
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses and structures of dimesitylphosphinite complexes of alkali metals and their catalytic activity in hydrophosphorylation reactions†
Benjamin E.
Fener
 ,
Philipp
Schüler
,
Phil
Liebing
,
Helmar
Görls
and
Matthias
Westerhausen
*
,
Philipp
Schüler
,
Phil
Liebing
,
Helmar
Görls
and
Matthias
Westerhausen
*
Friedrich Schiller University Jena, Institute of Inorganic and Analytical Chemistry, Humboldtstraße 8, D-07743 Jena, Germany. E-mail: m.we@uni-jena.de
First published on 30th October 2024
Abstract
Metalation of dimesitylphosphane oxide, Mes2P(O)H (1), with alkali metal reagents (nBuLi, NaH, and A(hmds); A = K, Rb, and Cs) in THF yields the corresponding dimesitylphosphinites of lithium (2-thf), sodium (3-thf), potassium (4-thf), rubidium (5-thf), and caesium (6). Their molecular structures exhibit a broad and fascinating variety. Dinuclear compounds 2-thf, 3-thf, and 5-thf have central four-membered A2O2 rings, whereas the potassium congener crystallises as a tetranuclear complex with an inner A4O4 heterocubane cage. The tetranuclear caesium congener precipitates without thf coligands and exhibits a quite unique structure in its crystalline state. Due to their catalytic activity in hydrophosphorylation reactions, we focus on the solvent–structure relationship of the potassium derivatives. In hydrocarbons, [K4(O-PMes2)4]2 (4) is formed, and bidentate Lewis bases like dme and tmeda are unable to deaggregate this tetranuclear cage compound, but bases with a higher denticity (diglyme, triglyme, and pmdeta) split this cage compound into dinuclear complexes with central K2O2 rings. In addition, very bulky P-bound aryl groups like 2,4,6-triisopropylphenyl in dinuclear 8-thf hinder the formation of tetranuclear cage compounds, whereas 2-methylnaphthyl substituents are not bulky enough and the tetranuclear cage compound 7-thf is stabilised. For the 2,4,6-triisopropylphenyl substituent, the rubidium and caesium congeners 11 and 12 crystallise with two central A2O2 rings interconnected by π-interactions. A heteroleptic potassium complex 9-hmds, containing hmds as well as phosphinite anions, represents a snapshot on the way from the starting K(hmds) to the phosphinite-based heterocubane congener. Finally, heterobimetallic [{(thf)K}2Mg(O-PMes2)4] (10-thf) with tetrahedrally coordinated Mg centres has been isolated.
Introduction
Phosphane oxides Ar2R′P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Ar = aryl) are valuable substrates for addition across alkynes and alkenes (R′ = H).1 However, this reaction requires a catalyst to overcome kinetic hindrance. s-Block organometallics such as alkali (A) and alkaline-earth (Ae) metal complexes like nBuLi, A(hmds) or Ae(hmds)2 [hmds = N(SiMe3)2] are suitable compounds to deprotonate secondary phosphane oxides yielding the corresponding alkali and alkaline-earth metal phosphinites A(O-PAr2) and Ae(O-PAr2)2 that are catalytically active.2–4 In addition, sodium can cleave a P–C bond of Ph3PO, leading to the formation of sodium diphenylphosphinite and phenylsodium.5 The THF adduct of lithium diphenylphosphinite crystallises as tetranuclear [(thf)Li–O-PPh2]4 with a central Li4O4 heterocubane cage,6 whereas bidentate 1,2-dimethoxyethane (dme) leads to the formation of dinuclear [(dme)Li–O-PPh2]2 with an inner four-membered Li2O2 ring.7 Homologous potassium diphenylphosphinite dismutates and hence [(18C6)K–O-PPh2] cocrystallises with the corresponding diphenylphosphinate congener [(18C6)K-O2PPh2].4 Therefore, many studies have been performed with dimesitylphosphane oxide to sterically suppress the dismutation of alkali metal diarylphosphinites. However, a comparable dismutation and sparing solubility of lithium dimesitylphosphinate lead to the precipitation of dinuclear [(thf)2Li(O2PMes2)]2 and the molecular structures of lithium diarylphosphinites remain unknown.8 In addition to these homoleptic diarylphosphinites, the molecular structures of heteroleptic derivatives have been authenticated by X-ray structure determination.9
O (Ar = aryl) are valuable substrates for addition across alkynes and alkenes (R′ = H).1 However, this reaction requires a catalyst to overcome kinetic hindrance. s-Block organometallics such as alkali (A) and alkaline-earth (Ae) metal complexes like nBuLi, A(hmds) or Ae(hmds)2 [hmds = N(SiMe3)2] are suitable compounds to deprotonate secondary phosphane oxides yielding the corresponding alkali and alkaline-earth metal phosphinites A(O-PAr2) and Ae(O-PAr2)2 that are catalytically active.2–4 In addition, sodium can cleave a P–C bond of Ph3PO, leading to the formation of sodium diphenylphosphinite and phenylsodium.5 The THF adduct of lithium diphenylphosphinite crystallises as tetranuclear [(thf)Li–O-PPh2]4 with a central Li4O4 heterocubane cage,6 whereas bidentate 1,2-dimethoxyethane (dme) leads to the formation of dinuclear [(dme)Li–O-PPh2]2 with an inner four-membered Li2O2 ring.7 Homologous potassium diphenylphosphinite dismutates and hence [(18C6)K–O-PPh2] cocrystallises with the corresponding diphenylphosphinate congener [(18C6)K-O2PPh2].4 Therefore, many studies have been performed with dimesitylphosphane oxide to sterically suppress the dismutation of alkali metal diarylphosphinites. However, a comparable dismutation and sparing solubility of lithium dimesitylphosphinate lead to the precipitation of dinuclear [(thf)2Li(O2PMes2)]2 and the molecular structures of lithium diarylphosphinites remain unknown.8 In addition to these homoleptic diarylphosphinites, the molecular structures of heteroleptic derivatives have been authenticated by X-ray structure determination.9
Alkaline-earth metal bis(diarylphosphinites) form preferably mononuclear molecular structures, and the free coordination sites are occupied by neutral coligands such as ethers.10 The knowledge of the coordination chemistry of alkali metal diarylphosphinites is very limited. There have been no molecular structures reported for Rb/Cs–O-PAr2 so far and for the homologous sodium congener, only the dinuclear tmeda adduct [(tmeda)Na–O-PMes2]2 has been described.11 The catalytically more active potassium complexes crystallise as tetranuclear molecules with central K4O4 heterocubane cages.3 Addition of [18]crown-6 leads to deaggregation and mononuclear potassium diarylphosphinites precipitate.4
Results and discussion
Synthesis of alkali metal dimesitylphosphinites
Lithium dimesitylphosphinite, Li–O-PMes2, is a very inefficient catalyst for the addition of diarylphosphane oxide across alkynes and therefore, the interest in this compound remains negligible.2d Its propensity to degrade via dismutation to dimesitylphosphinate and dimesitylphosphanide additionally lowered the attractiveness.8 Nevertheless, these properties raised our interest in understanding the allegedly disadvantageous behaviour. Dimesitylphosphane oxide (1) was metalated by commercially available n-butyllithium in a solvent mixture of THF and n-hexane yielding the thf adduct of lithium dimesitylphosphinite, [(thf)3(Li–O-PMes2)2] (2-thf), as a dinuclear complex as depicted in Scheme 1.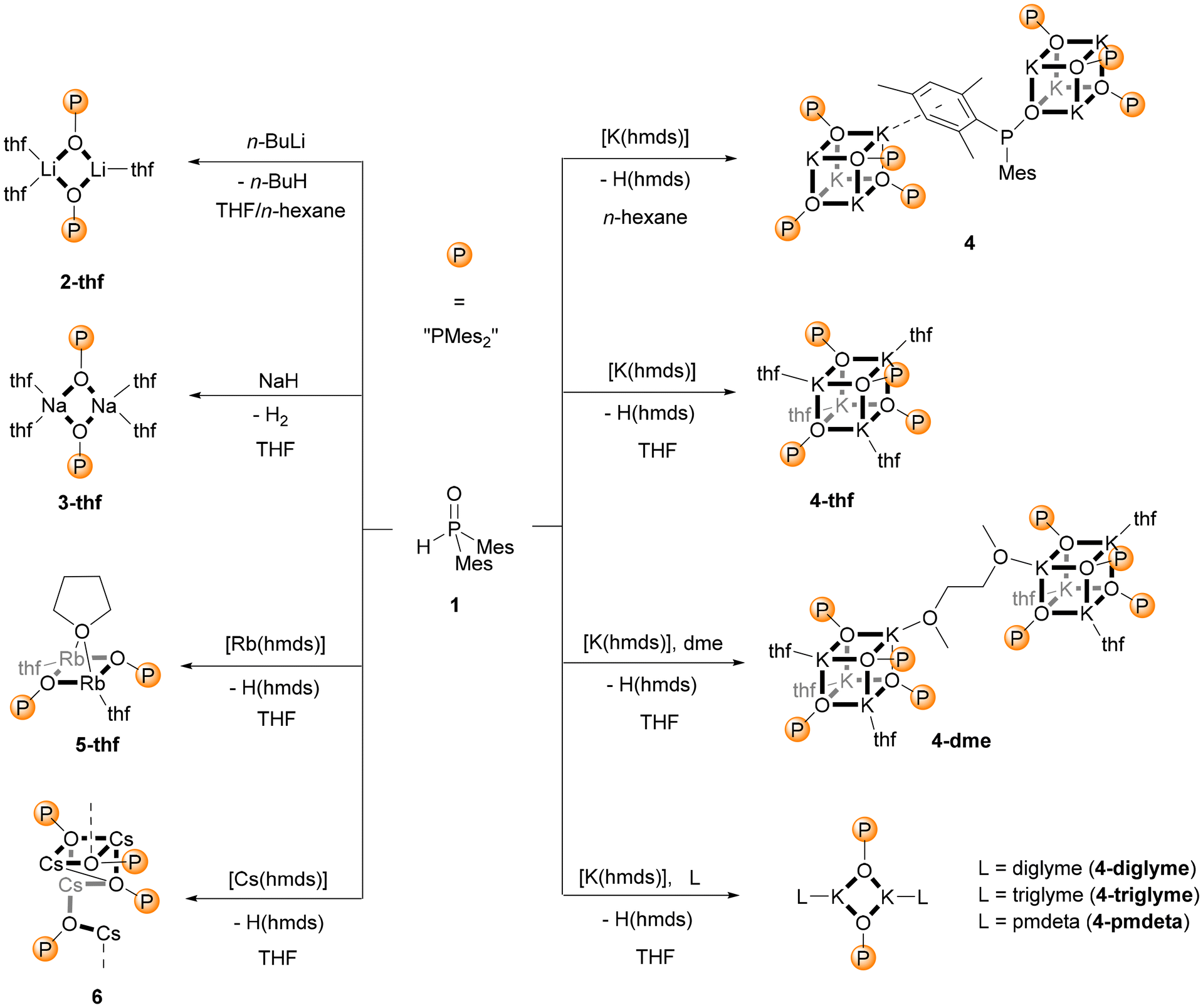 | ||
| Scheme 1 Metalation of dimesitylphosphane oxide (1) with n-butyllithium (2-thf), sodium hydride (3-thf), and A(hmds) of potassium (4-thf), rubidium (5-thf), and caesium (6) in THF as well as the synthesis of donor-free potassium phosphinite (4) in n-hexane. The syntheses of potassium phosphinite adducts with oligodentate amines (4-pmdeta) and ethers (4-dme, 4-diglyme, and 4-triglyme) are also depicted. | ||
Suitable metalation reagents of the heavier alkali metals were their bis(trimethylsilyl)amides, A(hmds) [hmds = N(SiMe3)2, hexamethyldisilazanide]. The reaction was performed in THF; however, the sodium congener crystallised only after the addition of stoichiometric amounts of 1,2-bis(dimethylamino)ethane (tmeda) as dinuclear [(tmeda)Na–O-PMes2]2 (3-tmeda) even after several attempts. The thf adduct of sodium dimesitylphosphinite could only be accessed by changing the metalation reagent to sodium hydride. As a result, it could be isolated as the dinuclear complex [(thf)4(Na–O-PMes2)2] (3-thf), in comparison with potassium dimesitylphosphinite which precipitated as a tetranuclear compound of the type [(thf)K–O-PMes2]4 (4-thf) with a central K4O4 heterocubane cage. Unexpected molecular structures were observed for the heaviest alkali metals rubidium and caesium. The structure of [(thf)3(Rb–O-PMes2)2] (5-thf) was quite similar to that of 2-thf but the larger Rb–O distances allowed one thf base to act as a bridging coligand above the Rb2O2 ring as depicted in Scheme 1. The tetranuclear caesium dimesitylphosphinite (6) precipitated from THF without neutral coligands and the basic cage was a cube with an opened edge. The metalation of Mes2P(O)H (1) with K(hmds) in n-hexane led to the formation of 4 with K4O4 heterocubane moieties. Two of such cages are interconnected by coordination of a mesityl π-system to a potassium ion of the neighbouring structural unit.
Bidentate Lewis bases such as dme and tmeda stabilised dinuclear complexes like [(dme)Li–O-PPh2]27 and [(tmeda)Na–O-PMes2]2 (3-tmeda). Attempts to deaggregate [(thf)K–O-PMes2]4 (4-thf) with these bases failed, and this cage compound did not form adducts with tmeda. Addition of 1,2-dimethoxyethane gave 4-dme where two tetranuclear molecules are interconnected by a bridging dme ligand, but the inner K4O4 heterocubane core did not open out. Breaking the cubane cage down to two molecules with four-membered K2O2 rings required Lewis bases with a higher denticity like diglyme, triglyme, and pmdeta yielding the corresponding potassium congeners 4-diglyme, 4-triglyme, and 4-pmdeta.
Deaggregation of the heterocubane cages was also possible with bulkier phosphinite anions. However, P-bound methylnaphthyl substituents were not demanding enough to hinder the formation of tetranuclear cages and [(thf)K–O–P(MeNaphth)2]47-thf crystallised from the reaction mixture (Scheme 2). The 2,4,6-triisopropylphenyl substituents (Tipp) were large enough to stabilise dinuclear [(thf)2K–O-PTipp2]2 (8-thf) with a central K2O2 ring as depicted in Scheme 3. Comparable structures were observed for the phosphinite complexes with the heavier homologous alkali metals rubidium (11) and caesium (12) as depicted in Scheme 3. However, strong π-interactions led to the interconnection of two central A2O2 rings, forming tetranuclear species. The reaction rate of the metalation of Mes2P(O)H with K(hmds) depended upon the solvent, with decelerated formation of the phosphinite products in non-donor solvents like hydrocarbons. An equimolar mixture of Mes2P(O)H with K(hmds) was heated to 100 °C in methylcyclohexane containing a very small amount of diglyme until a clear solution formed. Thereafter, the solution was cooled to room temperature and crystalline [(diglyme)K6(hmds)2(O-PMes2)4] (9-hmds) precipitated (Scheme 4). This complex, consisting of four K–O-PMes2 units and two K(hmds) moieties, represented a snapshot on the way from the substrates to potassium dimesitylphosphinite with a central heterocubane cage. One could already adumbrate here the heterocubane cage which would indeed form via substitution of the bis(trimethylsilyl)amide ligands by dimesitylphosphinite anions. The diglyme ligand, which ensures solubility and formation of a homogeneous reaction mixture, ligated to an outer potassium ion.
