
Synthesis, structure, oxygen evolution reaction (OER) and visible-light assisted organic reaction studies on A2M2TeB2O10 (A = Ba and Pb; M = Mg, Zn, Co, Ni, Cu, and Fe)†
Indrani G.
Shanmugapriya‡
,
Shreenibasa
Sa‡
and
Srinivasan
Natarajan
 *
*
Framework Solids Laboratory, Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore – 560012, India. E-mail: snatarajan@iisc.ac.in
First published on 4th December 2024
Abstract
Compounds with the general formula A2M2TeB2O10 (A = Ba and Pb; M = Mg, Zn, Co, Ni, Cu, and Fe) have been synthesised via solid-state techniques and characterised. The structure exhibits M2B2O10 layers connected by TeO6 octahedra giving rise to a three-dimensional structure with voids, where Ba2+ ions reside. Substitution of Mg by transition elements (M = Co, Ni, and Cu) in Ba2Mg2TeB2O10 and (Ba0.5Pb1.5)Mg2TeB2O10 gives rise to interesting colored compounds. NIR reflectivity studies indicated that white-colored compounds exhibited good NIR reflectivity, which was is comparable to that of TiO2. Dielectric studies indicated reasonable values with low dielectric loss at low frequencies. The cobalt-substituted compounds Ba2(MgCo)TeB2O10 and (Ba0.5Pb1.5)(MgCo)TeB2O10 were explored towards the oxygen evolution reaction (OER) in an alkaline medium. The compound (Ba0.5Pb1.5)(MgCo)TeB2O10 was found to be a good electrocatalyst for the OER with a faradaic efficiency of ∼96%. The Cu-substituted compound Ba2(Mg1.5Cu0.5)TeB2O10 was found to be a good photocatalyst for the formation of α-chloroketones under visible light in the presence of molecular oxygen.
Introduction
Compounds based on borates have been explored over the years for their many properties in glasses, catalysis, non-linear optical materials and as a host for luminescence.1 The oxyanions of boron have become one of the important constituents of solid-state chemistry.2 Studies carried out on borate-containing compounds illustrate the rich diversity in their structural arrangement and properties.3,4 It is becoming clear that borate compounds with their unique structures, some of which are related to minerals,5,6 could be harnessed towards new advanced functional materials. The growth of borate-based compounds is so rapid that it is important to understand their structure–property–function relationships to provide guidance for future research. In order to conduct such a study, we considered a recently reported borate compound, A2Mg2TeB2O10 (A = Ba and Pb).7Water splitting employing electrocatalysis has received much attention in recent years with many reviews summarizing its various aspects and highlighting its importance.8–10 Among the reactions, hydrogen evolution reactions (HERs) and oxygen evolution reactions (OERs) are the two most important reactions that are being actively pursued. Among the HER and OER, the OER involves a four-electron transfer and is the key process that governs overall electrochemical water splitting. Of many oxides that have been studied for OER catalysis, IrO2 and RuO2 are in the pole position.11,12 The overall cost of noble metals and their availability have prompted many researchers to look actively for alternatives that are based on non-precious elements and could be cost-effective.13–16 One such alternative could be compounds based on borates. It has been showed that borates could be good electrocatalysts for the OER.17–19
The borate compound A2Mg2TeB2O10 (A = Ba and Pb) appears to have an interesting structure.7 B is three coordinated and forms [BO3] isolated units, which link with octahedral Mg2+ ions forming [Mg2B2O10]10−∞ two dimensional layers. The layers are connected by [TeO6]6− octahedra, forming a three-dimensional structure with voids, where Ba2+/Pb2+ ions are located. Octahedral Mg2+ ions provide a good opportunity to substitute other transition elements, which can exhibit electrocatalytic behavior. In this paper, we explored the borate compound A2Mg2TeB2O10 (A = Ba and Pb) towards new colored materials as well as their photocatalytic20,21 and electrochemical behavior.
Experimental section
Stoichiometric quantities of the respective metal salts and oxides were weighed and mixed using a pestle and mortar. A 10% excess of boric acid was taken in the initial mixture to compensate for possible losses at the elevated temperature. The synthesis was carried out by employing the solid-state technique. The starting mixtures were heated in the temperature ranges of 800–900 °C for 12–24 h duration with many intermittent grindings.The prepared samples were characterised by powder X-ray diffraction (PXRD) (PANalytical Empyrean-X-ray diffractometer using nickel filtered Cu Kα (λ = 1.5417 Å) in the 2θ range of 10°–80° with a step size 0.02°). The observed PXRD patterns were compared with the simulated PXRD pattern of the Ba2Mg2TeB2O10 structure (Fig. 1).7 The compositional analysis was carried out using energy-dispersive X-ray (EDX) spectroscopy (SEM-EDX) and elemental mapping was carried out using a JEOL SEM IT300 instrument.
