
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How plutonium “brown” peroxo complex emerges from electrolysis experiments†
Richard
Husar
b,
Quentin
Hervy
 *a,
Thomas
Dumas
*a,
Philippe
Guilbaud
a,
Matthieu
Virot
c and
Philippe
Moisy
a
*a,
Thomas
Dumas
*a,
Philippe
Guilbaud
a,
Matthieu
Virot
c and
Philippe
Moisy
a
aCEA, DES, ISEC, DMRC, Univ Montpellier, Bagnols-sur-Ceze 30200, France. E-mail: Thomas.dumas@cea.fr
bDepartment Environmental and Radionuclide Analysis, VKTA Dresden, Bautzner Landstraße 400, 01328 Dresden, Germany
cICSM, University of Montpellier, CEA, CNRS, ENSCM, 30207 Bagnols-sur-Ceze, France
First published on 10th February 2025
Abstract
An original spectroelectrochemical approach was employed to probe in situ redox reactions with plutonium. Monitoring Pu electrolysis under aerated conditions revealed the formation of a transient species. Using chemometric methods, the associated spectroscopic signature was found to correspond with a dimeric peroxo-bridged Pu complex described 70 years ago, often referred to as the plutonium “brown” complex. A separate synthesis of this complex confirmed the presence of the transient species during electrolysis, challenging conventional concepts of Pu electrochemistry. The mechanistic role of the Pu “brown” complex during Pu electrolysis is also discussed.
Introduction
In acidic and non-complexing aqueous solution, plutonium can coexist under four oxidation states, with the solvated cations Pu3+, Pu4+, and the molecular ions PuO2+ and PuO22+. Such coexistence is possible because Pu(IV) can disproportionate into Pu(III) and Pu(VI), while Pu(V) disproportionates into Pu(IV) and Pu(VI). In the tetravalent state, Pu is also highly sensitive to hydrolysis reactions leading to the formation of hydroxides as well as polymeric or colloidal species at pH levels above 1.1,2 These phenomena complicate the redox reactions of plutonium in aqueous solutions by introducing additional side reactions. Additionally, it is important to note that the formation of the linear trans-dioxo ions AnO2+/2+ involves slow kinetics and overpotential to be applied during electrolysis experiments. The crossed standards potentials for the PuO22+/PuO2+, Pu4+/Pu3+, PuO22+/Pu3+, PuO22+/Pu4+, PuO2+/Pu3+ and PuO2+/Pu4+ redox couples (respectively 913, 982, 1022, 1043, 1077 and 1172 mV vs. Standard Hydrogen Electrode (SHE) in 1 M HClO4 solution1) often result in steady states where both oxidized and reduced plutonium species are simultaneously formed. Electrolysis can be considered as an interesting approach to gain more quantitative insights into the Pu redox reactions occurring in aqueous solution. However, the generation of intermediate species during electrolysis may interfere with the studied reactions, leading to undesired or disruptive phenomena. In particular, it has been reported that hydrogen peroxide can be formed under aerated conditions via a direct two electron Oxygen Reduction Reaction (ORR) (eqn (1)) at the catalytic electrode surface. This reaction is favoured by Pt electrode surfaces and is followed by the rapid decomposition of peroxide ions, either through 2e− oxidation (eqn (1)) or 2e− reduction (eqn (2)), depending on the pH and applied potential.3–5O2 + 2H+ + 2e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) H2O2 H2O2 | (1) |
| H2O2 + 2H+ + 2e− 2H2O | (2) |
Since platinum electrodes are widely used, especially in nitric acid solutions, improving the understanding of H2O2 formation on the surface of these electrodes and the resulting chemical reactions will be very useful for future electrochemistry experiments.
Unlike the well-established uranyl peroxide chemistry,6 the formation and characterization of oligomeric peroxo-bridged Pu(IV) species is rare and has been very scarcely reported in the literature.7–11 The addition of a small amount of hydrogen peroxide to Pu(IV) aqueous solutions has been reported to generate a deep brown soluble complex, which turns red with further addition of H2O2. Hypothetic linear and ring-like structures for these two complexes were proposed by Connick and McVey in 1949.7 In 1972, Ekstrom and McLaren8 proposed two distinct formation mechanisms based on competitive Pu(IV) hydrolysis and peroxide complexation to explain the formation of the Pu “brown” complex, also suggesting a linear or ring-like structure. Both the “brown” and “red” peroxo complexes, described respectively by the formulas Pu2(O2)OH5+ and Pu2(O2)24+, exhibit strong molar extinction coefficients for their main absorption bands at 495 (ε = ca. 266 M−1 cm−1) and 530 nm (ε = ca. 440 M−1 cm−1) respectively.7,12
From these assessments, it can be inferred that intermediate molecular peroxo ions may form in solution during electrolysis under air atmosphere. Understanding the potential contribution of H2O2 during Pu electrolysis is important for elucidating redox reaction mechanisms and for predicting parasitic side reactions. In particular, the contribution of parasitic side reactions may interfere in the determination of redox potential and limit our capacity to stabilize oxidation states by electrolysis. Individual plutonium (III, IV, V and VI) aqueous complexes present intense and distinguishable UV-vis-NIR spectrophotometric signals, making it relatively easy to identify the Pu redox states, but the potential occurrence of a transient species has to be taken into account in order to properly analyze the UV-vis-NIR spectrum variations. Additionally, a substantial amount of UV-vis-NIR spectral footprints are available for plutonium complexes with basic inorganic ligands.13–24 Hence, this work aimed to evaluate the potential contribution of Pu peroxo species during electrolysis cycle experiments under air atmosphere. Another original approach of the study is related to the in situ spectrophotometric characterization of the Pu system by using our recently developed set-up (ESI.1†), based on previous studies of plutonium spectroelectrochemistry in nitric acid.23,24 Spectral components were isolated using chemometric analysis, providing an innovative approach to explore plutonium speciation.
Experimental
Caution! 239Pu is an α-emitting element that poses serious health risks. Studies involving Pu require careful handling and trained personnel with appropriate radiation safety infrastructures.Plutonium stock solution was prepared by a standard anion-exchange method. The isotopic signature of the Pu solution was 96.9% 239Pu, 2.98% 240Pu, 0.04% 241Pu, and 0.06% 242Pu. Reagents were of analytical grade. Aqueous solutions were prepared with purified ultrapure water (resistivity higher than 18.2 MΩ cm at 25 °C).
