
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu-exchanged hydroxyapatites as green catalysts for CO oxidation†
Guillermo
Escolano-Casado
 ,
Sadaf
Fatima Jafri
,
Matteo
Signorile
,
Elisa
Borfecchia
and
Lorenzo
Mino
*
,
Sadaf
Fatima Jafri
,
Matteo
Signorile
,
Elisa
Borfecchia
and
Lorenzo
Mino
*
Department of Chemistry and Interdepartmental NIS Centre, University of Torino, Via P. Giuria 7, 10125 Torino, Italy. E-mail: lorenzo.mino@unito.it
First published on 26th May 2025
Abstract
Hydroxyapatite (HA) is gaining attention as a sustainable and efficient green catalyst due to its environmental compatibility, non-toxicity and peculiar surface acid–base properties. In this study, we synthesized HA with needle-like or plate-like morphologies, characterized by Ca-rich or P-rich surface terminations, respectively. Then, we functionalized them with increasing amounts of Cu through an ion-exchange procedure. After a thorough characterization of their structural, morphological and redox properties, combining X-ray diffraction, electron microscopy and X-ray absorption spectroscopy, the materials were tested in the CO oxidation reaction. Pristine needle and plate HAs showed poor CO conversion (<5%), while all Cu-functionalized samples were catalytically active, reaching complete CO conversion at 573 K. The complete set of results highlighted that doped Ca-rich HAs exhibit better catalytic activity despite a lower copper content. This is explained by the easier reducibility of Cu2+ to Cu+ in these materials, as evidenced by IR measurements of CO adsorption.
1. Introduction
Heterogeneous catalysis plays a pivotal role in the chemical and technological industries and modern research focuses on developing sustainable approaches which preserve process efficiency. In this context, hydroxyapatite (HA), a naturally occurring form of calcium phosphate with composition Ca10(PO4)6(OH)2, is emerging as a sustainable and efficient green catalyst due to its unique chemical properties.1–3 HA is biocompatible, non-toxic, and abundantly available, representing an environmentally friendly alternative to conventional catalysts. Biomimetic HA can be obtained by different synthetic procedures and crystallizes in the P63/m space group, exhibiting a morphology elongated in the c-axis direction and predominantly exposing {010} facets.4,5 Structurally, it can be described as a sequence of -ABA-ABA- layers along the [010] direction, where A and B correspond to Ca3(PO4)2 and Ca4(PO4)2(OH)2, respectively.6 Different interruptions during growth lead to distinct surface terminations: stoichiometric (-ABA-ABA-), phosphorus-rich (-ABA-ABA-A), or calcium-rich (-ABA-AB) surfaces.6,7 These terminations significantly influence surface acid–base properties, which are related to the molar Ca/P ratio.8 Thus, stoichiometric HA (Ca/P = 1.67) exhibits predominantly basic surfaces, while Ca-rich (Ca/P > 1.67) and P-rich (Ca/P < 1.67) variants show enhanced basicity and acidity, respectively.2,4HA can act as a catalyst directly by exploiting the acid–base properties described above or through functionalization, leveraging its structural flexibility and surface reactivity. Indeed, its surface can be functionalized by metal nanoparticles or proteins, and can exchange native ions, i.e. Ca2+, (PO4)3−, OH−, with others of similar size and charge.9 One interesting catalytic application of HA is CO oxidation, a reaction which plays a crucial role in different fields. It is widely used in catalytic converters to reduce CO emissions from vehicles and in air purification systems to ensure safe indoor environments.10 In fuel cells, it prevents CO poisoning of catalysts, improving efficiency, and is essential in syngas purification and industrial gas treatment.11 In this context, HA has been mainly studied as a support for nanoparticles, considering different metals (e.g. Au,12 Cu,13 Au–Cu,14 Pd,15 Pd–Cu–Fe16) which show a promising activity in CO oxidation. Recent research has also explored the possibility of employing ion-exchanged HAs, focusing on Ag-functionalized systems.17 In this regard, copper remains underexplored as an active metal in ion-exchanged HAs for CO oxidation, despite its remarkable redox versatility and significant catalytic potential.9,18–20
In this study we synthesized different biomimetic HA with distinct morphologies associated with Ca-rich (i.e. Ca/P > 1.67) or P-rich (i.e. Ca/P < 1.67) surface terminations and then we functionalized them with increasing amounts of Cu by an ion-exchange procedure. After a preliminary characterization of the chemical composition of the materials, their structure and morphology were investigated by X-ray diffraction and electron microscopy. Then, the Cu active sites were studied in detail combining X-ray absorption spectroscopy and infrared spectroscopy of adsorbed CO. Finally, the results of this thorough physico-chemical characterization were employed to rationalize their activity in the CO oxidation reaction.
