
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReprogramming pretrained language models for protein sequence representation learning†
Ria Vinod‡
a,
Pin-Yu Chen *b and
Payel Das*b
*b and
Payel Das*b
aDepartment of Computational and Molecular Biology, Brown University, USA. E-mail: ria.vinod@brown.edu
bIBM Research, USA. E-mail: pin-yu.chen@ibm.com; daspa@us.ibm.com
First published on 23rd May 2025
Abstract
Machine learning-guided solutions for protein learning tasks have made significant headway in recent years. However, success in scientific discovery tasks is limited by the accessibility of well-defined and labeled in-domain data. To tackle the low-data constraint, recent adaptions of deep learning models pretrained on millions of protein sequences have shown promise; however, the construction of such domain-specific large-scale models is computationally expensive. Herein, we propose representation reprogramming via dictionary learning (R2DL), an end-to-end representation learning framework in which we reprogram deep models for alternate-domain tasks that can perform well on protein property prediction with significantly fewer training samples. R2DL reprograms a pretrained English language model to learn the embeddings of protein sequences, by learning a sparse linear mapping between English and protein sequence vocabulary embeddings. Our model can attain better accuracy and significantly improve the data efficiency by up to 104 times over the baselines set by pretrained and standard supervised methods. To this end, we reprogram several recent state-of-the-art pretrained English language classification models (BERT, TinyBERT, T5, and roBERTa) and benchmark on a set of protein physicochemical prediction tasks (secondary structure, stability, homology, and solubility) as well as on a biomedically relevant set of protein function prediction tasks (antimicrobial, toxicity, antibody affinity, and protein–protein interaction).
Introduction
Recent advances in artificial intelligence (AI), particularly in deep learning, have led to major innovations and advances in many scientific domains, including biology. These deep learning models aim to learn a highly accurate and compressed representation of the biological system, which can be employed for a range of tasks. These include high-quality protein structure prediction from protein sequences,1–4 molecule optimization,5,6 and accurate prediction of protein properties7 to enabling novel and functional peptide discoveries.8,9 Many of these advances rely on developing deep learning models3,10,11 which are trained from scratch on massive amounts (on the order of billions of tokens) of data. However, labeled data in biology is scarce and sparse, which is also the case for many other real-world scenarios in the scientific domain. In the biological domain, label annotation can involve biological assays, high-resolution imaging, and spectroscopy, which are all costly and time-consuming processes.The technique of pretraining deep learning models was proposed to address this issue. Pretraining methods leverage large amounts of sequence data and can learn to encode features that can explain the variance seen in sequences across biological task-specific training samples. In the context of protein sequences, pretraining has enabled meaningful density modeling across protein functions, structures, and families.12 In this work, we reference two types of pretraining methods: (i) unsupervised pretraining, where all data are unlabeled, and (ii) self-supervised pretraining, where a model learns to assign labels to its unlabeled data. Large models then pretrain on massive amounts of unlabeled data, specifically biological sequences, which are available at scale. Once pretrained, these foundation models (FMs)13 are fine-tuned on smaller amounts of labeled data, which correspond to a specific downstream task. Interestingly, for the large-scale models pretrained on protein sequences, biological structure and function seem to emerge in the learned protein representation, even though such information was not included in model training.10
Although highly powerful, the training of those domain-specific foundation models from scratch is highly resource-intensive.14 For example, one training run of BERT (the language model considered in this work) learns 110 million parameters, costs up to $13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 USD and takes 64 days (without parallelized computing) and results in 0.7 tons of carbon emissions.15 A single training run of another popular language model, the T5 transformer, learns 11 billion parameters, costs up to $1.3 million USD, takes 20 days, and results in 47 tons of carbon emissions.16,17 Such pretrained language models and size variants are abundantly available with the advent of model libraries (e.g., Hugging Face18) which host pretrained models and datasets. The scale of data, computing, and financial resources required to train these models is not only available to a limited number of researchers but also infeasible for applications with limited labeled data. To this end, we propose a lightweight and more accurate method for large-scale pretraining from alternative domains. Specifically, we introduce a method to reprogram open-sourced, classification foundation models of high capacity that are trained on data from a different domain. This situation calls for innovations in cross-domain transfer learning, which is largely unexplored, particularly in scientific domains.
000 USD and takes 64 days (without parallelized computing) and results in 0.7 tons of carbon emissions.15 A single training run of another popular language model, the T5 transformer, learns 11 billion parameters, costs up to $1.3 million USD, takes 20 days, and results in 47 tons of carbon emissions.16,17 Such pretrained language models and size variants are abundantly available with the advent of model libraries (e.g., Hugging Face18) which host pretrained models and datasets. The scale of data, computing, and financial resources required to train these models is not only available to a limited number of researchers but also infeasible for applications with limited labeled data. To this end, we propose a lightweight and more accurate method for large-scale pretraining from alternative domains. Specifically, we introduce a method to reprogram open-sourced, classification foundation models of high capacity that are trained on data from a different domain. This situation calls for innovations in cross-domain transfer learning, which is largely unexplored, particularly in scientific domains.
One known fact is that biological sequences are similar to natural language, as they also contain long-range dependencies and follow Zipf's law.19 These sequences and their associated dependencies are crucial for determining their structural and functional properties. Such similarity has motivated the use of deep learning architectures and mechanisms that are widely used in natural language processing (NLP) to build protein sequence models from scratch. Work along this line of inquiry consists of training models jointly on protein and text sequences,20–22 by minimizing a contrastive loss on the representation of text obtained from an English language encoder and the representation of protein sequences obtained from an amino-acid encoder. However, this style of work relies on the availability of high-performance in-domain protein language models. In this work, we remove any reliance on pretrained protein models by exploring an alternative warm-start paradigm, i.e. how to effectively and efficiently reprogram an existing, fully trained large English language model to learn a meaningful (i.e., biomedically relevant) representation of protein sequences. Our proposal can attain better data efficiency because it only requires in-domain task-specific labeled data and spares the need for accessing in-domain unlabeled data for pretraining. The goal is to create a more carbon-friendly, resource-efficient, and broadly accessible framework to motivate different scientific domains toward democratizing the representation power of large AI models. This warm-start paradigm is defined by the framework's ability to achieve the performance of transformers that are pretrained on billions of tokens in alternative domains, with a lighter-weight training procedure that is similar to that of a standard supervised classifier trained from scratch. In particular, we consider highly specific biological and biomedical protein sequence datasets (illustrated in Fig. 1) which have much fewer samples than standard supervised language task datasets. Reprogramming thus provides a more data and resource-efficient approach to developing models to achieve deep representational capacity and performance for downstream protein tasks. Reprogramming has been previously explored as a cross-domain transfer learning methdology,26,27 reprogramming language models were explored for alternate text classification tasks,28 reprogrammed acoustic models were explored for time series classification,29 and reprogrammed ImageNet classification models were explored for alternate image classification tasks. However, none of these methods investigated mappings between domains that require a very high representational capacity (from natural language to biological sequence), which is the setting we require in the protein sequence domain.