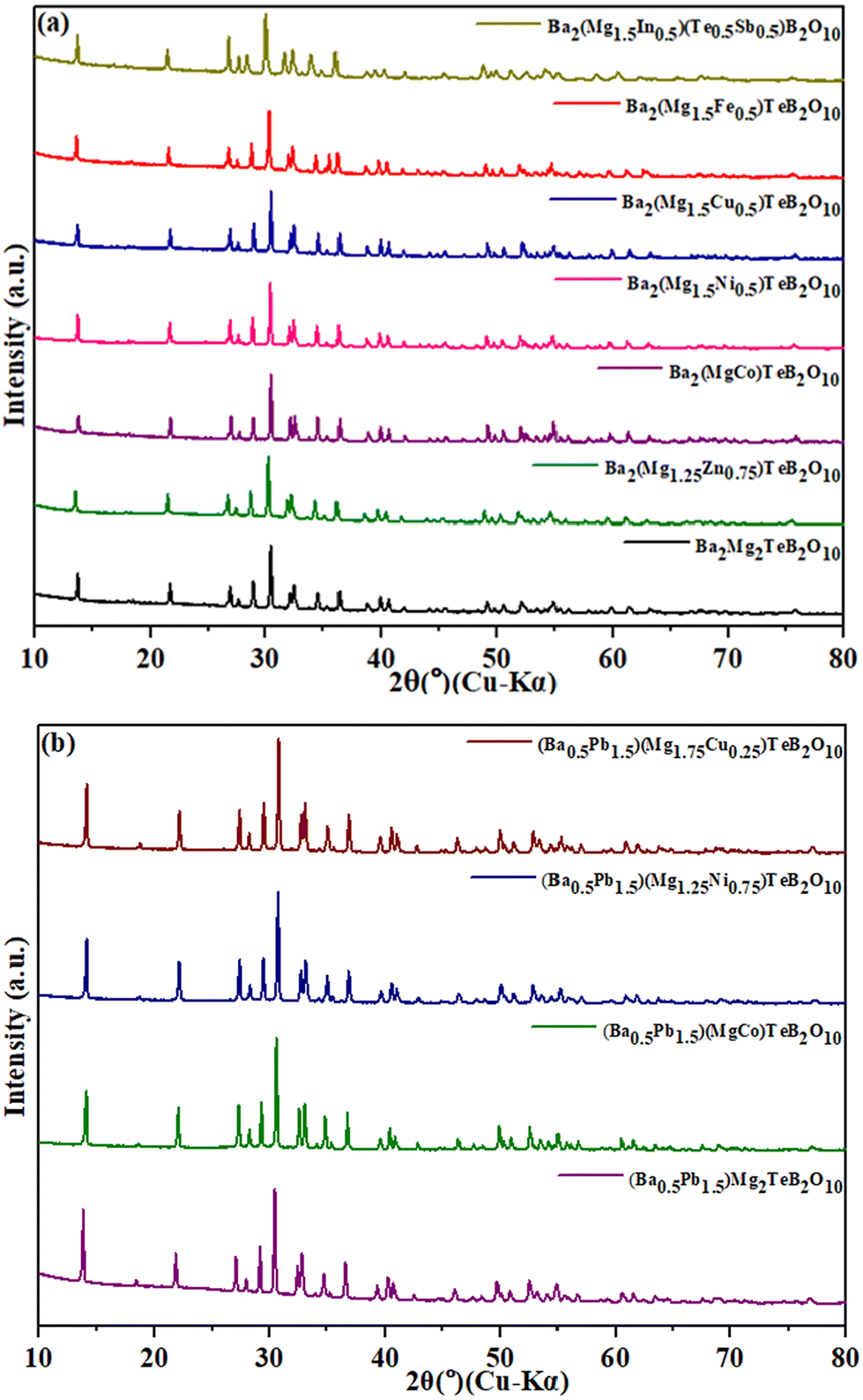 | ||
| Fig. 1 (a) PXRD patterns of the Ba2(Mg2−xMx)TeB2O10 (M = Co, Ni, Cu, Fe, and Zn) and (b) (Ba0.5Pb1.5)(Mg2−xMx)TeB2O10 (M = Co, Ni, and Cu) compounds. | ||
For the Rietveld refinement of the prepared compounds the PXRD data was collected at room temperature in the 2θ range of 10–120° with a step size of 0.02° and step duration of 50 s. The Rietveld refinements for the selected compounds were carried out using GSAS-II program.22,23 The lattice parameters, scale factors, background (Fourier polynomial background function), pseudo-Voigt (U, V, W, and X), and isothermal temperature factors (Uiso) were refined. Thermal parameters were constrained to be the same for the atoms occupying the same sites. The optical absorption and NIR reflectance for all the samples were recorded at room temperature (PerkinElmer Lambda 950 UV/vis double beam spectrometer, range of 200–2500 nm). X-ray photoelectron spectroscopy (XPS) was used to determine the oxidation states of the ions. Dielectric measurements [Novocontrol impedance analyzer (Alpha-A)] were carried out in the frequency range 105 Hz–0.1 Hz at room temperature. Raman spectroscopic studies (HORIBA Lab RAM HR Evolution) were carried out in the range of 50 to 1400 cm−1. 1H NMR spectra were recorded on a Bruker instrument. The chemical shifts were relative to the CDCl3.
The electrochemical performance of (Ba0.5Pb1.5)(MgCo)TeB2O10 was studied employing a PARSTAT analytical electrochemical workstation (PAR) using the three-electrode system. The glassy carbon along with the catalyst was employed as the working electrode, the Hg/HgO electrode was employed as the reference electrode and a graphite rod was employed as the counter electrode. The electrolyte was 0.5 M KOH. The electrocatalytic performance towards the OER was evaluated employing an overpotential 10 mA cm−2.24
The photocatalytic reactions were performed by employing a modified procedure.20,21 A 20 mg of the photocatalyst (Ba2(Mg1.5Cu0.5)TeB2O10), in acetonitrile (3 mL) along with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of vinyl arene and acetyl chloride were taken in a 10 mL round-bottom flask. The RB flask along with the oxygen balloon was placed under a 60 W LED bulb at room temperature and the reaction was carried out for 12 h under constant stirring. The progress of the reaction was monitored by thin-layer chromatography (TLC). After the completion of the reaction, the compound was isolated by column chromatography using silica gel and a mixture of hexane and ethyl acetate as the eluent.
:3 ratio of vinyl arene and acetyl chloride were taken in a 10 mL round-bottom flask. The RB flask along with the oxygen balloon was placed under a 60 W LED bulb at room temperature and the reaction was carried out for 12 h under constant stirring. The progress of the reaction was monitored by thin-layer chromatography (TLC). After the completion of the reaction, the compound was isolated by column chromatography using silica gel and a mixture of hexane and ethyl acetate as the eluent.
Results and discussion
Synthesis and structural characterization
The compounds, A2Mg2TeB2O10 (A = Ba, Pb) have been reported earlier (Fig. S1†). The main feature of the structure is the framework [Mg2TeB2O10]4− having voids where the Ba2+/Pb2+ ions reside. We made attempts to prepare newer analogues of the structure by substituting Mg2+ ions (octahedrally coordinated) with other transition elements, which may give rise to interesting-colored compounds under day light.All the compounds were prepared by solid-state methods and characterized by the PXRD method. The list of the prepared compounds is given in Table 1. The compound, Ba2(MgCo)TeB2O10, was refined using the Ba2Mg2TeB2O10 structure as the model (Fig. 2).7
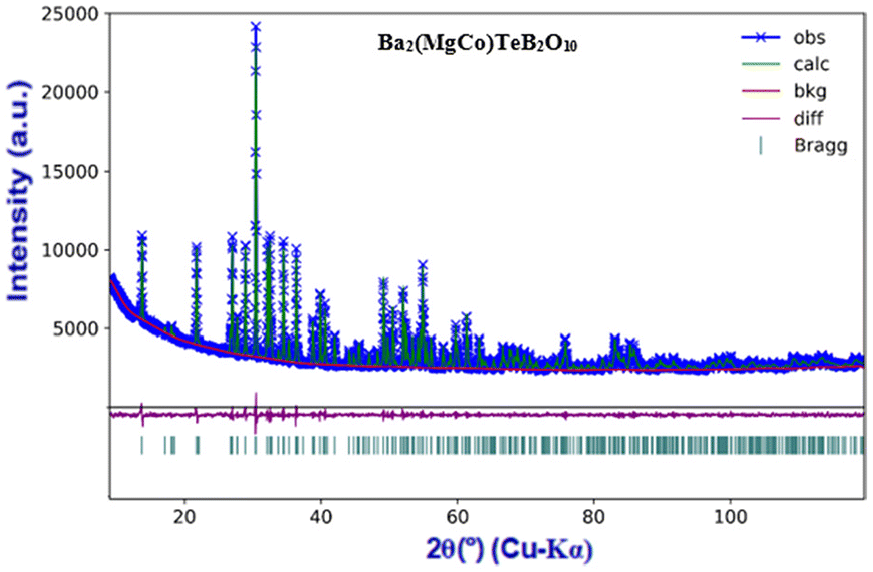 | ||
| Fig. 2 Rietveld refinement of Ba2(MgCo)TeB2O10 from the PXRD data. The observed (×), calculated (green line), and difference (bottom cyan line) profiles are shown. The vertical bars (|) indicate Bragg reflections. | ||
| Compounds | Temperature (°C)/time (h) | Color as seen under daylight |
|---|---|---|
| Ba2Mg2TeB2O10 | 800/12; 840/12 |