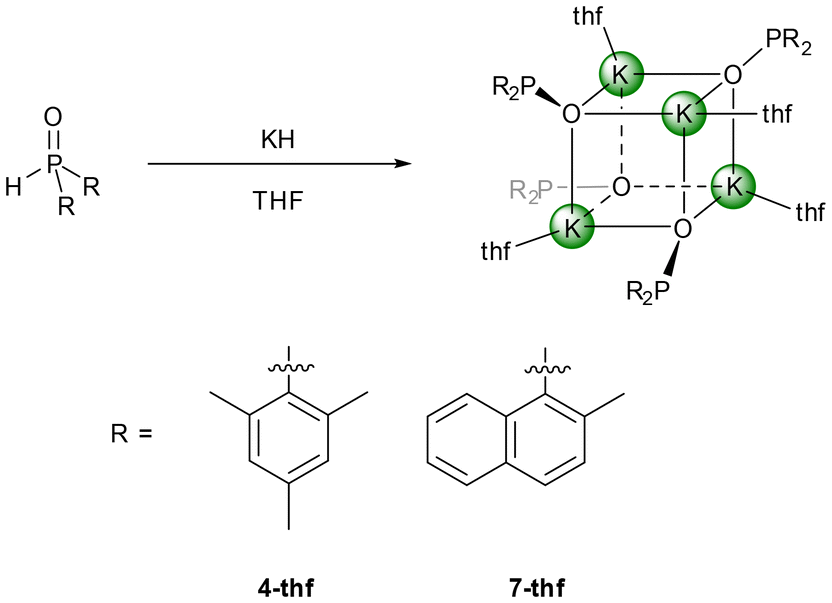 | ||
| Scheme 2 Synthesis of tetrameric potassium phosphinite complexes with mesityl (4-thf) and 2-methylnaphthyl (7-thf) substituents via metalation of the corresponding secondary phosphane oxide with potassium hydride in THF. | ||
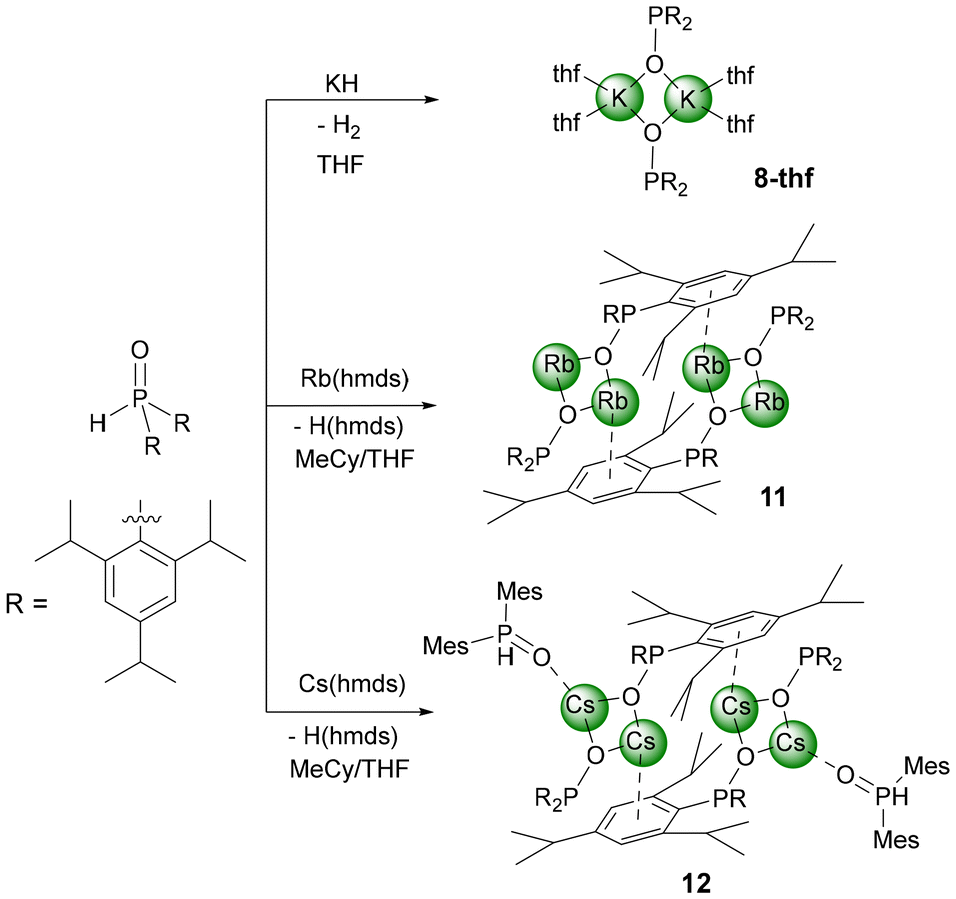 | ||
| Scheme 3 Synthesis of potassium (8-thf), rubidium (11), and caesium bis(2,4,6-triisopropylphenyl)phosphinite (12) via metalation of bis(2,4,6-triisopropylphenyl)phosphane oxide with potassium hydride and rubidium or caesium hexamethyldisilazanide. | ||
 | ||
| Scheme 4 Synthesis of heteroleptic octanuclear potassium phosphinite (9-hmds) via metalation of dimesitylphosphane oxide with potassium hexamethyldisilazanide in methylcyclohexane in the presence of small amounts of diglyme. | ||
Intrigued by the fascinating work of Hevia and coworkers,2e who prepared potassium magnesiates of comparable catalytic activity as heavy alkali metal phosphinites, we included this substance class in our investigation. The Hevia group metalated diphenylphosphane oxide with (pmdeta)2K2Mg(CH2SiMe3)4 in benzene and isolated crystalline [{(L)K}2Mg(O-PPh2)4] (L = Ph2P(O)H, [18]crown-6, [2,2,2]-cryptand). Under our reaction conditions, a mixture of K(hmds) and Bu2Mg was able to metalate Mes2P(O)H (1) and hitherto unknown [{(thf)K}2Mg(O-PMes2)4] (10-thf) with a tetra-coordinate Mg ion was obtained. The distorted tetrahedrally coordinated magnesium centre formed the central structural unit as also observed in Hevia's study.
NMR studies
A comparison of the 31P NMR chemical shifts of the herein presented phosphinites is depicted in Table 1. The 31P NMR resonances of the alkali metal dimesitylphosphinites are in the range of 90 to 95 ppm, significantly shifted to higher frequencies compared to the substrate dimesitylphosphane oxide (1, δ(31P) = 10.1 ppm). In addition, the 1J(P–iC) coupling constants of the phosphinite ions are much smaller than the value of phosphane oxide 1, whereas the signal of the ipso-carbon atom is strongly shifted toward the lower field. These differences are the consequences of different environments of the phosphorus atom. In phosphane oxide 1 the tetracoordinate P atom is in a distorted tetrahedral environment, whereas in all phosphinite complexes, the tricoordinate P atoms are in trigonal pyramidal environments.| Compound | δ(31P) | δ(i-13C)a | 1 J(P–iC)a | δ(1HoMe)b |
|---|---|---|---|---|
| a P-bound ipso-carbon atom iC. b Chemical shift of the ortho-bound methyl groups of the mesityl substituent. c Two major species are observed in solution. d Not observed due to very broad signals. e NMR spectra recorded in CDCl3. f NMR spectra recorded in [D8]toluene. | ||||
| 1 | 10.1 | 126.3 | 100.0 | 2.38 |
| 2-thf | 95.0 | n. o.d | n. o.d | 2.41 |
| 3-thf | 98.8 | 147.5 | 56.1 | 2.42 |
| 4-thf | 95.0 | n. o.d | n. o.d | 2.31 |
| 5-thf | 93.7 | 146.4 | 57.7 | 2.38 |
| 6 | 90.9 | n. o.d | n. o.d | 2.40 |
| 4 | 95.9 | 144.5 | 58.5 | 2.41 |
| 4-dme | 95.1 | 146.5 | 57.8 | 2.38 |
| 4-diglyme | 94.9 | 148.1 | 58.7 | 2.39 |
| 4-triglyme | 94.7 | 148.0 | 59.5 | 2.40 |
| 4-pmdeta | 95.0 | 146.5 | 56.3 | 2.38 |
| 7-thf | 94.8 | 148.3 | 65.3 | — |
| 8-thf | 94.8 | 146.9 | 58.3 | — |
| 10-thf | 95.4; 93.9 | 142.0; 144.5 | 45.6; 51.7 | 2.55; 2.59 |
| 11 | 93.6 | n. o.d | n. o.d | — |
Issleib and coworkers observed a slight high-field shift with increasing size of the alkali metal cation, as they investigated the 31P nuclear magnetic resonances of the dialkyl- and diarylphosphinites of alkali metals.12 It is noteworthy that the aggregation degree of the potassium phosphinites plays a negligible role in the NMR parameters. Furthermore, the coordination of oligodentate ethers and amines also has a negligible influence on the NMR parameters.
ECC-DOSY studies of the investigated s-block metal dimesitylphosphinites were performed to reveal their solution structures. In THF solution, potassium dimesitylphosphinite 4-thf was observed as a dimer with two [D8]THF molecules at each potassium ion (MWcalc = 969 g mol−1, MWfound = 913 g mol−1, and Δ = 6%). In [D8]toluene, in contrast, an equilibrium between the heterocubane and the dimeric structure was observed. Temperature-dependent DOSY NMR measurements allowed us to characterise this equilibrium precisely (Fig. 1). By plotting ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K versus T−1, the thermodynamic parameters of the reaction could be calculated. The reaction from the tetramer to the dimer is highly exergonic at room temperature (ΔRH = −80 kJ mol−1; ΔRS = 261 J K−1 mol−1; ΔRG(297 K) = −157 kJ mol−1). The high entropy value is consistent with the postulated splitting of the heterocubane.
K versus T−1, the thermodynamic parameters of the reaction could be calculated. The reaction from the tetramer to the dimer is highly exergonic at room temperature (ΔRH = −80 kJ mol−1; ΔRS = 261 J K−1 mol−1; ΔRG(297 K) = −157 kJ mol−1). The high entropy value is consistent with the postulated splitting of the heterocubane.
 | ||
| Fig. 1 By ECC-DOSY determined molecular masses of 4 depending on the temperature in [D8]toluene. | ||
Adding one more equivalent of dimesitylphosphane oxide (1) to a solution of [K–O-PMes2] (4) in [D8]THF resulted in a significantly smaller molar mass (MWfound = 684 g mol−1) which could be attributed to the monomeric species [(HPOMes2)K–O-PMes2([D8]thf)] (MWcalc = 690 g mol−1, Δ = 1%). A pronounced coalescence of 1H NMR signals that was assigned to the acid–base equilibrium between [K–O-PMes2] and HPOMes2 at 280 K allowed us to calculate the energy barrier to be 55 kJ mol−1 by means of the Gutowsky–Holm equation.13 This observation was comparable to that in a solution of [K–O-PMes2] (4) in [D8]toluene. Further addition of HPOMes2 did not alter the found molar masses, indicating that the coordination sphere of the potassium ion was already saturated with one molecule of HPOMes2.
Addition of polydentate ethers and amines (e.g., diglyme or pmdeta) to potassium phosphinite (4) in [D8]THF solution yielded mainly the thf saturated dimeric structure without coordination of the polydentate donor ([([D8]thf)4K–O-PMes2] = 644 g mol−1, MW(exp) = 623 g mol−1, Δ = 3%). The large excess of the strong donor THF outperformed the chelate effect of the polydentate ligands.
The 31P NMR spectrum of potassium magnesiate 10-thf in [D8]THF surprisingly showed three signals (δP = 100.3, 98.8, 96.1 ppm) which prompted us to investigate its solution structure. The magnesiate solution mainly (87%) consisted of solvent-separated ion pairs (see Table S1† and Scheme 5). The determined molecular masses indicated an equilibrium between the monopotassium (30%, Type IIa) and dipotassium (57%, Type IIIa) magnesiates in solution. Additionally, a small amount (13%) of phosphinite-bridged structure type III was found. To classify this observation in terms of the dependency of used metal ions, NMR samples with several combinations of Li or K with the alkaline-earth metals Mg–Ba were prepared and analysed by ECC-DOSY and 31P NMR. It is striking that separated ion pairs (type IIa and IIIa) were only observed for the potassium compounds with magnesium and barium, whereas with calcium and strontium only the bridging type III was found. Furthermore, going from Li–Ca to Li–Ba favors type III over type II.
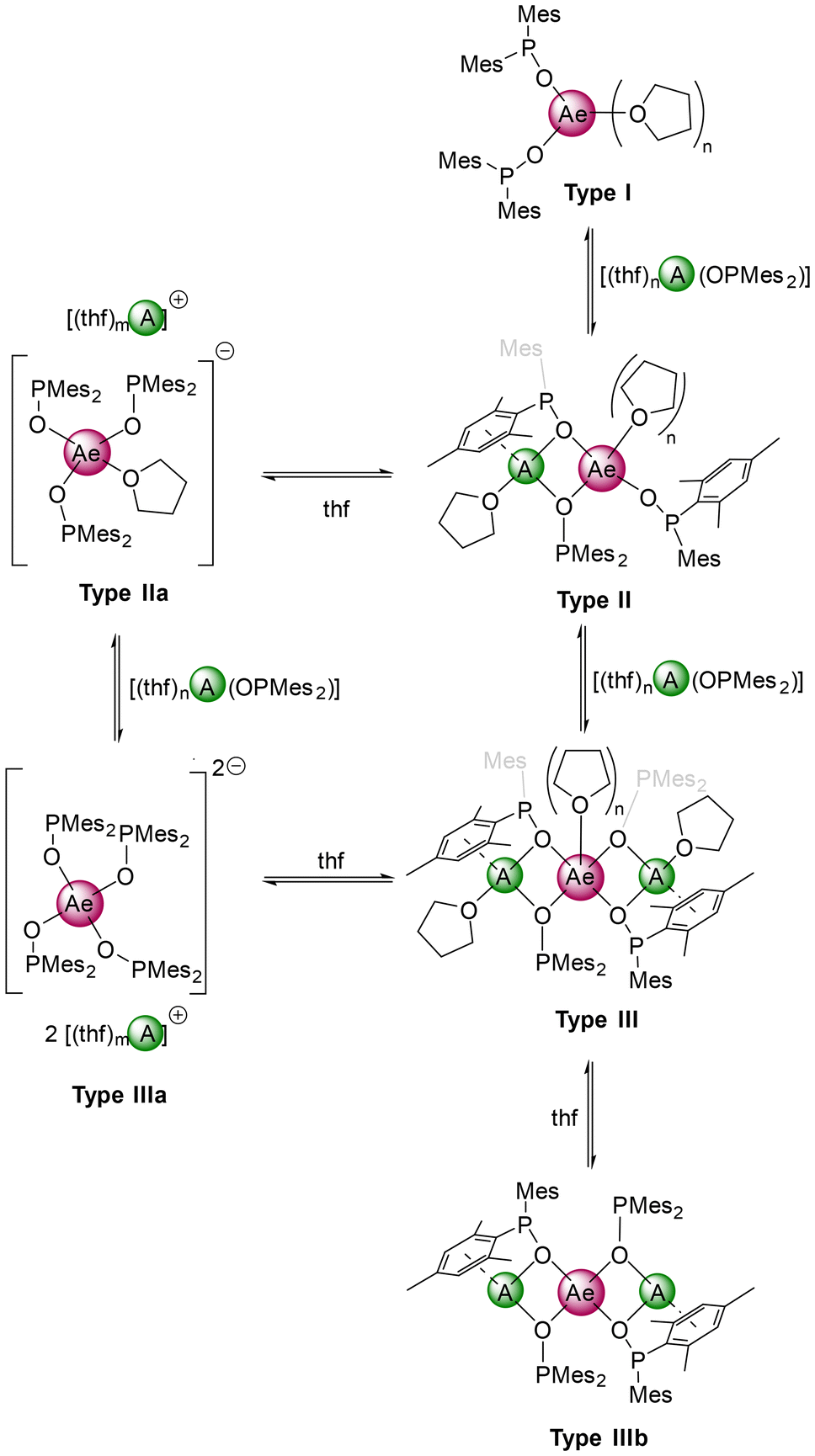 | ||
| Scheme 5 Postulated structure types of heterobimetallic s-block metal phosphinites deduced from ECC-DOSY NMR measurements. | ||
Molecular structures of the alkali metal dimesitylphosphinites
It had been recognised earlier that the lithium complex 2-thf easily dismutates in ethereal solution to the phosphinate and phosphanide.8 We were able to obtain the NMR parameters, and the pure complex did not dismutate in [D8]THF. However, already small amounts of Brønsted acids (including weak acids like phosphane oxides) initiate dismutation into dimesitylphosphinate and dimesitylphosphanide compounds, as reported earlier for thio- and selenophosphinites.14 Therefore, pure samples had to be recrystallised to isolate single crystals for X-ray diffraction experiments. Due to the bulky dimesitylphosphinite ligands, a dinuclear complex crystallised with the lithium atoms having different coordination numbers of three and four. The molecular structure and atom labelling scheme of 2-thf are depicted in Fig. 2.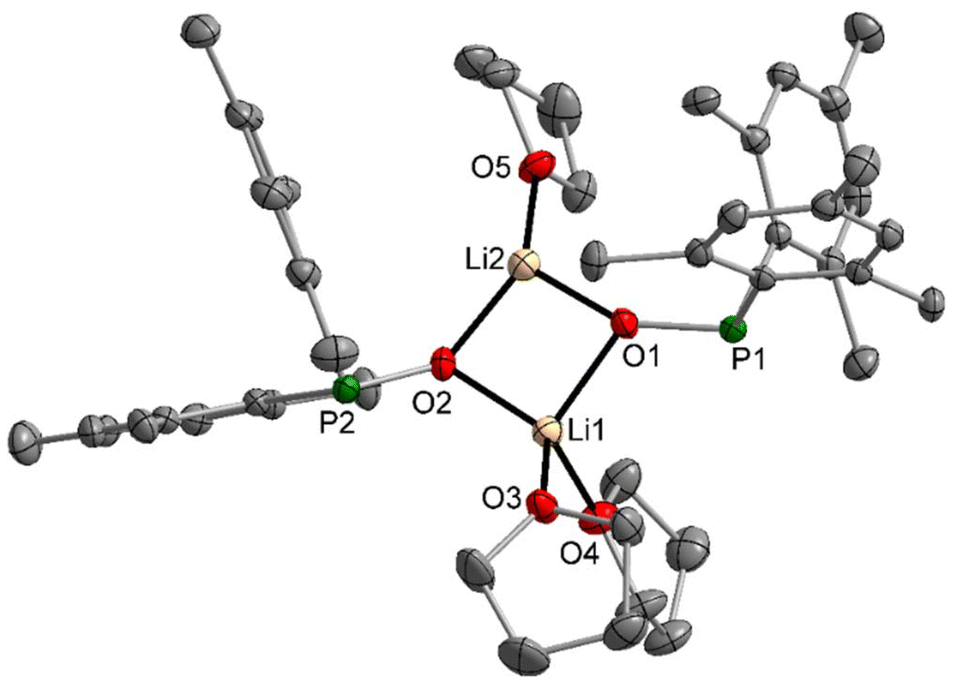 | ||
| Fig. 2 Molecular structure of 2-thf in the crystal. Displacement ellipsoids are drawn at the 50% probability level, H atoms and minor-occupancy orientations of disordered THF ligands are omitted for clarity reasons. Selected bond lengths (pm): Li1–O1 191.8(3), Li1–O2 192.3(3), Li1–O3 195.8(3), Li1–O4 199.0(3), Li2–O1 182.4(3), Li2–O2 184.7(3), Li2–O5 189.9(3), P1–O1 157.0(1), P1–C1 188.1(2), P1–C10 186.2(2), P2–O2 157.6(1), P2–C19 187.3(2), P2–C28 186.1(2); angles (°): P1–O1–Li1 122.9(1), P1–O1–Li2 149.3(1), Li1–O1–Li2 82.5(1), P2–O2–Li1 115.0(1), P2–O2–Li2 133.3(1), Li1–O2–Li2 81.8(1), O1–Li1–O2 94.8(1), O1–Li2–O2 100.8(2), O1–Li2–O5 122.0(2), O2–Li2–O5 124.0(2). | ||
The influence of the coordination number on the Li–O bond lengths is quite large, with significantly smaller distances to tricoordinate Li2 with a flattened trigonal pyramidal environment. Due to the tetrahedral environment of Li1, the P-bound mesityl groups are turned toward the side of three-coordinate Li2, enhancing steric repulsion between the aryl substituents and the Li2-bound thf molecule. This arrangement prevents the binding of a second ether base at Li2 and in addition, this intramolecular strain together with the trigonal pyramidal environment of the P atoms leads to significantly enhanced Li2–O1/2–P1/2 bond angles.