A specifically designed spectroelectrochemical micro-cell (quartz windows with optical path of 20 mm, see Fig. S1, ESI†) was used inside a glovebox, this cell was adapted for UV-vis-NIR spectroscopy from an X-ray absorption cell.25 This cell was connected to a Cary 6000 spectrophotometer (Agilent, USA) via optical fibers (Hellma, Germany) and a potentiogalvanostat PGStat101 (Metrohm, Germany) for potentiostatic electrolyses. Experiments were carried out under aerated (air) or Ar-bubbling atmospheres in 0.1 M HNO3 at room temperature. The electrode material was oriented within the cell to avoid interferences with the optical path. The working electrode compartment (Pt wire, A-2234, Bio-Logic, France) was separated from the auxiliary electrode (Pt wire, A-2233, Bio-Logic, France) by a ceramic diaphragm (micro-diaphragm 6.1240.00, Metrohm, Germany), while the redox potential in the solution was measured against an Ag/AgCl reference electrode (0.222 V vs. SHE, DriRef-2SH, World Precision Instruments, USA). Successive isolation of the Pu redox states was achieved via potentiostatic electrolysis, starting from an initial Pu(VI) aqueous solution which was exhaustively reduced to Pu(V), and subsequently to Pu(III). The resulting Pu(III) solution was then re-oxidized to Pu(IV).
Simultaneously, UV-vis-NIR absorption spectra were recorded online during electrolysis from 300 to 1300 nm (600 nm min−1). The duration of the bulk electrolysis was 1200 s. Absorption spectra were recorded as a function of the applied potential. Homogenization of the Pu solution was achieved by gentle and constant magnetic stirring throughout the experiment. The stirring speed can control the thickness of the diffusion layer on the electrode surface, which may influence the redox reactions. Nevertheless, preliminary experiment demonstrated that the quality of the signal recorded in UV-Vis happened to be affected by too high or too low stirring speed limiting our ability to study diffusion effects with this set-up. The electronic structures of the transient species were analyzed by spectral deconvolution using conventional procedures based on linear spectral simulation, principal component analysis and iterative transformation factor analysis.26–28 Finally, the Pu “brown” peroxo complex was obtained by dilution of a Pu(IV) aliquot (previously stabilized in nitric media) in a 5 mM aqueous solution of hydrogen peroxide under stirring. The final molar ratio H2O2/Pu was 0.5.
Results and discussion
At first, the successive stabilization of pure oxidation states Pu(III), Pu(IV), Pu(V) and Pu(VI) was achieved using potentiostatic electrolysis under moderate acidic conditions and low plutonium concentration: [Pu(VI)] = 4.3 × 10–4 M in 0.1 M HNO3. These conditions were selected as a compromise to ensure good stability of Pu(VI) mother solution, prevent too strong complexation by nitrate anions so that aquo species predominate, minimize Pu(IV) hydrolysis and slow the disproportionation reactions of Pu(IV) and Pu(V).29 Starting by the electrochemical reduction of the initial Pu(VI) solution to Pu(V) at 760 mV, the experiment was followed by reduction to Pu(III) at 110 mV and re-oxidation to Pu(IV) at 960 mV (all redox potential