
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper coordination chemistry of the patellamides – cyanobactins in the ascidian-Prochloron symbiosis†
Philipp
Baur
 *ab and
Peter
Comba
*bc
*ab and
Peter
Comba
*bc
aInstitute for Molecular Bioscience, The University of Queensland, 305 Carmondy Road, 4067 St Lucia, QLD, Australia. E-mail: philipp.baur@uq.edu.au
bAnorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: peter.comba@aci.uni-heidelberg.de
cInterdisciplinary Center for Scientific Computing (IWR), Heidelberg University, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
First published on 9th December 2024
Abstract
Prochloron didemni, an obligate symbiont of certain ascidians (sea squirts found in tropical areas), produces various cyclic pseudo-octapeptides in large quantities. These secondary metabolites have attracted the attention of medicinal chemists and, due to their four azol(in)e and four amide donor groups, coordination chemists have become interested in these molecules. The structures of the metal-free macrocycles and their dinuclear copper(II) complexes are known, and solution equilibria, spectroscopic properties and a range of biologically relevant reactions have been studied in detail. However, until recently, the properties of the patellamides and structures of the copper(II) complexes in living systems have not been known unambiguously. These are reviewed in the present Perspective and, as a result, it now is possible to discuss possible biological functions of these species.
Introduction
With the aim of identifying new medically interesting substances, a group of cyclic peptides found in large amounts in the ascidian Lissoclinum patella was discovered.1 Unlike many marine cyclic peptides, they did not show significant cytotoxicity or palatability changes towards any of the tested marine organisms.2 In drug discovery screens, they exhibited cytotoxicity against different human cancer cell lines,3 and patellamide D has been shown to act as a selective antagonist in multidrug-resistant leukemia cell lines.4 However, none of these properties seemed likely to be correlated to their natural function, as the effects were related to competitive binding to overexpressed transport proteins. All molecules from the patellamide family show a similar structure, being cyclized pseudo-octapeptides with two oxazoline and two thiazole heterocycles as well as hydrophobic side chains, with the two side chains next to the thiazoles showing inverted stereochemistry (see Chart 1 for the structure of ascidiacyclamide).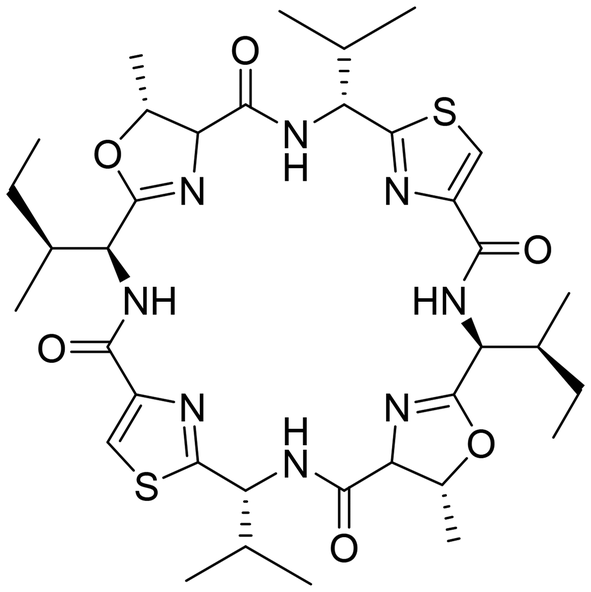 | ||
| Chart 1 Structure of ascidiacyclamide, one of the most abundant members of the patellamide family. | ||
As one would expect from the structure of the patellamides with an arrangement of amides alternating with the four heterocycles in a rigid cyclic scaffold, the pseudo-octapeptide macrocycles were soon discovered as versatile ligands for metal ions.5–7 Metal binding studies have shown that patellamides have the highest affinity towards copper(II) and that they can bind two CuII ions cooperatively.8–11 The resulting dinuclear CuII complexes of the patellamides have been shown to have high catalytic activity towards different substrates.12,13,14 While CuII patellamide complexes have not been identified unambiguously in natura, a 2017 study that used a fluorescence-tagged patellamide analogue showed the ability to bind CuIIin vivo.15 More recently, EXAFS studies on biological material clearly showed that two CuII ions are coordinated to ascidiacyclamide in living Prochloron cells.16
Notably, the evaluation of the genome of the ascidian as well as its obligate symbiont, the cyanobacterium Prochloron didemni, revealed that only the cyanobacterium has the genes required for the synthesis of the patellamides.17 Thus, the question arose, for which purpose the molecule is made, especially on its role in the symbiosis, and why it is exported to the host in exceedingly large amounts.18,19 While Prochloron didemni has proven to be an obligate symbiont for the ascidian host, no studies address whether the patellamides serve any function for the host or whether they are produced by the cyanobacterium merely for its own benefit.
In the current perspective we give a short overview on the biological background and medicinal properties and review synthetic aspects and the CuII coordination chemistry of the patellamides before discussing the state-of-the-art view on the biological function of the patellamides and their CuII complexes.
The biology behind the patellamides
Lissoclinum patella
The didemnid ascidian Lissoclinum patella (L.p.) is a colonial tunicate organism (Fig. 1), found near different reefs in the Western Pacific.20 Most ascidians are sea squirts that sustain themselves by filtering nutrients out of the surrounding water, and most didemnids contain a photosynthetic symbiont, in this case the cyanobacterium Prochloron didemni (P.d.). The symbionts assume important functions such as nitrogen fixation21,22 or photosymbiosis. Especially the presence of the photosynthetic symbiont seems to be essential for the organisms, as shading of an ascidian colony and the resulting drop in photosynthesis rates leads to slower growth of the ascidian.23 | ||
| Fig. 1 A colony of Lissoclinum patella growing on a coral (photo taken in 2022 at a depth of 1.2 m at the Blue Pools Dive Site, Heron Island, Australia). | ||
Each ascidian colony consists of multiple individual animals (“zooids”), embedded in a common tunic immobilized on the coral or rock the organism is growing on,20,24 and shares common colonial cloacal cavities, which are inhabited by P.d.25 Individual colonies can grow quite large to sizes of up to one square meter but more commonly are significantly smaller, in the range of 10–50 cm2, and are around 1 cm thick.20 They have a firm gelatinous, smooth, white surface with multiple opaque rounded ridges and transparent concave indentations in between, which contain the cloacal and zooid openings of sub-colonies of ascidian zooids (Fig. 2).
