
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A terpyridine-based copper complex for electrochemical reduction of nitrite to nitric oxide†
Jyotiprokash
Biswas
 a,
Sebastian
Sanden
a,
Prabhakar
Bhardwaj
b,
Daniel
Siegmund
ac,
Pankaj
Kumar
*b and
Ulf-Peter
Apfel
*ac
a,
Sebastian
Sanden
a,
Prabhakar
Bhardwaj
b,
Daniel
Siegmund
ac,
Pankaj
Kumar
*b and
Ulf-Peter
Apfel
*ac
aInorganic Chemistry I, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@ruhr-uni-bochum.de
bDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Tirupati 517507, India
cDepartment of Electrosynthesis, Fraunhofer UMSICHT, Osterfelder Str. 3, 46047 Oberhausen, Germany
First published on 4th December 2024
Abstract
In biological systems, nitrite reductase enzymes (NIRs) are responsible for reduction of nitrite (NO2−) to nitric oxide (NO). These NIRs have mostly Cu- or Fe-containing active sites, surrounded by amine-containing ligands. Therefore, mononuclear Cu complexes with N-donor ligands are highly relevant in the development of NIR model systems and in the mechanistic investigation of the nitrite reduction reaction. Herein, we report on a terpyridine-based CuII complex with square planar geometry for H+-assisted electrochemical reduction of NO2−. Through electrochemical measurements, spectroscopic characterization and isotope-labelling experiments we propose a mechanistic reaction pathway involving an unstable HNO2 state. The CuI intermediate, formed electrochemically, was isolated and its molecular structure was deduced, showing linkage isomerism of the nitrite ligand. Moreover, qualitative and quantitative product analysis by GC-MS shows N2O formed as a side product along with the main product NO. Furthermore, by obtaining single crystals and conducting structural analysis we were able to determine the structural arrangement and redox state of the complex after electrochemical treatment.
Introduction
Nitrogen metabolism and fixation occurs simultaneously in almost every form of life.1,2 This two-way process of supply and fixation spans from local to global ecological systems. Mostly due to human activity and exploitation, the environmental nitrogen cycle has been disturbed causing a nitrogen imbalance and eventually an increase in nitrogen content in several areas of the environment.3–5 The denitrification steps in the global nitrogen cycle have the pivotal function of releasing the aggregated nitrogen species back into the atmosphere. Among the denitrification steps performed by microorganisms, the transformation of nitrite (NO2−) to nitric oxide (NO) is very important and has been studied extensively for decades.6 Moreover, the reduced product of NO works as an important cell-signalling agent and vasodilator.7–11 Therefore, NO2− acts as a source of NO in the bloodstream under hypoxic conditions.12–14 It has been established that nitric oxide synthases (NOSs) are responsible for NO evolution in muscles and tissues by converting arginine to citrulline along with the generation of NO.15,16 Additionally, certain non-NOS groups of enzymes are also capable of performing the reduction of NO2− to NO, such as cytochrome c oxidase (CcO), xanthine oxidase (XO) and nitrite reductases (NIRs). Cytochrome c oxidase, present in the mitochondrial network, is a heterobimetallic (heme-Fe/Cu) system.17–19 On the other hand, mammalian xanthine oxidase (Mo)20–22 and microbial NIRs (Fe or Cu) have monometallic active sites.23–25In the denitrification process, the one-electron reduction step of NO2− to NO is mediated by NIRs and depending on the metal atom present at the active site they are called either CuNIRs (Cu-containing active site) or FeNIRs (Fe-containing active site). In CuNIRs, the main stage for the whole catalytic process, i.e. the active site, is a type II Cu atom, which is coordinated with three histidinyl nitrogens and a water molecule as reported in X-ray structure studies.26–30 NO2− coordinates with the CuII centre by substituting the water molecule followed by subsequent electron and proton transfer steps (Scheme 1).31,32
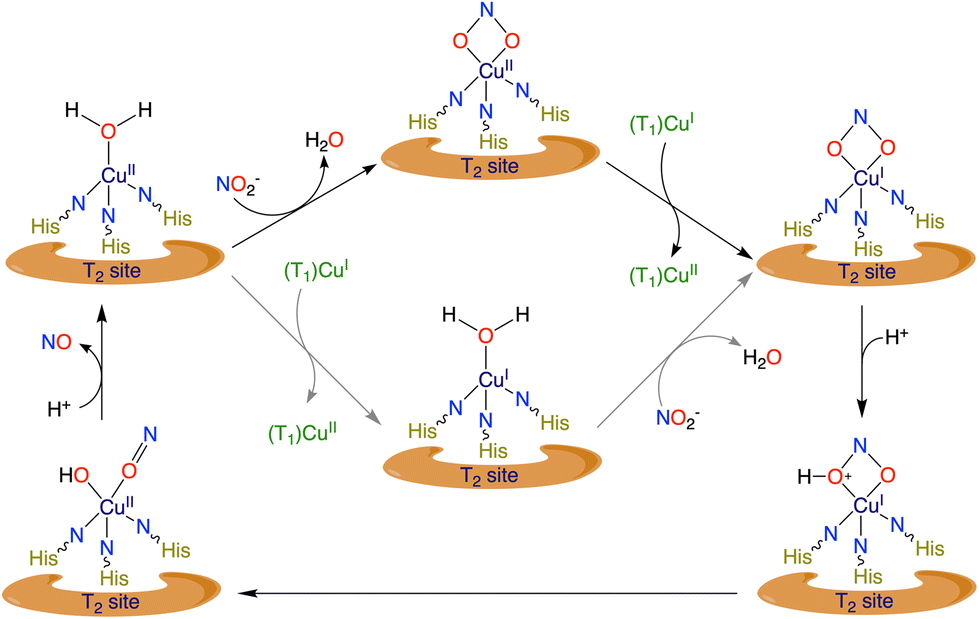 | ||
| Scheme 1 Simplified mechanistic illustration of reduction of NO2− to NO by CuNIRs.6,43 | ||
Several Cu- and Fe-based mononuclear complexes with amine-containing ligands have been reported as models of CuNIRs and FeNIRs, respectively, when exploring mechanisms and energetics of the nitrite reduction process. Tolman first reported a copper–nitrosyl complex in 1992,33 and subsequent studies, including work by Murphy,28,34 Fujisawa35 and Nam's group,36revealed intermediates such as {CuNO}10 and {FeNO}6 during nitrite reduction. Recently, we reported proton-assisted NO2− reduction, mediated by a heterobimetallic CuCo complex, highlighting the benefit of the bimetallic complex over a monometallic one.37
Furthermore, there has been significant development in the research of CuNIR model complexes that can perform nitrite reduction electrochemically. Since 1993, after Komeda et al. reported on electrochemical nitrite reduction for the first time,38,39 several electrochemical studies for nitrite reduction have been published. Meyerhoff and coworkers investigated electrochemical reduction of nitrite in aqueous solution using a series of mononuclear CuII complexes (Scheme 2a to d), aiming to develop demand-based biomedical devices for controlled NO release.40–42 Recently, the groups of Smith and Bren have reported buffer-assisted electrocatalytic nitrite reduction featuring CoIII-based (Scheme 2g)44 and FeIII-based (Scheme 2f)45 complexes, respectively. The buffer appeared to be highly crucial for the catalyst's stability. Moreover, some reports have demonstrated electrochemical nitrite reduction in organic solution as well. Symes and coworkers reported nitrite reduction in acetonitrile using a mononuclear CuII complex with a pendant carboxyl group (Scheme 2e), which enhances the proton relay mechanism and significantly improves activity toward nitrite activation.43 The electrochemical reactivity of CuNIR model complexes have shown crucial pH dependence, which is expected from the elemental reaction equation (eqn (1)):
| NO2− + 2H+ + e− → NO + H2O | (1) |
 | ||
| Scheme 2 Cu-, Co -and Fe-based molecular catalysts, reported for electrochemical nitrite reduction.40–45 | ||
Reduction of NO2− is believed to go through an {M–NO}n intermediate, which is generated from the decomposition of a very transient {M–ONOH} species.37,43,46 As a result, over the years, different research groups have tried to isolate metal-nitrosyl species and investigate their properties, stability and reactivity.47–51 There are few reports that dig deeply into the rate-determining steps46 or proton–electron transfer mechanisms.43 However, to the best of our knowledge, not much focus has been given to the post-electrolytic conditions of the catalyst or the model complex either. The very few reports that are available on this matter are mostly theoretical.43,46
Taking all the above-mentioned data and discussion into consideration, herein, we report on the synthesis and characterization of a CuII complex, [TerpyCu(CH3CN)](ClO4)2, (1) (Terpy = 2,2′:6′,2′′-terpyridine) (Scheme 3) and its activity towards electrochemical nitrite reduction. We conducted electrochemical nitrite reduction using CH3COOH as a proton source, identifying products via GC-MS and 1H-NMR. Notably, we also obtained the structural proof of linkage isomerism of the nitrite ligand during redox transitions.