2. Experimental
2.1 Hydroxyapatites preparation
Two different strategies for the synthesis of non-exchanged hydroxyapatites were adopted following previously developed co-precipitation methodologies.7,19,21 All reagents were purchased from Sigma-Aldrich. To obtain needle-like hydroxyapatite nanoparticles (hereafter labelled as “Needle”), the whole preparation was performed at 310 K by dropping (flux = 1 drop/second) a H3PO4 (80% pure) solution (1.26 M, 0.225 L) over a Ca(OH)2 (≥95% pure) solution (1.35 M, 0.375 L) under vigorous stirring. After the addition, the dispersion was kept under stirring for 24 hours. Conversely, the plate-like hydroxyapatite nanoparticles (hereafter labelled as “Plate”) were prepared at 298 K by adding a Na2HPO4 (>99% anhydrous) solution (0.35 M, 0.3 L) with the same drop flux (1 drop/second), over a Ca(CH3COO)2·0.5H2O (high purity grade) solution (0.21 M, 0.3 L). The pH was maintained below 10 by adding a 5 N solution of NH4 (>85%) as needed. As well as in the previous preparation, the obtained dispersion was maturated for 24 hours under stirring. Once the maturation time was completed, the materials obtained with both co-precipitation methodologies were isolated from the mother liquors by centrifugation (4500 rpm, 5 min, Jouan BR4i centrifuge). Then, they were rinsed three times with double-distilled water and, finally, lyophilized. Granular fractions with dimensions <75 μm were isolated by sieving the HA powders. Moreover, for the plate nanoparticles, one additional step after completing the maturation time and before starting the washing procedure is needed to remove the excess of acetates complexing the surface Ca2+ ions, thus allowing the subsequent exchange with the Cu2+ ions. It consists of suspending the plate HA powder in a NaOH (≥98%) (0.01 M, 0.5 L for 2.5 g) solution for 5 minutes at room temperature under stirring.2.2 Ion exchange procedure
Surface Ca2+ ions were exchanged by Cu2+ ions by immersing the HA powders in Cu(NO3)2·2.5H2O solutions (≥99.99% pure) (17 mL for 500 mg of HA) at room temperature and stirring for 15 minutes, as previously described elsewhere.19 Once the exchange procedure was completed, the samples were again washed three times with double-distilled water and lyophilized. A wide range of Cu(NO3)2 concentrations in solution were explored for both HA nanoparticles, needle and plate: 0.02 M, 0.05 M, 0.1 M, and 0.2 M. As result, HA nanoparticles with different copper uptakes as function of the initial Cu2+ amount in solution were obtained. The labelling of the materials refers to their morphology and to the final copper uptake measured by ICP-OES and expressed in weight percent (wt%). Therefore, the obtained HA nanoparticles presented in this work are labelled as follows: needle, needle_4, needle_6, needle_6′, needle_6′′; plate, plate_4, plate_11, plate_14 and plate_14′. The samples without number correspond to the non-substituted HA, used as control nanoparticles, and the samples containing the apostrophe (6′, 6′′ and 14′) correspond to the same final copper uptake.2.3 Specific surface area (SSA) determination
The SSA was determined with a Micromeritics ASAP 2020 by nitrogen adsorption at 77 K applying the Brunauer–Emmett–Teller (BET) model. Before performing the measurements, the powders (100 mg for each measurement) were outgassed at room temperature for 12 hours.2.4 Elemental analysis
The chemical analysis of the samples was performed at the Institute of Science, Technology and Sustainability for Ceramics (ISSMC) of the National Research Council (CNR) of Faenza, as described in previous publications.19,21 Briefly, 10 mg of the HA powders were dissolved in 50 mL of a HNO3 solution (1 wt%) and the elemental composition was quantified by inductively coupled plasma-optical emission spectrometry (ICP-OES) by using an Agilent 5100 spectrometer (Agilent Technologies 5100, Santa Clara, CA, USA).2.5 High-resolution transmission electron microscopy (HR-TEM)
The HA powders were deposited on Cu grids coated with a lacey carbon film. The images were obtained with a JEOL 3010-UHR operated at an acceleration potential of 300 kV. Particular attention was given to minimizing exposure time, as HA powders are prone to further crystallization and/or loss of bulk water under the electron beam.2.6 X-ray powder diffraction (XRPD)
The XRPD patterns were collected with a PW3050/60 X'Pert Pro powder X-ray diffractometer (Malvern Panalytical, Malvern, UK) in Bragg–Brentano geometry using a flat-quartz sample holder. The Cu Kα radiation (1.5406 Å) generated at 40 kV and 45 mA was employed for the collection of the patterns. The 2θ range varied from 10° to 60° with a step size (2θ) of 0.02° and a counting time of 90 seconds per point.2.7 Fourier transform infrared spectroscopy (FT-IR)
A Bruker INVENIO R instrument, equipped with a MCT detector, was used to acquire the FT-IR spectra at 4 cm−1 resolution. The number of scans was adjusted to 64 to obtain a good signal-to-noise ratio. The HA powders were pressed in self-supporting pellets and then placed in a custom IR quartz cell designed to perform measurements at low temperatures in transmission mode and equipped with KBr windows. The cell was connected to a conventional vacuum line (residual pressure <5 × 10−4 mbar) that allows adsorption–desorption experiments and in situ thermal treatments of the sample. Prior to the measurement, the sample was heated at 423 K in dynamic vacuum. Subsequently, the sample was cooled down by thermal contact with liquid N2 (ca. 100 K) and, finally, 30 mbar of CO were admitted to the HA and stepwise outgassed. The collected spectra were normalized by the optical density (weight of the pellet in mg per area of the pellet in cm2) to make the absorbance differences in the spectra independent from differences in the thickness of the pellets.2.8 X-ray absorption spectroscopy (XAS)
