
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pressure-induced phase transitions in a new luminescent gold(I)–arylacetylide†
Róża
Dziewiątkowska
 *a,
Joanna
Krzeszczakowska
a,
Marta
Głodek
b,
Michał
Łomzik
b,
Damian
Plażuk
bc and
Anna
Makal
*a
*a,
Joanna
Krzeszczakowska
a,
Marta
Głodek
b,
Michał
Łomzik
b,
Damian
Plażuk
bc and
Anna
Makal
*a
aChemistry Department, Biological and Chemical Research Centre, University of Warsaw, ul. Żwirki i Wigury 101, 02-089 Warszawa, Poland. E-mail: am.makal@uw.edu.pl
bDepartment of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland
cLaboratory of Molecular Spectroscopy, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland
First published on 9th January 2025
Abstract
Stimulus-responsive molecular materials are highly desirable because of the wide range of their potential applications. In particular, switching of physical properties opens application pathways for molecular materials as sensors or actuators. Property switching in solids can be achieved by inducing single-crystal-to-single-crystal (SCSC) phase transitions. Elucidating the mechanisms of such transformations and identifying the factors and (supra)molecular motifs that increase their probability is thus paramount to understanding property switching and materials design. Here, we present a new compound, (p-methoxy-phenylacetylide)(triethylphosphine)gold(I) (ArPEt), combining the photoluminescent core of gold(I) acetylide with triethylphosphine that should provide a rich conformational landscape and favor SCSC phase transitions due to low-energy interconversion pathways. We demonstrate its potential to undergo multiple pressure-induced SCSC transformations at ≈0.5 GPa and ≈2 GPa, the obtainable phases being dependent on the types of applied pressure-transmitting media. We describe structures of polymorphs of ArPEt and the mechanism of pressure-induced structural reorganizations based on very high-quality single-crystal X-ray diffraction data.
1. Introduction
Stimulus-responsive molecular materials are very important and highly desirable because of the wide range of their potential applications. As such, they constitute a hot topic in modern materials sciences.1–4 An interesting type of such solid molecular materials involves so-called molecular switches and molecular rotors, i.e., molecules where external factors can induce a conformational change or rotation of a whole molecule. Molecular rotations occurring in the crystalline matrix either independently in each molecular unit, or in a cooperative manner by means of some gearing mechanism, open possibilities for constructing molecular actuators5 and numerous other smart materials,6,7 including photoluminescence switches.8 Property switching in such crystalline materials is most often achieved due to a phase transition occurring without affecting the global ordering of the bulk, i.e., single-crystal-to-single-crystal (SCSC) phase transition. Preservation of crystallinity allows for studying in detail the structure of a material both before and after transformation and thus for elucidation of the phase transition mechanism. The latter is paramount to understanding property switching and new materials design.The chemical and physical properties of organo–gold(I) compounds create unfailing scientific interest. Very recent research covers their catalytic,9 cytotoxic10 and photoluminescence11–14 properties. They are promising and already-explored building blocks for stimuli-responsive materials15 including amphidynamic crystals.8,16 In particular, gold(I) alkynyl and isocyano compounds are known to display rich polymorphism with very distinct luminescence properties,17–20 with potential for temperature or pressure-switching in the solid state.
Triethylphosphine, a versatile ligand commonly used in organo–gold chemistry, with its three short aliphatic ‘arms’, can adopt several conformations that do not differ significantly either in energy or outward shape. The studies of Ellis et al.21 and de Silva et al.22 on the structures of metal–phosphine complexes have indeed shown that there are a few distinct, low-energy conformations of triethylphosphine, most preferably those realized in crystal structures. A study by Ellis et al. also suggested that transitions from one conformation to another would require non-negligible energy input and would preferably be achieved by single ‘flips’ of the –Et groups around the P–C bond. A few most-probable pathways for –PEt3 conformation switching have been proposed accordingly. That, as suggested by Castro et al.23 and as demonstrated in our former work,24 makes –PEt3 a viable component for molecular crystals prone to SCSC phase transitions.
Among various external stimuli that can be applied in physical property switching, exerting hydrostatic pressure is particularly efficient in affecting crystal structures in a controlled manner.25,26 With modern diamond anvil cells (DAC) enabling almost routine crystal structure determination at pressures of several GPa, intermolecular distances can be varied significantly and phase transitions induced that could not be achieved by other means. The same DAC-encased sample can be subjected to a variety of experimental exploration techniques, from Raman spectroscopy to crystal structure determination from single-crystal X-ray diffraction. This in turn allows for a detailed study of structural changes accompanying a phase transition, including conformation switching in relatively large molecular systems27 and phase transformation kinetics.28 In our quest to relate photoluminescence properties and crystal architectures of solid materials based on gold(I) acetylidenes, we prepared a new compound, (p-methoxyphenylacetylide)(triethylphosphine)gold(I), further denoted herein as ArPEt. It combines the photoluminescent core of gold(I) acetylide with two moieties that should provide a rich conformational landscape: triethylphosphine and p-methoxyphenylacetylide. Concerted pressure- or temperature-induced conformational switching in those ligands should in turn result in SCSC phase transformations. After presenting a straightforward synthetic procedure and basic characterization of the new compound, we demonstrate its potential to undergo multiple pressure-induced SCSC phase transitions and discuss their mechanism. We support our findings with detailed structural models of each new phase based on high-pressure X-ray diffraction data of exceptional quality.
2. Results and discussion
2.1. Synthesis
The gold(I) complex ArPEt was synthesized in 80% yield by modifying the formerly reported methodology29 (subsection 4.1 of the Experimental section). In short, the 4-ethynylanisole that was reacted with freshly prepared LiHMDS afforded the corresponding acetylide, which further reacted without isolation with ClAuPEt3 in THF and the product was isolated by crystallization (Fig. 1). The structure of the product was confirmed by NMR spectroscopy and MS analysis. In the 1H NMR spectrum of ArPEt in acetonitrile-d3, one set of signals assigned to the desired compound was present. In the 13C NMR spectrum, only the signals assigned to aromatic, aliphatic and one of the acetylene carbons (1H–13C HMBC spectra, Fig. S1.4†) were present. Due to the relativistic effects of the gold atom, we were unable to observe signals from the acetylide carbon directly bonded to the Au atom but this phenomenon is known and can be diminished by 13C-labeling of the compound.30 In the 31P NMR spectrum, only one singlet at 39.4 ppm was present. The ESI-MS analysis of ArPEt, however, was more complicated than expected. In the MS spectrum of ArPEt dissolved in MeCN (Fig. S1.5†), two types of signals were present: in the positive mode at m/z = 433.1 assigned to ((Et3P)2 Au+), while in the negative mode at m/z = 459.1 assigned to C18H14AuO2−. The formation of these ions can be attributed to the ligand scrambling occurring in ArPEt under ESI-MS conditions, with the formation of observable stable ions. | ||
| Fig. 1 (p-Methoxyphenylacetylide)(triethylphosphine)gold(I) synthesis. | ||
2.2. UV-vis absorption and photoluminescence
ArPEt is just barely photoluminescent in dilute polar solvents and noticeably luminescent in the solid state at room temperature.Absorption in two tested solvents features a well-visible oscillation structure, while photoluminescence shows broader curves with maxima at about 310 nm and very low (<1%) photoluminescence quantum yields (Table 1). According to tentative theoretical calculations (section S6.1 in the ESI†) the excitation band at about 290 nm could be attributed to the S0 → S1 (H → L) electronic excitation, mainly π–π* in character, with the ligand-to-metal charge transfer component arising from the gold(I) cation.
