
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and electrochemical properties of molybdenum nitrido complexes supported by redox-active NHC and MIC ligands†
Daniel
Leitner
 ,
Florian R.
Neururer
and
Stephan
Hohloch
*
,
Florian R.
Neururer
and
Stephan
Hohloch
*
Department of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: Stephan.Hohloch@uibk.ac.at
First published on 1st November 2024
Abstract
We report the synthesis of a series of molybdenum nitrido complexes supported by bis-phenolate N-heterocyclic and mesoionic carbenes (NHC & MIC). The reaction between MoN(OtBu)3 and the corresponding azolium salts [H3L1]Cl and [H3L2]Cl (with L1 = bis-phenolate triazolylidene and L2 = bis-phenolate benzimidazolylidene) gives clean access to the corresponding NHC/MIC complexes 1-Cl and 2-Cl. Electrochemical investigations of these complexes showed that they can be reversibly reduced at potentials of −1.13 and −1.01 V vs. Fc/[Fc]+ and the reduced complexes [1-Cl]− and [2-Cl]− can be cleanly isolated after chemical reduction with one equivalent of decamethylcobaltocene. Exchange of the halide atoms is furthermore reported to give a series of nitrido complexes supported by tert-butanolate (1-OtBu and 2-OtBu), perfluoro-tert-butanolate (1-OtBuF9 and 2-OtBuF9), tritylate (1-OCPh3 and 2-OCPh3), mesitolate (1-OMes and 2-OMes), thio-tert-butanolate (1-StBu), thiotritylate (1-SCPh3 and 2-SCPh3) and thiomesitolate complexes (1-SMes). The electrochemical properties of all complexes were evaluated and compared. All isolated complexes were characterized by multinuclear and multidimensional NMR spectroscopy and (if applicable) by EPR spectroscopy. Furthermore, the reactivity of 1-Cl and 2-Cl in the presence of protons and decamethylcobaltocene was investigated, which shows facile extrusion of ammonia, yielding diamagnetic bis-molybdenum(III) complexes 3 and 4.
Introduction
The effective conversion of nitrogen into ammonia under ambient conditions (1 atm N2 at room temperature) is one of the key challenges in modern chemistry. Surely, and despite its large energy consumption, this process is efficiently solved on an industrial scale, using the Haber–Bosch process.1 However, given the promising results of ammonia also acting as an energy/hydrogen storage system and potential future fuel,2 the development of delocalized ammonia generators is an important goal.3 This requires catalysts that operate under mild or, at best, ambient conditions, efficiently producing ammonia and related compounds, e.g. tris-trimethylsilylamines (N(SiMe3)3) or nitrogen-functionalized organic molecules.4 Within the past few decades, molybdenum-based catalysts, in particular, have been thoroughly studied in this context (Fig. 1),5 and also other metals such as titanium,6 vanadium,7 chromium,8 tungsten,9 rhenium,10 iron,11 cobalt,12 manganese13 or the lanthanides,14 the actinides15 and boron16 have been utilized to facilitate this reaction. For molybdenum, one of the most promising systems so far incorporated the use of tridentate PCP chelating N-heterocyclic carbene (NHC) ligands.17 Computational methods have further shown that the substitution pattern (and thus the electronic structure) on the NHC ligand seems to play a crucial role in this reaction.18 Given these results and the large variety of NHC ligands reported to date,19 it is particularly noteworthy that the number of NHC ligands investigated in this context is still very limited.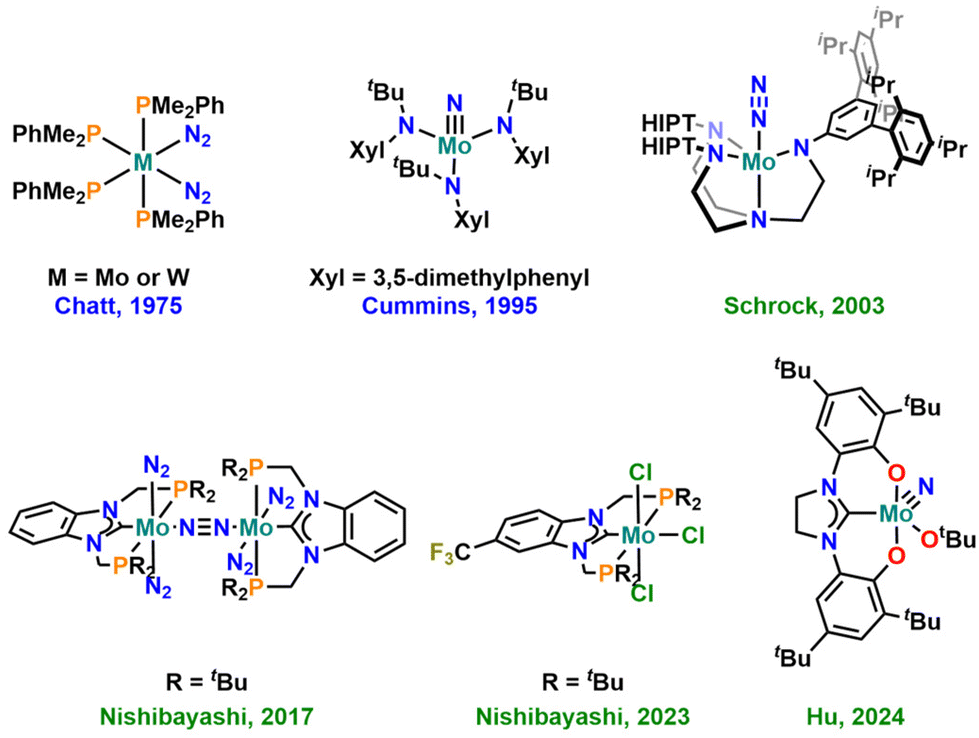 | ||
| Fig. 1 Selected milestones of molybdenum-based complexes for dinitrogen fixation and functionalisation. The green labels indicate that the molecule is catalytic in nitrogen functionalisation, while the blue labels indicate stoichiometric reactivity. | ||
Based on the seminal works by Kawaguchi,20 Grubbs,21 Bercaw22,23 and Bellemin-Laponnaz,24–26 we and others27 have recently started to investigate the chemistry of bis-phenolate-supported normal N-heterocyclic carbene (nNHC) and mesoionic carbene (MIC) ligands of the (benz)imidazolylidene- and 1,2,3-triazolylidene type in the chemistry of (early) transition metals.28–33 This revealed an unprecedented catalytic potential and metal–carbene stability of benzimidazolylidene-based systems in deoxygenation catalysis.28,30 Given the advantageous properties of triazolylidenes,34 combined with their modular synthesis,35–37,38 these ligands were often found to enhance the catalytic potential of metal complexes,39 in some cases outperforming nNHC congeners.37,40–42 Thus, we believe that both these ligands are valuable and promising candidates for the preparation of efficient and stable ammonia evolution catalysts. This is further supported by the recent appearance of a bis-phenolate nNHC molybdenum nitrido complex in the literature that shows unprecedented turnover numbers and selectivity in the catalytic silylation of dinitrogen using TMS–Cl and KC8.43 Here, we present the synthesis and in-depth characterisation of overall 18 new NHC- and MIC-supported molybdenum nitrido complexes by spectroscopic (NMR, EPR, UV-Vis-NIR, IR), electrochemical (CV) and structural means. Preliminary results show that these complexes can be reductively denitrogenated under protic conditions, leading to dimeric Mo(III) complexes with a direct Mo–Mo bond, which is indicative of their catalytic potential in ammonia evolution from dinitrogen.
Results and discussion
Synthesis of the triazolylidene and benzimidazolylidene complexes 1-Cl and 2-Cl was achieved following a protonolysis approach between the nitride precursor MoN(OtBu)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 and the corresponding azolium salts [H3L1]Cl29 and [H3L2]Cl22,25 in THF at room temperature (Scheme 1).24,31,33,43 After stirring the reaction mixture for 24 h, evaporation of the volatiles and washing the residue with hexanes, the target complexes 1-Cl and 2-Cl can be isolated as purple powders in yields of 86% and 83%, respectively. Successful formation of the carbene complexes is evident by the corresponding 1H and 13C NMR signatures. First, the absence of the benzimidazolium-2H/triazolium-5H signals in the low-field region in the corresponding 1H NMR spectra is an indicator for successful metalation (Fig. S1 and S6†). Furthermore, for complex 1-Cl, the shift of the triazolylidene-CH3 group from 3.3029 to 4.63 ppm is typical (Fig. S1†). Additionally, the 13C NMR spectra show resonances at 160.9 ppm for 1-Cl (Fig. S2†) and 191.0 ppm (Fig. S7†) for 2-Cl, which are typical for NHC/MIC complexes.30 The presence of the nitride ligand was indicated by IR spectroscopy, showing characteristic Mo–N stretching frequencies at 1035 cm−1 for complex 1-Cl and 1039 cm−1 for complex 2-Cl.44 Assignment of this band to the Mo–N stretching was confirmed by DFT calculations (see the ESI† for further information). The lower frequency obtained in complex 1-Cl compared to complex 2-Cl (1035 vs. 1039 cm−1) is in line with triazolylidenes being stronger donors compared to classical nNHC donors.30,35,36,40–42 Unambiguous proof was given by X-ray diffraction studies on single crystals grown from THF/hexane mixtures at −40 °C (Fig. 2). Both complexes crystallize in the orthorhombic system in the space group Cmce (1-Cl) and Pbca (2-Cl), with half a molecule of complex 1-Cl (with a pseudo-mirror plane going through the C1–Mo1–N10–Cl1 plane) and one molecule of complex 2-Cl in the asymmetric unit. The molybdenum carbene distances (M1–C1) are 2.145(4) Å for 1-Cl and 2.208(2) Å for complex 2-Cl. The shorter distance of the Mo1–C1 bond in the triazolylidene complex is in line with early examples of MIC complexes, showing shorter M–C distances compared to their nNHC analogs and reflects in the stronger donor character of MIC vs. the NHC donor.30,35,36,40–42 Despite the different donors (well reflected in the Mo–N stretching frequencies, vide supra and redox chemistry, vide infra) no difference in the Mo1–N10 bond length is visible at 1.642(4) Å and 1.647(2) Å in 1-Cl and 2-Cl. Furthermore, the yaw angle45 of the benzimidazolylidene complex 2-Cl at 15.55(1)° is much larger compared to that of the triazolylidene complex 1-Cl at 0.32(1)°. Similar yaw angles have also been seen in other benzimidazolylidene complexes of ligand L2 of vanadium, niobium or molybdenum.30,32,33