referenced in solution vs. SHE). After quantitative electrolysis, the absorption spectra of Pu ions were recorded in situ. These initial experiments provided access to the electronic transitions of Pu ions under strictly identical conditions (Fig. S2, ESI.2†), thereby establishing a self-referencing system for subsequent chemometric analyses, which is essential for accurate spectral deconvolution. The acquired spectra well agreed with those published under acidic conditions for Pu(VI), Pu(III), Pu(V) and Pu(IV).13–24,30The first in situ experiment focused on the oxidation of Pu(III) as a function of electrolysis time. Under these conditions, Pu(III) is not prone to hydrolysis. A straightforward bimolecular reaction with a one-electron-transfer, following the simple redox equilibrium described in eqn (3), was anticipated.20,23,24 Presumably, such a reaction would result in a two-component spectral system with isobestic points. The spectral changes observed during the oxidation of Pu(III) at 960 mV under both Ar (Fig. 1a) and aerated (Fig. 1b) atmospheres are presented below.
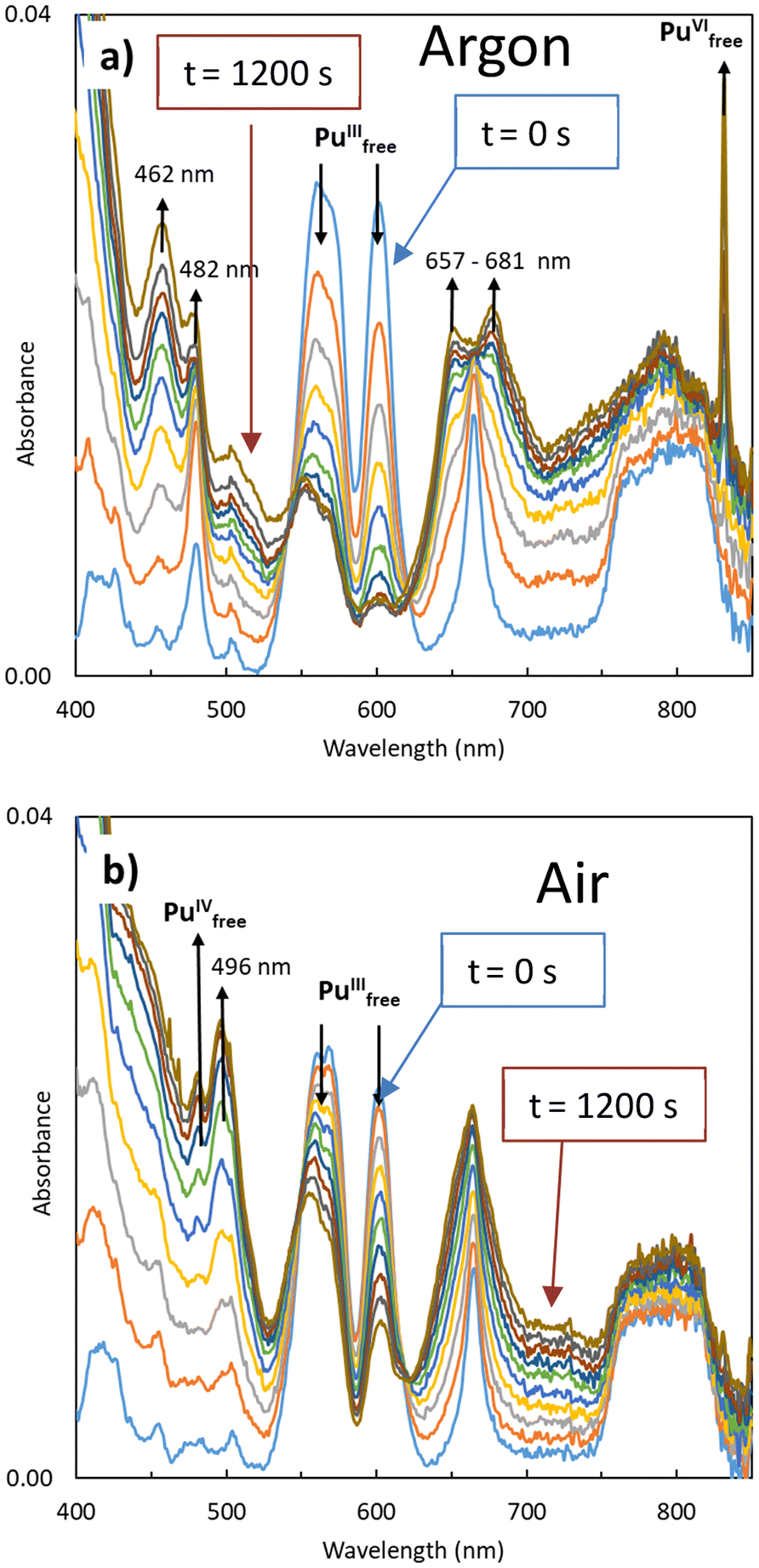 | ||
| Fig. 1 Spectrophotometric measurements during electrolysis experiments in 0.1 M HNO3 from t = 0 s to t = 1200 s: (a) initial 4.3 × 10−4 M Pu(III) (blue) electrolysis under Ar atmosphere at (+) 960 mV to Pu(IV). (b) Initial 3.5 × 10−4 M Pu(III) (blue) electrolysis under aerated atmosphere at (+) 960 mV to Pu(IV). | ||
Under Ar, the decrease of Pu(III) absorption bands located at λ = 562 and 602 nm is accompanied by an increase in Pu(VI) free ion absorption at λ = 830 nm, along with the appearance of two double bands at 462/482 nm and 657/681 nm. No Pu(IV) was observed. In contrast, under aerated atmosphere, the decrease of Pu(III) absorption bands is accompanied with by an increase at 475 nm, attributed to Pu(IV) free ion, along with a band located at λ = 496 nm. No Pu(VI) was observed under these aerated conditions.
| Pu3+ − e− Pu4+ | (3) |
The chemometric deconvolution (ESI.3†) of the spectra shown in Fig. 1a and b led to the identification of transient species for each experiment. The spectrum of the one observed under air atmosphere is presented in Fig. 2 alongside reference spectra, while the spectrum of the transient species observed under Ar is presented in Fig. S5 (ESI.3†).
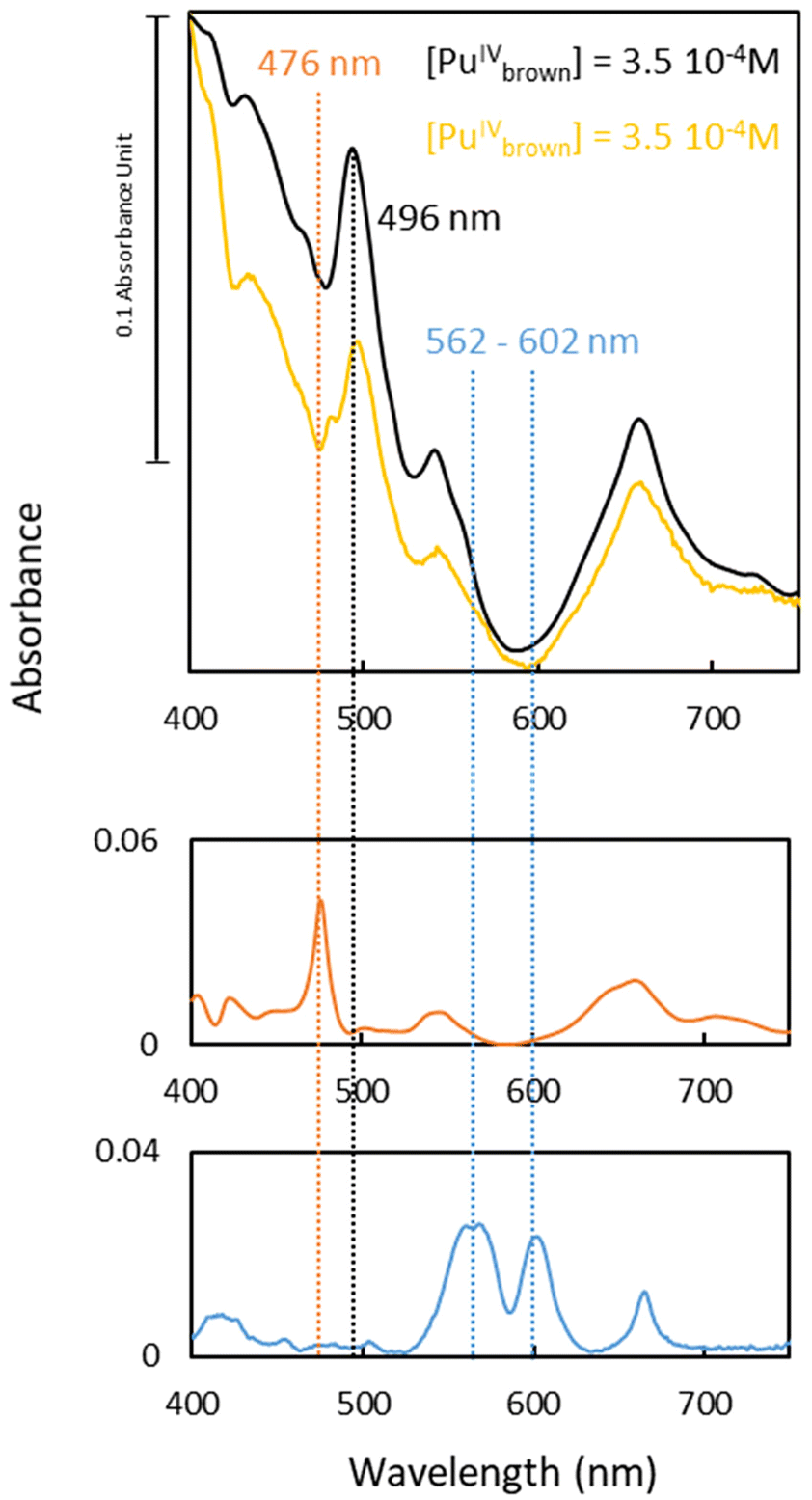 | ||
| Fig. 2 Molar electronic absorptivity of the transient species (Air atm.) (yellow) and references of trivalent (blue), tetravalent (orange) and plutonium peroxide brown complex (black) (3.5 × 10−4 M) in 0.1 M HNO3. The spectrum of the peroxide brown complex (black) was shifted for a better comparison. | ||
First, the transient species observed under Ar exhibits spectral characteristics previously observed by Cot-Auriol et al.31 in the process of plutonium hydrolysis, which may correspond to an hydrolysed form of Pu(IV), referred to as Pu(IV) hydroxo. Although this in situ electrolysis approach appears promising for studying Pu(IV) hydrolysis, we have set this topic aside for more detailed future research. The presence of this species is not surprising, given that at pH = 1, Pu(IV) formed by the oxidation of Pu(III) tends to hydrolyze.