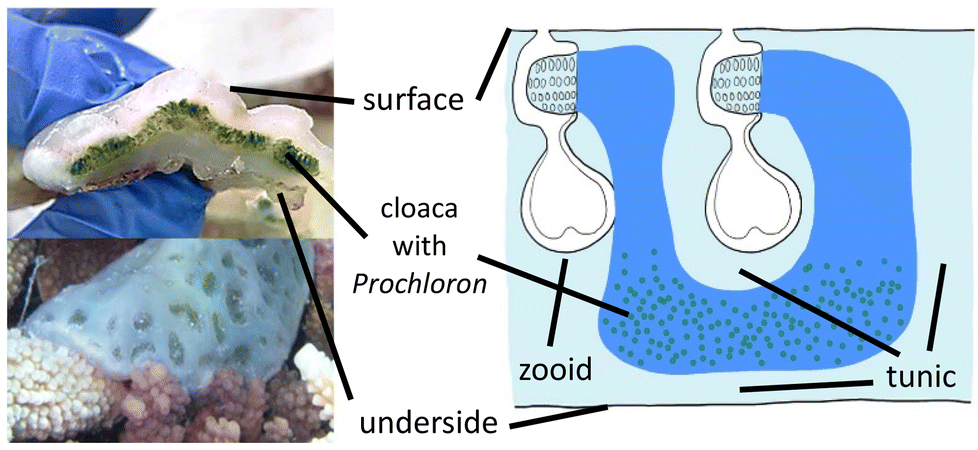 | ||
| Fig. 2 Left: Cross-section through a colony of Lissoclinum patella, and one colony in natura with well visible subcolony indentations. Right: Schematic representation of the ascidian anatomy.26 | ||
Prochloron didemni
Having evolved over 3 billion years,27 cyanobacteria are one of the oldest species on earth and key contributors to the great oxygenation event 2–2.4 billion years ago that initiated the evolution of higher species. In general, cyanobacteria only possess chlorophyll a and use phycobilisomes as antennae to increase the light-harvesting efficiency,28 which gives them a red-brown color. However, P.d. was discovered as a species in a new family of green-colored cyanobacteria, having both chlorophyll a and b but no phycobilisomes.29P.d. was initially discovered as symbiotic cyanobacterium associated with the surface of Didemnum colonies in the pacific and with Diplosoma virens near Hawaii29 but it was soon discovered in other species, including L.p.30,31The benefit of the symbiosis for L.p. is obvious as P.d. provides the host with photosynthesis products and intermediates32 to an extent that up to 60% of the host's demand for carbon is met.22P.d. was also found to participate in the recycling of nitrogen22,33,34 and possibly even in nitrogen fixation.35 Besides getting a habitat in a controlled environment, a possible export of nitrogen-containing waste from the host to the symbiont could be some of the benefits P.d. obtains from the symbiosis.
Besides assisting the host in the carbon- and nitrogen metabolism, P.d. produces large amounts of cyclic peptides, the patellamides, and exports them to the host.36 The cyclic peptides have been reported to be present in amounts of up to several percent of the animal's dry mass.17 The patellamides are classified as secondary metabolites, which generally are not involved in the growth, development or reproduction of an organism, and are relevant for long-term strategies such as control of the local environment or defense.37 This makes some initially proposed functions such as an enzymatic role in the primary metabolism of carbon or nitrogen unlikely.
Medicinal properties of the patellamides
Many marine cyclic peptides were discovered by medical researchers looking for potentially interesting new drugs – and generally most tests done with the molecules were toxicity assays.38 Therefore, in most cases the possible function discussed in literature is molecular defense. In case of the patellamides, these cyclic peptides do not have noteworthy toxicity against any microbial enemy or competitor of P.d.39 and do not seem to affect the feeding behavior of larger predators.2The patellamides were discovered by collecting large amounts of ascidians and homogenizing them, extracting, and separating the natural products. They were noticed due to their cytotoxicity against L1210 murine leukemia cells at 2–4 μg mL−1, and of patellamide A against the acute lymphoblastic leukemia cell line CeM with an IC50 value of 0.028 μg mL−1.1 A more striking activity is that patellamide D at 3.3 μM concentrations acts as a selective antagonist in multidrug-resistant CEM/VLB100 cell lines,4 where the IC50 value of different chemotherapeutics in some cases could be reduced from 1000 ng mL−1 to 110 ng mL−1.
A key issue with pharmaceutically interesting compounds from biological sources is that, while extraction can be done on a drug-discovery scale, for medical use a stable supply of a large amount in consistent quality is needed. Therefore, typically an efficient synthesis is required. There are various examples where, after a synthesis for the natural product was developed, the compound found use in clinical routine,40 while others are still being tested.41–44 In addition to the patellamides, several structurally similar cyclic peptides show pharmaceutical potential.45,46
Chemistry and biochemistry of the patellamides
Synthesis, biosynthesis and structural elements
When the patellamides originally were discovered in 1981,1 the assumed structure involved directly fused thiazole and oxazoline heterocycles. In the first synthetic attempt for different patellamides in 1985, it was noticed that the resulting compounds analytically differed from the natural patellamides, and consequently a corrected version of the patellamide structure was proposed.47,48 This was later confirmed by crystallography of the metal-free macrocycles,49 and crystallography as well as spectroscopy of CuII complexes.50,51 In the following years, different synthetic approaches, mostly combining contemporary peptide coupling approaches with modern, efficient heterocyclizations, have been developed, shifting away from tedious older heterocyclization strategies.47,48,52–54 An important new development was a building block-based approach that enabled a straightforward and cheap synthetic route to different analogues of the patellamides,55 and this was used to produce a series of patellamide analogues for functional studies, especially also derivatives containing 1,5-dimethylimidazole heterocycles instead of the natural ones,14,56–60 as well as the westiellamide-derived pseudo-hexapeptide macrocycles.61–63 Using Lawesson's reagent in this method for the synthesis of thiazole heterocycles, only 5-methylthiazole derivatives are available, as the starting material needs to have two secondary carbonyl groups. A new synthetic approach, also compatible with the mostly used Fmoc based peptide synthesis, solved this problem.64 Based on the building block approach and Fmoc-based peptide chemistry, a new synthetic scheme was developed that allows to prepare any desired natural or non-natural patellamide, while moving the potentially yield-determining oxazoline heterocyclization step to the third step of the respective building block synthesis.65As an alternative to the chemical approaches for the synthesis of patellamides, the biosynthetic route undertaken by the cyanobacteria was studied to understand and reproduce the synthesis. Firstly, the gene cluster “pat”, responsible for the synthesis of patellamides, was identified, isolated, amplified, and subsequently introduced into E. coli to successfully produce the patellamides in other organisms (Fig. 3).17 It was discovered that, apart from the eight amino acids used as precursor peptide for the biosynthesis, the majority of the gene encodes enzymes required for many of the modifications needed to obtain the correct product, such as a protease, a heterocyclase/cyclodehydratase, an oxidase, a macrocyclase, and possibly more enzymes in parts of the gene that are yet to be understood.68 The macrocyclase and the heterocyclase have been found to be quite flexible and powerful enzymatic tools for macrocyclizations66,69 and heterocyclizations70,71 of different substrates, making them interesting for different biosynthetic applications.
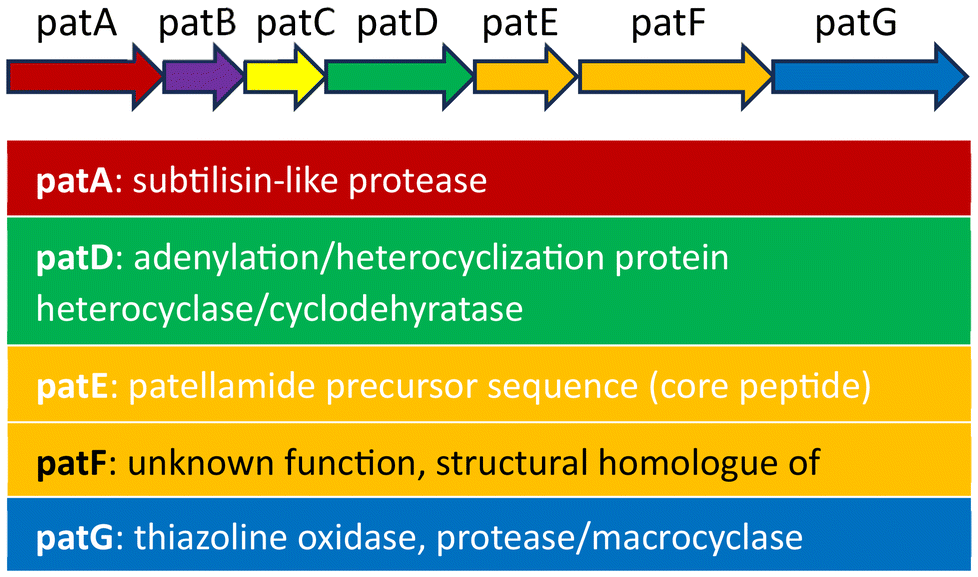 | ||
| Fig. 3 Contents of the pat gene, containing all the known enzymes and the precursor peptides required for the biosynthesis of the patellamides. The function of the genes patB, patC and pat F is unclear.66,67 | ||
Generally, the biosynthesis involves the synthesis of the enzymes encoded in the subgenes patA, patB, patC, patD, patF, patG and of large amounts of the precursor peptide contained in subgene patE. This precursor peptide contains the eight amino acids forming the final patellamides, cut from the precursor peptide with three additional amino acids that are removed in the macrocyclization step. After being cut out by the protease, the precursor peptide needs to undergo a heterocyclization step of the cysteines to the thiazolines and an oxidation of the thiazolines to the thiazoles,68 a heterocyclization of the threonines/serines to the oxazolines as well as the macrocyclization. The function of the different domains of unknown function within patA and patG is not fully understood, as is the exact order, in which these enzymatic reactions occur.72
The general structure of the patellamide family is given in Chart 2 and Table 1. The hydrophobic side chains next to the oxazolines have an S-stereocenter, while those next to the thiazoles always have R-configuration, although the corresponding free D-amino acids are present in the organism.
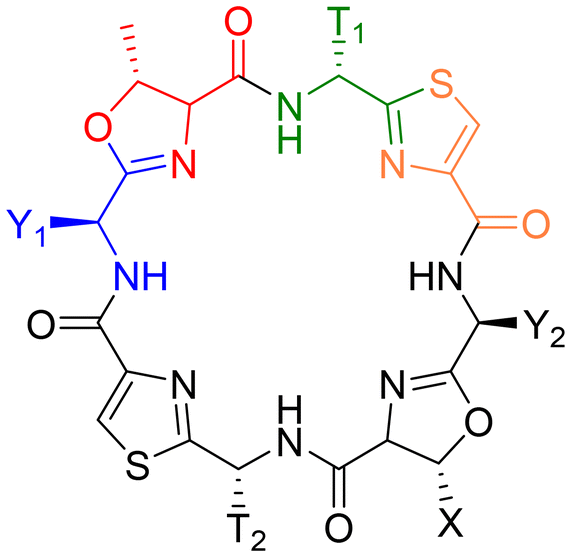 | ||
| Chart 2 Fundamental structure of the patellamides. All contain cysteine (yellow) heterocyclized with a hydrophobic amino acid with D-stereochemistry (green) and threonine (serine in case of Patellamide A), heterocyclized with a hydrophobic amino acid with L-stereochemistry (blue). Table 1 shows all known patellamides with the nature of their respective hydrophobic side chains (T near thiazole, Y near oxazoline) and the amino acid encoded in the sequence.73 | ||
| Patellamide | T1 | T2 | Y1 | Y2 | X |
|---|---|---|---|---|---|
| Ascidiacyclamide | iPr (V) | iPr (V) | sBu (I) | sBu (I) | Me (T) |
| A | iPr (V) | iPr (V) | sBu (I) | sBu (I) | H (S) |
| B | Me (A) | Bz (F) | iBu (L) | sBu (I) | Me (T) |
| C | Me (A) | Bz (F) | iPr (V) | sBu (I) | Me (T) |
| D | iPr (V) | Bz (F) | sBu (I) | sBu (I) | Me (T) |
| E | iPr (V) | Bz (F) | iPr (V) | sBu (I) | Me (T) |
| F | Me (A) | Bz (F) | iPr (V) | iPr (V) | Me (T) |
| G | iPr (V) | Bz (F) | sBu (I) | iBu (L) | Me (T) |
| Stereochemistry | D | L | L | ||
The structures of the patellamides have been extensively studied, both in solution and in the solid state.49,50,74–77 These form a 24-membered ring, which in its metal-free state commonly assumes a figure-of-eight conformation, in which the two thiazoles are parallel and close to each other, while the amide hydrogens are forming hydrogen bonds to proximate oxygen atoms from the peptide carbonyl groups and the heterocycles (Chart 3).75 In addition to the figure-of-eight conformer, a saddle/square-shaped conformer was identified, with hydrogen bonds between the amide hydrogens and the neighboring nitrogens from the heterocycles.75,78 Due to the structural restrictions from the macrocycle as well as the rigidity of the heterocycles, the nitrogen atoms of the heterocycles and the peptide bonds point in this conformation to the inside of the macrocycle, and the peptide side chains to the outside, making the molecule well suited for metal-binding.
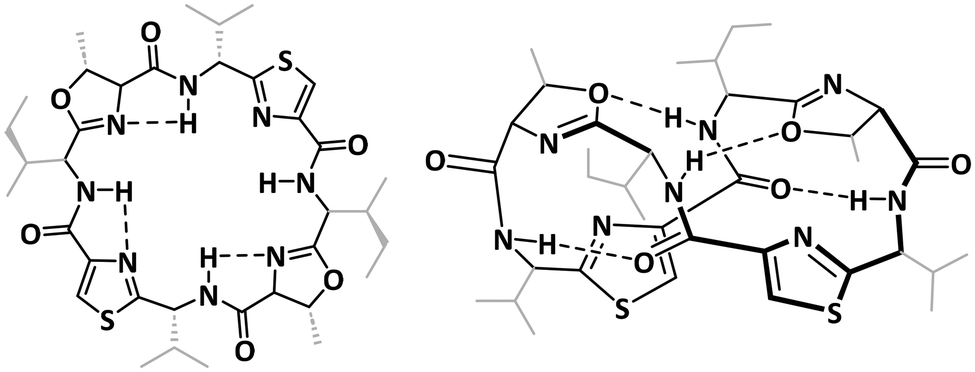 | ||
| Chart 3 Comparison of the saddle-shaped (square, left) and the figure-of eight configurations (right) of the fully protonated patellamides.26,78 | ||
In addition to the patellamides produced by P.d., there are several other cyclic molecules found in the ascidians that share some structural features with the patellamides. Among these are the lissoclinamides and ulicyclamide, which consist of seven amino acids instead of the eight found in the patellamides, with similar hydrophobic amino acids, such as phenylalanine and valine, but with three instead of four azol(in)es, two thiazoles and an oxazoline (instead of a second oxazoline, there is a proline pyrrolidine ring built into the backbone).46 The different members of the lissoclinamide and ulicyclamide families only differ in the hydrophobic side chains and the oxidation state of the thiazole rings, of which none, one or both can be thiazolines instead. Ulithiacyclamide, shares many of the structural characteristics with the patellamides, like the number and nature of heterocycles and macrocyclic ring size. However, the side chains next to the oxazoline are cysteines, which form a disulfide bridge through the middle of the macrocycle, hindering metal binding but increasing the rigidity and stability of the macrocycle.79 Lissoclinamides, ulicyclamides, and ulithiacyclamides are also produced by P.d., and the genes for their biosynthesis are in the same cluster as for the patellamides.80 In the ascidian Lissoclinum bistratum, P.d. produces a family of structurally similar cyclic hexapeptides, the bistratamides.
Other families of cyanobacteria are known to produce a wide range of cyclic peptides, many of them containing similar azol(in)e motifs (westiellamides from Westiellopsis prolifica,81 nostocyclamides from Nostoc 31, tenuecyclamides from Nostoc spongiaeforme,82 and raocyclamides from Oscillatoria raoi,83 to name just a few examples). To the majority of these azol(in)e containing cyclic peptides from marine sources usually a defensive function is attributed, as only cytotoxicity has been evaluated after or during the discovery process. This makes it possible if not likely, that many metal-related biological functions and possible applications have so far been underexplored.
Metal complexation
Based on the structural similarities to aza-crownether macrocycles and the structural properties discussed above, but also due to the observation of increased metal ion concentrations, specifically of copper, in ascidians, compared to the surrounding sea water,51,84 patellamides have captured the interest of coordination chemists in the last three decades.6–10,51,85 Recent studies indicate that the concentration of copper in the ascidians is on average around 0.3 ppm, while in the surrounding seawater it is around 0.2 ppb, i.e., there is an increase with respect to the surrounding seawater of around 1000–2000, 2000–3000 or 200–300 for the whole specimen, Prochloron and the cloacal cavity.51,84 Solution equilibria, extensively studied by UV-vis-NIR, CD, EPR spectroscopies and ESI-MS of the natural products and a series of synthetic analogues have produced a general scheme, with a number of structures of importance for reactivities and putative biological function (see Scheme 1).8,57 Complex stabilities with CuII and ZnII as well as with other metal ions (e.g. CaII) have been determined by CD, NMR, UV-vis spectroscopies and calorimetrically (see8 for a relevant recent compilation of complex stabilities), and the CuII stability constants with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K values around 5 generally are slightly higher than those for other metal ions. The coordination chemistry of patellamides with other metal ions than CuII has not been studied in great detail so far. However, in support of the suggestion that CuII might be more relevant than other metal ions is the observation from phosphatase kinetics that the Zn2+ and mixed Zn2+/Cu2+ species are slightly less reactive than the corresponding dinuclear Cu2+ complexes.14,56–60 This parallels the slightly larger stability constants with Cu2+ compared to Zn2+. The only other complex stability data available are with Ca2+, and these are also significantly lower than those with Cu2+.86 A structure of a K+ complex of partially hydrolyzed ascidiacyclamide has also been reported.87 Importantly, from some of the complexation studies as well as from solution spectroscopy and computational work, it appears that complexation of the first CuII center preorganizes the macrocycle for the second metal ion, i.e., there is cooperativity in the formation of the dicopper(II) complexes.8,56–58
K values around 5 generally are slightly higher than those for other metal ions. The coordination chemistry of patellamides with other metal ions than CuII has not been studied in great detail so far. However, in support of the suggestion that CuII might be more relevant than other metal ions is the observation from phosphatase kinetics that the Zn2+ and mixed Zn2+/Cu2+ species are slightly less reactive than the corresponding dinuclear Cu2+ complexes.14,56–60 This parallels the slightly larger stability constants with Cu2+ compared to Zn2+. The only other complex stability data available are with Ca2+, and these are also significantly lower than those with Cu2+.86 A structure of a K+ complex of partially hydrolyzed ascidiacyclamide has also been reported.87 Importantly, from some of the complexation studies as well as from solution spectroscopy and computational work, it appears that complexation of the first CuII center preorganizes the macrocycle for the second metal ion, i.e., there is cooperativity in the formation of the dicopper(II) complexes.8,56–58
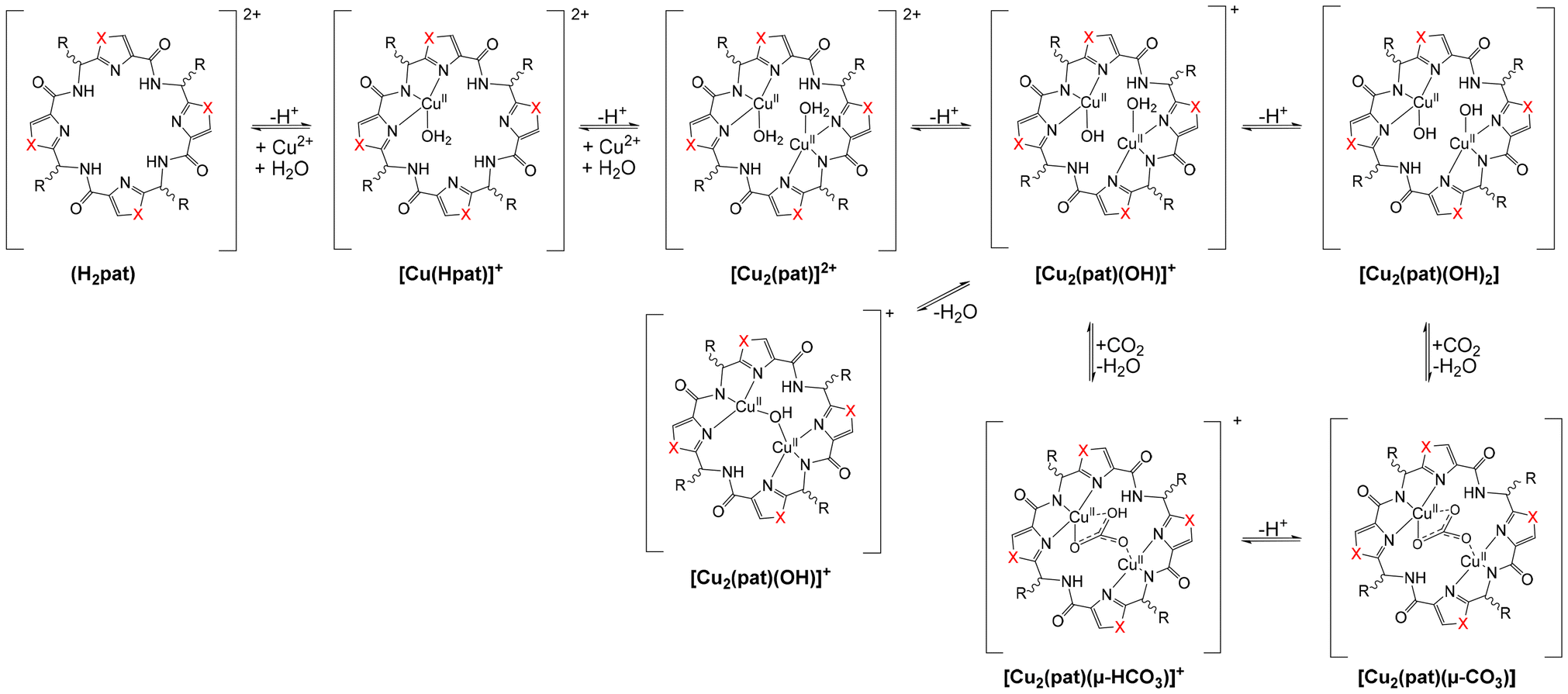 | ||
| Scheme 1 Solution equilibria of patellamides (general derivative pat with unspecified side chains and side-chain configurations) with CuII.8,57 | ||
There are some X-ray crystal structures from metal complexes of the patellamides,56,87,88 and EPR spectroscopy has been used extensively to obtain structural information, in particular of the dicopper(II) compounds, where dipole–dipole coupling of the two CuII centers leads to EPR transitions that can be simulated with the spin Hamiltonian parameters in addition to the CuII⋯CuII distance and the relative angular orientation of the two g tensors.89 The combination of (frozen) solution EPR spectroscopy, spectra simulation and molecular modeling,90 based on force field91,92 as well as DFT calculations, has been used extensively to obtain experimental solution structures of dicopper(II) complexes of the patellamides.8,56,57 Importantly, it has been shown that, where applicable, the MM-EPR (or DFT-EPR) solution structures are in good agreement with crystal structural data.56,57 Recent EXAFS studies on frozen Prochloron cells fully support this.16 Unfortunately, EPR spectroscopy on biological samples is hampered by a very low signal intensity and the exceedingly high intensity of signals of MnII species in natural samples. Therefore, the recent EXAFS studies are of importance as they clearly show that (i) two CuII ions are coordinated to ascidiacyclamide in Prochloron cells, (ii) that carbonate bridges the two copper centers, and (iii) that the structure is identical to the published crystal structure51 as well as the published solution structure.57
As many of the patellamides differ only in the size and structure of the hydrophobic side chains, the differences in metal binding affinities that have been observed for very similar molecules, must be influenced by differences in these side chains. In the case of patellamides A and C, the stability constants for mononuclear CuII complexes are 2.0 × 104 M−1 for patellamide A and 6.8 × 104 M−1 for patellamide C, respectively.5 The side chain differences are not expected to lead to a significantly different steric arrangement around the binding site but are expected to affect the bending angle and rigidity of the entire saddle-shaped structure. The azol(in)e type heterocycles are important for the binding properties, and this was demonstrated in a recently published study,93 showing that for a similar cyclic cyanobactin, the lissoclinamide, the ability to bind CuII is significantly decreased by interchanging the heteroatoms in the thiazole ring: when binding CuII, the backbone strongly deforms to allow coordination to the nitrogen donors. Apart from structural differences between flat, aromatic azoles and the bent, non-aromatic azolines, the difference in pKa between the different heterocycles contributes to the observed differences in the binding behavior (pKa, imidazole: 7.0, thiazole: 2.5, oxazoline: 4.75, oxazole: 0.865,94).
Fig. 4 is a plot of the crystal structure of the carbonato-bridged dicopper(II) complex of ascidiacyclamide. The hydrophobic side chains next to the thiazoles, due to the non-natural R-configuration, point outwards of the coordination plane, parallel to the plane of the CuII–CO32−–CuII site (blue planes in Fig. 4), while the hydrophobic side chains with L-configuration next to the oxazolines are oriented towards the inside of the saddle, posing a certain steric obstacle in that area (orange circle in Fig. 4).26 In carbonic anhydrase activity (see below), the valine-containing planes might therefore act as a pocket for the catalytically active site, helping to stabilize the intermediate and adding a certain amount of substrate preference.
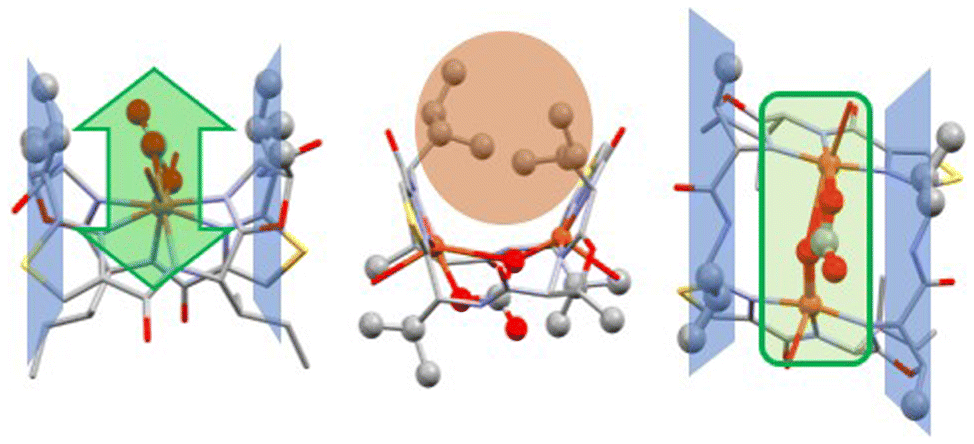 | ||
| Fig. 4 Crystal structure of the dinuclear CuII complex of ascidiacyclamide, bridged by carbonate (in green).86 The isobutyl side chains from the D-valines lie in a plane with the methyl group from the oxazolines (colored in blue), parallel to the plane containing the CuII–CO32−–CuII site. This preorganization allows for easy access to the dinuclear copper center of small, flat or linear molecules such as carbonate/CO2 from one side (highlighted in green).88 | ||
Reactivity and possible biological functions of the patellamides and their copper complexes
As the patellamides, like many cyanobactins, have been discovered when evaluating their cytotoxic properties (see above) the proposed function for the ascidians was defense.1 However, as no toxic properties or palatability changes were identified for the metal-free peptides2,95 the continued search for possible functions lead to the discovery of their interesting metal binding properties.51,88 This raised the question, whether the function of the patellamides might be related to their CuII binding properties. An interesting observation was that, whenever CuII patellamide complexes were exposed to air, carbonato-bridged complexes were formed,78,88 which instigated the evaluation of carbonic anhydrase reactivity of the complexes.78,96–98 The dicopper(II) complexes of the patellamides indeed have a remarkably high carbonic anhydrase activity, they are only two orders of magnitude slower than the enzymes, which all are mononuclear zinc complexes, and they are faster than any zinc-based model system.96,98 The reaction mechanism was thoroughly studied by stopped-flow kinetic measurements, isotope labeling and quantum-chemical calculations.14,57,99 Apart from carbonic anhydrase activity, the CuII patellamide complexes show several other catalytic activities, including phosphoesterase,12,60,65,100,101 glucosidase and β-lactamase.13 While the phosphodiesterase reaction like the carbonic anhydrase reaction occurs at high rates and physiologically relevant pH ranges of 6–7,12 the other activities have only been observed at low rates at high pH values (over 10), and are thus unlikely to be of relevance.13An important conclusion of a recent computational study of the carbonic anhydrase mechanism of patellamide-dicopper(II) complexes is that HCO3− product release, rather than the nucleophilic attack of OH−, coordinated to one of the CuII centers, on the carbon atom of CO2, interacting with the other CuII center, is rate limiting (see Fig. 5), and this is strongly pH dependent: at pH values above approx. 7, the carbonato bridge is deprotonated and inhibits catalysis. Therefore, carbonate transport from the ascidian to the photosynthetic symbiont Prochloron is proposed as biological function of the patellamides.99 This is supported by the observation that patellamides are produced in large quantities by Prochloron (see above).102
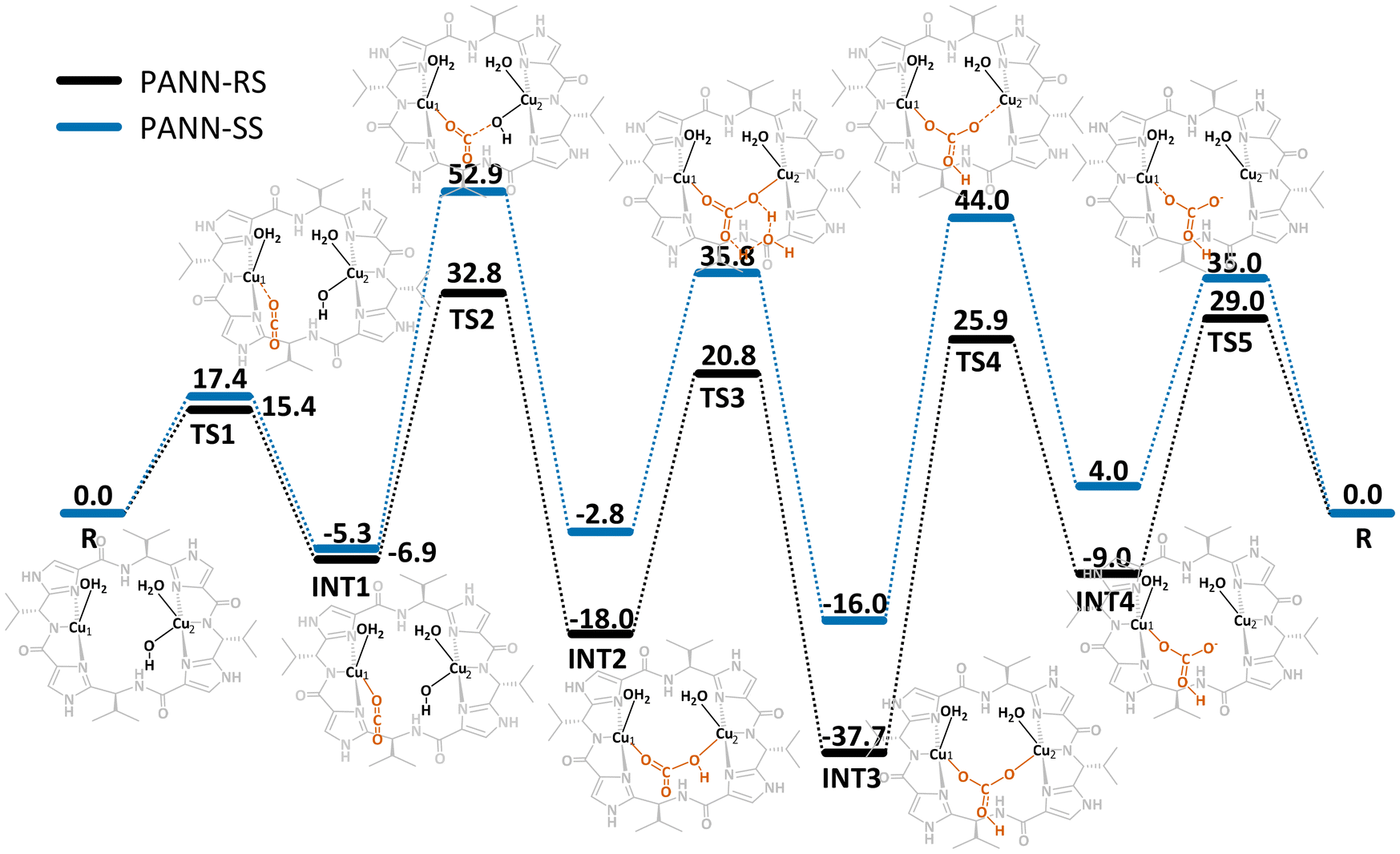 | ||
| Fig. 5 Free energy profile (kJ mol−1) for the CO2 hydration catalyzed by patellamide-dicopper(II) complexes. For the calculations two model analogues with the natural R,S stereochemistry and non-natural S,S stereochemistry were used to try to understand what advantages inverted stereochemistry in the natural patellamides might have.99 | ||
An additional observation from the computational study was that both the stereochemistry of the side chains and the type of azol(in)e rings in the patellamides affect the carbonate binding behavior significantly. Although it is not fully understood how the inversion of stereochemistry at the side chains occurs in the biosynthetic pathway,17 the alternating R,S stereochemistry of the side chains and the choice of thiazole and oxazoline heterocycles seems to be ideal for some possible functions. The patE region of the genome encodes the precursor amino acid sequence and leads to a quite large flexibility of the patellamides with 20 different versions found in 46 samples.103
Conclusions
Patellamides, secondary metabolites produced by P.d., an obligate symbiont of its host L.p., are intriguing molecules, specifically due to the quantities in that they are produced, their molecular structures and the properties of their CuII complexes. Over the last 30 years, coordination chemists have been drawn to their resemblance to other metal-binding macrocycles and their preference for CuII. A key feature of patellamides is cooperative binding of two CuII centers and the preorganization of the dicopper(II) complexes for bridging anions, specifically CO32−. One of the most remarkable observations is that dicopper(II)-patellamide complexes are very efficient carbonic anhydrase catalysts with turnover frequencies close to enzymes. However, at the pH in the cloaca of the ascidians, release of CO2 is inhibited. This, together with the exceedingly large concentration of the patellamides in the ascidians – much larger than expected for enzymatic activities – makes it unlikely that the catalytic hydrolysis of CO2 is of natural relevance, and we propose that CO2 transport to the cyanobacterium is the biological function. This is strongly supported by a DFT based computational in comparison with experimental reactivity studies of a range derivatives of the natural products.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful for financial support of our own work by the University of Heidelberg, the German Science Foundation (DFG), the Max Planck School Matter to Life, supported by the BMBF in collaboration with the Max Planck Society (grant no. M526300), and computational resources provided by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no. INST 40/575-1 FUGG (JUSTUS 2 cluster).References
- A. R. D. Chris, M. Ireland Jr, R. A. Newman and M. P. Hacker, J. Org. Chem., 1981, 1807–1811 Search PubMed.
- N. Lindquist, M. E. Hay and W. Fenical, Ecol. Monogr., 1992, 62, 547–568 CrossRef.
- B. M. Degnan, C. J. Hawkins, M. F. Lavin, E. J. McCaffrey, D. L. Parry, A. L. van den Brenk and D. J. Watters, J. Med. Chem., 1989, 32, 1349–1354 CrossRef CAS.
- R. S. J. Allen and B. Williams, Cancer Lett., 1993, 97–102 Search PubMed.
- L. A. Morris and M. Jaspars, Biodivers., 2000, pp. 140–166 Search PubMed.
- L. A. Morris, M. Jaspars, J. J. Kettenes-van den Bosch, K. Versluis, A. J. R. Heck, S. M. Kelly and N. C. Price, Tetrahedron, 2001, 57, 3185–3197 CrossRef CAS.
- C. J. Hawkins, Pure Appl. Chem., 1988, 60, 1267–1270 CrossRef CAS.
- P. Comba, N. Dovalil, L. R. Gahan, G. R. Hanson and M. Westphal, Dalton Trans., 2014, 43, 1935–1956 RSC.
- L. R. Gahan and R. M. Cusack, Polyhedron, 2018, 153, 1–23 CrossRef CAS.
- P. Comba and A. Eisenschmidt, Future Directions in Metalloprotein and Metalloenzyme Research, in Biological Magnetic Resonance, ed. G. Hanson and L. Berliner, Springer, Heidelberg, 2017, vol. 3 Search PubMed.
- P. Baur, M. Kühl, P. Comba and L. Behrendt, Mar. Drugs, 2022, 20, 119 CrossRef CAS PubMed.
- P. Comba, L. R. Gahan, G. R. Hanson and M. Westphal, Chem. Commun., 2012, 48, 9364–9366 RSC.
- P. Comba, A. Eisenschmidt, N. Kipper and J. Schiessl, J. Inorg. Biochem., 2016, 159, 70–75 CrossRef CAS PubMed.
- P. Comba, L. R. Gahan, G. R. Hanson, M. Maeder and M. Westphal, Dalton Trans., 2014, 43, 3144–3152 RSC.
- P. Comba, A. Eisenschmidt, L. R. Gahan, D. P. Herten, G. Nette, G. Schenk and M. Seefeld, Chem. – Eur. J., 2017, 23, 12264–12274 CrossRef CAS PubMed.
- P. Baur, V. Gunasekaran, K.-C. Wu, J. Lim, B. Degnan, B. M. Paterson, D. P. Fairlie and P. Comba, in preparation.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS.
- D. J. F. Christine and E. Salomon, J. Nat. Prod., 2002, 689–692 Search PubMed.
- E. W. Schmidt and M. S. Donia, Curr. Opin. Biotechnol., 2010, 21, 827–833 CrossRef CAS.
- P. Kott, Mem. Qd Mus., 1980, 20, 1–47 Search PubMed.
- M. Kühl, L. Behrendt, E. Trampe, K. Qvortrup, U. Schreiber, S. M. Borisov, I. Klimant and A. W. Larkum, Front. Microbiol., 2012, 3, 402 Search PubMed.
- I. Koike, M. Yamamuro and P. C. Pollard, Aust. J. Mar. Freshw. Res., 1993, 44, 173–182 CrossRef.
- R. R. Olson, Mar. Biol., 1986, 93, 437–442 CrossRef.
- P. Kott, J. Nat. Hist., 2004, 38, 2455–2526 CrossRef.
- G. Cox, New Phytol., 1986, 104, 429–445 CrossRef.
- P. Baur, M. Kühl, P. Comba and L. Behrendt, Mar. Drugs, 2022, 20, 119 CrossRef CAS.
- J. W. Schopf and B. M. Packer, Science, 1987, 237, 70–73 CrossRef CAS.
- A. R. Grossman, M. R. Schaefer, G. G. Chiang and J. L. Collier, Microbiol. Rev, 1993, 57, 725–749 CrossRef CAS.
- R. Lewin, Nature, 1976, 261, 697–698 CrossRef CAS.
- R. A. Lewin, Ann. N. Y. Acad. Sci., 1981, 361, 325–329 CrossRef CAS PubMed.
- E. Stackebrandt, E. Seewaldt, V. J. Fowler and K.-H. Schleifer, Arch. Microbiol., 1982, 132, 216–217 CrossRef.
- B. P. Kremer, R. Pardy and R. A. Lewin, Phycologia, 1981, 21, 258–263 CrossRef.
- M. S. Donia, W. F. Fricke, F. Partensky, J. Cox, S. I. Elshahawi, J. R. White, A. M. Phillippy, M. C. Schatz, J. Piel, M. G. Haygood, J. Ravel and E. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1423–E1432 CrossRef CAS.
- V. S. Odintsov, Endocyt. C. Res., 1991, 7, 253–258 Search PubMed.
- T. C. Kline and R. A. Lewin, Symbiosis, 1999, 26, 193–198 Search PubMed.
- E. W. Schmidt, Nat. Chem. Biol., 2008, 4, 466–473 CrossRef CAS.
- T. Stokes, Trends Plant Sci., 2000, 5, 366 Search PubMed.
- K. Sivonen, N. Leikoski, D. P. Fewer and J. Jokela, Appl. Microbiol. Biotechnol., 2010, 86, 1213–1225 CrossRef CAS.
- N. B. Lopanik and K. Clay, Funct. Ecol., 2014, 28, 328–340 Search PubMed.
- S. A. Dyshlovoy and F. Honecker, Mar. Drugs, 2015, 13, 5657–5665 Search PubMed.
- M. Jaspars, D. De Pascale, J. H. Andersen, F. Reyes, A. D. Crawford and A. Ianora, J. Mar. Biol. Assoc., 2016, 96, 151–158 CrossRef.
- M. S. Butler, A. A. Robertson and M. A. Cooper, Nat. Prod. Rep., 2014, 31, 1612–1661 RSC.
- R. K. Singh, S. P. Tiwari, A. K. Rai and T. M. Mohapatra, J. Antibiot., 2011, 64, 401–412 CrossRef CAS.
- A. Shahid, M. Khurshid, B. Aslam, S. Muzammil, H. M. Mehwish, M. S. R. Rajoka, H. F. Hayat, M. H. Sarfraz, M. K. Razzaq, M. A. Nisar and M. Waseem, J. Basic. Microbiol., 2022, 62, 1125–1142 CrossRef.
- J. Ogino, R. E. Moore, G. M. Patterson and C. D. Smith, J. Nat. Prod., 1996, 59, 581–586 CrossRef CAS.
- C. J. Hawkins, M. F. Lavin, K. A. Marshall, A. L. van den Brenk and D. J. Watters, J. Med. Chem., 1990, 33, 1634–1638 CrossRef CAS.
- Y. Hamada, S. Kato and T. Shioiri, Tetrahedron Lett., 1985, 26, 3223–3226 CrossRef CAS.
- M. S. Yasumasa Hamada and T. Shioiri, Tetrahedron Lett., 1985, 26, 5155–5158 CrossRef.
- F. J. Schmitz, M. B. Ksebati, J. S. Chang, J. L. Wang, M. B. Hossain, D. van der Helm, M. H. Engel, A. Serban and J. A. Silfer, J. Org. Chem., 1989, 54, 3463–3472 CrossRef CAS.
- G. Abbenante, D. P. Fairlie, L. R. Gahan, G. R. Hanson, G. K. Pierens and A. L. van den Brenk, J. Am. Chem. Soc., 1996, 118, 10384–10388 CrossRef CAS.
- A. L. van den Brenk, D. P. Fairlie, G. R. Hanson, L. R. Gahan, C. J. Hawkins and A. Jones, Inorg. Chem., 1994, 33, 2280–2289 CrossRef CAS.
- P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS.
- P. Comba, R. Cusack, D. P. Fairlie, L. R. Gahan, G. R. Hanson, U. Kazmaier and A. Ramlow, Inorg. Chem., 1998, 37, 6721–6727 CrossRef CAS PubMed.
- P. V. Bernhardt, P. Comba, D. P. Fairlie, L. A. Gahan, G. R. Hanson and L. Lötzbeyer, Chem. – Eur. J., 2002, 8, 1527–1536 CrossRef CAS PubMed.
- G. Haberhauer and F. Rominger, Eur. J. Org. Chem., 2003, 3209–3218 CrossRef CAS.
- P. Comba, N. Dovalil, G. R. Hanson and G. Linti, Inorg. Chem., 2011, 50, 5165–5174 CrossRef CAS PubMed.
- P. Comba, N. Dovalil, L. R. Gahan, G. Haberhauer, G. R. Hanson, C. J. Noble, B. Seibold and P. Vadivelu, Chem. – Eur. J., 2012, 18, 2578–2590 CrossRef CAS.
- P. Comba, N. Dovalil, G. Haberhauer, K. Kowski, N. Mehrkens and M. Westphal, Z. Anorg. Allg. Chem., 2013, 639(8–9), 1395–1400 CrossRef CAS.
- P. Comba, A. Eisenschmidt, N. Kipper and J. Schiessl, J. Inorg. Biochem., 2016, 159, 70–75 CrossRef CAS.
- P. Comba, A. Eisenschmidt, L. Gahan, G. Hanson, N. Mehrkens and M. Westphal, Dalton Trans., 2016, 45, 18931–18945 RSC.
- P. Comba, L. R. Gahan, G. Haberhauer, G. A. Hanson, C. J. Noble, B. Seibold and A. L. van den Brenk, Chem. – Eur. J., 2008, 14, 4393–4403 CrossRef CAS PubMed.
- P. Comba, N. Dovalil, G. Haberhauer, G. R. Hanson, Y. Karod and T. Taura, J. Biol. Inorg. Chem., 2010, 15, 1129–1135 CrossRef CAS PubMed.
- P. Comba, N. Dovalil, G. Hanson, J. Harmer, C. J. Noble, M. J. Riley and B. Seibold, Inorg. Chem., 2014, 53, 12323–12336 CrossRef CAS.
- P. García-Reynaga and M. S. VanNieuwenhze, Org. Lett., 2008, 10, 4621–4623 CrossRef PubMed.
- P. Baur, P. Comba and G. Velmurugan, Chem. – Eur. J., 2022, 28, e202200249 CrossRef CAS.
- J. Koehnke, A. Bent, W. E. Houssen, D. Zollman, F. Morawitz, S. Shirran, J. Vendome, A. F. Nneoyiegbe, L. Trembleau, C. H. Botting, M. C. Smith, M. Jaspars and J. H. Naismith, Nat. Struct. Mol. Biol., 2012, 19, 767–772 CrossRef CAS PubMed.
- National Library of Medicine (US), National Center for Biotechnology Information, Available from: https://www.ncbi.nlm.nih.gov/nuccore/AY986476.1, 1988.
- J. Koehnke, A. F. Bent, W. E. Houssen, G. Mann, M. Jaspars and J. H. Naismith, Curr. Opin. Struct. Biol., 2014, 29, 112–121 CrossRef CAS PubMed.
- E. Oueis, H. Stevenson, M. Jaspars, N. J. Westwood and J. H. Naismith, Chem. Commun., 2017, 53, 12274–12277 RSC.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 4089–4091 CrossRef CAS PubMed.
- J. A. McIntosh and E. W. Schmidt, ChemBioChem, 2010, 11, 1413–1421 CrossRef CAS PubMed.
- B. F. Milne, P. F. Long, A. Starcevic, D. Hranueli and M. Jaspars, Org. Biomol. Chem., 2006, 4, 631–638 RSC.
- W. E. Houssen and M. Jaspars, ChemBioChem, 2010, 11, 1803–1815 CrossRef CAS PubMed.
- B. F. Milne, L. A. Morris, M. Jaspars and G. S. Thompson, J. Chem. Soc., Perkin Trans. 2, 2002, 1076–1080 RSC.
- T. Ishida, Y. In, M. Doi, M. Inoue, Y. Hamada and T. Shiori, Biopolymers, 1992, 32, 131–143 CrossRef CAS PubMed.
- T. Ishida, Y. In, F. Shinozaki, M. Doi, D. Yamamoto, Y. Hamada, T. Shioiri, M. Kamigauchi and M. Sugiura, J. Org. Chem., 1995, 60, 3944–3952 CrossRef CAS.
- Y. In, M. Doi, M. Inoue, T. Ishida, Y. Hamada and T. Shioiri, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1994, 50(Pt 3), 432–434 CrossRef PubMed.
- P. Comba, N. Dovalil, L. R. Gahan, G. Haberhauer, G. R. Hanson, C. J. Noble, B. Seibold and P. Vadivelu, Chem. – Eur. J., 2012, 18, 2578–2590 CrossRef CAS PubMed.
- D. E. Williams, R. E. Moore and V. J. Paul, J. Nat. Prod., 1989, 52, 732–739 CrossRef CAS.
- M. S. Donia, J. Ravel and E. W. Schmidt, Nat. Chem. Biol., 2008, 4, 341–343 CrossRef CAS PubMed.
- M. R. Prinsep, R. E. Moor, I. A. Levine and G. M. L. Patterson, J. Nat. Prod., 1992, 55, 140–142 CrossRef CAS PubMed.
- R. Banker and S. Carmeli, J. Nat. Prod., 1998, 61, 1248–1251 CrossRef CAS PubMed.
- V. Admi, U. Afek and S. Carmeli, J. Nat. Prod., 1996, 59, 396–399 CrossRef CAS.
- P. Baur, P. Comba, L. R. Gahan and C. Scholz, Aust. J. Chem., 2022, 76, 44–48 CrossRef.
- J. P. Michael and G. Pattenden, Angew. Chem., Int. Ed. Engl., 1993, 1–23 CAS.
- R. M. Cusack, L. Grøndahl, G. Abbenante, D. P. Fairlie, L. R. Gahan, G. R. Hanson and T. W. Hambley, J. Chem. Soc., Perkin Trans. 2, 2000, 323–331 RSC.
- A. L. van den Brenk, D. P. Fairlie, L. R. Gahan, G. R. Hanson and T. W. Hambley, Inorg. Chem., 1996, 35, 1095–1100 CrossRef CAS PubMed.
- A. L. van den Brenk, K. A. Byriel, D. P. Fairlie, L. R. Gahan, G. R. Hanson, C. J. Hawkins, A. Jones, C. H. L. Kennard, B. Moubaraki and K. S. Murray, Inorg. Chem., 1994, 33, 3549–3557 CrossRef CAS.
- G. R. Hanson, C. J. Noble and S. Benson, Biol. Magn. Reson., 2009, 28, 105–173 CrossRef CAS.
- P. V. Bernhardt, P. Comba, T. W. Hambley, S. S. Massoud and S. Stebler, Inorg. Chem., 1992, 31, 2644–2651 CrossRef CAS.
- P. Comba, T. W. Hambley and M. Ströhle, Helv. Chim. Acta., 1995, 78, 2042–2047 CrossRef CAS.
- P. Comba, T. W. Hambley, G. Lauer and N. Okon, MOMEC97, a molecular modeling package for inorganic compounds, Heidelberg, 1997 Search PubMed.
- S. Xie, A. I. Savchenko, M. Kerscher, R. L. Grange, E. H. Krenske, J. R. Harmer, M. J. Bauer, N. Broit, D. J. Watters, G. M. Boyle, P. V. Bernhardt, P. G. Parsons, P. Comba, L. R. Gahan and C. M. Williams, Eur. J. Org. Chem., 2018, 1465–1476 CrossRef CAS.
- P. M. Dewick, Essentials of Organic Chemistry – For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry, John Wiley & Sons Inc., 2006 Search PubMed.
- R. R. Olson and R. McPherson, J. Exp. Mar. Biol. Ecol., 1987, 110, 245–256 CrossRef.
- P. Comba, L. R. Gahan, G. R. Hanson, M. Maeder and M. Westphal, Dalton Trans., 2014, 43, 3144–3152 RSC.
- P. Comba and A. Eisenschmidt, in Future Directions in Metalloprotein and Metalloenzyme Research (Eds.: G. Hanson, L. Berliner), Springer International Publishing, Cham, 2017, pp. 13–32 Search PubMed.
- P. Comba, N. Dovalil, L. R. Gahan, G. R. Hanson and M. Westphal, Dalton Trans., 2014, 43, 1935–1956 RSC.
- G. Velmurugan, P. Baur and P. Comba, Angew. Chem., Int. Ed., 2024, e202319530 CAS.
- M. Westphal, PhD thesis, Universität Heidelberg, 2013.
- N. Mehrkens, PhD thesis, Universität Heidelberg, 2015.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS.
- A. C. Jones, L. Gu, C. M. Sorrels, D. H. Sherman and W. H. Gerwick, Curr. Opin. Chem. Biol., 2009, 13.2, 216–223 CrossRef PubMed.
Footnote |
| † In part adapted from the PhD Thesis of PB (Heidelberg University, 2023). |
| This journal is © The Royal Society of Chemistry 2025 |