
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple synthesis of [Au(NHC)X] complexes utilizing aqueous ammonia: revisiting the weak base route mechanism†
Riku
Saito
 a,
Alberto
Prato
b,
Alessandro
Rubbi
b,
Laura
Orian
b,
Thomas
Scattolin
*b and
Steven P.
Nolan
*a
a,
Alberto
Prato
b,
Alessandro
Rubbi
b,
Laura
Orian
b,
Thomas
Scattolin
*b and
Steven P.
Nolan
*a
aDepartment of Chemistry and Center for Sustainable Chemistry, Ghent University, Krijgslaan 281 (S-3), 9000 Ghent, Belgium. E-mail: steven.nolan@ugent.be
bDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: thomas.scattolin@unipd.it
First published on 27th November 2024
Abstract
Aqueous ammonia has been examined as a new weak base for the synthesis of [Au(NHC)Cl] complexes, as well as for the activation of C–H, S–H, and N–H bonds. Its low cost and mild operational conditions (in air and using technical grade solvents) make it an attractive alternative for producing gold-NHC complexes. Synthetic pathways have been investigated in silico, assessing the role of the deprotonation and metalation steps within the reaction mechanisms.
N-heterocyclic carbenes (NHCs) have become widely used in organometallic catalysis and have been the subject of extensive research over the last few decades.1 Their widespread use as ligands in organometallic complexes is attributed to the remarkable stability of the bond between the carbenic carbon and the metal center. This stability results from their strong σ-donor ability towards transition metals, establishing them as privileged ligands in synthetic methodology.2
Among various metal-NHC complexes, those involving gold have attracted significant interest due to their effectiveness in catalyzing a wide range of organic reactions.3 These include processes such as the enyne cycloisomerization, the hydroamination of alkenes and alkynes, and the hydration of alkynes and nitriles, among numerous transformations.4 In addition to their catalytic capabilities, gold-NHC complexes have demonstrated potential in medicinal applications as anticancer agents5,6 and in materials applications in light of their luminescent properties.7,8 Among the various gold-NHC complexes, the [Au(NHC)Cl] family stands out due to its ability to be transformed into catalytic species in situ, usually facilitated by a chloride abstractor such as a silver(I) salt.9 These Au(I) complexes are particularly noteworthy because they can be used as pre-catalysts or, equally importantly, they serve as synthons10 to other gold-NHC species such as [Au(NHC)OH],11 [Au(NHC)(aryl)]12 and [Au(NHC)(carbazolyl)]13 complexes.
The appealing characteristics of [Au(NHC)Cl] complexes strongly motivate the development of easier, more accessible, and cost-effective synthetic methods. The traditional synthetic routes to [Au(NHC)Cl] complexes involve either the free carbene route or the transmetallation route using Ag- or Cu-NHC precursors14 (Scheme 1). While these methods are well-established and effective, they have notable limitations. First, both methods are two-step processes. Moreover, the free carbene method requires an inert atmosphere, anhydrous conditions, and the action of a strong base. The transmetallation method requires inert atmosphere, utilizes toxic solvents, and generates a significant amount of waste.1 In 2013, some of us3 and Gimeno15 independently demonstrated the use of a weak base for the synthesis of [Au(NHC)Cl] complexes (Scheme 1). This “weak base route” involves milder conditions, including operations under air, the use of a weak base such as K2CO3, and greener solvents such as acetone.16 This method not only facilitates the direct reaction between imidazolium salts and the gold(I) source but also enables the synthesis of the sterically more demanding [Au(IPr*)Cl] complex, which the classical transmetallation method could not produce.