 | ||
| Fig. 1 (Left) Descriptions of considered predictive tasks. We select the set of physicochemical property prediction tasks from the well-studied domains in ref. 11 and the biomedical function prediction tasks from studies with biomedically relevant small-sized labeled datasets.9,23,24 (Center) Comparison of R2DL to in-domain pretraining and standard supervised training methods. We refer to supervised methods as standard supervised classifiers that are trained from scratch from labeled data alone. Depending on how labeled and unlabeled data are used in pretraining, we consider in-domain pretraining to constitute unsupervised/supervised pretaining schemes using in-domain data. (Right) A comparative table showing the broad adaptability of the R2DL framework. In comparison to the existing gold standard methods, R2DL has a broader utility across different domains, sizes of training datasets, and data efficiency. We categorize supervised methods as cross-domain adaptable through various domain adaptation and transfer learning techniques.25 | ||
Toward this goal, we introduce R2DL (representation reprogramming via dictionary learning), a novel cross-domain transfer learning framework to reprogram an existing pretrained large-scale deep-learning model of the English language, namely an English language model such as BERT,15 to learn and predict physicochemical and biomedical properties of protein sequences. The success of pretraining in language models (LMs)15,17,30,31 is drawn from the principles of fine-tuning and in-context learning. With models of a large enough size, several studies have demonstrated emergent properties in language models.32 That is, knowledge about the language task emerges in learned representations of the models, even when there are very few in-context examples.30 Protein language models (PLMs) have demonstrated similar emergent properties, where learned representations exhibit knowledge about the structure and function of proteins, even when this information was not included in the training data. The success of LMs in the protein sequence domain can be attributed to the similarities in the structure of the modes of English and protein sequence data. Recent work demonstrates that there appear similarities between English and protein domain grammar.24 As language follows linguistic rules, referred to as “grammar”, only a subset of protein domains is viable as per evolution. These domains are determined by protein sequences, which are thus governed by some evolutionary rules, or “proteome grammar”. Recent work formalizes this concept, showing that with a linguistic probe analysis, English language (text sequences) and amino acid (protein sequences) both follow a Zipfian distribution and confirm the presence of a quasi-universal grammar.24 Under this paradigm, R2DL emerges as an intuitive method for cross-modal learning between the language and protein domains.
In step 3, the system maps the source task labels (e.g., positive/negative sentiments) to target task labels (e.g., toxic/non-toxic proteins) and optimizes the embedding mapping parameters based on the task-specific loss evaluation on a given protein sequence dataset. Finally, in step 4, the reprogrammed model is deployed for the test-time evaluation.
To the best of our knowledge, our work remains the first to address reprogramming in any biological, and more broadly, scientific domain. We posit reprogramming as an alternative to fine-tuning, as a method in which significantly less in-domain training data are required to achieve the same predictive performance across protein tasks. Further details on the model-data paradigm of fine-tuning versus reprogramming are provided in the ESI (Appendix 1†). In Fig. 1, we illustrate the set of protein physicochemical and functional property prediction tasks we consider, as well as the baseline methods against which we compare the R2DL performance to, and a brief description of R2DL's advantages compared to these existing methods. We test the reprogrammed model for a range of biomedically relevant downstream physicochemical property, structure and function prediction tasks, which include prediction of the secondary structure, homology, mutational stability, solubility, as well as antimicrobial nature, toxicity, antibody affinity, and protein–protein interaction of proteins. Each of these tasks involves learning on datasets that are limited to a few thousand labeled samples, at least an order of magnitude smaller needed to train an in-domain foundation model or a large in-domain protein language model.33 R2DL uses dictionary learning, a machine learning framework that finds the optimal sparse linear mapping between the English vocabulary embeddings and the amino acid embeddings. To do so, a protein property prediction task-specific loss is used to learn the optimal parameters of the reprogrammed model. We train R2DL in a supervised setting with the downstream protein prediction task datasets that are labeled and small in size (illustrated in Fig. 1). R2DL demonstrates consistent performance improvement from existing baselines across seven different physicochemical (e.g., up to 11% in stability), structural, and functional property prediction (e.g., up to 3% in toxicity) tasks of proteins. We estimate R2DL to be over 104 times more data-efficient than existing in-domain pretraining methods. We further demonstrate the performance robustness of R2DL when trained on a reduced-size version of the supervised protein datasets. In addition, we show that R2DL learns to encode physicochemical and biomedical properties in the learned representations, even in limited-data scenarios. This work thus blazes a path toward efficient and large-scale adaptation of existing foundation models toward different real-world learning tasks and accelerates scientific discovery, which naturally involves learning from limited real-world data.