|
| Ba2Mg1.25Zn0.75TeB2O10 | 800/12; 840/12 |

|
| Ba2MgCoTeB2O10 | 900/12 |

|
| Ba2Mg1.50Ni0.50TeB2O10 | 870/12; 900/12 |

|
| Ba2Mg1.50Cu0.50TeB2O10 | 870/12; 900/12 |

|
| Ba2Mg1.50Fe0.50TeB2O10 | 850/12; 920/12 |

|
| Ba2Mg1.50In0.50Te0.50Sb0.50B2O10 | 800/12; 840/12 |

|
| Ba0.50Pb1.50Mg2TeB2O10 | 800/12 |

|
| Ba0.50Pb1.50MgCoTeB2O10 | 820/12; 840/12 |

|
| Ba0.50Pb1.50Mg1.25Ni0.75TeB2O10 | 800/12; 840/12 |

|
| Ba0.50Pb1.50Mg1.75Cu0.25TeB2O10 | 800/12 |

|
The structure of Ba2(MgCo)TeB2O10 comprises (MgCo)B2O10 layers connected by TeO6 octahedra forming a three-dimensional structure with voids, wherein the Ba2+ ions are located (Fig. 3).
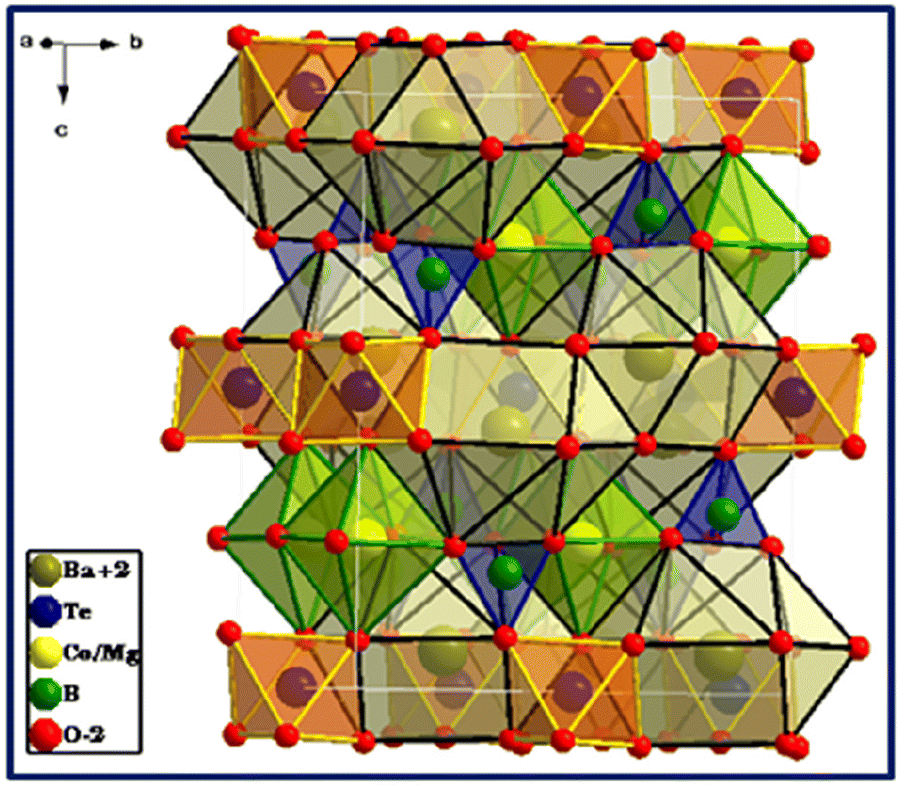 | ||
| Fig. 3 Crystal structure of Ba2(MgCo)TeB2O10 drawn from Rietveld refinement. | ||
All the bond distances are in the expected ranges (Table 2). We were successful in partially substituting Sb5+ ions in place of Te6+ ions (50%) by introducing In3+ ions in place of Mg2+ ions forming a Ba2(Mg1.5In0.5)(Te0.5Sb0.5)B2O10 compound. The SEM and EDX analysis indicated that the prepared compounds show expected composition of the samples (Fig. S2†).
| Atom | Site | x/a | y/b | z/c | S.O.F. | U iso |
|---|---|---|---|---|---|---|
| Space group: Cmca (64); a = 6.1997(5) Å; b = 10.4030 (4) Å; c = 12.9452 (4) Å; α = β = γ = 90°; Z = 4; V = 834.929 Å3. Reliability factors: Rp = 1.77%, Rwp = 2.38%, χ2 = 1.94. Bond length: Ba–O: 2.896 Å (avg.); Te–O:1.9698 Å (avg.); Mg/Co–O: 2.1209 Å (avg.); B–O: 1.3806 Å (avg.). Δ = 0.433 [Mg/Co]. Δ is the polyhedral distortion parameter defined by Δ = 1/N∑[{(ri − r)/r}2]103; where N is the number of bonds and ri and r are individual and average bond lengths, respectively. | ||||||
| Ba1 | 8 f | 0.5 | 0.1635(9) | 0.4348(1) | 1.0 | 0.0052(9) |
| Te1 | 4 b | 0.0 | 0.0 | 0.5 | 1.0 | 0.0033(2) |
| Mg1 | 8 e | 0.25 | 0.0866(3) | 0.75 | 0.5 | 0.0051(9) |
| Co1 | 8 e | 0.25 | 0.0866(3) | 0.75 | 0.5 | 0.0051(9) |
| B1 | 8 f | 0.0 | 0.1681(9) | 0.3021(3) | 1.0 | 0.0056(5) |
| O1 | 8 f | 0.5 | 0.0487(4) | 0.2528(3) | 1.0 | 0.0043(4) |
| O2 | 16 g | 0.2257 (9) | 0.0826(4) | 0.5812(9) | 1.0 | 0.0087(9) |
| O3 | 8 f | 0.0 | 0.1580(2) | 0.4105(5) | 1.0 | 0.0091(1) |
| O4 | 8 f | 0.0 | 0.2799(8) | 0.2561(6) | 1.0 | 0.0037(1) |
Raman spectroscopic studies
The prepared compounds were also characterised by Raman spectroscopic studies (Fig. 4). The main Raman bands were observed in the range of 130–1300 cm−1. The telluroborates and their derivatives crystallize in the orthorhombic crystal system, (Cmca (no. 64)). The presence of a center of symmetry in the space group indicates that the IR active vibration modes and the Raman active modes are mutually exclusive. In the structure of Ba2Mg2TeB2O10, the Ba2+ ions have dodecahedral geometry and Mg2+ and Te6+ ions have octahedral geometry and the B3+ ions have triangular geometry. For all the ions, due to their multiplicities, a total of 102 modes can be observed, out of which 48 modes are expected to be Raman active based on the nuclear site group analysis (Table S1†).25
| ΓRaman (48) = 14Ag + 10B1g + 9B2g + 15B3g |
 | ||
| Fig. 4 Raman spectra for (Ba2−xPbx)(Mg2−xMx)TeB2O10. | ||
The gerade symmetry modes, Ag, B1g, B2g, B3g totalling 48 are Raman active and the ungerade B1u, B2u, B3u symmetry modes (41 in total) are IR active, three ungerade B1u, B2u, and B3u symmetry modes are acoustic and the ten Au symmetry modes are optically inactive/silent modes. We did not observe all the expected Raman modes in our studies, which could be due to smaller intensity, overlapping of the modes and degeneracy.26
Raman spectra for the parent compound Ba2Mg2TeB2O10 and the transition metal substituted ones (Ba2(Mg2−xMx)TeB2O10, M = Fe, Co, Ni, Cu, Zn) indicate that the Raman bands are broader for the later (Fig. S3†). In addition, we observed a small shift in Raman bands compared to that in the parent compounds. The Raman spectra of the Ba2Mg2TeB2O10 compound were found to be sharper compared to that of the transition metal substituted ones (Ba2(Mg2−xMx)TeB2O10, M = Fe, Co, Ni, Cu, Zn). We observed the Ba–O stretching modes in the range of 724–770 cm−1.27
It is known that the isolated octahedra (MgO6 and TeO6) have six vibrational modes.28,29 Of these, three are Raman active (isolated TeO6:A1g ∼ 600–700 cm−1; Eg ∼ 743 cm−1; F2g ∼ 300–350 cm−1; MgO6:A1g ∼ 479–516 cm−1; Eg ∼ 572 cm−1; F2g ∼ 202 cm−1) and two are IR active and one is inactive towards both IR and the Raman. The Raman spectrum for Ba2Mg2TeB2O10 compounds has the bands for TeO6 octahedra in the range of 630–642 cm−1, which can be attributed to the symmetric stretching mode. The asymmetric stretching mode is observed at 724–770 cm−1. The bending vibration of TeO6 octahedra is observed at 383–412 cm−1. For MgO6, the symmetric stretching mode was observed at 440–461 cm−1, the asymmetric stretching mode at 669–683 cm−1 and the bending modes at 204–222 cm−1. The BO3 ion with a D3h symmetry, has four fundamental modes, of which three are Raman-active ( ; E′ ∼ 1250–1400 cm−1;
; E′ ∼ 1250–1400 cm−1; 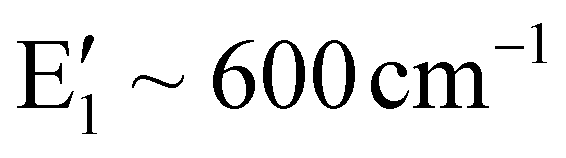 ) and one is infrared active.30,31 In the present compound, we observed the symmetric stretching mode in the range of 943–961 cm−1, the asymmetric stretching in the range of 1192–1208 cm−1 and the bending mode at 592–608 cm−1.
) and one is infrared active.30,31 In the present compound, we observed the symmetric stretching mode in the range of 943–961 cm−1, the asymmetric stretching in the range of 1192–1208 cm−1 and the bending mode at 592–608 cm−1.
When the transition elements were substituted in Ba2(Mg2−xMx)TeB2O10 (M2+ = Fe/Co/Ni/Cu/Zn), the Raman bands were found to be broader and shifted to higher wavenumbers (Table S2†).
For the compound, Ba2(Mg1.5In0.5)(Te0.5Sb0.5)B2O10, the Raman bands appeared broader along with a small shift. Here the octahedral Te6+ ion shares the position with the Sb5+ ion and the Mg2+ ion shares the position with the In3+ ion. The observed Raman bands for Sb5+ and In3+ ions are: Sb5+O6:A1g ∼ 769 cm−1; Eg ∼ 581 cm−1; F2g ∼ 381 cm−1; In3+O6:A1g ∼ 637 cm−1; Eg ∼ 540 cm−1; F2g ∼ 308 cm−1.32,33