The second unidentified species (air experiment, Fig. 2) is characterized by a main absorption band at 496 nm, along with additional bands located at 431, 540 and 658 nm (yellow spectrum in Fig. 2). These spectral features differ significantly from the typical absorption spectra of the well-known aqueous Pu ions, such as Pu(III) or Pu(IV). Neither previously described nitrate nor hydroxide complexes of Pu can account for these distinctive characteristics.12,14,15 However, the refined spectrum of the transient species observed under air electrochemical experiment, particularly the intense band observed at 496 nm and its large molar absorption coefficient (ε ≈ 230 L mol−1 cm−1), aligns well with the spectrum of the “brown” peroxo complex of Pu(IV) described by Connick and McVey in 1949.7 This species exhibits an absorption band at 495 nm with a shoulder at λ = 543 nm and an increased baseline below 600 nm. Such an absorption increase is characteristic of the “brown” complex and it is not related to the formation of Pu(IV) colloids. Indeed, an absorption band at 615 nm that is not observed in the current study characterizes the latter.31
The structure of the “brown” complex, described by the formula Pu2(O2)OH5+, remains unclear. Initially, it was proposed to have a ring-like structure involving a dimeric Pu(IV) species coordinated by bridging peroxide and hydroxo groups, or a linear peroxo-bridged structure.8 More recently, solid state evidence of an octahedral Pu2(O2)2 cage was reported in carbonate media. The structure involves two side-on coordinated peroxide ligands bridged to two Pu(IV) ions. The authors observed a shift in the monomeric Pu carbonate f–f transitions bands from 486 nm to 496 nm in the presence of peroxide groups, indicating the formation of Pu2(O2)n species. The intensified absorbance at 496 nm is again associated with increased absorbance below 600 nm. The nature of the electronic transitions in plutonium peroxide complexes is still unknown and the only structural characterizations available in carbonate media suggest that side-on peroxide coordination may dominate for peroxides in solution.
The “brown” peroxo complex was also prepared separately from the electrolysis experiments to further compare it with the transient species (air experiment). The synthesis involved a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Pu:H2O2 ratio leading to the immediate formation of a deep brown solution upon mixing (see ESI.4†). The UV-vis absorption spectrum of the synthesized complex was compared to the spectrum of the transient species observed under air and extracted from the chemometric analyses (Fig. 2). Apart from a minor signal at 483 nm and some discrepancies in overall extinction values, potentially related to the deconvolution procedure or slight variations in physicochemical conditions (pH, temperature) compared to the electrolysis experiment, the spectrum of the transient species (air, in yellow) closely resembles that of the synthetic Pu “brown” complex (black). This observation confirms that a plutonium peroxide interferes during electrolysis in aerated solutions.
:1, Pu:H2O2 ratio leading to the immediate formation of a deep brown solution upon mixing (see ESI.4†). The UV-vis absorption spectrum of the synthesized complex was compared to the spectrum of the transient species observed under air and extracted from the chemometric analyses (Fig. 2). Apart from a minor signal at 483 nm and some discrepancies in overall extinction values, potentially related to the deconvolution procedure or slight variations in physicochemical conditions (pH, temperature) compared to the electrolysis experiment, the spectrum of the transient species (air, in yellow) closely resembles that of the synthetic Pu “brown” complex (black). This observation confirms that a plutonium peroxide interferes during electrolysis in aerated solutions.
The chemometric analyses provided insights into the evolution of the plutonium species during electrolysis (Fig. 3). As expected for both oxidation reactions (Ar or air), the decrease of the concentration of Pu(III) aquo ion is accompanied with a concentration increase of Pu(IV) species of different nature. Under Ar atmosphere, Pu(IV) accumulates as a transient free ion before hydrolysis or disproportionation reactions take place. During Pu(III) oxidation, Pu(IV) hydroxo is the main product that accumulates, with traces of Pu(VI) also forming. A very low concentration of the “brown” Pu peroxide complex is also detected (Pu(IV)brown, <3%), possibly due to incomplete oxygen purging. In contrast, under aerated conditions, the Pu(IV) “brown” complex dominates over Pu(IV) free ion. Pu(IV) hydroxo appears later when a significant amount of Pu(IV) free is observed. The presence of Pu(VI) aquo ion is negligible. This suggests that under aerated conditions, the formation of the Pu(IV) “brown” complex is favored over both the Pu(IV) hydroxo and Pu(VI) aquo ions, despite competing processes like hydrolysis and disproportionation. Concerning the Pu(III) decrease kinetics order, the difference can be qualitatively explained: For (a), when the Pu(IV) is formed by the oxidation of Pu(III), it is directly consumed to form the plutonium(IV) hydroxide, which accelerates the plutonium(III) oxidation. In this case, the diffusion of Pu(III) at the electrode is the limiting phenomenon of the Pu(III) oxidation (eqn (3)), and the decrease kinetics order is one. The decrease of the concentration of Pu(III) seems to be exponential. For (b), the oxidation of Pu(III) is more complicated because of the “brown” complex which takes part as an intermediate. In this case, the electrochemical formation of peroxo species during the electrochemical reduction of O2 at the electrode surface (eqn (1)) is zero order due to the apparent constant O2 concentration in solution (at the solubility limit under air atmosphere). Then the concentration of Pu(III) decrease linearly with the time like the formation of Pu(IV). It should be noted that the follow-up to this experiment concerns only 50% of the conversion of Pu(III) to Pu(IV).
