
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced methods for the undecachlorination of [HCB11H11]− and syntheses of [XCB11Cl11]− from [HCB11Cl11]− (X = Cl, Br, I, NH2)†
Jack P.
Raker
 ,
Jovanny J.
Contreras
,
S. Olivia
Gunther
and
Oleg V.
Ozerov
*
,
Jovanny J.
Contreras
,
S. Olivia
Gunther
and
Oleg V.
Ozerov
*
Department of Chemistry, Texas A&M University, College Station, TX 77842, USA. E-mail: ozerov@chem.tamu.edu
First published on 28th March 2025
Abstract
Utilizing SbCl3/SbCl5 mixtures at reflux, or of TCCA in a solid–solid reaction at 200 °C (with properly described precautions) are described as reliable and convenient procedures for the conversion of [HCB11H11]− into [HCB11Cl11]−. Procedures for the derivatization of the carbon vertex of [HCB11Cl11]− to prepare [XCB11Cl11]− salts (X = Cl, Br, I, NH2) are also formulated.
Anions based on the 1-carba-closo-dodecaborate cage (hereafter: carboranes)1 are a class of weakly coordinating anions2 with many desirable properties.3,4 The parent carborane [HCB11H11]− can be substituted at the boron and carbon positions while maintaining cage integrity; these synthetic efforts have been thoroughly reviewed.3,4 Among the various substituted carboranes, [HCB11Cl11]− possesses an attractive combination of weak basicity, resistance to oxidation, robustness, and solubility.5,6 It has allowed isolation of one of the strongest possible purely Brønsted acids H[HCB11Cl11], which is stable to sublimation at 200 °C.7,8 It has also been used in the isolation of highly reactive carbocations, main group cations,9–13 as well as shown to be a competent counteranion in high-temperature, transition metal-catalyzed olefin polymerization.14 Accordingly, development of a reliable and convenient synthesis of [HCB11Cl11]− has attracted considerable attention.
The synthesis of [HCB11Cl11]− by Reed et al., originally reported in 1998, utilizes ICl as the chlorination agent in triflic acid as solvent at 220 °C (Fig. 1).7,15 It is consistently reproducible, but requires an expensive pressure-rated reactor, which also dramatically limits the reaction scale (to about 2 g in Reed's procedure). In addition, it is a lengthy process, taking weeks to the isolation of the pure product. An alternative method that can use non-gaseous reagents at atmospheric pressure and in regular glassware would clearly be advantageous.
 | ||
| Fig. 1 Previously reported methods for the synthesis of [HCB11Cl11]−. Details of workup and cation exchange not shown. | ||
In 2010, we reported two procedures that represented a significant improvement: reactions of Cs[HCB11H11] with either SO2Cl2 or SbCl5 at reflux.16 Both employed readily available and inexpensive liquid reagents as solvents. The SO2Cl2 procedure in particular seemed nearly perfect, because of the low volatility of SO2Cl2 itself and of the presumed chlorination by-products (SO2 and HCl). However, we were quickly hit with reports of irreproducibility after the publication of the paper, and soon encountered this issue ourselves. It turned out that, at least in our hands, only a particular batch of Cs[HCB11H11], and a particular brand of SO2Cl2 gave consistent undecachlorination, but otherwise only partial chlorination was achieved. We spent considerable effort to ascertain the origin of these discrepancies, but were ultimately unsuccessful. We did later show that SO2Cl2/MeCN at reflux is an effective way to convert [B12H12]2− into [B12Cl12]2−.17 Duttwyler et al. also reported the use of SO2Cl2 in the undecachlorination of [HOB12H11]−.18
The SbCl5 synthesis of [HCB11Cl11] has been used by others,19 although some have noted difficulty,20 and proved robustly reproducible in our hands. However, it has two disadvantages. First, because of the relatively high boiling points of SbCl5 (140 °C) and the chlorination by-product SbCl3 (224 °C), the workup is laborious. Second, although we originally reported undecachlorination with SbCl5 in as little as 1–3 days, the reaction sometimes takes up to a week21 for full conversion, and quality control by 11B NMR or mass-spectrometry is warranted before workup. Notably, the SbCl5 method was also successfully used for the undecachlorination of the related [H3NB12H11]− anion by Jenne et al.,22 and by our group in the synthesis of the [HCB11Cl10(OTf)]− anion.23
Subsequently, Shoji and coworkers reported complete undecachlorination of Cs[HCB11H11] using SO2Cl2 at 115 °C in a pressure reactor, although the reaction required 2 weeks to completion.24 Wehmschulte and coworkers reported a different variant of the SO2Cl2 procedure by utilizing UV irradiation to drive undechlorination to completion in an hour.25 Clearly, using SO2Cl2 under thermally or photochemically more forcing conditions leads to improved reactivity. However, these procedures again require specialized equipment (a pressure-rated reactor or a UV reactor plus quartz glassware), which limit availability, and may also limit the plausible laboratory scale. On balance, we continued to be interested in finding a more attractive, simpler procedure.
In considering improvements to the SbCl5 method, we surmised that performing the reaction at a higher temperature may result in faster and more reliable conversion. We further hypothesized that SbCl3 (bp = 224 °C) may be an appropriate co-solvent to raise the boiling point of the reaction mixture with SbCl5 (140 °C), without needing pressurized equipment. The melting point of SbCl3 (73 °C) also appeared appropriate. SbCl3 is a by-product of using SbCl5 as a chlorinating agent, so chemical compatibility was not a concern.
We first examined the reaction of Cs[HCB11H11] with SbCl3 alone as a control. Refluxing the mixture for 3 d led to only a modest amount of cage chlorination (Cl1 to Cl3, MS evidence). But when this mixture (after cooling) was treated with an excess of SbCl5 and then allowed to reflux for 16 h, MS evidence indicated near-quantitative conversion to [HCB11Cl11]−. Encouraged by this finding, we performed chlorinations of 0.5 g and of 6 g Cs[HCB11H11] with SbCl3/SbCl5 mixtures, overnight under reflux. In both cases, complete conversion to [HCB11Cl11]− was noted by MS and 11B NMR spectroscopy, and [Me3NH][HBC11Cl11] was isolated in 87% and 89% yield after standard workup (Scheme 1).
 | ||
| Scheme 1 New syntheses of [HCB11Cl11]−. Details of workup and cation exchange not shown. | ||
A separate experiment with 1 g of Cs[HCB11H11] and SbCl3/SbCl5 under reflux was monitored at more frequent intervals, and it was determined that full conversion to [HBC11Cl11]− was reached after only 4 h. Thus, “overnight” (ca. 18 h) treatment is probably unnecessary, but is a convenient period that is likely to always be sufficient, at least under normal atmospheric pressure conditions (Texas A&M University is at <100 m elevation from sea level). These experiments were run with 16–20 equivalents of SbCl5 and 70–100 equivalents of SbCl3 per Cs[HCB11H11], thus they generate a substantial amount of Sb waste.
Considering other options, we were attracted to VCl4 as a liquid, relatively inexpensive chlorinating agent with a boiling point (148 °C) that appeared appropriate. In addition, the notion of using a transition metal-based (paramagnetic!) solvent and reagent to functionalize a main group compound carried a certain contrarian appeal. Refluxing Cs[HCB11H11] in VCl4 for 48 h did lead to substantial cage chlorination, however it was a mixture of several different carboranes in the Cl5–Cl11 range. Therefore, VCl4 chemistry was not pursued further here.
We also decided to explore trichloroisocyanuric acid (TCCA) as an alternative chlorinating agent. TCCA is a very inexpensive common water treatment material (known as “pool chlorine” in the US). Mixing 100 mg of Cs[HCB11H11] and TCCA (16 equiv.) as solids, and heating them in a glass flask with stirring at 200 °C for 16 h resulted in complete chlorination of the boron positions and the conversion to [HCB11Cl11]− and [ClCB11Cl11]− (MS evidence). Treatment of this mixture with excess Na2SO3 served to cleave the C–Cl bond in the latter, and led to the isolation of [HNMe3][HCB11Cl11] in 91% yield upon further workup. However, we observed that the solid–solid reaction of TCCA with Cs[HCB11H11] tends to turn violent at the initial stages of mixing, or after heating is initiated, depending on the reaction mass and particle size. At the 100 mg scale or less, the violent nature of this event is tolerable for use in a fume hood with standard personal protective equipment. But at larger scales, the violence of the event becomes closer to what would be described as an “explosion”, and we strongly advise against attempts at >100 mg scale of Cs[HCB11H11]. Besides the obvious safety concern, larger scale reactions led to significantly lower yields of the isolated [HNMe3][HCB11Cl11]. It is not clear whether the lower yields are a result of chemical degradation in the runaway event, or also of the physical dispersal of the material. Furthermore, it is not clear to us that the observed violent events are a consequence of the runaway exothermic chlorination alone, or also of the triggered violent decomposition of TCCA. After all, the desired chlorination reaction is not conceived to generate gases which was clearly the case in the runaway event observations.
We explored the analogous use of other inexpensive N–Cl reagents N-chlorosuccinimide (NCS) and 1,3-dichloro-5,5-dimethlhydantoin (DCDMH). However, we observed only incomplete chlorination of the cage in these attempts.
Next, we attempted to obviate the undesired runaway event via initial chlorination of Cs[HCB11H11] in a solvent as a heat sink. Treatment of Cs[HCB11H11] with TCCA at ambient temperature in dichloromethane appeared to be safe, and did lead to partial chlorination of the cage. However, we observed that the subsequent evaporation of the solvent and treatment of the partially chlorinated residue with TCCA could still lead to the undesired runaway event. Cs[HCB11H11] was also only partially chlorinated with TCCA in boiling water. Moreover, on one occasion, a boiling water reaction detonated about an hour after being brought to reflux; we strongly advise against performing such reactions.
However, we were able to find success in scaling up TCCA reactions in the presence of ostensibly inert solids as ballast mass (Scheme 1). We safely performed the reaction a 1 g scale of Cs[HCB11H11] with 200 g NaCl and 13.5 g TCCA (16 equiv.) in the solid state at 200 °C and analogously, with 275 g of sand instead of NaCl. No runaway observations were noted. The reaction with sand produced a better yield (77%) of a purer [HNMe3][HCB11Cl11] upon workup than the one with NaCl. We surmise that the ballast mass serves as an inert heat sink that prevents a runaway thermal reaction.
The melting points of TCCA and of the presumed products derived from it (cyanuric acid, mono- and dichlorocyanuric acid) are above 200 °C, so the reaction is ostensibly proceeding in the solid. It is likely that the vapor pressure of TCCA at 200 °C is sufficient for some amount of mass transport taking place via the gas phase, in addition to the solid–solid contact.
Having observed C-chlorination with TCCA, we separately investigated the conversion of the C–H functionality in [HCB11Cl11]− to simple C–X groups under milder conditions (Scheme 2). [HCB11Cl11]− can be deprotonated, at least partly, by relatively weak bases on the order of alkoxide/hydroxide.26 We observed little difference in the 11B NMR spectra between solutions of Na[HCB11Cl11] alone in water, and in the presence of 1 equiv. of NaOH. However, even partial deprotonation is kinetically sufficient. We found that treatment of Na[HCB11Cl11] with NaOH (1.2 equiv.) in water, followed by TCCA (1 equiv.), resulted in full conversion to [ClCB11Cl11]− after 15 min (11B NMR evidence). Me3NH[ClCB11Cl11] was isolated in 81% yield upon workup. Similarly, NaOCl can be used instead of TCCA. When aqueous Na[HCB11Cl11]/NaOH was treated with H2NSO3H or with I2 (in EtOH), [H2NCB11Cl11]− and [ICB11Cl11]− were cleanly produced in situ, and isolated in high yield as Me3NH+ salts. The analogous reaction with Br2 did not lead to high conversion to [BrCB11Cl11]− and the reaction with H2O2 did not produce any C-hydroxylated derivative. However, we were able to isolate Me3NH[BrCB11Cl11] in high yield after workup from a reaction of Na[HCB11Cl11] with N-bromosuccinimide in acetonitrile at 70 °C. On the other hand, no reaction took place between Na[HCB11Cl11] and N-hydroxysuccinimide under the same conditions.
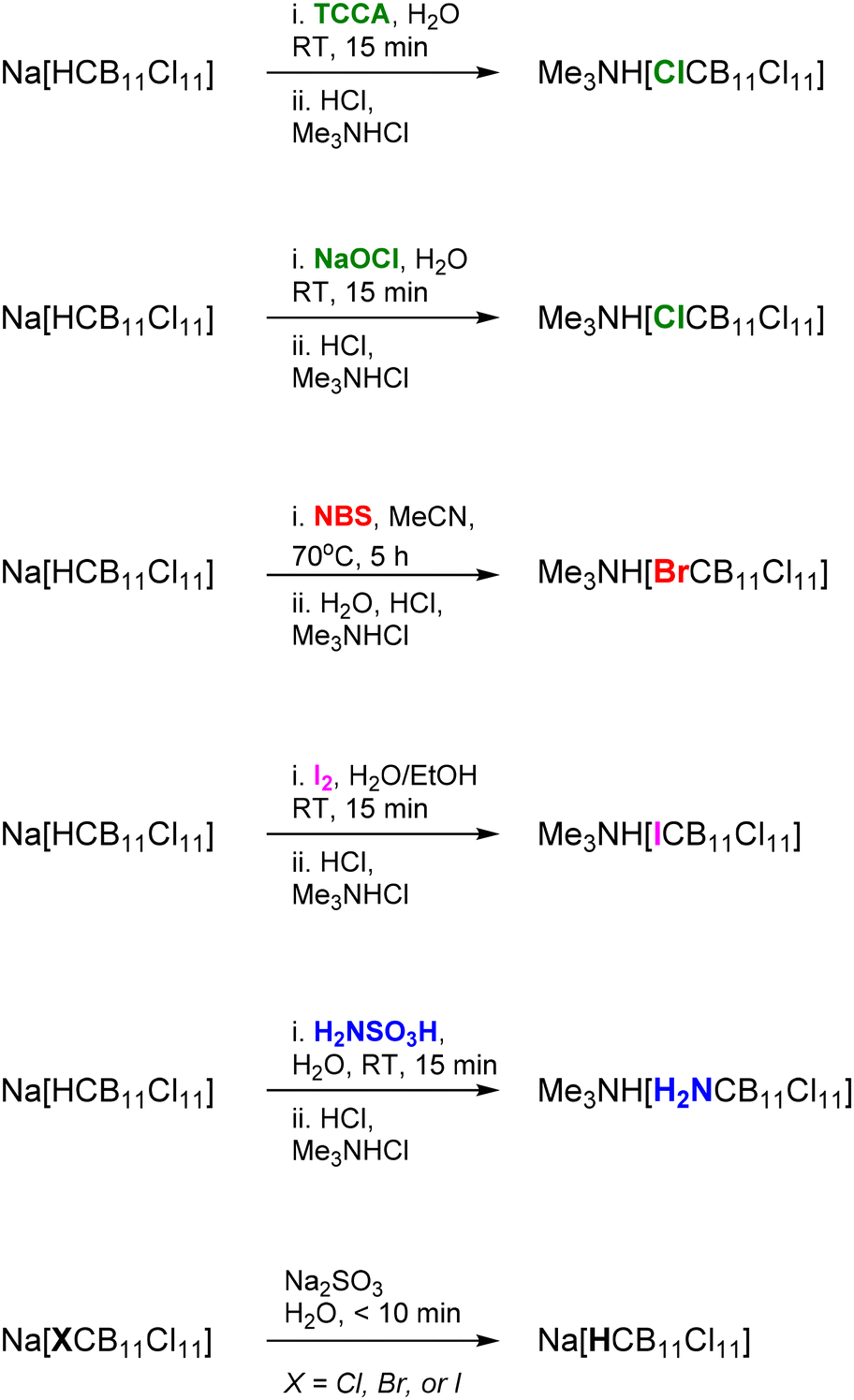 | ||
| Scheme 2 Elaboration of the carbon vertex in [HCB11Cl11]−. | ||
To the best of our knowledge, of the dodecahalogenated [XCB11X11]− anions (X = halogen), only [BrCB11Br11]− was previously reported, via sealed tube synthesis with HOTf/Br2 at 250 °C.27 A few carboranes of a general formula [H2N-CB11X11]− have been reported, but they were prepared via halogenation of an H2N-C containing carborane precursor.28–30 H2NSO3H has been used to aminate the [B12H12]2− cage.22
Dehalogenation of the isolated [HalCB11Cl11]− anions (Hal = Cl, Br, I) was easily accomplished in aqueous solution by treatment with sodium sulfite at ambient temperature. The carbon in [HalCB11Cl11]− is effectively playing a role of a more electronegative element, and of a good leaving group in the likely attack on the halogen by the sulfite or hydrosulfite anion.
In summary, we have demonstrated two new methods for the synthesis of the valuable [HCB11Cl11]− anion – using either a SbCl3/SbCl5 mixture, or TCCA in a solid–solid reaction. Compared to the previously reported methods, these offer improved convenience, reduced times, and greater reproducibility without requiring any equipment besides standard laboratory glassware. In particular, the TCCA method is arguably the most convenient to date, considering the simplicity of workup and the low cost of TCCA.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the support of this research by the Office of Science of the US Department of Energy, Basic Energy Sciences (Grant DE-SC0023280 to O. V. O.). We want to acknowledge Dr Samantha Yruegas and Dr Jillian J. Davidson for some initial screening experiments related to the carbon vertex substitution. We thank Dr Yohannes H. Rezenom for performing HRMS analysis. We also want to thank Dr Gregory P. Wylie and Dr Douglas W. Elliott for assistance in design and execution of long-duration NMR experiments.References
- C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 Search PubMed.
- L. Wang, Y. Jiang, S. Duttwyler, F. Lin and Y. Zhong, Coord. Chem. Rev., 2024, 516, 215974 CrossRef.
- C. Douvris and J. Michl, Chem. Rev., 2013, 113(10), PR179–PR233 Search PubMed.
- A. Wahab, C. Douvris, J. Klíma, F. Šembera, J. Ugolotti, J. Kaleta, J. Ludvík and J. Michl, Inorg. Chem., 2017, 56, 269–276 CrossRef PubMed.
- E. S. Stoyanov, K.-C. Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 8500–8508 CrossRef PubMed.
- M. Juhasz, S. Hofmann, E. S. Stoyanov, K. Kim and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 5352 CrossRef PubMed.
- M. Nava, I. V. Stoyanova, S. Cummings, E. S. Stoyanov and C. A. Reed, H[HCB11F11] is an even stronger Brønsted acid, but the preparation of the [HCB11F11]− anion requires the use of F2 gas, Angew. Chem., Int. Ed., 2014, 53, 1131–1134 CrossRef PubMed.
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 Search PubMed.
- S. O. Gunther, C.-I. Lee, E. Song, N. Bhuvanesh and O. V. Ozerov, Chem. Sci., 2022, 13, 4972–4976 Search PubMed.
- T. Kato and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 2908–2911 CrossRef CAS PubMed.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Muller and M. Oestreich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef CAS PubMed.
- S. O. Gunther, Q. Lai, T. Senecal, R. Huacuja, S. Bremer, D. M. Pearson, J. C. Demott, N. Bhuvanesh, O. V. Ozerov and J. Klosin, ACS Catal., 2021, 11, 3335–3342 CrossRef CAS.
- Z. Xie, C. Tsang, E. T. Sze, Q. Yang, D. T. W. Chan and T. C. W. Mak, Inorg. Chem., 1998, 37, 6444–6451 CrossRef CAS PubMed.
- W. Gu, B. J. McCulloch, J. H. Reibenspies and O. V. Ozerov, Chem. Commun., 2010, 46, 2820–2822 CAS.
- W. Gu and O. V. Ozerov, Inorg. Chem., 2011, 50, 2726–2728 CrossRef CAS.
- Y. Zhang, J. Liu and S. Duttwyler, Eur. J. Inorg. Chem., 2015, 5158–5162 CrossRef CAS.
- V. Lavallo, J. H. Wright II, F. S. Tham and S. Quinlivan, Angew. Chem., Int. Ed., 2013, 52, 3172–3176 CrossRef CAS PubMed.
- A. Hepp, R. Labow, F. Reiss, A. Schulz and A. Villinger, Eur. J. Inorg. Chem., 2018, 2905–2914 CrossRef CAS.
- H. Poleschner and K. Seppelt, Angew. Chem., Int. Ed., 2013, 52, 12838–12842 CrossRef.
- C. Bolli, J. Derendorf, C. Jenne, H. Scherer, C. P. Sindlinger and B. Wegener, Chem. – Eur. J., 2014, 20, 13783–13792 CrossRef PubMed.
- L. P. Press, B. J. McCulloch, W. Gu, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2015, 51, 14034–14037 RSC.
- N. Tanaka, Y. Shoji and T. Fukushima, Organometallics, 2016, 35, 2022–2025 Search PubMed.
- M. Saleh, D. R. Powell and R. J. Wehmschulte, Inorg. Chem., 2016, 55, 10617–10627 CrossRef PubMed.
- R. Ramírez-Contreras and O. V. Ozerov, Dalton Trans., 2012, 41, 7842–7844 Search PubMed.
- Z. Xie, C.-W. Tsang, E. T.-P. Sze, Q. Yang, D. T. W. Chan and T. C. W. Mak, Inorg. Chem., 1998, 37, 6444–6451 CrossRef PubMed.
- M. Finze, G. J. Reiss and M. Zähres, Inorg. Chem., 2007, 46, 9873–9883 CrossRef PubMed.
- R. R. Srivastava, D. K. Hamlin and D. S. Wilbur, J. Org. Chem., 1996, 61, 9041–9044 CrossRef PubMed.
- M. Finze and J. A. P. Sprenger, Z. Anorg. Allg. Chem., 2010, 636, 1538–1542 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, experimental descriptions. See DOI: https://doi.org/10.1039/d4dt03033h |
| This journal is © The Royal Society of Chemistry 2025 |