
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirhodium(II,II) catalyst optimisation for chemoselective hydroaminomethylation: towards efficient amine synthesis†
Stephen de Doncker ,
Gregory S. Smith* and
Siyabonga Ngubane*
,
Gregory S. Smith* and
Siyabonga Ngubane*
Department of Chemistry, University of Cape Town, Rondebosch, 7701, South Africa. E-mail: Gregory.Smith@uct.ac.za; Siyabonga.Ngubane@uct.ac.za
First published on 15th July 2025
Abstract
The optimisation of a suitable mixed ligand dirhodium(II,II) catalyst for chemoselective hydrogenation of imines and enamines was carried out using four previously synthesised heteroleptic dirhodium(II,II) acetato-bipyridyl complexes (1–4). As such, optimised reaction conditions incorporating changes in temperature, H2 pressure, catalyst loading, and reaction time were applied to model substrates to determine the selectivity towards target amine product(s). The title complexes were evaluated under the optimised conditions for the hydrogenation reaction with the trifluoromethyl substituted complex (3) showing the highest hydrogenation activity for the series. Varying the partial pressure of CO relative to H2 in the syngas mixture to optimise the conversion of olefin substrate to form the target amine products was then applied with catalyst precursor (3). The amine reactants were varied, and the use of aromatic amines resulted in low conversion to target products, while aliphatic amine substrates showed good to excellent production of both secondary and tertiary amines in combination with a range of olefins. With the optimised catalyst and reaction conditions in hand, the hydroaminomethylation of a suitable substrate using catalyst precursor 3 to afford two analogues of a known opioid analgesic, Tramadol® was carried out.
1. Introduction
Amines are a class of compounds with a variety of applications in industrial, pharmaceutical and academic research.1,2 Compounds containing the amine functionality for such applications are often derived from fossil fuel sources, however, bio-sourced amines are of great interest particularly for pharmaceutical purposes.3,4 The synthesis of amines is conventionally carried out by the reductive amination of alcohols, amination of aryl halides or through the reduction of nitriles.5–7 In recent times, the need for sustainable synthetic procedures is often met by the inclusion of catalytic processes or steps in the synthetic protocol tailored specifically toward the synthesis of desired products. Efforts toward the facile synthesis of compounds incorporating amine functionalities include Cu-catalysed coupling reactions, photoredox-mediated coupling reactions, Buchwald–Hartwig amination, hydroaminoalkylation with a variety of transition metals and Fe-catalysed reductive amination.8–13 Furthermore, the consumption of alkene feedstocks through carbonylation reactions such as hydroformylation offer a versatile means of synthesising aldehydes and corresponding carboxylic acids, alcohols, and amines through additional chemical processes.14 These extrinsic reaction steps often include stoichiometric amounts of harsh reagents, unfavourable reaction conditions, intensive purification procedures and the formation of unwanted or toxic by-products.14Hydroaminomethylation (HAM) involves the reaction of the aldehydes produced from a hydroformylation reaction with an amine reagent in situ, leading to the formation of intermediary imines or enamines.15 Subsequent catalytic reduction of these transient condensation products in the presence of the catalyst and H2 yields amines as major products (Scheme 1). This allows for a streamlined means of obtaining amines from suitable alkene feedstocks with a significant reduction in the environmental impact. The advantage of the atom economy of the hydroformylation reaction, which forms an integral part of HAM, allows for an efficient approach to obtaining amines directly from low-value industrial feedstock alkenes with the liberation of H2O.16,17
 | ||
| Scheme 1 An outline of the crucial steps in the tandem hydroaminomethylation process using a primary amine substrate. | ||
Since the condensation step liberates water as a by-product, the stability of the catalyst precursor to an aqueous environment may be required since deactivation of the active hydride species may hinder the catalytic steps, significantly reducing the yield. Ideally, catalyst precursors may be recycled by means of aqueous biphasic catalysis like hydroformylation reactions, where the catalyst precursor remains in the aqueous layer and the reaction occurs at the interface between the organic and aqueous phases, facilitated by rapid stirring and heating.18,19 Attempts at improving the selectivity, activity and recyclability of the catalyst species includes the incorporation of the active metal centre into large coordination assemblies or the use of micellar catalytic systems for HAM.20,21 The use of multifunction catalyst systems such as hetero-bimetallic rhodium/iridium and bimetallic rhodium/cobalt complexes has shown an increased efficiency compared to homo-metallic complexes.22–24 These catalyst systems utilise rhodium as the primary hydroformylation catalytic centre and iridium (or another transition metal) as the hydrogenation centre due to the affinity for each metal to carry out specific transformations.23–25 Despite these advances, the inclusion of additives such as cyclodextrins, HBF4, Zn(OTf)2 and H3PO4 or other ligand additives often result in higher condensation and hydrogenation efficiency for hydroaminomethylation, reported in conjunction with transition metal-based catalyst precursors or solvents which promote the reduction of enamines.26–31
The use of ligands and additives bearing donor atoms such as phosphorous, nitrogen and oxygen to enhance the reduction capabilities of transition metal-based catalysts in hydroaminomethylation are often reported.32,33 However, fewer reports on pre-formed nitrogen-containing rhodium-based complex catalysts exist.34,35 Recently, October et al. reported the synthesis and activity of Rh(I) based imino-pyridyl complexes with good activity and selectivity using aliphatic olefins.36 Comparable conversions were obtained with good regioselectivity under milder conditions to similar Rh(I)-based complexes at lower catalyst loading when compared to previous reports. In a subsequent study, suitable substrates were reported for obtaining products via hydrogenolysis of the appended benzyl groups to afford the target amines in good yields.36,37
Dirhodium(II,II) complexes containing bridging carboxylate-based ligands have shown incredible versatility, particularly in carbene and nitrene chemistry.38–40 As such, they are often employed as catalysts in catalytic amination reactions utilizing their broad scope via C–H insertion or aziridination, for example.41–44
To our knowledge, reports on substituent and/or counter-ion variation of the ancillary ligands in heteroleptic dirhodium(II,II) acetato-bipyridyl complexes and their effects are unexplored for homogeneous tandem catalytic reactions, particularly for hydroaminomethylation. The application of catalyst precursors previously showed no hydrogenation of either olefin or aldehyde when applied in the hydroformylation reaction,45 suggesting candidacy for the selective hydrogenation of imines under catalytic conditions.
Herein we describe the development and optimisation of a hydrogenation model reaction by altering gas composition, thermal parameters, counter-ion nature, bipyridyl substituent and catalyst loading to favour production of target amine products for hydroaminomethylation reactions. Furthermore, the versatility of the hydroaminomethylation reaction with the title catalyst precursors is demonstrated by application in the catalytic production of two analogues of a known active pharmaceutical ingredient (API), Tramadol®.
