
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Necrosis inducing tetranuclear Ru(II)–Re(I) metal complex for anticancer therapy†
Julia
Schleisiek‡
,
Eleni
Michaltsis‡
,
Stephan
Mayer
,
Nicolás
Montesdeoca
and
Johannes
Karges
 *
*
Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany. E-mail: johannes.karges@ruhr-uni-bochum.de; Tel: +49 2343224187 Web: https://www.kargesgroup.ruhr-uni-bochum.de
First published on 18th December 2024
Abstract
Chemotherapy is one of the most widely used anticancer treatments worldwide. However, despite its clinical effectiveness, most chemotherapeutic agents are associated with severe side effects. To address this limitation, there is an urgent need for the development of novel anticancer agents. Among the promising alternatives, Ruthenium and Rhenium complexes have garnered significant attention in the scientific literature. This study proposes combining these two metal moieties into a single tetranuclear complex, bridged by a 2,2′-bipyrimidine ligand. Cytotoxicity tests revealed broad activity of the novel metal complex against multiple cancer cell lines. Mechanistic studies suggested that the complex induces cell death by necrosis. Further analyses demonstrated its ability to eradicate colon carcinoma tumor spheroids at micromolar concentrations. To the best of our knowledge, this represents the first example of a Ru(II)–Re(I) tetranuclear metal complex as an anticancer agent.
Introduction
In recent decades, the search for new chemotherapeutic agents to combat cancer has been a significant challenge for scientists around the world. Chemotherapy, in combination with surgery, radiotherapy, and immunotherapy, forms part of a multimodal approach to cancer treatment.1 This combination aims to minimize the limitations of individual therapies and provide patients with the best possible chances of survival. Platinum complexes (i.e. cisplatin, oxaliplatin, carboplatin) are some of the most widely used chemotherapeutic drugs worldwide.2–4 Despite their clinical success, these compounds are associated with severe side effects (i.e., kidney damage, nerve damage, bone marrow suppression, drug resistance), limiting their application in the clinics.5–10 To overcome these drawbacks, there is an urgent need for novel chemotherapeutic agents. Among the most promising class of compounds, transition metal complexes are currently extensively studied.Coordinatively saturated and substitutionally inert Ru(II) complexes, first explored by Dwyer Tan et al. in the 1950s, were initially studied for their interactions with enzymes, bactericidal, and bacteriostatic properties. Early findings revealed that these complexes exhibited some bacteriostatic effects but were toxic to mice, likely due to inhibiting of the acetylcholinesterase enzyme, which plays a key role in neurotransmitter metabolism.11 However, due to the severe side effects, including neurotoxicity, further biological research on these complexes was largely abandoned for over 50 years. Interest in the potential anticancer properties of Ru(II) polypyridine complexes resurfaced in 2000 when Liu and Ji et al. demonstrated the cytotoxicity of certain Ru(II) polypyridine complexes was dependent on various factors such the ligand environment, the oxidation state of the metal, or the stability of the compound. Some metal complexes, although these compounds bound strongly to DNA, did not exhibit a cytotoxicity effect.12 Since then, numerous Ru(II) and Ru(III) complexes have been investigated for anticancer properties.13–24 Notably, Ru(III) complexes BOLD-100 (also known as IT-139 or NKP-1339) and NAMI-A are undergoing clinical trials as chemotherapeutic agents,25–27 while the Ru(II) complex TLD-1433 is being studied as a photosensitizer for photodynamic therapy.28,29
Recent studies have highlighted the potential of Re(I) tricarbonyl complexes as anticancer agents.30–33 Mao and Tan et al. demonstrated that these complexes can cause irreversible oxidative stress and disrupt glutathione metabolism by accumulating in the mitochondria.34 Simpson and Massi et al. designed Re(I) complexes capable of inducing cell death by inhibiting Aurora-A kinase phosphorylation.35 Visser, Gasser, and Schutte-Smith et al. reported on Re(I) 1,10-phenanthroline tricarbonyl complexes with low nanomolar cytotoxic activity toward prostate cancer cells.36 Wilson et al. reported a Re(I) isonitrile complex that triggered apoptosis through the unfolded protein response and showed anticancer activity in cancer cells and tumor-bearing mouse models.37,38 In further studies, the authors identified NUBP2, a scaffold protein involved in Fe–S cluster biogenesis, as a relevant target of the Re(I) tricarbonyl complex.39 Besides the application of Re(I) tricarbonyl complexes as cytotoxic agents, research efforts by Mascharak, Paira, Zhang, and Zobi have also been devoted towards their application as carbon monoxide releasing compounds or photosensitizers.40–43
Over the last years, only a few examples of Ru(II)–Re(I) dinuclear metal complexes have been reported for (light-activated) anticancer therapy. Bernardino de la Serna and Thomas et al. reported on the coordinative linkage of a Ru(II) polypyridine complex to a Re(I) tricarbonyl complex. The conjugate was found to selectively accumulate in the mitochondria and induce cell death in ovarian cancer cells upon irradiation for application in photodynamic therapy.44,45 Cariou and Gasser et al. demonstrated that the Re(I) core could be released from a dinuclear metal complex upon irradiation with light.46
Based on previous promising results from Ru(II) polypyridine and Re(I) tricarbonyl complexes as chemotherapeutic agents, herein, the combination of both classes of compounds into a single tetranuclear metal complex is proposed. The respective metal cores were bridged over a 2,2′-bipyrimidine ligand. The novel tetranuclear metal complex was found to be stable in phosphate-buffered saline and to be of lipophilic nature. Cytotoxicity studies revealed that the compound was broadly active against a variety of cancer cell lines at micromolar concentrations. Deeper analyses into the cell death mechanism suggested that the metal complex induced cell death by necrosis. The compound was able to eradicate colon carcinoma cancer multicellular tumor spheroids at micromolar concentrations. To the best of our knowledge, this study reports on the first example of a tetranuclear Ru(II)–Re(I) metal complex as an anticancer agent.
Results and discussion
Chemical synthesis and characterization
Ru(III)-chloride was reduced to Ru(II) using ethanol as solvent and reducing agent. The coordination sphere was saturated with dimethyl sulfoxide ligands, forming the metal precursor Ru(dimethyl sulfoxide)4(Cl)2. The resulting precursor and three equivalents of the bidentate ligand 2,2′-bipyrimidine were then dissolved in dimethylformamide and heated at reflux. The reaction solution yielded a mixture of different types of bis- or tris-coordinated Ru(II) polypyridine complexes as well as coordination polymers. Despite extensive efforts (i.e. recrystallization, column chromatography, reverse phase high-pressure column chromatography) in the isolation of the desired tris-coordinated species [Ru(2,2′-bipyrimidine)3][Cl]2 (Ru), this remained impossible in our hands. To reduce the number of possible products, the reaction was repeated with an excess of the ligand 2,2′-bipyrimidine and therefore ideally the rapid saturation of the coordination sphere, preventing the formation of coordination polymers. While this strategy resulted in a reduced number of side products, it was not possible in our hands to isolate the desired product purely. To overcome this limitation, the metal precursor [Ru(dimethyl sulfoxide)4(Cl)2] and silver(I) trifluoromethanesulfonate were dissolved in ethanol and heated at reflux for 3 h. The generated poorly soluble silver(I) chloride was removed by filtration and the solution of the Ru precursor was placed in a glass syringe. A solution of an excess of the ligand 2,2′-bipyrimidine in ethanol was separately prepared and heated at reflux. The Ru precursor solution was slowly added over a period of 20 h to the ligand solution using a syringe pump. After this time, sodium tetraphenylborate was added to the solution and the desired product Ru was isolated as a tetraphenylborate salt. Using the ion exchange resin Amberlite IR-410, the compound was converted into the chloride salt. Following this strategy, Ru was isolated in nearly quantitative yields. The generated Ru(II) polypyridine complex Ru was further reacted with the Re(I) precursor Re(CO)5Cl upon heating at reflux to form the tetranuclear Ru(II)–Re(I) metal complex (RuRe). While the preparation of the desired compound was verified by mass spectrometry, despite extensive efforts (i.e. recrystallization, column chromatography, reverse phase high-pressure column chromatography) it was not possible in our hands to isolate the desired product purely. Capitalizing on this, the synthetic strategy was changed and the bidentate ligand first coordinated to the Re(I) precursor. Re(CO)5Cl and 2,2′-bipyrimidine were suspended in equimolar amounts in toluene and heated at reflux for 4 h. The product [Re(2,2′-bipyrimidine)(CO)3(Cl)] (Re) was isolated in nearly quantitative yields by recrystallization. Analogous to the synthesis of Ru, [Ru(dimethyl sulfoxide)4(Cl)2] was mixed with silver(I) trifluoromethanesulfonate, the generated silver(I) chloride was removed by filtration, and the solution of the metal precursor was slowly added over a period of 20 h to a solution of Re using a syringe pump. Using column chromatography and counter ion exchange, the final product RuRe was obtained in high yields (Fig. 1). Using high-pressure liquid chromatography mass spectrometry the identity of the tetranuclear species was confirmed (Fig. S9†). All compounds were characterized by nuclear mass spectroscopy and mass spectrometry. The purity of the final compounds was further verified by elemental analysis and high-pressure liquid chromatography analysis (Fig. S1–S9†).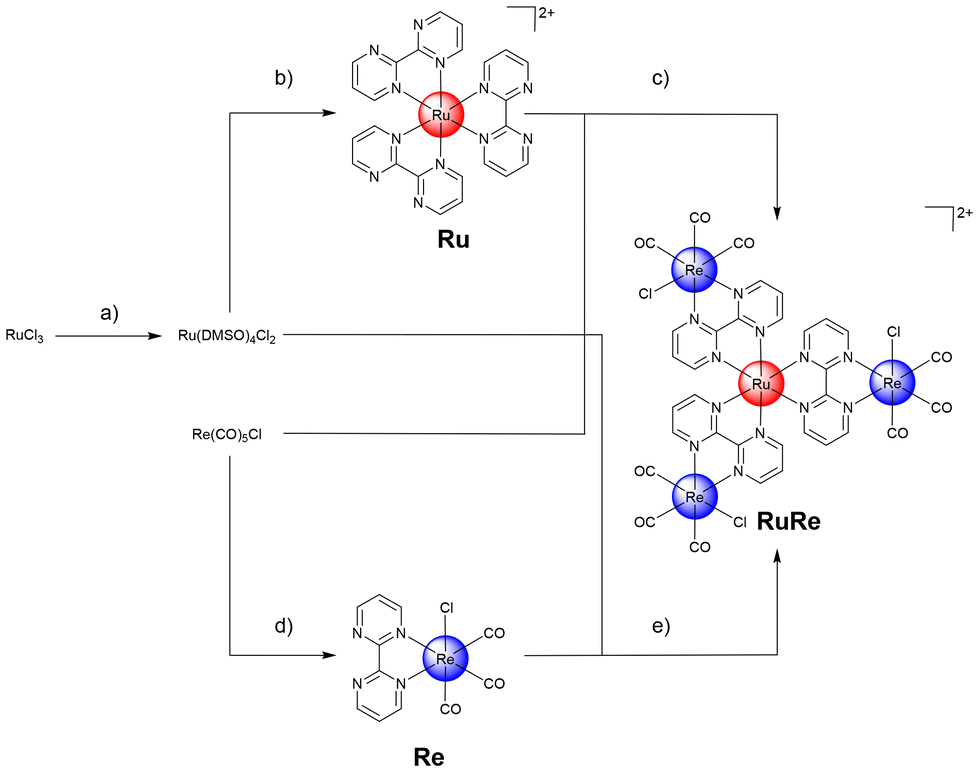 | ||
| Fig. 1 Schematic illustration of the strategy for the synthesis of the herein studied metal complexes. The complexes were isolated as chloride salts. (a) (1) Ethanol, reflux, 3 h; (2) dimethyl sulfoxide, 150 °C, 2 h; (b) (1) silver(I) trifluoromethanesulfonate ethanol, reflux, 3 h; (2) 2,2′-bipyrimidine, ethanol, reflux, syringe pump, 20 h; (c) ipropanol/octcanol, reflux, 5 h; (d) 2,2′-bipyrimidine, toluene, reflux, 4 h; (e) (1) silver(I) trifluoromethanesulfonate ethanol, reflux, 3 h; (2) [Re(2,2′-bipyrimidine)(CO)3(Cl)], isopropanol/octanol, reflux, syringe pump, 20 h. | ||
Physical properties
Based on the rich photophysical properties of Ru(II) polypyridine and Re(I) tricarbonyl complexes, the absorption and emission properties of the tetranuclear metal complex were investigated. The absorption spectrum of RuRe in water showed a strong absorption peak centered at approximately 250 nm and 350 nm with an absorption tail reaching into the red and infrared region (Fig. S10†). The ability of the tetranuclear metal complex to emit light in water upon excitation at 350 nm was studied. RuRe was found to be weakly emissive with an emission maximum at 622 nm (Fig. S10†). Upon comparison of the emission signal with the reference [Ru(2,2′-bipyridine)3][Cl]2, the emission quantum yield was determined. RuRe demonstrated to be more than an order of magnitude less emissive than [Ru(2,2′-bipyridine)3][Cl]2 with an emission quantum yield of approximately 0.2%. Contrary, Ru was found to be much stronger emissive with an emission quantum yield of approximately 2.9%. These findings indicate that the addition of the Re(I) moiety causes the quenching of the photophyiscal properties. The catalytic conversion of molecular oxygen into singlet oxygen upon excitation with light was assessed using the singlet oxygen probe 1,3-diphenylisobenzofuran as well as singlet oxygen sensor green. No significant amount of singlet oxygen production for RuRe was observed upon exposure to light.The stability of a compound under physiological conditions is essential for its biological application, ensuring a safe and effective therapeutic outcome, as unintended degradation could result in reduced efficacy and potential side effects.47 To assess the stability of the metal complex under these conditions, RuRe was incubated in phosphate-buffered saline (Fig. S11†) or DMEM cell media (Fig. S12†) for 48 h and the absorption profile monitored at various time points. No significant changes were detected in the absorption spectrum, indicating that the metal complex RuRe remained stable within an aqueous solution. Previous studies have shown that Ru(II) polypyridine complexes could undergo chemical transformations upon irradiation, causing the decomposition of the compound. To assess the stability of the metal complex upon light exposure, RuRe was incubated in phosphate-buffered saline and constantly irradiated for up to 30 min. Since no significant changes in the absorption profile (Fig. S13†) were observed, the photostability is indicated.
The lipophilicity of the metal complexes was assessed by determining their distribution coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) between the phosphate-buffered saline and octanol phases using the ‘shake-flask’ method. While Ru (logP = −1.5 ± 0.3) and Re (logP = −0.3 ± 0.2) were predominantly present in the aqueous phase, RuRe was found in the organic phase (logP = +1.3 ± 0.2), indicating its high lipophilicity.
P) between the phosphate-buffered saline and octanol phases using the ‘shake-flask’ method. While Ru (logP = −1.5 ± 0.3) and Re (logP = −0.3 ± 0.2) were predominantly present in the aqueous phase, RuRe was found in the organic phase (logP = +1.3 ± 0.2), indicating its high lipophilicity.
Cell membrane permeability was evaluated using a parallel artificial membrane permeability assay. Ru exhibited poor cellular permeability (0.007 ± 0.004 μm s−1), while Re (0.021 ± 0.006 μm s−1) and RuRe (0.018 ± 0.004 μm s−1) demonstrated moderate artificial cellular penetration rates.
Biological properties in monolayer cells
The cytotoxicity of the metal complexes Ru, Re, and RuRe in comparison to the clinically approved chemotherapeutic drug cisplatin in cancerous murine mouse colon carcinoma (CT-26), human breast adenocarcinoma (MCF-7), human pancreas adenocarcinoma (PT-45), and non-cancerous human fibroblasts (GM-5657) cells was investigated. The cells were incubated with the compounds for 48 h and the cell viability was assessed using the dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). The Ru(II) polypyridine complex Ru was found to be non-toxic in all investigated cell lines (IC50 > 100 μM). The Re(I) tricarbonyl complex Re showed a weak cytotoxic effect in CT-26 (IC50 = 71.7 ± 6.3 μM) and MCF-7 (IC50 = 54.1 ± 4.6 μM) cells and no therapeutic effect in PT-45 or GM-5657 cells (IC50 > 100 μM). In contrast, the tetranuclear metal complex RuRe demonstarted to be broadly active in the micromolar range in all cell lines. The compound was not able to differentiate between cancerous and non-cancerous cells. The therapeutic effect of RuRe was found to be in the same order of magnitude as cisplatin (Table 1). Based on the strongest cytotoxic effect, the following experiments were performed in CT-26 cells.| CT-26 | MCF-7 | PT-45 | GM-5657 | |
|---|---|---|---|---|
| Ru | >100 | >100 | >100 | >100 |
| Re | 71.7 ± 6.3 | 54.1 ± 4.6 | >100 | >100 |
| RuRe | 11.5 ± 1.8 | 12.3 ± 2.3 | 22.1 ± 3.3 | 17.4 ± 2.5 |
| Cisplatin | 18.1 ± 2.4 | 21.2 ± 1.8 | 24.7 ± 3.4 | 31.2 ± 2.7 |
The cell death mechanism induced by the metal complex was investigated. The cancer cells were preincubated with the specific autophagy (3-methyladenine), apoptosis (Z-VAD-FMK), paraptosis (cycloheximide), or necrosis (necrostatin-1) inhibitors, the cells were then treated with the IC50 value of RuRe (11.5 μM) for 48 h, and cell viability was measured using a MTT assay as previously reported.48 Preincubation with autophagy, apoptosis, and paraptosis, inhibitors did not significantly affect the cell survival, ruling out these pathways. Contrary, the preincubation with the necrosis inhibitor substantially increased the cell viability (Fig. 2), indicative that RuRe induced cell death by necrosis.
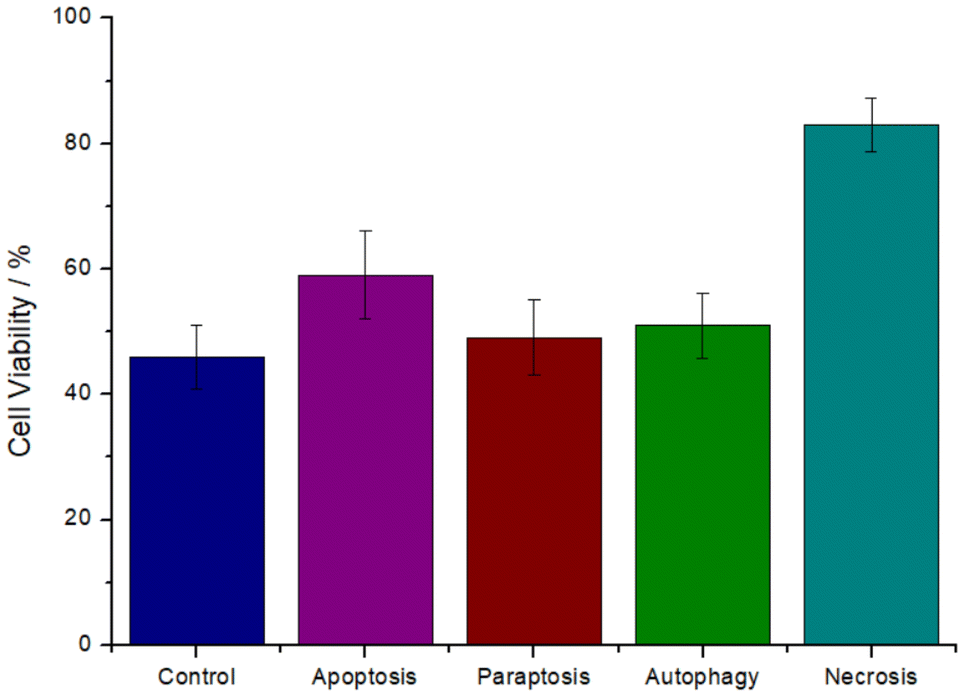 | ||
| Fig. 2 Changes in cell viability of CT-26 cells upon pre-incubation with autophagy (100 μM, 3-methyladenine), apoptosis (20 μM, Z-VAD-FMK), paraptosis (0.1 μM, cycloheximide), or necrosis (60 μM, necrostatin-1) inhibitors, that are suppressing the ability of the metal complex to induce the specific cell death mechanism, treated with the IC50 value of RuRe (11.5 μM). | ||
Previous studies have shown the essential role of receptor-interacting kinase 3 (RIP3) as a key regulator for necrosis. Capitalizing on this, the expression level of this protein within the cancerous cells upon treatment with RuRe was studied by Western Blot analysis. While barely any expression of RIP3 was observed in the untreated cells, a strong expression of RIP3 was monitored upon treatment with RuRe (Fig. 3). These findings confirm that RuRe induces cell death of the cancerous cells by necrosis.
 | ||
| Fig. 3 Western blot analysis of the enzymes receptor-interacting kinase 3 (RIP3) and glycerinaldehyd-3-phosphat-dehydrogenase (GADPH) as a reference in CT-26 cells upon treatment with RuRe. Full blot: Fig. S14.† | ||
Biological properties in multicellular tumor spheroids
Following the evaluation of the cytotoxicity in two-dimensional cancer cells, the therapeutic effects were further explored using three-dimensional multicellular tumor spheroids. Tumor spheroids serve as a tissue culture model that mimics the histological and pathological characteristics of solid tumors, including proliferation and nutrient gradients, as well as a hypoxic core. Many anticancer agents that show promise in two-dimensional cancer cell studies often fail to translate into successful animal model treatments, partly due to poor drug penetration into the tissue. Multicellular tumor spheroids can simulate the three-dimensional architecture of solid tumors. In this study, CT-26 multicellular tumor spheroids with an average diameter of approximately 600 μm were formed and cultivated. The therapeutic effect of the tetranuclear metal complex RuRe in comparison to cisplatin was studied using a luciferase-based luminescence assay to determine adenosine triphosphate levels of living cells. The results revealed that RuRe exhibited significantly higher cytotoxicity (IC50 = 43.8 ± 6.2 μM) than cisplatin (IC50 > 100 μM), which caused only a slight cytotoxic effect (Fig. 4).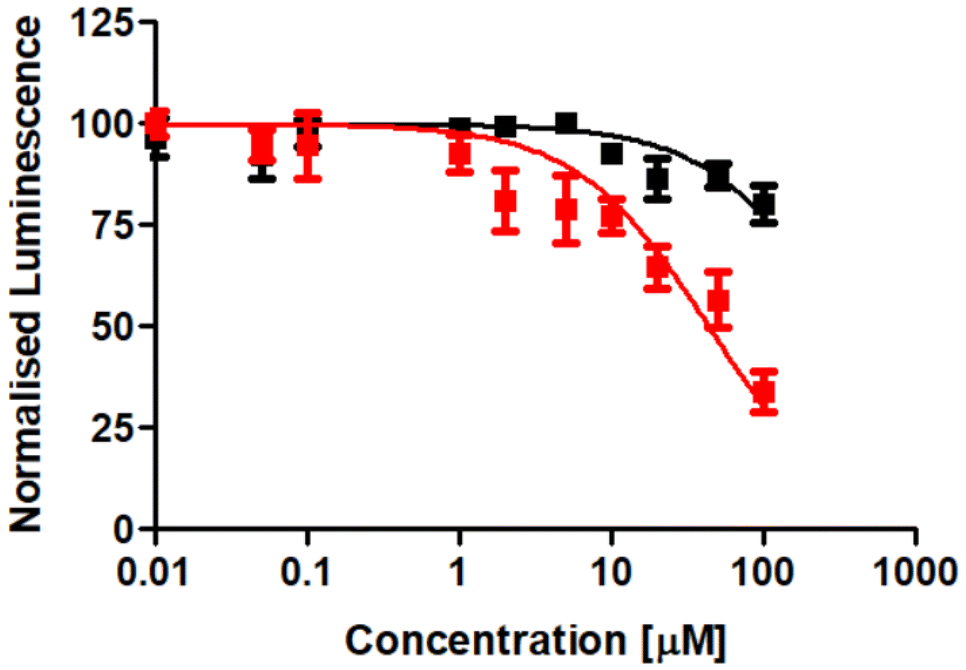 | ||
| Fig. 4 Drug response curves of cisplatin (black) and RuRe (red) upon treatment of CT-26 multicellular tumor spheroids with an approximate diameter of 600 μm. | ||
To gain further insight into the therapeutic effects, the size and morphology of the treated spheroids were examined using light microscopy. Cisplatin treatment had no observable effect on the size or morphology of the spheroids. In contrast, treatment with RuRe resulted in a marked reduction in tumor size and slight morphological damage (Fig. 5). These findings collectively demonstrate the ability of the tetranuclear metal complex to eradicate multicellular tumor spheroids.
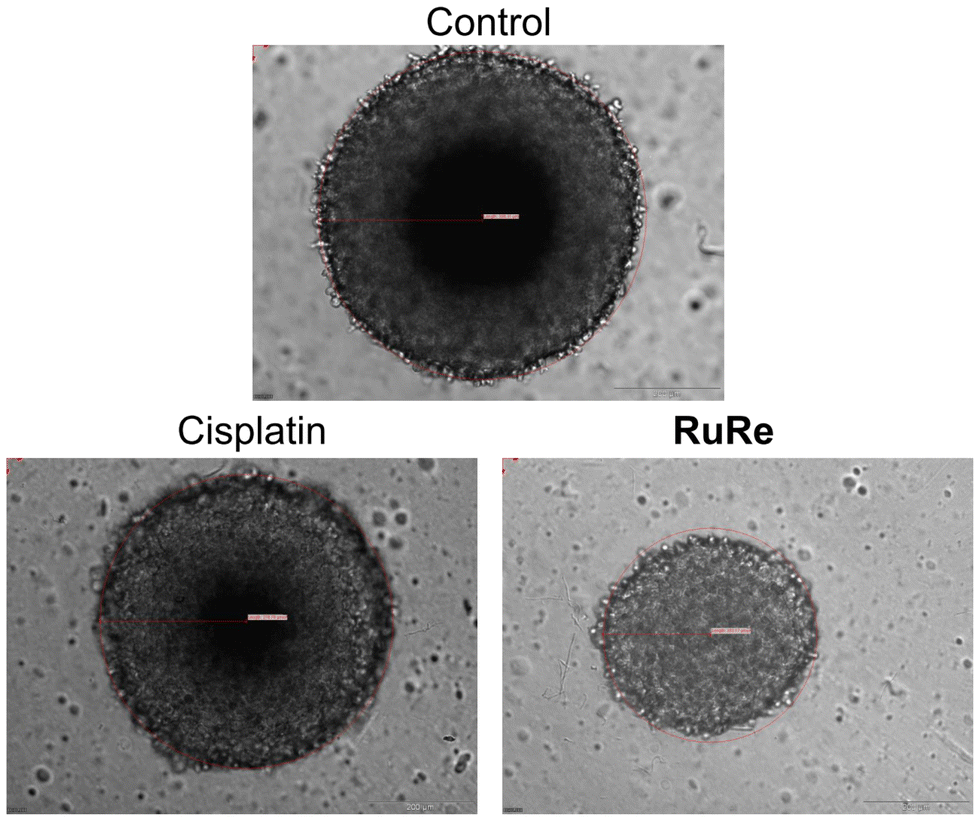 | ||
| Fig. 5 Light microscopy images of CT-26 multicellular tumor spheroids with an approximate diameter of 600 μm upon treatment with cisplatin or RuRe (100 μM). | ||
Conclusions
In summary, the chemical synthesis and biological evaluation of a tetranuclear Ru(II)–Re(I) metal complex for potential application in anticancer therapy is reported. Building on the previously reported promising outcomes of Ru(II) polypyridine and Re(I) tricarbonyl complexes as chemotherapeutic agents, this study aims to combine both classes into a single tetranuclear metal complex. The two metal centers are linked by a 2,2′-bipyrimidine ligand. Due to the synthetic challenging purification of the desired metal complex, the synthetic strategy was optimized. The bidentate ligand was first coordinated to Re(I) pentacarbonyl chloride and the monomeric species was isolated. Upon dropwise addition of a Re(II) precursor to the Re(I) complex over a period of 20 h, the desired tetranuclear complex was isolated in high yields by column chromatography and changes in solubility through counter ion exchange. The novel tetranuclear metal complex demonstrated stability in phosphate-buffered saline and displayed lipophilic characteristics. Cytotoxicity studies revealed broad-spectrum activity against several cancer cell lines at micromolar concentrations. Further analysis indicated that the complex primarily triggered cell death through necrosis. Further analyses demonstrated its ability to eradicate colon carcinoma tumor spheroids at micromolar concentrations. We are confident that this study could open new avenues toward the development of multinuclear metal complexes as anticancer agents.Experimental section
Materials and instruments
All chemicals were obtained from commercial sources and used without further purification. All syntheses were performed under an argon atmosphere in dried Schlenk flasks using dry solvents. Light-sensitive reactions were conducted in the absence of light by wrapping the apparatus in aluminum foil. Thin-layer chromatography was carried out using Merck TLC aluminum sheets. Column chromatography was performed with silica gel 60 M (0.04–0.063 mm, Macherey-Nagel) at atmospheric pressure. Thin-layer chromatography analysis, including substance detection, was performed using fluorescence detection under UV light (λ = 254 nm). 1H-NMR and 13C-NMR spectra were recorded using a Bruker Avance Neo 400 MHz at 298.15 K. Chemical shifts are reported in parts per million (ppm) relative to the 1H signals of the deuterated solvents.Synthesis of [Ru(dimethyl sulfoxide)4(Cl)2]
Ruthenium(III)-chloride hydrate (1 g, 3.82 mmol assuming that xH2O = 3) was partially dissolved ethanol (25 mL) in a round bottom flash equipped with an condenser. The mixture was stirred and heated to reflux for 3 h in which the compound dissolved completely and the colour changer from brown to deep green. After that the ethanol was removed with the rotary evaporator and the green oily residue that was left was dissolved in dimethyl sulfoxide (4 mL). The flask which is equipped with a condenser was placed in an oil bath heated at 150 °C and magnetically stirred for 2 h. Within the first minutes, the colour of the solution changed from dark green to bright orange and after 1 h a yellow solid was observed. To complete the precipitation, acetone (40 mL) was added and the flask was placed in the fridge overnight. The precipitate which is a bright yellow microcrystalline solid, was collected through filtration and washed with acetone. To increase the yield, the acetone in the filtrate was evaporated, added again to the mixture (30 mL), and the flask placed in the fridge for 3 h. The yellow precipitate that forms, was isolated by filtration and washed with acetone. This procedure was repeated three times. Yield: 1.46 g (3.03 mmol, 81%). 1H-NMR (400 MHz, D2O): δ = 3.52 (s, 6H), 3.50 (s, 6H), 3.42 (s, 6H), 2.75 (s, 6H) ppm; 13C-NMR (100 MHz, D2O): δ = 46.8, 46.6, 45.7, 45.2, 44.7, 44.4, 38.7 ppm.Synthesis of [Ru(2,2′-bipyrimidine)3][Cl]2 (Ru)
In a round-bottom flask equipped with a condenser, [Ru(dimethyl sulfoxide)4(Cl)2] (51.6 mg, 1 eq., 0.1 mmol) and silver(I) trifluoromethanesulfonate (27.1 mg, 1 eq., 0.1 mmol) were dissolved in ethanol (10 mL) and heated to reflux for 3 h. After cooling, the mixture was filtered and loaded into a 50 mL KDS glass syringe. In a separate round-bottom two-neck flask, 2,2′-bipyrimidine (164 mg, 10 eq., 1.0 mmol) was dissolved in ethanol (200 mL) and stirred vigorously. The mixture was then heated to reflux, and the content of the syringe was slowly added using a syringe pump over a time period of 20 h. Both the syringe and the two-neck flask were covered with aluminium foil. The reaction mixture was then transferred to a single-neck round-bottom flask, and the solvent was removed using a rotary evaporator. The residue was dissolved in a minimal amount of water (∼5 mL). The solution was filtered using a syringe filter to remove any undissolved precipitate. Subsequently, a saturated solution of sodium tetraphenylborate was added. Following the addition, immediately a brown precipitate formed. The solution was placed in a refrigerator at 4 °C for 3 h. The formed precipitate was isolated by centrifugation. The solid was washed with water (∼10 mL) and diethyl ether (∼10 mL) to obtain the product as a tetraphenylborate salt. The counter ion was exchanged to chloride by elution with acetonitrile from the ion exchange resin Amberlite IRA-410. Yield: 63 mg (0.098 mmol, 98%). 1H-NMR (400 MHz, DMSO-d6): δ = 9.22 (d, J = 1.9 Hz, 6H), 8.34 (d, J = 5.8 Hz, 6H), 7.71 (t, J = 5.3 Hz, 6H) ppm; 13C-NMR (100 MHz, DMSO-d6): δ = 162.3, 160.5, 158.4, 124 ppm; ESI-MS (pos. detection mode): calcd for C24H18N12Ru [M-2Cl]2+m/z 289.5; found: 289.6; RP-HPLC: tR = 8.0 min; elemental analysis calcd for C24H18Cl2N12Ru (%): C 44.59, H 2.81, N 26.00; found: C 44.93, H 2.97, N 25.73.Synthesis of [Re(2,2′-bipyrimidine)(CO)3(Cl)] (Re)
In a round-bottom flask, ReCO5Cl (235.7 mg, 1 eq., 0.65 mmol) and 2,2′-bipyrimidine (103.3 mg, 1 eq., 0.65 mmol) were dissolved in toluene (100 mL) and the mixture was heated at reflux for 4 h. After a few minutes, the color of the solution turned bright yellow. After this time, the toluene was removed using a rotary evaporator. The crude product was recrystallized from methanol. The microcrystalline solid was throughly washed with diethyl ether. Yield: 290 mg (0.62 mmol, 96%). 1H-NMR (400 MHz, DMSO-d6): δ = 9.40–9.30 (m, 4H), 7.93 (dd, J = 5.6, 4.8 Hz, 2H) ppm; 13C-NMR (100 MHz, DMSO-d6): δ = 161.4, 160.6, 160.2, 124.5 ppm; ESI-MS (pos. detection mode): calcd for C11H6N4O3Re [M − Cl]+m/z 429.0; found: 428.9; RP-HPLC: tR = 14.7 min; elemental analysis calcd for C11H6ClN4O3Re (%): C 28.48, H 1.30, N 12.08; found: C 28.22, H 1.24, N 11.94.Synthesis of [Ru(Re(2,2′-bipyrimidine)(CO)3(Cl))3][Cl]2 (RuRe)
In a round-bottom flask equipped with a condenser, [Ru(dimethyl sulfoxide)4(Cl)2] (65.6 mg, 1 eq., 0.13 mmol) and silver(I) trifluoromethanesulfonate (69.5 mg, 2 eq., 0.27 mmol) were dissolved in ethanol (50 mL) and heated at reflux for 3 h. After cooling to room temperature, the mixture was filtered and transferred to a 50 mL KDS glass syringe. In a separate round-bottom two-neck flask, [Re(2,2′-bipyrimidine)(CO)3(Cl)] (194.8 mg, 3.1 eq., 0.42 mmol) was dissolved in isopropanol/octanol (1:1, 100 mL) and stirred vigorously. The mixture was then heated to reflux, and the content of the syringe was slowly added using a syringe pump over a time period of 20 h. Both the syringe and the two-neck flask were covered with aluminium foil. The reaction mixture was then transferred to a single-neck round-bottom flask, and the solvent was removed using a rotary evaporator. The crude product was purified by column chromatography on silica gel with a acetonitrile/aqueous potassium nitrate (0.4 M) solution (10:1). The fractions containing the product were united and the solvent was removed. The residue was dissolved in acetonitrile and undissolved potassium nitrate was removed by filtration. The solvent was removed and the product was dissolved in water. Upon addition of ammonium hexafluorophosphate the product precipitated as a hexafluorophosphate salt. The solid was washed with water (∼10 mL) and diethyl ether (∼10 mL). The counter ion was exchanged to chloride by elution with acetonitrile from the ion exchange resin Amberlite IRA-410. Yield: 193.7 mg (0.12 mmol, 95%). 1H-NMR (400 MHz, DMSO-d6): δ = 9.32–9.29 (m, 12H), 7.88 (t, J = 5.2 Hz, 6H) ppm; 13C-NMR (100 MHz, DMSO-d6): δ = 161.5, 160.7, 160.3, 124.6, ppm; MALDI-TOF-MS (pos. detection mode): calcd for C33H18Cl4N12O9Re3Ru [M − Cl]+m/z 1528.7790; found: 1528.7763; RP-HPLC: tR = 19.6 min; elemental analysis calcd for C33H18Cl5N12O9Re3Ru (%): C 25.35, H 1.16, N 10.75; found: C 25.54, H 1.43, N 10.71.
Stability
The stability of the compound in phosphate-buffered saline was investigated by absorption spectroscopy. The complex was dissolved dimethyl sulfoxide and then diluted in phosphate-buffered saline reaching finally 1% dimethyl sulfoxide. The solution was stored at room temperature in the dark for 48 h. After various time points, the solution was analyzed by absorption spectroscopy using Jasco V-670 spectrophotometer.Distribution coefficient
The lipophilicity of the metal complex was determined by measuring its distribution coefficient between the octanol and phosphate-buffered saline phases using the ‘shake-flask’ method. For this procedure, the phases were pre-saturated with each other. The complex was initially dissolved in phase A, where it was predominantly present, with an absorbance of approximately 0.5 at 350 nm. This solution was then mixed with an equal volume of phase B, stirred at 300 rpm for 12 h, and allowed to equilibrate overnight. Phase A was carefully separated from phase B, and the concentration of the complex before and after mixing was measured using UV/Vis absorption spectroscopy at 350 nm with a Jasco V-670 spectrophotometer. The experiment was repeated three times, and the ratio between the organic and aqueous phases was calculated.Parallel artificial membrane permeability assay
The wells of the acceptor plate were filled with phosphate-buffered saline (300 μL). A 4% lecithin solution in dodecane (5 μL) was carefully applied to the membrane of the donor plate. The metal complex solution (200 μL, 1% DMSO) was added to the wells of the donor plate. The donor plate was then placed into the acceptor plate, and the combined setup was incubated in the dark at room temperature for 8 h. After this time, the donor plate was removed, and the absorbance at 350 nm in the acceptor plate wells was measured using a Jasco V-670 spectrophotometer. Commercially available reference compounds with known high (0.055 ± 0.005 μm s−1), medium (0.028 ± 0.004 μm s−1), and low (0.009 ± 0.002 μm s−1) cell permeability were used as controls. The permeability rate of the compound was calculated using the following equation:P = permeability rate in cm s−1, VD = donor volume (0.2 cm3), VA = acceptor volume (0.3 cm3), area = 0.24 cm2, time = incubation time in s, ODA = absorbance of acceptor solution, ODE = absorbance of equilibrated acceptor solution.
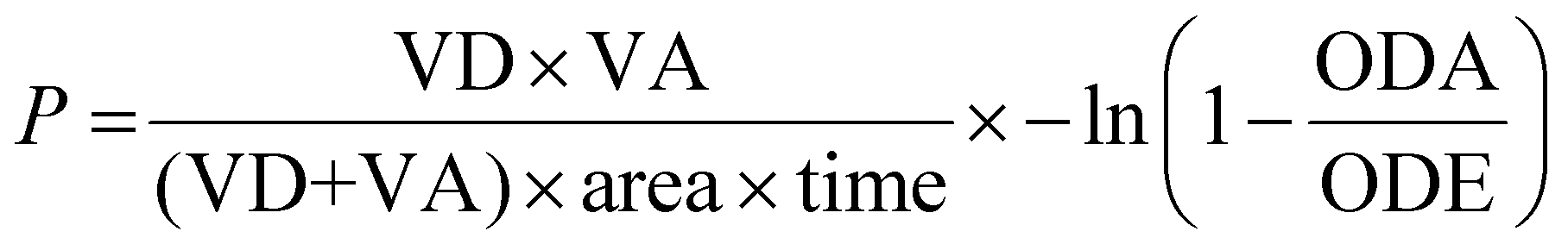
Cell culture
Murine mouse colon carcinoma cells (CT-26) cells were cultured in RPMI 1640 medium supplemented with 10% FBS Superior and 2% penicillin/streptomycin. Human breast adenocarcinoma cells (MCF-7), and human pancreas adenocarcinoma cells (PT-45) were cultured in DMEM medium (4.5 g L−1D-glucose, L-glutamine) supplemented with 10% FBS Superior, 2.4% penicillin/streptomycin, 1.2% GlutaMAX-I and 1% sodium pyruvate. Human fibroblasts (GM-5657) cells were cultured in DMEM medium (1 g L−1D-glucose) supplemented with 10% FBS Superior, 2.4% penicillin/streptomycin, 1.2% GlutaMAX-I and 1% sodium pyruvate. LGC Standards Ltd. provided all the cell lines, which were cultured at 37 °C with 10% CO2. Prior to each experiment, the cells were passaged at least 3 times.Cytotoxicity in monolayer cells
A total of 6 × 103 cells were seeded into 96-well plates and allowed to adhere overnight. The following day, the cells were treated with increasing concentrations of the metal complex, diluted in cell culture media to a final volume of 200 μL, and incubated for 48 h at 37 °C and 10% CO2. After the incubation period, the culture medium was replaced with phosphate-buffered saline containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at a final concentration of 12 μM. The cells were further incubated for 2 h at 37 °C and 10% CO2. Following this, the solution was replaced with 200 μL of dimethyl sulfoxide. The concentration of the formazan dissolved in dimethyl sulfoxide was measured using an Infinite M Nano Plus Microplate Reader (Tecan).Cell death mechanism
A total of 6 × 103 cells were seeded into 96-well plates and allowed to adhere overnight. The following day, the cells were pre-incubated with 3-methyladenine (100 μM) as an autophagy inhibitor, Z-VAD-FMK (20 μM) as an apoptosis inhibitor, cycloheximide (0.1 μM) as a paraptosis inhibitor, or necrostatin-1 (60 μM) as a necrosis inhibitor. The cells were treated with the IC50 value of the metal complex diluted in cell culture media to a final volume of 200 μL, and incubated for 48 h at 37 °C and 10% CO2. After the incubation period, the culture medium was replaced with phosphate-buffered saline containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at a final concentration of 12 μM. The cells were further incubated for 2 h at 37 °C and 10% CO2. Following this, the solution was replaced with 200 μL of dimethyl sulfoxide. The concentration of the formazan dissolved in dimethyl sulfoxide was measured using an Infinite M Nano Plus Microplate Reader (Tecan).Western blot
A total of 3 × 105 cells were seeded into 8-well plates and allowed to adhere overnight. The following day, the cells were treated with the metal complex, diluted in cell culture media to a final volume of 200 μL, and incubated for 24 h at 37 °C and 10% CO2. After this time, the media was removed and the cells were washed with phosphate-buffered saline buffer three-times. Lysis buffer, composed of cell lysis buffer, protease inhibitor cocktail, and 1 mM phenylmethylsulfonylfluoride, was added to each well, and the plates were shaken at 4 °C for 20 min. Cells were then scraped from the bottom of the plates, and the supernatant was collected after spinning down at 14000g at 4 °C for 10 min. The lysates were thawed on ice, and the total protein concentration was measured using a Pierce bicinchoninic acid protein assay. SDS-PAGE samples were prepared by combining 30–50 μg of the total protein and sample buffer composed of a non-reducing, fluorescent compatible sample buffer. After heating at 98 °C for 5 minutes and cooling to room temperature, the prepared samples were electrophoretically separated on a 4–20% TGX gel and transferred to a polyvinylidene fluoride membrane using a tris-glycine transfer buffer containing 20% methanol. The membrane was shaken in fluorescent blocking buffer at 4 °C for 1 h, followed by overnight incubation with antibodies (RIP3 polyclonal antibody, 1:1000) at 4 °C. The membrane was then washed three times with tris-buffered saline-Tween 20 (0.1% w/v Tween 20) for 1 h at room temperature and imaged.
Generation of multicellular tumor spheroids
A 0.75% agarose suspension in phosphate-buffered saline was heated in a high-pressure autoclave. The hot solution was then transferred into a 96-well plate (50 μL per well) and exposed to UV irradiation for 3 hours, allowing it to cool afterward. A cell suspension containing 1 × 104 cells was then seeded on top of the solidified agarose layer. Within two to three days, multicellular tumor spheroids formed from the cell suspension. The spheroids were cultured and maintained at 37 °C in a cell culture incubator with 5% CO2. The culture medium was refreshed every two days. The formation, structural integrity, diameter, and volume of the multicellular tumor spheroids were monitored regularly.Cytotoxicity in multicellular tumor spheroids
The cytotoxicity of the metal complexes in multicellular tumor spheroids was evaluated by measuring the adenosine triphosphate concentration. The spheroids were treated with increasing concentrations of the compound by replacing 50% of the culture media with drug-supplemented media. They were then incubated for 48 hours at 37 °C. Adenosine triphosphate levels were determined using the CellTiter-Glo 3D Cell Viability kit (Promega), and the resulting chemiluminescence was measured with an Infinite M200 PRO plate reader (Tecan).Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Karges gratefully acknowledges the financial support provided by the Liebig fellowship from the Chemical Industry Fund of the German Chemical Industry Association, the Life Sciences Bridge Award from the Aventis Foundation, and the Paul Ehrlich & Ludwig Darmstaedter Early Career Award 2024 – a prize awarded by the Paul Ehrlich Foundation, Germany.References
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, CA Cancer J. Clin., 2023, 73, 17–48 CrossRef.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- N. Montesdeoca, L. Johannknecht, E. Efanova, J. Heinen-Weiler and J. Karges, Angew. Chem., Int. Ed., 2024, 63, e202412585 CrossRef CAS PubMed.
- D. Gibson, Dalton Trans., 2016, 45, 12983–12991 RSC.
- G. Liang, T. Sadhukhan, S. Banerjee, D. Tang, H. Zhang, M. Cui, N. Montesdeoca, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202301074 CrossRef CAS PubMed.
- M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Med. Chem., 2005, 12, 2075–2094 CrossRef CAS.
- J. Karges, ChemNanoMat, 2023, 9, e202300295 CrossRef CAS.
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS.
- H. Wang, Y. Lai, D. Li, J. Karges, P. Zhang and H. Huang, J. Med. Chem., 2024, 67, 1336–1346 CrossRef CAS PubMed.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190–191 CrossRef CAS.
- J. Liu, X.-H. Zou, Q.-L. Zhang, W.-J. Mei, J.-Z. Liu and L.-N. Ji, Met.-Based Drugs, 2000, 7, 427618 Search PubMed.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- Pragti, B. K. Kundu and S. Mukhopadhyay, Coord. Chem. Rev., 2021, 448, 214169 CrossRef CAS.
- K. Qiu, Y. Chen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2019, 378, 66–86 CrossRef CAS.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- C. S. Allardyce and P. J. Dyson, Platinum Met. Rev., 2001, 45, 62–69 CrossRef CAS.
- A. Fennes, N. Montesdeoca, Z. Papadopoulos and J. Karges, Chem. Commun., 2024, 60, 10724–10727 RSC.
- R. F. Brissos, P. Clavero, A. Gallen, A. Grabulosa, L. A. Barrios, A. B. Caballero, L. Korrodi-Gregório, R. Pérez-Tomás, G. Muller, V. Soto-Cerrato and P. Gamez, Inorg. Chem., 2018, 57, 14786–14797 CrossRef CAS.
- V. Novohradsky, J. Yellol, O. Stuchlikova, M. D. Santana, H. Kostrhunova, G. Yellol, J. Kasparkova, D. Bautista, J. Ruiz and V. Brabec, Chem. – Eur. J., 2017, 23, 15294–15299 CrossRef CAS.
- M. R. Gill, H. Derrat, C. G. W. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877–880 CrossRef CAS.
- J. Shum, P. K.-K. Leung and K. K.-W. Lo, Inorg. Chem., 2019, 58, 2231–2247 CrossRef CAS.
- H. Zhang, N. Montesdeoca, D. Tang, G. Liang, M. Cui, C. Xu, L.-M. Servos, T. Bing, Z. Papadopoulos, M. Shen, H. Xiao, Y. Yu and J. Karges, Nat. Commun., 2024, 15, 9405 CrossRef CAS.
- G. Sava, I. Capozzi, K. Clerici, G. Gagliardi, E. Alessio and G. Mestroni, Clin. Exp. Metastasis, 1998, 16, 371–379 CrossRef CAS.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- D. Baier, T. Mendrina, B. Schoenhacker-Alte, C. Pirker, T. Mohr, M. Rusz, B. Regner, M. Schaier, N. Sgarioto, N. J.-M. Raynal, K. Nowikovsky, W. M. Schmidt, P. Heffeter, S. M. Meier-Menches, G. Koellensperger, B. K. Keppler and W. Berger, Adv. Sci., 2023, 10, 2301939 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS.
- M. S. Capper, H. Packman and M. Rehkämper, ChemBioChem, 2020, 21, 2111–2115 CrossRef CAS PubMed.
- H. S. Liew, C.-W. Mai, M. Zulkefeli, T. Madheswaran, L. V. Kiew, N. Delsuc and M. L. Low, Molecules, 2020, 25, 4176 CrossRef CAS.
- N. Montesdeoca, R. L. Borkar, M. Sathiyendiran and J. Karges, Chem. – Eur. J., 2024, 30, e202400217 CrossRef CAS.
- F.-X. Wang, J.-H. Liang, H. Zhang, Z.-H. Wang, Q. Wan, C.-P. Tan, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2019, 11, 13123–13133 CrossRef CAS.
- P. V. Simpson, M. Falasca and M. Massi, Chem. Commun., 2018, 54, 12429–12438 RSC.
- L. E. Enslin, K. Purkait, M. D. Pozza, B. Saubamea, P. Mesdom, H. G. Visser, G. Gasser and M. Schutte-Smith, Inorg. Chem., 2023, 62, 12237–12251 CrossRef CAS.
- A. P. King, S. C. Marker, R. V. Swanda, J. J. Woods, S.-B. Qian and J. J. Wilson, Chem. – Eur. J., 2019, 25, 9206–9210 CrossRef CAS PubMed.
- S. C. Marker, A. P. King, S. Granja, B. Vaughn, J. J. Woods, E. Boros and J. J. Wilson, Inorg. Chem., 2020, 59, 10285–10303 CrossRef CAS.
- B. Neuditschko, A. P. King, Z. Huang, L. Janker, A. Bileck, Y. Borutzki, S. C. Marker, C. Gerner, J. J. Wilson and S. M. Meier-Menches, Angew. Chem., Int. Ed., 2022, 61, e202209136 CrossRef CAS PubMed.
- I. Chakraborty, S. J. Carrington and P. K. Mascharak, ChemMedChem, 2014, 9, 1266–1274 CrossRef CAS PubMed.
- B. Kar and P. Paira, Chem. – Eur. J., 2024, e202401720 CrossRef.
- X. Yi, J. Zhao, J. Sun, S. Guo and H. Zhang, Dalton Trans., 2013, 42, 2062–2074 RSC.
- F. Zobi, A. Degonda, M. C. Schaub and A. Y. Bogdanova, Inorg. Chem., 2010, 49, 7313–7322 CrossRef CAS PubMed.
- H. K. Saeed, P. J. Jarman, S. Sreedharan, R. Mowll, A. J. Auty, A. A. P. Chauvet, C. G. W. Smythe, J. B. de la Serna and J. A. Thomas, Chem. – Eur. J., 2023, 29, e202300617 CrossRef CAS PubMed.
- H. K. Saeed, S. Sreedharan, P. J. Jarman, S. A. Archer, S. D. Fairbanks, S. P. Foxon, A. J. Auty, D. Chekulaev, T. Keane, A. J. H. M. Meijer, J. A. Weinstein, C. G. W. Smythe, J. Bernardino de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2020, 142, 1101–1111 CrossRef CAS PubMed.
- Y. Wang, P. S. Felder, P. Mesdom, O. Blacque, T. L. Mindt, K. Cariou and G. Gasser, ChemBioChem, 2023, 24, e202300467 CrossRef CAS.
- C. Bardin, A. Astier, A. Vulto, G. Sewell, J. Vigneron, R. Trittler, M. Daouphars, M. Paul, M. Trojniak and F. Pinguet, Ann. Pharm. Fr., 2011, 69, 221–231 CrossRef CAS PubMed.
- N. Montesdeoca, K. Ni and J. Karges, Chem. – Eur. J., 2024, 30, e202401988 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and figures referenced throughout the text. See DOI: https://doi.org/10.1039/d4dt02992e |
| ‡ The authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2025 |