44 and the corresponding azolium salts [H3L1]Cl29 and [H3L2]Cl22,25 in THF at room temperature (Scheme 1).24,31,33,43 After stirring the reaction mixture for 24 h, evaporation of the volatiles and washing the residue with hexanes, the target complexes 1-Cl and 2-Cl can be isolated as purple powders in yields of 86% and 83%, respectively. Successful formation of the carbene complexes is evident by the corresponding 1H and 13C NMR signatures. First, the absence of the benzimidazolium-2H/triazolium-5H signals in the low-field region in the corresponding 1H NMR spectra is an indicator for successful metalation (Fig. S1 and S6†). Furthermore, for complex 1-Cl, the shift of the triazolylidene-CH3 group from 3.3029 to 4.63 ppm is typical (Fig. S1†). Additionally, the 13C NMR spectra show resonances at 160.9 ppm for 1-Cl (Fig. S2†) and 191.0 ppm (Fig. S7†) for 2-Cl, which are typical for NHC/MIC complexes.30 The presence of the nitride ligand was indicated by IR spectroscopy, showing characteristic Mo–N stretching frequencies at 1035 cm−1 for complex 1-Cl and 1039 cm−1 for complex 2-Cl.44 Assignment of this band to the Mo–N stretching was confirmed by DFT calculations (see the ESI† for further information). The lower frequency obtained in complex 1-Cl compared to complex 2-Cl (1035 vs. 1039 cm−1) is in line with triazolylidenes being stronger donors compared to classical nNHC donors.30,35,36,40–42 Unambiguous proof was given by X-ray diffraction studies on single crystals grown from THF/hexane mixtures at −40 °C (Fig. 2). Both complexes crystallize in the orthorhombic system in the space group Cmce (1-Cl) and Pbca (2-Cl), with half a molecule of complex 1-Cl (with a pseudo-mirror plane going through the C1–Mo1–N10–Cl1 plane) and one molecule of complex 2-Cl in the asymmetric unit. The molybdenum carbene distances (M1–C1) are 2.145(4) Å for 1-Cl and 2.208(2) Å for complex 2-Cl. The shorter distance of the Mo1–C1 bond in the triazolylidene complex is in line with early examples of MIC complexes, showing shorter M–C distances compared to their nNHC analogs and reflects in the stronger donor character of MIC vs. the NHC donor.30,35,36,40–42 Despite the different donors (well reflected in the Mo–N stretching frequencies, vide supra and redox chemistry, vide infra) no difference in the Mo1–N10 bond length is visible at 1.642(4) Å and 1.647(2) Å in 1-Cl and 2-Cl. Furthermore, the yaw angle45 of the benzimidazolylidene complex 2-Cl at 15.55(1)° is much larger compared to that of the triazolylidene complex 1-Cl at 0.32(1)°. Similar yaw angles have also been seen in other benzimidazolylidene complexes of ligand L2 of vanadium, niobium or molybdenum.30,32,33
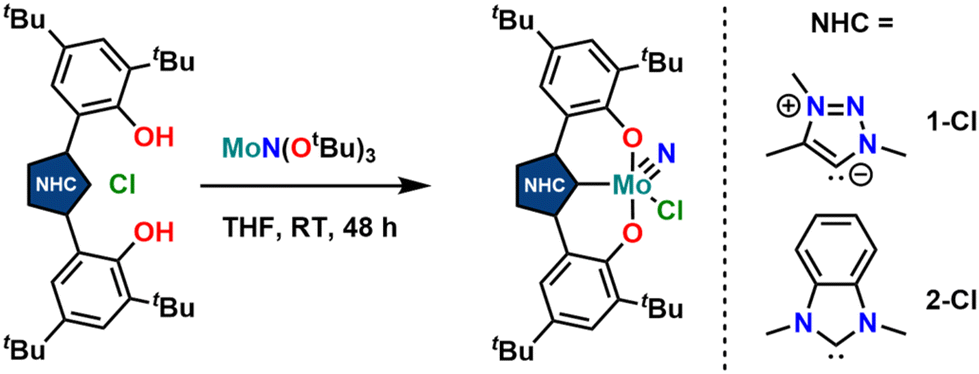 | ||
| Scheme 1 Synthesis of molybdenum nitride complexes featuring triazolylidene (1-Cl) and benzimidazolylidene ligands (2-Cl) via protonolysis between MoN(OtBu)3 and the corresponding azolium salts [H3L1]Cl and [H3L2]Cl. | ||
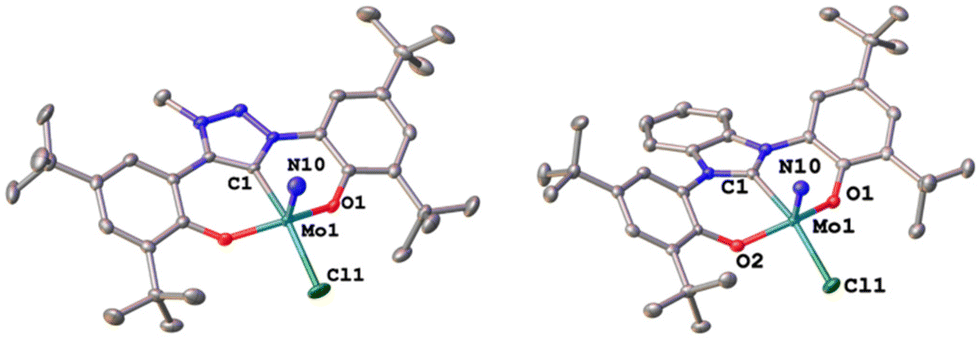 | ||
| Fig. 2 Molecular structures of the triazolylidene and benzimidazolylidene molybdenum nitride complexes 1-Cl and 2-Cl. Hydrogen atoms are omitted for clarity, and ellipsoids are shown at a probability level of 50%. | ||
Next, the electrochemical properties of the complexes were investigated with special emphasis on their reduction chemistry. The cyclic voltammograms of 1-Cl and 2-Cl showed several (reversible) redox processes (Fig. 3). The first reduction appears at −1.13 and −1.01 V vs. Fc/Fc+ for 1-Cl and 2-Cl, respectively. Given the stronger donor character of the MIC in 1-Clvs. the nNHC in 2-Cl, the reduction potential of 1-Cl is anodically shifted.31 This also explains why for the benzimidazoylidene complex 2-Cl, two further redox waves are present at −2.77 and −3.02 V, while for the triazolylidene complex 1-Cl, the third reduction is shifted beyond the solvent window and only one reduction can be observed at −2.59 V.
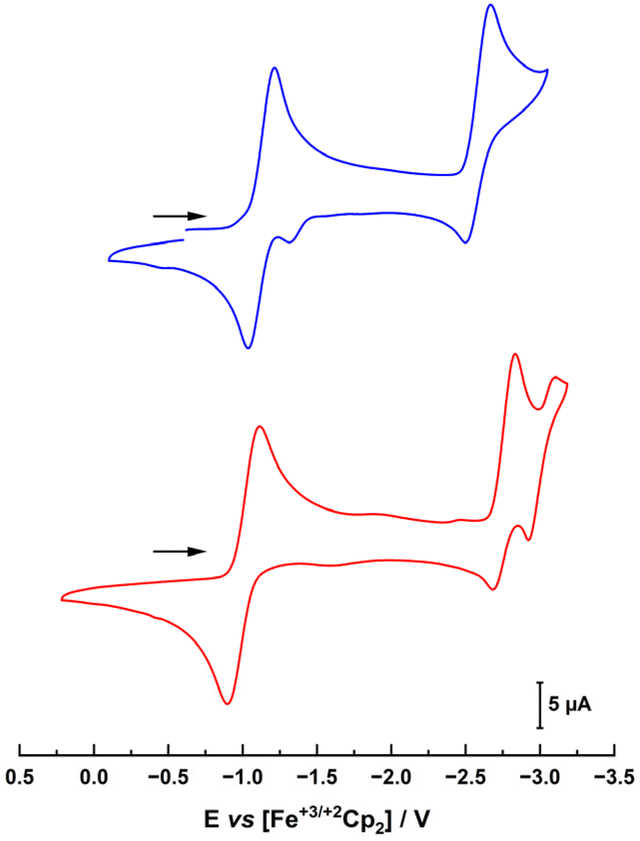 | ||
| Fig. 3 Cyclic voltammograms of complexes 1-Cl (blue trace, top) and 2-Cl (red trace, bottom) in THF with 0.15 M NBu4PF6 as a supporting electrolyte. Scan rate is shown at 100 mV s−1. | ||
Thus, we aimed for chemical reduction of the Mo(VI) chloride complexes 1-Cl and 2-Cl. Given the fact that the reduction of vanadium-oxo complexes with similar ligand scaffolds has worked well, using decamethylcobaltocene, we used this strategy to obtain Mo(V) complexes (Scheme 2). Mixing decamethylcobaltocene with 1-Cl or 2-Cl in dichloromethane at low temperature is accompanied by a fast colour change from purple to green, giving access to the Mo(V) NHC/MIC complexes [1-Cl]− and [2-Cl]− with a [(Cp*)2Co]+ counterion in yields of 62 and 75%, respectively.31 Due to the paramagnetic nature of these complexes, the 1H NMR spectra showed only broadened and unassignable features (Fig. S12 and S14†), but Evans’ method revealed a magnetic moment of 1.86 and 1.97μB, respectively (Fig. S11 and S13†), which is in agreement with a d1-configured metal centre. The EPR spectra show the typical Mo(V) seven-line pattern at room temperature. Unambiguous proof for the successful reduction of the complexes 1-Cl and 2-Cl was given by X-ray diffraction studies performed on single crystals obtained by the slow evaporation of dichloromethane from a hexane solution of the complexes (Fig. 4 and S120, S121;† note: the reduced complexes [1-Cl]− and [2-Cl]− are entirely insoluble in hexane/aliphatic solvents but dissolve well if dichloromethane is added to these suspensions). The complexes crystalize in the orthorhombic space group Pbca for complex [1-Cl]− and the monoclinic space group Cc for complex [2-Cl]− with one molecule of dichloromethane in the asymmetric unit. The Mo–C1 distances shorten from 2.145(4) Å in 1-Cl to 2.115(3) Å in [1-Cl]− and from 2.208(2) in 2-Cl to 2.173(3) Å in complex [2-Cl]−. Although this is counterintuitive to a metal-centred reduction process, the shortening of the Mo1–C1 bond could indicate some minor back-bonding effects.31,33 However, the shortening of the Mo–C bond could also be a result of ligand distortion caused by the elongated Mo–O bonds (vide infra), enforcing a shorter Mo–C distance of the central NHC/MIC core unit. The Mo1–O1/O2 distances increase from 1.9268(19)/1.9269(19) Å in 1-Cl to 2.0655(19)/2.0422(19) Å in [1-Cl]− and from 1.9203(18)/1.9217(18) Å in 2-Cl to 2.0585(18)/2.0477(19) in [2-Cl]−. Similarly, the Mo1–Cl1 distances increases by about 0.1 Å upon reduction. The Mo1–N10 distance is mostly unaffected, showing a distance of 1.654(3) and 1.657(2) Å in [1-Cl]− and [2-Cl]−, respectively.