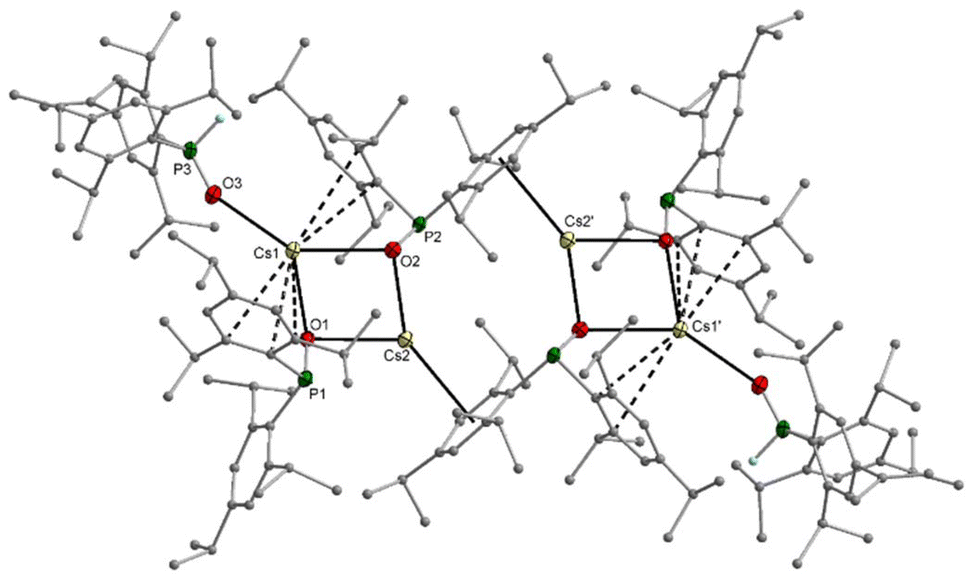
The sodium dimesitylphosphinite 3-thf crystallised as a dinuclear complex with a central Na2O2 unit whose structure could however only be determined by X-ray diffraction experiments after several crystallisation attempts (Fig. S69†). The potassium congener 4-thf contains a central K4O4 heterocubane cage (Fig. S71†). Addition of bidentate DME to [(thf)Li–O-PPh2]4 and of TMEDA to 3-thf led to the formation of dinuclear complexes with central A2O2 rings. Contrary to this reactivity, TMEDA was unable to substitute thf ligands in 4-thf, whereas DME linked two tetranuclear cages but was unable to deaggregate the heterocubane moieties. The molecular structure and atom labelling scheme of these interconnected heterocubane units are depicted in Fig. 3. This binding mode of the dme ligand does not enhance the steric strain within the heterocubane substructures, and no distortions of these fragments are induced.
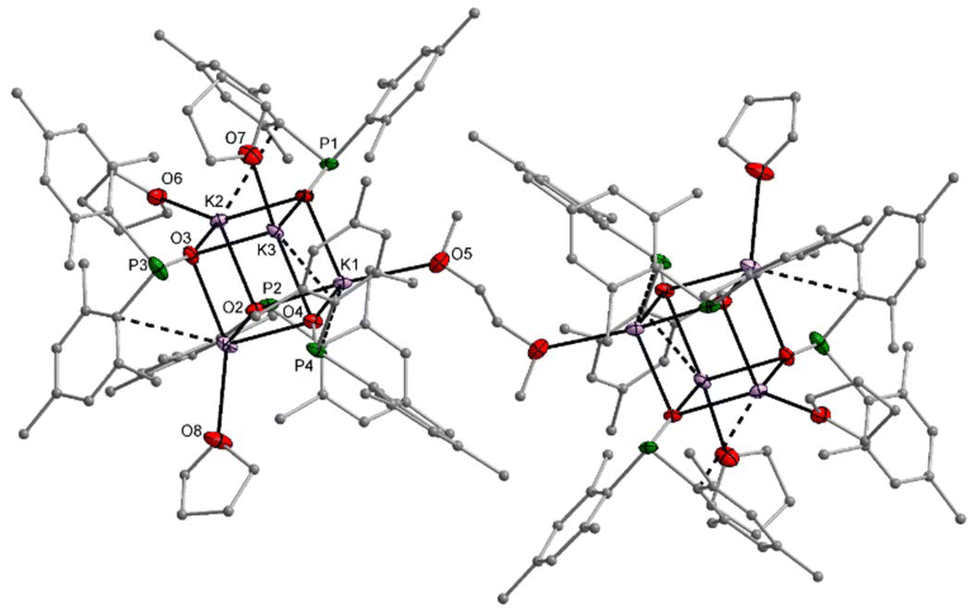 | ||
| Fig. 3 Molecular structure of 4-dme in the crystal. Displacement ellipsoids drawn at the 30% probability level and C atoms as spheres of arbitrary size; H atoms and minor-occupancy orientations of disordered aryl groups, THF ligands, and the DME ligand omitted for clarity. Selected bond lengths (pm): K1–O1 268.8(2), K1–O2 272.0(2), K1–O4 262.2(2), K1–O5 274.3(2), K2–O3 259.6(2), K2–O6 259.9(7), K2–O2 270.1(2), K2–O1 270.6(2), K3–O1 266.4(2), K3–O3 268.9(2), K3–O7 274.1(2), K3–O4 279.7(3), K4–O2 259.3(2), K4–O8 265.3(2), K4–O4 267.2(2), K4–O3 272.1(3), P1–O1 156.6(2), P1–C1 187.9(4), P1–C10 187.3(4), P2–O2 156.3(2), P2–C19 187.2(4) P2–C28 188.0(4), P3–O3 156.2(2), P3–C46 211.2(13), P3–C46A 165.5(11), P3–C37 187.6(4), P4–O4 157.4(2), P4–C64 186.9(4), P4–C55 187.5(3); angles (°): O4–K1–O1 92.52(7), O4–K1–O2 87.07(7), O1–K1–O2 89.38(7), O1–P1–C10 109.46(14), O3–K2–O2 87.91(8), O2–K2–O1 89.36(7). | ||
Enhancement of the denticity of the ligated ether and amine ligands broke down the heterocubane structures and allowed isolation of the dinuclear complexes 4-diglyme (Fig. S73†) and 4-triglyme (Fig. 4) as well as 4-pmdeta (Fig. S75†) with inner K2O2 rings. In 4-diglyme the tridentate ether ligands each bind to one potassium atom leading to a coordination number of five. The additional oxygen donors of the triglyme ligands act as bridging ligands, leading to coordination numbers of seven for the K atoms in 4-triglyme. This binding mode leads to an inner octahedral K2O4 core (see Fig. 4). The pmdeta ligand in 4-pmdeta shows a coordination mode that is very similar to tridentate diglyme.
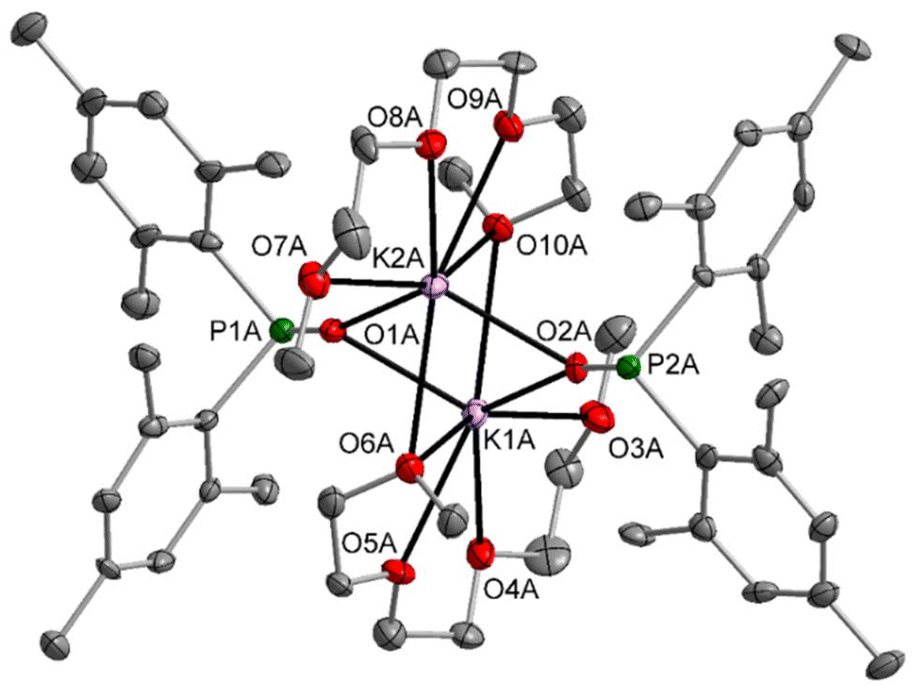 | ||
| Fig. 4 Molecular structure of molecule A of 4-triglyme in the crystal (one of two molecules in the asymmetric unit). Displacement ellipsoids are drawn at the 50% probability level, with H atoms omitted for clarity. Selected bond lengths (pm): K1A–O1A 268.9(6), K1A–O2A 259.0(5), K1A–O3A 292.0(6), K1A–O4A 282.9(5), K1A–O5A 286.7(5), K1A–O6A 328.7(5), K1A–O10A 298.2(5), K2A–O1A 258.7(5), K2A–O2A 268.8(6), K2A–O6A 294.7(5), K2A–O7A 294.6(6), K2A–O8A 285.9(5), K2A–O9A 285.7(5), K2A–O10A 327.4(5), P1A–O1A 155.9(5), P1A–C1A 187.7(8), P1A–C10A 185.4(8), P2A–O2A 154.8(5), P2A–C19A 188.2(8), P2A–C28A 188.5(7). | ||
Selected structural data of these dinuclear potassium complexes are summarised in Table 2. The angle sum ∑P at the phosphorus atom is quite similar for these complexes, but steric strain caused by the ether and amine ligands leads to slightly different C–P–C bond angles. The larger coordination number of the K atoms in 4-triglyme distorts the quadratic K2O2 ring toward a diamond shape, but the K–OP bond lengths are not elongated.
| 4-diglyme | 4-triglyme | 4-pmdeta | |
|---|---|---|---|
| a OP oxygen atom of the dimesitylphosphinite ligand. b Angle sum of the phosphorus atom of the dimesitylphosphinite ligands. | |||
| Av. K–OPa | 267.2 | 263.9 | 261.8 |
| K–OP(max)a | 274.0(2) | 269.2(6) | 266.9(1) |
| K–OP(min)a | 261.5(2) | 258.8(5) | 256.7(1) |
| Av. P–OPa | 156.0 | 155.3 | 155.6(1) |
| Av. P–C | 188.1 | 187.4 | 187.6 |
| K–OP–Ka | 90.2 | 78.5 | 93.36(4) |
| OP–K–OPa | 89.8 | 101.5 | 86.64(4) |
| C–P–C | 101.5 | 99.4 | 97.83(6) |
| ∑Pb | 313.8 | 312.8 | 313.1 |
The dimesitylphosphinite complexes of the heaviest alkali metals rubidium and caesium show quite peculiar solid-state structures. The molecular structure and atom labelling scheme of 5-thf are depicted in Fig. 5. Despite the fact that Rb is significantly larger than potassium, a dinuclear compound with a bridging thf ligand crystallises from the THF solution. This binding mode leads to a distorted trigonal Rb2O3 bipyramid. For the soft Rb cation the aryl π-system can compete with the harder ethereal Lewis bases and aggregation polymers form in the solid state via such interactions with neighbouring molecules.