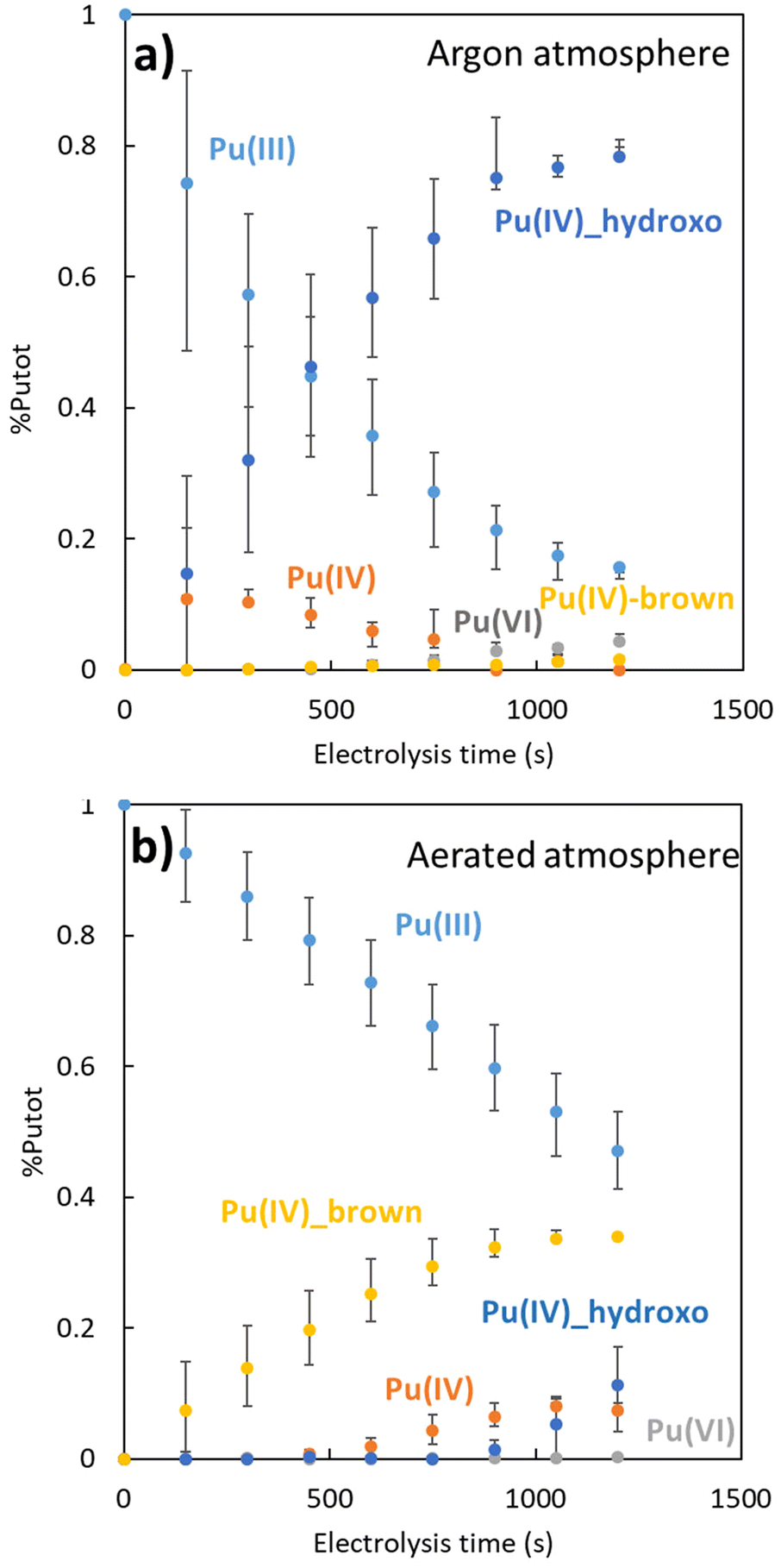 | ||
| Fig. 3 Speciation of the plutonium in function of the electrolysis time: (a) under argon atmosphere with initial 4.3 × 10−4 M Pu(III). (b) Under aerated atmosphere with initial 3.5 × 10−4 M Pu(III). | ||
The appearance of a peroxide complex of Pu(IV) in this experiment is unexpected, raising intriguing questions about its formation mechanism and stability in bulk solution. Under our experimental conditions, the potential formation of hydrogen peroxide through water α-radiolysis32–37 can be easily dismissed due to the low Pu concentration (4.3 × 10−4 M) and the low radioactivity of the used plutonium isotopic mixture. Indeed, with a time of 1200 s for each experiment and using the isotopic mixture of the plutonium used, we estimated an amount of approximatively 20 nM of H2O2 generated by alpha radiolysis. Alternatively, peroxide ions have been reported to possibly form in aerated solutions through a-two-electron Oxygen Reduction Reaction (ORR) at the catalytic Pt electrode surface (eqn (1)). This species is thought to react rapidly during electrolysis, either through reduction (E < 960 mV, eqn (2)) or back oxidation (E > 960 mV, eqn (1)).3–5 In addition, the maximum concentration of Pu observed for the “brown” complex is 1.75 × 10–4 M while the concentration of O2 which is dissolved in water is 2.5 × 10–4 M. As a reminder, the electrochemical cell is open, so the air is at a constant pressure during electrolysis (imposed by the air pressure in the glove box). This means that the quantity of oxygen is not limiting for the stability and the formation of the “brown” complex.
Since the “brown” complex (transient species formed under aerated conditions) forms independently of the applied electrolysis conditions (310 mV, 960 mV vs. SHE), we emphasize that hydrogen peroxide generated at the catalytic electrode interface rapidly reacts with Pu(IV) and becomes stabilized before undergoing further reduction (Fig. 4) or oxidation (eqn (1) and (2)). Fig. 4 illustrates the electrochemical reduction of a Pu(IV) solution under aerated conditions. The formation and stabilization of the “brown” peroxo complex are observed before its decomposition. This indicates that the electrochemical reduction of Pu(IV) also generates hydrogen peroxide. The formation of the “brown” peroxo complex of Pu(IV) enables to evidence H2O2 formation. Because of its total decomposition, due to the reduction of Pu(IV) into Pu(III), it is impossible, in our conditions, to know if H2O2 is still produced during electrolysis.