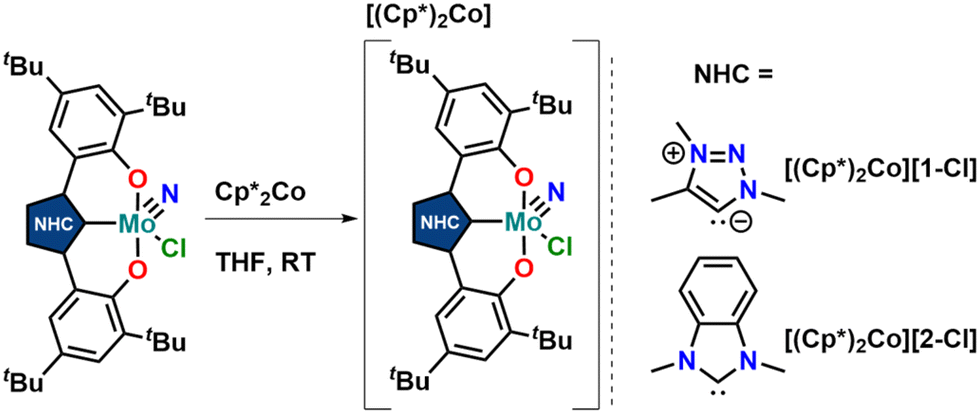 | ||
| Scheme 2 Synthesis of one-electron reduced complexes [1-Cl]− and [2-Cl]− using decamethylcobaltocene as a reductant. | ||
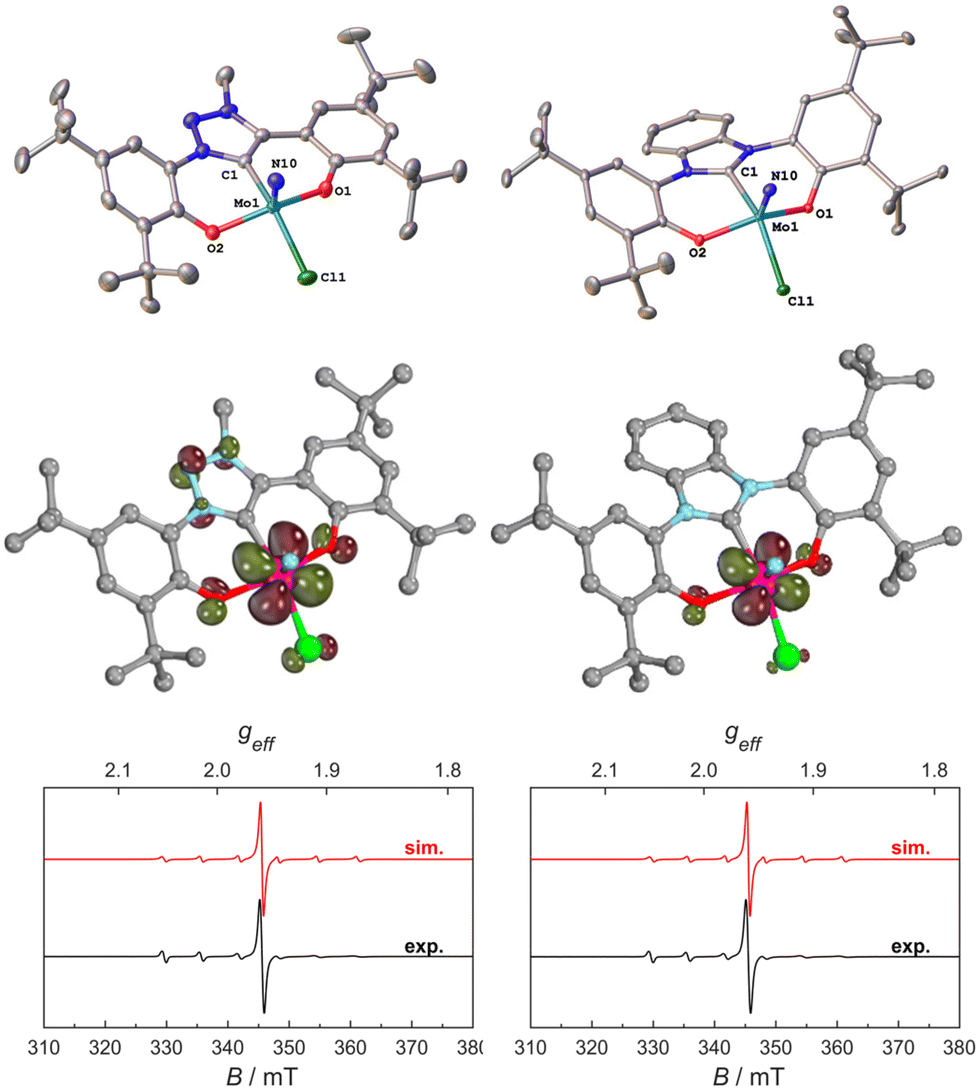 | ||
| Fig. 4 Molecular structures of the anionic Mo(V) complexes [1-Cl]− and [2-Cl]− (top). Hydrogen atoms, decamethyl cobaltocenium counter ions and solvent lattice molecules are omitted for clarity. Ellipsoids are shown at a probability level of 50%. Spin density plots of the two anionic complexes [1-Cl]− and [2-Cl]− showing the unpaired electrons residing in the 3d(yz) orbitals (QROs, middle). X-band EPR spectra of [1-Cl]− and [2-Cl]− of 5 mM solution in CH2Cl2 at 300 K; the black traces show the experimentally observed spectra and the red traces show the corresponding simulations (bottom). | ||
Unfortunately, using KC8 as a potential reductant fails on the chloride complexes and complicated mixtures are observed. This was also observed in vanadium complexes bearing the same ligands.31,33 Since molybdenum nitride complexes are potential precursors for homogeneous nitrogen fixation and functionalisation, we attempted the stoichiometric reduction of complexes 1-Cl and 2-Cl under protic conditions to see if ammonia could be evolved. Upon addition of five equivalents of lutidinium triflate and four equivalents of decamethylcobaltocene, we found the formation of ammonium after aqueous workup (Scheme 3). Quantification of the ammonium salts shows that in the case of complex 1-Cl, almost quantitative amounts of ammonium can be detected (96%, Fig. S17†), while in the case of 2-Cl, only 17% of ammonium could be isolated (Fig. S18†). Concerning the fate of the molybdenum complexes, we saw a moderately clean formation of a single species after recrystallisation from DCM/toluene for the triazolylidene complex 1-Cl (Fig. 5 and Fig. S15,†vide infra) in a yield of approx. 20%, while in the benzimidazolylidene case, several (paramagnetic) products are observed (Fig. S16†). After work-up/crystallisation from dichloromethane/diethylether, we were able to isolate complex 4 in single crystalline yields (<2%). In the triazolylidene case, the yield of the Mo dimer species is notably higher in the crude mixture, but the low yield results from the loss of material during crystallisation. This is in line with the observed ammonium formation (vide supra). Both complexes 3 and 4 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a partially occupied solvent molecule (toluene in the case of 3 and diethyl ether in the case of 4) and one complex molecule in the asymmetric unit (Fig. 5). Given the presence of one chlorido ligand, alongside the dianionic carbene ligands and the presence of a direct molybdenum–molybdenum interaction, we determined a +III oxidation state for the molybdenum centres. The Mo1–Mo1A distances of 2.2707(8) and 2.2726(10) Å in 3 and 4 are thereby in the range of previously reported (unsupported, non-bridged) Mo(III)–Mo(III) triple bonds.46 The Mo–Mo triple bond also explains the diamagnetic nature of the molecules, as observed by NMR spectroscopy (vide infra). Similar to the one-electron reduced complexes, the Mo1–C1 distances slightly decrease from 2.145(4) Å in 1-Cl and 2.208(2) in 2-Cl to 2.142(6) Å in 3 and to 2.177(7) Å in 4.31,33 The Mo1–O2/O2 distances remain largely unaffected by the reduction from Mo(VI) to Mo(III), changing from 1.9268(19)/1.9269(19) Å in 1-Cl to 1.929(5)/1.929(5) Å in 3 and from 1.9203(18)/1.9217(18) Å in 2-Cl to 1.921(5)/1.918(5) Å in 4.
with a partially occupied solvent molecule (toluene in the case of 3 and diethyl ether in the case of 4) and one complex molecule in the asymmetric unit (Fig. 5). Given the presence of one chlorido ligand, alongside the dianionic carbene ligands and the presence of a direct molybdenum–molybdenum interaction, we determined a +III oxidation state for the molybdenum centres. The Mo1–Mo1A distances of 2.2707(8) and 2.2726(10) Å in 3 and 4 are thereby in the range of previously reported (unsupported, non-bridged) Mo(III)–Mo(III) triple bonds.46 The Mo–Mo triple bond also explains the diamagnetic nature of the molecules, as observed by NMR spectroscopy (vide infra). Similar to the one-electron reduced complexes, the Mo1–C1 distances slightly decrease from 2.145(4) Å in 1-Cl and 2.208(2) in 2-Cl to 2.142(6) Å in 3 and to 2.177(7) Å in 4.31,33 The Mo1–O2/O2 distances remain largely unaffected by the reduction from Mo(VI) to Mo(III), changing from 1.9268(19)/1.9269(19) Å in 1-Cl to 1.929(5)/1.929(5) Å in 3 and from 1.9203(18)/1.9217(18) Å in 2-Cl to 1.921(5)/1.918(5) Å in 4.