As a part of the study, we partially replaced Ba2+ ions with Pb2+ ions. The compound, Pb2Mg2TeB2O10, has Pb2+ ions in a 5-coordinated position.7 When Pb2+ ions are substituted at the Ba2+ ions (11 coordinated) in Ba2Mg2TeB2O10, the Raman bands for the Ba–O were observed in the region of 140–145 cm−1 and the region of 286–291 cm−1 for Pb–O.
Dielectric measurements
We investigated the dielectric behavior of Ba2Mg2TeB2O10, (Ba0.5Pb1.5)Mg2TeB2O10, Ba2(Mg1.5In0.5)(Te0.5Sb0.5)B2O10 compounds. The frequency dependence of the dielectric constant, ε′, and dielectric loss, tanδ, of the prepared compounds were measured at room temperature (Fig. 5).34
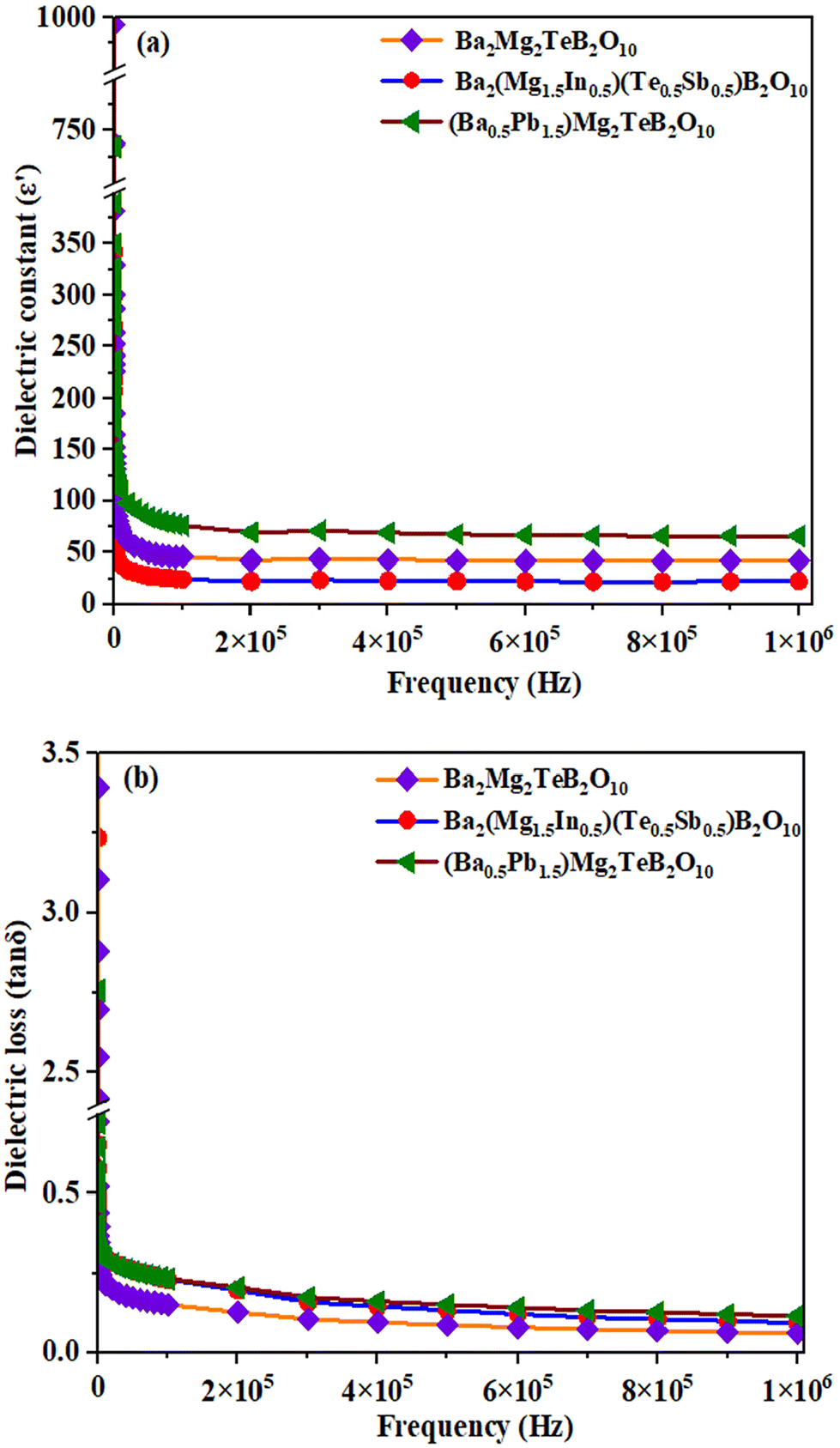 | ||
| Fig. 5 (a) The dielectric constant and (b) dielectric loss versus frequency plots for the compounds at room temperature. | ||
We observed a dielectric constant value of 993 and tanδ of 3.244 for Ba2Mg2TeB2O10 at low frequencies and the values start to decrease as the frequency is increased. The substitution of Pb2+ ions in place of Ba2+ ions in (Ba0.5Pb1.5)Mg2TeB2O10 gives rise to a slightly higher dielectric constant value of 1184 and tanδ value of 2.755.35 The substitution of Sb5+ ions in place of Te6+ ions in Ba2(Mg1.5In0.5)(Te0.5Sb0.5)B2O10, resulted in a decrease in the overall dielectric behavior. Similar behavior has been observed before.36
Normally, the dielectric behavior depends on the various polarizations in the compound such as the ionic, electronic, dipolar, and space charge. At lower frequencies, all the different components would contribute to the total polarizations and result in a larger value for the dielectric constant. As the applied field is increased, the different polarizations are relaxed out, which results in a decrease in the overall dielectric constant. This type of behavior is common in many ceramic oxides.37 The present compounds, however, exhibited smaller dielectric loss values, which may be beneficial towards their use in communication technologies.38
Optical studies
The optical behavior of the compounds was investigated by employing UV-vis and near-IR spectroscopic studies. The transition metal substituted compounds exhibited colors that are expected for the transition elements in octahedral coordination (Table S3 and Fig. S4†).The Co2+ substituted compound, Ba2(Mg2−xCox)TeB2O10 (0 < x ≤ 1) exhibits purple color under daylight. The optical absorption spectra exhibited one main absorption maxima at ∼2.27 eV and another weak absorption at ∼1.67 eV (Fig. 6).
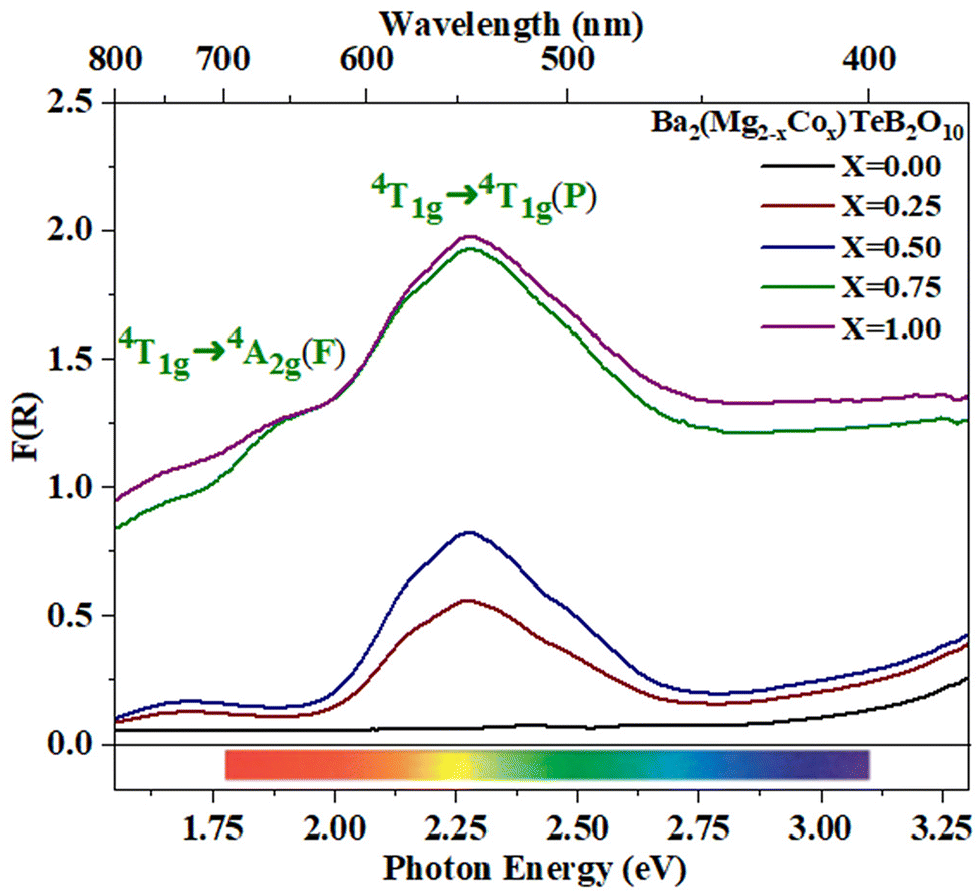 | ||
| Fig. 6 Optical absorption spectra for Ba2(Mg2−xCox)TeB2O10 (0 < x ≤ 1). | ||
The Co2+ ions in an octahedral coordination environment normally exhibit three spin-allowed transitions (1) 4T1g → 4T2g(F), (2) 4T1g → 4A2g(F), and (3) 4T1g → 4T1g(P).39,40 Of these, the 4T1g → 4A2g(F) and 4T1g → 4T1g(P) transitions are observed in the visible region. The main absorption at ∼2.27 eV (546 nm) may be due to the 4T1g → 4T1g(P) transition and the absorption at ∼1.67 eV (742 nm) corresponds to the 4T1g → 4A2g(F) transition. In addition to the main absorption peaks at 2.27 eV, we observed two valleys on either side of the absorption band at ∼1.9 eV (652 nm) and ∼2.75 eV (450 nm). The combination of the absorption bands along with the valleys in the absorption spectra results in a purple color for the cobalt-substituted compounds.
The Ni2+ ions substituted Ba2(Mg2−xNix)TeB2O10 (0 < x ≤ 0.5) exhibits a broad absorption at ∼2.82 eV (Fig. 7). In addition, we also observed weak absorptions at ∼2.40 eV, ∼1.74 eV and ∼1.52 eV. The Ni-substituted compounds have a yellow-lime green color and the absorption spectra exhibit a deep valley of no absorption at ∼2.1 eV (600 nm). The combination of the broad absorption band along with the valley gave rise to the yellow-lime-green color of the compounds. The Ni2+ ions in an octahedral environment have three transitions. (1) 3A2g(3F) → 3T2g(3F), (2) 3A2g(3F) → 3T1g(3F), and (3) 3A2g(3F) → 3T1g(3P).39,40 Of these, the 3A2g(3F) → 3T2g(3F) transition occur in the IR region. The main absorption at ∼2.82 eV (439 nm) can be assigned to the 3A2g(3F) → 3T1g(3P) transition and the weak absorption at ∼1.52 eV (815 nm) could be due to the 3A2g(3F) → 3T1g(3F). The shoulders at 2.40 eV (516 nm) and 1.74 eV (712 nm) could be due to the spin-forbidden transitions from 3A2g(3F) → 1A1g(1G)41 and 3A2g(3F) → 1Eg(1D).42
 | ||
| Fig. 7 Optical absorption spectra of Ba2(Mg2−xNix)TeB2O10 (0 < x ≤ 0.5). | ||
The Cu2+ ions substituted Ba2(Mg2−xCux)TeB2O10 (0 < x ≤ 0.50) compounds exhibit different shades of green under daylight (Fig. S5†). The absorption spectra have a broad absorption at ∼1.75 eV (708 nm). This absorption corresponds to the 2Eg → 2T2g (Cu2+-d9) transition.43 In addition, one can note a valley of low absorption in the region 2.2–2.5 eV. A combination of these results in the observed color for the Cu-substituted compounds. Similar behavior has been observed before.44
The substitution of Fe2+ ions in Ba2(Mg2−xFex)TeB2O10 (0 < x ≤ 0.5) results in a brownish-red colored compound (Table S3†). The UV-vis spectra exhibit a broad absorption in the range of 2.00–2.50 eV with a peak centered at 2.25 eV (Fig. S6†). The Fe2+ ions in an octahedral environment normally exhibit one spin-allowed transition 5T2g → 5Eg in the visible region.45 The broad band centered at 2.25 eV (551 nm) corresponds to the 5T2g → 5Eg transition.
As mentioned before, we have substituted the Pb2+ ions in place of Ba2+ ions in the compounds. It has been shown that the Pb2+ ion-containing compounds exhibited stronger and deeper colored compounds when transition elements are substituted.46 Thus, the cobalt-substituted compounds (Ba0.5Pb1.5)(Mg2−xCox)TeB2O10 (0 < x ≤ 1) were found to have a deeper purple color under daylight (Table S3†). Similar to the Ba2(MgCo)TeB2O10 compounds, the optical absorption spectra exhibited one main absorption maxima centered at 2.29 eV along with a shoulder at ∼1.80 eV (Fig. 8). The main absorption at ∼2.29 eV (541 nm) could be assigned to the 4T1g → 4T1g(P) transition and the shoulder at ∼1.80 eV (689 nm) corresponds to the 4T1g → 4A2g(F) transition.