| In solution | |||
|---|---|---|---|
| UV-vis | PL | ||
| λ max [nm] (ε [dm3 mol−1 cm−1]) | λ max_em [nm]/QY [%] | ||
| MeCN | DCM | MeCN | DCM |
291 (21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 680 ± 44) 680 ± 44) |
295 (12344 ± 866) |
306/0.2 | 314/0.8 |
| 280 (30611 ± 67) |
281 (14955 ± 1208) |
||
| 268 (24369 ± 55) |
270 (12237 ± 857) |
||
| 234 (16720 ± 53) |
|||
| Crystalline powder | ||
|---|---|---|
| Excitation | PL | |
| λ max_excit [nm] | λ max_em [nm] | QY [%] |
| 407 | 425 | 19.4 |
| 392 | 446 | |
| 380 | 456 | |
| 369 | 467 | |
| 492 | ||
In the solid state under standard conditions, the ArPEt exhibits moderate blue fluorescence covering a range from ≈430 to 530 nm with a tail beyond 600 nm, featuring a well-visible oscillation structure and the main maximum at about 470 nm (Table 1). The notable red Stokes shift in emission maxima (over 120 nm) with respect to DCM solution (Fig. 2) accompanied by a dramatic increase in the fluorescence quantum yield (over 19%) illustrates a substantial aggregation-induced emission enhancement, most probably due to restriction of molecular motions in the crystalline environment. Blue emission in the solid state is in agreement with trends observed for many gold(I) acetylides and cyanides in the absence of significant aurophilic interactions.15,18,19,31 Notably, single-crystal emission spectra show a more pronounced broadening at longer wavelengths, most probably attributable to the fact that the sample was excited with a slightly longer wavelength (Fig. 2b).
 | ||
| Fig. 2 Top: normalized UV-vis and PL spectra of ArPEt in solution with TDDFT-predicted electronic excitations represented as vertical black lines. Bottom: excitation and PL spectra from crystalline ArPEt-I. | ||
2.3. The structure at ambient pressure and temperature
Under atmospheric conditions, ArPEt crystallizes in the Pbca space group (Table 2) with one independent molecule in the asymmetric unit. As expected, the coordination of the gold atom is almost linear, with a C–Au–P angle above 178°. The structure is disordered (Fig. 3) with 69% of the major conformer (conf1) and 31% of the minor one (conf2), indicating notable mobility of the disordered fragments. Conf1 has the C (gauche+, anti, gauche+) conformation and conf2 the F (g+ag−) conformation of triethylphosphine, which are the two most popular triethylphosphine conformations in CSD.21 Conformers also differ in their aromatic part. The methoxy group in conf1 is in the plane of the aromatic ring, while in conf2 the group is slightly rotated out of that plane. The angle between phenyl ring planes of conf1 and conf2 equals 15.7°. The conformational flexibility of ArPEt and its sensitivity to external stimuli are supported by the presence of multiple and apparently reversible temperature-induced phase transitions in DSC analysis (section S1.3 in the ESI†).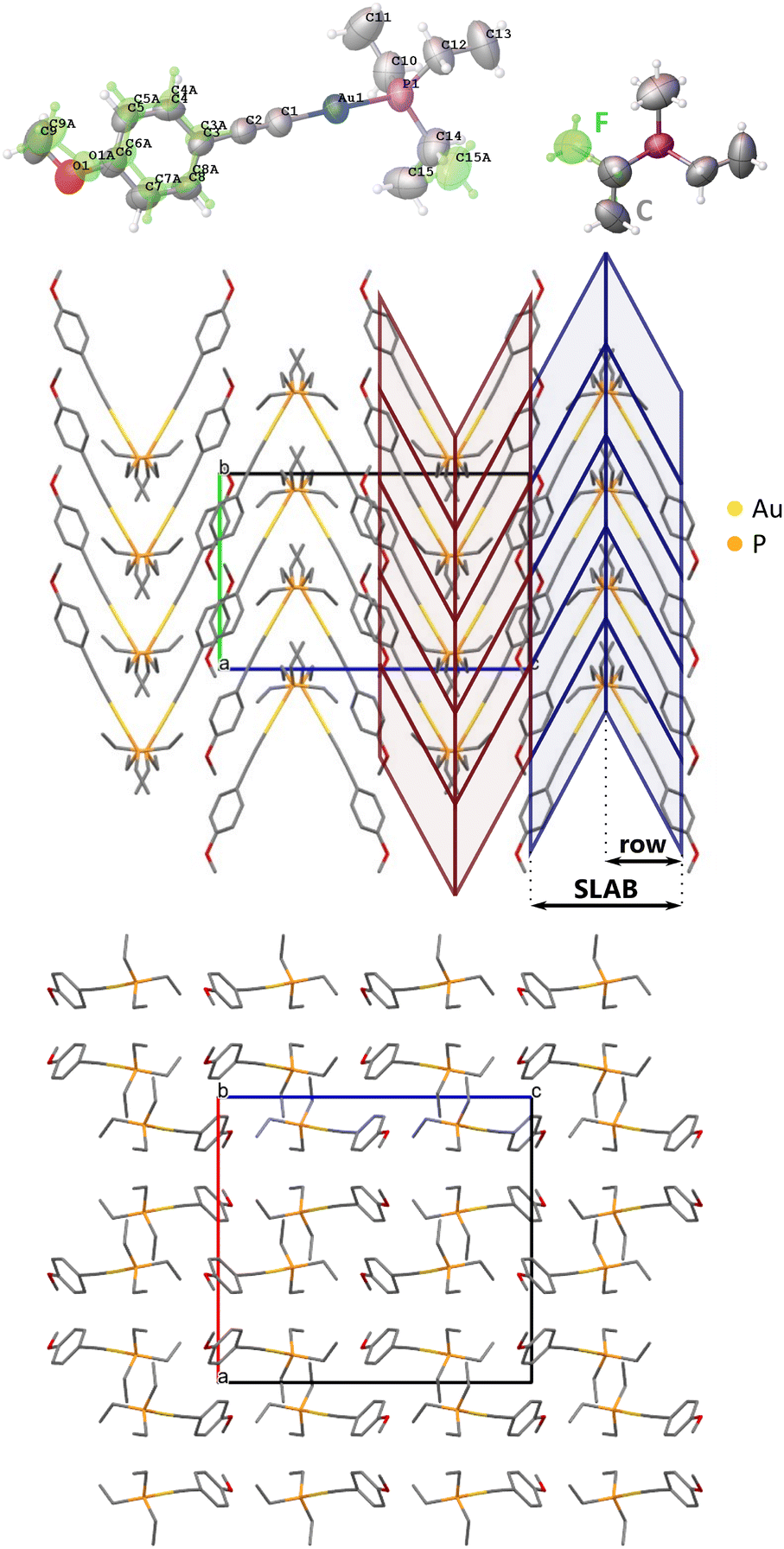 | ||
| Fig. 3 Structure of ArPEt at 295 K and under ambient pressure with its atom numbering scheme (upper left). The green part refers to the minor fraction of disorder; atomic displacement parameters represented at a 50% probability level; and triethylphosphine conformations in disordered variants (upper right). Herringbone-like motif in the crystal structure of ArPEt: view along the [100] direction highlighting the most important packing motifs (middle) and view along [010] (down). | ||
| Formula | C15H22AuOP | |||||
|---|---|---|---|---|---|---|
| Weight [g mol−1] | 446.26 | |||||
| Series | In-house | Synchrotron | ||||
| Pressure medium | n/a | Silicone oil | Pentanesa | Daphne-7575 | ||
| Pressure [GPa] | n/a | 0.26 | 0.89 | 2.13 | 2.59 | 3.30 |
|
a
n-Pentane/i-pentane 1:1 mixture.