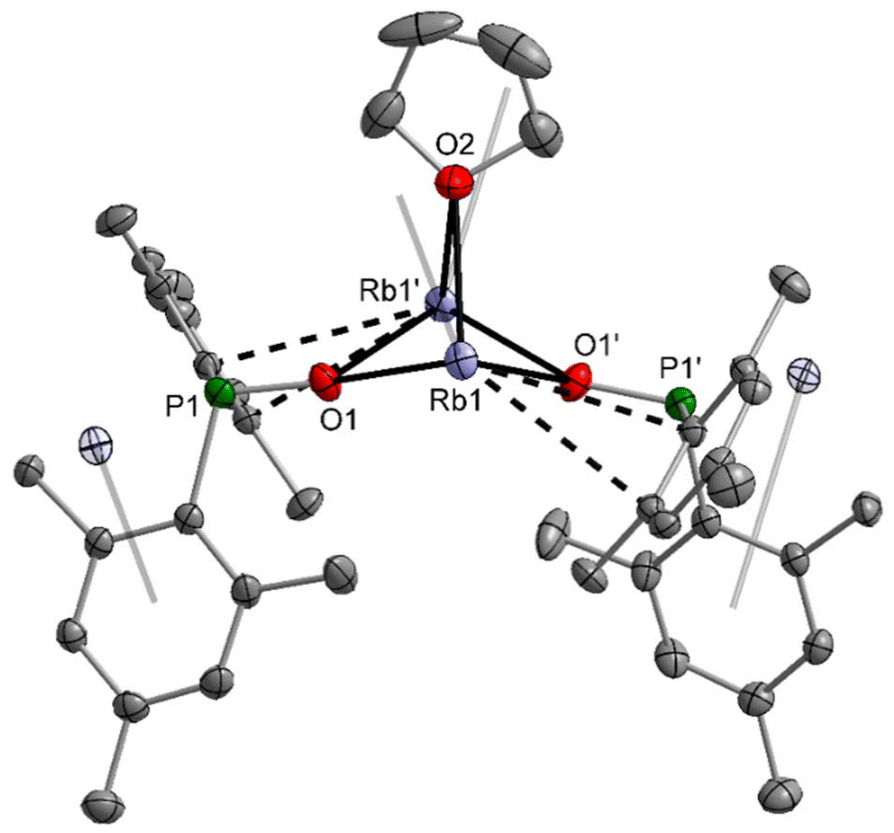 | ||
| Fig. 5 Molecular structure of 5-thf in the crystal. Displacement ellipsoids drawn at the 50% probability level, H atoms and disorder of the THF ligand omitted for clarity. Selected bond lengths (pm): Rb1–O1 271.1(1), Rb1–O1′ 275.6(1), Rb1–O2 292.6(2), P1–O1 155.4(1), P1–C1 187.9(2), P1–C10 187.9(2); angles (°): O1–Rb1–O1′ 83.47(5), O1–Rb1–O2 76.01(4), O1′–Rb1–O2 75.34(4), P1–O1–Rb1 146.88(8), P1–O1–Rb1′ 121.77(7), Rb1–O1–Rb1′ 88.19(4). | ||
Crystallisation of the caesium dimesitylphosphinite from the THF solution leads to the formation of the thf-free tetranuclear complex 6. Its molecular structure and atom labelling scheme are depicted in Fig. 6. The inner Cs4O4 core can be regarded as a severely distorted heterocubane cage, with one Cs corner (Cs3) removed and turned to the outside of this cage substructure. The very soft Cs cations are shielded via coordination to the soft aryl π-systems, and due to the large radius of this cation it fits better into the diameter of the aryl ring.
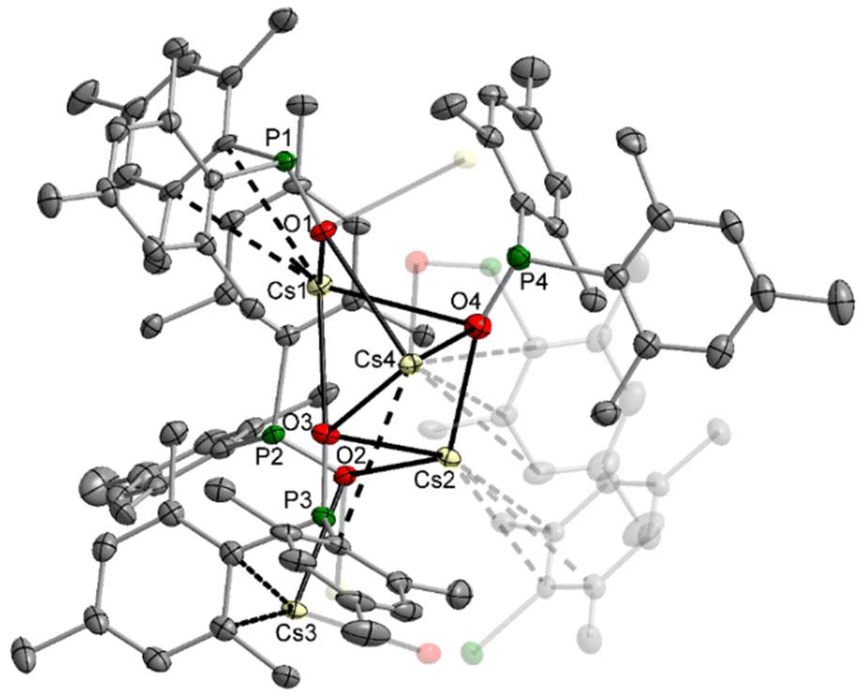 | ||
| Fig. 6 Molecular structure of 6 in the crystal. Displacement ellipsoids drawn at the 30% probability level, H atoms and minor-occupancy orientations of disordered aryl groups omitted for clarity. Selected bond lengths (pm): Cs1–O1 300.6(3), Cs1–O3 299.6(3), Cs1–O4 309.2(4), Cs2–O2 310.6(3), Cs2–O3 322.9(4), Cs2–O4 296.4(4), Cs3–O1 319.2(3), Cs3–O2 299.7(3), Cs4–O1′ 300.3(3), Cs4–O2′ 338.5(3), Cs4–O3 300.3(3), Cs4–O4 316.7(4), P1–O1 155.5(3), P2–O2 155.8(3), P3–O3 155.5(3), P4–O4 154.3(4); angles (°): O1–Cs1–O3 88.62(9), O1–Cs1–O4 85.96(9), O3–Cs1–O4 71.4(1), O2–Cs2–O3 95.98(9), O2–Cs2–O4 128.59(9), O3–Cs2–O4 69.87(9), O2–Cs3–O1′ 73.59(8), O1–Cs4–O2′ 70.72(8), O1–Cs4–O3 88.57(9), O1–Cs4–O4 84.70(9), O3–Cs4–O4 70.26(9), O3–Cs4–O2′ 149.89(9), O4–Cs4–O2′ 126.72(9). | ||
The thf adducts of the alkali metal dimesitylphosphinites are compared in Table 3 to elucidate the influence of the alkali metal on selected structural parameters. Expectedly, the A–OP distances increase with the radius of the alkali ion, but the P–OP bond lengths decrease. This finding can be explained by an increase in the ionicity of the complexes from Li to Cs due to a decrease in the electronegativity values of the heavier cations. The influence of the electronegativity and hardness of the cations on the P–C bond lengths is negligible. The smaller alkali metal complexes 2-thf, 3-thf, and 4-thf have strictly planar A2O2 rings, whereas the larger alkali ions allow folding of the four-membered rings and both A–OP–A and OP–A–OP bond angles are smaller than 90°. Again, the phosphorus atoms in the dimesitylphosphinite ligands have a trigonal pyramidal environment.
| 2-thf | 3-thf | 4-thf | 5-thf | 6 | |
|---|---|---|---|---|---|
| a OP oxygen atom of the dimesitylphosphinite ligand. b Angle sum of the phosphorus atom of the dimesitylphosphinite ligands. | |||||
| A | Li | Na | K | Rb | Cs |
| A–OPa | 187.8 | 224.4 | 267.6 | 273.4 | 306.9 |
| P–OPa | 157.3 | 154.5 | 156.5 | 155.4(1) | 155.3 |
| P–C | 186.9 | 186.7 | 188.2 | 187.9(2) | 187.8 |
| A–OP–Aa | 82.2 | 88.5(2) | 91.0 | 88.19(4) | 88.1 |
| OP–A–OPa | 97.8 | 89.8 | 89.0 | 83.47(5) | 87.1 |
| C–P–C | 99.9 | 101.4 | 98.8 | 98.47(5) | 98.9 |
| ∑Pb | 312.7 | 312.0 | 311.8 | 311.8 | 314.9 |
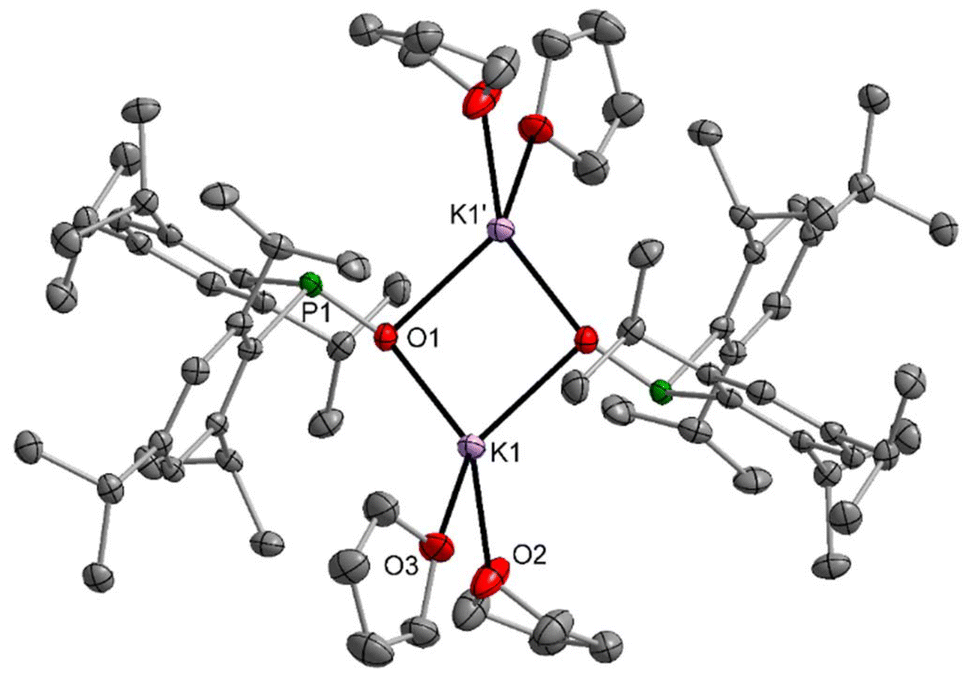
Blocking the coordination sites at the Lewis acidic alkali cation can avoid the formation of heterocubane cage compounds. Alternatively, enhancement of the steric requirements of the P-bound substituents can also hinder aggregation and hence can stabilise dinuclear complexes. While methylnaphthyl groups are not bulky enough for this purpose as shown for 7-thf (Fig. S78†), 2,4,6-triisopropylphenyl substituents hinder dimerisation of a dinuclear complex effectively; the coordination number four for the potassium ion is maintained by coordination to two thf ligands. The molecular structure and atom labelling scheme of 8-thf are depicted in Fig. 7. The centrosymmetric K2O2 ring is distorted toward a rectangle with slightly different K1–O1 and K1–O1′ bond lengths. Due to smaller electrostatic attraction, the K1–Othf distances to the thf ligands are larger than to the phosphinite bases.
 | ||
| Fig. 7 Molecular structure of 8-thf in the crystal. Displacement ellipsoids drawn at the 40% probability level, H atoms and minor-occupancy orientations of disordered THF ligands omitted for clarity. Selected bond lengths (pm): K1–O1 250.5(2), K1–O1′ 261.1(2), K1–O2 263.7(2), K1–O3 269.5(2), P1–O1 155.2(2), P1–C1 187.8(2), P1–C16 187.8(3); angles (°): P1–O1–K1141.5(1), P1–O1–K1′ 101.40(9), K1–O1–K1′ 94.30(6). | ||
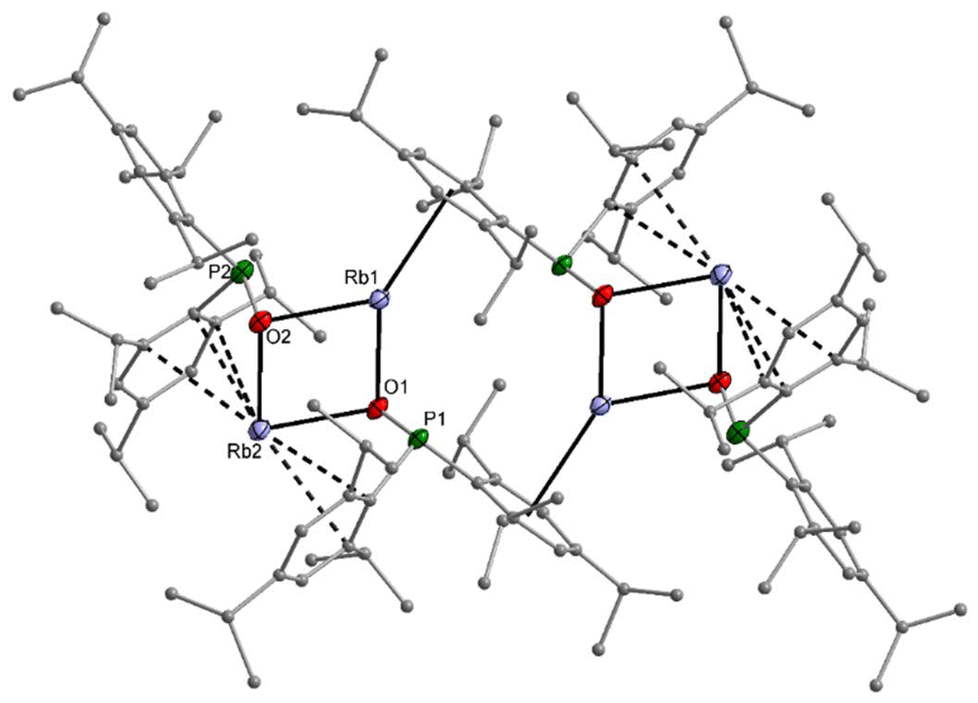
Comparable structures are observed for the heavier homologous rubidium (11) and caesium compounds (12). Despite the synthesis in THF solution, both compounds crystallise thf-free, but 12 contains one Tipp2P(H)O ligand per caesium phosphinite dimer. The rubidium complex 11 has a central diamond-shaped Rb2O2 ring. The molecular structure and atom labelling scheme are depicted in Fig. 8. The lack of ligated thf bases leaves vacant coordination sites at the alkali metal atoms, which are occupied by π-interactions with the side-on coordinated aromatic systems.
 | ||
| Fig. 8 Molecular structure of 11 in the crystal. Displacement ellipsoids drawn at the 30% probability level and C atoms as spheres of arbitrary sizes, H atoms and minor-occupancy orientations of disordered aryl groups omitted for clarity. Selected bond lengths (pm): Rb1–O1 282.2(3), Rb1–O2 275.0(3), Rb2–O1 273.4(3), Rb2–O2 272.9(3), P1–O1 154.8(3), P2–O2 155.2(3), P1–C1 186.9(4), P1–C16 187.6(4); angles (°): P1–O1–Rb1 93.51(13), P1–O1–Rb2 115.00(14), Rb2–O1–Rb1 96.55(8), Rb2–O2–Rb1 98.38(8), O2–Rb1–O1 81.49(8), O2–Rb2–O1 83.47(8), C1–P1–C16 100.21(18). | ||
The Rb2 atom is shielded intramolecularly by two Tipp substituents, and due to steric requirements, the interactions with the ipso- and ortho-carbon atoms are significantly stronger. In contrast, Rb1 binds in an η6-manner to an aryl group of the neighbouring rubidium phosphinite dimer, leading to a centrosymmetric tetrameric unit without the formation of a heterocubane cage. While Rb1 has slightly different Rb1–O1 and Rb1–O2 bond lengths, the bonds Rb2–O1 and Rb2–O2 have the same value. This structure type is maintained for the caesium congener 12, but the larger radius of the alkali metal opens the coordination sphere. The molecular structure and atom labelling scheme are depicted in Fig. 9. While the aggregation of the two dimers occurs very similarly to that observed for the rubidium compound 11, the outer caesium atoms Cs1 and Cs1′ bind an additional bis(2,4,6-triisopropylphenyl)phosphane oxide with a rather short Cs1–O3 bond of 290.3(5) pm, even slightly shorter than the distances between Cs2 and the bridging phosphinite oxygen bases.
 | ||
| Fig. 9 Molecular structure of 12 in the crystal. Displacement ellipsoids drawn at the 30% probability level and C atoms as spheres of arbitrary size; H atoms and minor-occupancy orientations of disordered isopropyl groups omitted for clarity. Selected bond lengths (pm): Cs1–O1 287.0(4), Cs1–O2 297.0(5), Cs1–O3 290.3(5), Cs2–O1 291.8(4), Cs2–O2 293.6(5), P1–O1 153.6(5), P2–O2 153.8(5), P3–O3 148.5(5), P1–C1 190.4(7), P1–C16 189.4(8), P3–C61 182.7(8), P3–C76 183.5(7); angles (°): P1–O1–Cs1 118.1(2), P1–O1–Cs2 93.0(2), Cs1–O1–Cs2 99.51(14), Cs2–O2–Cs1 96.86(14), O1–Cs1–O2 81.89(13), O1–Cs2–O2 81.66(13), C16–P1–C1 105.7(3), C61–P3–C76 108.7(3). | ||
In hydrocarbons, the formation of potassium phosphinite is quite slow. In methylcyclohexane with a very small amount of diglyme, single crystals of 9-hmds precipitated which can be considered as a snapshot on the way from the starting [KN(SiMe3)2]2 to the end product [K–O-PMes2]4. The first reaction step is metalation of one dimesitylphosphane oxide (1) and the formation of a dinuclear heteroleptic unit consisting of one KN(SiMe3)2 unit and one K–O-PMes2 fragment. The resulting four-membered K2NO rings can be recognised as isolated structural motifs for K1–N1–K2–O2 and K7–N4–K8–O4. Aggregation of two such structural elements via K–O bonds yields the central tetranuclear substructure, which can be considered as a heterocubane cage with an open face. The basicity of the bis(trimethylsilyl)amido ligands is very low and hinders the formation of K3–N3 and K6–N2 bonds. The potassium ions are additionally shielded by coordination to the mesityl π-systems. A diglyme ether binds to K1 and hinders the formation of an aggregation polymer (Fig. 10).