The XAS data were collected at the Cu K-edge in transmission mode on the Swiss-Norwegian beamline (BM31) of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) using a water-cooled double-crystal Si(111) monochromator. Three ionization chambers filled with a He/Ar mixture were used to detect the incident X-ray intensity (I0), transmitted intensity (I1), and reference (Cu foil) transmitted intensity (I2). The XAS spectra were acquired with an acquisition time of 3 min per scan. The powdered samples were prepared in the form of self-supporting pellets with 5 mm diameter. Masses were optimized for XAS data collection in transmission mode at room temperature (i.e. edge jump Δμx = 0.3–0.8 and total absorption after the edge μx = 2.5).All the XAS spectra were energy calibrated to the Cu foil, background subtracted and normalized to the edge jump using Athena software from Demeter.22 EXAFS fitting for the HA samples was performed in R space, in the ΔR = 1.0–4 Å range, on the FT-EXAFS spectrum obtained applying the Fourier transform on the k2-weighted χ(k) functions in the 2.2–10.0 Å−1 k-range. Scattering amplitudes f(k) and phase-shifts δ(k) were calculated by FEFF6 code using the Artemis software from the Demeter package.22
2.9 Catalytic tests
Before the catalytic tests, the material powders underwent a pressing and sieving procedure. The collected fractions fell within the 500–250 μm range to ensure homogeneity and appropriate agglomerate size. Subsequently, 100 mg of the HA agglomerates were placed in a tubular plug-flow quartz reactor (inner diameter: 6 mm, length: 300 mm). Throughout all the catalytic tests, a constant flow rate of 15 mL min−1 was maintained using pure helium as inert gas. Initially, pure He was fluxed for 10 minutes at room temperature. Then, the temperature was raised to 423 K and held for 120 minutes for cleaning the materials surfaces from adsorbed water molecules. Afterwards, CO and O2 gases (both at 10 % v/v in He) were continuously fluxed at rates of 10 and 5 mL min−1, respectively, leading to a feed with He![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO:O2 molar composition of 13.5:1.0:0.5. This resulted in a gas hourly space velocity of 9000 h−1. The catalytic study was conducted at 423, 523 and 573 K with a stabilization step of 30 minutes at each temperature. The ramp temperature used was 5 K min−1. An Agilent 8960 Gas Chromatograph (GC) equipped with sequential H-PLOT and MOLSIEVE columns and with a thermal conductivity detector (TCD) was coupled with the plug-flow reactor to evaluate the CO conversion (XCO) during subsequent catalytic steps, using the following formula:
:CO:O2 molar composition of 13.5:1.0:0.5. This resulted in a gas hourly space velocity of 9000 h−1. The catalytic study was conducted at 423, 523 and 573 K with a stabilization step of 30 minutes at each temperature. The ramp temperature used was 5 K min−1. An Agilent 8960 Gas Chromatograph (GC) equipped with sequential H-PLOT and MOLSIEVE columns and with a thermal conductivity detector (TCD) was coupled with the plug-flow reactor to evaluate the CO conversion (XCO) during subsequent catalytic steps, using the following formula:where C0 is the CO concentration at the reactor inlet and C is the CO concentration downstream the reactor after 30 minutes of stabilization at a given temperature.

3. Results and discussion
3.1 Elemental and textural analysis
As the first step of our study, we investigated the elemental composition for the two different samples morphologies as a function of the concentration of the Cu(NO3)2 solution employed for the ion exchange procedure (see experimental section). As visible in Fig. 1, as expected, there is a decrease in the Ca wt% correlated with an increase in the Cu wt% for progressively higher concentrations of Cu(NO3)2 in the starting solution. The maximum copper uptake for the HA needle series is 6 wt% and is reached for a 0.05 M Cu(NO3)2 solution (needle_6 sample). Conversely, for the HA plate series, the maximum copper content is 14 wt% (plate_14 sample), obtained using a 0.1 M Cu(NO3)2 solution. The different behaviour of the two series of HA can be explained considering the corresponding specific surface area values, which are ca. 100 and 200 m2 g−1 for pristine needle and plate HA, respectively (Fig. 1A′ and B′), resulting in a different surface cation exchange capacity.23,24 For more concentrated initial Cu(NO3)2 solutions (samples needle_6′, needle_6′′ and plate_14′), the copper uptake does not increase, but we observe a slight decrease in the phosphates content (Fig. 1A and B) and in the SSA (Fig. 1A′ and B′), which can be ascribed to a partial HA dissolution due to the lower pH of the solutions employed during the exchange procedure.19 For this reason, these samples will not be further discussed in the following sections. | ||
| Fig. 1 Top panel: Weight percent composition measured by ICP for the needle (A) and plate (B) HAs. Bottom panel: (Ca + Cu)/P ratio (orange bars) and BET SSA values (black bars) for the needle (A′) and plate (B′) HAs. The molar concentration of the Cu(NO3)2 solution employed for the ion exchange is also shown for each sample. The complete list of numerical values with their corresponding uncertainties is reported in Table S1.† | ||
Finally, we can focus on the (Ca + Cu)/P ratio, which should be 1.67 for stoichiometric HA. This value is higher for the needle samples (orange bars in Fig. 1A′), highlighting the presence of Ca-rich surface terminations, and lower for the plate samples (orange bars in Fig. 1B′), indicating the predominance of P-rich surfaces. We can also note that (Ca + Cu)/P ratio is almost constant in all the needle series confirming that the copper doping is predominantly occurring by a Ca2+ → Cu2+ ion exchange mechanism. This statement is further confirmed by analysis of the solution after the exchange procedure, performed in a previous study on similar samples, which revealed an almost perfect 1:1 exchange between Cu2+ and Ca2+, paired with a negligible phosphate release.19 Conversely, the plate samples show an increase of the (Ca + Cu)/P ratio, particularly evident for the plate_14. This observation suggests that at higher Cu2+ concentration also other secondary Cu2+ incorporation mechanisms are probably present, possibly including surface complexation, hydrogen substitution in structural hydroxyls forming oxocuprate ions and/or formation of copper oxide nanoclusters.25–27 This hypothesis is further supported by the Cu2+ surface density in more concentrated samples, which reaches approximately 7 Cu nm−2 for plate_14, exceeding the surface concentration of exchangeable Ca2+ ions (∼5 Ca nm−2).28