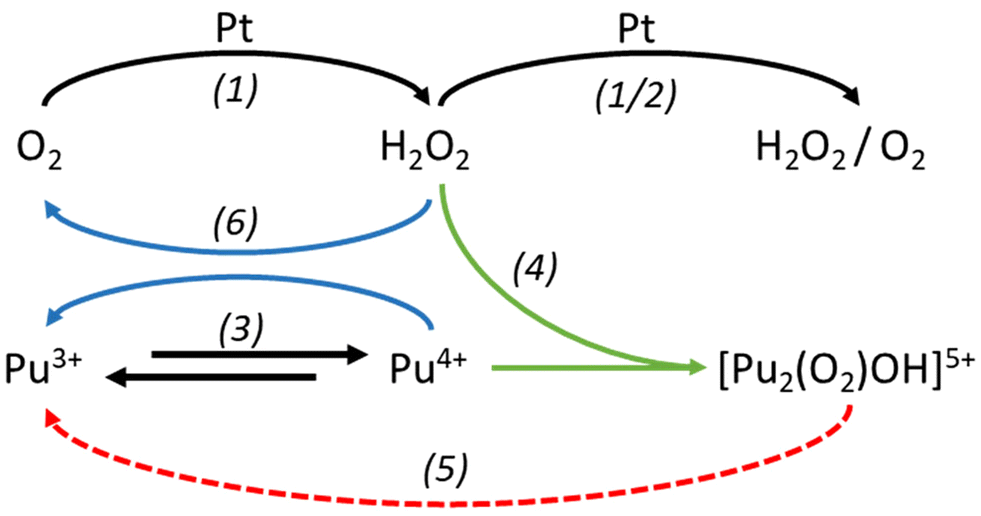 | ||
| Fig. 4 Possible reaction mechanism in Pu(IV)/Pu(III) electrolysis involving the formation of the Pu(IV) “brown” complex as an intermediate species. Black arrows indicate electro-oxidation and -reduction reactions at the Pt electrode surface, blue arrows are for redox reactions occurring in solution, green arrows represent complexation processes while the red one deals with decomposition of the “brown” complex. Numbers in brackets correspond to reaction equations. | ||
Since Ekstrom and McLaren8 determined an equilibrium constant of 9 × 106 for the formation of the Pu “brown” peroxo complex, which occur with rapid kinetics, we can infer that its formation may take place near the electrode surface under our experimental conditions in agreement with the reaction eqn (4). A combination of the reactions described in eqn (1)–(4) is likely to result in the formation of the Pu “brown” complex, as proposed in the scheme in Fig. 4, from both Pu(III) oxidation and Pu(IV) reduction experiments (Fig. 5).
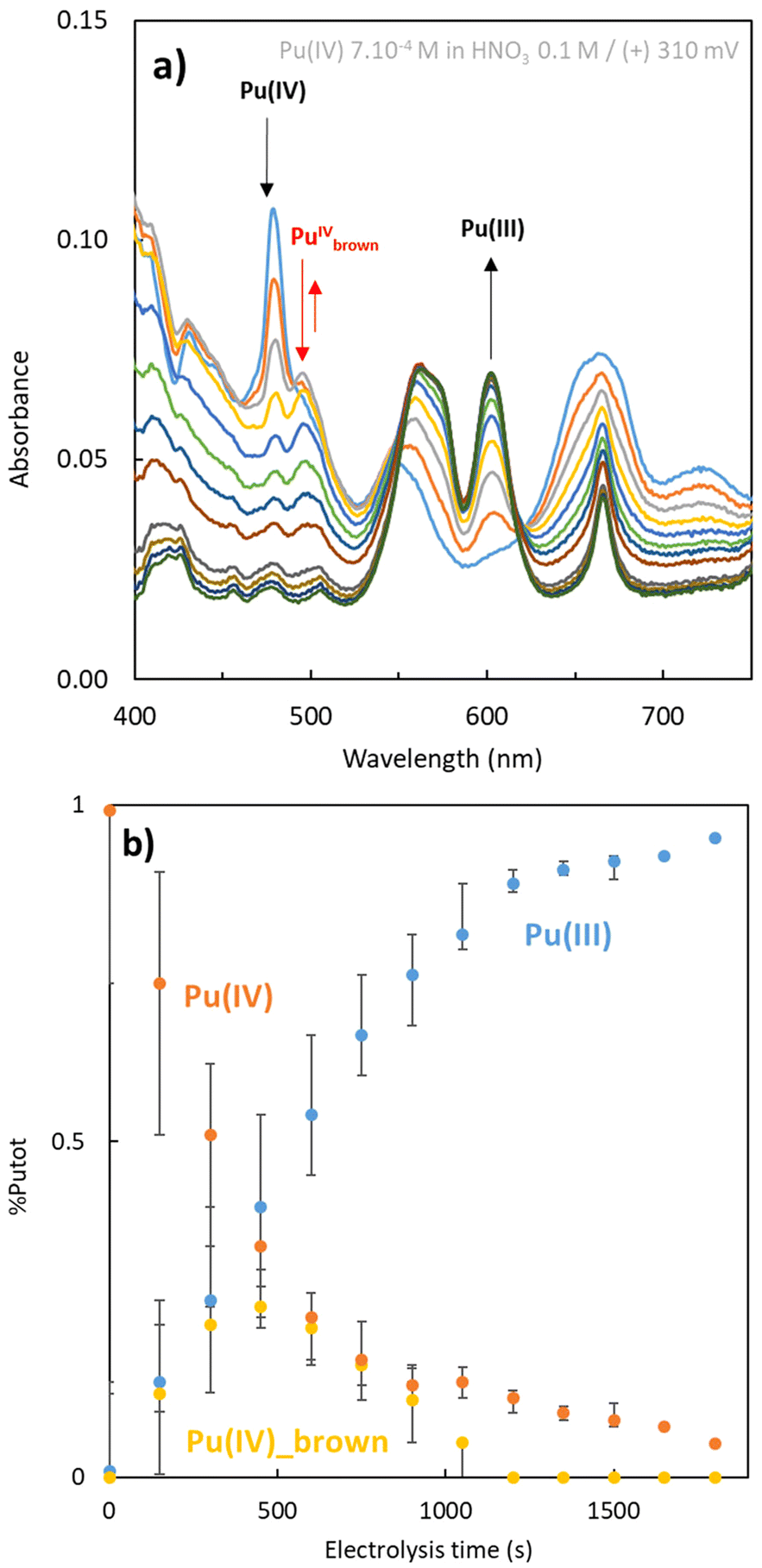 | ||
| Fig. 5 (a) Spectrophotometric measurements during electrolysis experiment: initial 7 × 10−4 M Pu(IV) (blue) electrolysis under aerated atmosphere at (+) 310 mV to Pu(III). (b) Speciation of the plutonium in function of the electrolysis time during reduction under aerated atmosphere. | ||
However, the formation of the peroxo complex may be limited by the molecular oxygen amount available in solution. This parameter was not controlled during our electrolysis experiments. Moreover, although both electrolysis experiments (i.e. oxidation and reduction) lead to the formation of the same Pu “brown” peroxo complex, it is produced at different rates depending on the applied potential (quantitative comparison from spectral deconvolution described in ESI.3†). Overall, while the high stability of the Pu(IV) peroxo complex is notable, its degradation and reactivity during electrolysis requires further investigations. One hypothesis involves its decomposition at the Pt electrode or its reaction with secondary transient oxygen species like OOH* (“*” is used to represent an unoccupied active site) which is the intermediate species generated during the reduction of O2.38 Its decomposition has also been reported to occur at low acidity, as shown in the reaction eqn (5), leading to the net reaction eqn (6).9
| 2 Pu4+ + H2O2 + H2O [Pu2(O2)OH]5+ + 3H+ | (4) |
| [Pu2(O2)OH]5+ + H+ 2 Pu3+ + O2 + H2O | (5) |
| 2 Pu4+ + H2O2 2 Pu3+ + O2 + 2H+ | (6) |
Conclusions
The observation of the peroxide “brown” complex in bulk solution contrasts with recent findings conducted under acidic conditions using conventional spectroscopic setups (e.g. short optical paths of 1 mm) to trace actinide redox reactivity on mesh electrode surfaces.23,24 Its formation within reasonable spectroscopic times in the bulk solution may be attributed to the moderate acidity and the improved spectral resolution provided by our 2 cm cell (setup details in ESI.1†). While establishing consistent bulk electrolysis conditions alongside high-quality spectroscopic measurements, this new approach highlights the importance of advanced redox-controlled in situ techniques. The observation of the “brown” peroxo complex during electrolysis under aerated conditions opens new avenues for the structural characterization of polynuclear Pu peroxide complexes. Additionally, the contribution of other polynuclear peroxo species such as the “red” or the “green” ones during electrolysis cannot be excluded during electrolysis. From an electrochemical perspective, Pu(IV) ions may play a crucial role in peroxide stability or at least, provide a reliable means for in situ detection, as their spectral signature emerges clearly distinguishes them from other components.Author contributions
Richard Husar – experimentation, analysis, electrochemical cell development, writing, original draft. Quentin Hervy – experimentation, analysis, writing, review & editing. Thomas Dumas – experimentation, analysis, investigation, electrochemical cell development, review & editing. Philippe Guilbaud – methodology, interpretation, validation, supervision, review & editing. Matthieu Virot – experimentation, interpretation, validation, review & editing. Philippe Moisy – methodology, interpretation, validation, supervision, review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support by FP7 Marie Sklodowska-Curie COFUND Program “Enhanced Eurotalents” (Nr.600382) and French Atomic Energy and Alternative Energies Commission (CEA ).References
- J. J. Katz, G. T. Seaborg and L. R. Morss, The chemistry of the Actinide Elements, Chapman and Hall, London, 1986 Search PubMed.