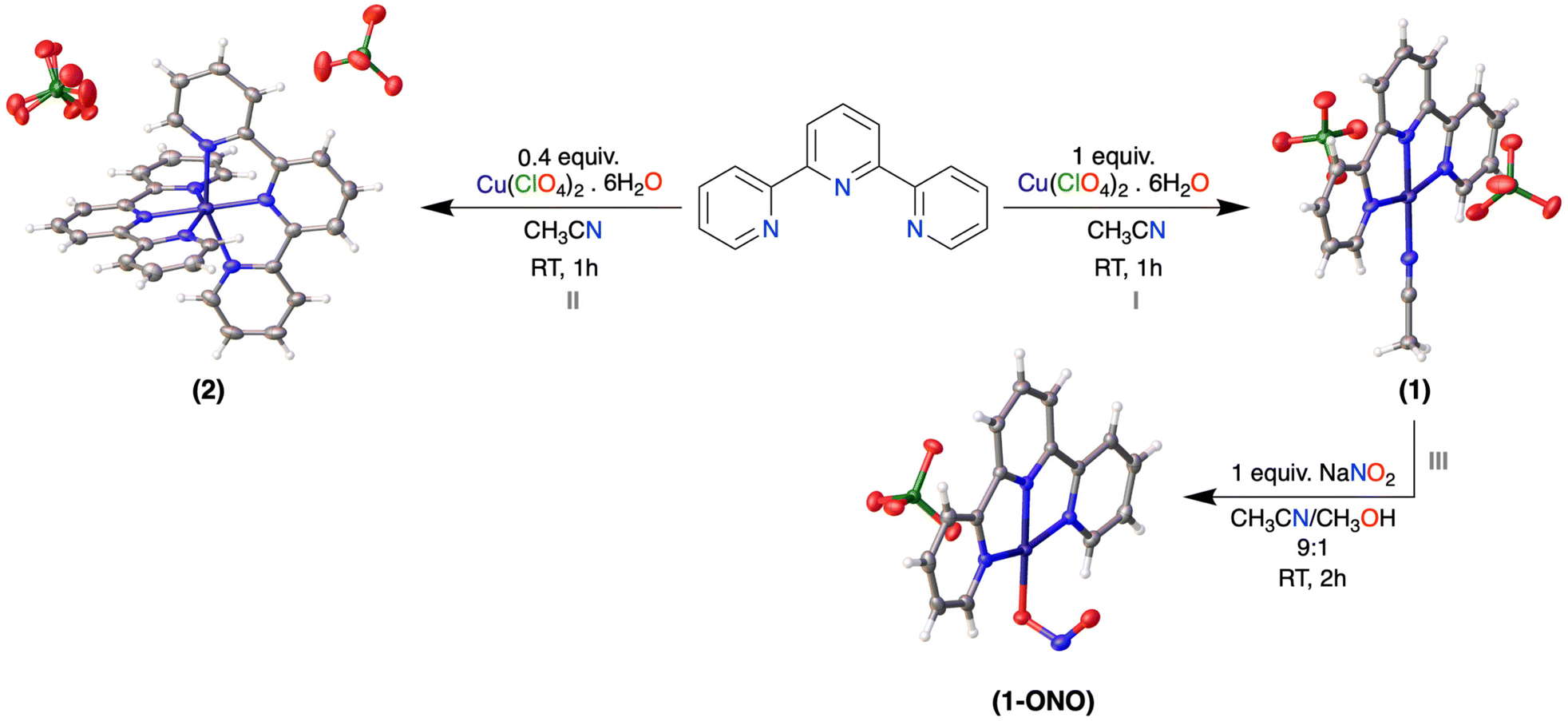 | ||
| Scheme 3 Synthetic scheme of [TerpyCu(CH3CN)](ClO4)2, (1), [(Terpy)2Cu](ClO4)2, (2) and [TerpyCu(ONO)]ClO4, (1-ONO), showing their corresponding molecular structures, deduced from SXRD analysis. Solvent molecules are omitted for clarity. Colour code: C grey, H white, N blue, O red, Cu violet, Cl green. | ||
Results and discussion
Synthesis and characterizations of the Cu complexes
Complex 1 was prepared by a single-step reaction between Terpy and Cu(ClO4)2·6H2O in acetonitrile at room temperature (Scheme 3, Step I). In the UV-Vis spectrum, upon addition of the CuII salt to the ligand solution a new band appears at 626 nm, which corresponds to the d–d transition (Fig. 1A). ESI-MS showed distinct m/z peaks at 296.0 and 395.0 corresponding to [TerpyCu]+ and [TerpyCu(ClO4)]+ (Fig. S8†). Additionally, the magnetic moment of 1 was calculated by Evans’ method using 1H-NMR spectroscopy (Fig. S14†). The magnetic moment of 1 was found to be 1.69 BM, which falls in line with the +II oxidation state of the Cu centre. We obtained single crystals for complex 1 and deduced its molecular structure via single-crystal X-ray diffraction (SXRD), which shows coordination of CuII with the ligand and an additional CH3CN molecule (Scheme 3). Notably, the molecular structure shows that the oxygen atoms of the perchlorate moiety are oriented toward the metal centre. The distances between the perchlorate oxygen atoms and the CuII centre range from 2.5 to 2.7 Å, suggesting the presence of weak interactions between the perchlorate oxygen and the metal centre. Additionally, the IR stretching frequency of the perchlorate anion (1113 cm−1, Fig. S1†) in complex 1 aligns with the stretching frequency of perchlorates in a terpyridine-N-oxide copper complex previously reported by Murphy and coworkers,52 who also highlighted weak interactions between perchlorate and Cu centres and included the ClO4− in describing the overall geometry of the complexes. Taking all the data and information into consideration, it can be inferred that there are weak interactions between the metal centre and the perchlorate ions and that the structure around the CuII centre in complex 1 could also be described as a tetragonally distorted octahedron with two very long-distance Cu–O interactions (involving oxygen atoms of the perchlorates). However, due to the large distance between the CuII centre and the perchlorate oxygen atoms, the interaction can be considered transient in solution with the complex being a cationic species.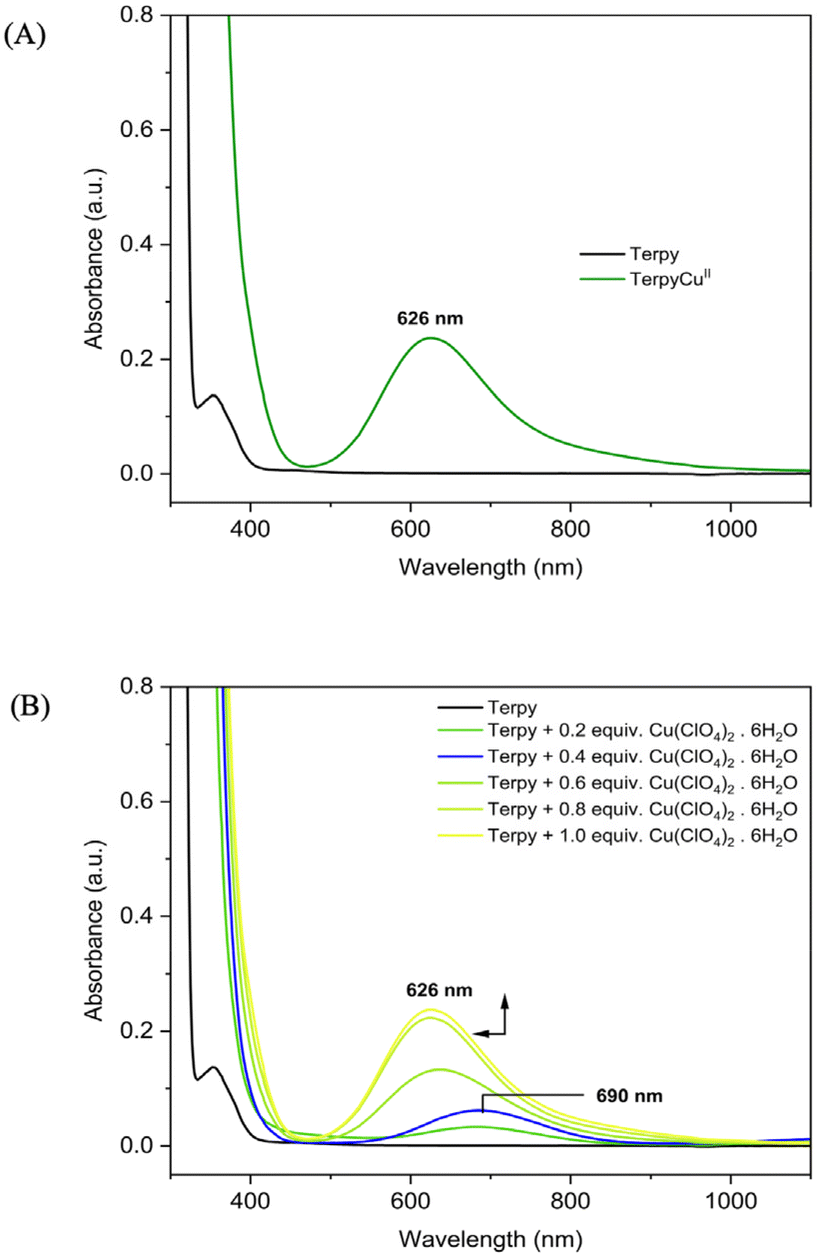 | ||
| Fig. 1 (A) UV-Vis spectrum of 1, showing a d–d transition band upon addition of CuII salt. (B) Change in UV-Vis spectra upon stepwise addition of CuII salt to the ligand solution (concentration: 2 mM; solvent: acetonitrile). | ||
To closely monitor the coordination of CuII with the terpyridine ligand, a stepwise addition of a CuII salt to the ligand solution was carried out, with the progress of the reaction being tracked using UV-Vis spectroscopy. Initially, the addition of the CuII salt up to 0.4 equiv., resulted in the appearance of a distinct absorption band at 690 nm in the UV-Vis spectrum (Fig. 1B, blue line). However, as more CuII salt was added beyond 0.4 equiv., a blue shift of the band at 690 nm to 626 nm was observed (Fig. 1B). Based on this result, we assumed that there could be a different Cu-containing species at the initial stage of the measurement. To gain more insight, a complexation reaction was set up with 1 equiv. of ligand and 0.4 equiv. of CuII salt (Scheme 3, Step II), and the product was isolated and characterized by UV-Vis, ESI-MS, FT-IR and SXRD. The UV-Vis spectrum of the prepared complex (Fig. S18A†) shows an identical band pattern to the spectrum (blue line in Fig. 1B) observed upon addition of 0.4 equiv. of CuII salt in the ligand