2. Experimental
2.1. Materials
All chemicals/reagents were purchased from Merck and used without purification unless stated otherwise. Solvents of analytical grade were used as received or freshly distilled, where necessary, and stored over molecular sieves. Column chromatography was carried out using Fluka Silica Gel 60 (40–63 microns), with the specified solvent system. All reactions were carried out under either Ar or N2 inert conditions, using standard Schlenk line techniques. Complexes 1–4 reported herein were synthesized as previously described.452.2. Equipment and instrumentation
Nuclear magnetic resonance (NMR) spectra were recorded on either a Bruker Nano 300 (1H: 300.08 MHz) or a Bruker Ultrashield X400 (1H: 400.22 MHz) spectrometer and were recorded using tetramethylsilane (TMS) as the internal standard. Coupling constants are reported in Hz and chemical shifts are reported in ppm relative to residual solvent signals. Analysis of the hydrogenation and hydroaminomethylation reactions were carried out on a Perkin Elmer Clarus 580 GC equipped with a flame-ionisation detector (FID) and/or an analytical Agilent HPLC 1260 equipped with an Agilent infinity diode array detector (DAD) 1260 UV-vis detector, with an absorption wavelength range of 210–640 nm. HPLC analysis of the compounds were carried out using a mixture of solvent A (10 mM NH4OAc/H2O) and solvent B (10 mM NH4OAc/MeOH) at a flow rate of 0.9 mL min−1. The gradient elution conditions were as follows: 10% solvent B from 0–1 min, 10–95% solvent B from 1–3 min, 95% solvent B from 3–5 min. Mass spectrometry was carried out using electron impact (EI) on a JEOL GCmatell instrument or using a Waters Synapt G2 equipped with an ESI probe with data recorded in positive mode. The catalytic reaction products were confirmed in relation to authenticated standards using GC-FID and/or LC-MS.2.3. General methods for hydrogenation and hydroaminomethylation reactions
Hydrogenation and hydroaminomethylation reactions were carried out in stainless steel pipe reactors (90 mL) equipped with a Teflon-coated magnetic stirrer bars. For hydrogenation reactions, each reactor was charged with toluene (5 mL), either one of the following aldehydes: (benzaldehyde, 4-nitrobenzaldehyde or 4-methoxy benzaldehyde (7.2 mmol)), n-propylamine (7.2 mmol), internal standard n-decane (1.26 mmol) and catalytic precursor (2.87 × 10−3 mmol). The pipe reactor was purged with nitrogen three times, followed by purging with hydrogen (40 bar) three times followed by heating to the required temperature. All reactions were performed in duplicate or triplicate and are recorded as an average of three identical experiments.Hydroaminomethylation reactions were carried out as follows: a 90 mL pipe reactor was charged with toluene (5 mL), olefin substrate (7.2 mmol), amine substrate (7.2 mmol), internal standard n-decane (1.26 mmol) and catalytic precursor (5.74 × 10−3 mmol). The pipe reactor was purged with nitrogen three times, followed by purging with syngas corresponding to a partial pressure ratio of (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, CO:H2) three times. The reactors were then heated to the required temperature whilst stirring for 4 hours. All reactions were performed in duplicate and are recorded as an average of two identical experiments.
:3, CO:H2) three times. The reactors were then heated to the required temperature whilst stirring for 4 hours. All reactions were performed in duplicate and are recorded as an average of two identical experiments.
2.4. Synthesis of reference imine and amine compounds for the hydrogenation model reaction and the synthesis of 3,4-dihydro-[1,1′-biphenyl]-1(2H)-ol
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1542 (C Ar–N). Purity: 98.86% by LC (tR = 0.57 min). MS (EI, m/z): 148.1 (35%, [M]+), calculated 148.0.C). Purity: 95.19% by LC (tR = 0.915 min). MS (EI, m/z): 157.2 (100%, [M–OH]+), calculated 157.1.
N), 1542 (C Ar–N). Purity: 98.86% by LC (tR = 0.57 min). MS (EI, m/z): 148.1 (35%, [M]+), calculated 148.0.C). Purity: 95.19% by LC (tR = 0.915 min). MS (EI, m/z): 157.2 (100%, [M–OH]+), calculated 157.1.3. Results and discussion
Recently we explored the synthesis and applicability of dirhodium(II,II) complexes containing acetate, diphenylformamidinate and bipyridine-based ligands in hydroformylation reactions.45,48 Both the homo- and heteroleptic dirhodium(II,II) complexes evaluated as catalyst precursors showed no appreciable formation of alkanes or alcohols from the hydrogenation of alkenes or aldehyde products under hydroformylation conditions.45,48 This may be exploited toward increasing the selectivity of a tandem reaction such as hydroaminomethylation, provided suitable conditions are determined for the hydrogenation of imine and/or enamine intermediates.3.1. Optimisation of imine hydrogenation reactions with complex 1 as catalyst precursor
The model reaction for evaluating the hydrogenation capabilities of the complexes was carried out by reacting benzaldehyde with n-propylamine in toluene in the presence of catalyst precursor (1) at a catalyst loading of 2.87 × 10−3 mmol (0.04%). This allows confirmation of the effects of the catalyst precursor on the condensation and hydrogenation steps in a one-pot reaction. Complex 1 (Scheme 2) was used as a model catalyst precursor under conditions for hydroformylation reactions (85 °C, 40 bar syngas (CO:H2 1:1) pressure and 4-hour reaction time) with benzaldehyde and n-propylamine in a 1:1 molar ratio (7.2 mmol) (Scheme 2).
 | ||
| Scheme 2 Outline of the model catalytic hydrogenation reaction using complexes 1–4 as catalyst precursors. | ||
Analysis of the reaction mixture by gas chromatography display signals corresponding to unreacted benzaldehyde (tR = 24.63 min) and the corresponding imine (tR = 30.32 min), with an average substrate conversion of 85%, and low amine yield (<5%). These signals were verified against authenticated standards of both substrates and intermediates synthesised via reported methods and characterised prior to catalytic evaluation.46,49 The toluene was removed under reduced pressure before further analysis of the reaction mixture by NMR spectroscopy (Fig. S4†). The relative integration of each signal confirms the reaction of benzaldehyde and n-propylamine to form the imine intermediate, with the relative aldehyde (H1, 9.91 ppm) to imine proton (H2, 8.17 ppm) signal integration observed in a 0.28:1 ratio. This corresponds to approximately 80% conversion to the imine, in agreement with the conversion calculated from the GC-FID analysis.