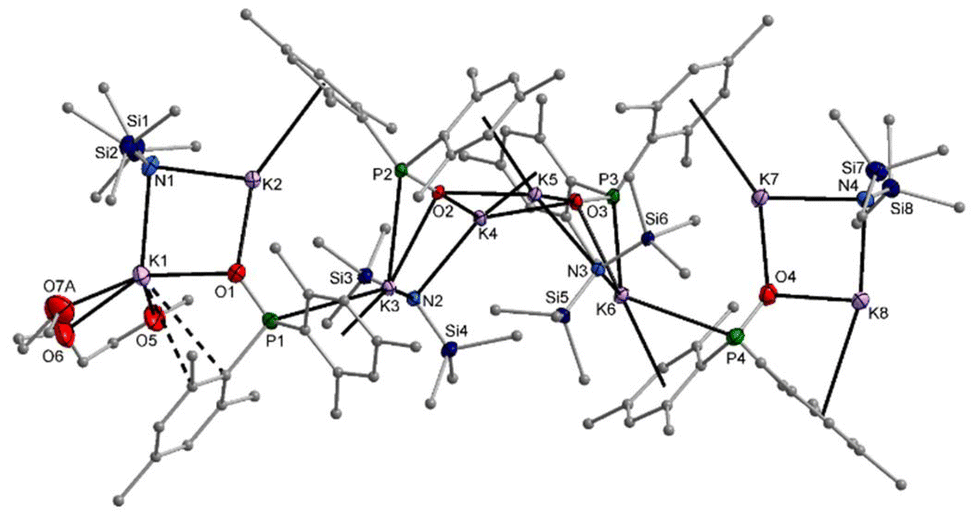 | ||
| Fig. 10 Molecular structure of 9-hmds in the crystal. Displacement ellipsoids drawn at the 30% probability level and C atoms as spheres of arbitrary size; H atoms and minor-occupancy orientations of disordered aryl groups, SiMe3 groups, and the diglyme ligand omitted for clarity. Selected bond lengths (pm): K1–O1 263.8(4), K1–N1 290.3(6), K2–O1 261.2(5), K2–N1 273.9(5), K3–O2 283.7(4), K3–N2 278.2(5), K4–O2 263.3(4), K4–O3 274.1(4), K5–O2 275.1(4), K5–O3 262.7(4), K6–O3 282.6(4), K6–N3 274.2(5), K7–O4 257.0(5), K7–N4 281.1(5), K8–O4 259.2(4), K8–N4 270.5(5), P1–O1 154.1(4), P2–O2 156.7(3), P3–O3 156.4(4), P4–O4 152.8(5); angles (°): K2–O1–K1 95.24(14), O1–K1–N1 85.28(14), K2–N1–K1 86.81(14), O1–K2–N1 89.21(15), P1–O1–K1 121.1(2), P1–O1–K2 131.7(2). | ||
In heterobimetallic K/Mg complexes, the phosphinite ions bind to the more electronegative magnesium atom, yielding the tetrakis(dimesitylphosphinito)magnesiate ion with potassium counterions. The molecular structure and atom labelling scheme of the contact ion pair [{(thf)K}2Mg(OPMes2)4] (10-thf) are depicted in Fig. S81.† The magnesium centre is embedded in a distorted tetrahedral coordination sphere, the K atoms each bind to two oxygen atoms and one P base of three phosphinite ligands.
Catalytic studies
Earlier investigations revealed that alkali metal phosphinites are outstanding catalysts for the hydrophosphorylation of aryl alkynes.2c–d,15A product mixture of (E)- and (Z)-isomers of alkenyl phosphane oxides as well as a dihydrophosphindole oxide generated by a subsequent ring closure reaction could be observed. Intrigued by these findings, we wanted to investigate both the influences of heterobimetallic phosphinites and multidentate ligands on the selectivity of the hydrophosphorylation reaction of phenylacetylene. For this purpose, we loaded an NMR tube with 1 equivalent of dimesitylphosphane oxide (1) and 1.1 equivalents of phenylacetylene in [D8]THF and initiated the reaction by adding 15 mol% of [A/Ae(hmds)x] (x = 1 for Li, Na, and K; x = 2 for Ca). We monitored the reaction via31P NMR spectroscopy. Triphenylphosphane was used as an internal standard. The conversion of dimesitylphosphane oxide and the (E)/(Z) ratio of the alkenyl phosphane oxide after 30, 60, and 180 minutes are listed in Table 4 (Table S2†). Furthermore, the table shows that the doubly hydrophosphorylated addition product 12 (intermediate of (E)/(Z) isomerisation) and dihydrophosphindole oxide 13 were present in the reaction mixture (Scheme 6).
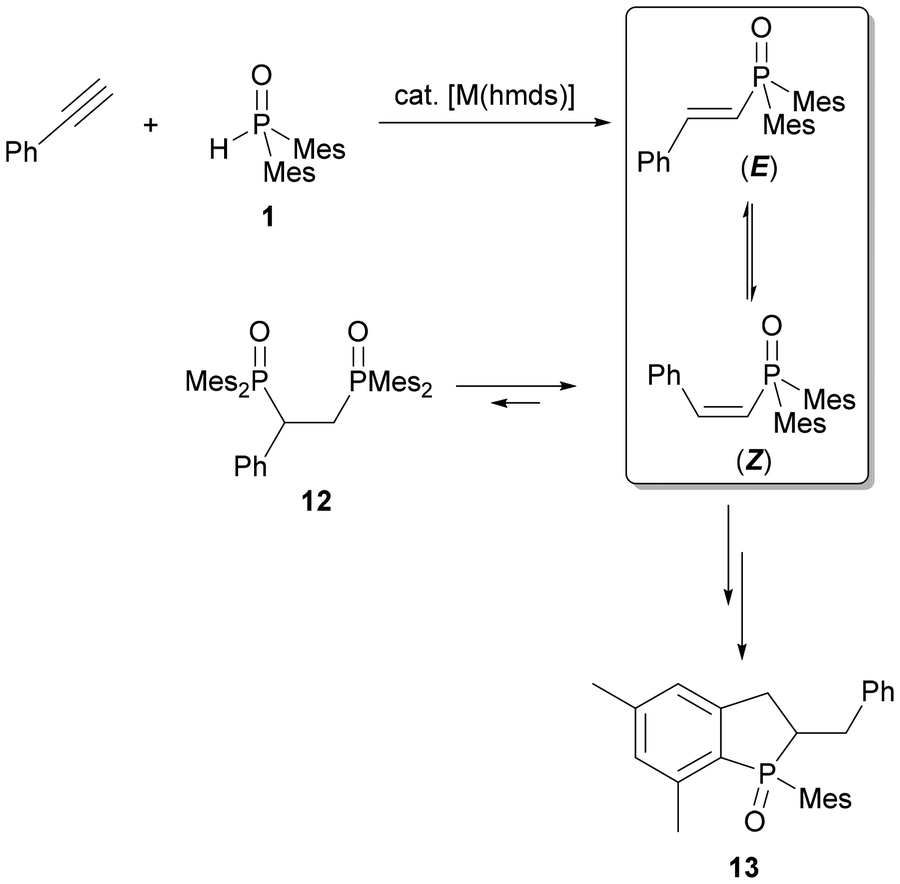 | ||
| Scheme 6 s-Block-metal-mediated addition of dimesitylphosphane oxide (1) across phenylacetylene yielding the (E)- and (Z)-isomers of the alkenylphosphane oxides that interconvert due to intermediately formed bis(phosphane) oxide 12 which itself can undergo base-catalysed elimination. Dihydrophosphindole oxide 13 as a product of subsequent cyclisation is shown as well. | ||
| Entry | M1 | M2 | M1/M2 | L | χ 30 | Y 30 | X 60 | Y 60 | X 180 | Y 180 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a 2.2 equivalents of phenylacetylene are applied. The full table is shown in the ESI (Table S2†). | ||||||||||||
| 1 | Li | — | — | — | 17 | 1/0 | 37 | 1/0 | 72 | 1/0 | — | — |
| 2 | Na | — | — | — | 76 | 4.9/1 | <99 | 20.3/1 | <99 | 1/0 | ✓ | — |
| 3 | K | — | — | — | <99 | 5.8/1 | <99 | 14/1 | <99 | 1/0 | ✓ | ✓ |
| 4 | Ca | — | — | — | 7 | 1.6/1 | 11 | 2.3/1 | 30 | 11.7/1 | — | — |
| 5 | Ca | Li | 1/1 | — | 46 | 1/0 | 68 | 1/0 | <99 | 1/0 | — | — |
| 6 | Ca | Li | 1/2 | — | 34 | 1/0 | 58 | 1/0 | <99 | 1/0 | — | — |
| 7 | Ca | Na | 1/1 | — | 44 | 3.5/1 | 74 | 10.3/1 | <99 | 1/0 | — | — |
| 8 | Ca | Na | 1/2 | — | 52 | 3/1 | 86 | 8.4/1 | <99 | 48/1 | — | — |
| 9 | Ca | K | 1/1 | — | 66 | 1.7/1 | <99 | 4/1 | <99 | 16.2/1 | — | — |
| 10 | Ca | K | 1/2 | — | <99 | 2.7/1 | <99 | 3.8/1 | <99 | 9.1/1 | — | ✓ |
| 11 | Ca | K | 1/4 | — | <99 | 3.5/1 | <99 | 3.9/1 | <99 | 6.5/1 | ✓ | ✓ |
| 12 | Na | Li | 1/1 | —— | 59 | 42.3/1 | 93 | 1/0 | <99 | 1/0 | — | — |
| 13 | K | Li | 1/1 | — | 92 | 33.7/1 | <99 | 1/0 | <99 | 1/0 | — | — |
| 14 | K | — | — | pmdeta | <99 | 6.5/1 | <99 | 15/1 | <99 | 1/0 | ✓ | ✓ |
| 15 | K | — | — | benzo-18C6 | <99 | 1/0 | <99 | — | <99 | — | — | ✓ |
| 16 | K | — | — | diglyme | <99 | 3.5/1 | <99 | 5.3/1 | <99 | 10.9/1 | ✓ | — |
| 17 | K | — | — | diglymea | <99 | 1.2/1 | <99 | 1.3/1 | <99 | 1.6/1 | — | — |
| 18 | K | — | — | dme | <99 | 5.8/1 | <99 | 14.6/1 | <99 | 1/0 | — | — |
As expected, it could be shown that the conversion rate was highly dependent on the metal ion as it increased tremendously within the main group (χ30 = 17% (Li), 76% (Na), and <99% (K)). This could be attributed to the increasing softness and polarizability from lithium to potassium. Moreover, dihydrophosphindole oxide 13 was only formed using the heavier homologues (K, Rb, and Cs). It was striking that the doubly hydrophosphorylated product 12 could only be detected in notable quantities using Na(hmds) as the precatalyst. Regarding the (E)/(Z) ratio, it became apparent that using lithium, the reaction was highly selective, as only the thermodynamically favoured (E)-isomer was formed, whereas with the other metals, both isomers could be observed, with the (E)-isomer always being preferred. Using Ca(hmds)2 as a precatalyst resulted in a significantly slower reaction (χ30 = 7%). The combination of the two metals Li and Ca led to an increased reaction rate while maintaining excellent (E)/(Z)-selectivity. However, using this heterobimetallic catalyst system, the reaction rate appeared to be conspicuously independent of the ratio of Ca and Li. We tested 1:1, 1:2, and 2:1 ratios of Ca and Li and noticed that the conversion rates differed only minimally. It was remarkable that the reaction with the homometallic precatalysts of lithium and calcium was slower than with their heterobimetallic counterpart, notwithstanding that the amide concentration was kept unaltered.
Furthermore, the presence of Li ions influenced the hydrophosphorylation reactions catalysed by sodium or potassium hexamethyldisilazanides. The added Li(hmds) markedly increased the selectivity in favour of the (E)-isomer (diastereomeric excess de30 = 66% (Na), 71% (K), 95% (Na/Li), and 94% (K/Li)). The reaction rate appeared to be determined by the heavier alkali metal. This observation denoted a synergistic effect of the heterobimetallic catalyst system. A further asset could be deduced from the available data by looking at the possible side products 12 and 13. While the potassium-catalysed reaction produced them both, the addition of a lithium base completely suppressed their formation, and hence, at the end of the reaction, the pure (E)-isomer was present in the solution.
The influence of oligodentate donor ligands (pmdeta, diglyme, and dme) on the hydrophosphorylation of phenylacetylene was also investigated. These Lewis bases were added to the NMR tube prior to the addition of the precatalytic bis(trimethylsilyl)amides. Again, the reaction was monitored via31P NMR spectroscopy. At first, it could be ascertained that the addition of the ligands did not hamper the hydrophosphorylation reaction. Surprisingly, kinetic measurements showed that the applied polydentate ligands did not influence significantly the reaction rate. Considering the isomer ratio, it became evident that the diastereomeric excess was only subject to minor deviations (56–73%), either. These findings of our catalytic testing were corroborated by the ECC-DOSY measurements we performed to verify that in THF solutions oligodentate ligands such as diglyme and pmdeta were substituted by THF molecules and thus, their catalytic activity appeared to be comparable to the thf complexes.
However, the application of dibenzo-18-crown-6 as a complexation reagent results in a faster reaction, which leads exclusively to the formation of dihydrophosphindole oxide 13 already within one hour.
The importance of doubly hydrophosphorylated bis(phosphane oxide) 12 on the isomerisation of alkenylphosphane oxides could be demonstrated by doubling the amount of phenylacetylene in the reaction mixture. This impeded the formation of 12 and as a result, the diastereomeric excess diminished distinctly (from 56 to 9%). Even after 180 minutes, the value rose only to 23%, compared to 81% for the reaction with 1.1 equivalents of phenylacetylene.