- M. Virot, T. Dumas, M. Cot-Auriol and P. Moisy, Synthesis and Multi-Scale Properties of PuO2 Nanoparticles: Recent Advances and Open Questions, Nanoscale Adv., 2022, 4, 4938–4971, 10.1039/d2na00306f.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. Ulrich Biedermann, A. A. Auer and K. J. J. Mayrhofer, Hydrogen Peroxide Electrochemistry on Platinum: Towards Understanding the Oxygen Reduction Reaction Mechanism, Phys. Chem. Chem. Phys., 2012, 14(20), 7384–7391, 10.1039/C2CP40616K.
- G. A. Attard, A. Brew, J.-Y. Ye, D. Morgan and S.-G. Sun, Oxygen Reduction Reaction Activity on Pt111 Surface Alloys, ChemPhysChem, 2014, 15(10), 2044–2051, DOI:10.1002/cphc.201402271.
- G. A. Attard and A. Brew, Cyclic Voltammetry and Oxygen Reduction Activity of the Pt{110}-(1×1) Surface, J. Electroanal. Chem., 2015, 747, 123–129, DOI:10.1016/j.jelechem.2015.04.017.
- J. Qiu and P. C. Burns, Clusters of Actinides with Oxide, Peroxide, or Hydroxide Bridges, Chem. Rev., 2013, 113(2), 1097–1120, DOI:10.1021/cr300159x.
- R. E. Connick and W. H. McVey, The Peroxy Complexes of Plutonium(IV), J. Am. Chem. Soc., 1949, 1534 CrossRef CAS.
- A. Ekstrom and A. McLaren, The Kinetic and Mechanism of the Formation of the Brown Complex of Plutonium(IV) and Hydrogen Peroxide, J. Inorg. Nucl. Chem., 1972, 1009–1016 CrossRef CAS.
- C. Maillard and J.-M. Adnet, Plutonium(IV) Peroxide Formation in Nitric Medium and Kinetics Pu(VI) Reduction by Hydrogen Peroxide, Radiochim. Acta, 2001, 89(8), 485–490, DOI:10.1524/ract.2001.89.8.485.
- W. Runde, L. F. Brodnax, G. S. Goff, S. M. Peper, F. L. Taw and B. L. Scott, Synthesis and Structural Characterization of a Molecular Plutonium(IV) Compound Constructed from Dimeric Building Blocks, Chem. Commun., 2007,(17), 1728–1729, 10.1039/B617878B.
- L. E. Sweet, J. F. Corbey, F. Gendron, J. Autschbach, B. K. McNamara, K. L. Ziegelgruber, L. M. Arrigo, S. M. Peper and J. M. Schwantes, Structure and Bonding Investigation of Plutonium Peroxocarbonate Complexes Using Cerium Surrogates and Electronic Structure Modeling, Inorg. Chem., 2017, 56(2), 791–801, DOI:10.1021/acs.inorgchem.6b02235.
- J. Margate, S. Bayle, T. Dumas, E. Dalodière, C. Tamain, D. Menut, P. Estevenon, P. Moisy, S. I. Nikitenko and M. Virot, Chronicles of Plutonium Peroxides: Spectroscopic Characterization of a New Peroxo Compound of Pu(Iv), Chem. Commun., 2024, 60(49), 6260–6263, 10.1039/D4CC01186D.
- D. Kirk Veirs, C. A. Smith, J. M. Berg, B. D. Zwick, S. Frederic Marsh, P. Allen and S. D. Conradson, Characterization of the Nitrate Complexes of Pu(IV) Using Absorption Spectroscopy, 15N NMR, and EXAFS, J. Alloys Compd., 1994, 213–214, 328–332, DOI:10.1016/0925-8388(94)90924-5.
- S. D. Reilly, W. Runde and M. P. Neu, Solubility of Plutonium(VI) Carbonate in Saline Solutions, Geochim. Cosmochim. Acta, 2007, 71(11), 2672–2679, DOI:10.1016/j.gca.2007.02.020.
- C. Walther, J. Rothe, B. Brendebach, M. Fuss, M. Altmaier, C. M. Marquardt, S. Büchner, H.-R. Cho, J.-I. Yun and A. Seibert, New Insights in the Formation Processes of Pu(IV) Colloids, Radiochim. Acta, 2009, 97(4–5), 199–207, DOI:10.1524/ract.2009.1595.
- S. I. Sinkov and E. I. Bozhenko, Complexation Behavior of Pu(IV) and Pu(VI) with Urea in Nitric Acid Solution, J. Alloys Compd., 1998, 271–273, 809–812, DOI:10.1016/S0925-8388(98)00223-0.
- M. A. Brown, A. Paulenova and A. V. Gelis, Aqueous Complexation of Thorium(IV), Uranium(IV), Neptunium(IV), Plutonium(III/IV), and Cerium(III/IV) with DTPA, Inorg. Chem., 2012, 51(14), 7741–7748, DOI:10.1021/ic300757k.