Results
Fig. 2 illustrates the proposed representation reprogramming via dictionary learning (R2DL) framework, which learns to embed a protein sequence dataset of interest by training on the representations of a transformer-based model that is pretrained on an English text corpus. A one-to-one label mapping function is assigned for each downstream protein prediction task for cross-domain machine learning, and a class label or a regression value is predicted using R2DL for each protein sequence during testing. Below, we discuss details of the general framework (tasks described in Fig. 1). | ||
| Fig. 2 System illustration of the representation reprogramming via dictionary learning (R2DL) framework. In step 1, R2DL loads a pretrained language model (source), obtains the source vocabulary embeddings, and specifies the protein tokens (target). In step 2, R2DL learns a sparse linear mapping between the source and target embeddings via dictionary learning, to represent a target token embedding as a sparse linear combination of source token embeddings. This is done via a k-SVD approximation of the dictionary weights, Θ (see the Method section for more details). | ||
R2DL framework formulation
The objective of R2DL is to reprogram a source model (here, a pretrained English language model) to be able to correctly classify, or predict the regression values of, protein sequences (for a target prediction task). We primarily use pretrained instances of BERT, a bidirectional transformer (termed the source model), which has been fine-tuned separately for different language tasks (e.g., sentiment classification, named entity recognition).15,34 We also study the effect of different source models presented in the ESI (Appendix 7†).For a protein sequence classification task, we use the source model trained on an alternative-domain language task for which there are ns sentence output classes (e.g., “positive” and “negative” for sentiment classification) and nt protein sequence classes (e.g., “toxic” and “non-toxic”), where ns ≥ nt. The output-label mapping h is then a simple one-to-one surjective correspondence between the source task labels and the target task labels, which ensures that every target class is assigned with a unique source class (e.g., positive → toxic and negative → non-toxic). For a regression task, R2DL uses a mapping between the regression values in the protein sequence feature space and the classification probability values in the source model embedding space. It does so by learning optimal thresholds of regression values that map to the source model class labels. The input data of the source English language model are tokenized at the word level. These tokens form the atoms for our dictionary representation of VS, a matrix with its rows corresponding to embedding vectors of source tokens. The input data to the target task, protein sequences, are tokenized on a character level with only 20 distinct tokens (corresponding to a set of 20 discrete natural amino acid characters). R2DL obtains VS from the learned embeddings of the source model and learns to represent VT, the matrix of the target token embedding, as a weighted combination of the English token embeddings. We propose token reprogramming by approximating a linear mapping between VS and VT. That is, we aim to find a transformation of the latent representation of the protein sequences, such that it can be embedded in the pretrained language model's latent space and enable R2DL to leverage these re-embedded tokens for learning. Specifically, we learn the linear map Θ by approximating a dictionary using a k-SVD solver.35 That is, we want to approximate VT = ΘVS. The k-SVD solver guarantees a task-specific level of sparsity in the coefficients when linearly combining English token embeddings to represent a protein sequence token embedding. In other words, it helps select k English tokens and use their linearly combined embeddings as the embedding of a target token. Additionally, with a one-to-one label mapping function of the English text label to the protein sequence label, we are able to use the pretrained language model for inference on the embedded protein dataset, VT. We thus design an end-to-end reprogramming framework for any arbitrary protein sequence classification or regression task.
R2DL training and optimization procedure
We are given a pretrained classifier C (which has been pretrained on a source-task dataset with source tokens denoted as and a target-task dataset with target tokes denoted as
and a target-task dataset with target tokes denoted as  ). The embedding matrices are VS and VT, respectively. We can encode an output label mapping function translating between source and target labels. In Fig. 2, we show how R2DL aims to find a linear mapping function Θ that learns the optimal coefficients for our atoms in VT to be represented as a sparse encoding of the dictionary VS such that VT = ΘVS. The map Θ is used to reprogram C to be able to correctly classify the protein sequences through the transformation h(C(θt, t)) where t is a protein sequence from a protein task and θt is the linear weights associated with the protein sequence t in Θ. We note that for each of the downstream protein property prediction task, R2DL only trains a corresponding token mapping function Θ while keeping the pretrained classifier C intact. Therefore, the number of trainable parameters in R2DL is simply the size of the matrix Θ, which is usually much smaller compared to the number of parameters in the pretrained deep neural network classifier C.
). The embedding matrices are VS and VT, respectively. We can encode an output label mapping function translating between source and target labels. In Fig. 2, we show how R2DL aims to find a linear mapping function Θ that learns the optimal coefficients for our atoms in VT to be represented as a sparse encoding of the dictionary VS such that VT = ΘVS. The map Θ is used to reprogram C to be able to correctly classify the protein sequences through the transformation h(C(θt, t)) where t is a protein sequence from a protein task and θt is the linear weights associated with the protein sequence t in Θ. We note that for each of the downstream protein property prediction task, R2DL only trains a corresponding token mapping function Θ while keeping the pretrained classifier C intact. Therefore, the number of trainable parameters in R2DL is simply the size of the matrix Θ, which is usually much smaller compared to the number of parameters in the pretrained deep neural network classifier C.
To approximate the dictionary, we use a k-SVD solver to optimize model weights with the cross entropy loss for updates to Θ. The critical settings that determine the approximate convergence of the k-SVD algorithm are the initialization of the dictionary and the choice of k, the number of non-zero coefficients. We initialize the dictionary with the results of k-means clustering on VS, a common approach. To adequately capture the underlying data distribution, we perform a grid search to identify the best choice of k. For each task-specific instance of R2DL, we train our model for the number of k-SVD iterations until convergence. The number of iterations used for each model can be found in Appendix 4 (ESI†).
We then apply the assigned label mapping h for protein classification tasks, or thresholding for regression tasks, and train the mapping function Θ using gradient-based optimization evaluated on the task-specific cross-entropy loss. Details for the R2DL training procedure are given in the Method section.
Benchmark tasks and evaluation
We consider four physicochemical structure and property prediction tasks from a well-established protein benchmark from ref. 11 (represented in Fig. 1). Secondary structure prediction involves predicting the secondary structure y ∈ {helix, strand, other} for each amino acid x in a given protein sequence. Solubility prediction considers mapping an input protein sequence x to a label of y ∈ {membrane-bound, water soluble}. Homology detection is a sequence classification task, where each input protein x is mapped to a label y ∈ {1,…, 1195}, representing different possible protein folds. Stability prediction is a regression task. We further consider 4 biomedically relevant function prediction tasks, which are sequence classification tasks (represented in Fig. 1). Using R2DL, we predict for a given sequence x, its binary class label y ∈ {AMP, non-AMP} for antimicrobial-nature prediction9 or y ∈ {toxic, non-toxic} for toxicity prediction.9 We also predict antigen and non-specific binding of antibody variant sequences from ref. 23: given a sequence x, the task is to predict y ∈ {on-target, off-target}. Finally, we predict protein–protein interactions: given a pair of amino acid sequences, x, x′ from the benchmark dataset in ref. 24, for each residue, we want to predict y ∈ {interaction, no interaction}. Further details on the protein tasks and datasets are in the Method section. The sizes of the individual datasets vary between 1000 and 50000 (see the ESI† for details on data sizes and train-test splits). Data efficiency is defined as the ratio of the R2DL prediction accuracy to the number of biological sequences used during pretraining and fine-tuning. We use data efficiency as a metric to compare the performance of R2DL to established benchmarks for the protein tasks in ref. 9, 11 and 23. For classification tasks, we evaluate the prediction accuracy with a top-1 accuracy, where each task has [1, n] possible classes, where n is the number of classes in the protein sequence classification task. For regression tasks, we evaluate prediction accuracy with Spearman's correlation. We conducted our experiments for 5 independent runs and reported the average performance. The standard deviation is reported in the ESI (Appendix 7†). The train and test splits that we train R2DL on are the same splits used by the original task-specific baseline model and are commonly used in benchmarking for protein sequence models. We do not introduce any new datasets. This allows for fair comparison to the protein language model benchmarks. Additionally, since R2DL does not use the protein sequence data for pretraining, there is less risk of data contamination, leakage, or bias, as compared to large-scale in-domain pretraining methods. We report R2DL performance compared to a simple supervised baseline of an LSTM trained from scratch on the task-specific protein sequence datasets and more complex unsupervised baselines of large-scale pretrained protein language models.