Under hydrogen atmosphere at 40 bar pressure and 85 °C (Table 1), the conversion to imine intermediate and amine yield increased to 99 and 15% respectively (Table 1, entry 1). This indicates that the system may be further optimised for maximal amine production.
| Entry | Catalyst precursor | Catalyst loading | Temperature | Conversion of aldehyde | Imine | Secondary amine | Tertiary amine |
|---|---|---|---|---|---|---|---|
| (× 10−3 mmol) | (°C) | (%) | (%) | (%) | (%) | ||
| Hydrogenation reactions carried out in toluene (5 mL), 40 bar H2 pressure, 4 h reaction time, 7.2 mmol benzaldehyde, varying the temperature with an initial catalyst loading of 2.87 × 10−3 mmol, equivalent to a Rh metal loading of 5.74 × 10−3 mmol. GC conversions were obtained using an internal standard of n-decane. Reactions were carried out in triplicate with average error values of 4.72%. NC – not calculated.a Benzaldehyde substrate.b Reaction carried out for 8 hours.c Reaction carried out in the absence of catalyst precursor.d 4-Nitrobenzaldehyde substrate.e 4-Methoxybenzaldehyde substrate. | |||||||
| 1a | 1 | 2.87 | 85 | 99 | 85 | 15 | <1 |
| 2a | 1 | 2.87 | 95 | 99 | 69 | 31 | <1 |
| 3a | 1 | 2.87 | 105 | 99 | 59 | 41 | <1 |
| 4a | 1 | 2.87 | 115 | 99 | 69 | 31 | <1 |
| 5b | 1 | 2.87 | 105 | 99 | 51 | 49 | <1 |
| 6c | — | — | 105 | 99 | >99 | <1 | <1 |
| 7a | 1 | 5.74 | 105 | 99 | 39 | 61 | <1 |
| 8d | 1 | 5.74 | 105 | 22 | 14 | <1 | 86 |
| 9e | 1 | 5.74 | 105 | 99 | <1 | 4 | 96 |
| 10 | 2 | 2.87 | 105 | 99 | 42 | 58 | <1 |
| 11a | 2 | 5.74 | 105 | 99 | 38 | 62 | <1 |
| 12a | 3 | 5.74 | 105 | 99 | 32 | 68 | <1 |
| 13a | 4 | 5.74 | 105 | 99 | 69 | 28 | 3 |
Temperature optimisation for the hydrogenation reaction was carried out under 40 bar hydrogen pressure in 10 °C increments from 85 up to 115 °C (Table 1, entries 1–4). The results obtained show the consumption of the substrate remains near quantitative (>99%) across the tested temperature range. This suggests that the thermal requirements for the condensation of benzaldehyde with n-propylamine are satisfied at 85 °C under H2 atmosphere. Additionally, a substantial increase in the amine products is observed at 95 and 105 °C with amine product production of 31 and 41% respectively (Table 1, entries 2 and 3). Increasing the reaction temperature to 115 °C resulted in a decrease in the amine production to 31% (Table 1, entry 4). This decrease is ascribed to the temperature limit of the toluene solvent (ca. 110 °C). 1H-NMR spectral analysis of the mixture of products obtained from reactions carried out at each temperature increment agrees with the data obtained from GC analysis (Fig. S5–S7†). The obtained data shows that the optimised temperature where maximum amines are produced (105 °C) would be used for subsequent optimisation experiments. Increasing the reaction time to 8 hours yielded a marginal improvement in the amine product obtained at 49% (Table 1, entry 5).
This disproportionate increase of 8% amine relative to the reaction time under identical conditions suggests that time is not the limiting factor for the hydrogenation reaction under these conditions. A reaction carried out without catalyst precursor under 105 °C and 40 bar pressure shows near quantitative consumption of the substrates with a quantitative amount of Schiff-base (>99%) detected (Table 1, entry 6), confirming that the presence of the catalyst is required for the hydrogenation reaction to form the target amine. Increasing the catalyst loading to 0.08 mol% resulted in the most significant increase in amine production at 61% (Table 1, entry 7). The combined optimisation data dictates that further evaluation of the catalyst precursors be carried out under reaction conditions of 105 °C, 40 bar H2 pressure and 4-hour reaction time at a 0.08 mol% catalyst loading.
3.2. The effect of electron-withdrawing and electron-donating substituents on hydrogenation
The effects of electron-withdrawing and electron-donating groups on the hydrogenation activity were explored by carrying out the hydrogenation reaction using either 4-nitrobenzaldehyde or 4-methoxybenzaldehyde with n-propylamine under the optimised conditions. The results obtained for the reaction with 4-nitrobenzaldehyde shows low substrate consumption (22%) compared to that obtained for the reaction with benzaldehyde (Table 1, entries 7 and 8). This is attributed to the strong electron-withdrawing effect of the nitro-substituent, hindering the formation of condensation products in the presence of the Lewis acid catalyst through lower stability of the imine intermediate.24,50 Additionally, the major product observed is the corresponding tertiary amine with no secondary amine products detected in the reaction mixture. This indicates that the hydrogenation reaction is largely uncompromised by the presence of the electron-withdrawing nitro substituent, however the hydrogenation reaction of n-propylamine with 4-nitrobenzaldehyde, occurs with lower efficiency.Conversely, when substituted with a methoxy group (Table 1, entry 9) the consumption of substrate increases to near quantitative amounts (99%). This occurs due to favourable electron donation from the methoxy group in addition to the presence of the Lewis' acid catalyst allowing for unhindered Schiff-base condensation, followed by a reduced hydrogenation rate due to a less electrophilic imine. This facilitates condensation reactions between the more nucleophilic secondary amine and the aldehyde to form enamines, which undergoes hydrogenation to form tertiary amine products. The combination of these data suggests that sensitivity of the proposed catalytic hydrogenation reaction to changes in the appended aldehyde functional group depends primarily on the substituents ability to inhibit the Schiff base formation.
3.3. The evaluation of complexes 2–4 as catalyst precursors for the hydrogenation of imines
Under the optimised reaction conditions, the effects of counter-ion type and bipyridyl substituent were explored by evaluating each catalyst precursor (2–4) for applicability in the model hydrogenation reaction (Table 1, entries 10–13). The influence of the counter-ion was explored by evaluation of the hexafluorophosphate congener (complex 2) and comparing the data to that obtained for complex 1. The results show a significant increase in the production of the target amine product (58%) for complex 2 (Table 1, entry 10). This observation is ascribed to the combination of the steric and electronic influence of the hemi-labile acetate, as concluded from the reported hydroformylation study.45 This is a consequence of competitive binding between the acetate counter-ion and the imine product to the bimetallic core, or the presence of the basic acetate group negatively influencing the catalytic hydrogenation. For verification, the catalyst loading was increased from 0.04 to 0.08 mol% for catalyst precursor 2 under the optimised conditions (Table 1, entry 11). The results show a marginal increase in the formation of amine product (62%).To maximise product yield, further evaluation of the title dirhodium(II,II) bipyridyl chelate complexes was carried out at 0.08 mol% catalyst loading, equivalent to 0.16 mol% rhodium metal loading. The typical catalyst loading reported for efficient hydroaminomethylation using rhodium-based catalyst precursors is usually between 0.5–1 mol%.34,51–53 The proposed catalytic system thus represents a substantial improvement toward efficient rhodium catalysed hydroaminomethylation reactions without the addition of ligands or additives.29,30 The influence of the bipyridyl substituent on the hydrogenation reaction was elucidated by evaluating hexafluorophosphate complexes 3 and 4 under the optimised conditions and comparing the data to that obtained for complex 2. The results show that a strong dependence on the catalytic reduction relative to the substituent exists, with trifluoromethyl-containing complex 3 presenting the highest amine yield of 68% (Table 1, entry 12).