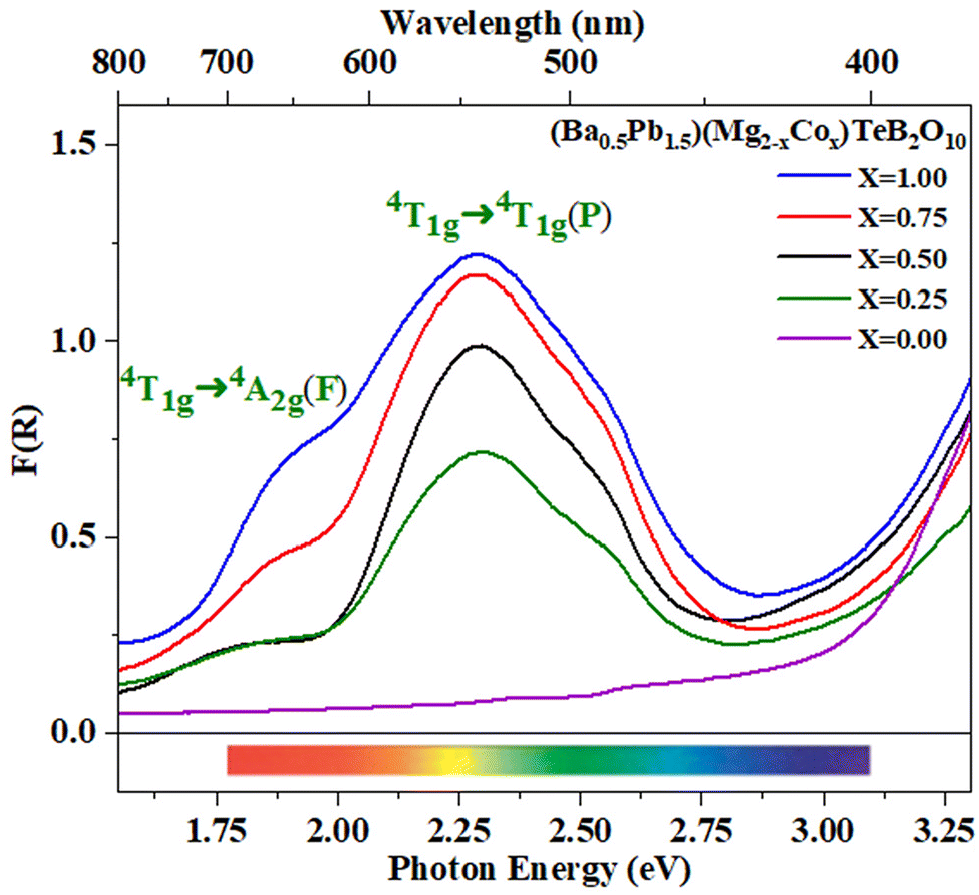 | ||
| Fig. 8 Optical absorption spectra for (Ba0.5Pb1.5)(Mg2−xCox)TeB2O10 (0 < x ≤ 1). | ||
The Ni-substituted (Ba0.5Pb1.5)(Mg2−xNix)TeB2O10 (0 < x ≤ 0.75) compounds exhibit a broad absorption bands at ∼2.91 eV and weak bands at ∼2.47 eV, ∼1.76 eV and ∼1.57 eV (Fig. S7†). The compounds exhibited deeper yellow-lime green color. The main absorption at ∼2.91 eV (426 nm) can be assigned to 3A2g(3F) → 3T1g(3P) transition and the absorption at ∼1.57 eV (789 nm) would be due to 3A2g(3F) → 3T1g(3F). The shoulders at 2.47 eV (502 nm) and 1.76 eV (704 nm) may be due to the spin-forbidden transition from 3A2g(3F) → 1A1g(1G) and 3A2g(3F) → 1Eg(1D).
The Cu2+ ions substituted compounds (Ba0.5Pb1.5)(Mg2−xCux)TeB2O10 (0 < x ≤ 0.25), exhibit different shades of green color under daylight and the absorption spectra have close similarity to the Cu-substituted Ba2(Mg1.5Cu0.5)TeB2O10 compound (Fig. S8†).
We examined the NIR reflectance of the white-colored compounds (Fig. S9†).47 All compounds, Ba2Mg2TeB2O10, (Ba0.5Pb1.5)Mg2TeB2O10, Ba2(Mg1.5In0.5)(Te0.5Sb0.5)B2O10 and Ba2(Mg1.25Zn0.75)TeB2O10 exhibited good near IR reflectivity in the range of ∼75%, which is comparable to that of TiO2.
We also investigated the stability of the color of the prepared compounds by soaking them in hot water as well as 2 N HNO3 for 24 h at room temperature. The tested samples did not exhibit any appreciable changes in the color, the PXRD patterns, and the UV-vis spectra (Fig. S10–S12†). This suggests that the compounds retain the color and are also stable.
Electrocatalytic studies
There has been a considerable surge in the development of electrocatalysts towards water oxidation studies in recent years.48 In the present compounds, we successfully substituted Co2+ ions at the octahedral position in Ba2(MgCo)TeB2O10 and (Ba0.5Pb1.5)(MgCo)TeB2O10 compounds. We explored the possible electrocatalytic behavior of these two compounds towards the OER reaction under alkaline conditions.The compound, Ba2(MgCo)TeB2O10, was added with acetylene carbon (4:1 ratio) and used as the electrode material. The OER reaction was investigated in the alkaline medium (0.5 M KOH), employing a three electrodes set-up where C_Ba2(MgCo)TeB2O10-coated glassy carbon electrode (GC) acts as the working electrode, and a graphite rod and mercury/mercuric oxide (MMO) are used as the counter and reference electrodes, respectively (Fig. S13†). Prior to the electrochemical studies, the alkaline solution was purged for about 30 min by employing argon gas to remove any dissolved oxygen. The linear sweep voltammetry (LSV) studies were carried out at a scan rate of 10 mV s−1 (Fig. 9a). The same experimental procedure was also followed for the (Ba0.5Pb1.5)(MgCo)TeB2O10 compound.
 | ||
| Fig. 9 (a) Linear sweep voltammogram (LSV) of cobalt substituted (Ba0.5Pb1.5)(MgCo)TeB2O10. (b) Comparison of the LSV plots of C_(Ba0.5Pb1.5)(MgCo)TeB2O10 and IrO2. | ||
The OER activity of C_Ba2(MgCo)TeB2O10 and C_(Ba0.5Pb1.5)(MgCo)TeB2O10 compounds was investigated. A current density of 10 mA cm−2 along with the applied overpotential of 700 mV for C_Ba2(MgCo)TeB2O10 was required, whereas an overpotential of 479 mV was sufficient for C_(Ba0.5Pb1.5)(MgCo)TeB2O10 compounds for observing the OER activity. The relatively smaller overpotential for the C_(Ba0.5Pb1.5)(MgCo)TeB2O10 compound suggested that the C_(Ba0.5Pb1.5)(MgCo)TeB2O10 compound is better for the electrocatalytic behavior compared to the C_Ba2(MgCo)TeB2O10 compound. Thus, we concentrated our studies on (Ba0.5Pb1.5)(MgCo)TeB2O10. The OER activity of the C_(Ba0.5Pb1.5)(MgCo)TeB2O10 compound was also compared with IrO2 (Fig. 9b). It appears that the OER activity of the compound is comparable with IrO2 and also to many other known compounds (Table S4†). The onset potential for C_(Ba0.5Pb1.5)(MgCo)TeB2O10 compound is ∼1.53 V vs. RHE.
The Tafel slope gives an idea about the OER behavior. The Tafel polarization curve was measured in the low over-potential region and fitted to the Tafel equation (Fig. 10a). The Tafel slope was found to be ∼73 mV dec−1, which is comparable to that observed for IrO2 (∼71 mV dec−1). The low value of the Tafel slope suggested good activity towards the OER reaction.49
 | ||
| Fig. 10 (a) Tafel plot for the electrocatalyst derived from LSV plot. (b) Electrochemical impedance studies for the catalyst. (c) Chronoamperometric studies for stability for the catalyst. (d) LSV before and after chronoamperometric studies. | ||
The electrochemical AC impedance studies were carried out at the onset DC potential in the frequency range from 10 MHz to 100 kHz. The electrochemical impedance spectroscopy (EIS) was performed to learn about the charge transfer across the electrode–electrolyte interface (Fig. 10b).50–52 The experimental data of EIS were fitted with the equivalent circuit containing the solution resistance (Rs) and two parallel RC pairs along with the Warburg element (Fig. S14†).53Rs is the solution resistance, which has a contribution from the electrode and the electrolyte. The constant phase elements (CPE1 and CPE2), imply the presence of an imperfect capacitor in the system. The Rint and Rct are the resistance from the polymeric film on the surface of the electrode and charge transfer resistance, respectively. The Warburg element (W) is required due to the diffusion of OH− ion at the electrode–electrolyte interface. The charge transfer kinetics is inversely proportional to the charge transfer resistance (Rct) and thus lower Rct value indicates faster charge transfer. The Rct measured from the Nyquist plot was found to be 94 Ω, which suggests rapid charge transfer kinetics during the electrochemical process.