Model baselines and data
The baseline models we consider in this work are of two types. Firstly, we consider a simple baseline – models trained in a supervised manner – by training standard sequence long-range short-term memory (LSTM) models from scratch. For each downstream peptide or protein classification task, we have labeled (supervised) datasets. The results of these models are reported in Fig. 3(a). Secondly, we consider a complex baseline – state-of-the-art models that are pretrained in an unsupervised manner – on protein sequence data and fine-tuned for a particular downstream protein task. These baselines consist of reported performance in the relevant work. We report the performance of the complex baselines in Table S21,† which consist of the widely cited models such as ESM1b10 and TAPE,11 pretrained on all of Pfam.36 We adopt ESM1b37 as the unified model, which is pretrained on in-domain data of protein sequences and fine-tuned on each downstream task. This model was benchmarked against and outperforms several other baselines, such as UniRep38 [Review 1.1.5]. Simpler baselines such as one hot encoding and alignment, naive transformer, and deep learning approaches39 are established in the TAPE leaderboard11 and reported in Table S3† for ease of reference. This framing parallels how R2DL is a unified framework fine-tuned on in-domain data samples. The choice of the source model that corresponds to the best performing reprogrammed R2DL instance for each task is reported in Table S1.† Pretraining methods that do not use labeled data pose an advantage, as those models can then learn from a significantly larger number of data samples. In the cases of toxicity and antimicrobial prediction tasks, the baseline model we compare to has been pretrained on a subset of UniProt database where sequences are limited to being 50 residues long.40 The pretraining corpus size is then 1.7 million peptide sequences. Using unlabeled data for pretraining is thus much more advantageous than pretraining in a supervised scheme. Of these 1.7 million sequences, only 9000 are labeled (0.005% of sequences). The model is a Wasserstein Autoencoder, which is a generative model that undergoes unsupervised pretraining on the subset of UniProt data. The WAE embeddings of the labeled sequences are then used to train a logistic regressor model on the labeled dataset to obtain a binary classifier for antimicrobial/non-antimicrobial (6489 labeled samples) or for toxic/non-toxic (8153 labeled samples) label prediction. For the physicochemical property prediction tasks, the baseline model we consider is pretrained on the Pfam corpus.36 This corpus consists of 31 million protein domains and is widely used in bioinformatics pipelines. Sequences are grouped by protein families which are categorized by evolutionarily related sequences. In contrast, the downstream physicochemical tasks of structure, homology, stability, and solubility prediction have labeled datasets that range from 5000 to 50000 samples which the model can be fine-tuned on. In-domain pretraining thus poses the advantage of modeling the density over a range of protein families and structures but stipulates that there must be sequence datasets that contain structural and functional information about the downstream task datasets and typically be of a size on the order of millions of sequences. R2DL eliminates this requirement by repurposing existing pretrained English language models and leveraging transferrable information from models that are not conditioned on protein sequence information. We also find that without R2DL, standard fine-tuning of a pretrained English language model for the considered protein sequence learning tasks does not lead to competitive results, which demonstrate the importance of R2DL in cross-domain learning. Please refer to Appendix 7 in the ESI† for more details.
 | ||
| Fig. 3 (a) Prediction accuracy and (b) data efficiency of R2DL, pretraining methods, and standard supervised training methods (trained from scratch). The prediction accuracy is the top-1 accuracy for each task-specific instance of R2DL. Each downstream task has a different number of classes, n, for the n-way classification tasks. Data efficiency is defined as the ratio of prediction accuracy to the number of in-domain samples used in training (including pretraining and fine-tuning methods). R2DL attains comparable accuracy to in-domain pretraining methods while attaining high data efficiency. Error bar values and variance are reported in Table S19.† Model and training details on the reported performance of each downstream task are available in the ESI.† | ||
Data efficiency and accuracy of reprogramming
We report the performance of R2DL for a set of 8 protein predictive tasks and their corresponding baselines in Fig. 3. We benchmark R2DL performance with the choice of the pretrained source model that results in the best downstream task performance. We provide a summary of the results of different source model choices in Appendix 7 of the ESI.† Baselines for the physicochemical prediction tasks are established by a transformer from ref. 11 that has been pretrained in an unsupervised setting on the Pfam pretraining corpus.41 Baselines for the antimicrobial and toxicity prediction tasks are established in ref. 9, where Das et al. pretrained a Wasserstein Autoencoder on the peptides from the UniProt corpus40 using unsupervised training and then used the latent encodings from an autoencoder to train the property classifiers. Baselines for the antibody affinity task are established in ref. 23 where they train a linear discriminant analysis model in a supervised setting. Baselines for the protein–protein interaction task are established by ref. 24, wherein ProtT5 descriptors42 are used to train a predictive network. Each physicochemical and biomedical function prediction task then has a relatively small, supervised dataset which we split into training and testing sets to train the R2DL framework and evaluate its performance on the test set. Henceforth, we refer to these baselines as task-specific baselines, whereas the baseline model we compare R2DL to varies with the downstream protein prediction task and the best-performing model available (see the ESI† for details on task-specific baselines). We show that, for 6 out of 8 tasks, we achieve a higher (or similar) test accuracy with R2DL than with the corresponding task-specific baseline model when both models are trained on the fully labeled dataset. R2DL shows performance improvement up to 11.2% when compared to the pretrained models and up to 29.3% performance when compared to a standard, supervised LSTM that is trained from scratch on the same dataset. However, R2DL needs a pretrained source model and only a small-sized, labeled protein sequence dataset as the input. Therefore, the size of the R2DL training set is limited to the number of samples in the downstream protein prediction dataset. On the other hand, in-domain pretrained models require a large amount of protein sequence data for pretraining, on the order of 106 samples, in addition to the downstream supervised protein task sequence data that the in-domain pretrained model is fine-tuned on. In Table S19 of the ESI,† we show the number of training samples and the corresponding accuracy metric (see the Method section for details) of the R2DL, pretrained, and supervised models. In Fig. 3(a) and (b), we compare their test accuracy and the data efficiency, i.e., the ratio of the downstream protein task accuracy to the number of in-domain training samples (including all labeled and unlabeled in-domain data used in pretaining and fine-tuning). We show that R2DL is a maximum of 104 times more data efficient, as in the case of the homology prediction task. This is due to the very large number of in-domain pretraining data samples required relative to the downstream protein task dataset. In Appendix 2 (ESI†), we also observe the improved performance of R2DL over baseline methods in the reduced-data training settings.Correlation between learned embeddings and evolutionary distances