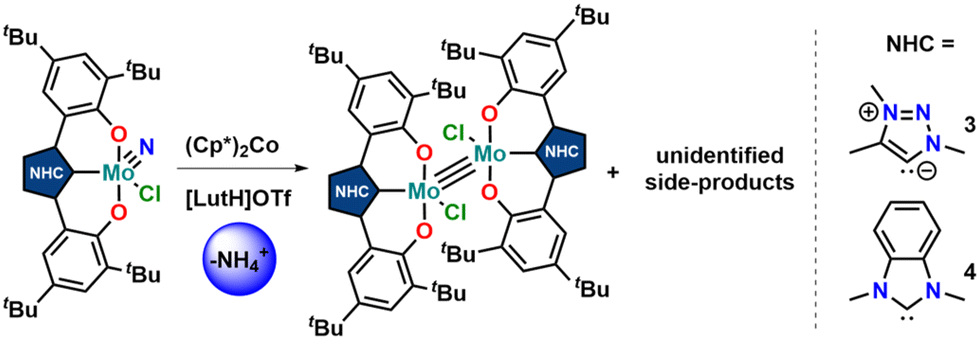 | ||
| Scheme 3 (Attempt) synthesis of molybdenum(III) dimers 3 and 4 by reduction of the nitrido complexes 1-Cl and 2-Cl under protic conditions. | ||
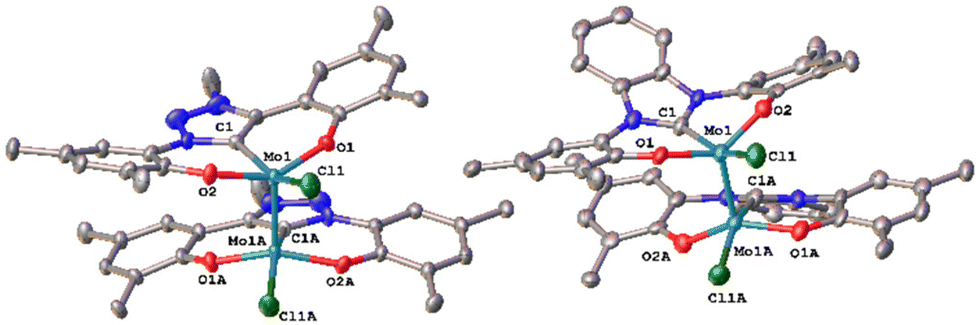 | ||
| Fig. 5 Molecular structures of the dimolybdenum(III) complexes 3 and 4 with a Mo–Mo triple bond connecting the two molybdenum centers. Hydrogen atoms, lattice solvent molecules and tBu groups are omitted for clarity. Ellipsoids are shown at a probability level of 50%. | ||
Finally, we aimed for further derivatisation of 1-Cl and 2-Cl (Scheme 4) with two main targets: (I) identifying how the reduction potentials change in dependence of other co-ligands (e.g. alkoxides, thiolates or (thio-)phenolates) and (II) if dimerization reactions under protic reduction conditions are suppressed by more steric bulk, blocking potential deactivation pathways in catalysis. Both complexes 1-Cl and 2-Cl allow for a range of functionalisations. The reaction between the corresponding lithium, sodium or potassium alkoxides, thiolates or (thio-)phenolates gives clean access to the anticipated functionalized complexes. We were able to synthesize tert-butanolate complexes 1-OtBu and 2-OtBu, the corresponding nona-fluoro-tert-butanolate complexes 1-OtBuF9 and 2-OtBuF9, tert-butanthiolate complex 1-StBu, tritylate complexes 1-OCPh3 and 2-OCPh3, thiotritylate complexes 1-SCPh3 and 2-SCPh3, mesitolate complexes 1-OMes and 2-OMes as well as the thiomesitolate complex 1-SMes. While the yields for 1-StBu and 1-SMes are significantly lower than their alkoxide congeners, the tert-butanthiolate complex 2-StBu and the thiomesitolate complex 2-SMes could not be isolated in pure form. 13C NMR spectroscopy confirmed the presence of a C–Mo carbene interaction by showing the characteristic low-field signals at 165.3 ppm for 1-OtBu, 196.7 ppm for 2-OtBu, 163.4 ppm for 1-OtBuF9, 193.2 ppm for 2-OtBuF9, 160.3 ppm for 1-StBu, 164.7 ppm for 1-OCPh3, 197.6 ppm for 2-OCPh3, 161.1 ppm for 1-SCPh3, 193.6 ppm for 2-SCPh3, 165.9 ppm for 1-OMes, 197.1 ppm for 2-OMes and 161.8 ppm for 1-SMes. From these values, certain trends are observable: for all functionalized complexes, the 13C resonances seem to be shifted to lower fields, indicating a stronger donation compared to the chloride ligand. Furthermore, oxygen-based donor systems cause a more pronounced low-field shift compared to sulfur-based systems, which agrees with the donor strength further corroborated by X-ray diffraction experiments and cyclic voltammetry (vide infra).
 | ||
| Scheme 4 Synthesis of a large variety of functionalized molybdenum nitride complexes supported by triazolylidene and benzimidazolylidene ligands to further study the influence of carbene and the co-ligands on the redox variability of the complexes, entries written in parentheses (namely 2-StBu and 2-SMes) have been attempted, but no clean products have been observed; therefore, only limited (crystals for 2-SMes) or no (crystals for 2-StBu) characterisation data are reported. | ||
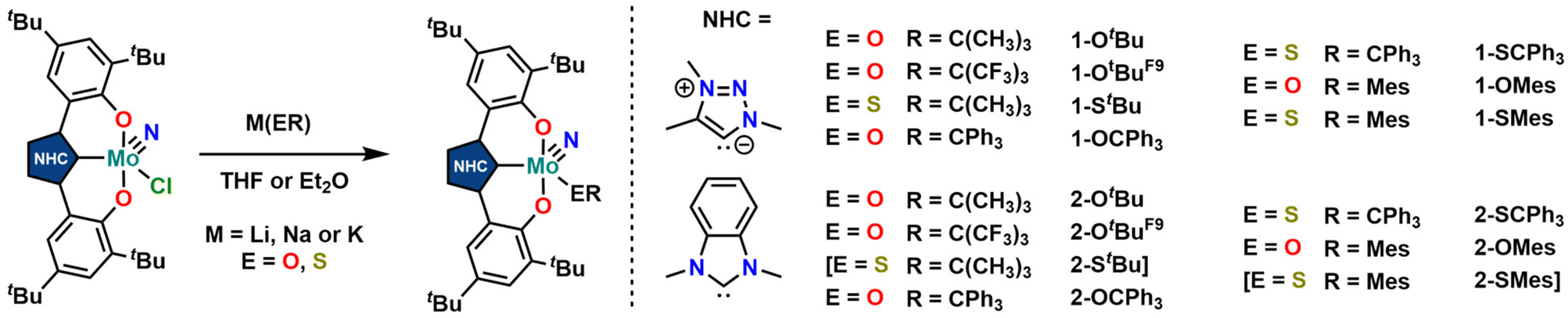
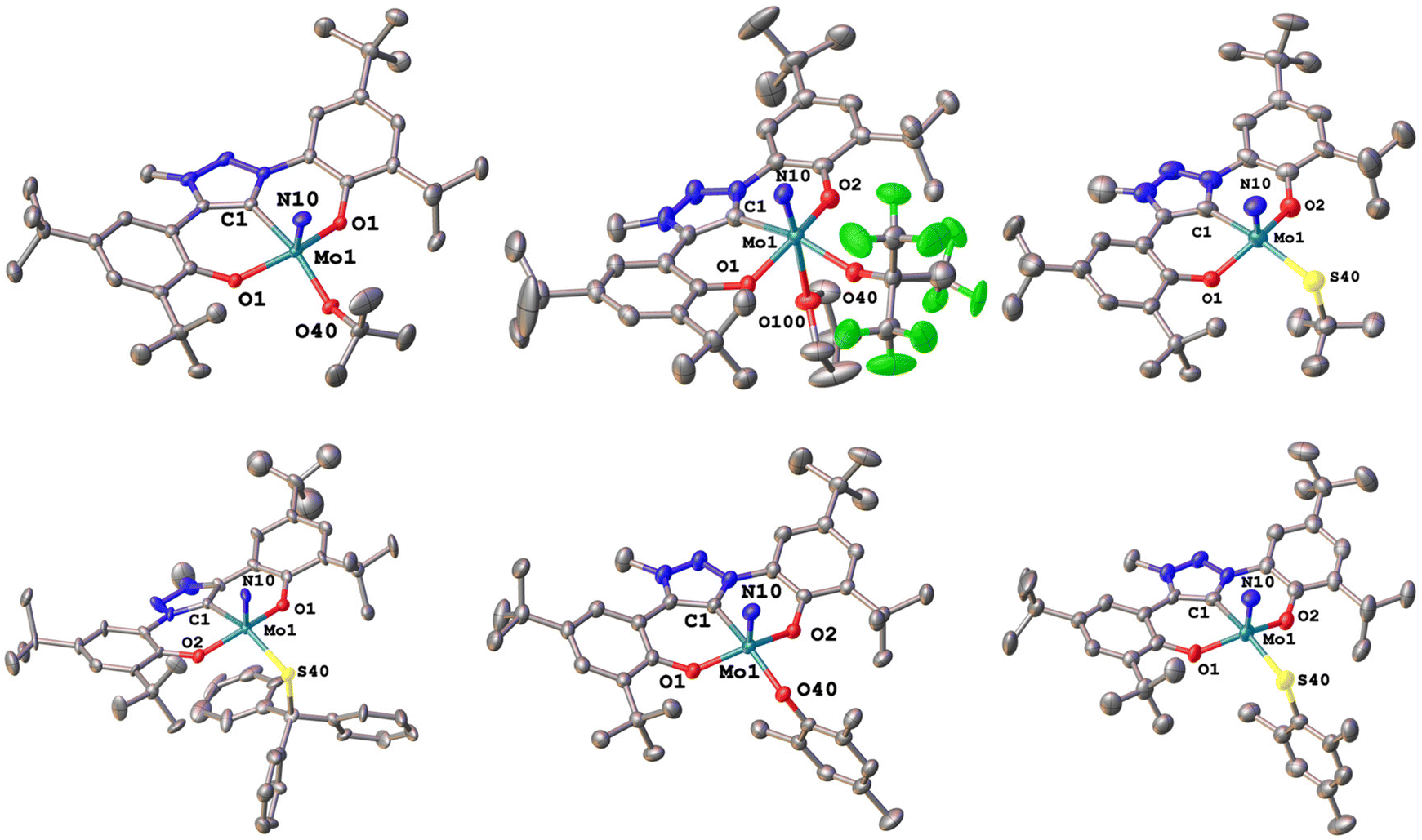
We were able to grow single crystals for all complexes, except for 2-StBu, 1-OCPh3 and 2-SCPh3 (Fig. 6 and 7). Given the structural similarity of all complexes, only the metal–carbene and metal–ligand bonds will be discussed in the following and further bond metrics can be found in the ESI (Tables S1–S3†). In all complexes, the additional donor ligand is situated in a trans-position to the carbene center,31–33 which elucidates its large influence on the 13C NMR carbene resonances described above (vide supra). Focussing on the triazolylidene complexes, the metal ligand distances (Mo1–O40 and Mo1–S40) are 1.8987(17) Å in 1-OtBu, 1.997(2) Å in 1-OtBuF9, 2.360(2) Å in 1-StBu, 2.369(3) Å in 1-SCPh3, 1.9378(17) Å in 1-OMes and 2.3767(17) Å in 1-SMes. The longer distances of the Mo–S bonds compared to those of the Mo–O bonds are in line with the size of the donor atoms. The perfluorinated tert-butanolate ligand in 1-OtBuF9 shows an about 0.1 Å longer Mo1–O40 distance (1.997(2) Å), compared to the non-fluorinated system 1-OtBu (1.8987(17) Å), while this distance in the arylated complex 1-OMes lies in between these two values (1.9378(17) Å), in line with the donor strength of the three alkoxide/phenolate ligands (OtBu > OMes > OtBuF9). This influence is also reflected in their electrochemical properties (vide infra). Interestingly, the Mo–S–C angles also seem to be more bent compared to the Mo–O–C angles. While for the complexes 1-OtBu, 1-OtBuF9 and 1-OMes, Mo1–O40–C40 angles of 141.87°, 152.0(2)° and 138.89(17)° are observed, and the Mo1–S40–C40 angles are 112.2(3)°, 107.5(3)° and 118.1(2)° in 1-StBu, 1-SCPh3 and 1-SMes. This effect is also well reflected in the line widths of the ligand groups in 1H-NMR, indicating significantly different rotational barriers. While 1-StBu displays a much broader tert-butyl signal than 1-OtBu (Fig. S19 and S41†), 1-SMes shows notably sharper signals than 1-OMes (Fig. S66 and S76†). The molybdenum–triazolylidene carbon interaction remains largely unaffected by the donor variations, displaying values between 2.154(3) and 2.188(2) Å with 1-OtBu showing the longest and 1-OMes showing the shortest metal carbene distance. For the benzimidazolylidene congeners, the above-mentioned trends are reproduced with the only difference that the Mo1–C1 distances are slightly longer, ranging from 2.201(4) Å in 2-OMes to 2.256(11) Å in 2-OtBu.28,30 This is in line with the stronger donor character of triazolylidenes compared to benzimidazolylidenes, leading to stronger metal–carbene interactions.40–42 Further information on the structural parameters of the functionalized complexes can be found in the ESI, Tables S1–S3.†
 | ||
| Fig. 6 Molecular structures of the functionalized triazolylidene complexes 1-OtBu, 1-OtBuF9, and 1-StBu (top) and 1-OCPh3, 1-OMes, and 1-SMes (bottom). Hydrogen atoms and lattice solvent molecules are omitted for clarity. Ellipsoids are shown at a probability level of 50%. | ||
 | ||
| Fig. 7 Molecular structures of the functionalized triazolylidene complexes 2-OtBu, 2-OtBuF9, and 2-OCPh3 (top) and 2-OMes, and 2-SMes (bottom). Hydrogen atoms and lattice solvent molecules are omitted for clarity. Ellipsoids are shown at a probability level of 50%. | ||
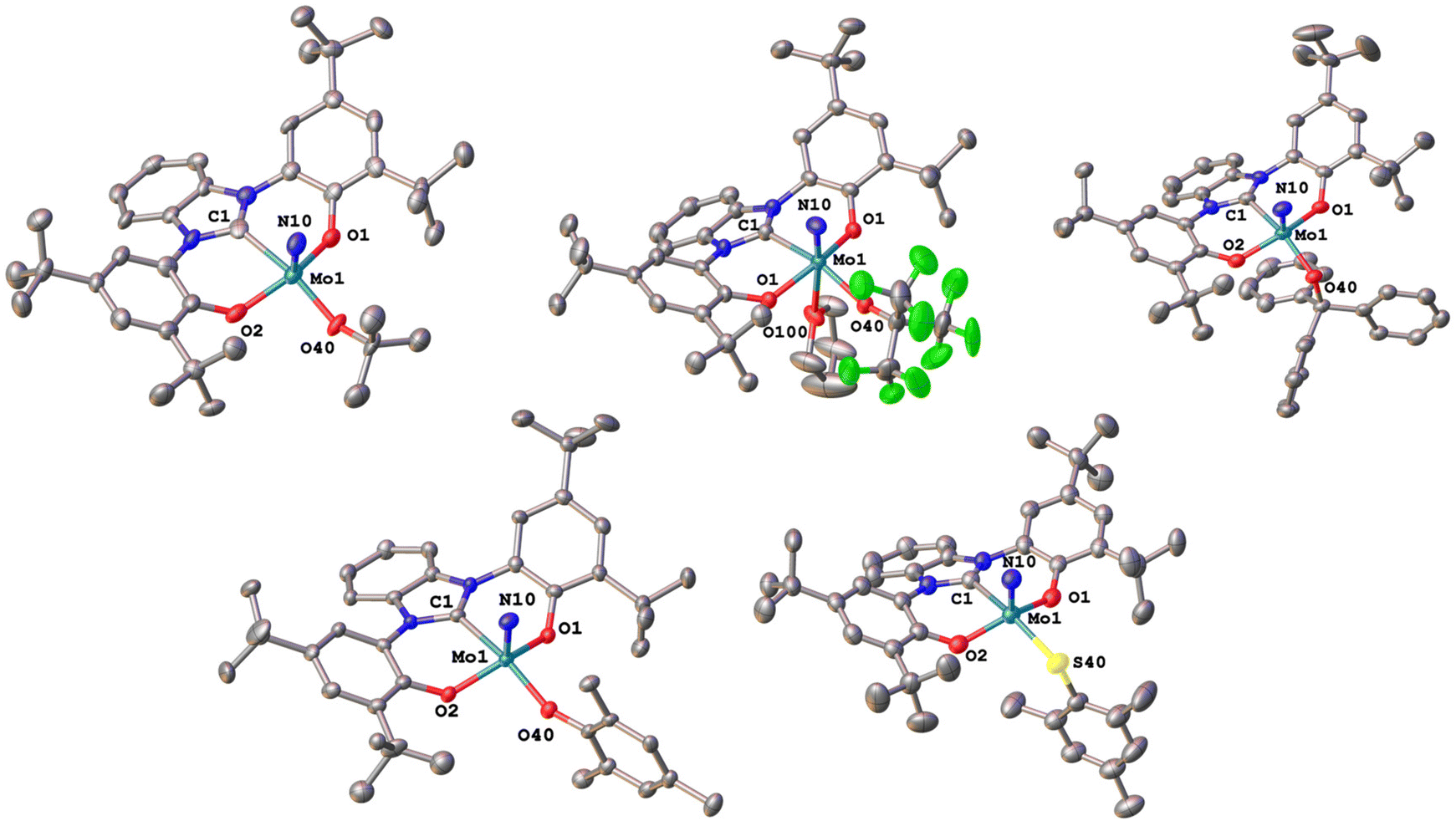