In order to further investigate the kinetics of the s-block metal-base-mediated hydrophosphorylation reaction, the addition of dimesitylphosphane oxide (1) across phenylacetylene in the presence of K(hmds) as a precatalyst was selected as a benchmark system. Toluene was found to be the most suitable solvent for this purpose, as previous studies had revealed that in toluene no subsequent reactions to bis(phosphane oxide) 12 and dihydrophosphindole oxide 13 occur, but only an (E)/(Z) mixture of dimesityl(2-ethenylphenyl)phosphane oxide formed (Fig. 11).2d
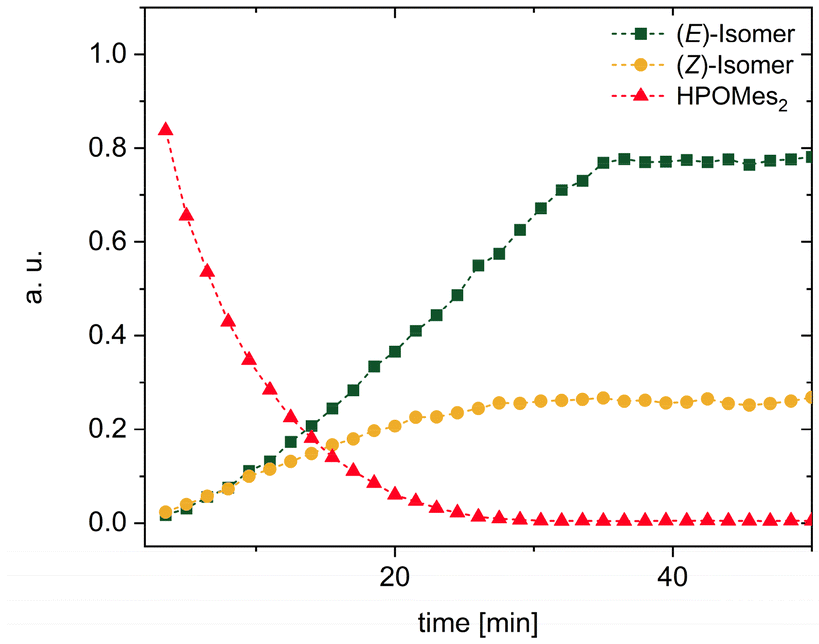 | ||
| Fig. 11 31P NMR kinetics of the hydrophosphorylation of phenylacetylene with dimesitylphosphane oxide (1) in toluene yielding a mixture of (E)- and (Z)-styryl dimesitylphosphane oxide. | ||
The progress of the reaction was monitored using 31P NMR spectroscopy. Firstly, we tried to determine the reaction order in terms of the phenylacetylene concentration by maintaining constant concentrations of HPOMes2 and K(hmds). The measured reaction rates were directly proportional to the added amount of phenylacetylene verifying first-order kinetics regarding the alkyne component (Fig. 12a). The same method was applied to examine the dependence of the reaction rate on the amount of pre-catalytically active K(hmds). Here, too, the data revealed a direct proportionality of the reaction rate and the catalyst concentration, and hence, first-order kinetics could be presumed (Fig. 12b). In contrast, the variation in the amount of HPOMes2—keeping the concentrations of PhCCH and K(hmds) constant—showed an intriguingly different trend. Contrary to expectations, the reaction rate decreased with increasing amounts of dimesitylphosphane oxide. In addition, the correlation could not be fitted sufficiently well with a linear function; hence, an exponential fit is depicted in the diagram (Fig. 12c). It is well known that phosphane oxides could act as strong ligands for s-block-metals, thus shielding them. Nevertheless, based on a KPOMes2/HOPMes2 ratio of 1:10, it was not expected that further deceleration of the reaction would occur at even higher concentrations of HPOMes2 due to increased shielding of the catalytically active potassium ion. We assumed that there were additional equilibria being influenced by the alteration of the concentration of HPOMes2. Comparing the pKa values of diarylphosphane oxide (pKa (HPOPh2) = 20.7) and phenylacetylene (pKa (PhCCH) = 21–23.2 depending on the solvent), we could assume that acid–base equilibrium played a key role in the formation of potassium phenylacetylide which was considered the actual catalytically active species. Adding K(hmds) to a mixture of dimesitylphosphane oxide (1) and phenylacetylene at −40 °C, and subsequent hydrolysis with deuterium chloride, led to the formation of d1-HPOMes2 and d1-phenylacetylene supporting our assumption. The same observation had previously been made with trimethylsilyl acetylene as an alkyne component.16 Thus, an increase in the concentration of HPOMes2 would shift this equation to the phosphinite species being the less active catalyst resulting in a slower reaction (Scheme 7).
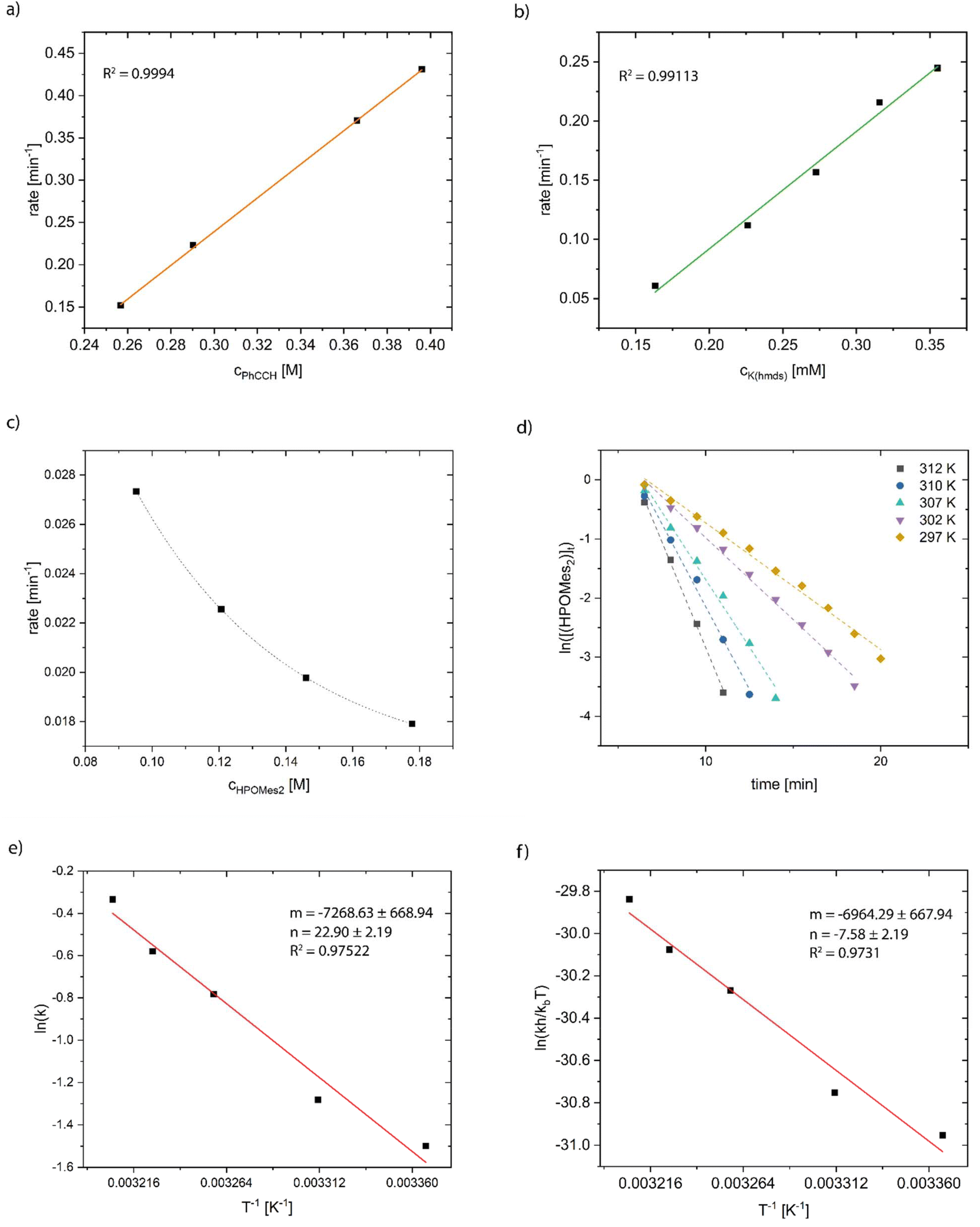 | ||
| Fig. 12 (a) Plot of the reaction rate versus concentration of phenylacetylene; (b) plot of the reaction rate versus concentration of K(hmds); (c) plot of the reaction rate versus concentration of dimesitylphosphane oxide; (d) plot of ln(HPOMes2) versus time at different temperatures; (e) Arrhenius plot; and (f) Eyring plot. | ||
 | ||
| Scheme 7 Acid–base-type equilibrium of KOPMes2 with phenylacetylene. | ||
Furthermore, the reaction rate of the hydrophosphorylation reaction was determined at different temperatures (Fig. 12d). Applying an Arrhenius plot (ln(r) versus 1/T) to the recorded data, the activation energy Ea was calculated to be 60 ± 5 kJ mol−1 (Fig. 12e). The thermodynamic parameters of activation were determined by means of Eyring analysis (plot ln(rh/kbT) versus 1/T) to ΔH‡ = 58 ± 6 kJ mol−1, ΔS‡ = −63 ± 18 J (kmol)−1, and ΔG‡(297 K) = 77 ± 6 kJ mol−1 (Fig. 12f).
Conclusions
Metalation of dimesitylphosphane oxide, Mes2P(O)H (1), with n-butyllithium, sodium hydride and the alkali metal bis(trimethylsilyl)amides, AN(SiMe3)2 with A = K, Rb, and Cs, yields the corresponding alkali metal dimesitylphosphinites, A–O-PMes2. In THF solution, the thf adducts form readily for the lighter alkali metal complexes, whereas the caesium congener crystallises without ligated thf molecules. The dimesitylphosphinite anions are rather bulky and stabilise dinuclear [(thf)3(Li–O-PMes2)2]2 (2-thf) with a four-membered Li2O2 ring in contrast to the tetranuclear lithium diphenylphosphinite with an inner heterocubane core. Such a tetranuclear complex of the type [(thf)A–O-PMes2]4 precipitates for potassium (4-thf). The larger and softer alkali metal congeners show unique molecular structures. The larger size of the alkali metal ions leads to increased softness and allows a more effective interaction with the aryl π-systems and hence, the soft mesityl π-systems can compete with ethereal Lewis bases. Therefore, the Cs compound 6 precipitates without ligated thf molecules.Tetranuclear [(thf)Li–O-PPh2]4 can be deaggregated by bidentate 1,2-dimethoxyethane yielding dinuclear [(dme)Li–O-PPh2]2. To deaggregate tetranuclear [(thf)K–O-PMes2]4 (4-thf) ethers with a larger denticity are required. Thus, DME only leads to a linkage between two tetranuclear cage complexes, whereas diglyme and triglyme can stabilise dinuclear potassium derivatives in 4-diglyme and 4-triglyme. While 4-thf does not react with TMEDA, tridentate PMDETA deaggregates 4-thf yielding dinuclear 4-pmdeta with an inner four-membered K2O2 ring. The absence of strong Lewis bases like THF, diglyme, triglyme, and pmdeta decelerates the metalation reaction significantly. This finding opens the opportunity to crystallise a heteroleptic compound of the type [(diglyme){K2(N(SiMe3)2)(OPMes2)}4] (9-hmds) from an alkane solution containing very small amounts of diglyme. Stabilisation of dinuclear potassium diarylphosphinites succeeds also by bulky P-bound aryl groups like 2,4,6-triisopropylphenyl substituents as in 8-thf.
The P atoms of the phosphinite ions are in trigonal pyramidal environments. The C–P–C angle is slightly influenced by intramolecular steric strain, whereas the P–C bond lengths are not affected. Increasing ionic bond character due to decreasing electronegativity of the alkali metal leads to shortening of the P–O distances.
The heavier dimesitylphosphinites of K, Rb, and Cs represent suitable catalysts for the addition of dimesitylphosphane oxide across alkynes, heterocumulenes and nitriles (Pudovik reaction). The lithium derivative itself is a very poor catalyst, but as an additive to heavier alkali metal hmds precatalysts, the advantages of heterobimetallic catalyst systems in terms of selectivity and rate become clearly evident.
Experimental
General information
All experiments were carried out under an inert nitrogen atmosphere using standard Schlenk techniques, if not otherwise noted. The solvents were dried over KOH and subsequently distilled over sodium/benzophenone under a nitrogen atmosphere prior to use. All substrates were purchased from Alfa Aesar, abcr, Sigma Aldrich or TCI and used without further purification. Dimesitylphosphane oxide was prepared according to a literature procedure.2d The yields given are not optimized. The purity of the compounds was verified by NMR spectroscopy. Deuterated solvents were dried over sodium, distilled, degassed, and stored under nitrogen over sodium. 1H and 13C{1H} NMR spectra were recorded using Bruker Avance III 400 (BBO, BBFO probes), Avance II HD 500 (BBO Prodigy probe) or Avance neo 500 (BBFO Prodigy probe) spectrometers. Chemical shifts are reported in parts per million relative to SiMe4 as an external standard referenced to the solvent residual proton signal using the xiref AU program for 13C and 31P. DOSY NMR spectra were recorded using the convection compensated standard pulse sequence dstebpgp3s (64 increments, d = 0.75 s, D = 2.5 ms). Molar masses in solution were calculated using the Stalke-ECC-DOSY method (standard: adamantane or residual signal of the solvent).17 ASAP-HSQC-spectra and ASAP-HSQC-DEPT-spectra were recorded using the published pulse sequences.18 The elemental analyses gave no reliable results due to the loss of ligated ether ligands during handling and carbonate formation during combustion. IR spectra were recorded using a Bruker Alpha IR spectrometer with an ATR unit. Intensities are given with the descriptors: vw – very weak, w – weak, m – moderate, s – strong, and vs – very strong. The NMR data of compound 4-thf corresponds to the published data.11Synthesis of [(thf)3Li2(OPMes2)2] 2-thf
Dimesitylphosphane oxide (0.58 g, 2.03 mmol, 1 eq.) was dissolved in 3 mL of THF and the solution was cooled to −20 °C. Then, n-BuLi (2.43 mL, 2.43 mmol, 1 M in n-hexane, 1.2 eq.) was added dropwise. Then, the dark yellow solution was warmed up to room temperature. The volume of the solution was reduced in vacuo to one half and upon storage at room temperature the phosphinite 2-thf crystallised as yellow blocks. The crystals were collected and dried carefully in vacuo (240 mg, 0.82 mmol, 41%).1H NMR (300.19 MHz, [D8]THF, 297 K) δ: 6.52 (br, 4H, CHMes), 2.41 (br, 12H, ortho-CH3), 2.12 (br, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 140.7 (d, 2JP–C = 13.2 Hz, ortho-CMes), 134.8 (br, para-CMes), 129.4 (br, meta-CMes), 21.4 (d, 3JP–C = 16.0 Hz, ortho-CH3), 20.8 (s, para-CH3) ppm, ipso-CMes not observed due to broad resonances; 31P NMR (121.52 MHz, [D8]THF, 297 K) δ: 95.0 ppm; IR (ATR) ν: 2916w, 2875w, 2725w, 1601vw, 1546vw, 1459w, 1448w, 1370w, 1048m, 916m, 847m cm−1.
Synthesis of [(tmeda)Na-μ-(OPMes2)2-Na(tmeda)] 3-tmeda
Dimesitylphosphane oxide (0.96 g, 3.3 mmol, 1 eq.) was dissolved in 40 mL of THF. The colourless solution formed was added dropwise to a suspension of NaH (0.1 g, 4.2 mmol, 1.25 eq.) and NaNH2 (0.18 g, 4.6 mmol, 1.38 eq.) in THF (ca. 20 mL) leading to gas evolution. After stirring at room temperature for 36 h the yellow solution was separated from excess NaH and NaNH2 by filtration through diatomaceous earth. TMEDA (0.75 mL, 5.0 mmol, 1.5 eq.) was added to the clear filtrate. The mixture was stirred for 0.5 h at room temperature. Then, the volume of the solution was reduced to 5 mL leading to precipitation of an amorphous yellow solid. Heating of the suspension to 60 °C and storage of the solution for three days led to the formation of monoclinic yellow crystals of sodium phosphinite 3-tmeda. The crystalline solid was collected by filtration, washed with cold n-hexane (2 × 3 mL) and carefully dried under reduced pressure (1.09 g, 1.3 mmol, 77%).1H NMR (400.22 MHz, [D8]THF, 297 K) δ: 6.74 (s, 4H, CHMes), 2.68 (br, 12H, ortho-CH3), 2.19 (s, 6H, para-CH3), 1.96 (s, 12H, TMEDA), 1.91 (s, 6H, TMEDA) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 146.5 (br, ipso-CMes), 140.2 (d, 2JP–C = 14.3 Hz, ortho-CMes), 135.4 (br, para-CMes), 130.0 (br, meta-CMes), 57.7 (s, TMEDA), 45.9 (s, TMEDA), 21.6 (d, 3JP–C = 16.1 Hz, ortho-CH3), 21.0 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 101.5 ppm; IR (ATR) ν: 2916m, 2857w, 2827w, 1604m, 1553w, 1452s, 1409m, 1171s, 1026vs, 954s, 846s cm−1.