The former may be corroborated by the result obtained for complex 4, with the lowest catalytic conversion of the imine corresponding to the methoxy-substituted complex (Table 1, entry 13). Similar trends were noted in the previous hydroformylation study, whereby the complex with the lowest reduction potential was the most active for hydroformylation.45 Furthermore, since the hydrogenation reaction proceeds under 40 bar H2 atmosphere and it is necessary that CO will need to be included in the reaction atmosphere, the reaction total pressure would be carried out at 50 bar for further experiments and the model hydroaminomethylation reaction.
3.4. Optimisation of partial gas pressures (CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :H2) hydroaminomethylation of cyclohexene with complex 3
:H2) hydroaminomethylation of cyclohexene with complex 3
Cyclohexene is a prime substrate for the hydroaminomethylation model reaction due to the optimized temperature for the hydrogenation reaction (105 °C) and the previously reported hydroformylation data regarding substrate scope. This is exemplified by the formation of one aldehyde product from hydroformylation and no extraneous formation of hydrogenated products, as observed from the data previously obtained.45 The model reaction was carried out as depicted in Scheme 3, with the results reported in Table 2.
 | ||
| Scheme 3 Outline of the model hydroaminomethylation reaction and possible product formation using complex 3 as a catalyst precursor. | ||
| Entry | CO:H2 ratio |
Olefin conversion | Aldehyde | Imine/enamine | Amines | 2° amine | 3° amine |
|---|---|---|---|---|---|---|---|
| (%) | (%) | (%) | (%) | (%) | (%) | ||
| Hydroaminomethylation of cyclohexene in toluene (5 mL), 50 bar total pressure, 105 °C, reaction time of 4 h and catalyst loading of 5.74 × 10−3 mmol. GC conversions were obtained using an internal standard of n-decane. Total amines are composed of secondary and tertiary amines in a mixture with a percentage sum of 100. Reactions carried out in duplicate with an average error value of 5.12%.a 1.5 equivalents of n-propylamine. | |||||||
| 1 | 0:1 |
— | — | — | — | — | — |
| 2 | 1:4 |
66 | <1 | 53 | 47 | 99 | 1 |
| 3 | 2:3 |
98 | <1 | 35 | 65 | 90 | 10 |
| 4 | 1:1 |
74 | <1 | 36 | 64 | >99 | <1 |
| 5a | 2:3 |
69 | <1 | <1 | >99 | >99 | <1 |
Under the optimised hydrogenation reaction conditions, no aldehyde, imine or amine products were obtained for the reaction carried out at 50 bar H2 atmosphere only (Table 2, entry 1). Since no aldehyde is produced under these conditions, no downstream intermediates such as imines or enamines and their corresponding amine products from the hydrogenation step are detected. Furthermore, under the higher pressure (50 bar) of H2 no cyclohexane was observed, indicating that under the mildly elevated reaction temperature and pressure, no hydrogenation of the alkene occurs.
The addition of CO to the reaction was carried out by increasing the partial pressure of CO relative to H2 to a 1:4 ratio (Table 2, entry 2). This resulted in 66% conversion of cyclohexene through the hydroformylation reaction; however, no aldehyde product was detected. This is ascribed to the condensation reaction between the aliphatic aldehyde and the amine substrate occurring efficiently, supported by the detection of intermediary imine/enamine products and amine products at 53 and 47% respectively. Interestingly, the secondary to tertiary amine distribution was observed to be 99:1 under these conditions. The formation of tertiary amines from the hydroaminomethylation reaction of an alkene with a primary amine is a consequence of the condensation reaction occurring between the initially formed secondary amine product with the aldehyde forming an enamine (Scheme 3).
Increasing the CO:H2 partial pressure ratio to 2:3 results in the near quantitative conversion of alkene substrate to 98% (Table 2, entry 3). An increase in the amount of amine products is observed (65%) under these conditions with an increase in the amount of tertiary amine detected relative to the reaction carried out under the 1:4 CO:H2 ratio. This is ascribed to the availability of the catalyst precursor for each catalytic reaction step, assuming that the active species cannot be involved in both reactions simultaneously. Evidence for this phenomenon may be shown by the presence of amide products formed from the reductive elimination of the amine and an acyl intermediate. These absence of amide products indicate that the hydroformylation and hydrogenation reactions do not occur simultaneously.
The higher proportion of tertiary amine produced (10%) under these conditions suggests a higher relative rate of the hydroformylation reaction compared to the condensation reaction. A lower consumption of substrate is observed for the reaction carried out under a CO:H2 ratio of 1:1 (Table 2, entry 4) with similar amounts of imine and amine produced. In this case, only secondary amine product was detected as compared to the reaction previously described (Table 2, entry 3). However, the comparable production of imine and amine observed suggests that the hydrogenation reaction proceeds in a similar manner under these conditions. Notably, catalyst precursor 3 showed a pronounced increase in the production of amine compared to 1 under the syngas (CO:H2 1:1) atmosphere, likely due to enhanced reactivity at the increased temperature.
The exclusive production of secondary amine under these conditions indicates that up to a certain level, the relative hydrogenation rate is slowed, possibly due to the higher concentration of CO in the reaction medium. The lower substrate consumption (74%) under these conditions is therefore ascribed to some suppression in activity of the hydroformylation catalyst, likely due to the faster formation of amines which may coordinate to the metal centre and hinder the reaction. An increase in the equivalents of the amine substrate (Table 2, entry 5) resulted in a decrease in the conversion to 69% with excellent conversion to specifically secondary amine product. The combined data from the optimisation of the partial pressure of CO relative to H2 suggests that for efficient hydroaminomethylation, the ratio of CO:H2 for further experiments should be 2:3. The optimised conditions and complex precursor 3 were further evaluated in the hydroaminomethylation reaction with cyclohexene while varying the amine substrate.