solution during the UV-Vis titration. After completion of the reaction, we observed a clear m/z peak at 264.6 in the ESI-MS analysis (Fig. S9†), corresponding to a previously reported bis-terpyridine copper complex,53,54 [(Terpy)2Cu]2+ (2), which was further confirmed by the molecular structure derived from SXRD (Scheme 3). One of the perchlorate ions is modelled as being disordered in the molecular structure. In contrast to complex 1, all the coordination sites are occupied by the ligands in complex 2, hence, the perchlorate counterion is hindered from coordinating with the metal centre. Therefore, the d–d transition band at 690 nm appears in the early stage of the measurement due to the formation of 2 in the solution. As we continue to add more CuII salt, the ligand-to-metal 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction is favoured, eventually giving a band at 626 nm, which is similar to that observed for complex 1.
:1 reaction is favoured, eventually giving a band at 626 nm, which is similar to that observed for complex 1.
When NaNO2 was added to the solution of 1, the blue-coloured solution turned green, indicating the coordination of nitrite with the CuII centre, resulting in [TerpyCuONO]+ (1-ONO) (Scheme 3, Step III). The UV-Vis spectrum revealed the appearance of a new LMCT band at 372 nm corresponding to the nitrite coordination. Additionally, a red shift of the d–d transition band from 626 nm to 640 nm was observed (Fig. 2). FT-IR spectroscopy of 1-ONO showed signals at 1268 cm−1 (νsym, NO2) and 1366 cm−1 (νasym, NO2), which shifted to 1251 cm−1 (νsym, 15NO2) and 1352 cm−1 (νasym, 15NO2), respectively, when 15N-labelled Na15NO2 was used, resulting in 1-O15NO (Fig. 2, Inset III and Fig. S3, S4†). In the ESI-MS analysis, an m/z peak at 342.1 for [TerpyCuONO]+ was observed and this peak was shifted to m/z 343.1 in the case of the 15N-labelled compound (Fig. S10 and S11†). The molecular structure of 1-ONO derived from SXRD analysis (Scheme 3) shows a four-coordinated CuII centre where the acetonitrile molecule is substituted by nitrite, which binds through one of the oxygens (–ONO, nitrito binding mode). Similar to complex 1, the perchlorate ion in 1-ONO also exhibits weak interaction with the metal centre, as indicated by the long Cu–O distance (2.4 Å) and IR stretching frequency (νs-ClO4−, 1111 cm−1; Fig. S3†). Due to this very weak interaction between the perchlorate ion and CuII, 1-ONO can be considered a cationic compound as well. We have also calculated the binding constant (Kb) for nitrite coordination by using the Benesi–Hildebrand equation.55,56 The slope (1/Kb) of the Benesi–Hildebrand plot (Fig. 2, Inset II) was found to be 0.4528 mM. Therefore, Kb for complex 1 is 2.21 × 103 M−1.
 | ||
| Fig. 2 UV-Vis spectral change upon addition of NaNO2. Grey line, 0.5 mM Terpy; teal blue line, 0.5 mM 1. Stepwise addition of NaNO2 is shown by the light green lines. Dark green line, spectrum after addition of 1 equiv. NaNO2. Inset I: rise of the band at 372 nm corresponding to NO2− coordination. Inset II: Benesi–Hildebrand plot for the binding constant calculation. Slope = 1/Kb = 0.4528 mM. Inset III: FT-IR spectra of 1-ONO (violet line) and 1-O15NO (green line) in KBr. | ||
Electrochemical NO2− reduction
Additionally, when NO2− was added to the complex solution we observed a shift of 200 mV for the CuII/CuI couple towards less cathodic potentials (Fig. 4C). In the presence of NO2− the E1/2 for CuII/CuI is −0.62 V (vs. Fc+/Fc). Upon the addition of nitrite, the solution turned green. Cyclic voltammograms were then recorded for varying equivalents of NO2− in the presence of CH3COOH as the proton source. A new reductive catalytic signal was observed at −1.27 V, which corresponds to NO2− reduction. As we increased the amount of nitrite in the solution, higher current values were observed at −1.27 V (Fig. 4D).