|
||||||
| Space group | Pbca | Pbca | Pb21a | P11a | Pbca | Pbca |
| a [Å] | 16.3373(7) | 16.1780(6) | 17.3429(4) | 16.7021(6) | 16.7686(6) | 16.5856(7) |
| b [Å] | 11.3081(5) | 11.1922(3) | 9.9201(2) | 9.8830(3) | 9.8173(2) | 9.7494(2) |
| c [Å] | 17.9484(8) | 17.8018(8) | 16.9287(5) | 16.8029(4) | 16.1828(17) | 16.0285(4) |
| γ [°] | 90 | 90 | 90 | 93.865(3) | 90 | 90 |
| Volume [Å3] | 3315.9(3) | 3223.3(2) | 2912.47(12) | 2767.29(14) | 2664.1(3) | 2591.80(13) |
| Z | 8 | 8 | 8 | 8 | 8 | 8 |
| ρ calc [g cm−3] | 1.788 | 1.839 | 2.035 | 2.142 | 2.225 | 2.287 |
| Wavelength [Å] | 0.56087 | 0.56087 | 0.56087 | 0.56087 | 0.56087 | 0.410 |
| R int | 0.0341 | 0.0791 | 0.0618 | 0.0547 | 0.1009 | 0.0479 |
| Resolution [Å] | 0.6313 | 0.7800 | 0.6171 | 0.7834 | 0.6470 | 0.5575 |
| Completeness | 0.984 | 0.929 | 0.912 | 0.651 | 0.639 | 0.811 |
| Data; rest.; param. | 6050; 0; 239 | 3269; 224; 251 | 10223; 1; 334 |
7038; 410; 666 | 2732; 6; 167 | 4215; 0; 167 |
| R 1(I > 2σ(I)) | 0.0464 | 0.0328 | 0.0382 | 0.0432 | 0.0464 | 0.0363 |
| wR2 (all data) | 0.1036 | 0.0572 | 0.0564 | 0.0711 | 0.0747 | 0.0862 |
| Δρmax [e Å−3] | 0.794 | 0.563 | 1.194 | 0.926 | 1.166 | 1.177 |
| Δρmin [e Å−3] | −0.589 | −0.476 | −1.026 | −0.971 | −1.014 | −1.120 |
Molecules related by the 21[010] axis form two inversely oriented rows, creating a herringbone-like pattern (Fig. 3). Two adjacent rows constitute a motif further referred to as a SLAB.
2.4. Pressure-induced changes
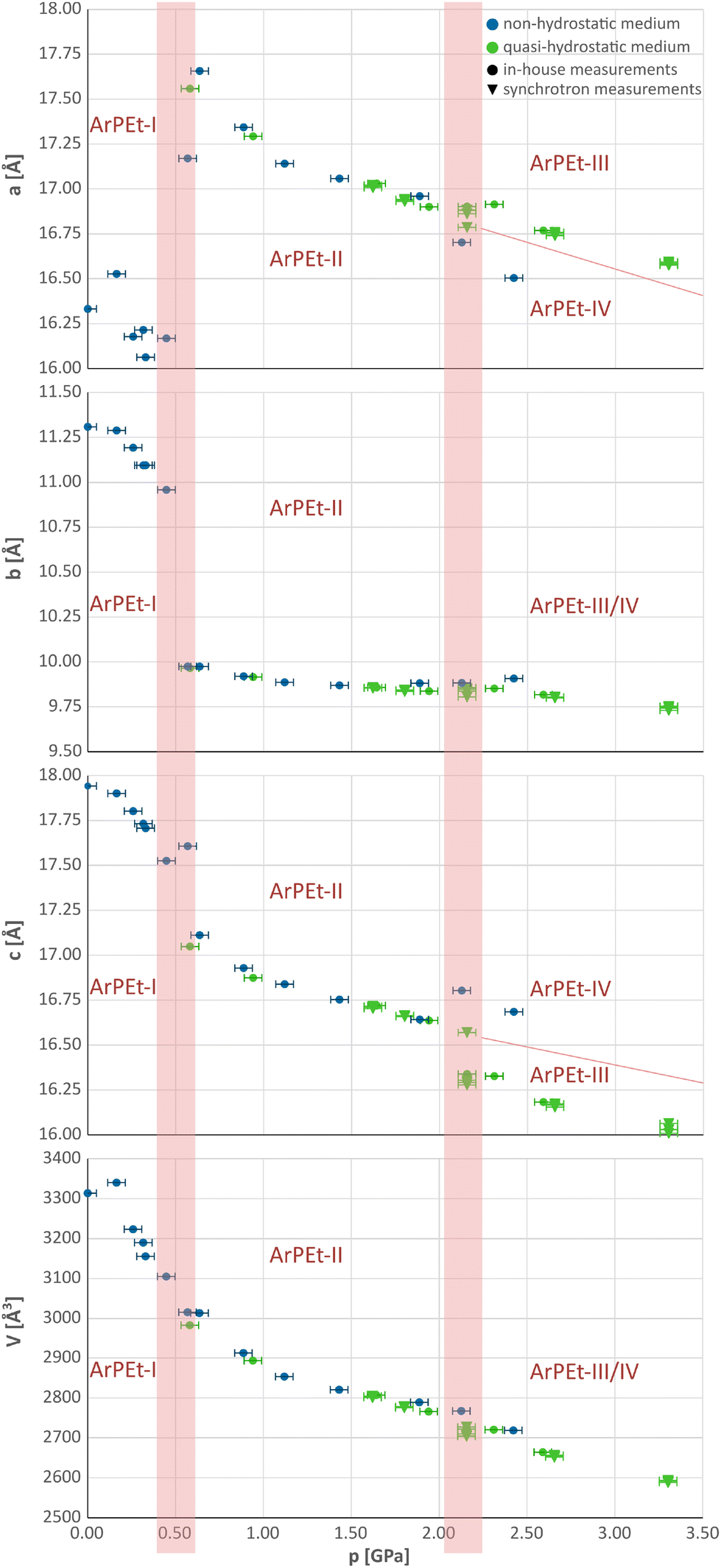 | ||
| Fig. 4 Evolution of the unit cell parameters with pressure in crystalline ArPEt based on the in-house and synchrotron measurements. Silicone oil is marked as a non-hydrostatic medium (blue), while the n-pentane/i-pentane 1:1 mixture and Daphne-7575 are marked as quasi-hydrostatic media (green). Uncertainties of pressures approximated as 0.05 GPa; experimental uncertainties of unit cell parameters are smaller than the drawn points. | ||
At 0.5 GPa, there is an increase of the a parameter of about 1.5 Å (≈9%), accompanied by a decrease of the b parameter of about 1 Å (≈9%) and a drop in the length of the c parameter by less than 0.5 Å (≈3%). Above 2.0 GPa, changes in unit cell parameters are less pronounced and depend on the pressure-transmitting medium (PTM) used. In the n-pentane/i-pentane mixture and Daphne-7575, which in the studied pressure range are known to be quasi-hydrostatic,32 the a parameter retains the same value with a slight pressure increase while c shrinks by about 1.5%. In silicone oil, which above 2 GPa may act as a non-hydrostatic medium32,33 (see section S2.2 in the ESI†), the changes are opposite: the a parameter decreases, while c increases by about 1.2%. The resultant new phases will be denoted as ArPEt-III and ArPEt-IV, subsequently.
Under quasi-hydrostatic conditions, systematic extinctions indicating the presence of the c glide plane and hence the increase in symmetry, reoccur at 2.16 GPa in the h0l layer (see Fig. S4.15 and section S4.3† for more details concerning the kinetics of this transition).
A different situation occurs in the case of a non-hydrostatic medium. At 2.13 GPa, reflections contradicting the presence of the b glide plane occur in the 0kl layer (Fig. S4.16†), leading to a pattern consistent with the P11a space group with a γ unit cell angle of 93.9°. All phase transitions follow group–subgroup or group–supergroup relations and can thus be characterized as type II phase transitions.34
In the new ArPEt-II phase, two independent molecules, formerly related by the c[010] glide plane, constitute the asymmetric unit as a result of symmetry lowering in the Pb21a space group. Change in symmetry is caused by conformational differentiation between molecules. The labeling scheme derived from ArPEt-I for this structure is presented in Fig. 6a.