3.5. The effects of primary or secondary alkyl or aryl amine substrates
The effects of varying alkyl amines were explored by reacting cyclohexene under the optimised catalytic conditions with secondary amines (piperidine and dibenzylamine) for comparison with the results obtained for n-propylamine. The results obtained for the hydroaminomethylation of cyclohexene with either n-propylamine or piperidine (Table 3, entries 1 and 2) show a decrease in the conversion from 98 to 81%, while the reaction with dibenzylamine shows 97% conversion of substrate (Table 3, entry 3). The hydroaminomethylation of cyclohexene and piperidine has been reported by Iqbal in 1971, with a similar yield (80%) using Rh2O3, although under high gas pressure (140 atm) and high temperature (170 °C).54 This reaction represents a significant improvement in not only the catalyst loading, but also the reaction conditions required for the hydroaminomethylation of these substrates.| Entry | Olefin substrate | Amine substrate | Olefin conversion | Aldehyde | Imine/enamine | Secondary amine | Tertiary amine |
|---|---|---|---|---|---|---|---|
| (%) | (%) | (%) | (%) | (%) | |||
| Hydroaminomethylation of olefin substrate with amine substrate in toluene (5 mL), 50 bar total pressure, 105 °C, reaction time of 4 h and catalyst loading of 5.74 × 10−3 mmol. GC conversions were obtained using an internal standard of n-decane. Reactions carried out in duplicate with an average error value of 4.31%.a Reaction mixture analyzed by LC-MS and GC-FID. | |||||||
| 1 (ref. 55) |  |
 |
98 | <1 | 35 | 90 | 10 |
| 2 (ref. 26, 54 and 56) |  |
 |
81 | <1 | <1 | — | 100 |
| 3a (ref. 57) |  |
 |
97 | <1 | <1 | — | 100 |
| 4 |  |
 |
99 | 97 | <1 | — | — |
| 5a (ref. 55) |  |
 |
57 | 91 | <1 | 9 | <1 |
| 6a (ref. 57) |  |
 |
98 | 86 | <1 | — | 14 |
| 7 |  |
 |
99 | <1 | <1 | <1 (lin) | — |
| >99 (br) | |||||||
| 8 (ref. 33, 34 and 58) |  |
 |
99 | <1 | <1 | — | 15 (lin) |
| 85 (br) | |||||||
| 9 (ref. 56) |  |
 |
91 | <1 | 5 | — | 100 (α-product) |
| 10a |  |
 |
99 | <1 | Mixture of difunctionalised enamine and amine products | ||
| 11a |  |
 |
5 | <1 | <1 | — | <1 |
| 12a |  |
 |
32 | <1 | <1 | — | 32 |
| 13 (ref. 34) |  |
 |
>99 | <1 | <1 | — | 40 (lin) |
| 60 (br) | |||||||
| 14a |  |
 |
>99 | <1 | <1 | — | 45 (lin) |
| 55 (br) | |||||||
The binding of amine moieties to metal catalysts often results in the reduction or complete deactivation in certain reactions and as such, may be considered as poisons in selected catalytic reactions.51,52 Since some binding between the amine substrate and the catalyst precursor may occur, the lower observed conversion for piperidine is attributed to a combination of ligation and the cyclic structure providing steric interference with the cyclohexene substrate in the hydroformylation reaction step. The results obtained with dibenzylamine substrate show an increase in conversion due to free rotation of the aromatic groups. No imine formation is possible when secondary amine reactants are used in HAM, therefore, exclusive production of enamines and tertiary amine products is expected for both dibenzylamine and piperidine (Table 3, entries 2 and 3). The near quantitative consumption of the enamine in both cases confirms that the hydrogenation of enamine intermediates proceeds efficiently under the optimised conditions. 1HNMR spectral analysis agrees with the structure of the products formed (Fig. S9 and S10†).
The hydroaminomethylation reaction carried out with dimethylammonium chloride shows no formation of intermediary or target amine products (Table 3, entry 4). The applicability of the catalyst for the hydroaminomethylation of primary and secondary aryl amines was evaluated by reacting cyclohexene with either aniline or diphenylamine. The results obtained from the reaction containing aniline substrate (Table 3, entries 5 and 6) shows a substantial reduction in the olefin conversion (57%) and the hydrogenation capability, with 9% amine product formed. An increase in the conversion (98%) of olefin is observed for the reaction carried out with diphenylamine substrate (Table 3, entry 6), with 86% aldehyde observed. This result is ascribed to a further decrease in nucleophilicity coupled with the steric implications of both phenyl groups. This is further supported by the observed formation of the proposed tertiary amine product, at a low level of 14%.
3.6. Performance of the optimised hydroaminomethylation catalytic system with various olefins
The olefin substrate scope was extended to styrene, cyclohex-2-en-1-one, cyclohex-2-en-1-ol, 1-octene and (E)-1,2-diphenylethylene (Table 3), to determine substrate sensitivity. The hydroformylation study previously reported showed the formation of both the linear and branched aldehyde products under the optimised reaction conditions for the styrene substrate.45 It is expected that increasing the number of intermediary aldehydes results in the formation of multiple intermediates (imines/enamines) formed and their associated hydrogenation products, particularly for primary amine substrates. The results obtained show near quantitative conversion of styrene with high production of amine products, indicating a highly efficient overall transformation of the substrates into the target amine products (Table 3, entries 7 and 8). Interestingly, the reaction which utilised n-propylamine as the amine substrate showed the greatest regioselectivity, with near quantitative conversion of branched product detected (Table 3, entry 7). Although it is well reported that the use of Rh-based catalysts favours branched aldehyde products with styrene, the higher proportion of branched product produced under these conditions is attributed to the higher reaction temperature.19,36,45,48The near quantitative consumption of the aldehyde and the selectivity for branched secondary amine product can be attributed to a balance in the formation of the imine with the branched aldehyde relative to the hydrogenation step. In this example, a product containing the phenylethylamine motif is obtained. Further analysis and verification were carried out by 1HNMR spectroscopy (Fig. S11†) where the expected proton signals corresponding to the branched secondary amine was observed. The obtained mass spectral data shows a base peak at m/z = 178.2, corresponding to the [M + H]+ protonated molecular ion (Fig. S12†). Conversely, the reaction carried out with piperidine (Table 3, entry 8) shows lower regioselectivity with an observed 85% formation of the branched tertiary amine product. This may be due to the increased steric influence of ligated piperidine or the reactivity differences of imine vs. enamine formation or a combination of these factors. The combined data shows that the product distribution of the hydroaminomethylation of styrene may be influenced by the nature of the amine and the reaction conditions to produce target amine products in good to excellent yields. Subsequent spectral analysis shows the expected signals in the 1HNMR spectra collected for both the linear and branched products (Fig. S13 and S14†), with base peaks observed at m/z = 204.2 in the LC-MS spectra for each respective product corresponding to the [M + H]+ protonated molecular ion in each case (Fig. S15 and S16†).
Further investigation into the effect of the substrate nature on the product distribution was carried out by applying the catalytic system to substrates cyclohex-2-en-1-one and cyclohex-2-en-1-ol in hydroaminomethylation reactions with piperidine. Due to the unsymmetrical nature of the alkenes in this case, addition of the formyl group in the hydroformylation step is expected to produce two possible aldehydes, thereby introducing the possibility of regioisomers. High conversion of the olefin was observed for the reaction carried out with cyclohex-2-en-1-ol (91%) with near quantitative consumption of the cyclohex-2-en-1-one substrate observed at 99% (Table 3, entries 9 and 10). No observed aldehyde was detected for the reactions of either substrate. However, the results for cyclohex-2-en-1-ol present a conversion to 95% amine product and 100% α- substituted product formed (Table 3, entry 9). This is supported by the obtained 1H-NMR and mass spectral data (Fig. S17 and S18†), the latter showing a base peak at m/z = 198.2 corresponding to the [M + H]+ protonated molecular ion. The regioselectivity can be rationalised by the presence of the hydroxyl group, which may results in directing the formation of the α-metal-acyl intermediate. Conversely, the reaction carried out with cyclohex-2-en-1-one showed multiple products formed, with the mass spectral analysis indicating the mixture of condensation products occurring at the ketone and aldehyde functionalities under catalytic conditions (Table 3, entry 10). The combined data shows that the regioselectivity may be influenced by directing groups through speculated interactions of the hydroxyl group interacting with the bimetallic core.