Beyond comparing the R2DL model against the individual protein task benchmarks, we demonstrate that the R2DL framework shows interpretable correspondences between the learned embeddings in the latent space and the specific protein property. We show this result for the antibody affinity, secondary structure, and toxicity prediction tasks. Fig. 4(a)–(c) show the t-SNE projection of task-specific R2DL embeddings VT = ΘVS of protein sequences for secondary structure, toxicity, and antibody affinity prediction tasks. Clear separation between different protein classes is evident. | ||
| Fig. 4 Biologically relevant correlations learned by R2DL. (a–c) Clustering of R2DL learned embeddings for secondary structure prediction, toxicity prediction, and antibody affinity prediction tasks. When tagged by protein property classification, we see high correspondence between the cluster and protein sequences with the same physicochemical or biomedical property classification. Rives et al.37 shows that “biochemical properties of amino acids are represented in the Transformer model's output embeddings”. We use their result as ground truth annotations to draw parallels with our finding of token representations in R2DL embeddings – leucine (L) and proline (P) are both polar; cysteine (C) and methionine (M) are both polar; tryptophan (T) and isoleucine (I) are both hydrophobic. We find these biochemical similarities between amino acids consistent with our results. (d) For the antibody affinity prediction task, we observe a high correlation coefficient along the diagonal. This shows that the representation learned by R2DL is highly similar to empirical observations of pairwise residue correlations. See Appendix 7 (ESI†) for comparisons to in-domain pretraining. | ||
We further calculate the similarity in the Euclidean distance between the latent representations at the last layer for each amino acid embedding and compare it to the pairwise evolutionary distance with the BioPython module. In Fig. 4(d), we show the Euclidean distances between the latent embeddings learned in the R2DL model and the pairwise evolutionary distances between protein sequences, as estimated using the BLOSUM62 matrix implemented in the pairwise function of the BioPython module. The matrix shows a correlation of close to 1.0 along the diagonal, showing a perfect correspondence between the learned representation and the empirical observations of amino acid relatedness. R2DL thus captures the underlying structure of the linear sequence of amino acid residues in protein sequences in the context of the protein task reprogrammed.
Discussion and conclusion
We propose a new framework, R2DL, to reprogram large language models for various protein tasks. R2DL demonstrates powerful predictive performance across tasks that involve evolutionary understanding, structure prediction, property prediction and protein engineering. We thus provide a strong alternative to pretraining large language models on up to 106 protein sequences. With only a pretrained natural language model (which are abundantly available at the time of writing), a small-sized labeled protein data set of interest, and a small amount of cross-domain fine-tuning, we can achieve better performance for each protein prediction task with interpretable correspondences between features. Beyond improvements in predictive performance, we show that the ratio of performance improvements to pretraining and training samples involved in the R2DL framework makes R2DL up to 104 times more data-efficient than any current methods. This work opens many doors to biological prediction tasks that can acquire very few labeled, high quality data samples. We emphasize the results of the data-efficiency of R2DL, when applied to biomedically relevant protein predictions, which are critical to advancing scientific understanding and discovery but have been unsuccessful until now.While R2DL does make gradient updates in the framework, the data and resource requirements of the R2DL method are much lower than any unsupervised or self-supervised pretraining approach for protein sequence modeling. Though R2DL has the same data and resource requirements as any standard supervised training approach, R2DL demonstrates much higher task accuracy across a broad and diverse range of property prediction tasks. We claim that R2DL is able to do this because of efficient cross-domain model adaptation via reprogramming, which standard supervised models cannot achieve without an unjustifiably large number of parameters. R2DL is thus more efficient than the existing baseline models in the following aspects: (i) R2DL only requires a pretrained transformer (trained on English language data) and a small-sized, labeled protein sequence data set of interest. We do not make any updates to the pretrained model itself, unlike traditional transfer learning methods. Rather, we make updates to the R2DL model during a supervised training process that optimizes over class-mapped labels. (ii) R2DL does not require large-scale un/self-supervised pretraining on millions of unlabeled protein sequences, as in ref. 9–11. (iii) Furthermore, R2DL does not require any large-scale supervised pretraining, which has been found to be beneficial in protein-specific tasks11 as well as in computer vision.43 Labeling protein sequences at scale, particularly for biomedical functions, is almost infeasible for the size of dataset that is required for supervised pretraining. With these three considerations in mind, we pose R2DL as a data-efficient alternative to pretraining methods for protein prediction tasks of biological and biomedical relevance. To the best of our knowledge, R2DL is the first framework without explicit pretraining that facilitates accurate predictions across a general suite of protein prediction tasks and provides interpretable correspondences between amino acid features that are very closely aligned with domain knowledge (evolutionary distances). The success of R2DL can be attributed to its representational power to encode a sparse representation by leveraging the natural language modeling entailed in large language models for efficient learning on protein structure and function prediction tasks, as both English and protein sequences follow Zipf's law.19 We first demonstrate the effectiveness of R2DL on a set of physicochemical structure and property prediction tasks and then on a set of biomedically relevant function prediction tasks for protein sequences. We show predictive performance improvements against pretrained methods (up to 11% in stability) and standard supervised methods (up to 3.2% in antibody affinity). Similarly, on the remaining tasks, we show performance improvements over the best reported baseline in structure prediction (4.1%), homology (2.3%), solubility (7.1%), antibody affinity (3.2%), toxicity (2.4%), and PPI (1.6%). R2DL thus shows the capability to learn a general representation of protein sequences that can be efficiently adopted to different downstream protein tasks. These powerful representation capabilities are evidenced by its ability to achieve high performance across protein datasets with a highly varied number of task-specific training samples. The performance of R2DL across protein tasks show the potential to repurpose and develop powerful models that can learn from small, curated, and function-specific datasets. This mitigates the need to train large pretrained models for peptide learning tasks. We thus provide an alternative method to pretraining that is cheaper to run and more accurate and therefore adaptable to broader researcher communities who may not have access to large-scale computing. This potential is critical for many applications, such as the discovery of new materials, catalysts, as well as drugs.