Synthesis of [(thf)2Na(OPMes2)]23-thf
Dimesitylphosphane oxide (2.39 g, 8.33 mmol, 1 eq.) and sodium hydride (0.4 g, 16.7 mmol, 2 eq.) were placed in a Schlenk flask and THF (8 mL) was added. The yellow solution was stirred at room temperature overnight and the slightly cloudy solution formed was filtered. The solvent of the clear filtrate was reduced in vacuo to 1 mL. Storage at −20 °C led to the precipitation of yellow-coloured crystals that were suitable for X-ray diffraction experiments. The crystals were collected and dried carefully in vacuo (0.453 g, 1.47 mmol, 18%).1H NMR (400.13 MHz, C6D6, 297 K) δ: 6.68 (s, 4H, CHMes), 3.57 (m, 4H, THF), 2.44 (br, 12H, ortho-CH3), 2.18 (s, 6H, para-CH3), 1.40 (m, 4H, THF) ppm; 13C{1H} NMR (100.62 MHz, C6D6, 297 K) δ: 143.7 (d, 1JP–C = 49.2 Hz, ipso-CMes), 140.02 (d, 2JP–C = 14.1 Hz, ortho-CMes), 136.5 (s, para-CMes), 130.5 (s, meta-CMes), 68.1 (s, THF), 25.7 (s, THF), 21.5 (d, 3JP–C = 15.7 Hz, ortho-CH3), 21.0 (s, para-CH3) ppm; 31P NMR (161.98 MHz, C6D6, 297 K) δ: 101.4 ppm; 1H DOSY NMR (400.13 MHz, C6D6, 297 K): D(ref, C6D6) = 1.265 × 10−9 m2 s−1, D(3-thf) = 2.63 × 10−10 m2 s−1, MW(calc, [(thf)NaOPMes2]3) = 1141 g mol−1, MW(exp) = 1141 g mol−1, Δ = 0%; 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.55 (s, 4H, CHMes), 2.42 (br, 12H, ortho-CH3), 2.15 (s, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 147.1 (d, 1JP–C = 56.1 Hz, ipso-CMes), 140.4 (d, 2JP–C = 14.2 Hz, ortho-CMes), 134.7 (s, para-CMes), 129.4 (s, meta-CMes), 21.3 (d, 3JP–C = 16.4 Hz, ortho-CH3), 20.9 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 98.9 ppm; IR (ATR) ν: 2972w, 2917w, 2870w, 1604w, 1553w, 1451w, 1410w, 1198w, 1170w, 1053m, 957m, 847s cm−1.
Synthesis of [K4(OPMes2)4]24
Dimesitylphosphane oxide (351 mg, 1.23 mmol, 1 eq.) and potassium hexamethyldisilazanide (245 mg, 1.23 mmol, 1 eq.) were placed in a Schlenk flask and 1 mL of n-hexane was added. Heating the yellow suspension formed to 70 °C and subsequent slow cooling to room temperature led to the formation of bright yellow crystals of 4 that were suitable for X-ray diffraction experiments. The crystals were collected on a frit and dried carefully in vacuo (306 mg, 0.94 mmol, 76%).1H NMR (400.13 MHz, [D8]Tol, 297 K) δ: 6.64 (s, 4H, CHMes), 2.41 (br, 12H, ortho-CH3), 2.16 (s, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 144.5 (d, 1JP–C = 58.5 Hz, ipso-CMes), 139.3 (d, 2JP–C = 13.9 Hz, ortho-CMes), 135.6 (br, para-CMes), 129.8 (br, meta-CMes), 21.1 (d, 3JP–C = 15.6 Hz, ortho-CH3), 20.6 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]Tol, 297 K) δ: 95.9 ppm; IR (ATR) ν: 2957w, 2921w, 2857vw, 1605w, 1554vw, 1457w, 1250m, 1172m, 1088m, 1026m, 952m, 847m cm−1.
Synthesis of the donor adducts [(L)K2(OPMes2)2]24-L
Dimesitylphosphane oxide (120 mg, 0.42 mmol, 2 eq.) was added to a solution of K(hmds) in THF (1.5 mL, 0.28 M, 2 eq.) resulting in an immediate colour change of the solution to an intense yellow. Then a multidentate ligand (2 eq.) was added and the mixture was stirred for 10 minutes at room temperature followed by reduction of the volume in vacuo to one half. Storage at room temperature overnight yielded yellow crystals which were suitable for X-ray diffraction experiments. The crystals were collected, washed with n-pentane (1 mL), and carefully dried in vacuo. The dimeric adducts were isolated as yellow crystalline compounds.[(Diglyme)K(OPMes2)]24-diglyme
0.42 mmol scale HPOMes2, yield: 112 mg, 0.12 mmol, 59%; 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.52 (s, 8H, CHMes), 3.53–3.51 (m, 8H, diglyme), 3.44–3.41 (m, 8H, diglyme), 3.27 (s, 12H, diglyme), 2.39 (s, 24H, ortho-CH3), 2.13 (s, 12H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 148.1 (d, 1JP–C = 58.7 Hz, ipso-CMes), 140.7 (d, 2JP–C = 13.7 Hz, ortho-CMes), 134.9 (s, para-CMes), 129.8 (s, meta-CMes), 73.0 (s, diglyme), 71.5 (s, diglyme), 59.1 (s, diglyme), 21.5 (d, 3JP–C = 15.9 Hz, ortho-CH3), 21.2 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 94.9 ppm; 1H DOSY NMR (400.13 MHz, [D8]THF, 297 K): D(ref, THF) = 2.36 × 10−9 m2 s−1, D(4-diglyme) = 6.95 × 10−10 m2 s−1, MW(calc, (([D8]thf)4KOPMes2)) = 644 g mol−1, MW(exp) = 623 g mol−1, Δ = 3%; IR (ATR) ν: 2915w, 1604w, 1556vw, 1449w, 1169m, 1085m, 1022m, 849m, 643m cm−1.[(Triglyme)K(OPMes2)]24-triglyme
0.85 mmol scale HPOMes2, yield: 305 mg, 0.3 mmol, 72%; 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.51 (s, 8H, CHMes), 3.53–3.50 (m, 16H, triglyme), 3.43–3.41 (m, 8H, triglyme), 3.27 (s, 12H, triglyme), 2.40 (s, 24H, ortho-CH3), 2.13 (s, 12H, para-CH3) ppm; 13C{1H} NMR (75.49 MHz, [D8]THF, 297 K) δ: 148.0 (d, 1JP–C = 59.5 Hz, ipso-CMes), 140.4 (d, 2JP–C = 13.7 Hz, ortho-CMes), 134.4 (s, para-CMes), 129.4 (s, meta-CMes), 72.6 (s, triglyme), 71.2 (s, triglyme), 71.0 (s, triglyme), 58.7 (s, diglyme), 21.2 (d, 3JP–C = 16.0 Hz, ortho-CH3), 20.8 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 94.7 ppm; 1H DOSY NMR (400.13 MHz, [D8]THF, 297 K): D(ref, THF) = 1.75 × 10−9 m2 s−1, D(4-triglyme) = 5.12 × 10−10 m2 s−1, MW(calc, (([D8]thf)4KOPMes2)) = 644 g mol−1, MW(exp) = 634 g mol−1, Δ = 2%; IR (ATR) ν: 2917m, 2873m, 1605m, 1451m, 1410w, 1378w, 1289w, 1159m, 1088vs, 1026s, 647vs, 488vs cm−1.[(pmdeta)K(OPMes2)]24-pmdeta
0.87 mmol scale HPOMes2, yield: 265 mg, 0.27 mmol, 61%; 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.55 (s, 32H, CHMes), 2.44–2.40 (m, 8H, pmdeta), 2.38 (s, 24H, ortho-CH3), 2.33–2.29 (m, 8H, pmdeta), 2.19 (s, 6H, pmdeta), 2.153 (s, 24H, pmdeta), 2.146 (s, 12H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 146.5 (d, 1JP–C = 56.3 Hz, ipso-CMes), 139.5 (br, ortho-CMes), 134.1 (s, para-CMes), 128.8 (s, meta-CMes), 57.9 (s, pmdeta), 56.4 (s, pmdeta), 45.2 (s, pmdeta), 42.3 (s, pmdeta), 20.4 (d, 3JP–C = 15.4 Hz, ortho-CH3), 20.0 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 95.0 ppm; IR (ATR) ν: 3674vw, 2971m, 2902m, 1604w, 1556vw, 1452w, 1408w, 1251w, 1171w, 1066m, 1053m, 955m, 846m cm−1.Synthesis of [{K4(OPMes2)4}-μ-dme-{K4(OPMes2)4}] 4-dme1.08 mmol scale HPOMes2, yield: 126 mg, 0.046 mmol, 34%; 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.53 (s, 4H, CHMes), 3.43 (s, 4H, dme), 3.27 (s, 6H, dme), 2.38 (s, 96H, ortho-CH3), 2.14 (s, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 146.5 (d, 1JP–C = 57.8 Hz, ipso-CMes), 139.5 (d, 2JP–C = 13.6 Hz, ortho-CMes), 134.0 (s, para-CMes), 128.8 (s, meta-CMes), 71.7 (s, dme), 57.9 (s, dme), 20.4 (d, 3JP–C = 15.9 Hz, ortho-CH3), 20.0 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 95.1 ppm; IR (ATR) ν: 2918w, 1604w, 1556vw, 1448w, 1376vw, 1238vw, 1171m, 1086w, 1027w, 846w cm−1.
Synthesis of [(thf)Rb-μ-(thf),(OPMes2)2-Rb(thf)]∞5-thf
Rb(hmds) (664 mg, 2.7 mmol, 1 eq.) was dissolved in methylcyclohexane (5 mL), dimesitylphosphane oxide (773 mg, 2.7 mmol, 1 eq.) was added in one portion and the mixture was slowly heated to 60 °C. THF (1 mL) was added dropwise. During the addition, a yellow precipitate formed. The mixture was cooled to room temperature, the supernatant was discarded, and the residue was washed with n-pentane (2 mL) and dissolved in a small amount of THF (ca. 3 mL) at 40 °C. Storage of the solution at room temperature for 24 h led to the formation of large yellow crystalline blocks of rubidium phosphinite 5-thf. The crystalline solid formed was collected by filtration and carefully dried under reduced pressure (631 mg, 1.7 mmol, 63%).1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.54 (s, 4H, CHMes), 2.38 (s, 12H, ortho-CH3), 2.15 (s, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 146.4 (d, 1JP–C = 57.7 Hz, ipso-CMes), 139.5 (d, 2JP–C = 13.6 Hz, ortho-CMes), 134.1 (s, para-CMes), 128.8 (s, meta-CMes), 20.5 (d, 3JP–C = 15.7 Hz, ortho-CH3), 20.0 (s, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 93.7 ppm; 1H DOSY NMR (400.13 MHz, [D8]THF, 297 K): D(ref, adamantan) = 1.380 × 10−9 m2 s−1, D(5-thf) = 4.895 × 10−10 m2 s−1, MW(calc, (([D8]thf)RbOPMes2)2) = 901 g mol−1, MW(exp) = 914 g mol−1, Δ = −1%; IR (ATR) ν: 2959w, 2930w, 2859w, 1605vw, 1459vw, 1378vw, 1180vw, 1151w, 1069m, 1022w, 910w, 849m, 648w cm−1.
Synthesis of [Cs4(OPMes2)4]∞6
Cs(hmds) (884 mg, 3 mmol, 1 eq.) was dissolved in n-hexane (5 mL), dimesitylphosphane oxide (863 mg, 3 mmol, 1 eq.) was added in one portion and the mixture was slowly heated to 40 °C. THF (ca. 2 mL) was added to the turbid mixture until the solid was completely dissolved. The solution was slowly cooled to room temperature. The reaction mixture was layered with n-pentane (2 mL) and stored at room temperature for 3 days. The yellow crystalline phosphinite 6 was separated by filtration and carefully dried under reduced pressure (434 mg, 1.04 mmol, 34%).1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.55 (s, 4H, CHMes), 2.40 (s, 12H, ortho-CH3), 2.14 (s, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 139.7 (d, 2JP–C = 13.4 Hz, ortho-CMes), 134.1 (s, para-CMes), 128.7 (s, meta-CMes), 20.5 (d, 3JP–C = 15.5 Hz, ortho-CH3), 20.0 (s, para-CH3) ppm, ipso-CMes not observed due to broad resonances; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 90.9 ppm; 133Cs NMR (52.48 MHz, [D8]-THF, 297 K) δ: 46.8 ppm; 1H DOSY NMR (400.13 MHz, [D8]THF, 297 K): D(ref, adamantan) = 1.606 × 10−9 m2 s−1, D(6) = 5.692 × 10−10 m2 s−1, MW(calc, (([D8]thf)CsOPMes2)2) = 916 g mol−1, MW(exp) = 915 g mol−1, Δ = 0%; IR (ATR) ν: 2957w, 2922w, 2857w, 1602vw, 1457w, 1375vw, 1178w,1024w, 947m, 847m, 642w cm−1.
Synthesis of [{(thf)K}2Mg(OPMes2)4] 10-thf
Method A:K(hmds) (104 mg, 0.52 mmol, 2 eq.) and dimesitylphosphane oxide (299 mg, 1.04 mmol, 4 eq.) were placed in a Schlenk flask and a solution of nBusecBuMg in n-hexane (0.37 mL, 0.7 M, 0.26 mmol, 1 eq.) was added. The suspension changed colour to yellow and was stirred at room temperature for 10 minutes before THF (0.2 mL) was added. Storage of the clear solution at room temperature overnight led to small light-yellow crystals suitable for X-ray diffraction experiments. The crystals were collected, washed with n-pentane (1 mL) and carefully dried in vacuo, yielding 10-thf (102 mg, 0.07 mmol, 28%).
Species A: 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.54 (s, 4H, CHMes), 2.35 (s, 12H, ortho-CH3), 2.13 (s, 6H, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 97.3 ppm;
Species B: 1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.44 (s, 4H, CHMes), 2.19 (s, 12H, ortho-CH3), 2.14 (s, 6H, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 95.7 ppm;
IR (ATR) ν: 2956w, 2924w, 2859w, 1602vw, 1461w, 1378vw, 1235w, 1175vw, 933w, 876w, 844m cm−1.
Method B:
Dimesitylphosphane oxide (25.0 mg, 0.087 mmol, 4 eq.), K(hmds) (8.7 mg, 0.043 mmol, 2 eq.), and Mg(hmds)2 (7.53 mg, 0.022 mmol, 1 eq.) were dissolved in a mixture of [D]8Tol and THF (0.55 mL; 0.95/0.05) in an NMR tube.
Species C: 1H NMR (400.13 MHz, [D8]Tol, 297 K) δ: 6.64 (s, 4H, CHMes), 2.55 (s, 12H, ortho-CH3), 2.12 (s, 6H, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]Tol, 297 K) δ: 98.8 ppm; 13C{1H} NMR (100.62 MHz, [D8]toluene, 297 K) δ: 142.0 (d, 1JP–C = 45.6 Hz, ipso-CMes), 140.5 (d, 2JP–C = 15.7 Hz, ortho-CMes), 135.8 (s, para-CMes), 129.5 (s, meta-CMes), 21.7 (d, 3JP–C = 16.6 Hz, ortho-CH3), 20.5 (s, para-CH3) ppm;
Species D: 1H NMR (400.13 MHz, [D8]Tol, 297 K) δ: 6.67 (s, 4H, CHMes), 2.59 (s, 12H, ortho-CH3), 2.14 (s, 6H, para-CH3) ppm; 31P NMR (161.98 MHz, [D8]Tol, 297 K) δ: 96.1 ppm; 13C{1H} NMR (100.62 MHz, [D8]toluene, 297 K) δ: 144.5 (d, 1JP–C = 51.7 Hz, ipso-CMes), 140.1 (d, 2JP–C = 15.1 Hz, ortho-CMes), 135.1(s, para-CMes), 129.4 (s, meta-CMes), 21.3 (d, 3JP–C = 16.6 Hz, ortho-CH3), 20.5 (s, para-CH3) ppm.