After phase transition, one molecule (Au1) changes the triethylphosphine conformation from C (g+ag+) to E (g+g−a) by changing the position/torsion angle of two ethyl groups. The second molecule retains the C conformation but with rotated ethyl groups in the triethylphosphine moiety. Aromatic rings from both molecules are no longer parallel and the angle between their planes equals 32–36°.
An overlay of the structures of ArPEt-I and ArPEt-II highlights the additional lateral shift of the (Au1) molecule along [010] by 1.5 Å (Fig. 6a) actuated by rotating –Et3 fragments ‘pushing’ or ‘climbing’ one over another.
Those changes result in the formation of two distinct SLABs: SLAB1 consists of molecules with the original conformation C, while SLAB2 consists of molecules with conformation E. Structural reorganization can be described more effectively by relative distances from other molecules within a SLAB or between SLABs represented by positions of Au atoms and their changes (Fig. 5) as well as inclinations of phenyl rings with respect to the a ([100]) direction.
 | ||
| Fig. 5 Calculated distances between neighboring gold atoms – overall (first), along [001] (second) and along [010] (third). The angle between aryl moieties and [100] in the unit cell (fourth). Definitions of distances are given in section S5 in the ESI.† | ||
During the first phase transition, overall distances between gold atoms from neighboring SLABs increase. This is mostly a result of the above-mentioned shifting of molecules Au1 in the [010] direction by about 1.5 Å. Because SLABs interact with each other through aromatic moieties, the movement of molecules also induces the rotation of phenyl rings – over 10° in SLAB1 and about 5° in SLAB2.
Under quasi-hydrostatic conditions at about 2.2 GPa, the structure of ArPEt regains the original Pbca space group and higher symmetry by conformational unification of molecules. The triethylphosphine moiety conformation in SLAB1 changes from C (g+ag+) to E (g+g−a), switching positions of all ethyl groups and rotating the –PEt3 moiety. Overlaying with the ArPEt-II form shows that there are conformational changes in both the –PEt3 moiety in the second molecule (Au2) and the methoxyphenyl group in the first one (Fig. 6b).
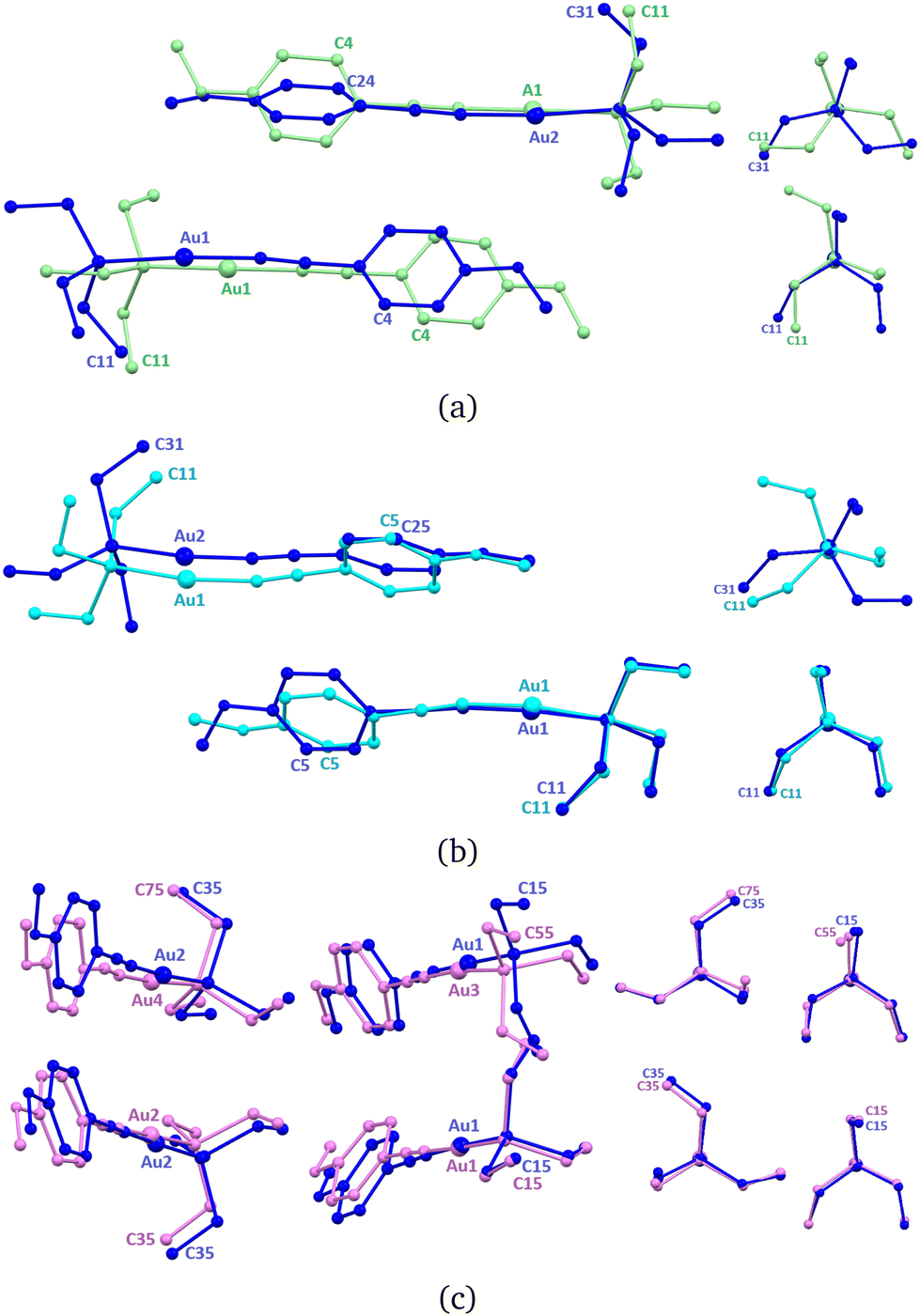 | ||
| Fig. 6 Structure overlays of (a) ArPEt-I (green) and ArPEt-II (blue) forms, (b) ArPEt-II (blue) and ArPEt-III (cyan) forms and (c) ArPEt-II (blue) and ArPEt-IV (pink) forms. Naming convention on symmetry lowering is illustrated by selected atoms. Hydrogen atoms and minor disorder variants from ArPEt-I are omitted for clarity. | ||
The overall distance between gold atoms from neighboring SLABs slightly decreases as a result of molecule Au2 shifting along the [100] and [001] directions (Fig. 6b and 5).
A different situation occurs under non-hydrostatic conditions. The unit cell becomes oblique. With the b glide plane gone, four independent molecules constitute the asymmetric unit (Fig. 6c). Overlaying with ArPEt-II shows that all molecules shift slightly. A subtle conformational change occurs only in the molecule with Au3, i.e., the triethylphosphine moiety changes its conformation from E (g+g−a) to D (g−aa), involving movements of two ethyl groups.
All traced phase transitions with their most important conformational changes are shown in Fig. 7.
 | ||
| Fig. 7 Schematic representation of the phase-transition pathways and associated –PEt3 conformation changes described in this study. | ||
2.5. Relative stability of ArPEt crystal phases
Starting from the optimized geometries and unit cell parameters of ArPEt-I, ArPEt-II, and ArPEt-III, the variation of the crystal structures with pressure was theoretically simulated and fitted with the third-order Birch–Murnaghan equation of states (EOSs) (Fig. 8a). | ||
| Fig. 8 (a) Theoretical (solid lines) and experimental (SC-XRD) unit cell volumes as a function of pressure; theoretical predictions are noticeably underestimated; (b) theoretically predicted enthalpies for the selected ArPEt polymorphs; the three curves approach each other at about 2.7 GPa. | ||
Unit cell parameters obtained from XRD experiments differ from optimized ones by about 10%, for all crystal phases and the theoretically predicted unit cell volumes are significantly underestimated. This can be explained to a certain extent by the fact that calculations were performed effectively at 0 K, not taking into account temperature-induced molecular and lattice vibrations, and partly by the relatively unsophisticated computational approach. The calculations also underestimate the slope for unit cell volume decrease in ArPEt-I, most likely because they could not reproduce the effects related to the structural disorder. However, they show significant differences in unit cell volumes between the considered phases, confirming that observed phase transitions are driven by steric effects, minimizing the volume per molecule. The estimated Gibbs free energies at T = 0 K (coinciding with enthalpy) increase with pressure, as shown in Fig. 8b. The Gibbs free energy changes are remarkably similar for all three ArPEt polymorphs and, in the pressure range of 0.0–4.0 GPa, the differences do not exceed 150 kJ mol−1. As expected, ArPEt-I is the most stable form at ambient pressure, whereas ArPEt-III appears the most stable above 2.5 GPa. It must be stressed that the differences between enthalpies of three ArPEt phases are small and at 295 K, the entropic contributions must play an important role.