The long-term electrochemical stability of the catalyst was measured by employing chronoamperometry with the same applied potential (10 mA cm−2) (Fig. 10c).54 The studies indicated that the compound has good electrochemical stability for up to 18 hours. We carried out XPS investigations before and after the chronoamperometry studies. From the XPS studies, we observed that there is a partial oxidation of cobalt from the +2 to +3 state. After the chronoamperometry stability studies, we carried out the LSV measurements, which indicated a slight variation, which may be due to the partial oxidation of Co2+ to Co3+ in the compound (Fig. 10d). The electrochemical cyclability studies gave a value of Co3+/Co2+ ratio of 0.158 after 1000 cycles (Table S5, Fig. S15†).
The electrochemical active surface area of the catalyst was calculated from the double-layer capacitance (Cdl).55 This was achieved by cyclic voltammetry by cycling the potential in the non-faradaic region at different scan rates from 5 to 120 mV s−1.
The Cdl value is directly proportional to the conductivity and electrochemically effective surface area of the catalyst. The Cdl value was calculated from the slope of the plot between the scan rate and half the difference in the current density variation (ΔJ = (1/2) (Ja − Jc)) at 1.16 V vs. RHE (Fig. S16†). The ECSA value for the C_(Ba0.5Pb1.5)(MgCo)TeB2O10 is 8.383 cm2.
The surface concentration and turnover frequency (TOF) of C_(Ba0.5Pb1.5)(MgCo)TeB2O10 were calculated by linear scan voltammetric measurements with a scan rate of 10 mV s−1 in 0.5 M KOH solution (Fig. S17†).56 TOF is a measure of the conversion from the reactant to the product per catalyst site per unit time.57 The calculated TOF was found to be 4.673 s−1. Similar values have been observed before.56
The faradaic efficiency (FE) of the electrocatalyst was investigated by employing an inverted burette to quantitatively measure the evolved oxygen as a function of time under constant DC bias. To this end, we employed a H-shaped electrochemical setup (Fig. S18†). The faradaic efficiency was estimated from the ratio between the moles of the oxygen gas produced experimentally and theoretically. We observed an evolution of 270.9 μmol of O2 gas, which is in close agreement with the theoretical value (279.8 μmol). This indicates a faradaic efficiency of ∼96% (Fig. S19†).58
The stability of the electrocatalyst was determined by employing PXRD before and after the electrocatalytic studies (Fig. S20†). As can be noted, there is little change in the PXRD pattern suggesting that the compound did not undergo deterioration during the electrocatalytic studies.
Visible light-driven organic transformation
Mixed metal oxides have played an important role in the study of heterogeneous catalysis.59 The use of TiO2, ZnO and other oxides towards photocatalytic purposes has been well established over the years.60,61 Similarly transition metal substituted compounds have also been explored towards photocatalytic organic transformations.62We explored visible light-activated organic transformations employing BiCdVO5 and BiMgVO5 compounds.63 In the present study we observed a reduction in the band gap when transition metal ions are substituted for Mg2+ ions in Ba2Mg2TeB2O10. The compound, Ba2(Mg1.5Cu0.5)TeB2O10 has a band gap of ∼1.91 eV. It occurred to us that this compound may act as a photocatalyst. To investigate the photocatalytic activity, we attempted the oxidative halogenation reaction by employing visible light.64
The aromatic α-chloroketones are important in many pharmaceuticals.65–67 The well-known methods for the synthesis of chloroketones are the direct halogenation of ketones, the oxidative halogenation of alkyne/alcohols and halogen exchange reactions.68–70 The conventional synthesis of α-haloketones generally involve employing molecular halogens or organic halogens (CH2Cl2/N-halosuccinimide) along with a strong oxidant (ClO2/K2S2O8). In the present study, we attempted the halogenation employing acetyl chloride along with molecular O2 in the presence of visible light. This approach may be more advantageous as the Cu-centers can facilitate the formation of superoxide (˙OOH) radicals and halogen radicals (˙X), which may lead to good selectivity. In the photocatalytic oxochlorination of vinyl arenes, acetonitrile was employed as the solvent, which act to stabilize the coligand during the catalytic cycle.21
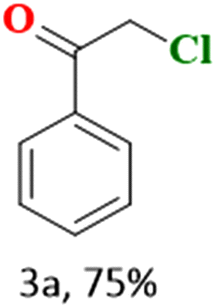
In the present study, we explored the oxidative halogenation by taking styrene and acetyl chloride as the substrate in acetonitrile in the presence of molecular oxygen under white light, 60 W LED, for 12 h. The photocatalytic reaction conditions were optimised with regard to reactants concentrations, solvent, catalyst quantity and the time of the reaction (Tables S6 and S7†). The reaction produced 2-chloro-1-phenylethan-1-one exclusively with a yield of ∼75% (Scheme 1).
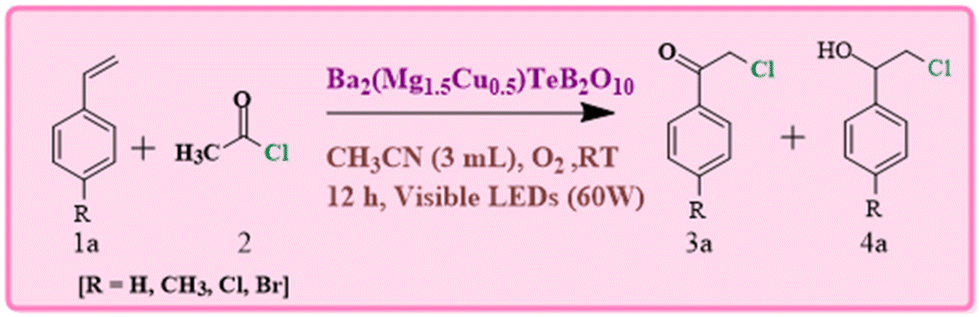 | ||
| Scheme 1 The optimized reaction of photocatalytic oxidative chlorination of vinyl arenes. | ||
We carried out control experiments to establish the catalytic nature of this reaction. To this end, we explored reactions without (i) catalyst, (ii) light source, (iii) oxygen and (iv) acetyl chloride. In all the cases, no desired product was observed (Table S8†).
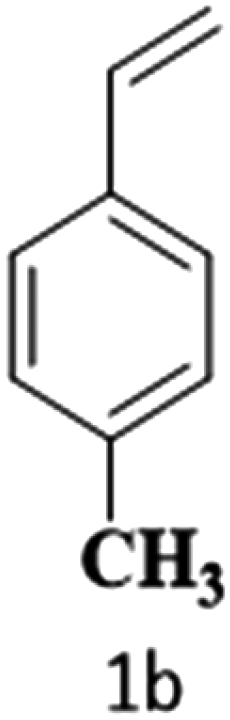
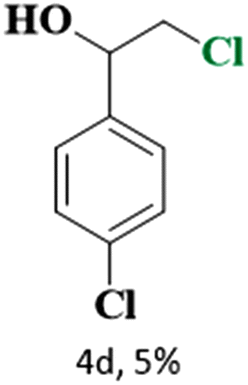
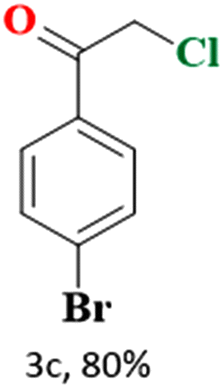
The catalytic reaction has expanded the scope of the study by exploring different substrates. The studies indicated that the electron-withdrawing groups give better yields compared to the electron-donating group (Table 3). It is likely that the electron-withdrawing group may stabilise the α-carbon radical, facilitating the reaction with the photogenerated oxygen radicals (˙OOH). Similar observations have been made before.64
| Entry | Reactant | Main product | Byproduct |
|---|---|---|---|
| 1 |