The signals of a third species E (δP = 100.3 ppm) could not be assigned properly due to low intensity and overlapping signals.
Synthesis of [(thf)4K4(OP(2-MeNaphth)2)4] 7-thf
Potassium hydride (91 mg, 2.27 mmol, 1.5 eq.) was suspended in THF (5 mL) and bis(2-methylnaphthalen-1-yl)phosphane oxide (500 mg, 1.51 mmol, 1 eq.) was added resulting in a dark red solution. The mixture was stirred at room temperature overnight and filtered to remove excess potassium hydride. The solvent of the filtrate was removed completely, and the dark red solid was recrystallised in a mixture of Et2O (3 mL) and THF (0.5 mL). The resulting dark red crystals were suitable for X-ray diffraction experiments. The crystals were collected and carefully dried in vacuo yielding 7-thf (383 mg, 1.04 mmol, 69%).1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 10.2 (s, 2H, CHNaphth), 7.70–7.64 (m, 2H, CHNaphth), 7.47 (d, 3JH–H = 8.2 Hz, 2H, CHNaphth), 7.27–7.19 (m, 4H, CHNaphth), 7.01 (d, 3JH–H = 8.2 Hz, 2H, CHNaphth), 2.41 (s, 6H, CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 148.3 (d, 1JP–C = 65.3 Hz, CHNaphth), 136.5 (br, CHNaphth), 136.2 (br, CHNaphth), 132.7 (s, CHNaphth), 129.2 (s, CHNaphth), 127.9 (s, CHNaphth), 127.8 (s, CHNaphth), 125.8 (s, CHNaphth), 123.4 (s, CHNaphth), 123.2 (s, CHNaphth), 21.2 (d, 3JP–C = 20.2 Hz, CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 94.8 ppm; IR (ATR) ν: 3044vw, 2971w, 2863w, 1614vw, 1504w, 1448vw, 1342vw, 1175w, 1057m, 982m, 811s, 778m, 745m cm−1.
Synthesis of [(thf)2KOPTipp2]28-thf
Bis(2,4,6-triisopropylphenyl)phosphane oxide (491 mg, 1.08 mmol, 1 eq.) was dissolved in THF (10 mL) and potassium hydride (52 mg, 1.3 mmol, 1.2 eq.) was added in one portion. Gas evolution and a colour change to bright yellow could be observed. The suspension was stirred at room temperature for 24 h. Then, the excess of insoluble potassium hydride was removed by filtration over diatomaceous earth. The volume of the clear yellow filtrate was reduced to a quarter of the original volume. Storage at room temperature overnight afforded 291 mg (0.59 mmol, 55%) of 8-thf.1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.79 (s, 4H, CHAr), 2.38 (s, 12H, ortho-CH3), 4.59 (br, 4H, o-(CH3)2CH), 2.72 (hept, 3JH–H = 6.9 Hz, 2H, p-(CH3)2CH), 1.15 (d, 3JH–H = 6.9 Hz, 12H, p-(CH3)2CH), 0.91 (d, 3JH–H = 6.8 Hz, 24H, o-(CH3)2CH) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 148.6 (d, 3JP–C = 12 Hz, ortho-C), 147.8 (d, 1JP–C = 58.3 Hz, ipso-C), 146.6 (s, para-C), 121.1 (s, meta-C), 34.9 (s, p-(CH3)2CH), 28.5 (d, 3JP–C = 19 Hz, o-(CH3)2CH), 24.9 (s, o-(CH3)2CH), 24.5 (s, o-(CH3)2CH), 24.4 (m, p-(CH3)2CH) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 94.8 ppm; IR (ATR) ν: 3040m, 2864w, 1601vw, 1553vw, 1460w, 1419vw, 1174w, 1160w, 1055m, 959w, 877w cm−1.
Synthesis of [{(diglyme)K-μ-(hmds,OPMes2)K}-μ-{K(hmds)-(OPMes2)}2-{K(hmds)(OPMes2)}] 9-hmds
K(hmds) (0.67 g, 3.38 mmol, 2 eq.) and dimesitylphosphane oxide (1.45 g, 5.06 mmol, 3 eq.) were placed in a Schlenk flask and MeCy (12 mL) was added. The suspension was heated up to 100 °C until all solids were dissolved. Storage of the clear yellow solution overnight led to the precipitation of the amorphous material and a few single crystals that were suitable for X-ray diffraction experiments delivering the structure of 9-hmds. Due to the small quantity of crystals, their yield could not be determined. The NMR measurements were carried out with the amorphous solid.1H NMR (400.13 MHz, [D8]Tol, 297 K) δ: 6.66 (s, 4H, CHMes), 2.40 (s, 12H, ortho-CH3), 2.17 (s, 6H, para-CH3) ppm; 13C{1H} NMR (100.62 MHz, [D8]Tol, 297 K) δ: 144.6 (d, 1JP–C = 52.1 Hz, ipso-CMes), 139.7 (d, 2JP–C = 13.8 Hz, ortho-CMes), 136.3 (s, para-CMes), 130.3 (s, meta-CMes), 21.4 (d, 3JP–C = 13.8 Hz, o-CH3), 21.0 (s, p-CH3) ppm; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 95.7 ppm; IR (ATR) ν: 2921m, 2852w, 1603vw, 1448w, 1374vw, 1236w, 1175w, 1087m, 1027w, 971w, 816w cm−1.
Synthesis of [Rb2(OPTipp2)2]211
Bis(2,4,6-triisopropylphenyl)phosphane oxide (886 mg, 1.95 mmol, 1 eq.) and Rb(hmds) (479 mg, 1.95 mmol, 1 eq.) were suspended in MeCy (3 mL). The mixture was heated to 40 °C and THF (0.5 mL) was added until a bright yellow coloured solution formed. The solution was cooled down to room temperature overnight which led to the formation of yellow crystalline needles which were suitable for X-ray diffraction experiments. The crystals of the clear yellow solution were collected and dried carefully yielding 385 mg (0.64 mmol, 33%) of 11.1H NMR (400.13 MHz, [D8]THF, 297 K) δ: 6.83 (br, 4H, CHAr), 4.57 (br, 4H, o-CH(CH3)2), 2.74 (br, 2H, p-CH(CH3)2), 1.17 (d, 3JH–H = 7.0 Hz, 12H, p-(CH3)2CH), 0.93 (d, 3JH–H = 7.0 Hz, 24H, o-(CH3)2CH) ppm; δ: 151.4 (br, ortho-C), 146.5 (s, para-CMes), 121.3 (s, meta-CMes), 34.9 (s, p-(CH3)2CH), 28.6 (br, o-(CH3)2CH), 24.7 (s, o-(CH3)2CH), 24.3 (s, p-(CH3)2CH) ppm, ipso-CAr not observed due to broad resonances; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 93.6 ppm; IR (ATR) ν: 2954m, 2923m, 2864w, 1598vw, 1458w, 1378w, 1360w, 950m cm−1.
Synthesis of [(HPOTipp2)Cs2(OPTipp2)2]212
Bis(2,4,6-triisopropylphenyl)phosphane oxide (921 mg, 2.03 mmol, 1 eq.) and Cs(hmds) (594 mg, 2.03 mmol, 1 eq.) were suspended in MeCy (3 mL). The mixture was heated to 40 °C and THF (1 mL) was added until a bright, yellow solution formed. The solution was cooled down to −20 °C leading to yellow crystalline needles that were suitable for X-ray diffraction experiments. Due to the small quantity of crystals, their yield could not be determined. The NMR measurements were carried out with the amorphous solid.1H NMR (300.19 MHz, [D8]THF, 297 K) δ: 6.88 (br, 4H, CHAr), 4.44 (br, 4H, o-CH(CH3)2), 2.76 (hept, 3JH–H = 7.0 Hz, 2H, p-CH(CH3)2), 1.18 (d, 3JH–H = 6.9 Hz, 12H, p-(CH3)2CH), 0.96 (d, 3JH–H = 6.9 Hz, 24H, o-(CH3)2CH) ppm; 13C{1H} NMR (100.62 MHz, [D8]THF, 297 K) δ: 151.5 (br, ortho-C), 147.4 (br, para-CMes), 121.5 (s, meta-CMes), 35.0 (s, p-(CH3)2CH), 28.7 (br, o-(CH3)2CH), 24.6 (br, o-(CH3)2CH), 24.3 (s, p-(CH3)2CH) ppm, ipso-CAr not observed due to very broad resonances; 31P NMR (161.98 MHz, [D8]THF, 297 K) δ: 90.6, 4.9 (d, 1JP–H = 479 Hz) ppm; 133Cs NMR (39.37 MHz, [D8]THF, 297 K) δ: 47.8 ppm; IR (ATR) ν: 2954m, 2922m, 2862w, 1598vw, 1548vw, 1458w, 1417vw, 1379w, 1360w, 1177w, 1101w, 958m, 877w cm−1.
Author contributions
B. E. F.: conceptualisation, synthesis and characterisation, data analysis and methodology, data acquisition, writing original draft and editing; P. S.: data analysis, methodology, and editing; P. L. and H. G.: X-ray structure determinations at single crystals and editing of the manuscript; and M. W.: conceptualisation and supervision, writing original draft and editing. All authors have given approval to the final version of the manuscript.Data availability
The crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC 2330417 (2-thf), 2383246 (3-thf), 2330418 (4), 2330419 (4-thf), 2330420 (4-dme), 2330421 (4-diglyme), 2330422 (4-triglyme), 2330423 (4-pmdeta), 2330424 (5-thf), 2330425 (6), 2330426 (7-thf), 2330427 (8-thf), 2330428 (9-thf), 2330429 (10-thf), 2383247 (11), and 2383248 (12).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the valuable support from the NMR service platform (https://www.nmr.uni-jena.de) of the Faculty of Chemistry and Earth Sciences of Friedrich Schiller University Jena, Germany. Parts of the equipment were provided by the German Research Foundation (DFG, INST 275/442-1FUGG) which we kindly acknowledge.References
- (a) K. Hirano and M. Miura, Tetrahedron Lett., 2017, 58, 4317–4322 CrossRef CAS; (b) S. Kotani and M. Nakajima, Tetrahedron Lett., 2020, 61, 151421 CrossRef CAS; (c) Z.-Y. Wang, Q. Guo, S. Xu and K.-K. Wang, Synthesis, 2021, 3683–3698 CrossRef CAS.
- (a) T. M. A. Al-Shboul, H. Görls, S. Krieck and M. Westerhausen, Eur. J. Inorg. Chem., 2012, 5451–5455 CrossRef CAS; (b) S. Härling, J. Greiser, T. M. A. Al-Shboul, H. Görls, S. Krieck and M. Westerhausen, Aust. J. Chem., 2013, 66, 1264–1273 CrossRef; (c) S. M. Härling, B. E. Fener, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2018, 37, 4380–4386 CrossRef; (d) B. E. Fener, P. Schüler, N. Ueberschaar, P. Bellstedt, H. Görls, S. Krieck and M. Westerhausen, Chem. – Eur. J., 2020, 26, 7235–7243 CrossRef CAS; (e) A. W. J. Platten, A. M. Borys and E. Hevia, ChemCatChem, 2022, 14, e202101853 CrossRef CAS; (f) L. Zhang, S. Bai and L. Zheng, Organometallics, 2023, 42, 2262–2268 CAS.
- S. M. Härling, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2017, 56, 10741–10750 CrossRef.
- S. M. Härling, S. Krieck, H. Görls and M. Westerhausen, Inorg. Chem., 2017, 56, 9255–9263 CrossRef PubMed.
- J.-Q. Zhang, J. Ye, T. Huang, H. Shinohara, H. Fujino and L.-B. Han, Commun. Chem., 2020, 3, 1 CrossRef CAS.
- (a) A. J. Hoskin and D. W. Stephan, Organometallics, 1999, 18, 2479–2483 CrossRef CAS; (b) C. Berthold, L. R. Thomas-Hargreaves, S. I. Ivlev and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2021, 76, 651–658 CrossRef CAS.
- M. Westerhausen, Monolithiumphosphid·DME und -arsenid·DME – wertvolle Edukte für die Synthese von Acylphosphanen und -arsanen und verwandten Verbindungen, Ph.D. thesis, Philipps University Marburg/Lahn, Germany, 1987 Search PubMed.
- M. A. Beswick, N. L. Cromhout, C. N. Harmer, J. S. Palmer, P. R. Raithby, A. Steiner, K. L. Verhorevoort and D. S. Wright, Chem. Commun., 1997, 583–584 RSC.
- (a) M. S. Hill, M. F. Mahon and T. P. Robinson, Chem. Commun., 2010, 46, 2498–2500 RSC; (b) J. Langer, T. M. A. Al-Shboul, F. M. Younis, H. Görls and M. Westerhausen, Eur. J. Inorg. Chem., 2011, 3002–3007 CrossRef CAS.
- (a) T. M. A. Al-Shboul, G. Volland, H. Görls, S. Krieck and M. Westerhausen, Inorg. Chem., 2012, 51, 7903–7912 CrossRef CAS; (b) M. Westerhausen, S. Krieck, J. Langer, T. M. A. Al-Shboul and H. Görls, Coord. Chem. Rev., 2013, 257, 1049–1066 CrossRef CAS; (c) B. E. Fener, P. Schüler, H. Görls, P. Liebing and M. Westerhausen, Z. Anorg. Allg. Chem., 2023, 649, e202200359 CrossRef CAS.
- S. M. Härling, s-Block-Metall-vermittelte Hydrophosphoranylierung von Heterokumulenen und Alkinen, Ph.D. thesis, Jena/Germany, 2018 Search PubMed.
- K. Issleib, B. Walther and E. Fluck, Z. Chem. (Zeitschrift für Chemie), 1968, 8, 67 CrossRef CAS.
- (a) H. Kessler, Angew. Chem., Int. Ed. Engl., 1970, 9, 219–235 CrossRef CAS; (b) G. Binsch and H. Kessler, Angew. Chem., Int. Ed. Engl., 1980, 19, 411–428 CrossRef; (c) R. G. Bryant, J. Chem. Educ., 1983, 60, 933–935 CrossRef CAS; (d) H. Günther, NMR-Spektroskopie, Georg Thieme, New York, 3rd edn, 1992 Search PubMed.
- R. C. C. Dorow, P. Liebing, H. Görls and M. Westerhausen, Dalton Trans., 2024, 53, 5711–5720 RSC.
- I. Banerjee, A. Harinath and T. K. Panda, Eur. J. Inorg. Chem., 2019, 2224–2230 CrossRef CAS.
- B. E. Fener, P. Schüler, F. E. Pröhl, H. Görls, P. Liebing and M. Westerhausen, Organometallics, 2024, 43, 1095–1109 CrossRef CAS.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364 RSC.
- (a) D. Schulze-Sünnighausen, J. Becker and B. Luy, J. Am. Chem. Soc., 2014, 136, 1242–1245 CrossRef; (b) D. Schulze-Sünnighausen, J. Becker, M. R. M. Koos and B. Luy, J. Magn. Reson., 2017, 281, 151–161 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2330417 (2-thf), 2383246 (3-thf), 2330418 (4), 2330419 (4-thf), 2330420 (4-dme), 2330421 (4-diglyme), 2330422 (4-triglyme), 2330423 (4-pmdeta), 2330424 (5-thf), 2330425 (6), 2330426 (7-thf), 2330427 (8-thf), 2330428 (9-thf), 2330429 (10-thf), 2383247 (11), and 2383248 (12). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02721c |
| This journal is © The Royal Society of Chemistry 2025 |