- D. Jebaraj Mahildoss and T. N. Ravi, Spectrophotometric Determination of Plutonium III, IV, and VI Concentrations in Nitric Acid Solution, J. Radioanal. Nucl. Chem., 2012, 294(1), 87–91, DOI:10.1007/s10967-012-1614-4.
- S. Georgette, S. Picart, C. Bouyer, J. Maurin, I. Bisel, S. Grandjean, J. Deseure and F. Lapicque, Study of the Plutonium(IV) Electrochemical Behavior in Nitric Acid at a Platinum Electrode. Application to the Cathodic Reduction of Pu(IV) in a Plate Electrolyzer, J. Electroanal. Chem., 2014, 727, 163–170, DOI:10.1016/j.jelechem.2014.06.015.
- A. Fallet, N. Larabi-Gruet, S. Jakab-Costenoble and P. Moisy, Electrochemical Behavior of Plutonium in Nitric Acid Media, J. Radioanal. Nucl. Chem., 2016, 308(2), 587–598, DOI:10.1007/s10967-015-4423-8.
- E. Acher, T. Dumas, C. Tamain, N. Boubals, P. Lorenzo Solari and D. Guillaumont, Inner to Outer-Sphere Coordination of Plutonium(IV) with N, N -Dialkyl Amide: Influence of Nitric Acid, Dalton Trans., 2017, 46(12), 3812–3815, 10.1039/C7DT00031F.
- E. Dalodière, M. Virot, V. Morosini, T. Chave, T. Dumas, C. Hennig, T. Wiss, O. Dieste Blanco, D. K. Shuh, T. Tyliszcak, L. Venault, P. Moisy and S. I. Nikitenko, Insights into the Sonochemical Synthesis and Properties of Salt-Free Intrinsic Plutonium Colloids, Sci. Rep., 2017, 7(1), 43514, DOI:10.1038/srep43514.
- A. M. Lines, S. R. Adami, A. J. Casella, S. I. Sinkov, G. J. Lumetta and S. A. Bryan, Electrochemistry and Spectroelectrochemistry of the Pu (III/IV) and (IV/VI) Couples in Nitric Acid Systems, Electroanalysis, 2017, 29(12), 2744–2751, DOI:10.1002/elan.201700465.
- A. M. Lines, S. R. Adami, S. I. Sinkov, G. J. Lumetta and S. A. Bryan, Multivariate Analysis for Quantification of Plutonium(IV) in Nitric Acid Based on Absorption Spectra, Anal. Chem., 2017, 89(17), 9354–9359, DOI:10.1021/acs.analchem.7b02161.
- R. Husar, T. Dumas, M. L. Schlegel and D. Schlegel, X-Ray Absorption Spectroscopy and Actinide Electrochemistry: A Setup Dedicated to Radioactive Samples Applied to Neptunium Chemistry, J. Synchrotron Radiat., 2022, 1–10 CrossRef CAS.
- C. Lucks, A. Rossberg, S. Tsushima, H. Foerstendorf, A. C. Scheinost and G. Bernhard, Aqueous Uranium(VI) Complexes with Acetic and Succinic Acid: Speciation and Structure Revisited, Inorg. Chem., 2012, 51(22), 12288–12300, DOI:10.1021/ic301565p.
- A. Roßberg, T. Reich and G. Bernhard, Complexation of Uranium(VI) with Protocatechuic Acid—Application of Iterative Transformation Factor Analysis to EXAFS Spectroscopy, Anal. Bioanal. Chem., 2003, 376(5), 631–638, DOI:10.1007/s00216-003-1963-5.
- S. Li, Editorial to the 3rd International Conference on “Asian Nuclear Prospects” (ANUP2012), Energy Procedia, 2013, 39, 1–2, DOI:10.1016/j.egypro.2013.07.185.
- I. Grenthe, A. V. Plyasunov and W. H. Runde, Second Update On The Chemical Thermodynamics Of Uranium, Neptunium, Plutonium, Americium And Technetium, OECD NEA, 2020 Search PubMed.
- E. Dalodière, M. Virot, T. Dumas and C. Berthon, Structural and Magnetic Susceptibility Characterization of Pu(V) Aqua Ion Using Sonochemistry as a Facile Synthesis Method, Inorg. Chem. Front., 2018, 100–111 RSC.
- M. Cot-Auriol, M. Virot, T. Dumas and O. Diat, First Observation of [Pu6(OH)4O4]12+ Cluster during the Hydrolytic Formation of PuO2 Nanoparticles Using H/D Kinetic Isotope Effect, R. Soc. Chem., 2022, 13147–13150, 10.1039/d2cc04990b.
- A. Damjanovic and V. Brusic, Electrode Kinetics of Oxygen Reduction on Oxide-Free Platinum Electrodes, Electrochim. Acta, 1967, 12(6), 615–628, DOI:10.1016/0013-4686(67)85030-8.
- A. Damjanovic, M. A. Genshaw and J. O. Bockris, The Role of Hydrogen Peroxide in the Reduction of Oxygen at Platinum Electrodes, J. Phys. Chem., 1966, 70(11), 3761–3762, DOI:10.1021/j100883a515.
- E. K. Dukes, Kinetics and Mechanisms for the Oxidation of Trivalent Plutonium by Nitrous Acid, J. Am. Chem. Soc., 1960, 82(1), 9–13, DOI:10.1021/ja01486a003.
- J. Fanning, The Chemical Reduction of Nitrate in Aqueous Solution, Coord. Chem. Rev., 2000, 199(1), 159–179, DOI:10.1016/S0010-8545(99)00143-5.
- G. R. Hall, D. Herniman and A. J. Walter, Spectrophotometric Studies of Pluronium in Nitric Acid Solution, 1951 Search PubMed.
- G. P. Horne, C. R. Gregson, H. E. Sims, R. M. Orr, R. J. Taylor and S. M. Pimblott, Plutonium and Americium Alpha Radiolysis of Nitric Acid Solutions, J. Phys. Chem. B, 2017, 121(4), 883–889, DOI:10.1021/acs.jpcb.6b12061.
- Y. Guo, X. Tong and N. Yang, Photocatalytic and Electrocatalytic Generation of Hydrogen Peroxide: Principles, Catalyst Design and Performance, Nano-Micro Lett., 2023, 15(1), 77, DOI:10.1007/s40820-023-01052-2.
Footnote |
| † Electronic supplementary information (ESI) available: Material, table data and deconvolution method. See DOI: https://doi.org/10.1039/d4dt02796e |
| This journal is © The Royal Society of Chemistry 2025 |