Further exploration
Although we establish the efficacy and efficiency of R2DL in a domain where pretrained large language models already exist, we hope that our work will pave the path to extending this approach to other domains where pretrained LLMs do not exist, such as polymers. The ability to tokenize any sequence representation of domain knowledge offers the opportunity to reprogram English LMs for cross-domain transfer learning. Thus, R2DL is a promising initial tool for processing SMILES representations of molecules or any other structure information.The success of the R2DL reprogramming framework is dependent on the available pretrained English language transformers and protein sequence datasets. In this work, we consider several standard BERT-style transformers due to the open-sourced codebases available at the time. However, the field has seen many advancements in developing large language models that are highly performing across several benchmarks but are not open sourced. It is possible that adversarially reprogramming newer generative models such as the GPT family or Gemini could improve cross-domain transfer learning to protein sequence tasks. Additionally, k-SVD is an approximation of the dictionary weights, Θ. While we choose hyperparameters from the appearance of the convergence of the k-SVD algorithm, it is a computationally expensive method which is feasible on the order of 109 million parameters. Attempting to reprogram larger models with R2DL could require a more computationally efficient approximation algorithm.
We report the performance of the R2DL approach trained with different source natural language models in Table S20.† These results show that different vocabularies, distributions of training data, model size, and training regimes of the source model can affect the performance of R2DL. While we include a preliminary analysis of this question, we leave a more exhaustive study of the choice of source model study to future work.
Method
Representation of tokens
In the R2DL framework, we use 2 input datasets, an English language text dataset (source dataset) and a protein sequence dataset (target dataset). The vocabulary size of a protein sequence dataset at a unigram level is 20, as proteins are composed of 20 different natural amino acids. We obtain a latent representation of the English text vocabulary, VS, by extracting the learned embeddings of the data from a pretrained language model (source model). The protein sequence data are embedded in the same latent space and are termed the target vocabulary, VT. For each task, the token embedding matrix has dimensions (n, m), where n is the number of tokens and m is the length of the embedding vectors. We use the same encoding scheme of VS and VT across all downstream tasks.Procedure description of the R2DL framework for a protein task
• Procedure inputs: pretrained English sentence classifier C, target model training data for task
for task  class mapping label function
class mapping label function  (if classification), where
(if classification), where 
• Procedure hyperparameters: maximum number of iterations T1 for updates to Θ, number of iterations T2 for k-SVD, and step size 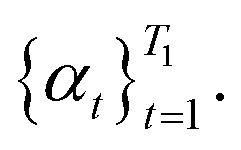
• Procedure initialization: random initialization of Θ, obtain the source token embedding matrix VS.
• Define objective function: objective function for k-SVD: ‖VT – ΘVS‖ ≤ ε.
• k-SVD approximation of Θ: If t1 ≤ T1, while t2 ≤ T2 use approximate k-SVD to solve VT ≈ ΘVS, t2 ← t2 + 1.
• Calculate the loss and perform gradient descent: 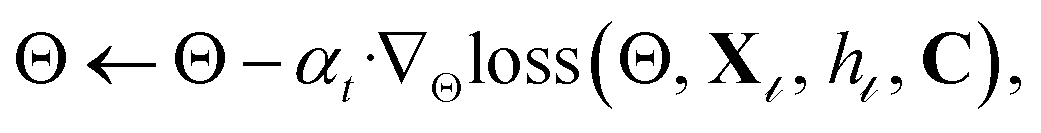 t1 ← t1 + 1 and return to the previous k-SVD step.
t1 ← t1 + 1 and return to the previous k-SVD step.
• Output protein sequence labels for protein sequence x of task 
We are given a pretrained English classifier, C, and a protein sequence target-task dataset  We denote the task with
We denote the task with  such that
such that  We also encode an output label mapping function
We also encode an output label mapping function  specifying the one-to-one correspondence between source and target labels. As shown in Fig. 2, the source vocabulary embedding, VS, is extracted from the pretrained model, C. The next objective is to learn Θ that approximates the embedding of tokens in
specifying the one-to-one correspondence between source and target labels. As shown in Fig. 2, the source vocabulary embedding, VS, is extracted from the pretrained model, C. The next objective is to learn Θ that approximates the embedding of tokens in 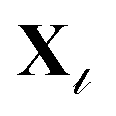 (denoted as VT) in the representation space of the source model. We aim to learn Θ ∈ Ra×b that finds the optimal coefficients {θt} for each of the target tokens t ∈ {1,…, a} in VT ∈ Ra×m to be represented as a sparse encoding of the dictionary, VS ∈ Rb×m, such that VT = ΘVS. For a given target protein sequence x from the
(denoted as VT) in the representation space of the source model. We aim to learn Θ ∈ Ra×b that finds the optimal coefficients {θt} for each of the target tokens t ∈ {1,…, a} in VT ∈ Ra×m to be represented as a sparse encoding of the dictionary, VS ∈ Rb×m, such that VT = ΘVS. For a given target protein sequence x from the  -th task, Θ is used to perform the target task through the transformation
-th task, Θ is used to perform the target task through the transformation  . While we do not make any modification to the parameters or architecture of C, we assume access to the gradient ∇Θloss(·) for loss evaluation and parameter updates during training.