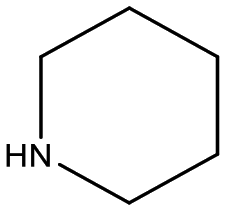
Reactions carried out with (E)-1,2-diphenyl ethylene shows low conversion of substrate with piperidine (Table 3, entry 11), whereas the reaction carried out with dibenzylamine shows a 32% conversion with tertiary amine product detected (Table 3, entry 12). The reaction utilizing 1-octene, a C8 aliphatic olefin, displayed regioselectivity follow the same trend as reported for the hydroformylation of alkenes utilizing these catalyst precursors, favouring iso-aldehyde products and in turn branched amines.45 Supporting mass spectral data for reactions carried out with 1-octene and (E)-1,2-diphenyl ethylene substrates are given in Fig. S19–S22.†
3.7. The application of the catalyst system toward the synthesis of analogues of Tramadol® via hydroaminomethylation
The data obtained from the optimisation and substrate variation of the hydroaminomethylation reaction suggest that the catalyst precursor and the conditions may be exploited for the synthesis of a suitable API. A synthetic pathway incorporating catalytic steps for obtaining known API's such as Fentanyl®, Chlorphenamine®, Tolterodine®, Tapentadol® and Tramadol® is advantageous for atom economy and waste generation (Fig. 1).53,59–62 | ||
| Fig. 1 Examples of the molecular structures of some API's which may be obtained by incorporating catalytic hydrogenation or hydroaminomethylation reactions as a key step. | ||
Tramadol® is a class IV scheduled opioid analgesic, administered as a racemate, used for short term relief from moderate to severe pain and is widely used due to a less addictive drug profile compared to codeine and morphine.60,63 Alvarado et al. reported a multistep synthetic route using harsh reagents, incorporating no catalytic reactions.60
A Mannich condensation reaction of the suitable amine to cyclohexanone is described, followed by addition of the methoxy benzene moiety via an organolithium reagent to the resulting ketone. If dimethylammonium hydrochloride is used as the amine reagent, the hydrochloride salt may be formed in fewer steps.59 Obtaining an active pharmaceutical in this class catalytically is economically and environmentally beneficial and significantly reducing by-product formation and the cost of production. An analogue of Tramadol®, 1-phenyl-2-(piperidin-1-ylmethyl)cyclohexan-1-ol, was retro-synthetically analysed and suitable synthons were identified to determine the applicability of the developed catalyst system as a proof of concept. The first disconnection shows that the hydroaminomethylation of a suitable substrate, 3,4-dihydro-[1,1′-biphenyl]-1(2H)-ol, with piperidine could be used as a final key step for obtaining a proposed tramadol analogue. Similar analysis shows that the required olefin substrate may be obtained in a Grignard reaction between bromobenzene and cyclohex-2-en-1-one. Furthermore, as was observed for the hydroaminomethylation of cyclohex-2-en-1-ol, the regioselectivity of this reaction should be influences by the presence of the hydroxy group to favour the α-substituted regioisomer.
 | ||
| Scheme 4 Reaction outline for the formation of 3,4-dihydro-[1,1′-biphenyl]-1(2H)-ol (S1) via a Grignard addition reaction of bromobenzene to cyclohex-2-en-1-one. | ||
Analysis of the obtained 1H-NMR spectrum (Fig. S3†) confirms the proposed structure, with the aromatic protons integrating for a total of 5H relative to the 1H integration for each olefin proton. The broad signals resonating in the 2.5 to 1.7 ppm region integrating for a combined 6H, agrees with the aliphatic protons in the proposed structure, with a broad signal resonating at around 1.6 ppm ascribed to residual H2O from the workup procedure. The former broadening of these signals is ascribed to conformational differences in the half-chair molecular structure, averaging out the axial and equatorial proton signals over the NMR timescale. The presence of the olefin proton signals in the 6 ppm region supports the chemoselectivity of the Grignard reaction for the ketone functionality, in which addition to the phenyl moiety to the carbonyl is observed. Further analysis by LC-MS indicates a purity of 95.19% and shows a base peak at m/z = 157.2 assigned to the [M–OH]+ molecular ion (Fig. S23†). This agrees with the 1H-NMR data and confirms the proposed structure for use as a substrate for the hydroaminomethylation reaction.
 | ||
| Fig. 2 Graphic representation of the results obtained from hydroaminomethylation reactions of 3,4-dihydro-[1,1′-biphenyl]-1(2H)-ol (S1) with piperidine and dibenzylamine. Regioisomer ratios are expressed as a percentage in favour of the α-substituted product. | ||
The HPLC chromatogram obtained for the S1 substrate shows a peak at a retention time of 0.915 min with the corresponding mass, which was used to determine the conversion by LC-MS analysis (Fig. S23†). The observed yields for reactions carried out with either piperidine or dibenzylamine indicates that under catalytic conditions, the known piperidine analogue66 was obtained in a 74% yield (m/z = 274.2), while the reaction with dibenzylamine yielded the target product in a lower 36% yield (m/z = 386.2) (Fig. S24 and S25†). In both cases, crystallisation of the products with HCl/ether and subsequent product analysis via LC-MS, exclusively reveals the expected signals at m/z = 274.2 and 386.2 for the [M + H]+ protonated molecular ions, confirming the data obtained from crude mixture analysed. Furthermore, mass spectral analysis indicates two base peaks with m/z corresponding to the target products at different retention times. This is attributed to the formation of regioisomers, calculated as a ratio of 4:1 and 3:1 in favour of the product substituted α- to the hydroxyl group, for reactions carried out with piperidine and dibenzylamine respectively. This suggests that the regioselectivity and product production is due to steric implications of the large dibenzyl group. This is likely due to unfavourable interaction with the phenyl substituent on the substrate, not seen for the reaction carried out with piperidine. Compared to the reaction carried out with piperidine and cyclohex-2-en-1-ol where only the α-substituted product was detected, this suggests that the presence of the phenyl group plays a significant role in directing the regioselectivity.
Considering the combined data from the hydroformylation,45 hydrogenation and hydroaminomethylation reactions carried out and the results reported herein, a proposed auto tandem mechanism, including hypothesized steps, for the overall conversion of alkenes to amines via hydroaminomethylation with these dirhodium(II,II) complexes is given in Scheme 5 with styrene and propylamine as substrates. Although complex 3 was determined as the best catalyst precursor, complex 2 is depicted in the scheme for simplicity. For both the homo- and heteroleptic complexes containing bridging acetato ligands reported as hydroformylation catalysts, we reported a trend whereby complexes presenting the lowest reduction potential showed the highest hydroformylation capability. This suggests that an important step in catalytic cycle is reductive elimination from a higher energy species to reproduce the catalyst precursor and the product. Furthermore, based on the accepted hydroformylation cycle for mono metallic rhodium catalyst precursors, activation of the catalyst precursor to a metal hydride is required to initiate the reaction.
 | ||
| Scheme 5 Proposed auto tandem hydroaminomethylation catalytic cycle utilising heteroleptic dirhodium(II,II) acetate-bipyridyl complexes as catalyst precursors. | ||
The catalysis may proceed across the bimetallic center, or at one Rh atom with the metal–metal bond imparting an additional electron sync, enhancing the catalytic abilities.67 The scheme depicts a catalytic cycle based on the latter, with each step in corresponding to an expected transformation as expected for Rh(I) based hydroformylation, where an Rh2(II,IV) intermediate is formed, which then undergoes reductive elimination. This regeneration is evident from the isolation of the catalyst precursor from the reaction mixture after recycling the catalyst as previously reported.45 Subsequent in situ Schiff base condensation provides the intermediary imines/enamines which undergo hydrogenation by the activated catalyst via the previously hypothesized Rh2(II,IV) intermediate, forming the amine product after which the catalyst precursor is regenerated.