2.6. Intermolecular and inter-slab interaction energies
Energies of the interaction between subsequent molecular dimers (Fig. 9) as well as between the SLABs were calculated in CRYSTAL17 based on the optimized geometries of ArPEt-I, ArPEt-II (at 0.6 GPa), and ArPEt-III (at 2.6 GPa). Only the most prominent interactions, with energies of −20 kJ mol−1 or more negative, will be discussed. | ||
| Fig. 9 Definition of the dimers in SLABs (interaction energy >20 kJ mol−1) and interaction energies [kJ mol−1] calculated for subsequent dimers on a schematic representation of ArPEt crystal packing along the [100] direction. | ||
Intermolecular interactions within the crystal structure of ArPEt-I are at a similar order of magnitude. The strongest (i), involving two nearest neighbors in a row and in a SLAB, shows interaction energy at −44 kJ mol−1, while the strongest intermolecular interaction between the SLABs (iv) shows energy at −31 kJ mol−1. The inter-SLAB intermolecular interactions are only slightly less stabilizing than the intra-SLAB ones. Nevertheless, the definition of a SLAB seems energetically justified in that the energy per SLAB is more stabilizing than the sum of inter-SLAB interactions (Table 3).
| kJ mol−1 | Energy (kJ mol−1) | ||
|---|---|---|---|
| ArPEt-I | ArPEt-II | ArPEt-III | |
| SLAB 1 | −1010 | −918 | — |
| SLAB 2 | — | −915 | −903 |
| Between SLABs | −731 | −721 | −799 |
At increased pressure in the ArPEt-II phase, the conformational C → E change in –PEt3 in every other SLAB significantly affects the strongest intermolecular interactions (i). The new E conformation in that dimer in SLAB-1 is evidently more energetically favorable (Fig. 9). Notably, the total energies of two distinct SLABs remain very similar and less stabilizing with respect to the atmospheric conditions, probably due to a significant contraction of the unit cell volume. Further quasi-hydrostatic compression results in conformational C → E changes in SLAB-2 and in the transformation from ArPEt-II to ArPEt-III. In the latter phase, the order of intermolecular interactions within a SLAB is reversed, with (iii) now being the most stabilizing. There is also a tendency for intermolecular interactions between the SLABs to become more stabilizing as the SLABs are compressed closer together.
2.7. Phase transition mechanism and kinetics
Comparison of the fast investigations performed at the synchrotron and necessarily slow experiments in-house sheds light on the kinetics of the observed phase transitions. From synchrotron measurements at 2.16 GPa, three structures of ArPEt-III and one structure of the ArPEt-II form were unambiguously determined (Fig. 10). The possibility of obtaining two co-existing phases in the course of synchrotron experiments suggests that the kinetics of the phase transformation are rather slow, with a non-negligible initiation barrier, and can take some time to complete. The experiment at about the same pressure performed in-house over the course of ≈50 hours yielded an ambiguous diffraction pattern (see section S4.3.1†). The structure based on such data could not be refined reliably, neither in Pb21a nor in Pbca space groups. It can be surmised that the data accumulated over time contained contributions from the initial lower-symmetry phase and the final high-symmetry state and all intermediate stages. All observed phase transitions involve substantial conformational changes in –PEt3 moieties. In addition, these phase transitions occur sequentially. At a time, they involve only every other SLAB, i.e. only a distinct supramolecular motif within which the –PEt3 fragments interlock (Fig. 11). We propose that the phase transition mechanism involves a structural change starting locally and propagating within a SLAB, analogous to one described formerly for (ferrocenylacetylide)(triethylphosphine)gold(I).24 An initial conformation change of one or a few ‘seeding’ triethylphosphines can generate enough steric strain or ‘chemical pressure’35,36 for the adjacent interlocked phosphines within a SLAB to be forced to adjust their conformations and ‘drag’ molecules to new positions. The phase transition can thus be viewed as a series of interlocked molecular switches or gears. Interlocking gears must rotate in opposite directions, and so must the interacting –PEt3 moieties, being mirror images of each other (the relationship is via the crystallographic a[001] glide plane, Fig. 11). The second phase transition under hydrostatic conditions, restoring the initial Pbca symmetry, requires that molecules in the hitherto unchanged layers undergo an analogous sequential molecular-gear-driven change. Concerted motions taking place within the crystal matrix allow the ArPEt material to qualify as a proper molecular actuator. | ||
| Fig. 10 Four crystal specimens of ArPEt as placed inside a DAC during X-ray diffraction experiments at the ESRF. Space groups at which data were indexed for each specimen at a given pressure indicate a particular ArPEt phase. A piece of gold foil (dark triangle) and ruby spheres are visible in the lower-right corner. | ||
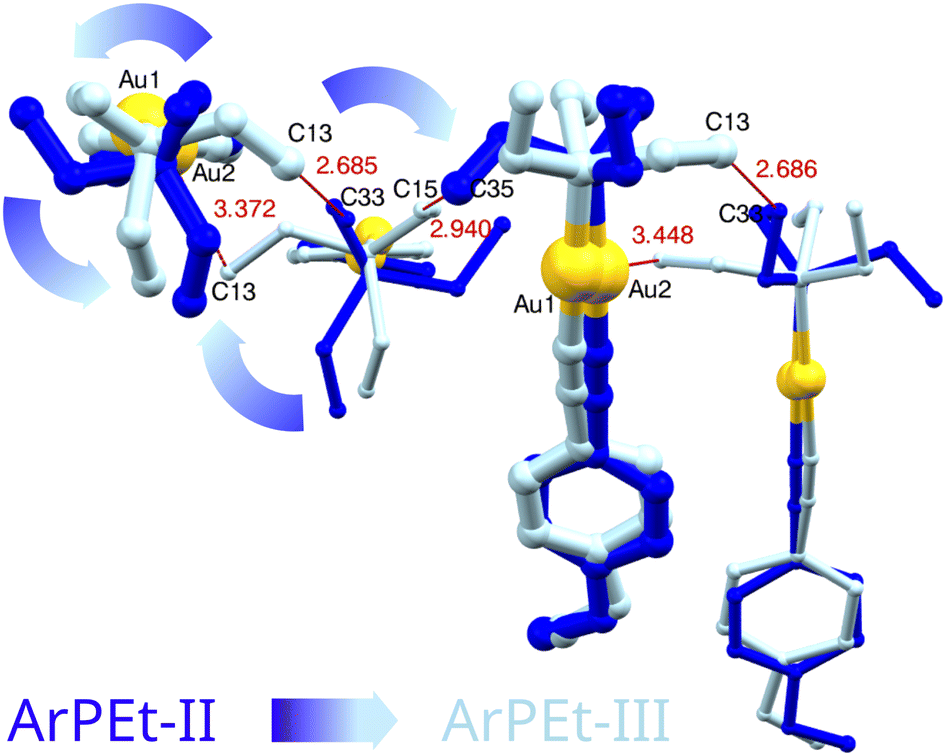 | ||
| Fig. 11 An overlay of ArPEt-II (blue) and ArPEt-III (cyan) structures, SLAB-2, viewed along [010]. Thinner lines indicate molecules placed below the projection plane (further from the viewer). Assuming a conformation change inherent to the ArPEt-II → ArPEt-III phase transition, namely a counterclockwise rotation and the –Et3 flips, had been triggered in a molecule on the left, the new conformer would be in steric conflict with its nearest neighbor in a SLAB (too-short hypothetical interatomic distances in Å represented in red). In order to avoid the clash, the adjacent molecule has to adjust its conformation by rotating clockwise and executing further –Et3 flips. | ||
In the context of potential applicability, it must be noted, that irrespective of the applied PTM, the conformational changes in –PEt3 in each instance follow low-energy conformation interconversion pathways as proposed by Ellis et al.,21C → E or C → E → D accordingly. This suggests that analogous changes can be predictable for other –PEt3-containing materials. However, the exact transformations involve not only a flip of a single –Et moiety, but concerted rotations of two or three ethyl moieties accompanied by shifts and rotations of whole molecules.