:1). In the chronoamperometric plot, we observed a current drop after the 5 h mark (Fig. 4E). We suspected the current drop could be due to some deposition on the electrode surface. After completion of the experiment, we indeed noticed some deposits on the electrode surface. The deposited material was analyzed by an SEM/EDX technique, and it was found that the material mainly consisted of metallic copper (Fig. S22†). This deposition is the reason for the anodic stripping signal observed at −0.82 V. Additionally, it was observed that the electrolyte solution, initially colourless during electrolysis, gradually turned blue over time when left standing or upon exposure to air, indicating the presence of copper species in the solution. Fortunately, we were able to obtain single crystals from this blue-coloured solution which, after SXRD analysis, turned out to be a Terpy-CuII complex with an acetate moiety and a water molecule coordinated with the CuII centre, namely [TerpyCuII(H2O)(OCOCH3)]PF6 (3) (Fig. 3). In the molecular structure there is minor disorder in the PF6 counterion, which was left unmodelled due to apparently low component occupancy and clear identity of the counterion. [For other spectroscopic characterization data of complex 3, please refer to the ESI (Fig. S5, S12 and S19†).]
 | ||
| Fig. 3 Molecular structure of [TerpyCuII(H2O)(OCOCH3)]PF6, (3) deduced from SXRD analysis. Colour code: C grey, H white, N blue, O red, Cu violet, P purple, F light green. | ||
 | ||
| Fig. 4 (A) Cyclic voltammograms of 1 (1 mM). Voltammograms over different potential ranges are shown in different colours. Purple line: potential range −0.58 V to −1.57 V. Black line: potential range −0.58 V to −1.07 V. (B) Cyclic voltammograms of 1 mM complex 1 (black line) and 1 mM complex 1 in the presence of 150 equiv. CH3COOH (red line). (C) Reversible redox wave of the CuII/CuI (E1/2 = −0.84 V) couple under cyclic voltammetry (teal blue line). The green traces show the shift of CuII/CuI (E1/2 ∼ −0.62 V) to the anodic side upon addition of NO2−. (D) Cyclic voltammograms in the presence of 1 mM complex 1, and 150 equiv. CH3COOH and NO2−, showing generation of a reductive catalytic signal at −1.27 V. Cyclic voltammograms upon addition of 5 equiv., 10 equiv. and 20 equiv. are shown by orange, violet and blue lines, respectively. Cyclic voltammogram of complex 1 is shown with a grey dotted line for reference. (E) Chronoamperometric (CA) plot for 2 mM 1, 150 mM CH3COOH, and 40 mM NO2− at −1.29 V, showing a drop in cathodic current after 5 h. Solvent: CH3CN; supporting electrolyte: TBAPF6 (0.1 M for CV and 0.2 M for CA). Electrodes: glassy carbon as working electrode, Pt wire as counter electrode, Ag wire as pseudo-reference electrode. All the potential values are presented with reference to the Fc+/Fc couple. | ||
Therefore, it can be concluded that after the electrolysis some of the catalyst was deposited on the electrode surface, while some remained in solution as complex 3.
Initially, when the LSV scan was initiated from −1.1 V, a voltammogram (purple trace, Fig. 5) that did not match the catalytic LSV profile of complex 1 (blue trace, Fig. 5) was obtained. This result indicates that the deposited Cu0 is not the catalytically active species.
 | ||
| Fig. 5 Linear sweep voltammograms (LSV) of 5 mM NaNO2 in solution (grey trace); rinse test LSV with starting potential (Eb) −1.10 V (purple trace) and Eb −0.65 V (brown trace) in the presence of 5 mM NaNO2; LSV of 2 mM 1 and 5 mM NaNO2 in solution. Solvent: MeCN/H2O (100:1); proton source: CH3COOH (150 mM); supporting electrolyte: 0.1 M nBu4PF6; electrodes: glassy carbon (WE), Pt wire (CE), Ag wire (RE). Potentials are given with reference to Fc+/Fc. | ||
Further experiments were performed by initiating the LSV scan from a less negative (more oxidative) potential of −0.65 V. In this case, the resulting voltammogram (brown trace, Fig. 5) displayed a current response similar to that observed for complex 1 in solution. These findings suggest that at more oxidative potentials, the deposited Cu0 is re-oxidized to CuII, which subsequently undergoes reduction to form CuI. The CuI species generated in this process facilitates the catalytic reaction, confirming it as the catalytically active species.
Product analysis and quantification
In the chromatogram, at 3.80 min (Rt), a peak of m/z 31 was observed, which can be attributed to 15NO (Fig. S23†). These results provide conclusive evidence of NO formation in the electrochemical NO2− reduction process. Moreover, two successive peaks of m/z 44 were observed in the chromatogram at 5.05 min and 5.37 min (Rt) in the case of the analysis of the gas mixture obtained from bulk electrolysis using NaNO2. Interestingly, the m/z 44 peak at 5.05 min (Rt) remained the same but the peak at 5.37 min (Rt) changed to m/z 46 when 15N-labelled Na15NO2 was used in the electrocatalytic process (Fig. S24†). This suggests that the m/z 44 peak at Rt 5.37 min is for nitrous oxide (N2O) and, correspondingly, the m/z 46 peak is for 15N2O. It is postulated that N2O originates from disproportionation of NO.57–60 The m/z 44 peak at 5.05 min corresponds to CO2 originating from atmospheric interference (Fig. S24†).
NO and N2O were then quantified via GC-MS (Fig. 6), obtained from electrolysis experiments of different durations. Separate bulk electrolysis experiments with 2 mM complex 1, 40 mM NaNO2, 150 mM CH3COOH and 0.2 M TBAPF6 at a fixed potential of −1.29 V were performed for 1 h, 3 h, 5 h and 7 h in the same cell and the headspace gas mixture was analyzed. We achieved a maximum FE of 81% for NO production (Fig. 6). Up to 5 h, an increasing trend in the concentration of NO and N2O accumulating in the headspace was observed. After 5 h a decrease in headspace NO concentration was observed, which is in accordance with the current drop in bulk electrolysis after 5 h. Interestingly, even though NO production abated, we still observed an increase in N2O concentration after 7 h. This result implies that after 5 h, NO production subsides due to the deposit formation on the electrode surface, but disproportionation of the formed NO continues.