|
|

|
| 2 |
|

|

|
| 3 |

|

|
|
| 4 |

|
|
|
The recyclability test for the photocatalyst was examined by repeating the experiment (Fig. 11a). We observed that the catalyst was stable for up to 4 cycles without losing much of the catalytic activity (Fig. S21†). We also carried out the hot filtration studies. For this, the catalyst was removed from the reaction mixture after 4 h by centrifugation and the filtrate was used to continue the reaction for 12 h under the same conditions. We found that the reaction did not proceed in the absence of the catalyst (Fig. 11b).
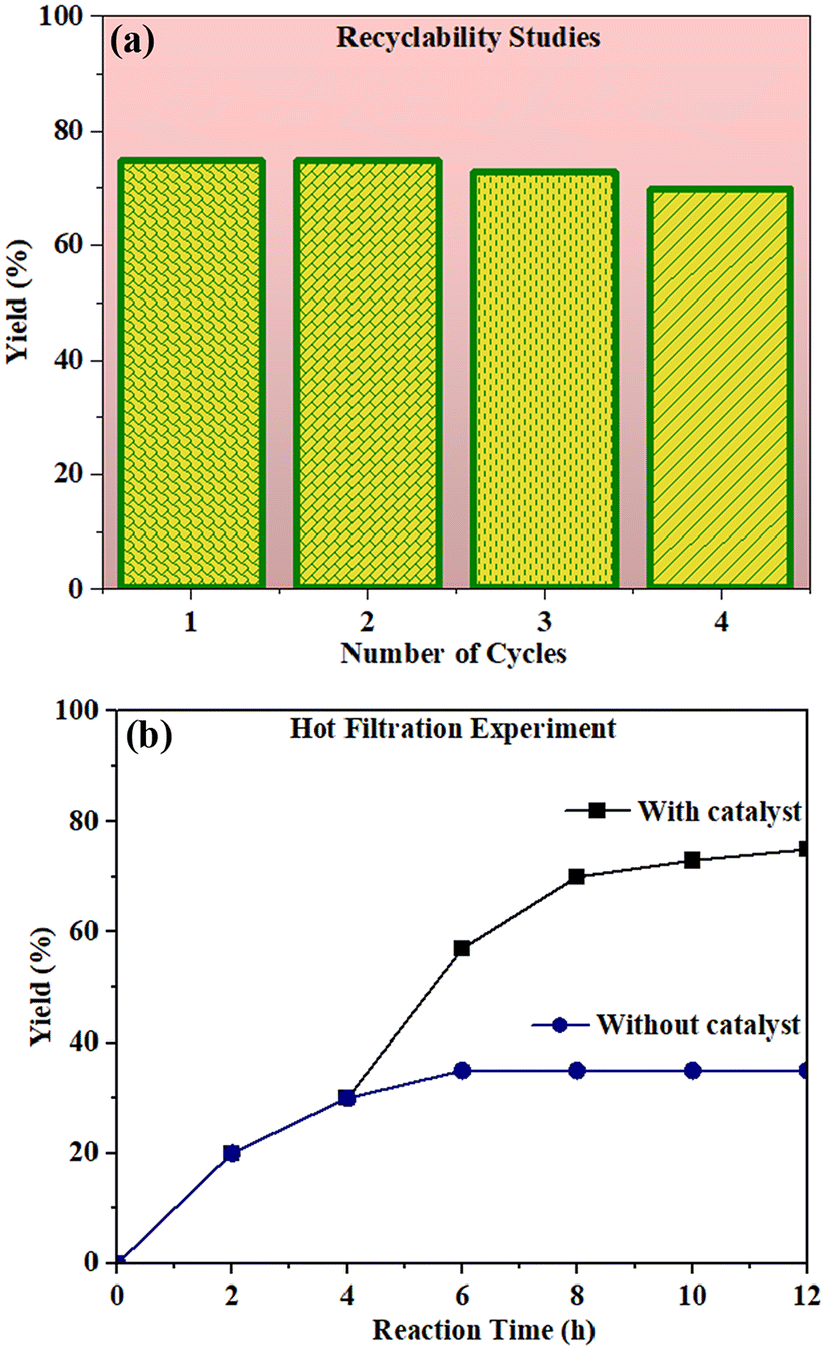 | ||
| Fig. 11 (a) The recyclability studies of photocatalyst for oxochlorination of vinyl arenes. (b) Hot filtration experiment of photocatalytic oxidative halogenation of vinyl arenes. (■) reaction progress in the presence of the photocatalyst. (●) the percentage yield when the photocatalyst was removed via centrifugation after 4 h. | ||
We proposed a possible pathway for the formation of the α-chloroketone derivatives, which is based on earlier observations64 (Fig. S22†). The photoactivation of the Cu-centers helps in the formation of chlorine radicals through light-induced homolysis. The formed chlorine radical then attacks the styrene forming the secondary benzylic radical. The benzylic radical reacts with the molecular oxygen to form the 2-chloroacetophenone. At the same time, the liberated proton from the reaction gives rise to water by the reduction of molecular oxygen. The overall photocatalytic activity of Ba2(Mg1.5Cu0.5)TeB2O10 appears to be comparable to some of the reported compounds (Table S9†).
XPS studies
X-ray photoelectron spectroscopic (XPS) studies were carried out to investigate the oxidation state of cobalt in the compound before and after the electrocatalytic studies (Fig. 12). The Co 2p spectra of the (Ba0.5Pb1.5)(MgCo)TeB2O10 exhibits Co 2p3/2 peak at ∼781.3 eV and Co 2p1/2 peak around 796.6 eV, along with the satellite features at ∼784.2 eV and ∼799.1 eV. These are typical values for Co2+ ions. After the electrocatalytic studies, the XPS studies indicated the presence of two additional peaks at ∼779.7 eV and ∼794.9 eV, which may be due to the Co 2p3/2 and Co 2p1/2 peaks, respectively, of Co3+ ions. A similar value for Co3+ ions has been noted earlier.71 | ||
| Fig. 12 X-ray photoelectron spectroscopy (XPS) for (Ba0.5Pb1.5)(MgCo)TeB2O10 before (a) and after (b) electrocatalysis. | ||
The oxidation state of copper in the Ba2(Mg1.5Cu0.5)TeB2O10 compound was examined before and after the photocatalyst studies (Fig. 13). The Cu 2p spectra exhibit a Cu 2p3/2 peak at ∼934.2 eV and a Cu 2p1/2 peak around ∼953.8 eV, along with the satellite features at ∼942.6 eV and ∼962.3 eV. These are typical values for Cu2+ ions.72 After the photocatalytic studies, the XPS studies did not exhibit any change in the Cu 2p spectra, which suggests that the catalyst was stable.
 | ||
| Fig. 13 XPS spectra of Ba2(Mg1.5Cu0.5)TeB2O10 before (a) and after (b) photocatalysis. The data points (○) are experimentally observed while the continuous light-yellow lines (—) are fitted curves. | ||
Conclusions
A series of telluroborates of the general formula, A2M2TeB2O10 (A = Ba, Pb; M = Mg, Zn, Co, Ni, Cu, Fe) were prepared and their properties investigated. The substitution at the bivalent octahedral positions by bivalent transition elements gave rise to compounds exhibiting interesting colors. The white-colored compounds exhibited NIR reflectivity values that are comparable to the commercial NIR compound, TiO2. The dielectric behavior of the white compounds exhibited reasonable values at low frequencies with low dielectric loss. The exploration of Co-substituted compounds towards OER activity in an alkaline medium was fruitful with (Ba0.5Pb1.5)(MgCo)TeB2O10 exhibiting OER activity that is comparable to IrO2. This compound exhibits 96% faradaic efficiency. The Ba2(Mg1.5Cu0.5)TeB2O10 compound, with a band gap of ∼1.91 eV, was found to be a good visible light-activated photocatalyst for the formation of α-chloroketones. The study highlights the utility of telluroborates towards many materials properties and more such investigations are presently underway.Author contributions
SS contributed to the synthesis of the compounds and performed PXRD measurements and UV-visible optical characterization. The electrochemical and photochemical studies were carried out by IGS. The overall scientific problem was conceived by SN.Data availability
This is to certify that the additional data that are connected with the above manuscript is available as part of the electronic ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. S. N. thanks the Science and Engineering Research Board (SERB), Government of India, for the award of a research grant as well as the J. C. Bose National fellowship. We are thankful to Prof. Rajeev Ranjan for his help with dielectric studies. We thank Dr H. C. Sudeeksha and the Horiba facility for help with the Raman studies. IGS thanks Prof. S. Sampath for help with the electrocatalytic studies. IGS also thanks the University Grants Commission (UGC), the Government of India and the Indian Institute of Science (IISc), for the research fellowship. Dr SS thanks DST-SERB for an NPDF (Grant No. PDF/2022/002873).References
- M. Mutailipu, K. R. Poeppelmeier and S. Pan, Chem. Rev., 2020, 121, 1130–1202 CrossRef PubMed.
- P. Becker, Adv. Mater., 1998, 10, 979–992 CrossRef CAS.
- E. L. Belokoneva, Crystallogr. Rev., 2005, 11, 151–198 CrossRef CAS.
- T. T. Tran, H. Yu, J. M. Rondinelli, K. R. Poeppelmeier and P. S. Halasyamani, Chem. Mater., 2016, 28, 5238–5258 CrossRef CAS.
- P. C. Burns, Can. Mineral., 1995, 33, 1167–1176 CAS.
- J. D. Grice, P. C. Burns and F. C. Hawthorne, Can. Mineral., 1999, 37, 731–762 CAS.
- M. Wen, H. Wu, C. Hu, Z. Yang and S. Pan, Inorg. Chem., 2019, 58, 11127–11132 CrossRef CAS PubMed.
- F. Liu, C. Shi, X. Guo, Z. He, L. Pan, Z. Huang, X. Zhang and J. Zou, Adv. Sci., 2022, 9, 2200307 CrossRef CAS PubMed.
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843–3876 RSC.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- S. Anantharaj and V. Aravindan, Adv. Energy Mater., 2020, 10, 1902666 CrossRef CAS.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329 CrossRef CAS.
- Y. Xiong and P. He, J. Mater. Sci., 2023, 58, 2041–2067 CrossRef CAS.