Conclusions
The extension of the applicability of dirhodium(II,II) acetato-bipyridyl chelate complexes to the hydroaminomethylation reaction was achieved through optimisation of the reaction conditions required for catalytic hydrogenation of imines. This optimisation was explored using a model reaction with complex 1 as a catalyst precursor showing that under catalytic conditions, the in situ formation of the model imine complex occurs readily between 85 and 115 °C. Additionally, a temperature dependent increase in the hydrogenation was observed resulting in a maximum occurring at 105 °C indicating the optimal reaction temperature. A reaction carried out in the absence of the catalyst precursor shows no detectable hydrogenation product with full consumption of the substrate, confirming presence of the catalyst as a requirement for hydrogenation. Exchange of the acetate counter-ion for the hexafluorophosphate congener (complex 2) resulted in a 17% increase in amine product production. Further evaluation of the complexes revealed the highest production of amine from the hydrogenation reaction at 68% at 105 °C, 40 bar total pressure and 4 h reaction time at 0.08 mol% catalyst loading, achieved with the trifluoromethyl substituted complex (3).The hydroaminomethylation model reaction carried out with cyclohexene and n-propylamine as substrates was further optimised by altering the partial pressure of CO relative to H2 in the syngas mixture. The data indicated the importance of the CO:H2 ratio on both the hydroformylation and hydrogenation capabilities, culminating in the highest amine production at a 2:3 ratio of CO:H2 with a total pressure of 50 bar. The evaluation of aliphatic primary and secondary amines showed good conversion, with low conversion of aromatic amines or ammonium salts to the target products. The olefin substrate scope was varied (styrene, cyclohex-2-en-1-one, cyclohex-2-en-1-ol, 1-octene and 1,2-diphenylethylene) and showing moderate good applicability and selectivity except in the reaction with cyclohex-2-en-1-one, which resulted in the formation of bis-substituted amine and enamine product mixtures. The application of the hydroaminomethylation reaction was then extended toward the synthesis of two analogues of Tramadol®, using the hydroaminomethylation reaction as a key step. The hydroaminomethylation of the synthesized 3,4-dihydro-[1,1′-biphenyl]-1(2H)-ol substrate with piperidine or dibenzylamine resulting in the target compounds formed in moderate (74%) and low (36%) yields respectively, with distributions in the formed regioisomers correlated to a combination of steric influence of the phenyl substituent and the presence of the hydroxyl group on the substrate.
Data availability
After publication, the data will be made available in the public domain as ESI.† The work emanating from the results from graduated students will be included in dissertations or theses, deposited and made available through OpenUCT. OpenUCT is the open access institutional repository of the University of Cape Town (UCT) that makes available and digitally preserves the scholarly outputs produces at UCT.Author contributions
Stephen de Doncker: investigation, writing – original draft, data analysis, curation, and validation. Gregory S. Smith: conceptualization, co-supervision, funding acquisition, writing – review and editing, resources. Siyabonga Ngubane: conceptualization, project management, funding acquisition, resources, writing – review and editing, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is based on the research supported in part by the National Research Foundation of South Africa (UID: 138086). Financial support from the University of Cape Town (UCT), Sasol Group Technology, the Department of Science and Technology of South Africa Centre of Excellence in Catalysis – c*change is gratefully acknowledged.References
- M. Pelckmans, T. Renders, S. Van de Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS.
- F. Albericio, M. Álvarez, C. Cuevas, A. Francesch, D. Pla and J. Tulla-Puche, in Molecular imaging for integrated medical therapy and drug development, Springer, Boston, MA, 2010, p. 237 Search PubMed.
- S. Chen, G. Ravindran, Q. Zhang, S. Kisely and D. Siskind, CNS Drugs, 2019, 33, 225–238 CrossRef PubMed.
- H. Liu, D. L. Wang, X. Chen, Y. Lu, X. L. Zhao and Y. Liu, Green Chem., 2017, 19, 1109–1116 RSC.
- S. Shekhar, P. Ryberg, J. F. Hartwig, J. S. Mathew, D. G. Blackmond, E. R. Strieter and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 3584–3591 CrossRef CAS PubMed.
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 4111–4122 CrossRef CAS PubMed.
- D. Ma, Y. Zhang, J. Yao, S. Wu and F. Tao, J. Am. Chem. Soc., 1998, 120, 12459–12467 CrossRef CAS.
- S. Tivari, P. K. Singh, P. P. Singh and V. Srivastava, RSC Adv., 2022, 12, 35221–35226 RSC.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- K. Natte, H. Neumann, R. V. Jagadeesh and M. Beller, Nat. Commun., 2017, 8, 1344 CrossRef PubMed.
- P. M. Edwards and L. L. Shafer, Chem. Commun., 2018, 54, 12543–12560 RSC.
- R. C. DiPucchio, S. Rosca and L. L. Schafer, J. Am. Chem. Soc., 2022, 144, 11459–11481 CrossRef CAS PubMed.
- H. Elsen, C. Färber, G. Ballmann and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 7156–7160 CrossRef CAS PubMed.
- W. Reppe and H. Vetter, Ann. Chem., 1953, 582, 133–163 CAS.
- M. J. Schneider, M. Lijewski, R. Woelfel, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2013, 52, 6996–6999 CrossRef CAS PubMed.
- P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed.
- E. A. Karakhanov and A. L. Maksimov, Russ. J. Gen. Chem., 2009, 79, 1370–1383 CrossRef CAS.
- S. Siangwata, N. J. Goosen and G. S. Smith, Appl. Catal., A, 2020, 603, 117736 CrossRef CAS.
- C. Qian, Q. Zheng, J. Chen, B. Tu and T. Tu, Green Chem., 2003, 25, 1368–1379 RSC.
- F. Migliorini, E. Monciatti, G. Romagnoli, M. L. Parisi, J. Taubert, M. Vogt, R. Langer and E. Petricci, ACS Catal., 2023, 13, 2702–2714 CrossRef CAS.
- H. Liu, D. Yang, Y. Yao, Y. Xu, H. Shang and X. Lin, Mol. Catal., 2020, 484, 1–8 Search PubMed.
- B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2372–2375 CrossRef CAS PubMed.