 | ||
| Fig. 6 GC-MS analysis of NO (left-hand y-axis, green columns) and N2O (inset, purple columns) produced from NO2− after running the electrolysis experiment for different time durations. Faradaic efficiency (FE) of the total NO in each case is shown along the right-hand y-axis (blue columns). | ||
Subsequent reduction of NO to NH3 in homogeneous electrocatalysis is rarely reported in the literature,61 as it necessitates the transfer of 5 H+. Using secondary amines as proton donors, Co-based macrocycles were able to perform the final reduction step to NH3 in aqueous solution.62 Alternatively, N–O cleavage was also achieved using a bimetallic Ru complex, which was able to stabilize an unusual μ-nitrido species between two Ru atoms.63
Mechanistic insight of NO2− reduction
In Scheme 4, a proposed mechanistic cycle is shown. First, nitrite coordinates with 1 through one of its oxygens (O-bound, nitrito binding mode), as evident from the molecular structure 1-ONO. Upon reduction of the CuII to CuI centre, the –ONO ligand undergoes linkage isomerism and binds through nitrogen (N-bound, nitro binding mode). Fortunately, we have been able to isolate and crystallize this CuI compound from the electrolytic solution. One could assume that this rearrangement might be initiated by the protonation of nitrite. To gain more information, terpyridine was reacted with a CuI salt and NaNO2. In both cases, i.e. from the electrolytic solution and from the reaction mixture with the CuI salt, the same product, a nitrite-bound di-copper complex, [TerpyCu(NO2)]2 (4), was obtained, which was characterized via SXRD analysis (Fig. 7A). From this result it can be conclusively stated that the rearrangement of the nitrite ligand is independent of the protons in the electrolytic solution. Maji et al. proposed a similar rearrangement of the nitrite ligand64 and, herein, our crystallographic studies support that claim with clear structural data. Fig. 7A shows the structure of the isolated CuI state, thereby confirming the linkage isomerism of the nitrite ligand. Generally, CuI preferentially adopts tetrahedral over planar geometry. As a result, when the CuII centre (having a square planar geometry) in 1 was reduced to CuI, the metal centre protrudes out of the square plane in order to attain tetrahedral geometry and ends up coordinating with two N atoms of one terpyridine molecule and with another terpyridine-N atom of another Terpy–CuI fragment, resulting in a homobimetallic CuI complex (Cu–Cu distance ∼3.0 Å) with nitrite bonded through its N-atom with the metal centre (Fig. 7A). Having d10 electronic configuration at CuI centres, complex 4 is diamagnetic in nature, which was further confirmed when a long-range 1H-NMR spectrum was recorded for 4 and no paramagnetically shifted peak was found (Fig S15†). Additionally, no shift of the reference peak was observed in the 1H-NMR spectrum, recorded by following Evans’ method (Fig. S16†). Stability of 4 was also monitored by UV-Vis spectroscopy. Under an inert atmosphere, a band at 370 nm was observed in the UV-Vis spectrum for a freshly prepared solution of 4 in acetonitrile (Fig. S18B†). Then the solution was allowed to come into contact with air by opening the cuvette and the change of the solution in the presence of air was monitored for 6 h via UV-Vis spectroscopy. In the UV-Vis spectra, we observed initial increase followed by gradual decrease in band intensity at 370 nm and the rise of a new d–d transition band at 670 nm, which indicates oxidation of the CuI centre of 4 (Fig. S18C†). | ||
| Scheme 4 Proposed mechanistic route for the electro-reduction of NO2− to NO by TerpyCu (1). | ||
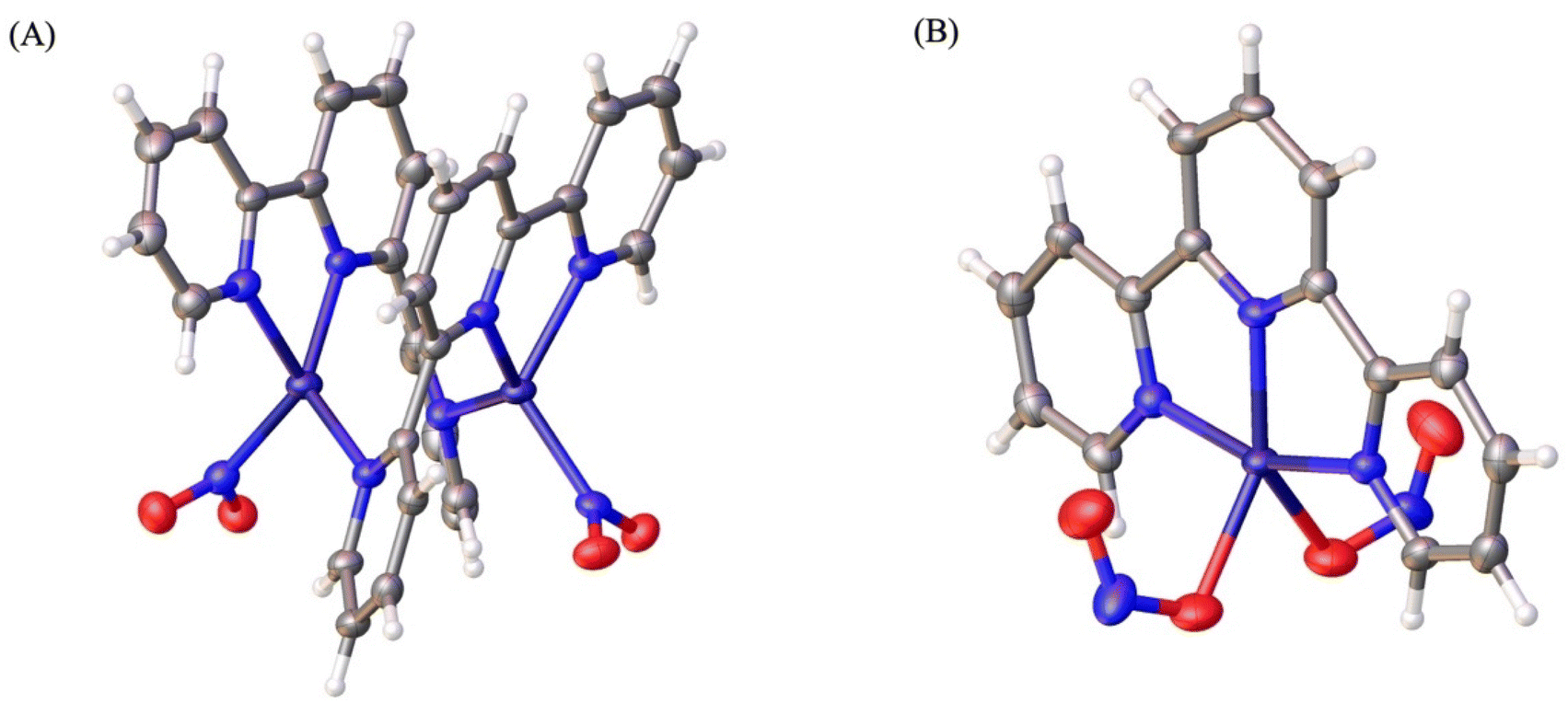 | ||
| Fig. 7 Molecular structure of (A) [TerpyCu(NO2)]2, (4) and (B) [TerpyCu(ONO)2] deduced from SXRD analysis. Colour code: C grey, H white, N blue, O red, Cu violet. | ||
Moreover, we observed very few green cuboid-shaped crystals in the crystallization vial of the complex 4 and, upon analyzing those green crystals by SXRD, they were found to be a CuII-dinitrito complex, [TerpyCu(ONO)2] (Fig. 7B). Although for the nitrite ligands the ‘nitrito’ mode (i.e. –ONO) was found to be predominant in the molecular structure, the isomeric linkage mode, i.e. ‘nitro’ mode (–NO2), was found to have an occupancy of ∼7% in the structure. The linkage isomerism was modelled accordingly in the structure (see the ESI, Fig. S29†). This CuII complex is a disproportionation product of complex 4.
NO2− reduction is believed to proceed via an unstable HNO2 intermediate with an abundance of protons. Cioncoloni et al. observed that the electron transfer and the protonation of nitrite happen simultaneously.43 This HNO2 intermediate further dissociates to form NO and H2O. We have already detected the formation of NO so, to prove the generation of H2O, 1H-NMR spectra of the electrolyte solution before and after electrolysis were recorded. In the 1H-NMR spectrum of the electrocatalytic solution (solvent CD3CN/D2O 100:1) before electrolysis we did not observe any peak for water but in the case of the post-electrolysis solution a new peak at 3.51 ppm was observed (Fig. 8). To confirm that the peak at 3.51 ppm is indeed originating from H2O, the 1H-NMR spectrum was recorded for an identical electrolyte solution with water externally added to it. A rise of the peak at 3.51 ppm was observed in the spectrum, which gives conclusive proof that the peak corresponds to the protons of water (Fig. S17†). Furthermore, the entire electrolysis process was repeated with the deuterated reagent acetic acid-d4 (CD3COOD) and this was tracked by 1H-NMR. In this 1H-NMR spectrum, no peak for water was found (Fig. 8). Therefore, it can be inferred that the protons of the water molecule come from acetic acid.
 | ||
| Fig. 8 1H-NMR spectra of the pre-electrolysis solution (brown trace), post electrolysis solution with CH3COOH (green trace) and with CD3COOD (blue trace). ‘*’ denotes the signal for the solvent acetonitrile. | ||
Conclusion
In summary, a new copper-based complex (1) was synthesized that is capable of reducing NO2− electrochemically to NO at −1.29 V vs. Fc+/Fc with FENO of 81%. We have managed to isolate and deduce molecular structures for the majority of the intermediate species involved in the mechanistic cycle. In particular, concrete structural proof for the linkage isomerism of NO2− upon reduction of CuII to CuI was acquired by isolating and crystallizing the intermediate TerpyCuI species. Additionally, we have successfully determined the post-electrolysis state of the catalyst and electrode via SXRD and SEM/EDX analysis, respectively. Through, isotope-labelling experiments using 15N-labelled Na15NO2 (the nitrite source) we have established that the N in NO and N2O indeed comes from the nitrite source. Likewise, using deuterated acetic acid, CD3COOD (the D+ source), it was demonstrated that acetic acid is the proton source during the entire conversion process and that the H atoms of the side-product water originate from acetic acid.These results give us important insight about the mechanistic route involving rearrangement from O-bound to N-bound nitrite. Our report showcases a wide canvas of NO2− conversion with great structural detail that may aid future designs of new molecular catalysts for conversion of NOx or other small molecules. It is believed that the intermediates could also be very handy for the activation of some other species.