3.2 Structural and morphological study
The general structural characterization and the phase identification of the HA nanoparticles was performed by X-ray powder diffraction. As visible in Fig. 2, the XRD patterns show broad diffraction peaks, typical of nanometric poorly crystalline materials,29 which can be described as a hydroxyapatite single phase (PDF no. 00-009-0432). There are no evident differences between the pristine and Cu-doped samples, confirming that the Cu exchange has occurred only at the surface, without significantly altering the crystal structure. For the pristine needle HA, the size of the crystalline domains, evaluated using the Scherrer equation, is 39 ± 1 nm along the c-axis (D(002)) and 12 ± 1 nm in the a-b plane (D(310)). As expected considering the BET SSA, these values are smaller for the plate HA being D(002) = 13 ± 1 nm and D(310) = 4 ± 1 nm. Also the Cu-doped samples show comparable crystalline domain dimensions with a D(002)/D(310) ratio ∼3, typical of an elongated morphology with preferentially exposed {010} facets as already discussed in previous investigations.19 | ||
| Fig. 2 XRD patterns of the needle HA nanoparticles (top panel in red) and plate HA nanoparticles (bottom panel in blue). The reference HA phase (PDF no. 00-009-0432) is also reported. | ||
Transmission electron microscopy was employed to better investigate the nanomaterial morphology. The low-magnification images in Fig. 3 provide a representative overview of the morphology and dimensions of the needle (A), plate (B), needle_6 (C) and plate_11 (D) HAs. Due to the strong tendency of HA nanoparticles to form agglomerates, analyzing isolated nanoparticles and accurately estimating their size is challenging. However, HA nanoparticles of nanometric size are evident in all samples and, at the edges of the agglomerates, elongated plate-like nanoparticles can be observed. While plate HA (Fig. 3B and B′) consists exclusively of these plate-like nanoparticles, the HA needle sample also contains several elongated nanoparticles with a needle-like shape (Fig. 3A and A′). Considering the evidence from the XRD analysis and previous literature,7 we can infer that plate-like nanoparticles likely predominantly expose their (010) surface to the electron beam with elongation occurring along the [002] direction. Conversely, the needle-like nanoparticles are likely exposing facets perpendicular to the [010] direction, as the electron beam passes through their less-developed facets. Needle HAs show bigger crystallographic domains along the c-axis, in agreement with the XRD results and with their lower specific surface area. Finally, the TEM images of needle_6 and plate_11 (panels C and D, respectively) confirm that the nanoparticles are not significantly modified by the ion exchange process.
 | ||
| Fig. 3 Representative TEM images of the needle (A), plate (B), needle_6 (C), and plate_11 (D) samples. Inset A′ and B′ evidence in more detail the nanoparticles morphology for HA needle and plate, respectively. | ||
3.3 Characterization of the Cu active sites
The characterization of the cations exposed at the HA surface was performed by FTIR of adsorbed CO, which is a versatile probe molecule sensitive to the oxidation state of the metal centers30,31 and it is also the reagent of the target reaction in this study. The measurements were performed at low temperature (ca. 100 K) for favouring the interaction between CO and the surface cations. The samples were previously heated at 423 K in dynamic vacuum to remove the adsorbed water without altering the HA structure.7 Nitrogen adsorption measurements at 77 K confirmed that the specific surface area does not change significantly as a result of the thermal treatment: for example, for the needle_6 sample, the BET specific surface area measured after outgassing at room temperature is 101 m2 g−1, while after activation at 423 K it is 99 m2 g−1. As shown in Fig. S1 in the ESI,† the samples exhibit a type IV isotherm characterized by a hysteresis loop indicative of irregular mesoporosity, likely of interparticle origin, and by negligible microporosity (micropore volume: 0.005 cm3 g−1).Considering the FTIR spectra of pristine needle and plate materials (Fig. 4A and B), we can note the presence of a single band at 2168 cm−1 assigned to CO adsorbed on Ca2+,7,19,32 which progressively shifts to 2175 cm−1 while decreasing CO coverage, owing to the fading out of CO–CO lateral interactions.33 This signal is more intense in the plate sample owing to its higher SSA (see also Fig. S2 and S3† for a specific comparison of the IR spectra at the highest CO coverage).
 | ||
| Fig. 4 FTIR spectra of CO adsorbed at ca. 100 K at different coverages starting from 30 mbar (colored curves) with stepwise outgassing till 1 × 10−3 mbar (black curves) for HA needle (A), needle_4 (A′), needle_6 (A′′), HA plate (B), plate_4 (B′) and plate_14 (B′′). The spectrum of the activated material has been subtracted from all spectra (see Fig. S2 and S3† for further details about the analysis procedure). | ||
Moving to Cu-doped samples containing the same metal amount (4 wt%), we can notice the appearance of two new bands (Fig. 4A′ and B′): (i) at 2155 cm−1, which shifts till 2157 cm−1 at low CO coverage, and can be assigned to Cu2+ species; (ii) at 2116–2113 cm−1, shifting till 2118 cm−1, which is less reversible upon outgassing and can be ascribed to Cu+.19,30,34 In both materials we can still highlight the presence of the signal at 2168 cm−1 assigned to Ca2+, which is more evident in the plate_4 sample since, owing to its higher SSA, at the same metal loading the fraction of surface Ca substituted by Cu is lower with respect to the needle_4 sample.