 | ||
| Scheme 1 Synthetic routes to [Au(NHC)Cl] complexes. | ||
Since the development of the weak base synthetic methodology, exploring and expanding the weak base repertoire (such as K2CO3,3,15 NEt3,17 or NaOAc9) has been of interest. Testing several bases with varied basicities has helped to propose the mechanism and limitations associated with this synthetic route. For instance, DFT investigation of the [Au(NHC)Cl] synthesis using NMe3 showed that the pKb of weak bases is not significantly influential in reaching the final product.17 A concerted bond-making and bond-breaking process has been proposed, in which the steric hindrance of a weak base is crucial for the simultaneous deprotonation of the imidazolium precursor and the formation of [Au(NHC)Cl] complexes.9,17
In this context, in the present report, ammonia was examined as a potential new weak base. Ammonia, if the reaction proved successful, could be added to the repertoire of weak bases, and represents a more economical and greener alternative for the synthesis of [Au(NHC)Cl] complexes. It would be initially interesting to explore the limitation of the weak base synthetic method as ammonia is ca. 10 times a weaker base than K2CO3 (pKb water = 3.67 for K2CO3 and pKb water = 4.75 for NH3).18 Secondly, ammonia is produced in large quantities (approximately 170 million metric tons annually) and stands as the most affordable weak base (ca. US$ 300 per ton).19–21 Moreover, increasing initiatives are focused on creating “greener” ammonia.22 Given its substantial societal demand (for use as fertilizer, chemical, and energy storage agent) and the substantial energy consumption and carbon emissions involved in its production,23 the development of greener ammonia production methods, such as utilizing green hydrogen,19 electro-24 or photocatalytic25 nitrogen reduction, and biomimetic synthesis,26 has become a pressing priority. When ammonia is produced only using green energy (green ammonia), its carbon footprint becomes minimal.
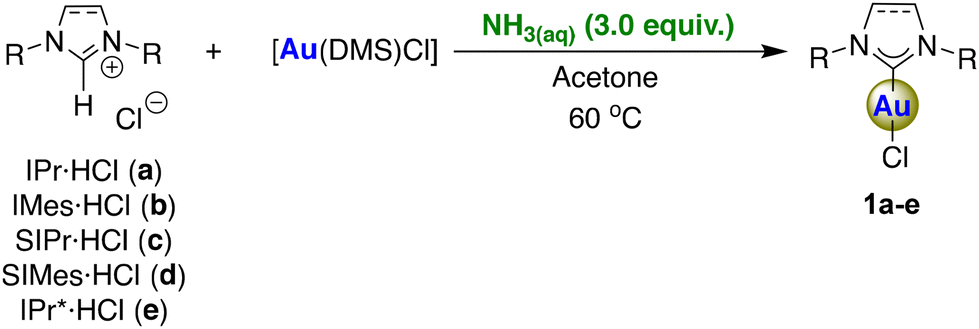
We have deployed aqueous ammonia as a cost-effective and environmentally friendly alternative base for use in the synthesis of [Au(NHC)Cl] complexes (Scheme 1). In initial experiments, aqueous ammonia was examined as the weak base for the synthesis of five different [Au(NHC)Cl] complexes including unsaturated and saturated azolium moieties with different bulkiness. The reactions between azolium salts and [Au(DMS)Cl] (DMS = dimethylsulfide) in the presence of aqueous ammonia (3.0 equiv.) in green acetone at 60 °C gave products 1a–e in comparable yields to the original weak base route using K2CO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 (Table 1). Notably, the complex containing the highly sterically hindered IPr* ligand (1e) was successfully isolated with an 84% yield, underscoring the broad applicability of this method. Notably, the workup consists of a simple extraction using water and an organic solvent (ethyl acetate or dichloromethane, we favour EtOAc in view of its more sustainable nature), followed by the removal of the latter under reduced pressure.
3 (Table 1). Notably, the complex containing the highly sterically hindered IPr* ligand (1e) was successfully isolated with an 84% yield, underscoring the broad applicability of this method. Notably, the workup consists of a simple extraction using water and an organic solvent (ethyl acetate or dichloromethane, we favour EtOAc in view of its more sustainable nature), followed by the removal of the latter under reduced pressure.
|
|
|||
|---|---|---|---|
| Entrya | NHC·HCl | Time [h] | Isolated yield [%] |
| a Reaction condition: NHC·HCl (100 mg), [Au(DMS)Cl] (1.0 equiv.), NH4OH aqueous solution (3.0 equiv. of NH3), 1.0 mL of acetone at 60 °C. b Large-scale reaction: 1 g of IPr·HCl, [Au(DMS)Cl] (1.0 equiv.), NH4OH aqueous solution (3.0 equiv. of NH3), 10 mL of acetone at 60 °C for 1 h. | |||
| 1 | IPr·HCl (a) | 1 | 95 (92)b |
| 2 | IMes·HCl (b) | 1 | 86 |
| 3 | SIPr·HCl (c) | 24 | 84 |
| 4 | SIMes·HCl (d) | 24 | 65 |
| 5 | IPr*·HCl (e) | 4 | 84 |
To test the scalability of this methodology, a larger-scale reaction using 1 g of IPr·HCl was performed. Gratifyingly, the reaction reached completion after 1 h without increasing the need to increase the number of equivalents of ammonia, affording the product in a 92% isolated yield.
Mechanistic studies were performed at the SMD-PBE0-D3(BJ)/6-31G(d,p)//PBE0-D3(BJ)/6-31G(d,p) level of theory to probe the mechanism at play in this specific conditions. Our computational investigation of the reaction of [IPr-H]+ with AuCl2− (which are experimentally observed following the removal of dimethylsulfide from the starting reagents) leads us to propose a deprotonation–metalation mechanism. In fact, even though imidazolium salts are weakly acidic, the deprotonation equilibrium with NH3 is even more unfavorable if a non-covalent adduct with AuCl2− is formed. Once the small fraction of carbene intermediate is formed, it reacts rapidly with AuCl2− with an energy barrier of 6.1 kcal mol−1. Overall, following the proton exchange, the transition state leading to metalation is destabilized by 28.3 kcal mol−1. The overall reaction energy of −5.3 kcal mol−1 makes the entire process exergonic and accounts for the good yield at higher temperatures (see Scheme 2A and ESI† for details).
 | ||
| Scheme 2 Revisitation of the weak base route mechanisms. | ||
Encouraged by these results, we examined the possibility of further functionalizing the [Au(IPr)Cl] complex by various bond activations assisted by aqueous ammonia. First, the reactivity of the acidic S–H bond (pKa water = 6.62 (ref. 27)) of thiophenol (PhSH) with 1a was assessed in the presence of aqueous ammonia because we hypothesized that based-assisted thiol deprotonation by ammonia would proceed smoothly due to the high acidity of S–H bond (Scheme 3). The desired thiolato complex [Au(IPr)(SPh)] (2a) was indeed formed under very mild condition (EtOH, RT, 1 h) in a high isolated yield (88%), showing the superiority of this methodology to the previously reported method involving a strong base (NaH).28 As expected, the computed reaction energy of the acid–base equilibrium between NH3 and PhSH is as low as 2.9 kcal mol−1. The ligand exchange of Cl− with thiophenolate (PhS−) yielding 2a is then energetically favored by 13.3 kcal mol−1. The coordination of PhS− and the elimination of chloride occur simultaneously with a slight energy barrier. The overall activation energy, with the inclusion of the initial proton exchange step, amounts to only 5.5 kcal mol−1, consistent with the efficacy of mild experimental conditions (see Scheme 2B and ESI† for details).
 | ||
| Scheme 3 Synthesis of [Au(IPr)(SPh)] (2a). | ||
The notable property of this class of compounds (NHC gold thiolato) is their anticancer activity and simple synthetic methodologies have been reported.29
To examine how general this NH3(aq) weak base can be deployed, we explored the reactivity of the C–H bond of phenylacetylene (pKa water = 23.2 (ref. 27)) with 1a (Scheme 4). The gold-alkynyl motif has received significant attention not only as a synthon29 but also due to its interesting photophysical properties.30 Historically, these complexes have been prepared by reactions using a strong base31 and NHC gold chloride complexes or using gold synthons such as gold hydroxide11 or gold aryl complexes,12 which are multistep reactions in non-green solvents such as toluene or benzene. We were pleased to observe that [Au(IPr)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)] (3a) can be isolated in a good yield through the reaction of [Au(IPr)Cl] with phenylacetylene in the presence of aqueous ammonia (6.0 equiv.) using EtOH as a green solvent (Scheme 4).
CPh)] (3a) can be isolated in a good yield through the reaction of [Au(IPr)Cl] with phenylacetylene in the presence of aqueous ammonia (6.0 equiv.) using EtOH as a green solvent (Scheme 4).
 | ||
| Scheme 4 Synthesis of [Au(IPr)(CCPh)] (3a). | ||
Differently from the other reactions, phenylacetylene is not involved in an acid–base equilibrium with NH3; however, gold complexes are able to bind alkynes and are employed as catalysts for the electrophilic activation of triple bonds.32
We have thus examined a stepwise mechanism for the reaction of phenylacetylene with 1a. From our calculations, it turns out that the formation of an intermediate species is very likely, as the tri-coordinated complex [AuCl(IPr)(η2-phenylacetylene)] lies at 6.0 kcal mol−1 in energy relative to the reactants. In this first step, the Cl atom is displaced from its position to accommodate the alkyne molecule, which binds to the metal centre. The elimination of a chloride ligand follows as a slightly endergonic step that leads to a charge separated species. At this point, the positively charged [Au(IPr)(η2-phenylacetylene)]+ complex can be deprotonated by NH3, forming 3a. While the initial coordination of 1a by phenylacetylene has an energy barrier of 10.4 kcal mol−1, the sequential elimination of Cl− and H+ are accompanied by a combined activation energy of 12.7 kcal mol−1, making it the rate-determining step along the reaction pathway. However, we are not able to exclude a priori the possibility that the elimination of Cl− and the deprotonation of the alkynyl ligand occur as a single concerted process (see Scheme 2C and ESI† for details).
In our final experiment, we have investigated the reactivity of the N–H bond of carbazole (CbzH) with compound 1a, using aqueous ammonia to potentially produce 4a (Scheme 5). Carbene–metal–amido (CMA) complexes containing coinage metals (Cu, Ag, Au) have recently gained considerable attention due to their intriguing photophysical properties.33,34 In fact, applications such as organic light-emitting diodes (OLEDs) have already been demonstrated using such complexes.35,36 We have been interested especially in gold NHC carbazolyl complexes and in exploring their use in various photocatalytic reactions.37–39 Consequently, we aimed to develop a cost-effective and more sustainable synthesis method leading to these complexes to facilitate further advancements and applications. To evaluate the reactivity of the N–H bond of carbazole, compound 1a was mixed with a slight excess of carbazole (1.05 equiv.) in EtOH and aqueous ammonia (10 equiv.) for 24 hours at 40 °C (Scheme 5). The reaction did proceed and permitted a 74% conversion (as gauged by NMR spectroscopy) but does not reach completion. Even after several attempts with different reaction conditions, the conversion could not be improved (see Table S1 in the ESI† for optimization details).
 | ||
| Scheme 5 Synthesis of [Au(IPr)(cbz)] (4a). | ||
Interestingly, although the acidity of carbazole is significantly higher than that of phenylacetylene (pKa DMSO = 19.9 (ref. 40) for carbazole and pKa DMSO = 28.7 for phenylacetylene41), this reaction did not reach completion. The reason behind this observed poorer reactivity is that, in the case of carbazole, the deprotonation precedes the reaction with 1a. Indeed, the computed reaction energy for the proton exchange between carbazole and NH3 is 21.4 kcal mol−1, whereas for the reactant complex [Au(IPr)Cl][CbzH] it is worth some 28.6 kcal mol−1. After the deprotonation step, the substitution of the chloride ligand with carbazolate proceeds rapidly to yield 4a, as the corresponding transition state is 1.5 kcal mol−1 lower in energy compared to the free reactants (see Scheme 2D and ESI† for details). The strongly unfavorable acid–base equilibrium, alongside the fact that the entire reaction is endergonic by 7.3 kcal mol−1, possibly explains why the conversion of 1a is incomplete.
In conclusion, a new weak base, aqueous ammonia, has been added to the repertoire of weak bases for the synthesis of [Au(NHC)Cl] complexes and represents a more cost-effective and greener carbon-free alternative. It provides mild reaction conditions and high yields for several key Au(NHC) complexes. Its use also demonstrated high efficiency for various bond activation reactions under mild conditions. We are currently conducting studies to expand this straightforward weak base protocol to a diverse range of related organometallic systems with this and related new weak bases.
The discussed reactions have relevant broad validity to inspire the scientific community engaged in the design and study of various applications of metal-NHC complexes. Indeed, the use of aqueous ammonia as a weak and inexpensive base can be extended to other metals of interest to both industry and academia. As a testimony to the versatility of the method, we report (see ESI†) the preparation of the highly sterically hindered copper complex [Cu(IPr*)Cl] (5a) using aqueous ammonia as weak base in an excellent 87% yield.
Notably, in some cases, such as in the synthesis of [RuCl2(p-cymene)(NHC)] or amino–cobalt–NHC complexes, the weak bases traditionally used in the literature (K2CO3 and NaOAc) do not allow for the direct formation of the desired products. These bases (in the form of carbonate or acetate ions) are sufficiently coordinating to replace chloride or amino ligands, in addition to promoting the formation of the metal–NHC bond. In the case of [RuCl2(p-cymene)(NHC)] complexes, this issue has been addressed by introducing an additional synthetic step in which a source of chloride ions (trimethylsilyl chloride) is added to the reaction mixture with the aim of isolating the target product.42 Conversely, the synthesis of metal-NHC complexes with early and middle transition elements (oxophilic metals) is still often carried out via the free carbene route.16 This approach is frequently imposed by the incompatibility of these metal centers with weak oxygen-based bases (K2CO3 and NaOAc), which can act as efficient chelating ligands and thus compete with the ligands in the coordination sphere of the metal precursors.
In addition to expanding the repertoire of weak bases that can be employed in the chemistry of metal–carbene complexes, this work successfully pursued a second key achievement: a revisitation of the intimate mechanism of the weak base route. Since this synthetic strategy is now used in more than half of the protocols for the preparation of late transition metal-NHC complexes,16 any advancement in knowledge in this area, particularly concerning the reaction mechanism, is of fundamental importance to fully exploit its potential and identify its intrinsic limitations.
Herein, we have proposed a refinement to the mechanism previously suggested in earlier contributions. The original mechanism involved the concerted deprotonation of the organic substrate (azolium salt, terminal alkyne, or a carbazole derivative) by the weak base, along with metallation of the same substrate at the metal center, with the loss of a chloride ligand.9,13,17
This mechanism is perfectly consistent with experimental data, where no intermediate species are observed (e.g. no free carbene species in the case of the formation of [Au(NHC)Cl] complexes). However, it is statistically very unlikely that three different species (substrate, base, and metal precursor) simultaneously participate in forming a transition state, evolving through the simultaneous breaking of two bonds and the formation of two new ones.
For this reason, in Scheme 2, we propose mechanisms that are still consistent with the experimental evidence but do not involve the interaction of more than two species in any single elementary step. Particularly important is the mechanism proposed here for the formation of [Au(NHC)Cl] complexes, which first involves the deprotonation of the azolium salt (the rate-determining step), followed by the metallation of the carbene species via reaction with the AuCl2− anion. It is noteworthy that this carbene species cannot exist in detectable concentrations, as it rapidly evolves into the final carbene complex.
We believe that the reaction mechanisms illustrated in Scheme 2 (green window) can be extended to other weak bases, such as those examined in previous contributions (e.g., K2CO3 and NaOAc), and thus can serve as reference mechanisms for these reactions moving forward.
Data availability
All data included and leading to conclusions presented in this manuscript are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The FWO is gratefully acknowledged for support of this work (G0A6823N). Umicore AG is thanked for their generous gifts of materials. All calculations were carried out on GALILEO100 @CINECA (Casalecchio di Reno, Italy), thanks to the ISCRA C SIM2 project (PI: Laura Orian).References
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS.
- A. Collado, A. Gómez-Suárez, A. R. Martin, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541 RSC.
- N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC.
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed.
- F. Guarra, A. Pratesi, C. Gabbiani and T. Biver, J. Inorg. Biochem., 2021, 217, 111355 CrossRef CAS PubMed.
- R. Visbal, I. Ospino, J. M. López-de-Luzuriaga, A. Laguna and M. C. Gimeno, J. Am. Chem. Soc., 2013, 135, 4712–4715 CrossRef CAS.
- A. Gómez-Suárez, D. J. Nelson, D. G. Thompson, D. B. Cordes, D. Graham, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2013, 9, 2216–2223 CrossRef PubMed.
- T. Scattolin, N. V. Tzouras, L. Falivene, L. Cavallo and S. P. Nolan, Dalton Trans., 2020, 49, 9694–9700 RSC.
- T. Scattolin, G. Tonon, E. Botter, S. G. Guillet, N. V. Tzouras and S. P. Nolan, Chem. – Eur. J., 2023, 29, e202301961 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- N. V. Tzouras, M. Saab, W. Janssens, T. Cauwenbergh, K. Van Hecke, F. Nahra and S. P. Nolan, Chem. – Eur. J., 2020, 26, 5541–5551 CrossRef CAS.
- N. V. Tzouras, E. A. Martynova, X. Ma, T. Scattolin, B. Hupp, H. Busen, M. Saab, Z. Zhang, L. Falivene, G. Pisanò, K. Van Hecke, L. Cavallo, C. S. J. Cazin, A. Steffen and S. P. Nolan, Chem. – Eur. J., 2021, 27, 11904–11911 CrossRef CAS.
- N-Heterocyclic Carbenes in Catalytic Organic Synthesis, ed. S. P. Nolan and C. S. J. Cazin, Thieme, Stuttgart, New York, 2017 Search PubMed.
- R. Visbal, A. Laguna and M. C. Gimeno, Chem. Commun., 2013, 49, 5642 RSC.
- T. Scattolin, A. A. Logvinov, N. V. Tzouras, C. S. J. Cazin and S. P. Nolan, Organometallics, 2023, 42, 2692–2730 CrossRef CAS.
- N. V. Tzouras, F. Nahra, L. Falivene, L. Cavallo, M. Saab, K. Van Hecke, A. Collado, C. J. Collett, A. D. Smith, C. S. J. Cazin and S. P. Nolan, Chem. – Eur. J., 2020, 26, 4515–4519 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Press, 97th edn, 2016 Search PubMed.
- H. S. Ahmed, Z. Yahya, W. Ali Khan and A. Faraz, Clean Energy, 2024, 8, 60–72 CrossRef.
- Green ammonia synthesis, Nat. Synth., 2023, 2, 581–582 Search PubMed.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- D. Ye and S. C. E. Tsang, Nat. Synth., 2023, 2, 612–623 CrossRef CAS.
- R. Manjunatha and A. Schechter, Electrochem. Commun., 2018, 90, 96–100 CrossRef CAS.
- A. Wu, J. Yang, B. Xu, X.-Y. Wu, Y. Wang, X. Lv, Y. Ma, A. Xu, J. Zheng, Q. Tan, Y. Peng, Z. Qi, H. Qi, J. Li, Y. Wang, J. Harding, X. Tu, A. Wang, J. Yan and X. Li, Appl. Catal., B, 2021, 299, 120667 CrossRef CAS.
- D. Yan, H. Li, C. Chen, Y. Zou and S. Wang, Small Methods, 2019, 3, 1800331 CrossRef.
- B. G. Cox, Acids and Bases: Solvent Effects on Acid-Base Strength, Oxford University Press, 2013 Search PubMed.
- V. Somasundaram, P. N. Gunawardene, A. M. Polgar, M. S. Workentin and J. F. Corrigan, Inorg. Chem., 2018, 57, 11184–11192 CrossRef CAS PubMed.
- M. Safir Filho, T. Scattolin, P. Dao, N. V. Tzouras, R. Benhida, M. Saab, K. Van Hecke, P. Lippmann, A. R. Martin, I. Ott and S. P. Nolan, New J. Chem., 2021, 45, 9995–10001 RSC.
- J. A. Garg, O. Blacque, J. Heier and K. Venkatesan, Eur. J. Inorg. Chem., 2012, 2012, 1750–1763 CrossRef CAS.
- L. Gao, D. V. Partyka, J. B. Updegraff, N. Deligonul and T. G. Gray, Eur. J. Inorg. Chem., 2009, 2009, 2711–2719 CrossRef.
- H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2–23 CrossRef CAS.
- G. U. Mahoro, J. Fernandez-Cestau, J. Renaud, P. B. Coto, R. D. Costa and S. Gaillard, Adv. Opt. Mater., 2020, 8, 2000260 CrossRef CAS.
- A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti and M. Bochmann, Chem. – Eur. J., 2017, 23, 4625–4637 CrossRef CAS PubMed.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Beliš, K. Van Hecke, C. S. J. Cazin and S. P. Nolan, Chem. Sci., 2022, 13, 6852–6857 RSC.
- Y. Zhao, V. A. Voloshkin, E. A. Martynova, B. Maity, L. Cavallo and S. P. Nolan, Chem. Commun., 2024, 60, 3174–3177 RSC.
- V. A. Voloshkin, M. Villa, E. A. Martynova, M. Beliš, K. Van Hecke, P. Ceroni and S. P. Nolan, Chem. Sci., 2024, 15, 4571–4580 RSC.
- F. G. Bordwell, G. E. Drucker and H. E. Fried, J. Org. Chem., 1981, 46, 632–635 CrossRef CAS.
- F. G. Bordwell, G. E. Drucker, N. H. Andersen and A. D. Denniston, J. Am. Chem. Soc., 1986, 108, 7310–7313 CrossRef CAS.
- X. Ma, S. G. Guillet, M. Peng, K. Van Hecke and S. P. Nolan, Dalton Trans., 2021, 50, 3959–3965 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02955k |
| This journal is © The Royal Society of Chemistry 2025 |