Experimental section
General techniques
All reactions were carried out under air unless otherwise stated. To conduct synthesis under an inert atmosphere, the standard Schlenk technique or an MBraun UNIlabECO glovebox was used with dry or degassed solvents. Reagents, e.g. the Terpy ligand, and metal salts were purchased from commercial vendors and used directly without further purification. Solvents were dried using an MBraun SPS solvent purification system or by following standard methods. 1H-NMR spectroscopy was conducted at room temperature with Bruker AVIII-400 and AVIII-300 spectrometers and chemical shifts are reported in ppm. Mass spectra of samples were recorded in acetonitrile solvent using a Bruker ESQ 3000 spectrometer. IR studies were done with a Shimadzu IRTracer-100 equipped with a Pike Miracle ATR unit and spectra are reported in wavenumbers (cm−1). UV-Vis measurements were done with a Shimadzu UV-1900i spectrometer at room temperature and are reported in nm. CHN elemental analysis was done with an Elementar Vario MICRO cube. Suitable single crystals were analyzed using a RigakuXTAlab Synergy diffractometer (Cu Kα, λ = 1.54184 Å) to which was attached a HyPix-6000HE detector. The recorded data were processed and analyzed with CrysAlisPro and Platon software.65 The structures were solved using SHELXT66 and refined against F2 (SHELXL).67 OLEX2 software was used to generate graphics and pictures.68 Detailed crystallographic data are provided in the ESI.†Synthetic procedures
:1) and filtered. The filtrate was then evaporated to obtain blue-coloured complex 1 with yield of 84%. Single crystals for SXRD analysis were obtained from layering an acetonitrile solution of 1 on top of a dichloromethane (CH2Cl2) layer at −6 °C. ESI-MS: calcd for [TerpyCu(ClO4)]+m/z 394.97, found 395.0; calcd for [TerpyCu]+m/z 296.02, found 296.0. FT-IR (KBr): 3033 (m) (νsArC–H); 1596 (m), 1576 (w) (νs C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 1499 (w), 1472 (s), 1449 (s) (νs CC); 1406 (w), 1383 (m), 1330 (m), 1303 (m), 1113 (s) (νs ClO4−); 1019 (m); 970 (w); 940 (w); 908 (w); 828 (w); 777 (s); 729 (m) cm−1. Magnetic moment (1H-NMR using Evans’ method): 1.69 BM. UV-Vis: λmax 626 nm (ε 113.6 L mol−1 cm−1). Elemental analysis (%): calcd C 38.0, H 2.6, N 10.4; found C 37.7, H 3.0, N 9.7.
N); 1501 (w), 1476 (s), 1452 (νs CC); 1404 (w), 1384(w), 1318(m), 1247 (m) (νs C–N); 1194 (w), 1167 (w), 1090 (s) (νs ClO4−); 1012 (w); 973 (w); 920 (w); 899 (w); 829 (w); 771 (s); 733 (w) cm−1. UV-Vis: λmax 690 nm (ε 65.0 L mol−1 cm−1). Elemental analysis (%): calcd C 49.0, H 3.1, N 11.4; found C 49.0, H 3.1, N 11.4.
N); 1499 (w), 1472 (s), 1449 (s) (νs CC); 1406 (w), 1383 (m), 1330 (m), 1303 (m), 1113 (s) (νs ClO4−); 1019 (m); 970 (w); 940 (w); 908 (w); 828 (w); 777 (s); 729 (m) cm−1. Magnetic moment (1H-NMR using Evans’ method): 1.69 BM. UV-Vis: λmax 626 nm (ε 113.6 L mol−1 cm−1). Elemental analysis (%): calcd C 38.0, H 2.6, N 10.4; found C 37.7, H 3.0, N 9.7.
N); 1501 (w), 1476 (s), 1452 (νs CC); 1404 (w), 1384(w), 1318(m), 1247 (m) (νs C–N); 1194 (w), 1167 (w), 1090 (s) (νs ClO4−); 1012 (w); 973 (w); 920 (w); 899 (w); 829 (w); 771 (s); 733 (w) cm−1. UV-Vis: λmax 690 nm (ε 65.0 L mol−1 cm−1). Elemental analysis (%): calcd C 49.0, H 3.1, N 11.4; found C 49.0, H 3.1, N 11.4.
1-ONO can also be synthesised by direct addition of NaNO2 in the reaction solution of 1 without isolating it. However, the yield (71%) of the product in this method was lower compared to that of the former. ESI-MS: calcd for [TerpyCu(ONO)]+m/z 342.02, found 342.1. FT-IR (KBr): 3042 (m) (νsArC–H); 1597 (s), 1575 (m) (νs CN); 1498 (m), 1473 (s), 1449 (s) (νs CC); 1405 (w), 1384 (w), 1366 (w) (νasym, NO2); 1329 (m), 1303 (m), 1268 (s) (νsym, NO2); 1111 (s) (νs ClO4−); 1034 (w); 1019 (m); 972 (w); 940 (w); 907 (w); 827 (w); 779 (s); 731 (m) cm−1. UV-Vis: λmax 372 nm (ε 364 L mol−1 cm−1) and λmax 640 nm (ε 54 L mol−1 cm−1). Elemental analysis (%): calcd C 40.7, H 2.5, N 12.7; found C 40.6, H 2.8, N 13.1.
N); 1500 (m), 1472 (s), 1450 (s) (νs CC); 1406 (w), 1384 (w), 1352 (νasym, 15NO2); 1332 (m), 1302 (m), 1251 (νsym, 15NO2); 1118 (s) (νs ClO4−); 1019 (w); 972 (w); 940 (w); 908 (w); 828 (w); 776 (w); 730 (m) cm−1.
N); 1471 (s), 1452 (s) (νs CC); 1421 (w), 1383 (w), 1325 (w) (νasym, NO2; 1295 for 15NO2); 1270 (νsym, NO2; 1243 for 15NO2); 1059 (s) (νs BF4−); 881 (w); 832 (w); 780 (s); 735 (w); 670 (w); 651 (w); 630 (w) cm−1. UV-Vis: λmax 370 nm (ε 900 L mol−1 cm−1).69 No paramagnetically shifted signal was observed in the long-range paramagnetic NMR. The 1H-NMR spectrum was recorded as described in Evans’ method;70,71 no shift in the acetonitrile peak was observed.