Finally, we can consider the samples at the highest copper uptake, needle_6 (panel A′′) and plate_14 (B′′), which show the same triplet of bands previously discussed. In this case the intensity of the components arising from CO–Ca2+ interactions are markedly reduced, especially for the needle_6 sample, in agreement with the presence of surfaces largely functionalized with copper. Moreover, as visible also in the materials with 4 wt% Cu, the intensity of the relative signal of the Cu+ species with respect to the Cu2+ ones is higher for the needle HA, suggesting an easier reducibility of copper ions for this HA particle morphology. This observation is of particular interest since Cu+ has been reported to be a crucial active species in the CO oxidation reaction.35,36
The electronic structure and local coordination environment of the active metal centres were also investigated by XAS measurements.29,37 Cu K-edge XANES and EXAFS spectra for needle and plate HA samples with different copper loadings are reported in Fig. 5. In the XANES region we can recognize a pre-edge peak at 8978 eV, which is characteristic of the 1s → 3d transition in Cu2+.38 Moreover, the intensity of the main peak (white line) does not show significant variations, strongly suggesting a similar Cu2+ coordination environment in all samples. This is further confirmed by considering the FT-EXAFS spectra, which are comparable for all materials.
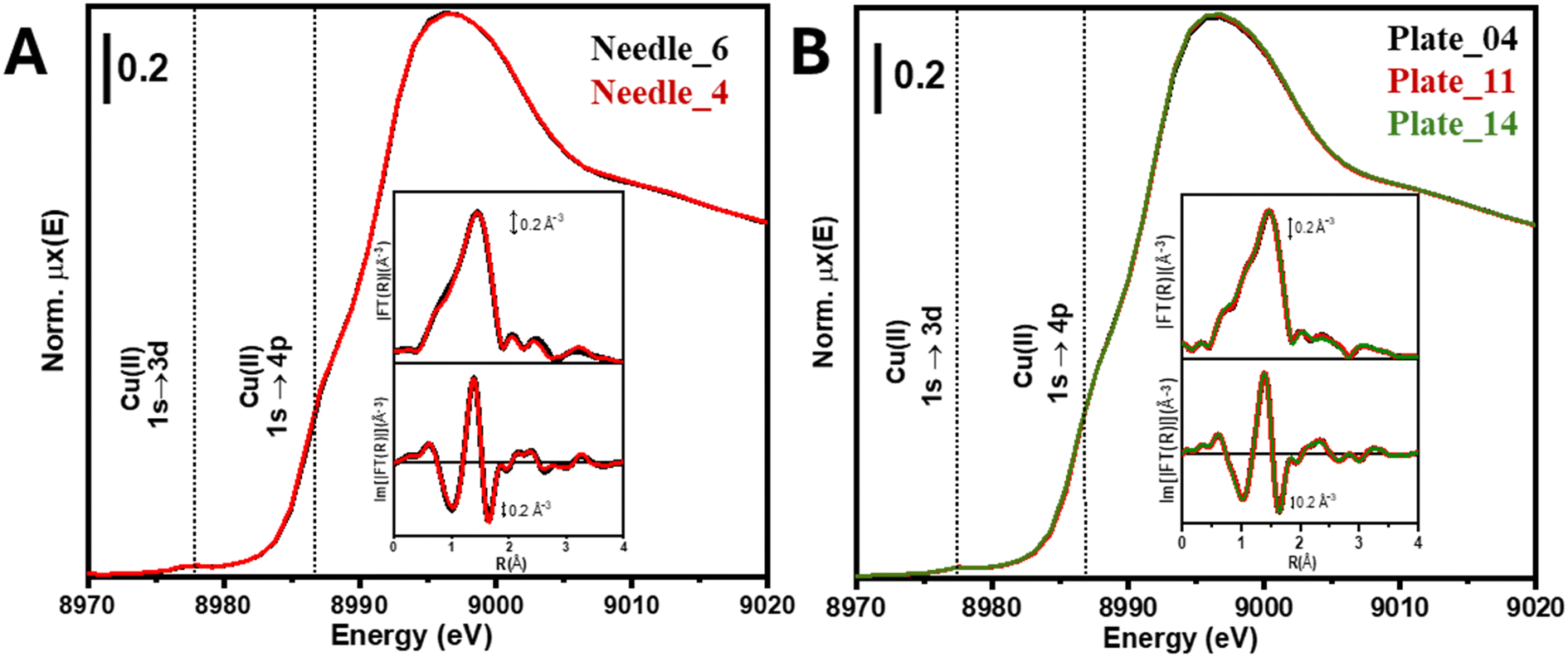 | ||
| Fig. 5 Cu K-edge XAS spectra of needle (A) and plate (B) HA samples with various copper loadings. The inset displays the modulus and the imaginary part of the phase-uncorrected Fourier transform of the k2-weighted χ(k) signal (see Fig. S5†) in the 2–10 Å−1 range. | ||
From the fit of FT-EXAFS spectra (Fig. 6) we can obtain more quantitative information. Owing to the high number of parameters to be refined, the amplitude (S02) and the Debye-Waller factors (σ2) for each scatterer were kept fixed during the fitting and only individual scattering paths for Cu–O, Cu–P and Cu–Ca were considered. From Table 1 we can note that the first shell peak is arising from Cu–O contributions at ∼2.0 Å, while the second and third shell peaks are ascribed to Cu–P and Cu–Ca at ∼3.0 Å and ∼3.9 Å, respectively. As highlighted also by previous investigations,38,39 the first shell bond distances are significantly shorter than the corresponding ones in undoped hydroxyapatites (i.e. Ca–O ∼ 2.4 Å40,41). This effect can be explained by the strong difference between the ionic radii of Ca2+ (0.99 Å) and Cu2+ (0.73 Å).39 The coordination numbers of the first shell are in line with those reported in previous studies on Cu-exchanged HAs39 and are lower than those reported for Ca in pure HAs, as expected since Cu is only located at the surface sites. Regarding the coordination numbers of the second and third shells, it is important to emphasize that they are heavily correlated with the Debye–Waller factors.29 Moreover, the inclusion in the model of only single scattering paths did not fully account for the effect of possible interferences of anti-phase backscattering waves stemming from multiple scattering contributions, which would result in an apparent lowering effect for the optimized coordination number.