2.8. Note on the photoluminescence under increased pressure
The variety of available solid phases is only partly reflected in their photoluminescence. The fluorescence spectra collected under several pressure values are compared in Fig. 12. | ||
| Fig. 12 Fluorescence of a single crystal of ArPEt placed inside a DAC in the n-pentane:i-pentane mixture (left) or in silicone oil (right) and their evolution with pressure. Spectra recorded during pressure increase are represented by thick lines, and during decrease, by thin lines. The colors represent pressure. The persistent spike at 444 nm represents the Raman peak of the diamonds in a DAC. All spectra were normalized to their maximum value and plotted with increments along the vertical axis for clarity. | ||
Irrespective of an applied PTM, the photoluminescence of ArPEt is hardly affected by increased pressure up to 1.0 GPa. The spectra retain their visible vibrational substructure, with the individual oscillation sub-maxima showing slight blue-shifts with increased pressure (detailed illustration in Fig. S3.13a in the ESI†). Such shifts would be consistent with a slight increase in the frequencies of internal molecular vibrations resulting from a more compressed, i.e. more restricted environment. Between 1.0 and 1.5 GPa (unambiguously the ArPEt-II phase), the photoluminescence characteristics change dramatically. The oscillation substructure gradually disappears and the spectra become broad, covering almost all visible ranges, with a constant pressure-insensitive maximum at about 520 nm and some additional broad features at about 600 nm, producing a net visual effect of a bright white emission. The latter features become dominant at higher pressures in the case of the sample enclosed in the mixture of pentanes (ArPEt-III phase, Fig. 12 left) differentiating them from the emission observed for the sample pressurized in Si-oil (ArPEt-IV). It must be stated, however, that these features were noticeably dependent on the site of the specimen exposed to the exciting UV beam as illustrated in Fig. S3.13(b–d) in the ESI.† Notably, the above-described changes were fully reversible with decreasing pressure.
3. Conclusions
In our quest to relate photoluminescence properties and crystal architectures of solid materials based on gold(I) acetylidenes, we obtained a new compound ArPEt ((p-methoxyphenylacetylide)(triethylphosphine)gold(I)) in very good yield. In this compound, we successfully combined the photoluminescent core of gold(I) acetylide with two moieties possessing rich conformational landscapes, namely triethylphosphine and p-methoxyphenyl. While barely photoluminescent in dilute polar solvents, ArPEt exhibited noticeable blue photoluminescence in the solid state under ambient conditions.As expected, concerted pressure-induced conformational switching of –PEt3 resulted in SCSC phase transitions occurring in sequence at ≈0.5 GPa and then at ≈2.2 GPa. The exact structural transformation pathways were documented by high-quality crystal structure models, obtained by unrestrained refinements against unique high-coverage single-crystal X-ray diffraction data. According to the theoretical calculations, all ArPEt phases exhibit strikingly similar estimates of Gibbs free energy with wide regions of overlap, which account for the ease of interconversion. Stabilization in the new phases appears to be a global effect and not reliant on any particular intermolecular interaction. The two distinct transformation pathways are dependent on the hydrostaticity of the applied pressure-transmitting media (Fig. 7). Under quasi-hydrostatic conditions, the transformations on pressure increase involve first symmetry lowering, differentiating between two layers containing two distinct C and E –PEt3 conformers, followed by symmetry increasing, with all molecules ending up with the E –PEt3 conformation Pbca ↔ Pb21a ↔ Pbca. Under non-hydrostatic conditions, the transformations on pressure increases involve systematic symmetry lowering, Pbca ↔ Pb21a ↔ P11a, first differentiating between two layers containing two distinct C and E –PEt3 conformers, and then all molecules ending up with C, E or D –PEt3 conformations. Our observations confirm that low hydrostaticity of PTM can lead not only to powderization/amorphization of a crystal, but also to new polymorphic forms, which would not have been obtainable otherwise.
Tentative photoluminescence studies showed somewhat disappointingly that pressure-induced structural transformations in ArPEt are not directly reflected in the material's luminescence in the solid state. Photoluminescence characteristics switch reversibly at about 1.0 GPa, with hardly any pressure-dependence below or above this point. Slight differences in the emission characteristics occurring above 2.0 GPa between ArPEt-III and ArPEt-IV will require further confirmation.
Irrespective of the applied PTM, the mechanism of phase transitions relies on the conformational changes in –PEt3, which in each instance follow low-energy conformation interconversion pathways as proposed by Ellis et al.21 However, the exact transformations involve not only a flip of a single –Et moiety, but concerted rotation of two or three ethyl moieties accompanied by shifts and rotations of the whole molecules. The slow kinetics of the observed phase transitions was confirmed by the coexistence of two phases at about 2.2 GPa, observed both in the course of slow in-house and fast synchrotron diffraction experiments.
Our study proved that –PEt3 as a ligand is indeed a good SCSC-inducing agent in the solid state. It also showcased that detailed structural analysis under high pressure conditions is nowadays attainable with a high level of accuracy even for larger, labile molecular systems, for which crystal structure modeling at a high level of theory, as advertised for simpler inorganic functional materials,37,38 is still unattainable.
4. Experimental section
4.1. Synthesis
The synthetic reaction was performed under an argon atmosphere. Commercially available solvents and reagents were used as received. Tetrahydrofuran (THF) was dried by refluxing with sodium in the presence of benzophenone as an indicator and distilled prior to use.1.0 mL of 2.5 M n-butyllithium solution in THF (2.5 mmol) was added dropwise to a solution of 0.408 g (2.5 mmol) of hexamethyldisilazane in 3.0 mL of anhydrous THF, which had been cooled to −30 °C. After removing the cooling bath, the resulting mixture was stirred for 10 minutes, and a solution of 0.265 g (2.0 mmol) of 4-ethynylanisole in 2.0 mL of anhydrous THF was added dropwise. After 10 minutes of stirring at room temperature (RT), a solution of 0.650 g (1.85 mmol) of ClAuPEt3 in 6.0 mL of anhydrous THF was added in one portion, and stirring was continued for the next 3 hours. The reaction was quenched by adding 30 mL of water, and the product was extracted with three portions of 25 mL of dichloromethane. The combined organic layers were washed with water, followed by brine, dried over magnesium sulfate, and evaporated to dryness. The obtained yellow oil was dissolved in approximately 10 mL of dichloromethane, and approximately 50 mL of n-pentane was added and evaporated. This procedure was repeated thrice, and the product was crystalized from the n-pentane–dichloromethane mixture at −28 °C to afford 0.659 g (80%) of the product as an off-white solid. Slow recrystallization in the same media at −28 °C resulted in colourless, thick needles.
1H, 13C{1H}, 31P{1H}, and 13C DEPT 135 NMR spectra were recorded at 300 K on a Bruker Neo 600 MHz spectrometer (spectrometer frequency: 600.1 MHz for 1H, 150.0 MHz for 13C and 243.0 MHz for 31P). Chemical shifts for the NMR spectra were referenced with respect to the residual signals in CD3CN (d = 1.94 ppm for 1H and d = 118.7 ppm for 13C). 1H NMR (CD3CN) δ 7.25 (d, J = 8.88 Hz, 2H, CHAr), 6.80 (d, J = 8.87 Hz, 2H, CHAr), 3.76 (s, 3H, OCH3), 1.88–1.85 (m, 6H, P(CH2CH3)3), 1.16 (dt, J = 18.3, 7.8 Hz, 9H, P(CH2CH3)3); 13C{1H} NMR (CD3CN) δ 159.3 (CAr), 133.8 (CHAr), 119.1 (CAr), 114.7 (CHAr), 103.0 (d, 3JC−P = 25.2 Hz C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAu), 55.9 (OCH3), 18.3 (d, J = 33.5 Hz, P(CH2CH3)3), 9.4 (P(CH2CH3)3); 31P{1H} NMR (CD3CN) δ 39.4. Full spectra are presented in section S1.1 in the ESI.†
CAu), 55.9 (OCH3), 18.3 (d, J = 33.5 Hz, P(CH2CH3)3), 9.4 (P(CH2CH3)3); 31P{1H} NMR (CD3CN) δ 39.4. Full spectra are presented in section S1.1 in the ESI.†
Mass spectrometry (MS) data were recorded on a Shimadzu HPLC-MS system equipped with an LCMS-2020 detector in positive and negative electrospray ionization (ESI) modes. The sample was dissolved in acetonitrile and injected directly into the LCMS-2020 detector to a volume of 0.1 μL. MS calculated for C15H22AuOP m/z = 446.1 [M]+, calculated for C18H14AuO2−m/z = 459.1 found m/z = 458.9 ((4-MeOC6H4CC)2Au−), and calculated for C12H30AuP2m/z = 433.1 found 433.1 ((Et3P)2Au+). Full spectra are presented in Fig. S1.5 in the ESI.†
The melting point was determined by DSC at 87.3(5) °C (section S1.3 in the ESI†).
4.2. Single-crystal X-ray diffraction
Clear, colorless blocks of ArPEt suitable for single-crystal X-ray diffraction experiments were obtained by recrystallization from an n-pentane:dichloromethane mixture at −28 °C. A few series of X-ray diffraction experiments were performed in order to (a) determine the crystal structure of the new compound under atmospheric conditions and (b) analyze the behavior of the new material under increased pressure and determine the mechanisms of SCSC phase transitions using either in-house equipment or synchrotron facilities.
:1 mixture and, in the case of synchrotron experiments, the Daphne-7575.
In order to collect reliable diffraction and photoluminescence data at increased pressure and ensure satisfactory X-ray data completeness during in-house diffraction experiments, a single crystal of ArPEt was placed in an Almax DACOne20 or Merill–Basset DAC oriented in such a way as to avoid alignment of the main crystallographic directions with the DAC axis in accordance with the procedure proposed by Tchoń et al.39 Appropriate maps of theoretically attainable data completeness as a function of crystal orientation, DAC type and X-ray wavelength (potency maps) are presented in section S2.1.2 of the ESI.† The placement was accomplished by propping the crystal on a small drop of glue against the diamond culet. In the case of diffraction experiments at the synchrotron, four tiny crystals were placed in different (random) orientations on a diamond culet in order to ensure sufficient data coverage by merging datasets.
The pressure in a DAC during all experiments was determined using the R1 fluorescence line from reference ruby spheres, based on the calibration curve determined by Piermarini et al.40 with Ragan's temperature correction.41
High-pressure diffraction synchrotron experiments were performed at the European Synchrotron Facility (ESRF) in Grenoble, France on the ID15B beamline. Data were collected for each crystal separately using a 6 μm X-ray beam (λ = 0.41 Å) on a single axis goniometer with an EIGER2 × 9 M CdTe detector. Data reduction was performed with CrysAlisPro42 and multi-scan absorption correction was applied using spherical harmonics with the SCALE3 ABSPACK algorithm on each dataset. At every pressure point, datasets from individual crystals were merged together, provided that they indexed in the same space group and they showed Rint statistics below 10%. Weighted average unit cell parameters for merged datasets were calculated using a number of reflections as a weight. Merging was done using the SORTAV module in WinGX43 with auto rejection of outliers. Merging results are summarized in Tables S2.12 and S2.13.†
All collected datasets are summarized in section S2.3 in the ESI.†
4.3. UV-vis absorption and photoluminescence studies
The UV-vis spectra were recorded on a Lambda 45 (PerkinElmer) spectrometer at 296 K at a concentration of approximately 2 × 10−5 M, while the ε values were calculated based on UV-vis spectra measured at five different concentrations, ranging from 1.58 × 10−5 to 7.86 × 10−5 M for the DCM solution, and from 1.51 × 10−5 to 7.53 × 10−5 for the MeCN solution. The photoluminescence (PL) spectra in the solution were recorded using a FluoroMax-4 (Horiba) spectrofluorometer equipped with a long-pass filter (cutoff at 290 nm) at a concentration of 5 × 10−6 M for both solutions. Photoluminescence quantum yields in solutions were determined by an indirect method according to the established protocol.49 An aqueous solution of tryptophan (QY of 12%) was used as a fluorescence standard in the measured range. The optical density of the solutions was below 0.05 at the excitation maximum. QYs were calculated separately at each excitation wavelength for a given solvent (Table 1) and averaged. The standard spectra were collected with the same excitation parameters and instrument settings.The photoluminescence and excitation spectra of polycrystalline samples were recorded using an Edinburgh FS5 equipped with an enhanced range photomultiplier detector (PMT-EXT) using front-face mode at room temperature. Photoluminescence quantum yields were measured using an integrating sphere (Edinburgh Instruments FS5) according to the established procedure.50 Solid samples were excited at 390 nm.
The PL spectra for a single crystal were recorded with a Labram HR800 spectrometer (Horiba Jobin Yvon) coupled with an Olympus BX61 confocal microscope and a diode-pumped Nd:YAG laser of 405 nm as the excitation source from a specimen (a) placed on a glass microscope slide under atmospheric conditions or (b) enclosed in a Merill–Basset DAC with a selected pressure-transmitting medium for high-pressure experiments. Sample mounting in a DAC followed the procedure described in subsection 4.2.1, except that no epoxy resin was used for affixing the crystal.
4.4. Theoretical calculations
In order to assess the relative stability of the polymorphs of ArPEt and establish the hierarchy of intermolecular interactions in their crystal structures, quantum-chemical calculations were carried out in CRYSTAL1751,52 dedicated to ab initio for periodic systems and utilizing atomic basis sets. Calculations were performed using the B3LYP-D* method, in which B3LYP is augmented with an empirical dispersion term,53,54 and dedicated basis sets as listed in Table S6.18.†Crystal structure optimizations of different ArPEt phases were started from experimental XRD geometries: ArPEt-I from the major variant of the disordered crystal structure determined at ambient pressure, ArPEt-II at 0.6 GPa and ArPEt-III at 2.6 GPa. The symmetry of each form was maintained during the optimization process. External pressures were set for high-pressure crystal phases. Full relaxation of both lattice parameters and atomic coordinates by means of analytical energy gradients was applied when possible (for details see section 6.2 of the ESI†).
The evolution of crystal structures with pressure was modeled using the Theoretical Equations of States (EOS) algorithm as implemented in CRYSTAL17 for ArPEt-I, ArPEt-II and ArPEt-III. Third-order Birch–Murnaghan equations of states (EOSs) were applied. In each case, a sequence of 8 periodic optimizations was performed, with constrained, systematically increasing or decreasing volumes of the unit cell. The crystal symmetry was maintained during the whole optimization process. A detailed summary of EOS settings is presented in Table S6.19.†
Energies of interactions were calculated between pairs of molecules (see Fig. 9 for an explanation on how a particular dimer was defined) as well as between the main supramolecular motifs (SLABs). The SLABs were defined according to herringbone rows as shown in Fig. 3c.