- A. P. Alonso, D. Pham Minh, D. Pla and M. Gómez, ChemCatChem, 2023, 15(13), 1867–3880 Search PubMed.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363–375 CrossRef CAS.
- Y. Sun, M. Ahmed, R. Jackstell, M. Beller and W. R. Thiel, Organometallics, 2004, 23, 5260–5267 CrossRef CAS.
- J. Meng, X. H. Li and Z. Y. Han, Org. Lett., 2017, 19, 1076–1079 CrossRef CAS PubMed.
- L. Wu, I. Fleischer, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2013, 135, 3989–3996 CrossRef CAS PubMed.
- K. U. Künnemann, D. Weber, C. Becquet, S. Tilloy, E. Monflier, T. Seidensticker and D. Vogt, ACS Sustainable Chem. Eng., 2021, 9, 273–283 CrossRef.
- T. Roth, R. Evertz, N. Kopplin, S. Tilloy, E. Monflier, D. Vogt and T. Seidensticker, Green Chem., 2023, 25, 3680–3691 RSC.
- A. Serrano-Maldonado, T. Dang-Bao, I. Favier, I. Guerrero-Ríos, D. Pla and M. Gómez, Chem. – Eur. J., 2020, 26, 12553 CrossRef CAS PubMed.
- X.-C. Chen, T. Lan, K.-C. Zhao, L. Guo, Y. Lu and Y. Liu, J. Catal., 2022, 411, 158–166 CrossRef CAS.
- S. Li, K. Huang, J. Zhang, W. Wu and X. Zhang, Org. Lett., 2013, 15, 3078–3081 CrossRef CAS PubMed.
- Z. Nairoukh and J. Blum, J. Org. Chem., 2014, 79, 2397–2403 CrossRef CAS PubMed.
- T. O. Vieira and H. Alper, Chem. Commun., 2007, 2710–2711 RSC.
- J. October and S. F. Mapolie, Catal. Lett., 2020, 150, 998–1010 CrossRef CAS.
- J. October and S. F. Mapolie, Tetrahedron Lett., 2021, 70, 153018 CrossRef CAS.
- R. Wu, Z. Xu, D. Zhu and S. Zhu, Acc. Chem. Res., 2025, 58, 799–811 CrossRef CAS PubMed.
- R. Wu, D. Zhu and S. Zhu, Org. Chem. Front., 2023, 10, 2849–2878 RSC.
- R. Hrdina, Eur. J. Inorg. Chem., 2021, 501–528 CrossRef CAS.
- K. W. Fiori, C. G. Espino, B. H. Brodsky and J. A. Du Bois, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS.
- D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 7558–7559 CrossRef CAS PubMed.
- N. D. Chiappini, J. B. C. Mack and J. Du Bois, Angew. Chem., 2018, 130, 5050–5053 CrossRef.
- J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Science, 2014, 343, 6–65 CrossRef PubMed.
- S. de Doncker, G. S. Smith and S. Ngubane, Appl. Catal., A, 2023, 667, 119440 CrossRef CAS.
- C. L. Bumgardner, E. L. Lawton and J. G. Carver, J. Org. Chem., 1972, 37, 407–409 CrossRef CAS.
- W. Zhang, P. C. Zhang, Y. L. Li, H. H. Wu and J. Zhang, J. Am. Chem. Soc., 2022, 144, 19627–19634 CrossRef CAS PubMed.
- S. de Doncker, A. Casimiro, I. A. Kotze, S. Ngubane and G. S. Smith, Inorg. Chem., 2020, 59, 12928–12940 CrossRef CAS PubMed.
- D. Giffard, E. Fischer-Fodor, C. Vlad, P. Achimas-Cadariu and G. S. Smith, Eur. J. Med. Chem., 2018, 157, 773–781 CrossRef CAS PubMed.
- R. Pich, S. Mallet-Ladeira, P. Lavedan, J. Lahitte, J. Remigy, D. Pla and M. Gómez, Adv. Synth. Catal., 2024, 366, 822–829 CrossRef CAS.
- E. B. Maxted and M. S. Biggs, J. Chem. Soc., 1957, 3844–3847 RSC.
- P. Marcazzan, B. O. Patrick and B. R. James, Organometallics, 2003, 22, 1177–1179 CrossRef CAS.
- F. C. Braga, T. O. Ramos, T. J. Brocksom and K. T. de Oliveira, Org. Lett., 2022, 42, 8331–8336 CrossRef PubMed.
- A. F. M. Iqbal, Helv. Chim. Acta, 1971, 54, 1440–1445 CrossRef CAS.
- L. Jiang, P. Zhou, Z. Zhang, Q. Chi and S. Jin, New J. Chem., 2017, 41, 11991–11997 RSC.
- R. H. K. Foster and A. J. Carman, J. Pharmacol. Exp. Ther., 1947, 91, 195–209 CrossRef CAS PubMed.
- J. An, Z. Gao, Y. Wang, Z. Zhang, J. Zhang, L. Li, B. Tang and F. Wang, Green Chem., 2021, 23, 2722–2728 RSC.
- A. M. Seayad, K. Selvakumar, M. Ahmed and M. Beller, Tetrahedron Lett., 2003, 44, 1679–1683 CrossRef CAS.
- Q. Zhang, J. F. Li, G. H. Tian, R. X. Zhang, J. Sun, J. Suo, X. Feng, D. Fang, X. R. Jiang and J. S. Shen, Tetrahedron: Asymmetry, 2012, 23, 577–582 CrossRef CAS.
- C. Alvarado, Á. Guzmán, E. Díaz and R. Patiño, J. Mex. Chem. Soc., 2005, 49, 324–327 CAS.
- M. Ahmed, C. Buch, L. Routaboul, R. Jackstell, H. Klein, A. Spannenberg and M. Beller, Chem. – Eur. J., 2007, 13, 1594–1601 CrossRef CAS PubMed.
- O. R. Thiel, C. Bernard, W. Tormos, A. Brewin, S. Hirotani, K. Murakami, K. Saito, R. D. Larsen, M. J. Martinelli and P. J. Reider, Tetrahedron Lett., 2008, 49, 13–15 CrossRef CAS.
- E. T. Jarvi, N. A. Grayson and R. E. Halvachs, Synthesis and Purification of (r*,r*)-2-[(dimethylamino) Methyl]-1-(3-methoxyphenyl) Cyclohexanol Hydrochloride, US6399829B1, 2002.
- D. Seyferth, Organometallics, 2009, 28, 1598–1605 CrossRef CAS.
- R. M. Peltzer, J. Gauss, O. Eisenstein and M. Cascella, J. Am. Chem. Soc., 2020, 142, 2984–2994 CrossRef CAS PubMed.
- A. L. Morrison and H. Rinderknecht, J. Chem. Soc., 1950, 1510–1513 RSC.
- T. S. Teets, T. R. Cook, B. D. McCarthy and D. G. Nocera, Inorg. Chem., 2011, 50, 5223–5233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00630a |
| This journal is © The Royal Society of Chemistry 2025 |