 | ||
| Fig. 6 Comparison between the experimental data (black lines) and corresponding best fits in the 1–4 Å range (red lines) for the modulus and the imaginary part of the phase-uncorrected Fourier transform of the k2-weighted χ(k) signal of the needle_6 (A) and plate_14 (B) HA samples. | ||
| Sample | S 0 2 | E 0 (eV) | N Cu–X | R Cu–X (Å) | σ 2 (Å2) | R factor |
|---|---|---|---|---|---|---|
| Needle_ 6 | 1 | −1.5 ± 0.8 | Cu–O: 3.32 ± 0.12 | Cu–O: 2.01 ± 0.05 |

|
0.013 |
| Cu–P: 0.53 ± 0.26 | Cu–P: 3.00 ± 0.04 |

|
||||
| Cu–Ca: 0.60 ± 0.35 | Cu–Ca: 3.94 ± 0.01 |

|
||||
| Plate_14 | 1 | −1.5 ± 0.8 | Cu–O: 3.33 ± 0.12 | Cu–O: 2.01 ± 0.05 |

|
0.010 |
| Cu–P: 0.56 ± 0.27 | Cu–P: 3.00 ± 0.04 |
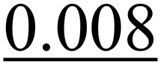
|
||||
| Cu–Ca: 0.54 ± 0.37 | Cu–Ca: 3.94 ± 0.01 |

|
The samples were also studied after being subjected to the CO oxidation catalytic tests (vide infra). An initial XRD analysis did not reveal evident changes in the crystalline structure (see Fig. S4†), while N2 adsorption measurements highlighted a small decrease of specific surface area after the catalytic run (e.g. from 99 to 82 m2 g−1 for the needle_6 sample, see Fig. S1†). XAS measurements were also carried out to compare needle_6 and plate_14 samples as prepared (fresh samples) and after (spent samples) the catalytic tests. Looking at Fig. S6 in the ESI,† we can notice a decrease in the white line intensity at 8996 eV for the spent sample associated with a change in the coordination number for some of the Cu sites. This conclusion is also supported by the slight decrease in the first shell peak intensity in the FT-EXAFS. In addition, in the XANES region a shoulder develops at 8987 eV, in the spectral range characteristic of 1s → 4p transitions in Cu+,38 further highlighting the conversion of a small percentage of Cu2+ into Cu+ in the spent catalyst. In order to estimate the Cu(II) and Cu(I) fractions, we applied XANES linear combination fit (LCF). As reference compounds, after considering different options (see Fig. S7†), we selected Cu2CO3(OH)2 (malachite) for the Cu(II) component and Cu2O for the Cu(I) component. As expected, the analysis revealed that fresh samples are overwhelmingly dominated by Cu(II), constituting nearly 100% of total copper. In spent samples, needle_6 shows a small but statistically significant Cu(I) contribution (∼3% of total Cu), while quantitative LCF results for plate_14 indicate only 1% Cu(I), which falls within the margin of fitting error (see Fig. S8–S9 and Table S2 with related discussion in the ESI†).