Crystallographic data
See the ESI, pages S18–S24.†Experimental details of the electrochemical experiments
Electrochemical experiments were performed with a standard three-electrode setup (glassy carbon working electrode, Pt wire counter electrode and Ag wire as pseudo-reference electrode) using a PalmSens4 potentiostat. The working electrode was prepared by polishing the glassy carbon with 1.0 and 0.3 μm alumina solution and then subsequently subjecting it to sonication in the applied solvent for 30 min before using. Tetrabutylammonium hexafluorophosphate (nBu4NPF6) (0.1 M in the case of CV and 0.2 M in the case of CA) was used as a supporting electrolyte. All the electrochemical measurements were done under an inert atmosphere inside a glovebox. Since Ag wire is a pseudo-reference electrode, the potentials of all the redox events were referenced against a ferrocene/ferrocenium (Fc+/Fc) redox couple.All CV experiments were performed in MeCN containing 1 mM 1, 150 mM CH3COOH and 0.1 M nBu4NPF6. A NaNO2 stock solution in water was prepared from which specific aliquots were extracted and added during different CV measurements. Cyclic voltammograms were recorded in the presence of 5 mM, 10 mM and 20 mM NO2−, with the total electrolyte volume maintained at 2 mL for all experiments. All scans were recorded at a scan rate of 100 mV s−1.
Bulk electrolysis (chronoamperometry) was performed with the same setup as mentioned above. The electrolysis was performed in a solvent mixture of MeCN/H2O (100:1) containing 2 mM 1, 150 mM CH3COOH, 40 mM NaNO2 and 0.2 M nBu4NPF6. Separate bulk electrolysis experiments were conducted with durations of 1 h, 3 h, 5 h and 7 h. For all the experiments the potential was fixed at −1.29 V.
Furthermore, to determine the catalytically active species, a rinse test was performed. First, a chronoamperometric experiment was carried out by holding the reductive potential at CuI → Cu0 resulting in the deposition of metallic copper on the electrode surface. Subsequently, the electrochemical cell was replenished with a fresh solution containing 5 mM NaNO2 and 150 mM CH3COOH, without altering the working electrode. Then, linear sweep voltammetry (LSV) was performed to monitor the current response of the deposited material. For comparison, an LSV scan was also recorded for a solution containing 2 mM compound 1, 150 mM CH3COOH and 5 mM sodium nitrite.
Spectroscopic detection of NO using CoTPP
A small, uncapped vial containing 1 mL of 40 μM CoTPP solution in THF, was attached to the inner wall of the electrochemical cell in such a way that the CoTPP solution does not come into contact with the electrolyte solution. The electrochemical cell was sealed under an inert atmosphere, and bulk electrolysis was performed under these conditions and setup. The vial of CoTPP was kept open so that the NO(g), which accumulated in the headspace, can diffuse into the CoTPP solution and coordinate with the CoII centre. The whole operation was carried out under an inert atmosphere. After that, the CoTPP solution was removed and diluted to 8 μM and UV-Vis spectra were recorded showing a shift of the 527 nm band of CoTPP to 535 nm.Experimental details of GC-MS analysis for NO and N2O detection
GC-MS analyses were done using a Shimadzu GC-MS-QP2010 Plus instrument with a flame ionized detector (FID) and a HP-Plot-Q column of 30 m length, inner diameter of 0.53 mm and a thickness of 40 μm.Starting temperature: 35 °C, hold time: 2 min; ramp: 25 °C min−1 to 240 °C, split ratio: 20.0; pressure: 30 kPa. Flow rate: 66.7 mL min−1; carrier gas: helium (He). Retention time: Rt (NO) = 3.78 min, Rt (N2O) = 5.37 min.
Data availability
All data supporting these findings can be found in the article and its ESI† or are available from the authors upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) for financial support under Germany's Excellence Strategy – (EXC-2033 390677874 – “RESOLV” and AP242/9-1). This work was further supported by the Fraunhofer Internal Programs under Grant No. Attract 097- 602175. Likewise, this work was also supported by Grants-in-Aid (Grant No. CRG/2021/003371, and EEQ/2021/000109) from SERB, and DAAD (ID: 57552334).References
- L. Y. Stein and M. G. Klotz, Curr. Biol., 2016, 26, R94–R98 CrossRef CAS PubMed.
- M. M. M. Kuypers, H. K. Marchant and B. Kartal, Nat. Rev. Microbiol., 2018, 16, 263–276 CrossRef CAS PubMed.
- W. Steffen, K. Richardson, J. Rockström, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sörlin, Science, 2015, 347, 1259855 CrossRef PubMed.
- K. Richardson, W. Steffen, W. Lucht, J. Bendtsen, S. E. Cornell, J. F. Donges, M. Drüke, I. Fetzer, G. Bala, W. von Bloh, G. Feulner, S. Fiedler, D. Gerten, T. Gleeson, M. Hofmann, W. Huiskamp, M. Kummu, C. Mohan, D. Nogués-Bravo, S. Petri, M. Porkka, S. Rahmstorf, S. Schaphoff, K. Thonicke, A. Tobian, V. Virkki, L. Wang-Erlandsson, L. Weber and J. Rockström, Sci. Adv., 2023, 9, eadh2458 CrossRef PubMed.
- D. Gerten, V. Heck, J. Jägermeyr, B. L. Bodirsky, I. Fetzer, M. Jalava, M. Kummu, W. Lucht, J. Rockström, S. Schaphoff and H. J. Schellnhuber, Nat. Sustain., 2020, 3, 200–208 CrossRef.
- L. B. Maia and J. J. G. Moura, Chem. Rev., 2014, 114, 5273–5357 CrossRef CAS.
- L. Lamattina, C. García-Mata, M. Graziano and G. Pagnussat, Annu. Rev. Plant Biol., 2003, 54, 109–136 CrossRef CAS PubMed.
- V. Calabrese, C. Mancuso, M. Calvani, E. Rizzarelli, D. A. Butterfield and A. M. G. Stella, Nat. Rev. Neurosci., 2007, 8, 766–775 CrossRef CAS.
- S. A. Omar and A. J. Webb, J. Mol. Cell. Cardiol., 2014, 73, 57–69 CrossRef CAS.
- J. Heinecke and P. C. Ford, Coord. Chem. Rev., 2010, 254, 235–247 CrossRef CAS.
- G. R. Navale, S. Singh and K. Ghosh, Coord. Chem. Rev., 2023, 481, 215052 CrossRef CAS.
- M. T. Gladwin, A. N. Schechter, D. B. Kim-Shapiro, R. P. Patel, N. Hogg, S. Shiva, R. O. Cannon, M. Kelm, D. A. Wink, M. G. Espey, E. H. Oldfield, R. M. Pluta, B. A. Freeman, J. R. Lancaster, M. Feelisch and J. O. Lundberg, Nat. Chem. Biol., 2005, 1, 308–314 CrossRef CAS PubMed.
- J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7, 156–167 CrossRef CAS.
- K. Cosby, K. S. Partovi, J. H. Crawford, R. P. Patel, C. D. Reiter, S. Martyr, B. K. Yang, M. A. Waclawiw, G. Zalos, X. Xu, K. T. Huang, H. Shields, D. B. Kim-Shapiro, A. N. Schechter, R. O. Cannon and M. T. Gladwin, Nat. Med., 2003, 9, 1498–1505 CrossRef CAS.
- R. G. Knowles and S. Moncada, Biochem. J., 1994, 298(Pt 2), 249–258 CrossRef CAS PubMed.
- T. B. McCall, N. K. Boughton-Smith, R. M. Palmer, B. J. Whittle and S. Moncada, Biochem. J., 1989, 261, 293–296 CrossRef CAS.
- S. Basu, N. A. Azarova, M. D. Font, S. B. King, N. Hogg, M. T. Gladwin, S. Shiva and D. B. Kim-Shapiro, J. Biol. Chem., 2008, 283, 32590–32597 CrossRef CAS PubMed.
- F. Cava, O. Zafra and J. Berenguer, Mol. Microbiol., 2008, 70, 507–518 CrossRef CAS PubMed.
- S. Shahid, M. Ali, D. Legaspi-Humiston, J. Wilcoxen and A. A. Pacheco, Biochemistry, 2021, 60, 2098–2115 CrossRef CAS PubMed.
- B. L. J. Godber, J. J. Doel, G. P. Sapkota, D. R. Blake, C. R. Stevens, R. Eisenthal and R. Harrison, J. Biol. Chem., 2000, 275, 7757–7763 CrossRef CAS PubMed.