. While we do not make any modification to the parameters or architecture of C, we assume access to the gradient ∇Θloss(·) for loss evaluation and parameter updates during training.
A target token embedding vt ∈ Rm can be represented as a sparse linear combination of the source token embeddings (rows) in VS, vt = θtVs. Here, vt is the representation of the protein token in the dictionary space and satisfies ‖vt − θtVs‖p ≤ ε, where ‖·‖p is an Lp norm and θt is made to be sparse by satisfying ‖θt‖0 ≤ k for all t. An exact solution vt = θtVS is computationally expensive to find and is subject to various convergence traps, so for the purpose of our efficient fine-tuning approach, we approximate vt ≈ θtVS using k-SVD. We first fix the dictionary VS, as extracted from C, and then find the optimal Θ according to the optimization problem, by minimizing the alternative objective  subject to ‖VT − ΘVS‖2F ≤ ε as explored in ref. 35. While algorithms exist to choose an optimal dictionary (an exact solution to k-SVD) that can be continually updated,35 we penalize computational expense over performance for the purpose of maintaining an efficient solution (at the cost of statistically insignificant improvements in accuracy) by using a predetermined number of iterations for k-SVD convergence, which is then used to evaluate the cross entropy loss on
subject to ‖VT − ΘVS‖2F ≤ ε as explored in ref. 35. While algorithms exist to choose an optimal dictionary (an exact solution to k-SVD) that can be continually updated,35 we penalize computational expense over performance for the purpose of maintaining an efficient solution (at the cost of statistically insignificant improvements in accuracy) by using a predetermined number of iterations for k-SVD convergence, which is then used to evaluate the cross entropy loss on  and update the mapping function Θ. The ESI† contains citations to ref. 44–51, including background materials for the relevant datasets and methods.
and update the mapping function Θ. The ESI† contains citations to ref. 44–51, including background materials for the relevant datasets and methods.
Data availability
In the ESI,† we have provided (1) a document for data and instructions and (2) a GitHub link https://github.com/riavinod/r2dl-proteins containing codes associated with this paper (DOI: 10.5281/zenodo.15262269). In the document, we have enumerated the protein task datasets with links to the original data files we used to train R2DL. We also have provided instructions on downloading and training source language models and the files used to train the R2DL model. The accompanying code is contained in the zip file in the ESI.† In what follows, we provide five biologically relevant downstream physicochemical property prediction tasks, adapted from ref. 11 to serve as benchmarks. We categorize these into property prediction, structure prediction, evolutionary understanding, and protein engineering tasks. The sizes of the individual datasets vary between 4000 and 5000 (see the ESI† for details on data sizes and train-test splits).Author contributions
Ria Vinod conducted the data collection, model training, and evaluation. All authors contributed to idea generation, experimental design, result analyses, and paper writing.Conflicts of interest
There are no conflicts to declare.References
- J. Xu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16856–16865 CrossRef CAS PubMed.
- J. Yang, I. Anishchenko, H. Park, Z. Peng, S. Ovchinnikov and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1496–1503 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, et al., Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch and R. D. Schaeffer, et al., Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- S. C. Hoffman, V. Chenthamarakshan, K. Wadhawan, P.-Y. Chen and P. Das, Nat. Mach. Intell., 2022, 4, 21–31 CrossRef.
- E. Lo and P.-Y. Chen, Digital Discovery, 2023, 2, 1380–1389 RSC.
- R. Singh, S. Sledzieski, B. Bryson, L. Cowen and B. Berger, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220778120 CrossRef CAS PubMed.
- S. Gelman, S. A. Fahlberg, P. Heinzelman, P. A. Romero and A. Gitter, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104878118 CrossRef CAS PubMed.
- P. Das, T. Sercu, K. Wadhawan, I. Padhi, S. Gehrmann, F. Cipcigan, V. Chenthamarakshan, H. Strobelt, C. Dos Santos and P.-Y. Chen, et al., Nat. Biomed. Eng., 2021, 5, 613–623 CrossRef CAS PubMed.
- J. Meier, R. Rao, R. Verkuil, J. Liu, T. Sercu and A. Rives, Adv. Neural Inf. Process. Syst., 2021, 34, 29287–29303 Search PubMed.
- R. Rao, N. Bhattacharya, N. Thomas, Y. Duan, P. Chen, J. Canny, P. Abbeel and Y. Song, Adv. Neural Inf. Process. Syst., 2019, 32, 9689–9701 Search PubMed.
- E. Nijkamp, J. A. Ruffolo, E. N. Weinstein, N. Naik and A. Madani, Cell Syst., 2023, 14, 968–978 CrossRef CAS PubMed.
- R. Bommasani, D. A. Hudson, E. Adeli, R. Altman, S. Arora, S. von Arx, M. S. Bernstein, J. Bohg, A. Bosselut, E. Brunskill, et al., arXiv, 2021, preprint, arXiv:2108.07258.
- B. Yuan, Y. He, J. Q. Davis, T. Zhang, T. Dao, B. Chen, P. Liang, C. Re and C. Zhang, arXiv, 2022, preprint, arXiv:2206.01288.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova, arXiv, 2018, preprint, arXiv:1810.04805.
- D. Patterson, J. Gonzalez, Q. Le, C. Liang, L.-M. Munguia, D. Rothchild, D. So, M. Texier and J. Dean, arXiv, 2021, preprint, arXiv:2104.10350.
- C. Raffel, N. Shazeer, A. Roberts, K. Lee, S. Narang, M. Matena, Y. Zhou, W. Li and P. J. Liu, J. Mach. Learn. Res., 2020, 21, 5485–5551 Search PubMed.
- T. Wolf, L. Debut, V. Sanh, J. Chaumond, C. Delangue, A. Moi, P. Cistac, T. Rault, R. Louf, M. Funtowicz, et al., arXiv, 2019, preprint, arXiv:1910.03771.
- M. E. Newman, Contemp. Phys., 2005, 46, 323–351 CrossRef.
- J. Su, X. Zhou, X. Zhang and F. Yuan, bioRxiv, 2024 DOI:10.1101/2024.05.30.596740.
- K. E. Wu, H. Chang and J. Zou, bioRxiv, 2024, 5 DOI:10.1101/2024.05.14.594226v1.