3.4 Catalytic activity in CO oxidation
The catalytic activity in CO oxidation to CO2 was evaluated by monitoring the CO conversion at 473, 523 and 573 K after a stabilization step of 30 minutes for each temperature. As shown in Fig. 7, poor catalytic activity (XCO < 5%) was observed for the non-functionalized materials, needle or plate, at any of the temperatures. On the contrary, all the copper-functionalised materials were catalytically active, showing an increase of the catalytic activity proportional to the temperature and evidencing that the CO conversion is activated by the surface Cu2+ ions. Firstly, at 423 K, all the materials exhibited low and similar CO conversion (ca. 10%), being slightly higher in the case of needle_4 (12%) and slightly lower in the cases of plate_11 and plate_14 (6%). The increase in temperature until 523 K entails a noticeable increase in the catalytic activity. Moreover, the HA needles exhibit differences as a function of their Cu2+ content, being higher the CO conversion for needle_6 (61%) than for needle_4 (55%). On the other hand, XCO is more homogeneous for the plate HA, being 47% for plate_4 and plate_11, and 46% for plate_14. Therefore, at 523 K, the needle HA show approximately a XCO 14% higher than the plate HA. Finally, at 573 K, the CO conversion is complete (ca. 99%), either for the needle or for the plate HA. Only the plate_4 sample exhibit a slightly lower XCO = 94%. Overall, the results highlight that the efficiency of CO conversion depends more on the HA morphology than on the Cu content. | ||
| Fig. 7 CO conversion (XCO) for the different HA materials at different temperatures under continuous flow of CO:O2 (1:0.5 molar ratio) diluted in He. | ||
4. Conclusions
We synthesized biomimetic HA with needle-like or plate-like morphologies, characterized by Ca-rich (i.e. Ca/P ratio of 1.8) or P-rich (i.e. Ca/P ratio of 1.48) surface terminations, respectively. Then, we functionalized both materials with increasing amounts of Cu through an ion-exchange procedure. The maximum copper uptake for the HA needle series was 6 wt% and was achieved with a 0.05 M Cu(NO3)2 solution. In contrast, the HA plate series reached a higher copper content of 14 wt% when using a 0.1 M Cu(NO3)2 solution. This difference can be attributed to the higher specific surface area of pristine plate HA (ca. 200 vs. 100 m2 g−1).XRD analysis revealed no noticeable differences between the pristine and Cu-doped samples, confirming that Cu exchange occurred exclusively at the surface without significantly affecting the HA crystal structure. XAS measurements confirmed a similar Cu2+ coordination environment in all samples, characterized by the presence of oxygen atoms at an average distance of 2.0 Å in the first coordination shell.
The materials were tested in the CO oxidation reaction and both pristine needle and plate HAs showed poor CO conversion (<5%), while all Cu-functionalized samples exhibited catalytic activity. At 523 K, needle HA with both 4 and 6 Cu wt% achieved higher CO conversion (up to 61%) than plate HA with 4, 11 or 14 Cu wt% (XCO ∼ 47%). At 573 K, nearly complete CO conversion (∼99%) was observed for all materials. We can thus conclude that the copper content is not the main factor determining the catalytic efficiency of the doped HAs. On the contrary, the different redox properties of the samples, which are influenced by their distinct morphology and surface acid–base properties, seem to play a crucial role. In particular, as highlighted by IR measurements of CO adsorption and XAS analysis on the spent catalysts, the surface Cu2+ in the needle-like samples is more reducible to Cu+ compared to the plate-like ones. This leads to more efficient catalytic activity despite the lower Cu weight content.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001). This work received financial support from MUR and the European Union – Next Generation EU, Mission 4, Component 1 through the PRIN Project PERFECT (CUP: P2022TK9B9). The authors acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities under proposal CH-6598 and thank the BM31 staff for assistance and support in using the beamline.References
- M. Gruselle, J. Organomet. Chem., 2015, 793, 93–101 CrossRef CAS
.
- A. Fihri, C. Len, R. S. Varma and A. Solhy, Coord. Chem. Rev., 2017, 347, 48–76 CrossRef CAS
- O. V. Kharissova, Y. P. Méndez, B. I. Kharisov, L. T. González and S. V. Dorozhkin, Particuology, 2025, 96, 203–217 CrossRef CAS
- M. Ibrahim, M. Labaki, J. M. Giraudon and J. F. Lamonier, J. Hazard. Mater., 2020, 383, 18 CrossRef PubMed