- L. Cui, W. Zhang, R. Zheng and J. Liu, Chem. – Eur. J., 2020, 26, 11661–11672 CrossRef CAS PubMed.
- M. Dincă, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef PubMed.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- X. Ji, L. Cui, D. Liu, S. Hao, J. Liu, F. Qu, Y. Ma, G. Du, A. M. Asiri and X. Sun, Chem. Commun., 2017, 53, 3070–3073 RSC.
- T. Mandal, N. Katta, H. Paps and O. Reiser, ACS Org. Inorg. Au, 2023, 3, 171–176 CrossRef CAS PubMed.
- A. S. Mereshchenko, P. K. Olshin, A. M. Karimov, M. Y. Skripkin, K. A. Burkov, Y. S. Tveryanovich and A. N. Tarnovsky, Chem. Phys. Lett., 2014, 615, 105–110 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, Powder Diffr., 2014, 29, S2–S6 CrossRef CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louër and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- H. L. Tuller, Mater. Renewable Sustainable Energy, 2017, 6, 1–16 CrossRef.
- D. L. Rousseau, R. P. Bauman and S. P. S. Porto, J. Raman Spectrosc., 1981, 10, 253–290 CrossRef CAS.
- Y.-Y. Sun and S. Zhang, J. Chem. Phys., 2016, 145, 021102 CrossRef PubMed.
- M. R. Chandana, B. R. R. Krushna, J. Malleshappa, K. Manjunatha, T.-E. Hsu, S. Y. Wu, S. C. Sharma, B. D. Prasad, B. Subramanian and H. Nagabhushana, Mater. Today Sustainability, 2023, 22, 100397 CrossRef.
- R. L. Frost, Spectrochim. Acta, Part A, 2009, 72, 903–906 CrossRef PubMed.
- S. Chandra, G. Kaur, S. Abhay, B. Shukla, V. Srihari, G. M. Bhalerao and R. Govindaraj, J. Raman Spectrosc., 2024, 55, 728–738 CrossRef CAS.
- N. Kononova, V. Shevchenko, A. Kokh, T. Nabeeva, D. Chapron, A. Maillard, A. Bolatov and B. Uralbekov, Mater. Res., 2016, 19, 834–838 CrossRef CAS.
- D. Kasprowicz, T. Runka, K. Jaroszewski, A. Majchrowski and E. Michalski, J. Alloys Compd., 2014, 610, 600–605 CrossRef CAS.
- R. Mukherjee, S. Saha, A. Dutta and T. P. Sinha, J. Alloys Compd., 2015, 651, 222–229 CrossRef CAS.
- H. Thauern and R. Glaum, Z. Anorg. Allg. Chem., 2004, 630, 2463–2467 CrossRef CAS.
- W. B. Westphal and A. Sils, Dielectric constant and loss data, MIT Tech. Report, Air Force Materials Laboratory, Air Force Systems Command, 1972, vol. 72 Search PubMed.
- S. A. Larrégola, J. A. Alonso, J. C. Pedregosa, M. J. Martínez-Lope, M. Algueró, V. De la Peña-O'shea, F. Porcher and F. Illas, Dalton Trans., 2009, 5453–5459 RSC.
- S. J. Mills, U. Kolitsch, R. Miyawaki, L. A. Groat and G. Poirier, Am. Mineral., 2009, 94, 1012–1017 CrossRef CAS.
- A. Klein, J. Am. Ceram. Soc., 2016, 99, 369–387 CrossRef CAS.
- M. T. Sebastian, Dielectric materials for wireless communication, Elsevier, 2010 Search PubMed.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 2nd edn, 1984, p. 557 Search PubMed.
- D. N. Sathyanarayana, Electronic absorption spectroscopy and related techniques, Universities Press, 2001 Search PubMed.
- M. Kozielski, I. Pollini and G. Spinolo, J. Phys. C: Solid State Phys., 1972, 5, 1253 CrossRef CAS.
- M. Llusar, E. García, M. T. García, V. Esteve, C. Gargori and G. Monrós, J. Eur. Ceram. Soc., 2015, 35, 3721–3734 CrossRef CAS.
- K. B. N. Sarma, B. J. Reddy and S. V. J. Lakshman, Phys. Lett. A, 1982, 92, 305–308 CrossRef.
- A. Bhim, J. Gopalakrishnan and S. Natarajan, Eur. J. Inorg. Chem., 2018, 2018, 2277–2284 CrossRef CAS.
- I. Fontana, A. Lauria and G. Spinolo, Phys. Status Solidi B, 2007, 244, 4669–4677 CrossRef CAS.
- A. Bhim, W. Zhang, P. S. Halasyamani, J. Gopalakrishnan and S. Natarajan, Inorg. Chem., 2019, 58, 8560–8569 CrossRef CAS PubMed.
- P. Jeevanandam, R. S. Mulukutla, M. Phillips, S. Chaudhuri, L. E. Erickson and K. J. Klabunde, J. Phys. Chem. C, 2007, 111, 1912–1918 CrossRef CAS.
- Y. Li, X. Du, J. Huang, C. Wu, Y. Sun, G. Zou, C. Yang and J. Xiong, Small, 2019, 15, 1901980 CrossRef PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- A. R. C. Bredar, A. L. Chown, A. R. Burton and B. H. Farnum, ACS Appl. Energy Mater., 2020, 3, 66–98 CrossRef CAS.
- E. Laouini, M. Hamdani, M. I. S. Pereira, J. Douch, M. H. Mendonça, Y. Berghoute and R. N. Singh, J. Appl. Electrochem., 2008, 38, 1485–1494 CrossRef CAS.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2009, 631, 62–70 CrossRef CAS.
- D. A. Harrington and P. Van Den Driessche, Electrochim. Acta, 2011, 56, 8005–8013 CrossRef CAS.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS.
- D. M. Morales and M. Risch, J. Phys.: Energy, 2021, 3, 34013 CAS.
- S. Anantharaj, P. E. Karthik and S. Kundu, Catal. Sci. Technol., 2017, 7, 882–893 RSC.
- Y. Yan, X. Ge, Z. Liu, J.-Y. Wang, J.-M. Lee and X. Wang, Nanoscale, 2013, 5, 7768–7771 RSC.
- V. Kiran, D. Mukherjee, R. N. Jenjeti and S. Sampath, Nanoscale, 2014, 6, 12856–12863 RSC.
- J. M. Thomas and W. J. Thomas, Principles and practice of heterogeneous catalysis, John Wiley & Sons, 2014 Search PubMed.
- O. M. Ishchenko, V. Rogé, G. Lamblin and D. Lenoble, Semiconductor Photocatalysis-Materials, Mechanisms and Applications, 2016, pp. 3–30 Search PubMed.
- K. Wetchakun, N. Wetchakun and S. Sakulsermsuk, J. Ind. Eng. Chem., 2019, 71, 19–49 CrossRef CAS.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- A. Bhim, S. Sasmal, J. Gopalakrishnan and S. Natarajan, Chem. – Asian J., 2020, 15, 3104–3115 CrossRef CAS PubMed.
- F. Han, D. Zhang, S. Salli, J. Ye, Y. Li, F. Rosei, X.-D. Wen, H. Niemantsverdriet, E. Richards and R. Su, ACS Catal., 2022, 13, 248–255 CrossRef PubMed.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- K. Pchalek, A. W. Jones, M. M. T. Wekking and D. S. Black, Tetrahedron, 2005, 61, 77–82 CrossRef CAS.
- A. W. Erian, S. M. Sherif and H. M. Gaber, Molecules, 2003, 8, 793–865 CrossRef CAS.
- J. C. Lee, J. Y. Park, S. Y. Yoon, Y. H. Bae and S. J. Lee, Tetrahedron Lett., 2004, 45, 191–193 CrossRef CAS.
- Y. Jing, C. G. Daniliuc and A. Studer, Org. Lett., 2014, 16, 4932–4935 CrossRef CAS PubMed.
- P. Klahn, H. Erhardt, A. Kotthaus and S. F. Kirsch, Angew. Chem., Int. Ed., 2014, 53, 7913–7917 CrossRef CAS PubMed.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal structure and SEM-EDX elemental mapping (Fig. S1 and S2), Raman studies (Tables S1 and S2, Fig. S3), optical studies (Fig. S4–S9, Table S3), stability studies (Fig. S10–S12), OER studies (Fig. S13–S20, Table S5), photocatalytic studies (Fig. S21–S26, Tables S6–S9). See DOI: https://doi.org/10.1039/d4dt02706j |
| ‡ These authors are equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |