H. Kuhlenbeck and B. R. Cuenya, Nat. Commun., 2023, 14, 4791 CrossRef CAS PubMed.
- B.-J. Kim, E. Fabbri, I. E. Castelli, M. Borlaf, T. Graule, M. Nachtegaal and T. J. Schmidt, Catalysts, 2019, 9, 263 CrossRef CAS.
- B.-J. Kim, E. Fabbri, I. E. Castelli, M. Borlaf, T. Graule, M. Nachtegaal and T. J. Schmidt, Catalysts, 2019, 9, 263 CrossRef CAS.
- L. J. Enman, M. B. Stevens, M. H. Dahan, M. R. Nellist, M. C. Toroker and S. W. Boettcher, Angew. Chem., Int. Ed., 2018, 57, 12840–12844 CrossRef CAS PubMed.
- S. Song, J. Zhou, X. Su, Y. Wang, J. Li, L. Zhang, G. Xiao, C. Guan, R. Liu, S. Chen, H.-J. Lin, S. Zhang and J.-Q. Wang, Energy Environ. Sci., 2018, 11, 2945–2953 RSC.
- Y. Zhu, H. A. Tahini, Z. Hu, Z.-G. Chen, W. Zhou, A. C. Komarek, Q. Lin, H.-J. Lin, C.-T. Chen, Y. Zhong, M. T. Fernández-Díaz, S. C. Smith, H. Wang, M. Liu and Z. Shao, Adv. Mater., 2020, 32, 1905025 CrossRef CAS PubMed.
- S. Y. Istomin, O. A. Tyablikov, S. M. Kazakov, E. V. Antipov, A. I. Kurbakov, A. A. Tsirlin, N. Hollmann, Y. Y. Chin, H. J. Lin, C. T. Chen, A. Tanaka, L. H. Tjeng and Z. Hu, Dalton Trans., 2015, 44, 10708–10713 RSC.
- C. Wu, Y. Sun, Z. Yang, J. Hu, T.-Y. Ding, J. Cheng and K. H. L. Zhang, Chem. Mater., 2022, 34, 10509–10516 CrossRef CAS.
- X. Li, Y. Sun, Q. Wu, H. Liu, W. Gu, X. Wang, Z. Cheng, Z. Fu and Y. Lu, J. Am. Chem. Soc., 2019, 141, 3121–3128 CrossRef CAS.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chem, 2017, 3, 812–821 CAS.
- M. Merz, P. Nagel, C. Pinta, A. Samartsev, H. V. Löhneysen, M. Wissinger, S. Uebe, A. Assmann, D. Fuchs and S. Schuppler, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 174416 CrossRef.
- J.-M. Chen, Y.-Y. Chin, M. Valldor, Z. Hu, J.-M. Lee, S.-C. Haw, N. Hiraoka, H. Ishii, C.-W. Pao, K.-D. Tsuei, J.-F. Lee, H.-J. Lin, L.-Y. Jang, A. Tanaka, C.-T. Chen and L. H. Tjeng, J. Am. Chem. Soc., 2014, 136, 1514–1519 CrossRef CAS.
- Z. Hu, H. Wu, M. W. Haverkort, H. H. Hsieh, H. J. Lin, T. Lorenz, J. Baier, A. Reichl, I. Bonn, C. Felser, A. Tanaka, C. T. Chen and L. H. Tjeng, Phys. Rev. Lett., 2004, 92, 207402 CrossRef CAS.
- A. Bergmann, T. E. Jones, E. M. Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- J.-Y. Zhang, Y. Yan, B. Mei, R. Qi, T. He, Z. Wang, W. Fang, S. Zaman, Y. Su, S. Ding and B. Y. Xia, Energy Environ. Sci., 2021, 14, 365–373 RSC.
- W. H. Lee, M. H. Han, Y.-J. Ko, B. K. Min, K. H. Chae and H.-S. Oh, Nat. Commun., 2022, 13, 605 CrossRef CAS PubMed.
- R. Zhang, L. Pan, B. Guo, Z.-F. Huang, Z. Chen, L. Wang, X. Zhang, Z. Guo, W. Xu, K. P. Loh and J.-J. Zou, J. Am. Chem. Soc., 2023, 145, 2271–2281 CrossRef CAS.
- C. Wang, C. Yu, B. Qian, Y. Ren, L. Wang, Y. Xie, X. Tan, X. He and J. Qiu, Small, 2024, 20, 2307349 CrossRef CAS PubMed.
- H. Wang, G. Peng, L. M. Miller, E. M. Scheuring, S. J. George, M. R. Chance and S. P. Cramer, J. Am. Chem. Soc., 1997, 119, 4921–4928 CrossRef CAS.
- J. Zhou, P. N. Duchesne, Y. Hu, J. Wang, P. Zhang, Y. Li, T. Regier and H. Dai, Phys. Chem. Chem. Phys., 2014, 16, 15787–15791 RSC.
- D. Aegerter, E. Fabbri, M. Borlaf, N. S. Yüzbasi, N. Diklić, A. H. Clark, V. Romankov, C. Piamonteze, J. Dreiser, T. Huthwelker, T. Graule and T. J. Schmidt, J. Mater. Chem. A, 2024, 12, 5156–5169 RSC.
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, I. Furó, M. S. G. Ahlquist and L. Sun, Nat. Catal., 2022, 5, 414–429 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- F. Zasada, W. Piskorz and Z. Sojka, J. Phys. Chem. C, 2015, 119, 19180–19191 CrossRef CAS.
- F. Zasada, W. Piskorz, P. Stelmachowski, A. Kotarba, J.-F. Paul, T. Płociński, K. J. Kurzydłowski and Z. Sojka, J. Phys. Chem. C, 2011, 115, 6423–6432 CrossRef CAS.
- J. Huang, N. Hales, A. H. Clark, N. S. Yüzbasi, C. N. Borca, T. Huthwelker, T. J. Schmidt and E. Fabbri, Adv. Energy Mater., 2024, 14, 2303529 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00056d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |