
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTipping the balance between twist and chair ligand conformers in multinuclear ethylzinc cyclophosphazenates†‡
Ramamoorthy
Boomishankar
 ab,
Philip I.
Richards
a and
Alexander
Steiner
*a
ab,
Philip I.
Richards
a and
Alexander
Steiner
*a
aDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: a.steiner@liverpool.ac.uk
bDepartment of Chemistry, Indian Institute of Science Education and Research, Pune, Dr Homi Bhabha Road, Pune-411008, India
First published on 27th December 2024
Abstract
Hexaanionic cyclophosphazenate ligands [(RN)6P3N3]6− provide versatile platforms for the assembly of multinuclear metal arrays due to their multiple coordination sites and highly flexible ligand core structure. This work investigates the impact of incrementally increasing the steric demand of the ligand periphery on the coordination behavior of ethylzinc arrays. It shows that the increased congestion around the ligand sites is alleviated by progressive condensation with the elimination of diethylzinc. The condensation is accompanied by a switch in ligand conformation: the phosphazene ring of the methyl derivative adopts the chair conformer in the dimeric complex (EtZn)12[(RN)6P3N3]2, while the more sterically demanding isobutyl derivative exhibits twist conformers in both the monomeric complex (EtZn)6[(iBuN)6P3N3] and the condensed dimer (EtZn)8Zn2[(iBuN)6P3N3]2. Ethyl and propyl derivatives mark a tipping point: the dimeric complex (EtZn)12[(RN)6P3N3]2 exhibits the chair conformation and is observed in solution and within the stabilising environment of a co-crystal. However, when crystallising in pure form, it transforms with the loss of one equivalent of Et2Zn into the collapsed dimer (EtZn)10Zn[(RN)6P3N3]2 which features the twist conformer.
Introduction
Cyclotriphosphazenes are prominent inorganic N-heterocycles featuring the extremely robust P3N3 ring system.1,2 A great variety of functionalised substituents have been attached to the phosphazene ring, resulting in a multitude of systems that offer a wide range of applications, for example in drug delivery,3 gas sorption,4,5 liquid crystals,6 sensors,7 phase transfer catalysis,8 ferroelectrics9 and chiral separation.10,11 When equipped with additional donor sites, they can serve as polydentate ligands that exhibit a rich coordination chemistry which has been studied extensively by Chandrasekhar and others.12–14Polyanionic cyclophosphazenates [(RN)x(RNH)6−xP3N3]x− provide highly flexible ligand platforms with multiple coordination sites that facilitate the accommodation of multinuclear metal arrays.15–23 They are obtained via the deprotonation of hexa(organoamino) cyclotriphosphazenes, (RNH)6P3N3. Progressive metalation leads to a greater distortion of the P3N9 core, which can be attributed to the increasing negative charge on deprotonated N(exo) atoms and the participation of N(ring) atoms in metal coordination.16 Various ring conformations have been encountered demonstrating the flexibility but also the robustness of the ligand core. The two principal ring conformers of the hexaanionic ligand [(RN)6P3N3]6− are the chair featuring a P3N9 core of C3v symmetry and the twist characterised by a C2 axis running through opposite phosphorus and nitrogen atoms in the phosphazene ring. The chair is found predominantly in lithium complexes,16,17 while the twist occurs in the presence of more acidic metal centres, such as aluminium18 and titanium19 (Fig. 1).
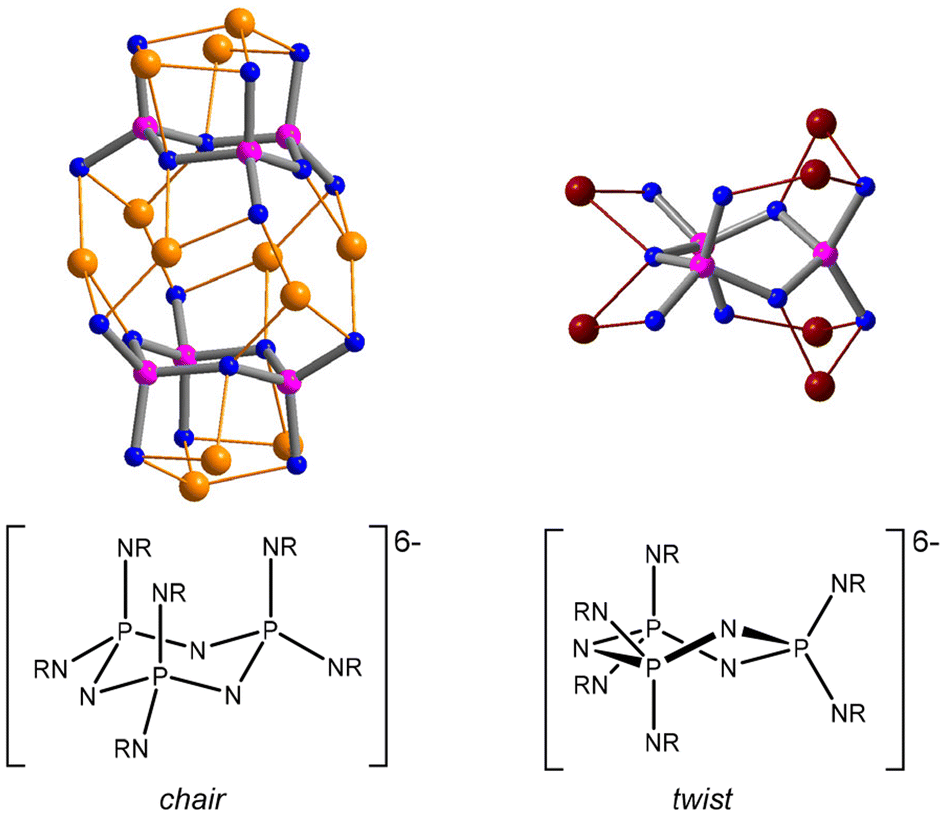 | ||
| Fig. 1 Principal conformers of phosphazenate ligands alongside depictions of the core metal–ligand structures of representative complexes: thf4Li12[(CyN)6P3N3]2 (chair)21 and (Me2Al)6[(iBuN)6P3N3] (twist).18 | ||
Ethylzinc complexes show a more ambivalent behaviour. The monomeric complex (EtZn)6[(CyN)6P3N3] (Cy = cyclohexyl) features the twist conformer,22 while dimeric complexes of the trianionic ligand [(RNH)3(RN)3P3N3]3− exhibit the chair conformer.23 This work investigates the steric impact of the alkyl substituents on the coordination of ethylzinc arrays with the aim of exploring the boundary between the two regimes. Diethylzinc was employed as the metalating agent. It is an important reagent in organic synthesis,24,25 for example, in the enantio-selective alkylation of carbonyls26 and imines27 and in cyclopropanation reactions.28 Furthermore, it is used as a volatile precursor in metalorganic chemical vapor deposition (MOCVD) for the preparation of wide band gap II–VI semiconducting films and nanostructures.29
Results and discussion
The hexaprotic precursors (RNH)6P3N3 used in this study included methyl (1), ethyl (2), propyl (3) and isobutyl (4) derivatives.30 They were treated with 6.1 equivalents of diethylzinc to generate complexes of the fully deprotonated ligands [(RN)6P3N3]6−. Reactions were monitored with 31P NMR and products were analysed by single crystal X-ray diffraction. Scheme 1 lists the coordination modes that were observed in the crystal structures. Scheme 2 displays the reactions that led to the formation and subsequent condensation of ethylzinc complexes discussed here. | ||
| Scheme 1 Coordination modes observed in crystal structures. Dashed lines indicate long Zn–N interactions (>2.3 Å). | ||
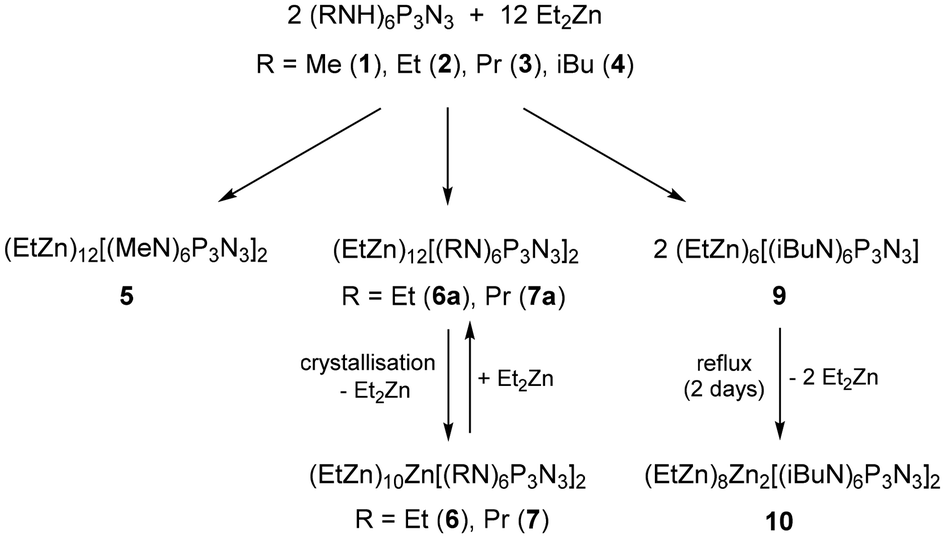 | ||
| Scheme 2 Formation and subsequent condensation reactions of ethylzinc phosphazenates. | ||
The reaction of the methyl derivative 1 with Et2Zn was conducted in thf/hexane due to its poor solubility in pure hydrocarbon solvents. A clear solution was obtained after a short reflux (15 min). The 31P NMR taken from the reaction solution indicates three phosphorus environments at δ 44.9, 37.7 and 26.8. Crystals suitable for X-ray structure analysis were obtained from a concentrated solution in hexane. The crystal structure consists of a dimeric complex of composition (EtZn)12[(MeN)6P3N3]2 (Fig. 2). Its molecular symmetry is characterised by a local C2 axis that runs between the two ligands. The ligands exhibit chair conformation and are arranged in a similar way to that in the corresponding lithium complexes.16,17 They are connected via six EtZn groups, each forming a bridge between an N(ring) and an equatorial N(exo) site. The remaining EtZn groups are bonded to the axial N(exo) sites: for each ligand, there is one EtZn group that caps the three axial N(exo) sites in tridentate type VIII (Zn4, Zn10), one that binds to an axial N(exo) site in monodentate type I (Zn6, Zn12) and one that is chelated in a bidentate fashion to two axial N(exo) sites with an additional long interaction with an N(ring) site (type IVa) (Zn5, Zn11).
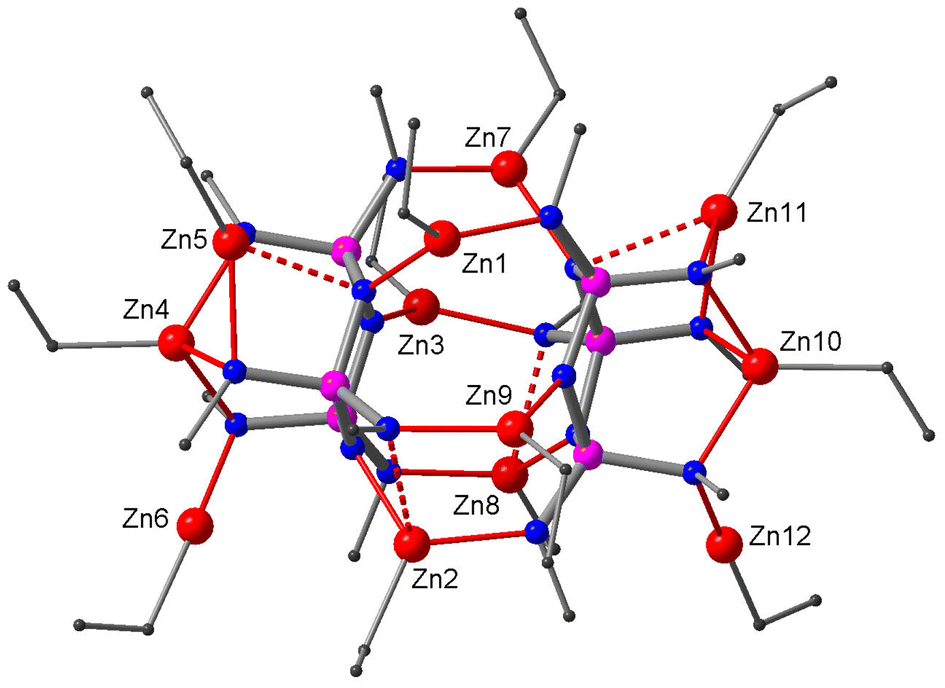 | ||
| Fig. 2 Crystal structure of the dimeric complex 5. Zn, red; N, blue; P, purple; C, grey. Hydrogen atoms are omitted for clarity. | ||
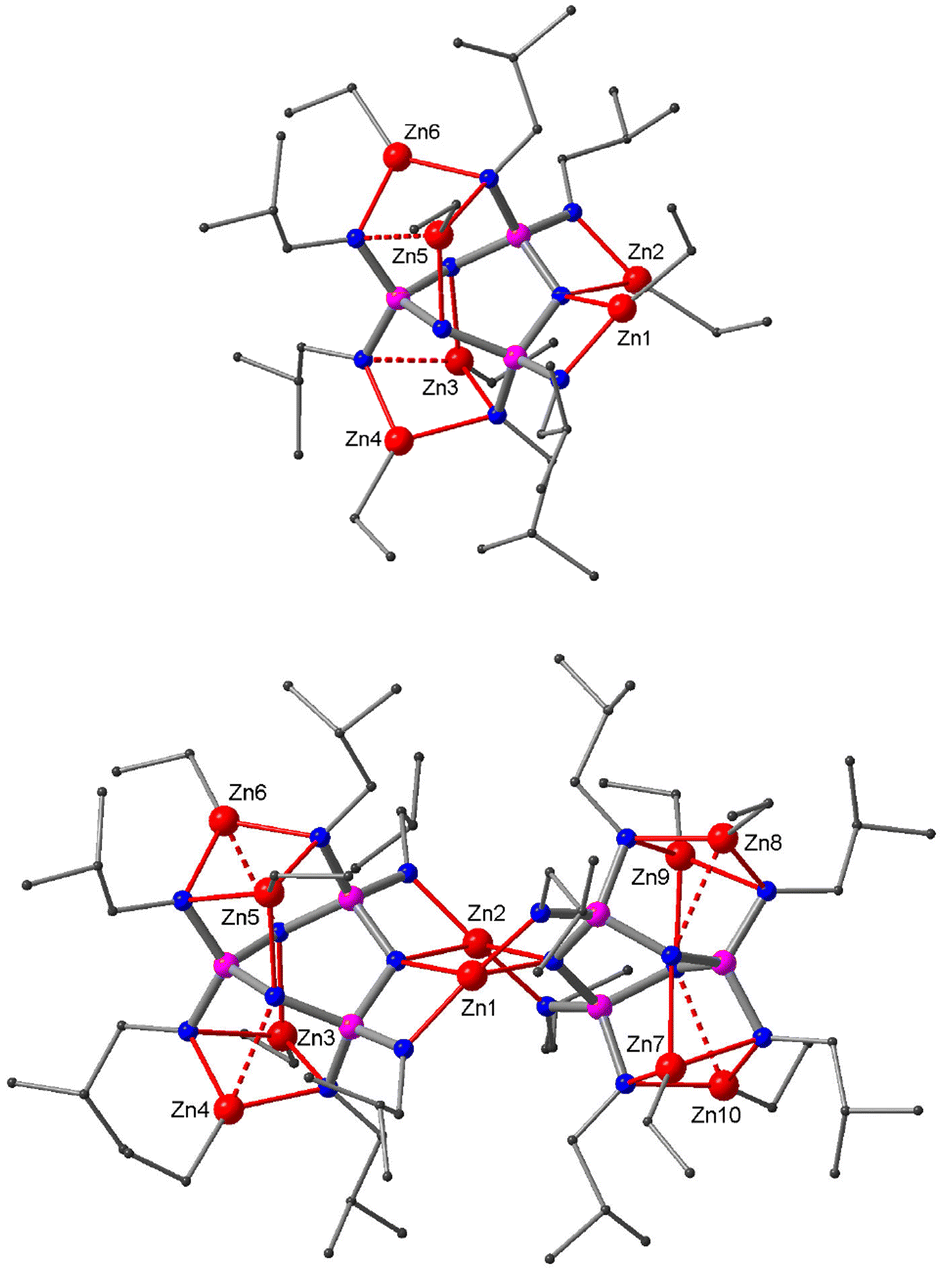
Reactions of ethyl (2) and propyl derivatives (3) with six equivalents of Et2Zn were carried out in hexane. Clear solutions were obtained after a short reflux (15 min). The 31P NMR spectra of the reaction mixtures show a singlet (δ 39.9 for the reaction of 2, δ 40.4 for 3), indicating the chemical equivalence of the three P-atoms in the phosphazene ring. The X-ray structure analyses of the crystals grown from the reaction solutions reveal crystal structures of composition (EtZn)10Zn[(RN)6P3N3]2 (R = Et (6), Pr (7)) (Fig. 3). The complexes can be described as ‘collapsed’ dimers that contain ten EtZn groups and one Zn centre (Zn1) that does not bear an ethyl group. The two ligands show different coordination patterns and ring conformations: one adapts the characteristic twist form, while the other sits somewhere between twist and boat according to its ring puckering parameters (see the ESI‡). Zn1 occupies a central position between the two ligands where it is chelated by the twist ligand in bidentate type III and by the twist–boat ligand in bidentate type IV, resulting in a distorted tetrahedral coordination environment. Furthermore, there are two bridging EtZn groups (Zn2, Zn7), each of which is chelated by one ligand in type III and coordinates the other in type I. In addition, each ligand hosts four non-bridging EtZn groups. The twist ligand accommodates two EtZn groups in type IV (Zn3, Zn5) and two in type Va sites (Zn4, Zn6), while the twist–boat ligand exhibits four distinct coordination modes of types III (Zn9), IIIa (Zn11), IV (Zn10) and V (Zn8) (see also ESI Fig. S13‡).
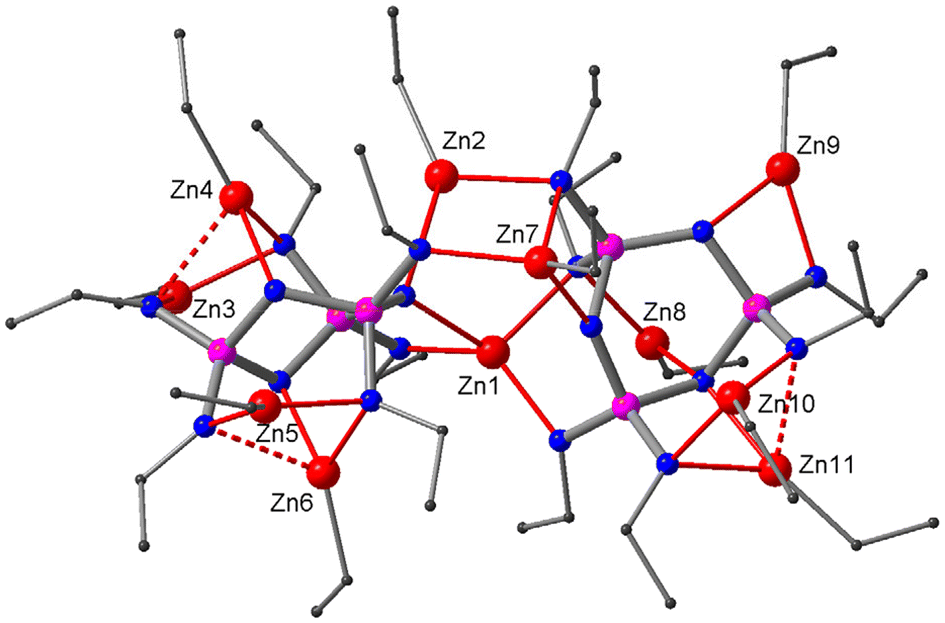 | ||
| Fig. 3 Crystal structure of the collapsed dimer 6. Zn, red; N, blue; P, purple; C, grey. Hydrogen atoms are omitted for clarity. | ||
To our surprise, the 31P NMR spectra of the redissolved crystals of 6 and 7 are different from those recorded prior to crystallization. They consist of two sharp signals with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (6: δ 44.7, 39.5; 7: δ 45.2, 38.9), which can be attributed to two inequivalent sets of P-nuclei. When the crystals are redissolved and treated with one equivalent of Et2Zn, the 31P NMR spectra revert to the same singlet recorded before crystallisation. This implies that the species that initially formed (here referred to as 6a and 7a) must contain twelve EtZn groups but lose one equivalent of Et2Zn during crystallisation.
:2 (6: δ 44.7, 39.5; 7: δ 45.2, 38.9), which can be attributed to two inequivalent sets of P-nuclei. When the crystals are redissolved and treated with one equivalent of Et2Zn, the 31P NMR spectra revert to the same singlet recorded before crystallisation. This implies that the species that initially formed (here referred to as 6a and 7a) must contain twelve EtZn groups but lose one equivalent of Et2Zn during crystallisation.
In our quest of uncovering the identity of species 6a and 7a, we were aided by a fortunate accident: a reaction mixture of 3 and excess Et2Zn that must have been contaminated with moisture yielded, amongst an indistinct solid residue, a few crystals that were suitable for X-ray structure analysis. It revealed a co-crystal (8) containing the complexes {(EtZn)12[(PrN)6P3N3]2} (8a) and {(EtZn)10Zn3O3[PrNH(PrN)5P3N3]2} (8b), both exhibiting crystallographic C2 symmetry (Fig. 4). Complex 8b consists of two moieties (EtZn)5[PrNH(PrN)5P3N3] that sandwich a planar Zn3O3 ring in a similar way to that in the previously reported complex (EtZn)6Zn3O3[(PrNH)3(PrN)3P3N3]2.23 However, more relevant to our study is complex 8a as it shows the same coordination pattern and the same molecular symmetry as the dimeric complex 5 that hosts twelve EtZn groups.
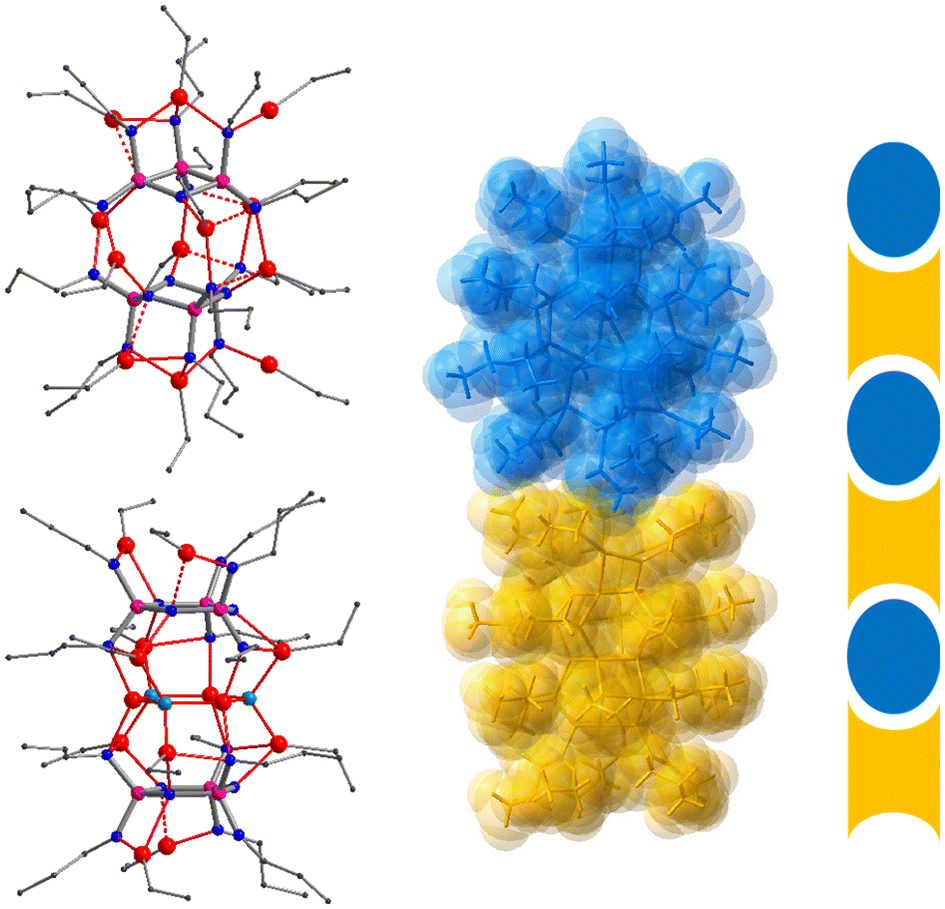 | ||
| Fig. 4 Left: crystal structure of the co-crystal 8 consisting of complexes 8a (top) and 8b (bottom). Zn, red; N, blue; P, purple; C, grey; O, green. Hydrogen atoms are omitted for clarity. Right: superimposition with the space-filling model to illustrate the ball-and-socket packing of 8a (blue) and 8b (orange) in the co-crystal. | ||
We conclude that 7a, the species that exists in solution prior to crystallization, is identical to 8a. This raises the question of why it appears in the co-crystal of 8 but undergoes condensation when crystallising in pure form. A close inspection of the crystal packing of 8 reveals that 8a has an ellipsoidal shape, while 8b is roughly cylindrical with bowl-shaped cavities at either end. In the crystal, 8a and 8b are stacked in an alternate fashion forming a kind of ball-and-socket arrangement whereby the tip of the ellipsoid of 8a rests in the cavity of 8b (Fig. 4). Evidently, this arrangement must have a stabilising effect on 8a. In the absence of a suitable socket such as 8b, it will undergo internal condensation with the loss of one molecule of Et2Zn yielding the collapsed dimer 7 which may pack more efficiently in the crystal than 7a.
The reaction of the isobutyl derivative 4 with six equivalents of Et2Zn in hexane produces after a short reflux a singlet signal in the 31P NMR spectrum (δ 42.6). Upon continued refluxing, an additional AX2 signal pattern emerges (δ 43.4(t), 41.4(d)) which becomes the sole species after heating the mixture for two days. We were able to crystallise both compounds and analyse their X-ray structures. The crystal structure obtained from the initial reaction consists of a monomeric complex of composition (EtZn)6[(iBuN)6P3N3] (9) (Fig. 5). It has the same core structure as the corresponding cyclohexyl derivative (EtZn)6[(CyN)6P3N3] reported earlier.22 The complex is approximately C2 symmetric and adopts the twist conformation. Two EtZn groups (Zn1, Zn2) occupy type III chelates that share the same N(ring) site, a further two are in type IV sites (Zn4, Zn6) and another two in type Va sites (Zn3, Zn5).
 | ||
| Fig. 5 Crystal structure of the monomeric complex 9 (top) and the condensed dimer 10 (bottom). Zn, red; N, blue; P, purple; C, grey. Hydrogen atoms are omitted for clarity. | ||
The crystal structure of the product obtained after prolonged reflux consists of the dimeric complex (EtZn)8Zn2[(iBuN)6P3N3]2 (10) (Fig. 5). The presence of two Zn atoms bearing no ethyl groups (Zn1, Zn2) confirms that 10 is the product of the condensation of two complexes of 9 with the elimination of two equivalents of Et2Zn. The molecular symmetry of 10 is close to the point group D2; both of its ligands exhibit the twist conformation. Zn1 and Zn2 bridge the ligands via type III chelates. Furthermore, each ligand accommodates four EtZn groups: two in type IVa sites (Zn4, Zn6, Zn8, Zn10) and two in tridentate type VII coordination sites (Zn3, Zn5, Zn7, Zn9). Both monomeric 9 and dimeric 10 feature the typical coordination pockets of twist conformers but show slight variations in the coordination modes (see also Fig. S13 in the ESI‡).
In line with other metal phosphazenates, the metalation leads to a marked distortion of the ligands due to the enhanced negative charge on the deprotonated N(exo) sites and the involvement of N(ring) sites in metal coordination. On average, the P–N(exo) bonds become shorter, the P–N(ring) bonds become longer, the exocyclic N–P–N angles become wider and the ring angles become sharper. Furthermore, bond lengths and angles show a much greater distribution in the metal complexes than in the precursor molecules (see also Fig. S15 and S16 in the ESI‡).
The marked distortion of the ligand core causes the P3N3 rings in the metal complexes to pucker. The two principal ring conformers that were observed are chair and twist. Both were identified by their characteristic distribution of ring torsion angles and their ring puckering parameters31 (see Fig. S17 and Table S2 in the ESI‡). The chair conformation is adopted in dimers 5 and 8a, while the twist conformation occurs in the collapsed dimers 6 and 7 as well as in the monomeric complex 9 and its condensed dimer 10. In line with other complexes of hexaanionic phosphazenates, the chair conformer is found only in dimers that accommodate twelve metal centres, six connecting the two ligands via equatorial N(exo) and N(ring) sites and six coordinating the axial N(exo) sites. The twist conformer, on the other hand, appears in monomeric and dimeric complexes hosting fewer than twelve metal centres. It offers two adjacent bidentate type III chelates and four coordination pockets of either bidentate type IV, tridentate types VI and VII or some intermediate form. In addition to the chair and twist conformers, a third conformer was observed which is less symmetrical and sits, according to its ring puckering parameters, somewhere between twist and boat. It appears in the collapsed dimers 6 and 7 alongside the twist conformer. Its existence may be attributed to the presence of only one bridging Zn atom bearing no ethyl group (Zn1 in Fig. 3), which forces the complex into an asymmetric arrangement, thereby preventing one of the ligands from adopting the more common twist conformation. Complex 10 demonstrates that indeed two such bridging Zn atoms are required to maintain a symmetrical dimer with two twist conformers (see also ESI Fig. S19‡).
The Zn–N bond lengths span a wide range. The shortest bond lengths measure 1.893(9) and 1.902(9) Å and are found in the two linear Et–Zn–N(exo) moieties of 5 that are coordinated in a monodentate fashion (Zn6 and Zn12 in Fig. 2); their C–Zn–N angles measure 175.3(6)° and 177.2(6)°. Two-coordinated linear Et–Zn–N arrangements are very rare. The only other example that has been reported is stabilised by a bulky amide ligand.32 In 5, however, the steric demand seems to be imposed by the crowding of EtZn groups around ligand sites. Short Zn–N bonds also appear in the type III chelates of 9 and 10 (Zn1 and Zn2 in Fig. 5); their Zn–N(exo) bonds are much shorter (1.920(3)–1.945(4) Å) than their Zn–N(ring) bonds (2.201(4)–2.248(3) Å). The bonds of other bidentate chelates lie somewhere between the two extremes. Longer Zn–N bonds tend to involve N-sites that coordinate two metal atoms. Zinc atoms of EtZn groups residing in bidentate chelates have a planar coordination environment. A deviation from planarity indicates that there is an additional weak interaction with another N-site. Coordination modes with weak interactions are signified by types IIIa, IVa, and Va. These are drawn with dashed lines in Scheme 1 and in the crystal structures. A more evenly balanced tridentate coordination of type VIII exists in 5 that involves the three axial N(exo) sites of the chair conformer. A tridentate coordination was also observed with a twist conformer in the form of type VII in 10.
Bond lengths in EtZn phosphazenates are comparable to those found in zinc complexes of neutral phosphazenes equipped with donor side arms33–36 and in complexes containing anionic ligands featuring N–P–N37 or N–P–N–P–N38 chelates. Zn–C bond lengths, on the other hand, show a much tighter margin, averaging 1.95 Å, which is similar to the bond distance observed in the crystals of diethylzinc.39
The wide range of Zn–N distances indicates that the pathways between the coordination modes depicted in Scheme 1 are rather smooth and require only subtle shifts of EtZn groups aided by some conformational readjustment of the ligand. This flexibility facilitates dynamic behaviour and may explain the difference in the chemical equivalences of phosphorus atoms encountered in solution and in the crystal structures. Only in the cases of 5 and 10 does the multiplicity of 31P NMR signals observed in solution reflect the molecular symmetry in the crystal. In all other instances, it indicates a higher symmetry for the solution species. For example, the monomeric complex 9 exhibits only one chemical shift in the 31P NMR spectrum, but its crystal structure indicates molecular C2 symmetry with two distinct sets of P-atoms.
To shed some light on the fluxional behaviour, we conducted a variable temperature study of 9 in hexane.§31P NMR spectra were recorded at intervals from room temperature down to 178 K (Fig. 6). Upon cooling, the singlet broadens and splits into an AX2 signal pattern (δ 42.7(d), 42.4(t)). This indicates a dynamic process at room temperature that renders the three P-atoms of the ligand equivalent, while at lower temperature, the two inequivalent P-environments mirror the molecular C2 symmetry of the crystal structure. It would be rather speculative to deliberate an exact mechanism of the dynamic process. Nonetheless, it must involve at least a sufficiently rapid exchange between ligand conformers and a fast enough movement of EtZn groups between N-sites to equalise the three P-atoms on the NMR timescale. A similar case has been reported where four EtZn groups engage in fluxional behaviour around a tetraanionic amidinate ligand.40
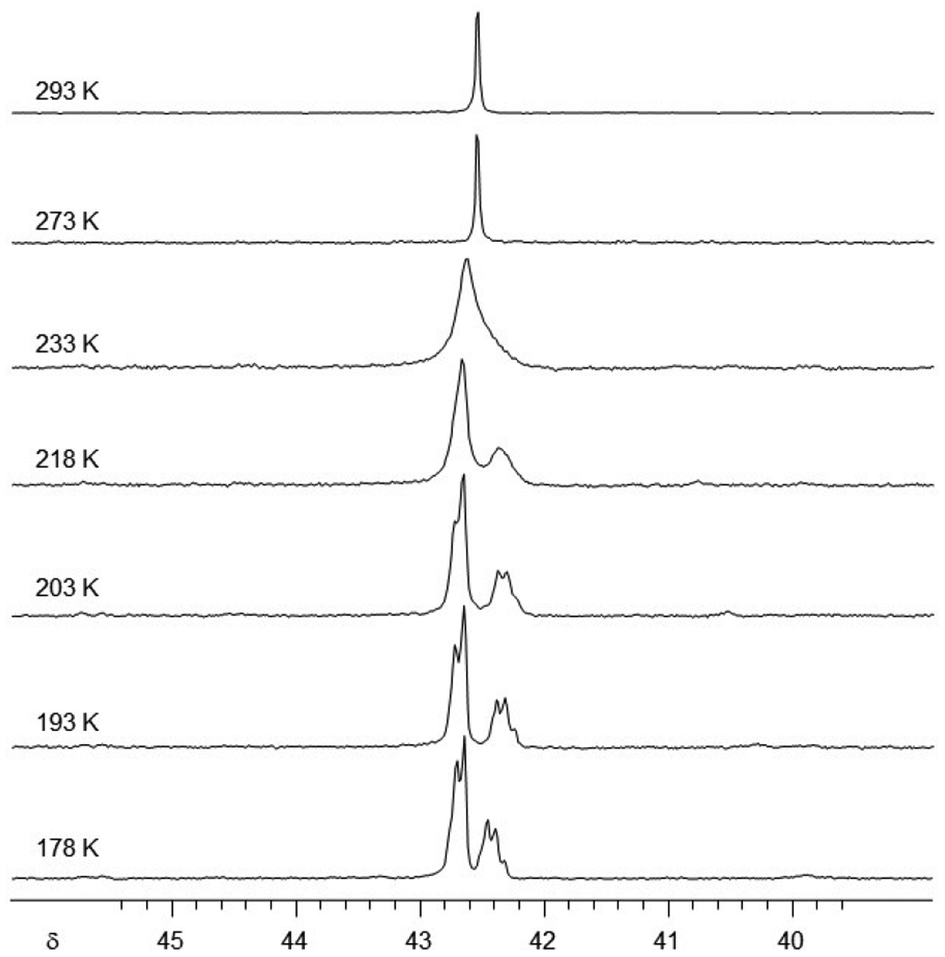 | ||
| Fig. 6 Variable temperature 31P NMR of 9 in hexane. | ||
The condensation reactions of ethylzinc phosphazenates are somewhat reminiscent of Schlenk equilibria.41 The increased congestion around sterically more demanding ligands is alleviated through condensation with the elimination of Et2Zn, which reduces the number of metal centres in the complex. The methyl derivative 5 has still sufficient space for hosting twelve EtZn groups in a dimeric complex. Ethyl and propyl derivatives form the corresponding dimers (6a and 7a) only in solution and in the stabilising environment of a co-crystal. However, they condense upon crystallisation with the elimination of one equivalent of Et2Zn to form collapsed dimers 6 and 7. The isobutyl ligand, on the other hand, is sufficiently bulky to support the monomeric complex 9. It aggregates into the dimeric complex 10 only after prolonged heating, in which case two equivalents of Et2Zn are eliminated.
Conclusion
This study shows that hexaanionic cyclophosphazenates offer robust but at the same time highly flexible ligand platforms for multinuclear assemblies of ethylzinc. The coordination behaviour of the ligand is controlled by the steric demand of the alkyl substituents. Overcrowding can be mitigated through condensation with the elimination of Et2Zn, which lowers the metal count in the complex. Unlike other metal phosphazenates, EtZn complexes feature both chair and twist conformers. The methyl derivative displays the chair conformer that is characteristic of dimers hosting twelve EtZn groups, while isobutyl complexes contain the twist conformer that appears in monomeric complexes and condensed dimers. Complexes of ligands equipped with ethyl and propyl groups mark the tipping point between the two conformers, exhibiting the chair conformer in solution or in the stabilising environment of a co-crystal but the twist conformer in the crystals of the pure complex. This work demonstrates that the ligand system can be finely tuned by simple modifications of its periphery to provide tailored platforms for multinuclear metal arrays.Data availability
The data supporting this article have been included as part of the ESI.‡ Crystallographic data have been deposited at the CCDC under 2383654 (9), 2383655 (5), 2383656 (8), 2383657 (6), 2383658 (7) and 2383659 (10).‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC.References
- H. R. Allcock, Chem. Rev., 1972, 72, 315 CrossRef CAS.
- V. Chandrasekhar and V. Krishnan, Adv. Inorg. Chem., 2002, 53, 159 CrossRef CAS.
- G. Casella, S. Carlotto, F. Lanero, M. Mozzon, P. Sgarbossa and R. Bertani, Molecules, 2022, 27, 3523 CrossRef PubMed.
- H. R. Allcock, Acc. Chem. Res., 1978, 11, 81 CrossRef CAS.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1741 CrossRef.
- J. Barberá, M. Bardají, J. Jiménez, A. Laguna, M. P. Martínez, L. Oriol, J. L. Serrano and I. Zaragozano, J. Am. Chem. Soc., 2005, 127, 8994 CrossRef PubMed.
- A. Uslu, S. O. Tümay and S. Yeşilot, J. Photochem. Photobiol., C, 2022, 53, 100553 CrossRef CAS.
- M. Craven, R. Yahya, E. Kozhevnikova, R. Boomishankar, C. M. Robertson, A. Steiner and I. Kozhevnikov, Chem. Commun., 2013, 349 RSC.
- S. Deswal, R. Panday, D. R. Naphade, P.-A. Cazade, S. Guerin, A. Steiner, S. Ogale, T. D. Anthopoulos and R. Boomishankar, Small, 2023, 19, 2300792 CrossRef CAS PubMed.
- A. Uslu and S. Yeşilot, Coord. Chem. Rev., 2015, 291, 28 CrossRef CAS.
- C. Jose, N. Sradha and R. Boomishankar, Inorg. Chem., 2024, 63, 18788 CrossRef CAS PubMed.
- V. Chandrasekhar, P. Thilagar and B. M. Pandian, Coord. Chem. Rev., 2007, 251, 1045 CrossRef CAS.
- V. Chandrasekhar and S. Nagendran, Chem. Soc. Rev., 2001, 30, 193 RSC.
- V. Chandrasekhar and B. M. Pandian, Acc. Chem. Res., 2009, 42, 1047 CrossRef CAS PubMed.
- A. Steiner, S. Zacchini and P. I. Richards, Coord. Chem. Rev., 2002, 227, 19 CrossRef.
- F. Rivals, G. T. Lawson, M. A. Benson, P. I. Richards, S. Zacchini and A. Steiner, Inorg. Chim. Acta, 2011, 372, 304 CrossRef CAS.
- G. T. Lawson, F. Rivals, M. Tascher, C. Jacob, J. F. Bickley and A. Steiner, Chem. Commun., 2000, 341 RSC.
- P. I. Richards, G. T. Lawson, J. F. Bickley, C. M. Robertson, J. A. Iggo and A. Steiner, Inorg. Chem., 2019, 58, 3355 CrossRef CAS PubMed.
- R. Boomishankar, P. I. Richards, A. K. Gupta and A. Steiner, Organometallics, 2010, 29, 2515 CrossRef CAS.
- F. Rivals and A. Steiner, Chem. Commun., 2001, 1426 RSC.
- A. Steiner and D. S. Wright, Angew. Chem., Int. Ed. Engl., 1996, 35, 636 CrossRef CAS.
- G. T. Lawson, C. Jacob and A. Steiner, Eur. J. Inorg. Chem., 1999, 1881 CrossRef CAS.
- R. Boomishankar, P. I. Richards and A. Steiner, Angew. Chem., Int. Ed., 2006, 45, 4632 CrossRef CAS PubMed.
- L. Stahl, in Comprehensive Organometallic Chemistry, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, 3rd edn, 2006, vol. 2, p. 309 Search PubMed.
- E. Erdik, Organozinc Reagents in Organic Synthesis, CRC Press, Boca Raton, Fl, 1996 Search PubMed.
- L. Pu and H.-B. Yu, Chem. Rev., 2001, 101, 757 CrossRef CAS PubMed.
- K. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874 CrossRef CAS PubMed.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 102, 977 CrossRef PubMed.
- J. Wang and M. Isshiki, in Springer Handbook of Electronic and Photonic materials, ed. S. Kasab and P. Capper, Springer, New York, 2006, p. 325 Search PubMed.
- J. F. Bickley, R. Bonar-Law, G. T. Lawson, P. I. Richards, F. Rivals, A. Steiner and S. Zacchini, Dalton Trans., 2003, 1235 RSC.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354 CrossRef CAS.
- M. J. C. Dawkins, E. Middleton, C. E. Kefalidis, D. Dange, M. M. Juckel, L. Maron and C. Jones, Chem. Commun., 2016, 52, 10490 RSC.
- E. W. Ainscough, A. M. Brodie, P. J. B. Edwards, G. B. Jameson, C. A. Otter and S. Kirk, Inorg. Chem., 2012, 51, 10884 CrossRef CAS PubMed.
- Y. Byun, D. Min, J. Do, H. Yun and Y. Do, Inorg. Chem., 1996, 35, 3981 CrossRef CAS PubMed.
- V. Chandrasekhar, B. M. Pandian and R. Azhakar, Inorg. Chem., 2006, 45, 3510 CrossRef CAS PubMed.
- V. Chandrasekhar, V. Krishnan, A. Steiner and J. F. Bickley, Inorg. Chem., 2004, 43, 166 CrossRef CAS PubMed.
- B. Goswami, T. J. Feuerstein, R. Yadav, S. Lebedkin, P. J. Boden, S. T. Steiger, G. Niedner-Schatteburg, M. Gerhards, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2021, 27, 15110 CrossRef PubMed.
- S. K. Pandey, A. Steiner, H. W. Roesky and D. Stalke, Inorg. Chem., 1993, 32, 5444 CrossRef CAS.
- J. Bacsa, F. Hanke, S. Hindley, R. Odedra, G. R. Darling, A. C. Jones and A. Steiner, Angew. Chem., Int. Ed., 2011, 50, 11685 CrossRef CAS PubMed.
- B. Gutschank, S. Schulz, U. Westphal, D. Bläser and R. Boese, Organometallics, 2010, 29, 2093 CrossRef CAS.
- J. Boersma and J. G. Noltes, Tetrahedron Lett., 1966, 7, 1521 CrossRef.
Footnotes |
| † Dedicated to Prof. Vadapalli Chandrasekhar in celebration of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2383654–2383659. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03004d |
| § Variable temperature measurements of 6, 6a, 7 and 7a were hampered by poor solubility at lower temperature. |
| This journal is © The Royal Society of Chemistry 2025 |