Next, we turned to the investigation of their electrochemical properties in comparison to the halide complexes 1-Cl and 2-Cl (Table 1, Fig. 8). As expected, the exchange of the halide with chalcogenide ligands shifts the reduction potential anodically. As such, the tert-butanolate complexes 1-OtBu and 2-OtBu show pronounced shifts from −1.13 V and −1.01 V in 1-Cl and 2-Cl to −1.78 V and −1.67 V vs. Fc/[Fc]+. Similar to the halide complexes, the triazolylidene-supported system has a potential approx. 0.1 V more negative than the benzimidazolylidene-supported system. Perfluorination of the tert-butanolate ligand in 1-OtBuF9 and 2-OtBuF9 results in the potential shifting cathodically to −1.26 V and −1.13 V compared to the non-fluorinated tert-butanolate complexes 1-OtBu and 2-OtBu. Replacing tert-butanolate with tritylate residues gives redox potentials of −1.65 V in 1-OCPh3 and −1.53 V in 2-OCPh3. Switching from alkoxide to phenolate, e.g. mesitolate changes the redox potentials to −1.47 V and 1.38 V in 1-OMes and 2-OMes. Another way to tune the redox potential is the exchange of oxygen for sulfur-based ligands. Compared to their oxygen congeners, the sulfur-based systems shift the redox potentials less anodically, taking 1-Cl and 2-Cl as reference points. For example, we found the Mo(VI)/Mo(V) reduction at −1.78 V vs. Fc/[Fc]+ for 1-OtBu while for the sulfur congener, 1-StBu, the reduction appears at −1.52 V vs. Fc/[Fc]+. Similarly, the reduction potentials for 1-SCPh3 and 1-SMes are found at −1.43 V and −1.23 V vs. Fc/[Fc]+ (compare 1.65 V for 1-OCPh3 and −1.47 V for 1-OMes). These trends are also observed in the benzimidazolylidene complex 2-SCPh3vs.2-OCPh3 (see Table 1 for comparison).
 | ||
| Fig. 8 Graphical comparison of the Mo(VI)/Mo(V) reduction potentials of the complex series 1-L (top) and 2-L (bottom). (L = Cl, OtBu, OtBuF9, StBu, OCPh3, SCPh3, OMes and SMes.) | ||
| Compound | E 1,red | E 2,red | E 3,red |
|---|---|---|---|
| a n.o. = not observed. b Irreversible process, value is given as Epc. | |||
| 1-Cl | −1.13 | −2.59 | n.o.a |
| 1-SMes | −1.23 | −2.82b | n.o.a |
| 1-OtBuF9 | −1.26 | −2.71b | n.o.a |
| 1-SCPh3 | −1.43 | −2.76b | −3.12b |
| 1-OMes | −1.47 | −3.02b | n.o.a |
| 1-StBu | −1.52 | −2.77b | n.o.a |
| 1-OCPh3 | −1.65 | −2.93b | n.o.a |
| 1-OtBu | −1.78 | −2.82b | n.o.a |
| 2-Cl | −1.01 | −2.77 | −3.02 |
| 2-OtBuF9 | −1.13 | −2.89 | n.o.a |
| 2-SCPh3 | −1.23 | n.o.a | n.o.a |
| 2-OMes | −1.38 | −2.96 | n.o.a |
| 2-OCPh3 | −1.53 | −2.90 | n.o.a |
| 2-OtBu | −1.67 | −2.91 | n.o.a |
Given the fact that the first reduction is metal centred in all complexes 1-L and 2-L, the different redox potentials observed can be directly correlated to the donor strength of the corresponding co-ligands and allow us to sort their donor properties in the following order: OtBu > OCPh3 > StBu > OMes ≈ SCPh3 > OtBuF9 ≈ SMes (compare also Table 1 and Fig. 8).47 Since efficient nitrogen reduction chemistry is a complex interplay between sterics and reduction potentials (more positive reduction potentials are favoured as they allow the use of “weaker” reductants), this series suggests thiomesitolate, as well as the perfluorinated tert-butanolate ligands to be promising candidates for reductive nitrogen functionalization.
Schrock as well as Hu and co-workers have already successfully demonstrated that the use of tert-butanolate ligands has advantageous effects on catalysis, leading to higher TONs and efficiencies.43,48
Conclusions
In summary, we have presented the synthesis of sixteen new molybdenum nitrido complexes 1-L and 2-L as well as two new dimeric molybenium(III) complexes 3 and 4, with a direct molybdenum(III)–molybdenum(III) triple bond supported by NHC and MIC bis-phenolate ligands. While the former sixteen complexes represent potential catalysts for the catalytic functionalization of dinitrogen, the latter two show potential catalyst deactivation routes, which need to be suppressed. Addressing this issue, we identified the thiomesitolate complexes 1-Mes and 2-Mes as well as the nonafluoro-tert-butanolate complexes 1-OtBuF9 and 2-OtBuF9 to be promising candidates, providing low reduction potentials, while offering enough steric bulk to prevent the dimerization reactions.Experimental section
General considerations
Unless stated otherwise, all operations were performed in an argon filled glovebox (H2O and O2 < 1 ppm) or using high-vacuum standard Schlenk techniques under an argon atmosphere. Solvents were dried using an MBraun SPS and stored over 3 Å molecular sieves at least 3 days prior to use. THF was distilled over sodium/benzophenone and stored over 3 Å molecular sieves. CD2Cl2 and C6D6 were degassed with an argon stream and stored over 3 Å molecular sieves for at least three days prior to use. The metal precursor MoN(OtBu)344 and proligands [H3L1]Cl29 and [H3L2]Cl22,25 were synthesized according to literature-known procedures. Lithium and potassium salts were obtained by deprotonation of the alcohol or thiol in hexane with n-BuLi or KHDMS, respectively. All other reagents were used as received from commercial sources. NMR spectra were recorded on a 400 MHz Bruker Avance 4 Neo spectrometer. 1H and 13C NMR chemical shifts (δ) are reported in ppm and were calibrated to residual solvent peaks. IR spectra were recorded on a Bruker Alpha spectrometer using the ATR method. UV/Vis/NIR spectra were recorded on an Avantes spectrometer using deuterium and halogen light sources and a CMOS detector. Elemental analysis was performed using an Elementar Vario Micro cube instrument. Single-crystal X-ray diffraction was performed on a Bruker D8 Quest diffractometer and data were collected using the ApexIV software package. Structures were solved using SHELXT49 and refined using the OLEX 2 software package.50 All non-hydrogen atoms were refined anisotropically, and hydrogen atoms were included at the geometrically calculated positions and refined using a riding model using SHELXL.51 For heavily disordered solvent molecules, the SQUEEZE algorithm was applied.52 EPR measurements were performed on a Magnettech MS5000 X-band spectrometer equipped with a temperature control unit in 3 mm o.d. J-Young style fused silica tubes. Spectra were processed using the EasySpin package for Matlab®.53 Cyclic voltammograms were recorded with a Gamry Interface 1010B potentiostat using a three-electrode setup (a glass carbon WE, an Ag wire RE, and a Pt wire CE) in 0.15 M NBu4PF6 solution in THF. Potentials are referenced to the Fc/Fc+ couple.
General procedure for the synthesis of complexes 1 and 2
The molybdenum precursor MoN(OtBu)3 and the corresponding proligand were mixed in a 100 mL Schlenk flask and 50 mL THF was added. The reaction mixture was stirred for 24 hours, during which the color changed to a deep purple. The solvent was removed in vacuo, hexane (20 mL) was added and the resulting solution was stirred for two hours. The product precipitated as dark purple powder was filtered off, washed with hexane and dried under high vacuum. When thoroughly dried, both complexes are pale purple.430 L mol−1 cm−1), 352 (ε = 20570 L mol−1 cm−1), 511 (ε = 2410 L mol−1 cm−1). IR (cm−1): 2955, 2906, 2869, 1605, 1533, 1484, 1466, 1431, 1413, 1388, 1362, 1303, 1245, 1211, 1153, 1127, 1088, 1047, 1035, 970, 921, 876, 849, 800, 774, 759, 723, 712, 692, 643, 559, 480, 453, 416. Elemental analysis (%) calc'd for C31H43ClMoN4O2·0.25CH2Cl2: C 57.19, H 6.68, N 8.54; found: C 56.6, H 6.74, N 8.18.
710 L mol−1 cm−1), 484 (ε = 4580 L mol−1 cm−1). IR (cm−1): 2961, 2906, 2869, 1580, 1482, 1468, 1431, 1386, 1362, 1333, 1290, 1254, 1239, 1200, 1162, 1121, 1070, 1039, 988, 921, 880, 857, 810, 800, 759, 747, 696, 639, 612, 567, 541, 486, 478, 449, 439. Elemental analysis (%) calc'd for C35H44ClMoN3O2: C 62.73, H 6.62, N 6.27; found: C 62.75, H 6.72, N 6.02.
General procedure for the one-electron reduction of complexes 1-Cl and 2-Cl
The corresponding MoVI complex 1-Cl or 2-Cl was dissolved in THF (5 mL) in a 20 mL scintillation vial and cooled to −40 °C. Solid (Cp*)2Co was added in small portions under stirring during which the color of the reaction changed to a dark brownish-green. The reaction mixture was allowed to warm to room temperature and after two hours, the solvent was removed in vacuo. The product was purified by crystallization from dichloromethane.General procedure for the (attempted) synthesis of dimers 3 and 4
The corresponding MoVI complex 1-Cl or 2-Cl was dissolved in THF (5 mL) in a 20 mL scintillation vial and cooled to −40 °C. In two separate scintillation vials, (Cp*)2Co and [LutH][OTf] were dissolved in 2 mL THF each. Keeping the reaction mixture at −40 °C, solutions with the reactants were simultaneously added dropwise to the complex upon which the mixture turned black. After the addition was complete, the reaction mixture was allowed to warm to room temperature and stirred for 16 hours. Yellow [(Cp*)2Co][OTf] was filtered off and the solvent was removed in vacuo.General procedure for the ammonia quantification experiment
Inside an argon filled glovebox, the respective complex (1 equiv.) and [LutH][OTf] (5 equiv.) were placed in a Schlenk flask and dissolved in 2 mL dry THF. A dropping funnel was attached and charged with a THF solution (approx. 10 mL) of Cp*2Co (4 equiv.). The apparatus was removed from the glovebox and the flask was cooled to −78 °C. The Cp*2Co solution was added dropwise over a period of 15 minutes and the apparatus was warmed to room temperature over the course of 2 hours. 2 mL of HCl in diethyl ether was added quickly through a dropping funnel and the mixture was stirred for an additional one hour. All subsequent steps were performed under air. The reaction mixture was filtered to remove yellow decamethyl cobaltocenium triflate and the solvent was removed in vacuo. The residue was suspended in dichloromethane, filtered and washed thoroughly with DCM. The remaining solid was extracted with water, evaporated to dryness and 17.7 mg (0.020 mmol) NaBArF24 was added. NMR spectra were recorded in DMSO-d6 and the yield was calculated relative to the NaBArF24 standard.For 1-Cl: From 25.0 mg (0.039 mmol, 1 equiv.) 1-Cl, 50.6 mg (0.197 mmol, 5 equiv.) [LutH][OTf] and 51.9 mg (0.157 mmol, 4 equiv.) Cp*2Co. Isolated yield of NH4+: 96% (0.038 mmol).
For 2-Cl: From 25.0 mg (0.037 mmol, 1 equiv.) 2-Cl, 48.0 mg (0.187 mmol, 5 equiv.) [LutH][OTf] and 49.2 mg (0.149 mmol, 4 equiv.) Cp*2Co. Isolated yield of NH4+: 17% (0.0065 mmol).
General procedure for the synthesis of salt metathesis products 1-ER and 2-ER
The respective chloride complex 1-Cl or 2-Cl was dissolved in THF (5 mL) in a 20 mL scintillation vial and cooled to −40 °C. A solution of the alkoxide or thiolate in THF (5 mL) was added dropwise at −40 °C, followed by slow warming to room temperature. The reaction mixture was stirred for 16 hours during which the color slowly changed from deep purple to between bright yellow and dark red, depending on the reactant. The solvent was removed in vacuo and the residue was dissolved in 5 mL of hexane. The complexes bearing OC(CF3)3, OCPh3 and SCPh3 ligands are barely soluble in hexane, therefore 1 mL diethyl ether or dichloromethane was added. The resulting suspension was filtered and the solution was concentrated in vacuo. Unless stated otherwise, an analytically pure product was precipitated upon cooling to −40 °C which was filtered off and washed with a minimal amount of cold hexane. The washed product was concentrated and the process was repeated for a total of three times.840 L mol−1 cm−1), 362 (ε = 25170 L mol−1 cm−1), 368 (ε = 25230 L mol−1 cm−1). IR (cm−1): 2957, 2903, 2867, 1533, 1480, 1429, 1417, 1386, 1358, 1294, 1258, 1237, 1202, 1174, 1129, 1090, 1074, 1060, 1019, 955, 874, 849, 802, 780, 757, 717, 692, 643, 572, 547, 492, 476, 445. Elemental analysis (%) calc'd for C35H52MoN4O3: C 62.49, H 7.79, N 8.33; found: C 62.35, H 7.66, N 8.15.
630 L mol−1 cm−1). IR (cm−1): 2950, 2903, 2867, 1572, 1482, 1466, 1431, 1382, 1358, 1333, 1292, 1270, 1256, 1229, 1202, 1176, 1055, 1041, 1025, 960, 921, 878, 855, 800, 786, 766, 745, 690, 637, 614, 594, 557, 492, 445. Elemental analysis (%) calc'd for C39H53MoN3O3: C 66.18, H 7.55, N 5.94; found: C 66.00, H 7.46, N 5.86.
010 L mol−1 cm−1), 499 (ε = 790 L mol−1 cm−1). IR (cm−1): 2959, 2908, 2871, 1776, 1535, 1482, 1433, 1390, 1362, 1294, 1262, 1239, 1178, 1131, 1080, 1055, 1035, 970, 921, 878, 847, 802, 774, 757, 727, 694, 643, 553, 492, 476, 447. Due to the fluorine present in the sample, no reproducible and reliable elemental analysis could be achieved.
210 L mol−1 cm−1), 464 (ε = 3970 L mol−1 cm−1). IR (cm−1): 2955, 2908, 2871, 1480, 1431, 1384, 1360, 1337, 1323, 1264, 1243, 1180, 1117, 1070, 1051, 1039, 1023, 970, 921, 892, 876, 855, 810, 782, 761, 727, 690, 647, 637, 610, 553, 537, 486, 445. Due to the fluorine present in the sample, no reproducible and reliable elemental analysis could be achieved.
200 L mol−1 cm−1). IR (cm−1): 2955, 2903, 2865, 1517, 1480, 1445, 1429, 1415, 1392, 1360, 1296, 1256, 1200, 1153, 1131, 1078, 1051, 1031, 921, 874, 853, 815, 798, 768, 757, 714, 692, 637, 576, 545, 502, 461, 449, 431, 408. Elemental analysis (%) calc'd for C35H52MoN4O2S·C6H6: C 64.21, H 7.62, N 7.31; found: C 64.60, H 7.45, N 6.90.
140 L mol−1 cm−1), 351 (ε = 42730 L mol−1 cm−1), 431 (ε = 4780 L mol−1 cm−1). IR (cm−1): 2959, 2906, 2871, 1527, 1480, 1448, 1431, 1417, 1394, 1362, 1292, 1256, 1213, 1156, 1131, 1080, 1043, 1033, 1009, 931, 902, 876, 847, 786, 770, 743, 727, 704, 696, 676, 643, 627, 549, 506, 480, 445, 412. Elemental analysis (%) calc'd for C50H58MoN4O3: C 69.91, H 6.81, N 6.52; found: C 70.25, H 7.11, N 6.28.
810 L mol−1 cm−1), 342 (ε = 35430 L mol−1 cm−1), 357 (ε = 37800 L mol−1 cm−1), 422 (ε = 7370 L mol−1 cm−1). IR (cm−1): 2955, 2865, 1482, 1429, 1382, 1360, 1331, 1290, 1270, 1256, 1231, 1184, 1151, 1117, 1043, 996, 923, 904, 876, 855, 800, 780, 763, 749, 731, 704, 678, 643, 625, 610, 557, 512, 486, 443, 425. Elemental analysis (%) calc'd for C54H59MoN3O3·0.33CH2Cl2: C 70.75, H 6.52, N 4.56; found: C 70.70, H 6.81, N 4.54.
510 L mol−1 cm−1). IR (cm−1): 2955, 2903, 2867, 1601, 1525, 1480, 1443, 1417, 1394, 1362, 1292, 1254, 1202, 1186, 1153, 1127, 1074, 1033, 921, 874, 847, 800, 757, 737, 696, 672, 639, 618, 547, 490, 471, 445, 420. Elemental analysis (%) calc'd for C50H58MoN4O2S·0.33CH2Cl2: C 66.92, H 6.55, N 6.20; found: C 66.66, H 6.71, N 6.25.
500 L mol−1 cm−1), 398 (ε = 127610 L mol−1 cm−1). IR (cm−1): 2955, 2867, 1597, 1480, 1441, 1429, 1382, 1360, 1331, 1290, 1266, 1256, 1229, 1205, 1182, 1160, 1133, 1082, 1068, 1031, 988, 921, 876, 855, 800, 761, 745, 737, 698, 672, 645, 623, 610, 553, 476, 443, 420. Elemental analysis (%) calc'd for C54H59MoN3O2S: C 71.27, H 6.53, N 4.62; found: C 71.26, H 6.80, N 4.59.
590 L mol−1 cm−1), 444 (ε = 67400 L mol−1 cm−1). IR (cm−1): 2955, 2869, 1523, 1478, 1445, 1417, 1388, 1362, 1290, 1233, 1205, 1156, 1127, 1088, 1074, 1055, 1037, 957, 921, 874, 849, 800, 772, 757, 745, 733, 712, 692, 643, 608, 557, 541, 500, 478, 447, 416. Elemental analysis (%) calc'd for C40H54MoN4O3·C4H8O: C 65.49, H 7.74, N 6.94; found: C 65.80, H 7.83, N 6.91.
740 L mol−1 cm−1), 348 (ε = 73530 L mol−1 cm−1), 459 (ε = 112790 L mol−1 cm−1). IR (cm−1): 2955, 2906, 2865, 1472, 1431, 1386, 1360, 1339, 1288, 1266, 1237, 1158, 1133, 1117, 1070, 1037, 988, 957, 919, 874, 853, 800, 763, 743, 731, 694, 676, 639, 610, 582, 559, 549, 488, 447, 437. Elemental analysis (%) calc'd for C44H55MoN3O3: C 68.65, H 7.20, N 5.46; found: C 68.07, H 7.22, N 5.40.
830 L mol−1 cm−1), 492 (ε = 149130 L mol−1 cm−1). IR (cm−1): 2955, 2906, 2869, 1605, 1525, 1480, 1445, 1417, 1394, 1362, 1292, 1254, 1202, 1153, 1127, 1086, 1074, 1053, 1037, 921, 874, 849, 800, 772, 757, 743, 712, 688, 678, 641, 551, 490, 476, 447, 418. Elemental analysis (%) calc'd for C40H54MoN4O2S·0.5C6H6: C 65.38, H 7.27, N 7.09; found: C 65.39, H 7.41, N 6.89.
Data availability
The spectral data (NMR, IR) as well as electrochemical and computational data supporting this article are available in the ESI† of this article. Crystallographic data have been deposited at the CCDC (for CCDC numbers, see ESI, Tables S1 and S2†) and can be obtained from https://www.ccdc.cam.ac.uk/strucutres.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded in whole or in part by the Austrian Science Fund (FWF); Grant-DOI: 10.55776/P34626. For open access purposes, the author has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission. We are furthermore thankful to the University of Innsbruck as well as the University of Paderborn for partial funding of this work. The computational results presented here have been achieved (in part) using the LEO HPC infrastructure of the University of Innsbruck. F. R. S. Purtscher is kindly acknowledged for help in setting up the theoretical calculations.Notes and references
- (a) R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed; (b) J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- (a) https://cen.acs.org/business/petrochemicals/ammonia-fuel-future/99/i8 ; (b) M. B. Bertagni, R. H. Socolow, J. M. P. Martirez, E. A. Carter, C. Greig, Y. Ju, T. Lieuwen, M. E. Mueller, S. Sundaresan, R. Wang, M. A. Zondlo and A. Porporato, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2311728120 CrossRef CAS PubMed; (c) S. Chen, J. Jelic, D. Rein, S. Najafishirtari, F.-P. Schmidt, F. Girgsdies, L. Kang, A. Wandzilak, A. Rabe, D. E. Doronkin, J. Wang, K. Friedel Ortega, S. DeBeer, J.-D. Grunwaldt, R. Schlögl, T. Lunkenbein, F. Studt and M. Behrens, Nat. Commun., 2024, 15, 871 CrossRef CAS PubMed.
- L. Hollevoet, M. de Ras, M. Roeffaers, J. Hofkens and J. A. Martens, ACS Energy Lett., 2020, 5, 1124–1127 CrossRef CAS.
- (a) Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2019, 389, 73–93 CrossRef CAS; (b) Z.-J. Lv, J. Wei, W.-X. Zhang, P. Chen, D. Deng, Z.-J. Shi and Z. Xi, Natl. Sci. Rev., 2020, 7, 1564–1583 CrossRef CAS PubMed; (c) Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2022, 472, 214783 CrossRef CAS; (d) D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed; (e) S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed; (f) M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed; (g) Y. Tanabe and Y. Nishibayashi, Angew. Chem., Int. Ed., 2024, e202406404 CAS; (h) S. Suginome and Y. Nishibayashi, ChemCatChem, 2023, 15, e202300850 CrossRef CAS; (i) L. Alig, M. Finger and S. Schneider, Inorganic Chemistry in Germany, Elsevier, 2023, vol. 82, pp. 1–40 Search PubMed; (j) J. Sun, J. Abbenseth, H. Verplancke, M. Diefenbach, B. de Bruin, D. Hunger, C. Würtele, J. van Slageren, M. C. Holthausen and S. Schneider, Nat. Chem., 2020, 12, 1054–1059 CrossRef CAS PubMed; (k) T. Schmidt-Räntsch, H. Verplancke, J. N. Lienert, S. Demeshko, M. Otte, G. P. van Trieste III, K. A. Reid, H. H. Reibenspies, D. C. Powers, M.C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2022, 61, e202115626 CrossRef.
- (a) J. Chatt, A. J. Pearman and R. L. Richards, Nature, 1975, 253, 39–40 CrossRef CAS; (b) J. Chatt and R. L. Richards, J. Organomet. Chem., 1982, 239, 65–77 CrossRef CAS; (c) C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CrossRef CAS PubMed; (d) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed; (e) R. R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512–5522 CrossRef CAS; (f) J. Junge, T. A. Engesser and F. Tuczek, Chem. – Eur. J., 2023, 29, e202202629 CrossRef CAS; (g) T. A. Engesser, A. Kindjajev, J. Junge, J. Krahmer and F. Tuczek, Chem. – Eur. J., 2020, 26, 14807–14812 CrossRef CAS PubMed; (h) S. Hinrichsen, A.-C. Schnoor, K. Grund, B. Flöser, A. Schlimm, C. Näther, J. Krahmer and F. Tuczek, Dalton Trans., 2016, 45, 14801–14813 RSC; (i) F. Studt and F. Tuczek, Angew. Chem., Int. Ed., 2005, 44, 5639–5642 CrossRef CAS PubMed; (j) G. A. Silantyev, M. Förster, B. Schluschaß, J. Abbenseth, C. Würtele, C. Volkmann, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2017, 56, 5872–5876 CrossRef CAS.
- (a) P. Ghana, F. D. van Krüchten, T. P. Spaniol, J. van Leusen, P. Kögerler and J. Okuda, Chem. Commun., 2019, 55, 3231–3234 RSC; (b) H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS; (c) A. Wong, M. Hernandez, T. Ochiai, G. Rao, D. Britt, N. Kaltsoyannis and P. Arnold, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-ph3qr; (d) T. Shima, Q. Zhuo, X. Zhou, P. Wu, R. Owada, G. Luo and Z. Hou, Nature, 2024, 632, 307–312 CrossRef CAS.
- (a) W. Huang, L.-Y. Peng, J. Zhang, C. Liu, G. Song, J.-H. Su, W.-H. Fang, G. Cui and S. Hu, J. Am. Chem. Soc., 2023, 145, 811–821 CrossRef CAS; (b) L. Wang, Y. Liu, H. Wang, T. Yang, Y. Luo, S. Lee, M. G. Kim, T. T. T. Nga, C.-L. Dong and H. Lee, ACS Nano, 2023, 17, 7406–7416 CrossRef CAS; (c) D. King, J. Catal., 1965, 4, 253–259 CrossRef CAS; (d) Y. Cao, A. Saito, Y. Kobayashi, H. Ubukata, Y. Tang and H. Kageyama, ChemCatChem, 2021, 13, 191–195 CrossRef CAS.
- Y. Ashida, A. Egi, K. Arashiba, H. Tanaka, T. Mitsumoto, S. Kuriyama, K. Yoshizawa and Y. Nishibayashi, Chem. – Eur. J., 2022, 28, e202200557 CrossRef CAS.
- (a) F. Tuczek, A.-M. Vogt, T. A. Engesser, J. Krahmer, N. Michaelis, M. Pfeil, J. Junge, C. Näther and N. Le Poul, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-02bp6; (b) J. Junge, S. Froitzheim, T. A. Engesser, J. Krahmer, C. Näther, N. Le Poul and F. Tuczek, Dalton Trans., 2022, 51, 6166–6176 RSC.
- (a) F. Meng, S. Kuriyama, A. Egi, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Organometallics, 2023, 42, 1065–1076 CrossRef CAS; (b) F. Meng, S. Kuriyama, H. Tanaka, A. Egi, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., 2021, 133, 14025–14031 CrossRef.
- (a) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed; (b) S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105–1115 CrossRef CAS; (c) S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef CAS PubMed; (d) H. Broda and F. Tuczek, Angew. Chem., Int. Ed., 2014, 53, 632–634 CrossRef CAS.
- (a) T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 10782–10783 CrossRef CAS PubMed; (b) Y. Jiang, R. Takashima, T. Nakao, M. Miyazaki, Y. Lu, M. Sasase, Y. Niwa, H. Abe, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2023, 145, 10669–10680 CrossRef CAS PubMed; (c) S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 14291–14295 CrossRef CAS PubMed.
- S. Kim, H. Zhong, Y. Park, F. Loose and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 9518–9524 CrossRef CAS.
- A. Wong, F. Y. Lam, M. Hernandez, J. Lara, T. M. Trinh, R. P. Kelly, T. Ochiai, G. Rao, R. D. Britt, N. Kaltsoyannis and P. L. Arnold, Chem Catal., 2024, 4, 100964 CrossRef CAS.
- P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS.
- (a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed; (b) M.-A. Légaré, G. Bélanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 12, 1076–1080 CrossRef; (c) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef.
- (a) Y. Ashida, T. Mizushima, K. Arashiba, A. Egi, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Nat. Synth., 2023, 2, 635–644 CrossRef CAS; (b) A. Eizawa, K. Arashiba, H. Tanaka, S. Kuriyama, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2017, 8, 14874 CrossRef CAS PubMed.
- A. Egi, H. Tanaka, T. Nakamura, K. Arashiba, Y. Nishibayashi and K. Yoshizawa, Bull. Chem. Soc. Jpn., 2024, 97, uoae041 CrossRef.
- D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- H. Aihara, T. Matsuo and H. Kawaguchi, Chem. Commun., 2003, 2204–2205 RSC.
- (a) A. El-Batta, A. W. Waltman and R. H. Grubbs, J. Organomet. Chem., 2011, 696, 2477–2481 CrossRef CAS; (b) G. M. Miyake, M. N. Akhtar, A. Fazal, E. A. Jaseer, C. S. Daeffler and R. H. Grubbs, J. Organomet. Chem., 2013, 728, 1–5 CrossRef CAS; (c) A. W. Waltman and R. H. Grubbs, Organometallics, 2004, 23, 3105–3107 CrossRef CAS.
- E. Despagnet-Ayoub, L. M. Henling, J. A. Labinger and J. E. Bercaw, Dalton Trans., 2013, 42, 15544–15547 RSC.
- D. R. Weinberg, N. Hazari, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 89–100 CrossRef CAS.
- S. Bellemin-Laponnaz, R. Welter, L. Brelot and S. Dagorne, J. Organomet. Chem., 2009, 694, 604–606 CrossRef CAS.
- E. Borré, G. Dahm, A. Aliprandi, M. Mauro, S. Dagorne and S. Bellemin-Laponnaz, Organometallics, 2014, 33, 4374–4384 CrossRef.
- (a) S. Dagorne, S. Bellemin-Laponnaz and C. Romain, Organometallics, 2013, 32, 2736–2743 CrossRef CAS; (b) C. Romain, L. Brelot, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2010, 29, 1191–1198 CrossRef CAS; (c) C. Romain, S. Choua, J.-P. Collin, M. Heinrich, C. Bailly, L. Karmazin-Brelot, S. Bellemin-Laponnaz and S. Dagorne, Inorg. Chem., 2014, 53, 7371–7376 CrossRef CAS; (d) C. Romain, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2014, 33, 5730–5739 CrossRef CAS; (e) C. Romain, K. Miqueu, J.-M. Sotiropoulos, S. Bellemin-Laponnaz and S. Dagorne, Angew. Chem., Int. Ed., 2010, 49, 2198–2201 CrossRef CAS; (f) C. Romain, D. Specklin, K. Miqueu, J.-M. Sotiropoulos, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2015, 34, 4854–4863 CrossRef CAS.
- G. A. Gurina, A. V. Markin, A. V. Cherkasov, I. A. Godovikov, A. M. Ob’edkov and A. A. Trifonov, Eur. J. Inorg. Chem., 2023, e202300392 CrossRef CAS.
- S. Liu, J. I. Amaro-Estrada, M. Baltrun, I. Douair, R. Schoch, L. Maron and S. Hohloch, Organometallics, 2021, 40, 107–118 CrossRef CAS.
- M. Baltrun, F. A. Watt, R. Schoch, C. Wölper, A. G. Neuba and S. Hohloch, Dalton Trans., 2019, 48, 14611–14625 RSC.
- M. Baltrun, F. A. Watt, R. Schoch and S. Hohloch, Organometallics, 2019, 38, 3719–3729 CrossRef CAS.
- F. R. Neururer, S. Liu, D. Leitner, M. Baltrun, K. R. Fisher, H. Kopacka, K. Wurst, L. J. Daumann, D. Munz and S. Hohloch, Inorg. Chem., 2021, 60, 15421–15434 CrossRef CAS PubMed.
- F. R. Neururer, K. Huter, M. Seidl and S. Hohloch, ACS Org. Inorg. Au, 2023, 3, 59–71 CrossRef CAS.
- F. R. Neururer, D. Leitner, S. Liu, K. Wurst, H. Kopacka, M. Seidl and S. Hohloch, Eur. J. Inorg. Chem., 2023, e202300180 CrossRef CAS.
- (a) A. Pavun, R. Niess, L. A. Scheibel, M. Seidl and S. Hohloch, Dalton Trans., 2024, 53, 2749–2761 RSC; (b) F. A. Watt, B. Sieland, N. Dickmann, R. Schoch, R. Herbst-Irmer, H. Ott, J. Paradies, D. Kuckling and S. Hohloch, Dalton Trans., 2021, 50, 17361–17371 RSC; (c) B. Wittwer, N. Dickmann, S. Berg, D. Leitner, L. Tesi, D. Hunger, R. Gratzl, J. van Slageren, N. I. Neuman, D. Munz and S. Hohloch, Chem. Commun., 2022, 58, 6096–6099 RSC; (d) B. Wittwer, D. Leitner, F. R. Neururer, R. Schoch, M. Seidl, J. Pecak, M. Podewitz and S. Hohloch, Polyhedron, 2024, 250, 116786 CrossRef CAS; (e) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (f) R. Maity and B. Sarkar, JACS Au, 2022, 2, 22–57 CrossRef CAS; (g) W. Stroek and M. Albrecht, Chem. Soc. Rev., 2024, 53, 6322–6344 RSC; (h) P. Dierks, A. Kruse, O. S. Bokareva, M. J. Al-Marri, J. Kalmbach, M. Baltrun, A. Neuba, R. Schoch, S. Hohloch, K. Heinze, M. Seitz, O. Kühn, S. Lochbrunner and M. Bauer, Chem. Commun., 2021, 57, 6640–6643 RSC; (i) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- L. Hettmanczyk, S. J. P. Spall, S. Klenk, M. van der Meer, S. Hohloch, J. A. Weinstein and B. Sarkar, Eur. J. Inorg. Chem., 2017, 2112–2121 CrossRef CAS.
- S. Hohloch, S. Kaiser, F. L. Duecker, A. Bolje, R. Maity, J. Košmrlj and B. Sarkar, Dalton Trans., 2015, 44, 686–693 RSC.
- S. Hohloch, L. Suntrup and B. Sarkar, Inorg. Chem. Front., 2016, 3, 67–77 RSC.
- (a) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 3164–3171 CrossRef CAS; (b) L. Suntrup, S. Hohloch and B. Sarkar, Chem. – Eur. J., 2016, 22, 18009–18018 CrossRef CAS; (c) D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Chem. Commun., 2014, 50, 2431–2433 RSC; (d) P. Pinter, C. M. Schüßlbauer, F. A. Watt, N. Dickmann, R. Herbst-Irmer, B. Morgenstern, A. Grünwald, T. Ullrich, M. Zimmer, S. Hohloch, D. M. Guldi and D. Munz, Chem. Sci., 2021, 12, 7401–7410 RSC.
- (a) R. Maity, A. Verma, M. van der Meer, S. Hohloch and B. Sarkar, Eur. J. Inorg. Chem., 2016, 111–117 CrossRef CAS; (b) M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2016, 46, 86–95 RSC; (c) W. Stroek, N. A. V. Rowlinson, M. Keilwerth, D. M. Pividori, K. Meyer and M. Albrecht, Organometallics, 2024, 43, 1386–1392 CrossRef CAS; (d) W. Stroek, M. Keilwerth, D. M. Pividori, K. Meyer and M. Albrecht, J. Am. Chem. Soc., 2021, 143, 20157–20165 CrossRef CAS PubMed; (e) S. Friães, C. S. B. Gomes and B. Royo, Organometallics, 2023, 42, 1803–1809 CrossRef.
- S. Hohloch, D. Scheiffele and B. Sarkar, Eur. J. Inorg. Chem., 2013, 3956–3965 CrossRef CAS.
- S. Hohloch, B. Sarkar, L. Nauton, F. Cisnetti and A. Gautier, Tetrahedron Lett., 2013, 54, 1808–1812 CrossRef CAS.
- S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067–3075 CrossRef CAS.
- Z. Li, C. Liu, J. An, X. Wang and S. Hu, ACS Catal., 2024, 14, 6558–6564 CrossRef CAS.
- D. M. T. Chan, M. H. Chisholm, K. Folting, J. C. Huffman and N. S. Marchant, Inorg. Chem., 1986, 25, 4170–4174 CrossRef CAS.
- (a) C. H. Leung, C. D. Incarvito and R. H. Crabtree, Organometallics, 2006, 25, 6099–6107 CrossRef CAS; (b) J. Petit, P.-A. Pavard and C. Camp, Mendeleev Commun., 2021, 31, 51–53 CrossRef CAS.
- (a) C. Bittner, H. Ehrhorn, D. Bockfeld, K. Brandhorst and M. Tamm, Organometallics, 2017, 36, 3398–3406 CrossRef CAS; (b) M. H. Chisholm, K. Folting, J. C. Huffman and A. L. Ratermann, Inorg. Chem., 1984, 23, 613–618 CrossRef CAS; (c) M. H. Chisholm, D. L. Clark and J. C. Huffman, Polyhedron, 1985, 4, 1203–1211 CrossRef CAS; (d) M. H. Chisholm, K. Folting, W. E. Streib and D.-D. Wu, Inorg. Chem., 1999, 38, 5219–5229 CrossRef CAS; (e) M. H. Chisholm, C. E. Hammond, M. Hampden-Smith, J. C. Huffmann and W. G. van der Sluys, Angew. Chem., Int. Ed. Engl., 1987, 26, 904–906 CrossRef; (f) M. H. Chisholm, F. A. Cotton, C. A. Murillo and W. W. Reichert, Inorg. Chem., 1977, 16, 1801–1808 CrossRef CAS; (g) M. H. Chisholm, J.-H. Huang, J. C. Huffman and I. P. Parkin, Inorg. Chem., 1997, 36, 1642–1651 CrossRef CAS PubMed; (h) M. H. Chisholm, J. C. Huffman and R. J. Tatz, J. Am. Chem. Soc., 1983, 105, 2075–2077 CrossRef CAS; (i) T. W. Coffindaffer, I. P. Rothwell and J. C. Huffman, Inorg. Chem., 1983, 22, 2906–2910 CrossRef CAS; (j) T. M. Gilbert, C. B. Bauer, A. H. Bond and R. D. Rogers, Polyhedron, 1999, 18, 1293–1301 CrossRef CAS; (k) K. L. Fujdala and T. D. Tilley, Chem. Mater., 2004, 16, 1035–1047 CrossRef CAS; (l) T. M. Gilbert, A. M. Landes and R. D. Rogers, Inorg. Chem., 1992, 31, 3438–3444 CrossRef CAS; (m) T. M. Gilbert, J. C. Littrell, C. E. Talley, M. A. Vance, R. F. Dallinger and R. D. Rogers, Inorg. Chem., 2004, 43, 1762–1769 CrossRef CAS; (n) S. Krackl, J.-G. Ma, Y. Aksu and M. Driess, Eur. J. Inorg. Chem., 2011, 1725–1732 CrossRef CAS; (o) S. Krackl, C. I. Someya and S. Enthaler, Chem. – Eur. J., 2012, 18, 15267–15271 CrossRef CAS; (p) U. Piarulli, D. N. Williams, C. Floriani, G. Gervasio and D. Viterbo, J. Organomet. Chem., 1995, 503, 185–192 CrossRef CAS.
- Please note that this order is deduced from the reduction potentials in this specific system and does not display a general order of the donor strength of these ligands in other complexes.
- L. A. Wickramasinghe, T. Ogawa, R. R. Schrock and P. Müller, J. Am. Chem. Soc., 2017, 139, 9132–9135 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2364759–2364775. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02405b |
| This journal is © The Royal Society of Chemistry 2025 |