- L. B. Maia and J. J. G. Moura, J. Biol. Inorg. Chem., 2011, 16, 443–460 Search PubMed.
- L. B. Maia and J. J. G. Moura, Redox Biol., 2018, 19, 274–289 CrossRef CAS.
- B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- S. Horrell, D. Kekilli, R. W. Strange and M. A. Hough, Metallomics, 2017, 9, 1470–1482 CrossRef CAS.
- J. W. Godden, S. Turley, D. C. Teller, E. T. Adman, M. Y. Liu, W. J. Payne and J. LeGall, Science, 1991, 253, 438–442 CrossRef CAS PubMed.
- F. E. Dodd, S. S. Hasnain, Z. H. Abraham, R. R. Eady and B. E. Smith, Acta Crystallogr., Sect. D:Biol. Crystallogr., 1997, 53, 406–418 CrossRef CAS PubMed.
- M. E. P. Murphy, S. Turley and E. T. Adman, J. Biol. Chem., 1997, 272, 28455–28460 CrossRef CAS PubMed.
- M. J. Boulanger and M. E. P. Murphy, Protein Sci., 2003, 12, 248–256 CrossRef CAS PubMed.
- S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12041–12046 CrossRef CAS.
- S. Ghosh, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 277–288 CrossRef CAS.
- I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef CAS.
- S. M. Carrier, C. E. Ruggiero, W. B. Tolman and G. B. Jameson, J. Am. Chem. Soc., 1992, 114, 4407–4408 CrossRef CAS.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. P. Murphy, Science, 2004, 304, 867–870 CrossRef CAS.
- K. Fujisawa, A. Tateda, Y. Miyashita, K. Okamoto, F. Paulat, V. K. K. Praneeth, A. Merkle and N. Lehnert, J. Am. Chem. Soc., 2008, 130, 1205–1213 CrossRef CAS.
- S. Hong, J. J. Yan, D. G. Karmalkar, K. D. Sutherlin, J. Kim, Y.-M. Lee, Y. Goo, P. K. Mascharak, B. Hedman, K. O. Hodgson, K. D. Karlin, E. I. Solomon and W. Nam, Chem. Sci., 2018, 9, 6952–6960 RSC.
- J. Biswas, K. Kulbir, P. Bhardwaj, S. Ghosh, S. C. Sahoo, U.-P. Apfel and P. Kumar, Chem. – Eur. J., 2024, e202402295 CrossRef CAS PubMed.
- N. Komeda, H. Nagao, G. Adachi, M. Suzuki, A. Uehara and K. Tanaka, Chem. Lett., 1993, 22, 1521–1524 CrossRef.
- N. Komeda, H. Nagao, Y. Kushi, G. Adachi, M. Suzuki, A. Uehara and K. Tanaka, Bull. Chem. Soc. Jpn., 1995, 68, 581–589 CrossRef CAS.
- H. Ren, J. Wu, C. Xi, N. Lehnert, T. Major, R. H. Bartlett and M. E. Meyerhoff, ACS Appl. Mater. Interfaces, 2014, 6, 3779–3783 CrossRef CAS.
- Y. Qin, J. Zajda, E. J. Brisbois, H. Ren, J. M. Toomasian, T. C. Major, A. Rojas-Pena, B. Carr, T. Johnson, J. W. Haft, R. H. Bartlett, A. P. Hunt, N. Lehnert and M. E. Meyerhoff, Mol. Pharmaceutics, 2017, 14, 3762–3771 CrossRef CAS PubMed.
- K. K. Konopińska, N. J. Schmidt, A. P. Hunt, N. Lehnert, J. Wu, C. Xi and M. E. Meyerhoff, ACS Appl. Mater. Interfaces, 2018, 10, 25047–25055 CrossRef.
- G. Cioncoloni, I. Roger, P. S. Wheatley, C. Wilson, R. E. Morris, S. Sproules and M. D. Symes, ACS Catal., 2018, 8, 5070–5084 CrossRef CAS.
- S. E. Braley, H.-Y. Kwon, S. Xu, E. Z. Dalton, E. Jakubikova and J. M. Smith, Inorg. Chem., 2022, 61, 12998–13006 CrossRef CAS PubMed.
- J. R. Stroka, B. Kandemir, E. M. Matson and K. L. Bren, ACS Catal., 2020, 10, 13968–13972 CrossRef CAS.
- P. H. van Langevelde, S. Engbers, F. Buda and D. G. H. Hetterscheid, ACS Catal., 2023, 13, 10094–10103 CrossRef CAS PubMed.
- Kulbir, S. Das, T. Devi, M. Goswami, M. Yenuganti, P. Bhardwaj, S. Ghosh, S. C. Sahoo and P. Kumar, Chem. Sci., 2021, 12, 10605–10612 RSC.
- Kulbir, S. Das, T. Devi, S. Ghosh, S. C. Sahoo and P. Kumar, Chem. Sci., 2023, 14, 2935–2942 RSC.
- Kulbir, A. Keerthi C. S, S. Beegam, S. Das, P. Bhardwaj, M. Ansari, K. Singh and P. Kumar, Inorg. Chem., 2023, 62, 7385–7392 CrossRef CAS PubMed.
- S. Das, Kulbir, S. Ray, T. Devi, S. Ghosh, S. S. Harmalkar, S. N. Dhuri, P. Mondal and P. Kumar, Chem. Sci., 2022, 13, 1706–1714 RSC.
- K. Ghosh, A. A. Eroy-Reveles, B. Avila, T. R. Holman, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2004, 43, 2988–2997 CrossRef CAS PubMed.
- A. J. Amoroso, M. W. Burrows, S. J. Coles, R. Haigh, R. D. Farley, M. B. Hursthouse, M. Jones, K. M. A. Malik and D. M. Murphy, Dalton Trans., 2008, 506–513 RSC.
- J. Karges, K. Xiong, O. Blacque, H. Chao and G. Gasser, Inorg. Chim. Acta, 2021, 516, 120137 CrossRef CAS.
- A. Meyer, G. Schnakenburg, R. Glaum and O. Schiemann, Inorg. Chem., 2015, 54, 8456–8464 CrossRef CAS PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- A. K. Purohit, S. K. Padhan, J. R. Mohanty and P. K. Kar, Photochem. Photobiol. Sci., 2018, 17, 815–821 CrossRef CAS.
- A. Dey, T. Albert, R. Y. Kong, S. N. MacMillan, P. Moënne-Loccoz, K. M. Lancaster and D. P. Goldberg, Inorg. Chem., 2022, 61, 14909–14917 CrossRef CAS PubMed.
- S. Metz, Inorg. Chem., 2017, 56, 3820–3833 CrossRef CAS PubMed.
- C. E. Ruggiero, S. M. Carrier and W. B. Tolman, Angew. Chem., Int. Ed. Engl., 1994, 33, 895–897 CrossRef.
- C. J. Hoerger, H. S. L. Pierre, L. Maron, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2016, 52, 10854–10857 RSC.
- X. Zhang, Y. Wang, Y. Wang, Y. Guo, X. Xie, Y. Yu and B. Zhang, Chem. Commun., 2022, 58, 2777–2787 RSC.
- S. Xu, H.-Y. Kwon, D. C. Ashley, C.-H. Chen, E. Jakubikova and J. M. Smith, Inorg. Chem., 2019, 58, 9443–9451 CrossRef CAS PubMed.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, J. Am. Chem. Soc., 2018, 140, 842–847 CrossRef CAS.
- R. C. Maji, S. K. Barman, S. Roy, S. K. Chatterjee, F. L. Bowles, M. M. Olmstead and A. K. Patra, Inorg. Chem., 2013, 52, 11084–11095 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. Jökel, F. Nyßen, D. Siegmund and U.-P. Apfel, Dalton Trans., 2021, 50, 14602–14610 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- J. Loliger and R. Scheffold, J. Chem. Educ., 1972, 49, 646 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2376874–2376878 and 2376880. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02777a |
| This journal is © The Royal Society of Chemistry 2025 |