Author contributions
AM: conceptualization, data analysis, supervision, acquisition of funding and resources. RD: conceptualization, investigation – X-ray diffraction studies, data curation and analysis, and preparation of the first draft. MG, MŁ, and DP: investigation – chemical synthesis, purification, PL and UV-vis study in solution. JK: investigation – DFT theoretical calculations, and data analysis. All authors contributed to and reviewed the final manuscript.Data availability
Crystallographic data for all presented compounds have been deposited at the CCDC under numbers 2370854–2370943 (detailed assignment are given in Table S3.6 in the ESI†) and can be obtained from https://www.ccdc.cam.ac.uk/structures/. Further data supporting this manuscript have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research comes from the NCN OPUS grant DEC-2021/41/B/ST4/02760. The access to ESRF was financed by the Polish Ministry of Science and Higher Education, decision 2021/WK/11. Daniel Tchoń is acknowledged for providing a script for simultaneous analysis of multiple structural parameters from CIF files. Paulina Marek-Urban is acknowledged for the assistance in collecting PL and excitation data in the solid state. We acknowledge the ESRF for provision of synchrotron radiation facilities under proposal number CH-6630 and we would like to thank Tomasz Poręba for assistance and support in using beamline ID15B.References
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef.
- S. K. Park and Y. Diao, Chem. Soc. Rev., 2020, 49, 8287–8314 RSC.
- Q. Guan, Y. Fang, X. Wu, R. Ou, X. Zhang, H. Xie, M. Tang and G. Zeng, Mater. Today, 2023, 64, 138–164 CrossRef.
- Z. Mao Png, C.-G. Wang, J. C. Chuan Yeo, J. J. Cheng Lee, N. Erdeanna Surat'man, Y. Lin Tan, H. Liu, P. Wang, B. Hoon Tan, J. Wei Xu, X. Jun Loh and Q. Zhu, Mol. Syst. Des. Eng., 2023, 8, 1097–1129 RSC.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- L. Catalano and P. Naumov, CrystEngComm, 2018, 20, 5872–5883 RSC.
- I. Liepuoniute, M. J. Jellen and M. A. Garcia-Garibay, Chem. Sci., 2020, 11, 12994–13007 RSC.
- M. Jin, T. S. Chung, T. Seki, H. Ito and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 18115–18121 CrossRef PubMed.
- A. Sorroche, S. Moreno, M. Elena Olmos, M. Monge and J. M. López-de Luzuriaga, Angew. Chem., Int. Ed., 2023, 62, e202310314 CrossRef PubMed.
- T. Lin, Y. Lin, J. Yao, Z. Wu, N. Fu and Q. Wei, Eur. J. Inorg. Chem., 2024, 27, e202400059 CrossRef.
- Z. Chen, Y. Yin, S. Pu and S. H. Liu, Dyes Pigm., 2021, 184, 108814 CrossRef.
- L. Luciani, N. Sargentoni, C. Graiff, M. Monge, M. Rodríguez-Castillo, J. M. López-de Luzuriaga and R. Galassi, RSC Adv., 2023, 13, 25425–25436 RSC.
- S. Petrovskii, A. Paderina, A. Sizova and E. Grachova, Inorg. Chem., 2023, 62, 5123–5133 CrossRef PubMed.
- A. C. Brannan, H.-H. Cho, A.-P. M. Reponen, S. Gorgon, N. L. Phuoc, M. Linnolahti, N. C. Greenham and A. S. Romanov, Adv. Mater., 2024, 2404357 CrossRef PubMed.
- N. Mirzadeh, S. H. Privér, A. J. Blake, H. Schmidbaur and S. K. Bhargava, Chem. Rev., 2020, 120, 7551–7591 CrossRef PubMed.
- R. Ando, A. Sato-Tomita, H. Ito and M. Jin, Angew. Chem., Int. Ed., 2023, 62, e202309694 CrossRef.
- W. Lu, N. Zhu and C.-M. Che, J. Am. Chem. Soc., 2003, 125, 16081–16088 CrossRef PubMed.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef.
- A. Furoida, M. Daitani, K. Matsumoto, K. Hisano and O. Tsutsumi, Mater. Adv., 2024, 5, 5052–5062 RSC.
- M. Głodek, S. PawlÈ©dzio, A. Makal and D. Plażuk, Chem. – Eur. J., 2019, 25, 13131–13145 CrossRef.
- D. D. Ellis, M. F. Haddow, A. G. Orpen and P. J. Watson, Dalton Trans., 2009, 10436 RSC.
- N. De Silva, F. Zahariev, B. P. Hay, M. S. Gordon and T. L. Windus, J. Phys. Chem. A, 2015, 119, 8765–8773 CrossRef.
- M. Castro, L. R. Falvello, E. Forcén-Vázquez, P. Guerra, N. A. Al-Kenany, G. Martínez and M. Tomás, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 731–742 CrossRef.
- A. Makal, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 427–435 CrossRef PubMed.
- B. A. Zakharov and E. V. Boldyreva, CrystEngComm, 2019, 21, 10–22 RSC.
- A. Katrusiak, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 918–926 CrossRef PubMed.
- S. Moreno, N. Casati, M. Rodríguez-Castillo, M. Monge, M. E. Olmos and J. M. López-de Luzuriaga, Inorg. Chem., 2023, 62, 10307–10316 CrossRef.
- M. Szafrański and A. Katrusiak, J. Phys. Chem. Lett., 2017, 8, 2496–2506 CrossRef PubMed.
- M. Głodek, A. Makal, P. Paluch, M. Kadziołka-Gaweł, Y. Kobayashi, J. Zakrzewski and D. Plażuk, Dalton Trans., 2018, 47, 6702–6712 RSC.
- M. Głodek, A. Makal, P. Paluch, M. Kadziołka-Gaweł, Y. Kobayashi, J. Zakrzewski and D. Plażuk, Dalton Trans., 2018, 47, 6702–6712 RSC.
- K. T. Chan, G. S. M. Tong, W.-P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352–2364 RSC.
- S. Klotz, J.-C. Chervin, P. Munsch and G. Le Marchand, J. Phys. D: Appl. Phys., 2009, 42, 075413 CrossRef.
- R. J. Angel, M. Bujak, J. Zhao, G. D. Gatta and S. D. Jacobsen, J. Appl. Crystallogr., 2007, 40, 26–32 CrossRef CAS.
- A. G. Christy, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1995, 51, 753–757 CrossRef.
- T. Luty and R. Fouret, J. Chem. Phys., 1989, 90, 5696–5703 CrossRef.
- N. M. Peachey and C. J. Eckhardt, J. Phys. Chem., 1993, 97, 10849–10856 CrossRef.
- E. Zurek, Reviews in Computational Chemistry, John Wiley & Sons, Ltd, 2016, pp. 274–326 Search PubMed.
- K. P. Hilleke, T. Bi and E. Zurek, Appl. Phys. A, 2022, 128, 441 CrossRef.
- D. Tchoń and A. Makal, IUCrJ, 2021, 8, 1006–1017 CrossRef.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef.
- D. D. Ragan, R. Gustavsen and D. Schiferl, J. Appl. Phys., 1992, 72, 5539–5544 CrossRef.
- Rigaku Oxford Diffraction, 2022 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1360 Search PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D’Arco, M. Llunell, M. Causà, Y. Noël, L. Maschio, A. Erba, M. Rerat and S. Casassa, CRYSTAL17 User's Manual, University of Torino, Torino, 2017 Search PubMed.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details concerning used methodology, additional tables and figures describing investigated structures, observed phase transitions and intermolecular interactions. CCDC 2370854–2370859, 2370873–2370878, 2370882–2370887, 2370902–2370910, 2370914–2370921 and 2370936–2370943. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03036b |
| This journal is © The Royal Society of Chemistry 2025 |