- P. Ivanchenko, G. Escolano-Casado, L. Mino, L. Dassi, J. F. Fernandez-Sánchez, G. Martra and J. Gómez-Morales, Colloids Surf., B, 2022, 217, 7 CrossRef PubMed
- C. A. Ospina, J. Terra, A. J. Ramirez, M. Farina, D. E. Ellis and A. M. Rossi, Colloids Surf., B, 2012, 89, 15–22 CrossRef CAS PubMed
- Y. Sakhno, P. Ivanchenko, M. Iafisco, A. Tampieri and G. Martra, J. Phys. Chem. C, 2015, 119, 5928–5937 CrossRef CAS
- S. Diallo-Garcia, M. B. Osman, J. M. Krafft, S. Casale, C. Thomas, J. Kubo and G. Costentin, J. Phys. Chem. C, 2014, 118, 12744–12757 CrossRef CAS
- S. Campisi, M. G. Galloni, F. Bossola and A. Gervasini, Catal. Commun., 2019, 123, 79–85 CrossRef CAS
- J. P. Ngorot Kembo, J. Wang, N. Luo, F. Gao, H. Yi, S. Zhao, Y. Zhou and X. Tang, New J. Chem., 2023, 47, 20222–20247 RSC
- K. Wei, X. Wang and J. Ge, Chem. Soc. Rev., 2024, 53, 8903–8948 RSC
- Z. Boukha, J. R. González-Velasco and M. A. Gutiérrez-Ortiz, Appl. Catal., B, 2021, 292, 120142 CrossRef CAS
- Z. Boukha, J. L. Ayastuy, M. Cortés-Reyes, L. J. Alemany, J. R. González-Velasco and M. A. Gutiérrez-Ortiz, Int. J. Hydrogen Energy, 2019, 44, 12649–12660 CrossRef CAS
- J. Guo, H. Yu, F. Dong, B. Zhu, W. Huang and S. Zhang, RSC Adv., 2017, 7, 45420–45431 RSC
- Z. Boukha, J. L. Ayastuy, J. R. González-Velasco and M. A. Gutiérrez-Ortiz, Appl. Catal., B, 2017, 201, 189–201 CrossRef CAS
- X. Li, L. Xing, W. Zhao, Y. Wang and Y. Zhao, Int. J. Hydrogen Energy, 2021, 46, 29940–29950 CrossRef CAS
- H. Martínez-Hernández, J. A. Mendoza-Nieto, H. Pfeiffer, J. Ortiz-Landeros and L. Téllez-Jurado, Chem. Eng. J., 2020, 401, 125992 CrossRef
- J. Jemal, H. Tounsi, K. Chaari, C. Petitto, G. Delahay, S. Djemel and A. Ghorbel, Appl. Catal., B, 2012, 113, 255–260 CrossRef
- G. Escolano-Casado, P. Ivanchenko, G. Paul, C. Bisio, L. Marchese, A. M. Ashrafi, V. Milosavljevic, L. Degli Esposti, M. Iafisco and L. Mino, Appl. Surf. Sci., 2022, 595, 153495 CrossRef CAS
- C. Wen, Y. Y. Cui, X. Chen, B. N. Zong and W. L. Dai, Appl. Catal., B, 2015, 162, 483–493 CrossRef CAS
- G. Escolano-Casado, C. Fusacchia, L. Degli Esposti, M. Cacaci, D. Squitieri, F. Bugli, M. Iafisco and L. Mino, Surf. Interfaces, 2025, 62, 106179 CrossRef CAS
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- O. Rosskopfova, M. Galambos, J. Ometakova, M. Caplovicova and P. Rajec, J. Radioanal. Nucl. Chem., 2012, 293, 641–647 CrossRef CAS
- L. Yang, W. H. Zhong, J. Cui, Z. G. Wei and W. Wei, J. Dispersion Sci. Technol., 2016, 37, 956–968 CrossRef CAS
- A. S. Karpov, J. Nuss, M. Jansen, P. E. Kazin and Y. D. Tretyakov, Solid State Sci., 2003, 5, 1277–1283 CrossRef CAS
- M. A. Zykin, A. V. Vasiliev, L. A. Trusov, R. E. Dinnebier, M. Jansen and P. E. Kazin, J. Solid State Chem., 2018, 262, 38–43 CrossRef CAS
- S. Campisi, C. Castellano and A. Gervasini, New J. Chem., 2018, 42, 4520–4530 RSC
- Y. Sakhno, L. Bertinetti, M. Iafisco, A. Tampieri, N. Roveri and G. Martra, J. Phys. Chem. C, 2010, 114, 16640–16648 CrossRef CAS
- C. Giannini, V. Holy, L. De Caro, L. Mino and C. Lamberti, Prog. Mater. Sci., 2020, 112, 100667 CrossRef CAS
- K. Hadjiivanov and H. Knozinger, Phys. Chem. Chem. Phys., 2001, 3, 1132–1137 RSC
- L. Mino, M. Signorile, V. Crocellà and C. Lamberti, Chem. Rec., 2019, 19, 1319–1336 CrossRef CAS PubMed
- F. Chiatti, M. Corno, Y. Sakhno, G. Martra and P. Ugliengo, J. Phys. Chem. C, 2013, 117, 25526–25534 CrossRef CAS
- S. Pantaleone, F. Pellegrino, V. Maurino, M. Corno, P. Ugliengo and L. Mino, J. Mater. Chem. A, 2024, 12, 4325–4332 RSC
- J. Schumann, J. Krohnert, E. Frei, R. Schlogl and A. Trunschke, Top. Catal., 2017, 60, 1735–1743 CrossRef CAS
- A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. Jin, C. Yang, A. Nefedov, C. Wöll, Y. Wang and W. Shen, Nat. Catal., 2019, 2, 334–341 CrossRef CAS
- J. Liu, M. Jing, R. Tao, W. Song, H. Cheng, H. Li, Z. Zhao, J. Liu, W. Zhu and S. Dai, Fuel, 2023, 333, 126303 CrossRef CAS
- L. Mino, D. Gianolio, F. Bardelli, C. Prestipino, E. S. Kumar, F. Bellarmine, M. Ramanjaneyulu, C. Lamberti and M. S. R. Rao, J. Phys. Condens. Matter, 2013, 25, 385402 CrossRef PubMed
- T. Bazin, M. Gaudon, E. Champion, I. Julien, C. Prestipino, S. J. Figueroa, M. Duttine and A. Demourgues, Inorg. Chem., 2024, 63, 22181–22193 CrossRef CAS PubMed
- A. Corami, F. D'Acapito, S. Mignardi and V. Ferrini, Mater. Sci. Eng., B, 2008, 149, 209–213 CrossRef CAS
- J. E. Harriest, D. W. L. Hukins and S. S. Hasnain, J. Phys. C: Solid State Phys., 1986, 19, 6859–6872 CrossRef
- S. Sugiyama, T. Moriga, H. Hayashi and J. B. Moffat, Bull. Chem. Soc. Jpn., 2001, 74, 187–192 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00370a |
| This journal is © The Royal Society of Chemistry 2025 |