- M. Xu, X. Yuan, S. Miret and J. Tang, International Conference on Machine Learning, 2023, pp. 38749–38767 Search PubMed.
- E. K. Makowski, P. C. Kinnunen, J. Huang, L. Wu, M. D. Smith, T. Wang, A. A. Desai, C. N. Streu, Y. Zhang and J. M. Zupancic, et al., Nat. Commun., 2022, 13, 1–14 Search PubMed.
- Z. Hou, Y. Yang, Z. Ma, K.-c. Wong and X. Li, Commun. Biol., 2023, 6, 73 CrossRef CAS PubMed.
- F. Zhuang, Z. Qi, K. Duan, D. Xi, Y. Zhu, H. Zhu, H. Xiong and Q. He, Proc. IEEE, 2020, 109, 43–76 Search PubMed.
- P.-Y. Chen, AAAI, 2024 Search PubMed.
- P. Neekhara, S. Hussain, S. Dubnov and F. Koushanfar, arXiv, 2018, preprint, arXiv:1809.01829.
- C.-H. H. Yang, Y.-Y. Tsai and P.-Y. Chen, International Conference on Machine Learning, 2021, pp. 11808–11819 Search PubMed.
- G. F. Elsayed, I. Goodfellow and J. Sohl-Dickstein, International Conference on Learning Representations, 2019 Search PubMed.
- T. Brown, B. Mann, N. Ryder, M. Subbiah, J. D. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry and A. Askell, et al., Adv. Neural Inf. Process. Syst., 2020, 33, 1877–1901 Search PubMed.
- Y. Liu, M. Ott, N. Goyal, J. Du, M. Joshi, D. Chen, O. Levy, M. Lewis, L. Zettlemoyer and V. Stoyanov, arXiv, 2019, preprint, arXiv:1907.11692.
- J. Wei, Y. Tay, R. Bommasani, C. Raffel, B. Zoph, S. Borgeaud, D. Yogatama, M. Bosma, D. Zhou, D. Metzler, et al., arXiv, 2022, preprint, arXiv:2206.07682.
- R. Bommasani, D. A. Hudson, E. Adeli, R. Altman, S. Arora, S. von Arx, M. S. Bernstein, J. Bohg, A. Bosselut, E. Brunskill, et al., arXiv, 2021, preprint arXiv:2108.07258.
- X. Jiao, Y. Yin, L. Shang, X. Jiang, X. Chen, L. Li, F. Wang and Q. Liu, arXiv, 2019, preprint, arXiv:1909.10351.
- M. Aharon, M. Elad and A. Bruckstein, IEEE Trans. Signal Process., 2006, 54, 4311–4322 Search PubMed.
- S. El-Gebali, J. Mistry, A. Bateman, S. R. Eddy, A. Luciani, S. C. Potter, M. Qureshi, L. J. Richardson, G. A. Salazar and A. Smart, et al., Nucleic Acids Res., 2019, 47, D427–D432 CrossRef CAS PubMed.
- A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin, J. Liu, D. Guo, M. Ott, C. L. Zitnick and J. Ma, et al., Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2016239118 CrossRef CAS PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Nat. Methods, 2019, 16, 1315–1322 CrossRef CAS PubMed.
- T. Bepler and B. Berger, Cell Syst., 2021, 12, 654–669 CrossRef CAS PubMed.
- U. Consortium, Nucleic Acids Res., 2019, 47, D506–D515 CrossRef PubMed.
- M. Punta, P. C. Coggill, R. Y. Eberhardt, J. Mistry, J. Tate, C. Boursnell, N. Pang, K. Forslund, G. Ceric and J. Clements, et al., Nucleic Acids Res., 2012, 40, D290–D301 CrossRef CAS PubMed.
- A. Elnaggar, M. Heinzinger, C. Dallago, G. Rihawi, Y. Wang, L. Jones, T. Gibbs, T. Feher, C. Angerer, M. Steinegger, et al., arXiv, 2020, preprint, arXiv:2007.06225.
- A. Dosovitskiy, L. Beyer, A. Kolesnikov, D. Weissenborn, X. Zhai, T. Unterthiner, M. Dehghani, M. Minderer, G. Heigold, S. Gelly, et al., arXiv, 2020, preprint, arXiv:2010.11929.
- B. E. Suzek, H. Huang, P. McGarvey, R. Mazumder and C. H. Wu, Bioinformatics, 2007, 23, 1282–1288 CrossRef CAS PubMed.
- A. L. Maas, R. E. Daly, P. T. Pham, D. Huang, A. Y. Ng and C. Potts, Proceedings of the 49th Annual Meeting of the Association for Computational Linguistics: Human Language Technologies, Portland, Oregon, USA, 2011, pp. 142–150 Search PubMed.
- Y. Gu, R. Tinn, H. Cheng, M. Lucas, N. Usuyama, X. Liu, T. Naumann, J. Gao and H. Poon, ACM Transactions on Computing for Healthcare (HEALTH), 2021, vol. 3, pp. 1–23 Search PubMed.
- N. Brandes, D. Ofer, Y. Peleg, N. Rappoport and M. Linial, Bioinformatics, 2022, 38, 2102–2110 CrossRef CAS PubMed.
- M. S. Klausen, M. C. Jespersen, H. Nielsen, K. K. Jensen, V. I. Jurtz, C. K. Soenderby, M. O. A. Sommer, O. Winther, M. Nielsen and B. Petersen, et al., Proteins: Struct., Funct., Bioinf., 2019, 87, 520–527 CrossRef CAS PubMed.
- J. J. Almagro Armenteros, C. K. Sønderby, S. K. Sønderby, H. Nielsen and O. Winther, Bioinformatics, 2017, 33, 3387–3395 CrossRef PubMed.
- G. J. Rocklin, T. M. Chidyausiku, I. Goreshnik, A. Ford, S. Houliston, A. Lemak, L. Carter, R. Ravichandran, V. K. Mulligan and A. Chevalier, et al., Science, 2017, 357, 168–175 CrossRef CAS PubMed.
- J. Hou, B. Adhikari and J. Cheng, Bioinformatics, 2018, 34, 1295–1303 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00195h |
| ‡ The majority of this work was done during Ria Vinod's internship at IBM Research. |
| This journal is © The Royal Society of Chemistry 2025 |